

Abstract
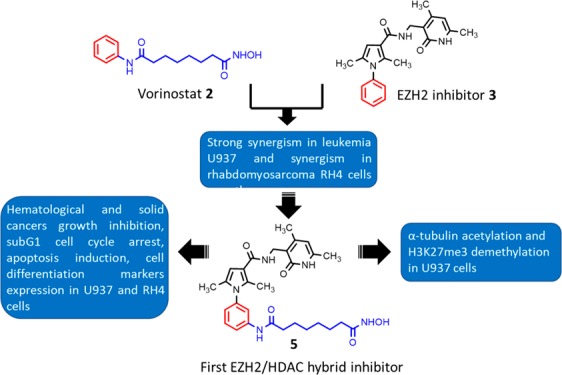
Since the histone modifying enzymes EZH2 and HDACs control a number of epigenetic-dependent carcinogenic pathways, we designed the first-in-class dual EZH2/HDAC inhibitor 5 displaying (sub)micromolar inhibition against both targets. When tested in several cancer cell lines, the hybrid 5 impaired cell viability at low micromolar level and in leukemia U937 and rhabdomyosarcoma RH4 cells provided G1 arrest, apoptotic induction, and increased differentiation, associated with an increase of acetyl-H3 and acetyl-α-tubulin and a decrease of H3K27me3 levels. In glioblastoma U87 cells, 5 hampered epithelial to mesenchymal transition by increasing the E-cadherin expression, thus proposing itself as a useful candidate for anticancer therapy.
Keywords: Drug discovery, histone deacetylase, histone methyltransferase, dual-target inhibitor, anticancer agent
The recent epigenetic polypharmacology strategy of hybrid drugs acting on different biological pathways is emerging as a new pioneristic road toward modern cancer therapy. The simultaneous inhibition of two or more targets by drug combination or by a single “hybrid molecule” is currently providing improvement in therapeutic efficacy and overcoming resistance.1 Particularly, the hybrid inhibitors acting on epigenetic targets have achieved promising results in preclinical2 and clinical3 settings. Because of their ability to activate multiple anticancer pathways such as extrinsic or intrinsic apoptosis, inflammation/immune response, growth arrest, mitotic and autophagic cell death, senescence, and antiangiogenic networks,4,5 histone deacetylase inhibitors (HDACi) emerged as very effective tools in cancer research and pharmacology. Consistently, five of them have been approved by the FDA [vorinostat (1, Figure 1), romidepsin, belinostat, and panobinostat] or by the Chinese FDA (chidamide) for the treatment of hematological malignancies.6 The coadministration of HDACi with other anticancer drugs showed improved anticancer effects and motivated the design of HDACi-based hybrid molecules,1,7−9 mainly targeting epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGFR), cyclin dependent kinases (CDKs), phosphoinositide 3-kinase (PI3K), and Janus kinase (JAK), in addition to HDAC. Noteworthy, the multitarget EGFR/HER2/HDAC and PI3Ks/HDAC hybrid inhibitors, CUDC-101 and CUDC-907, overcame the cancer resistance to receptor tyrosine kinase inhibitors and strongly improved the poor efficacy of HDAC inhibitors in solid tumors, hence deserving the clinical trials arena.10,11
Figure 1.
Rational design of dual EZH2/HDAC inhibitors 4 and 5.
Another epigenetic enzyme, the catalytic unit of the Polycomb Repressive Complex 2 (PRC2), i.e., the histone 3 lysine 27 (H3K27) methyltransferase Enhancer of Zeste Homologue 2 (EZH2), achieved importance due to its ability to silence many genes involved in cell proliferation, cell-cycle, cell differentiation, and self-renewal5 and has been found overexpressed or mutated in several human cancers.5 A number of 2-pyridone-based EZH2 inhibitors have been described, and notably some of them are under clinical investigation (GSK126, CPI-1205) or have just been approved (tazemetostat) for cancer treatment.12−14
Recently, our research group reported a series of 2-pyridone pyrazole-containing EZH2 inhibitors, and among them 2 (Figure 1), when tested in cancer cells, induced breast MDA-MB231, leukemia K562, and neuroblastoma SK-N-BE cells growth arrest, along with reduction of the H3K27me3 levels and induction of markers of apoptosis and autophagy.15 In human medulloblastoma stemlike cells belonging to the Sonic Hedgehog subgroup (SHH MB-SLCs), 2 impaired SHH MB cells proliferation and self-renewal and induced apoptosis when tested in vitro and in orthotopic xenografted MB-SLCs nude mice.16 Prompted by these findings, we designed a series of new compounds by replacing the pyrazole with the pyrrole nucleus. Among them, the 2-pyridone pyrrole 3 (Figure 1) is the prototype and two analogues, 3a and 3b (Figure 1) bearing at C3 of the N-phenyl ring a piperidine or morpholine moiety, respectively, reduced the H3K27me3 and increased the p21 and p27 expression levels, impairing primary glioblastoma (GBM) as well as U-87 GBM cell viability in a dose- and time-dependent manner similarly to tazemetostat.17 In combination with Temozolomide, 3a,b and tazemetostat displayed a comparable stronger effect on cell viability. Notably, 3a,b attenuated the GBM malignant phenotype by reduction of VEGFR1/VEGF expression, reversion of the epithelial-mesenchymal transition (EMT) progression, inhibition of cell invasion, and decrease of inflammatory cytokines levels.17
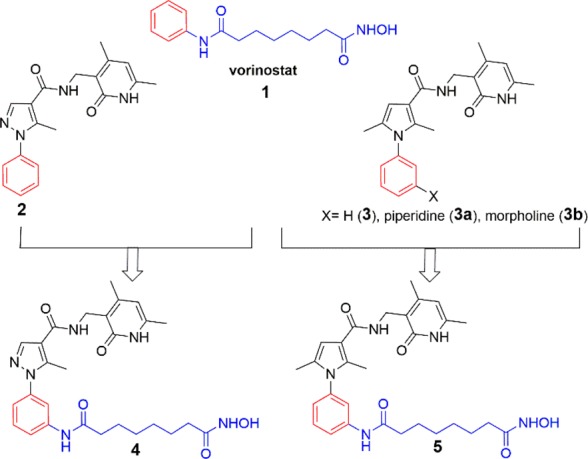
Recently, the combination of EZH2 and HDAC inhibitors has been reported to increase apoptosis and DNA damage in patient-derived brain-tumor initiating cell lines18 and to have synergistic effects on lymphoma,19 multiple myeloma,20 small cell carcinoma of the ovary, hypercalcemic type,21 and nonsmall-cell lung cancer,22 in addition to additive effects on triple negative breast cancers.23 These findings prompted us to design two hybrid prototypes (compounds 4 and 5, Figure 1) as dual EZH2/HDAC inhibitors by merging the 2 or 3 structure with the vorinostat 1 structural motif.
The cyclocondensation of (3-nitrophenyl)hydrazine with tert-butyl (Z)-2-((dimethylamino)methylene)-3-oxobutanoate in dry ethanol at 80 °C provided the tert-butyl 5-methyl-1-(3-nitrophenyl)-1H-pyrazole-4-carboxylate 6. Alternatively, the known 2,5-dimethyl-1-(3-nitrophenyl)-1H-pyrrole24 underwent Friedel–Crafts acylation with trichloroacetyl chloride in dry dichloroethane at 70 °C and successive hydrolysis with 2 N potassium hydroxide to give the corresponding carboxylic acid 7, which afterwards was protected as tert-butyl ester (8) by reaction with N,N-dimethylformamide di-tert-butyl acetal in dry toluene at 90 °C. Both the meta nitro functions of 6 and 8 were reduced with metal zinc and ammonium chloride in 1,4-dioxane:water 1:1 at 50 °C to afford the corresponding anilines 9 and 10, later acylated by the methyl 8-chloro-8-oxooctanoate to furnish the intermediate compounds 11 and 12. The carboxylic acids 13 and 14 were prepared by cleavage of the tert-butyl function with trifluoroacetic acid in dry dichloromethane. The coupling reaction, carried out on the amine 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one17 with O-benzotriazol-tetramethyluronium tetrafluoroborate (TBTU) and triethylamine in dry N,N-dimethylformamide, led to the corresponding amides 15 and 16. Methyl ester hydrolysis with lithium hydroxide in a water and tetrahydrofuran (1:1) mixture gave the acids 17 and 18 which were then converted into the corresponding hydroxamates 4 and 5 by reaction with O-(tetrahydro-2H-pyran-2yl)hydroxylamine, TBTU, and triethylamine in dry N,N-dimethylformamide followed by acidic cleavage with a 4 M hydrogen chloride solution in dioxane (Scheme 1).
Scheme 1. Synthesis of Hybrid Compounds 4 and 5.
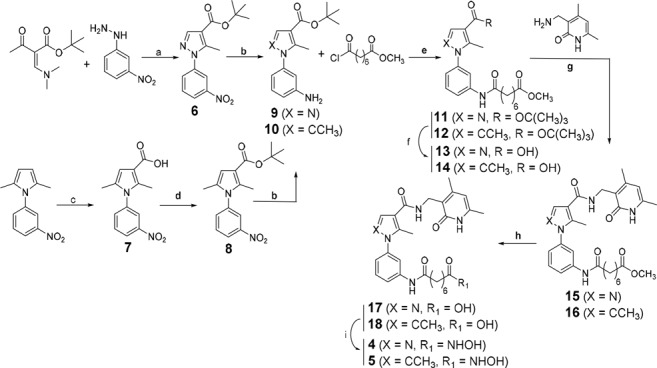
Reagents and conditions: a) triethylamine, dry ethanol, 80 °C; b) Zn(0), NH4Cl, H2O:1,4-dioxane 1:1, 50 °C; c) 1) trichloroacetyl chloride, dry dichloroethane, 70 °C; 2) 2 N KOH aqueous solution, ethanol/tetrahydrofuran 1:1; d) N,N-dimethylformamide di-tert-butyl acetal, dry toluene, 90 °C; e) triethylamine, dry dichloromethane, RT; f) trifluoroacetic acid, dry dichloromethane, RT; g) triethylamine, TBTU reagent, dry N,N-dimethylformamide, RT; h) LiOH(aq), tetrahydrofuran, RT; (i) 1) triethylamine, TBTU reagent, O-(tetrahydro-2H-pyran-2-yl)hydroxylamine, dry N,N-dimethylformamide, RT; 2) 4 N hydrogen chloride solution in 1,4-dioxane, dry tetrahydrofuran, RT.
The newly synthesized compounds 4 and 5 have been screened in a 10-dose IC50 mode with a 2-fold serial dilution starting from 200 μM solutions, in an in vitro enzymatic assay against a human five-component PRC2 complex containing EZH2, embryonic ectoderm development (EED), suppressor of zeste 12 (SUZ12), RbAp48, and adipocyte enhancer-binding protein (AEBP2), to evaluate their ability to inhibit the EZH2 catalytic activity. The assay was performed using core histone and 3H-SAM as substrate and cosubstrate, respectively. S-Adenosyl-l-homocysteine (SAH), 2, and 3 were used as reference compounds. The new hybrid molecules 4 and 5 were also screened against human recombinant HDAC1-6, -8 to assess their capability to inhibit some representative members of deacetylases as well. For this purpose, the fluorescent AMC-K(Ac)GL (for HDAC1-3, -6, and -8) or AMC-K(trifluoroAc)GL (for HDAC4, -5) substrate has been used, and 1 was employed as reference drug.
The biochemical data (Table 1) highlighted that the pyrazole-based hydroxamate 4 lost the EZH2 inhibiting activity (4% inhibition at 100 μM) shown by the corresponding prototype 2. Against the tested HDACs, 4 displayed submicromolar IC50 against HDAC1 (0.83 μM) and single-digit micromolar IC50 against the other class I HDACs (HDAC2, -3, and -8: 2.36, 1.48, and 2.08 μM, respectively) and inhibited HDAC6 (class IIb HDAC) at a nanomolar level (IC50: 0.01 μM), while being inactive against the class IIa HDAC4 and -5. Surprisingly, the replacement of the heterocyclic nucleus of the hybrid compound from pyrazole to pyrrole led to 5 which retained the biochemical inhibition against EZH2/PRC2 with similar potency as the corresponding single-target inhibitor 3. Against HDACs, the hybrid 5 exhibited a similar general inhibition profile as 4 but was more potent. Indeed, with respect to 4, 5 displayed a 2-fold greater potency against HDAC1 (IC50: 0.43 μM), -2 (IC50: 1.33 μM), and -6 (IC50: 0.005 μM), over a 3-fold higher inhibition against HDAC3 (IC50: 0.45 μM), and an almost 20-fold better efficacy against HDAC8 (IC50: 0.11 μM). Versus HDAC4 and -5, 5 displayed weak inhibition (IC50 values: 36.8 and 16.6 μM, respectively).
Table 1. Biochemical Data of 1–5 against EZH2/PRC2 and HDAC1-6, -8 Isoforms.
| IC50, μM |
||||||||
|---|---|---|---|---|---|---|---|---|
| hrHDAC
isoforms |
||||||||
| compd | EZH2/PRC2 | 1 | 2 | 3 | 4 | 5 | 6 | 8 |
| 1 | ND | 0.26 | 0.92 | 0.35 | 0.49 | 0.38 | 0.03 | 0.24 |
| SAH | 34.7 | ND | ND | ND | ND | ND | ND | ND |
| 2 | 15.4 | ND | ND | ND | ND | ND | ND | ND |
| 3 | 5.78 | ND | ND | ND | ND | ND | ND | ND |
| 4 | 4% at 100 μM | 0.83 | 2.36 | 1.48 | NA | 101 | 0.01 | 2.08 |
| 5 | 7.37 | 0.43 | 1.33 | 0.45 | 36.8 | 16.6 | 0.005 | 0.11 |
Before testing the dual EZH2/HDAC inhibitor 5 in cancer contexts, we studied the effects of coadministration of 1 and 3 on human U937 leukemia and alveolar rhabdomyosarcoma (RMS) RH4 cells, using 1 from 0.05 to 0.25 μM against both cell types and 3 from 5 to 25 μM (U937 cells) or from 10 to 50 μM (RH4 cells) (Figure 2). Single treatments of U937 cells with 1 or 3 dose-dependently reduced cell viability by 45% (1) or 25% (3) after 72 h at the highest doses (Figure 2A). Noteworthy, the combination of the two inhibitors nicely provided a synergistic effect (Figure 2A) by reducing around 70% of the U937 cell viability using 0.1 μM 1 plus 10 μM 3 and over 90% at 0.25 μM 1 plus 25 μM 3. The combination index (CI) (Table 2) and the isobologram graph (Figure 2C) proved a very strong synergism between the two compounds in U937 cell death induction, providing a CI value of 0.14 with the first cotreatment. When tested in RH4 cells, 1 and 3 tested alone displayed similar antiproliferative potency reducing over 30% the cell viability, while the combination exhibited over 60% of viability impairment at the highest tested doses (Figure 2B), with a synergistic effect confirmed by the calculated CI values (Table 2) and by isobologram analysis (Figure 2D).
Figure 2.
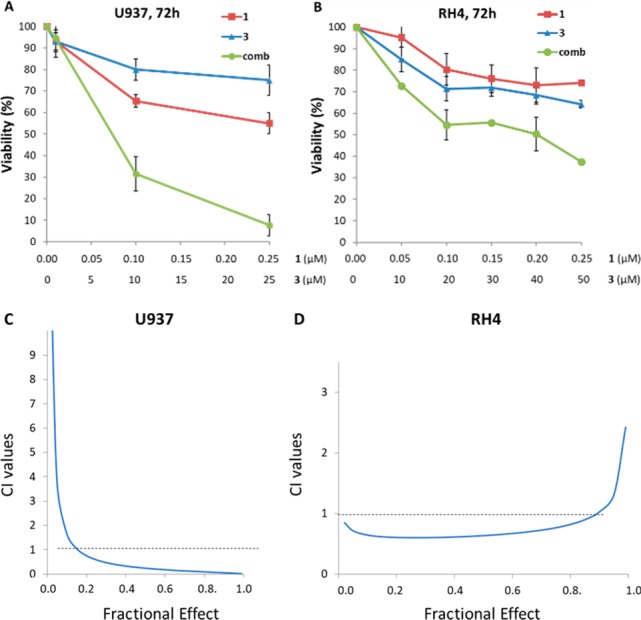
Cell viability of U937 (A) and RH4 (B) cells treated with 1 and 3 as single agents or in combination after 72 h at indicated increasing concentrations. Isobologram analyses of 1/3 combination in U937 (C) and RH4 (D) cells are reported.
Table 2. Combination Index Determination for 1 and 3 Cotreatment in U937 and RH4 Cells.
| U937 |
RH4 |
||||||
|---|---|---|---|---|---|---|---|
| μM |
μM |
||||||
| 1 | 3 | FAa | CIb | 1 | 3 | FAa | CIb |
| 0.01 | 1 | 0.05 | 3.34 | 0.05 | 10 | 0.27 | 0.64 |
| 0.1 | 10 | 0.68 | 0.14 | 0.1 | 20 | 0.45 | 0.49 |
| 0.25 | 25 | 0.92 | 0.04 | 0.25 | 50 | 0.63 | 0.54 |
FA, fraction affected.
CI, combination index.
Targets engagement by 1, 3, and the 1/3 combination in both U937 and RH4 cell lines has been demonstrated by Western blot analyses (Figure 3). The 1/3 combination in U937 cells gave an increase of Ac-H3 levels at 96 h, with partial loss of effect at 120 h, a time point in which the expected decrease of H3K27me3 levels was detected. With a different timing, RH4 cells treated with the 1/3 combination exhibited increased levels of Ac-tubulin and strong demethylation of H3K27me3 after 72 h, which was maintained at 96 h.
Figure 3.
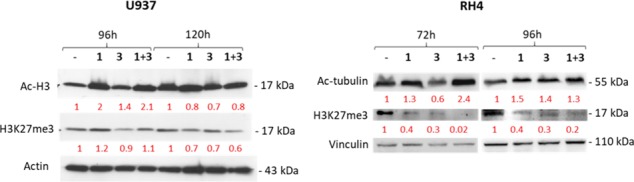
Effects of 1 and 3 as single agents and in combination in U937 (1 at 0.5 and 3 at 50 μM) and RH4 (1 at 0.15 and 3 at 50 μM) cells. Actin (U937) and vinculin (RH4) are used as equal loading. Densitometries for Ac-H3, Ac-tubulin, and H3K27me3 levels, normalized on an internal loading control and then on a vehicle (DMSO), are reported in red.
The synergy observed after 1/3 cotreatment in leukemia U937 and rhabdomyosarcoma RH4 cells prompted us to evaluate the dual EZH2/HDAC inhibitor 5 in the same cell lines at different time-points (48, 72, and 96 h) at increasing doses (at 5 to 25 μM in U937 and at 5 to 35 μM in RH4 cells).
After the longest treatment time, 5 reduced viability in U937 cells around 60% at 10 μM and 90% at 25 μM (Figure 4A) and in RH4 cells over 60% at 20 μM and 90% at 35 μM (Figure 4B).
Figure 4.
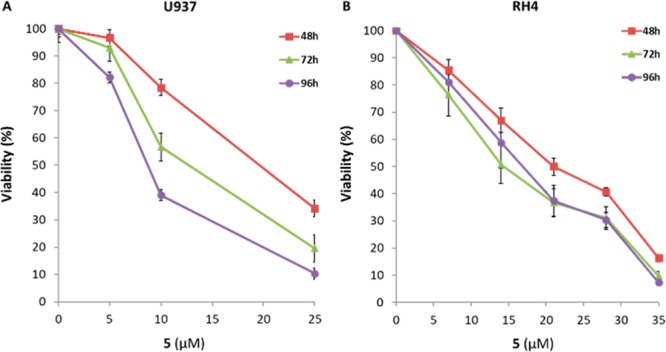
Cell viability of U937 (A) and RH4 (B) cells treated with compound 5 after 48–96 h of treatment at the indicated increasing concentrations.
Next, 5 was additionally tested in human acute myeloid leukemia THP1, neuroblastoma SH-N-SK, and glioblastoma U87 cell lines, exhibiting antiproliferative IC50 values ranging from 9 (U937) to 25 μM (SH-N-SK) after 72 h of treatment (Table 3).
Table 3. Antiproliferative Activity against Human U937, RH4, THP1, SH-N-SK, and U87 Cancer Cell Linesa.
| IC50, μM ± SDb |
|||||
|---|---|---|---|---|---|
| compd | U937 | RH4 | THP1 | SH-N-SK | U87 |
| 5 | 9 ± 1.5 | 13 ± 2.7 | 12 ± 2.2 | 25 ± 4.1 | 20 ± 3.6 |
Data are the means of three independent experiments measured in triplicate.
SD, standard deviation.
Afterward, the ability of 5 to modulate both the EZH2 and HDAC targets in U937 and RH4 cells was evaluated (Figure 5). In U937 cells, 5 increased the acetylation levels of histone H3 as well as α-tubulin, the specific HDAC6 substrate, up to 72 and 96 h of treatment, respectively (Figure 5A). At these time points and indicated concentrations, negligible modulation of H3K27me3 was observed (Figure 5A). The same cells, treated with 5 at a higher dose (50 μM) and longer time (from 72 to 144 h), clearly showed a very strong induction on α-tubulin acetylation up to 96–120 h, much less evident at 144 h and, in parallel, a reduced H3K27me3 level only at 144 h (Figure 5A), maybe due to Jumonji domain 3 (JmjD3) or ubiquitously transcribed tetratricopeptide repeat X chromosome (UTX) inhibition, as already reported for HDAC inhibitors.25 In RH4 cells, 5 induced a strong induction of Ac-tubulin at 72 h, weaker after 96 h, and a clear reduction of H3K27me3 levels at both 72 and 96 h of treatment (Figure 5B).
Figure 5.
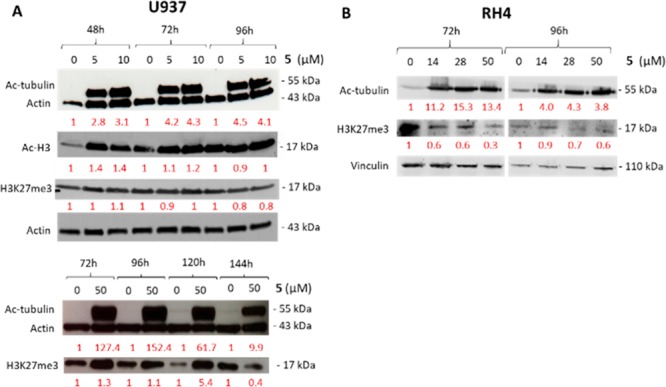
Western blot analysis. (A) Effect of dual hybrid compound 5 on histone H3 and α-tubulin acetylation and H3K27 trimethylation in U937 cells. (B) Effect of 5 on acetyl-tubulin and H3K27me3 levels in RH4 cells. Densitometries for Ac-H3, Ac-tubulin, and H3K27me3 levels, normalized on an internal loading control and then on a vehicle (DMSO), are reported in red.
In order to characterize the antiproliferative activity of the dual EZH2/HDAC inhibitor 5, the cell cycle was analyzed in leukemia U937 and rhabdomyosarcoma RH4 cells. After 72 h of treatment with 5 [10 or 25 μM in U937 (Figure 6A) and 14 or 28 μM in RH4 cells (Figure 6C)], the percentage of cells in the subG1 phase was dose-dependently increased in both cell lines suggesting the induction of apoptosis by 5. The analysis of Annexin V expression confirmed this hypothesis with an increase of the percentage of Annexin V positive cells up to 23.3% in U937 (Figure 6B) and to 26.5% in RH4 5-treated cells (Figure 6D).
Figure 6.
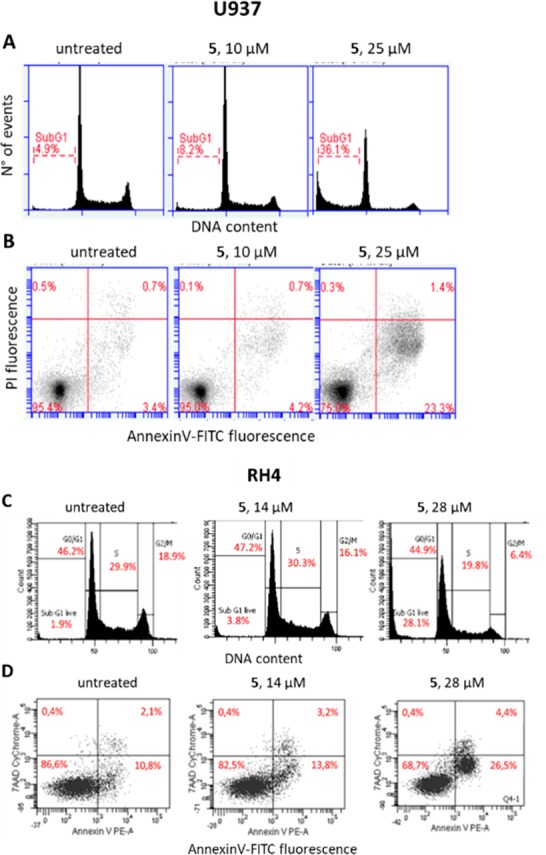
Cell cycle analysis (A, C) and annexin-V-FITC staining (B, D) after 72 h of treatment of U937 and RH4 cells with 5 at the indicated concentrations. Control cells (untreated) were treated with a vehicle (DMSO).
Additionally, the cytodifferentiation effect induced by 5 was evaluated in U937 and RH4 cells. In U937 cells the dual inhibitor 5, tested at 10 and 25 μM for 72 h, increased by 2- and 2.7-fold the cell surface adhesive receptor CD11b expression (Figure 7A), a marker of cell differentiation. In RH4 cells, 5 (7 μM, 96 h) increased the Myogenin (MYOG) levels, indicating an activation of the myogenic differentiation program (Figure 7B). This result is in agreement with a recent paper26 that not only demonstrates the role of HDAC6 in inhibition of RMS differentiation but also well correlates with the EZH2 involvement in suppressing the transcription of myogenic marker genes, such as MYOG.27 Taken together, these data suggest that the dual EZH2/HDAC inhibitor 5 could be useful in RMS treatment, in which cellular differentiation and myogenesis induction represent a potential therapeutic approach.
Figure 7.
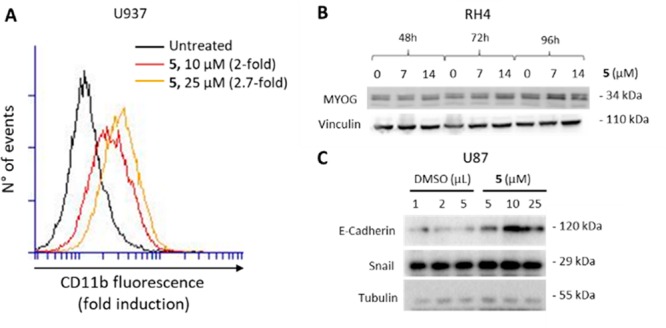
(A) CD11b expression upon a 72 h treatment of U937 cells with 5 at 10 and 25 μM. (B) Effect of 5 on MYOG in RH4 cells after 48, 72, and 96 h of treatment at 7 and 14 μM. Vinculin was used as a loading control. (C) Effects of 5 on E-cadherin and Snail in U87 cells after 24 h of treatment at 5, 10, and 25 μM. Tubulin was used as a loading control.
Since both EZH2 and HDAC are involved in EMT and either HDAC or EZH2 inhibitors reverted EMT in cancers17,28,29 upregulating the epithelial markers, we treated glioblastoma U87 cells with different amounts of 5 for 24 h using DMSO as a control, to check the effect of 5 on the levels of E-cadherin and Snail, two typical markers of epithelial and mesenchymal cells. The Western blots shown in Figure 7C highlighted that 5 provided a remarkable increase in the E-cadherin protein expression, whereas in the same conditions the Snail protein level was not affected. These data indicate that the dual inhibition of EZH2 and HDAC by 5 allows the rescue of the epithelial features in U87 cancer cells independently from Snail and are in accordance with previous evidence.30,31
In conclusion, we have described the discovery of the first dual hybrid molecule 5 able to inhibit at the same time the HDAC and EZH2 targets. When tested in several cancer cells, including hematological malignancies such as leukemia U937 and THP1 as well as solid cancers such as rhabdomyosarcoma RH4, neuroblastoma SH-N-SK, and glioblastoma U87, 5 displayed inhibition of proliferation at low micromolar concentration. In U937 and RH4 cells, 5 was able to modulate HDAC- and EZH2-dependent histone markers, leading to cell cycle arrest in the subG1 phase, apoptosis induction, and increased expression of cell differentiation markers. Furthermore, 5 hampered the EMT process in U87 cells by positively modulating the epithelial E-cadherin expression. This study highlights the basis for future biological investigation on the effects of EZH2/HDAC simultaneous inhibition in cancer.
Acknowledgments
The authors dedicate the work to the memory of Prof. Maurizio Botta, a distinguished scientist who will be always a special friend for us. This work was supported by funds from PRIN 2015 (prot.20152TE5PK) (A.M.), AIRC-19162 (A.M.), AIRC-15312 (R.R.), and AIRC-18560 (D.D.B.), Ministry of Health Ricerca Finalizzata PE-2013-02355271 (R.R. and A.M.), and NIH no. R01GM114306 (A.M.). S.P. is a recipient of Fondazione Umberto Veronesi 2019 fellowship. C.P. is a recipient of a doctoral fellowship by the Ministry of Research (MIUR). Myogenin (Wright WE) antibody was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by Department of Biology, The University of Iowa, Iowa City, IA 52242.
Glossary
Abbreviations
- AEBP2
adipocyte enhancer-binding protein
- CDK
cyclin dependent kinase
- EED
embryonic ectoderm development
- EGFR
epidermal growth factor receptor
- EMT
epithelial-mesenchymal transition
- EZH2
enhancer of zeste homologue 2
- GBM
glioblastoma
- H3K27
histone 3 lysine 27
- HDACi
histone deacetylase inhibitors
- JAK
janus kinase
- JmjD3
jumonji domain 3
- PI3K
phosphoinositide 3-kinases
- PRC2
polycomb repressive complex 2
- RMS
alveolar rhabdomyosarcoma
- SAH
S-adenosyl-l-homocysteine
- SHH MB-SLCs
stemlike cells belonging to the sonic hedgehog subgroup
- SUZ12
suppressor of zeste 12
- TBTU
O-benzotriazol-tetramethyluronium tetrafluoroborate
- UTX
ubiquitously transcribed tetratricopeptide repeat, X chromosome
- VEGFR
vascular endothelial growth factor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00014.
Synthetic procedures and chemical and physical data for compounds 4–18; elemental analyses for final compounds 4 and 5; biochemical assays, cell lines and culture conditions, cell proliferation assay, Western blot, and flow cytometric analysis of cell cycle, apoptosis, and cell differentiation (PDF)
Author Contributions
∇ The manuscript was written through contributions of all authors. These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Luan Y.; Li J.; Bernatchez J. A.; Li R. Kinase and Histone Deacetylase Hybrid Inhibitors for Cancer Therapy. J. Med. Chem. 2019, 62 (7), 3171–3183. 10.1021/acs.jmedchem.8b00189. [DOI] [PubMed] [Google Scholar]
- Stazi G.; Fioravanti R.; Mai A.; Mattevi A.; Valente S. Histone deacetylases as an epigenetic pillar for the development of hybrid inhibitors in cancer. Curr. Opin. Chem. Biol. 2019, 50, 89–100. 10.1016/j.cbpa.2019.03.002. [DOI] [PubMed] [Google Scholar]
- Tomaselli D.; Lucidi A.; Rotili D.; Mai A. Epigenetic polypharmacology: A new frontier for epi-drug discovery. Med. Res. Rev. 2020, 40 (1), 190–244. 10.1002/med.21600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W. S.; Parmigiani R. B.; Marks P. A. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007, 26 (37), 5541–52. 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- Bolden J. E.; Peart M. J.; Johnstone R. W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discovery 2006, 5 (9), 769–84. 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- McClure J. J.; Li X.; Chou C. J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. 10.1016/bs.acr.2018.02.006. [DOI] [PubMed] [Google Scholar]
- de Lera A. R.; Ganesan A. Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin. Epigenet. 2016, 8, 105. 10.1186/s13148-016-0271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti R.; Conte M.; Iside C.; Altucci L. Epigenetic-based therapy: From single- to multi-target approaches. Int. J. Biochem. Cell Biol. 2015, 69, 121–31. 10.1016/j.biocel.2015.10.016. [DOI] [PubMed] [Google Scholar]
- Hesham H. M.; Lasheen D. S.; Abouzid K. A. M. Chimeric HDAC inhibitors: Comprehensive review on the HDAC-based strategies developed to combat cancer. Med. Res. Rev. 2018, 38 (6), 2058–2109. 10.1002/med.21505. [DOI] [PubMed] [Google Scholar]
- Cohen R. B. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat. Rev. 2014, 40 (4), 567–77. 10.1016/j.ctrv.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Younes A.; Berdeja J. G.; Patel M. R.; Flinn I.; Gerecitano J. F.; Neelapu S. S.; Kelly K. R.; Copeland A. R.; Akins A.; Clancy M. S.; Gong L.; Wang J.; Ma A.; Viner J. L.; Oki Y. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. Lancet Oncol. 2016, 17 (5), 622–31. 10.1016/S1470-2045(15)00584-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C.; Chen S.; Zhang C.; Hu G.; Zhang W.; Li L.; Chen Y. Z.; Tan C.; Jiang Y. Synthesis and investigation of novel 6-(1,2,3-triazol-4-yl)-4-aminoquinazolin derivatives possessing hydroxamic acid moiety for cancer therapy. Bioorg. Med. Chem. 2017, 25 (1), 27–37. 10.1016/j.bmc.2016.10.006. [DOI] [PubMed] [Google Scholar]
- Gulati N.; Beguelin W.; Giulino-Roth L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk. Lymphoma 2018, 59 (7), 1574–1585. 10.1080/10428194.2018.1430795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravanti R.; Stazi G.; Zwergel C.; Valente S.; Mai A. Six Years (2012–2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2-Pyridone Compounds. Chem. Rec. 2018, 18 (12), 1818–1832. 10.1002/tcr.201800091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellini P.; Marrocco B.; Borovika D.; Polletta L.; Carnevale I.; Saladini S.; Stazi G.; Zwergel C.; Trapencieris P.; Ferretti E.; Tafani M.; Valente S.; Mai A. Pyrazole-based inhibitors of enhancer of zeste homologue 2 induce apoptosis and autophagy in cancer cells. Philos. Trans. R. Soc., B 2018, 373 (1748), 20170150. 10.1098/rstb.2017.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele E.; Valente S.; Alfano V.; Silvano M.; Mellini P.; Borovika D.; Marrocco B.; Po A.; Besharat Z. M.; Catanzaro G.; Battaglia G.; Abballe L.; Zwergel C.; Stazi G.; Milite C.; Castellano S.; Tafani M.; Trapencieris P.; Mai A.; Ferretti E. The histone methyltransferase EZH2 as a druggable target in SHH medulloblastoma cancer stem cells. Oncotarget 2017, 8 (40), 68557–68570. 10.18632/oncotarget.19782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stazi G.; Taglieri L.; Nicolai A.; Romanelli A.; Fioravanti R.; Morrone S.; Sabatino M.; Ragno R.; Taurone S.; Nebbioso M.; Carletti R.; Artico M.; Valente S.; Scarpa S.; Mai A. Dissecting the role of novel EZH2 inhibitors in primary glioblastoma cell cultures: effects on proliferation, epithelial-mesenchymal transition, migration, and on the pro-inflammatory phenotype. Clin. Epigenet. 2019, 11 (1), 173. 10.1186/s13148-019-0763-5. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Grinshtein N.; Rioseco C. C.; Marcellus R.; Uehling D.; Aman A.; Lun X.; Muto O.; Podmore L.; Lever J.; Shen Y.; Blough M. D.; Cairncross G. J.; Robbins S. M.; Jones S. J.; Marra M. A.; Al-Awar R.; Senger D. L.; Kaplan D. R. Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget 2016, 7 (37), 59360–59376. 10.18632/oncotarget.10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lue J. K.; Prabhu S. A.; Liu Y.; Gonzalez Y.; Verma A.; Mundi P. S.; Abshiru N.; Camarillo J. M.; Mehta S.; Chen E. I.; Qiao C.; Nandakumar R.; Cremers S.; Kelleher N. L.; Elemento O.; Amengual J. E. Precision Targeting with EZH2 and HDAC Inhibitors in Epigenetically Dysregulated Lymphomas. Clin. Cancer Res. 2019, 25 (17), 5271–5283. 10.1158/1078-0432.CCR-18-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding T.; Swanson J.; Van Ness B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget 2018, 9 (31), 21930–21942. 10.18632/oncotarget.25128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Chen S. Y.; Colborne S.; Lambert G.; Shin C. Y.; Santos N. D.; Orlando K. A.; Lang J. D.; Hendricks W. P. D.; Bally M. B.; Karnezis A. N.; Hass R.; Underhill T. M.; Morin G. B.; Trent J. M.; Weissman B. E.; Huntsman D. G. Histone Deacetylase Inhibitors Synergize with Catalytic Inhibitors of EZH2 to Exhibit Antitumor Activity in Small Cell Carcinoma of the Ovary, Hypercalcemic Type. Mol. Cancer Ther. 2018, 17 (12), 2767–2779. 10.1158/1535-7163.MCT-18-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashina T.; Kinoshita I.; Kikuchi J.; Shimizu Y.; Sakakibara-Konishi J.; Oizumi S.; Nishimura M.; Dosaka-Akita H. Combined inhibition of EZH2 and histone deacetylases as a potential epigenetic therapy for non-small-cell lung cancer cells. Cancer Sci. 2016, 107 (7), 955–62. 10.1111/cas.12957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. P.; Ling K. EZH2 and histone deacetylase inhibitors induce apoptosis in triple negative breast cancer cells by differentially increasing H3 Lys(27) acetylation in the BIM gene promoter and enhancers. Oncol. Lett. 2017, 14 (5), 5735–5742. 10.3892/ol.2017.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein P. J.; Chomet M.; Metaxas A.; Christiaans J. A.; Kooijman E.; Schuit R. C.; Lammertsma A. A.; van Berckel B. N.; Windhorst A. D. Synthesis, radiolabeling and evaluation of novel amine guanidine derivatives as potential positron emission tomography tracers for the ion channel of the N-methyl-d-aspartate receptor. Eur. J. Med. Chem. 2016, 118, 143–60. 10.1016/j.ejmech.2016.04.022. [DOI] [PubMed] [Google Scholar]
- Lillico R.; Sobral M. G.; Stesco N.; Lakowski T. M. HDAC inhibitors induce global changes in histone lysine and arginine methylation and alter expression of lysine demethylases. J. Proteomics 2016, 133, 125–133. 10.1016/j.jprot.2015.12.018. [DOI] [PubMed] [Google Scholar]
- Chen E.; Pham T. Q.; Robinson K.; Xu L.; Skapek S. X. HDAC6 promotes self-renewal and migration/invasion of rhabdomyosarcoma. bioRxiv 2019, 823864. 10.1101/823864. [DOI] [Google Scholar]
- Wang S.; Zuo H.; Jin J.; Lv W.; Xu Z.; Fan Y.; Zhang J.; Zuo B. Long noncoding RNA Neat1 modulates myogenesis by recruiting Ezh2. Cell Death Dis. 2019, 10 (7), 505. 10.1038/s41419-019-1742-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y.; Zheng Y.; Dai M.; Wang X.; Wu J.; Yu B.; Zhang H.; Cui Y.; Kong W.; Wu H.; Yu X. G9a and histone deacetylases are crucial for Snail2-mediated E-cadherin repression and metastasis in hepatocellular carcinoma. Cancer Sci. 2019, 110 (11), 3442–3452. 10.1111/cas.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T.; Wang Y.; Hu Q.; Wu W.; Wu Y.; Wei W.; Han D.; You Y.; Lin N.; Liu N. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget 2017, 8 (58), 98348–98359. 10.18632/oncotarget.21311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistelli C.; Sabarese G.; Santangelo L.; Montaldo C.; Gonzalez F. J.; Tripodi M.; Cicchini C. The lncRNA HOTAIR transcription is controlled by HNF4α-induced chromatin topology modulation. Cell Death Differ. 2019, 26, 890. 10.1038/s41418-018-0170-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi L.; Battistelli C.; de Turris V.; Noce V.; Zwergel C.; Valente S.; Moioli A.; Manzione A.; Palladino M.; Bordoni V.; Domenici A.; Mene P.; Mai A.; Tripodi M.; Strippoli R. HDAC1 inhibition by MS-275 in mesothelial cells limits cellular invasion and promotes MMT reversal. Sci. Rep. 2018, 8 (1), 8492. 10.1038/s41598-018-26319-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.