

Abstract
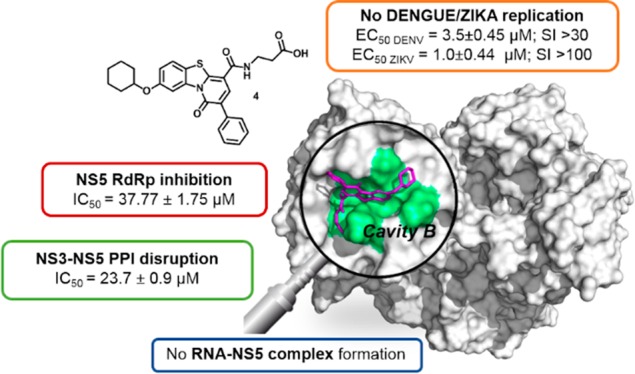
Treatment of dengue virus (DENV) and other flavivirus infections is an unmet medical need. The highly conserved flaviviral NS5 RNA-dependent RNA polymerase (RdRp) is an attractive antiviral target that interacts with NS3 and viral RNA within the replication complex assembly. Biochemical and cell-based evidence indicate that targeting cavity B may lead to dual RdRp and NS5–NS3 interaction inhibitors. By ligand-based design around 1H-pyrido[2,1-b][1,3]benzothiazol-1-one (PBTZ) 1, we identified new potent and selective DENV inhibitors that exert dual inhibition of NS5 RdRp and NS3–NS5 interaction, likely through binding cavity B. Resistance studies with compound 4 generated sequence variants in the 3′-untranslated region of RNA while further biochemical experiments demonstrated its ability to block also RNA-NS5 interaction, required for correct RNA synthesis in cells. These findings shed light on the potential mechanism of action for this class of compounds, underlying how PBTZs are very promising lead candidates for further evaluation.
Keywords: Antivirals, Dengue inhibitors, Zika inhibitors, NS5 RdRp inhibitors, protein−protein interaction inhibitors
The dengue virus, constituted by four serotypes (DENV1–4), causes the most common arboviral disease1 and represents a significant global health threat in 2019.2 Indeed, worldwide spread of DENV is constantly growing due to the adaptability of Aedes aegypti mosquito vectors to urban developments.3 The same mosquito vector also transmits flaviviruses such as Zika (ZIKV), West Nile (WNV), and Yellow fever viruses (YFV). Over 300 million DENV infections occur annually, of which 25% of cases manifest clinical symptoms ranging from mild flu-like illness to severe dengue hemorrhagic fever and shock syndrome, associated with high hospitalization rate and mortality. No antiviral drugs are available to treat these infections and the live-attenuated tetravalent vaccine Dengvaxia (CYD-TDV, Sanofi-Pasteur), approved in 20 endemic countries,4 shows limited efficacy and safety. Indeed, from 2018 the vaccine administration has to follow specific guidelines: it is limited to 9–45 years old people, and it is strongly discouraged in seronegative individuals because of the possible increased risk of severe dengue, through the antibody-dependent enhancement.5 Therefore, new therapeutics against DENV are needed. Moreover, broad-spectrum antiflaviviral compounds would be particularly desirable and their identification could be possible due to structural and functional similarities among all genus members.
DENV is a small enveloped virus whose genome is formed by a positive single-strand RNA (≈11 kb), with nearly 70% of sequence identity among the four serotypes (DENV1–4), and consisting of a single open reading frame (ORF), flanked by 5′ and 3′ untranslated regions (UTRs) with a type I cap at the 5′ terminus.6 The UTRs cover structural and functional roles forming secondary and tertiary RNA structures involved in the switching between replication and translation of the genome.7 The ORF encodes for a polyprotein precursor (≈3300 aa) that is cleaved by host and viral proteases into three structural and seven nonstructural (NS) proteins.6 Some NS proteins exert enzymatic functions essential for viral replication: NS3 exerts protease, ATPase/helicase, and RNA 5′-triphosphatase activities while NS5 exerts both RNA-dependent RNA polymerase (RdRp) and methyltransferase activities.8 Moreover, NS5 and NS3 interact and colocalize in host cells for the correct assembly of replication complex (RC), a macromolecular machine that includes viral transient double-strand RNA, NS and host proteins in tight association. Although the exact RC components are not yet well understood, interactions of NS3 with NS5 which in turn interact with the UTRs are essential for RNA replication and infectious virions production.9−12 To date, the NS3–NS5 3D structure is still not available, although the NS5 cavity B and an area of NS3 have already been reported as the main surfaces responsible for this interaction.10,12 In particular, Leu327, Leu328, Lys330, Thr858, Trp859, Asn862, Ile863, and Ala866 of cavity B from DENV2 RdRp have been identified as hot spots for the protein–protein interaction (PPI) and essential for viral replication.10,12 Those residues are well conserved across DENV1–4 (100% identity), and high identity was also observed in WNV and ZIKV (Table S1). Alanine replacement of Leu328, Trp859, and Ile863 abrogated the de novo RNA synthesis while Lys330Ala mutant was still able to synthesize RNA but it cannot bind to NS3 and impaired virus replication.12 Thus, cavity B provides a site for RdRp allosteric inhibitors and/or for blockers of NS3–NS5 PPI. Nucleoside and non-nucleoside RdRp inhibitors have made a huge impact on the treatment of hepatitis C virus infections, a related member of the Flaviviridae family. However, no effective small molecules targeting NS5 through binding to cavity B and thus showing potent inhibition of RdRp and/or NS3–NS5 interaction have been identified. Only a purine derivative able to weakly inhibit the NS3–NS5 interaction has been recently reported.13 A few years ago, we reported a 1H-pyrido[2,1-b][1,3]benzothiazol-1-one (PBTZ) (1) (see Table 1 for the structure), that was able to inhibit DENV3 (IC50DENV3 = 1.5 ± 0.2 μM) and WNV RdRps in the μM range also showing broad-spectrum antiflavivirus activity.14
Table 1. DENV2 RdRp Biochemical Assay and Antiviral Evaluation in HuH7 Cells of Compounds 2–11.


% inhibitory activity against RdRp in de novo (primer independent) and elongation (primer dependent) assays at single 30 μM concentration.
Elong. IC50 is the concentration that inhibits 50% of RdRp activity in elongation; values represent average ± SD from a single experiment carried out with duplicates.
CC50 is the cytotoxic concentration that affects 50% of cell viability as determined by CellTiter-Glo Luminescent Assay (Promega).
EC50 is the effective concentration that inhibits 50% virus replication as determined by plaque assay against DENV2 EDEN 3295 (GenBank accession: EU081177.1) after 48 h treatment and ZIKV H/PF/2013 (GenBank accession: KJ776791.2) after 24 h treatment; values represent average ± SD from two independent experiments.
SI is the selectivity index calculated as the ratio CC50/EC50.
ND: not determined due to poor or no activity in the corresponding assay.
More recently, starting from compound 1 as template, we designed a C-2/C-8 modified PBTZ series. These compounds displayed antiviral activity against DENV1–4, WNV, YFV, and other flaviviruses whereas they were inactive against other RNA viruses.15 Moreover, the antiviral effect did not rely on the reduction of viral RNA synthesis or virion release but rather to the reduced infectivity of viral particles, suggesting a viral factor as possible target. Nevertheless, we were unable to identify the antiviral mode of action (MoA) of PBTZs at the molecular level due to the failure to select resistant mutants.15
In this contribution, in order to elucidate the MoA of our compounds and to extend the ligand-based strategy, we explored a further C-8 modification (compound 2), the variation of the C-4 amide side chain (compounds 3–8), and a core size reduction of the PBTZ scaffold (compounds 9–11) (Table 1). Target PBTZ derivatives 2–9 were synthesized (Scheme 1) through functionalization of the PBTZ core according to a procedure already reported by us.16 The chemistry section, experimental details, and analytical data are reported in the Supporting Information. Pyridones 10 and 11 were prepared by following the synthetic route depicted in Scheme 2, and also in this case all the details are described in the Supporting Information.
Scheme 1.

Reagents and conditions: (i) Cycloheptanol, PPh3, DIAD, dry THF, 0 °C to rt, ultrasounds; (ii) NaH 60%, diethylcarbonate, dry THF, reflux; (iii) DMF-DMA, dry DMF, 80 °C; (iv) phenylacetic anhydride, 110 °C, neat; (v) 10% NaOH, MeOH, 70 °C; (vi) R-NH2, TBTU, DIPEA, dry DMSO, rt; (vii) 1 N LiOH, dioxane, rt; (viii) ethanolamine, reflux; (ix) ethylene glycol, Dowtherm A, MW irradiation, 250 °C.
Scheme 2.

Reagents and conditions: (i) (3-methoxyphenyl)boronic acid, Cu(OAc)2·H2O, Py, 4 Å MS, dry CH2Cl2, rt; (ii) Pd(PPh3)4, phenylboronic acid, K2CO3 2 M, DME, MW, 100 °C; (iii) BBr3 1 M in CH2Cl2, dry CH2Cl2, 0 °C; (iv) Cyclohexanol, PPh3, DIAD, dry THF, 0 °C to rt, ultrasound; (v) 2 N NaOH, MeOH/THF, rt; (vi) Tyr methyl ester, TBTU, DIPEA, dry DMSO, rt; (vii) 1 N LiOH, dioxane, rt.
All the synthesized compounds 2–11 were subjected to in vitro DENV2 NS5 polymerase assays (both de novo initiation and elongation assays) as previously described17 using purified recombinant full-length protein (Table 1 and Figure 1A). In vitro antiviral efficacy assays against DENV2 infection in human hepatoma cells (HuH7) were initially carried out using an inhibitory concentration of 10 μM in order to select compounds for subsequent detailed dose–response testing from 0.01 to 100 μM to calculate the effective concentration that results in 50% reduction in infective virus particles (EC50). The compound toxicity was determined using the commercial CellTiter-Glo luminescence assay to obtain CC50 values, and then the selectivity index (SI; CC50/EC50) was evaluated (Table 1 and Figure 1B). Compound 1, which was shown to inhibit both DENV3 RdRp and viral replication in our previous study,14 was included for comparative purpose, notably retaining a similar overall biological profile despite different experimental conditions. Moreover, the chemically unrelated nucleoside analogue NITD-00818 was used as positive control in the in vitro cell-based infection assays (Table 1).
Figure 1.

Inhibition curves of selected PBTZs against DENV2 full-length NS5 RdRp elongation activity and cell-based DENV2 infection assay. (A) Concentration-dependent inhibitory effect of PBTZs (i) 1; (ii) 2; (iii) 4; (iv) 5 on RdRp elongation activity (black curve).17 The compound only control was included to determine its autofluorescence signal (red). Data are obtained from one experiment in duplicate. (B) Dose dependent viral inhibition assay in HuH7 infected cells measured at 48 h post treatment with PBTZs (i) 1; (ii) 2; (iii) 4; (iv) 5 (black curve) by plaque assay and cell viability (red). Data are obtained from three independent experiments.
In biochemical assays, C-4 aminoacyl PBTZs 2–5 showed good inhibition (>70%) of RdRp elongation activity at 30 μM with IC50’s ranging from 9.2 to 37.8 μM, while they were less effective or even inactive in the initiation phase, similar to parent compound 1. The presence of a bigger cycloheptyl ether at the C-8 position gave the equally potent compound 2 with respect to hit compound 1. Changing the amino acidic residue at the C-4 amide side chain did not have a heavy effect on the RdRp inhibitory potency, with the nonaromatic Ser 3 and β-Ala 4 derivatives showing only a 2- and 4-fold increase in IC50 values, respectively; on the other hand, the shifting of the hydroxyl group of the phenyl moiety from the para (1) to the meta (5) position gave almost equal inhibitory activity with respect to hit 1. The C-4 amide PBTZs 6 and 7, lacking the amino acidic moiety but still maintaining the aromatic portion were totally inactive. Compound 8, presenting an amidic residue whose chemical features are distinct from the Tyr moiety of compound 1, was a weak inhibitor of RdRp polymerase elongation activity. This indicates a crucial role for the carboxylic moiety and in turn for the aminoacyl nature of the C-4 side chain that results in a pharmacophore requirement for RdRp binding and inhibition. This is further supported by the fact that removal of the C-4 amide side chain as in compound 9 gave an inactive derivative against RdRp elongation activity. Moreover, the core size reduction reached by the opening of the flat tricyclic core to more flexible pyridone 10 resulted in about 10-fold potency decrease in comparison to 1. This data demonstrates that the presence of the planar PBTZ core is essential to properly orientate the substituents in order to inhibit the RdRp activity. In parallel, the N-methyl pyridone 11 which is a simpler and smaller analogue of compound 10 showed no inhibition of RdRp activity, confirming the importance of the cycloalkyl aryl ether.
In the initial single point (10 μM) in vitro cell-based infection assays, most of the PBTZs tested resulted in >80% viral titer reduction (data not shown), and they were therefore profiled for their antiviral potency and cytotoxicity. In particular, compounds 2, 4–6, and 8 were found to be low μM inhibitors (EC50s < 4 μM) against DENV2 with low cytotoxicity showing good SI values (from 30 to >100), similarly to parent 1 and NITD-00818 (Table 1). Regarding the new C-4 aminoacyl PBTZs, the comparable profile of Tyr derivatives 2 and 1 indicated the cycloheptyl moiety as a suitable replacer of the cyclohexyl group and preferred over a smaller cyclopentyl, that induced higher cytotoxicity as previously reported.15 The similar activity/selectivity of compounds 4 and 5 indicates that also nonconventional amino acids such as β-Ala and (m–OH)Phe, respectively, may provide an antiviral effect. Interestingly, the absence of the aromatic feature in derivative 4 did not influence the antiviral activity but was accountable for a 4-fold reduction in the NS5 RdRp inhibitory activity. Conversely, Ser derivative 3 was not effective in the cell-based assay despite the modest inhibition in biochemical elongation activity, probably due to high compound polarity that may limit its cell permeability. Compounds 6 and 8, having nonamino acidic polar substituents (p-hydroxybenzyl and ethanol, respectively) at the amide side chain, showed potent anti-DENV2 efficacy but no RdRp inhibition in the biochemical assay, suggesting that these derivatives may have a different MoA than PBTZs 1–5. On the other hand, the insertion of the apolar benzylamide (7) and the removal of the C-4 amide side chain (9) provided inactive compounds also in cell-based assays. Finally, size reduction to pyridones 10 and 11 was very detrimental, thus indicating the PBTZ tricyclic core as an essential chemical requirement also for the antiviral activity.
To investigate the broad spectrum inhibitory activity, the compounds were also tested against a ZIKV isolate (French Polynesian isolate; HPF). Notably, the compounds able to inhibit DENV2 replication showed even better potency against ZIKV infection with EC50 in the sub μM range (Table 1). Additionally, the inhibitory activity against the remaining DENV serotypes (DENV1: EU081230.1; DENV3: EU081190.1; DENV4: GQ398256.1) was evaluated for compounds 4 and 5, selected for their more favorable selective inhibition (highest SIs, Table 1). These compounds retained low μM antiviral potencies against all serotypes, similarly to compound 1 (Table 2). These data together with anti-ZIKV activity point to the potential broad antiflavivirus activity of this chemical class, in agreement with our previous findings on related compounds.15 Intrigued by the positive results for compounds 2, 4, and 5, we argued that these inhibitors could interact with the NS5 RdRp through the binding to cavity B, a pocket of the thumb subdomain suitable for allosteric inhibitors that potentially could also block NS3–NS5 interaction.12
Table 2. Cell-Based Efficacy of 1, 4, and 5 against DENV1-4 Serotypes.
| EC50 (μM)a |
||||
|---|---|---|---|---|
| Cpds | DENV1 | DENV2 | DENV3 | DENV4 |
| 1 | 2.8 ± 0.60 | 2.1 ± 0.22 | 2.8 ± 0.51 | 1.7 ± 0.24 |
| 4 | 7.0 ± 3.50 | 3.5 ± 0.45 | 7.8 ± 1.67 | 6.9 ± 1.95 |
| 5 | 3.2 ± 0.72 | 2.9 ± 0.35 | 4.3 ± 2.68 | 5.9 ± 2.65 |
EC50 is the effective concentration that inhibits 50% virus replication as determined by plaque assay against DENV1–4; data is presented as average ± SD from two independent experiments.
Since targeting PPI is considered a promising approach in antiviral chemotherapy,19 the identification of compounds able to disrupt flavivirus PPI would be a promising avenue for novel inhibitors. It was previously demonstrated that NS3–NS5 interaction-defective mutants can impair infectious virus production, viral protein synthesis, and RNA replication to varying degrees, which is likely dependent on the presence of the key amino acids involved in NS3–NS5 interaction.10 This finding together with the evidence that our previous PBTZs produced noninfective virions15 prompted us to assay the best compounds 2, 4, and 5 together with the reference compound 1 as NS3–NS5 PPI inhibitors. Before compound evaluation, we wanted to support our hypothesis carrying out in silico studies on cavity B.
At first, we performed AutoDock20 docking experiments of aminoacyl PBTZs against cavity B of the DENV3RdRp (PDB ID: 2J7U), defined by residues Leu326, Leu327, Lys329, Thr858, Trp859, Asn862, Ile863, and Ala866 (corresponding to Leu327, Leu328, Lys330, Thr858, Trp859, Asn862, Ile863, and Ala866 residues of DENV2).
The obtained binding modes were evaluated in terms of ligand binding energy (LBE) and numbers in cluster (NiC), a measure of the reliability of the virtual pose (Table S2). The results highlighted that the analyzed derivatives showed a reliable binding mode, with LBE values ranging from −7.78 to −9.56 kcal/mol and NiC > 20 (Table S2). Docking pose of the top-ranked compound 5, taken as representative example, placed the phenol ring in a small hydrophobic region defined by Leu327, Trp859, Ile863, and Ala866 of cavity B, where the hydroxyl group formed H-bonds with Leu326 and Lys329 backbone, while the aromatic moiety was involved in an edge-to-face stacking interaction with the Trp859 side chain (Figure 2). Interestingly, the hydroxyl group overlapped a crystallographic water molecule (Figure 2) originally bridging Leu326, Lys329 and Trp859, suggesting that the binding-site water could be displaced upon ligand recognition. Finally, the acid function of derivative 5 established a salt bridge with the positively charged Lys329 whereas the ether linker formed an additional H-bond with Lys325. Similar binding modes were generated for derivatives 1, 2, and 4 (Figure S1). As mentioned in the introduction, Leu327 (Leu326 in DENV3 RdRp), Trp859, and Ile863 (magenta residues in Figure 2) are key residues in the de novo RNA synthesis,12 while Lys330 (Lys329 in DENV3 RdRp, cyan residue in Figure 2) is the main player of the NS5–NS3 PPI.12 Thus, the predicted ligand–protein interactions could be consistent with the anti-RdRp activity of PBTZ derivatives and support their potential ability to inhibit the NS5–NS3 PPI.
Figure 2.

(A) 2D representation of the predicted interactions between the representative PBTZ 5 and DENV3 RdRp cavity B. (B) Docking pose of compound 5. Magenta: key residues for RdRp activity; cyan: key residue for NS3–NS5 interaction; red sphere: crystallographic water molecule.
To further validate the latter hypothesis, we tested the RdRp inhibitors 1, 2, 4, and 5 in a competitive NS3–NS5 ELISA interaction assay, as previously described10 (Figure 3A). Interestingly, the four compounds blocked NS3–NS5 interaction showing IC50 values from 18 to 42 μM, with derivative 5 resulting as the best inhibitor (Figure 3B). Thus, PBTZs 1,142, 4, and 5 represent the most potent NS3–NS5 PPI inhibitors, which likely bind cavity B, so far identified. Although a purine derivative has been recently reported as weak NS3–NS5 PPI inhibitor (33% inhibition at 50 μM),13 the compound showed a good antiviral effect that did not correlate with the poor activity in the biochemical assay. On the contrary, the potency of aminoacyl PBTZs 1, 2, 4, and 5 against flavivirus infection is consistent with the dual inhibition of RdRp activity and NS5–NS3 PPI, achieved by targeting cavity B, as suggested by in silico predictions.
Figure 3.
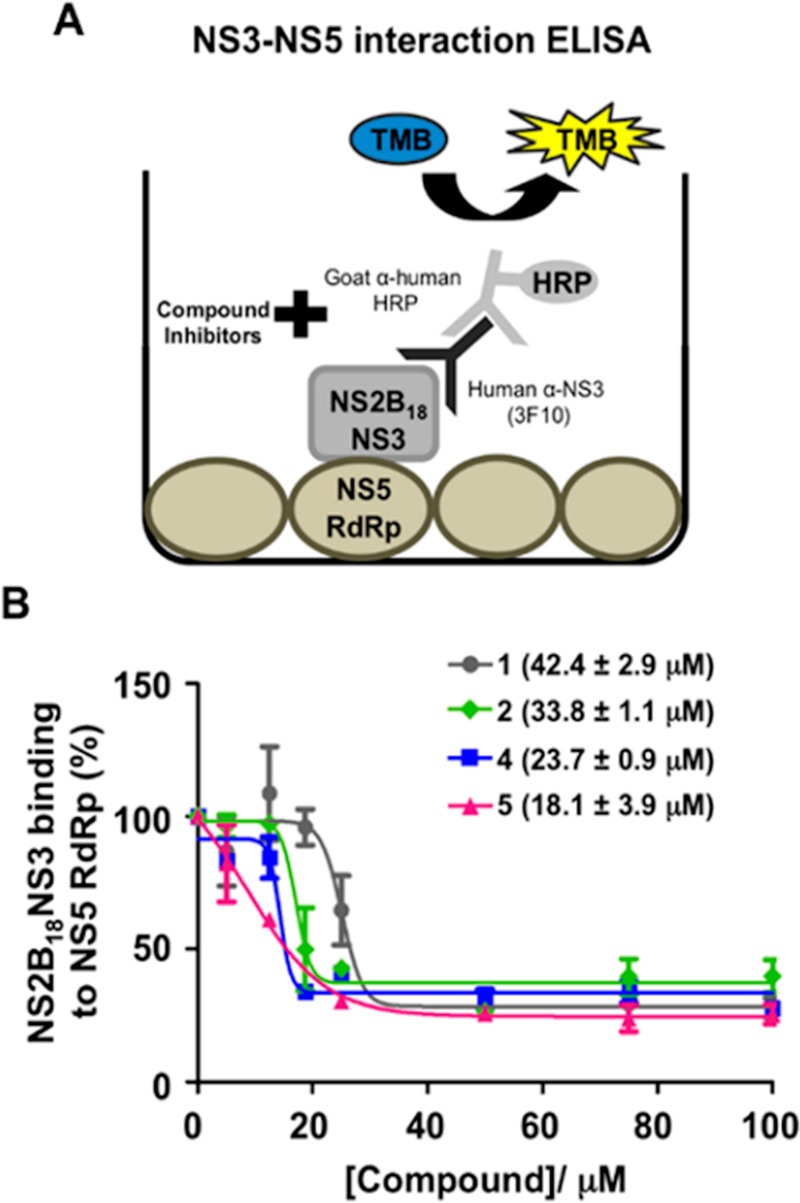
Functional evaluation of PBTZs as DENV NS3–NS5 PPI inhibitors. (A) Schematic diagram of the in vitro DENV NS3–NS5 ELISA interaction assay as previously described.10 (B) Concentration-dependent inhibition of NS3–NS5 interaction by PTBZ 1, 2, 4, and 5. The IC50 values were presented in parentheses as average ± SD obtained from a single experiment in duplicate.
To gain further mechanistic insight into the MoA of the aminoacyl PBTZs, we next performed compound resistance selection (Figure 4A) using compounds 1, 4, and 5 (CC50 > 100 μM, SI > 30). Compound 2 was excluded from this experiment because of its slight toxicity (CC50 ∼ 100 μM). DENV2-infected HuH7 cells were treated with increasing concentrations of the compounds for 10 passages, and the resultant P10 viruses were expanded in C6/36 insect cells before evaluating the antiviral activity to determine the virus susceptibility toward the compounds (Figure 4A). Interestingly, only the P10 virus from the compound 4 treatment (referred to as 4_R) showed resistance, with only a 20% reduction in virus production compared to 80% reduction for the control wild-type DENV2 that was similarly passaged (Figure 4B). The resistance of the 4_R virus was further indicated by the 2-fold rightward shift in the EC50 of compound 4 as compared to wild-type DENV2 (Figure 4C). Additionally, treatment of the 4_R (P10) resistant virus with other known antivirals of different mode-of-action showed similar level of viral reduction as the control wild-type DENV2 (Figure S2), suggesting that the observed resistance by compound 4 may be specific. Therefore, we performed full genome Sanger sequencing of the 4_R (P10) resistant virus and found the appearance of quasispecies variants at multiple positions in the 3′UTR region corresponding to nucleotide positions ranging from 10410 to 10500 (Figure S3). The precise mechanism of inhibition requires further studies, but it is conceivable that the PTBZ 4 may be disrupting NS5–RNA interactions since the 3′UTR consists of important elements recognized by NS5 RdRp to initiate viral RNA replication.21 Thus, compound 4 likely impairs RNA–NS5 interaction resulting in the antiviral effect (Figure 1A(iii)), and the RNA-Electrophoretic Mobility Shift Assay (REMSA) that detects the shift in RNA band upon complex formation with NS522 was carried out to provide preliminary experimental evidence in support of this hypothesis. First, the addition of NS5 to DENV2 3′UTR (454nt) caused an upward shift of the band compared to the RNA only lane (lane 2), indicating the formation of NS5–RNA complex (Figure 4D). However, the addition of compound 4 to the NS5/3′UTR RNA mixture led to the appearance of unbound RNA in the lane (lane 7), that is most likely related to the disruption of the protein–RNA interaction (Figure 4D).
Figure 4.

Resistance mutant selection, drug susceptibility test, and REMSA. (A) Schematic workflow showing the process of compound resistance selection. DENV2 infected HuH7 cells were treated with increasing concentrations of PBTZs for 72 h until P10. (B) Antiviral activity evaluation of compound 4 (at 10 μM) against DENV2 or 4_R clones; the bar graph is plotted as average ± SD from two independent experiments with duplicates. (C) Dose-dependent viral inhibition of P10 DENV2 or 4_R virus in HuH7 cells treated with 4 for 48 h; data are obtained from a single experiment with duplicates. (D) REMSA gel showing the shift of RNA band upon complex formation with DENV NS5 in the presence and absence of the compound (compound: 25 μM; NS5: 1.28 μM; RNA: 0.16 μM; molar ratio RNA/NS5 is 1:8; t = 37 °C; order of addition: compound, then NS5, then RNA). Lanes 1–7 are indicated above the image of the gel.
Interestingly, the disruption of NS5–RNA interaction relies on NS5 binding, as compounds 6 and 9, that were inactive against RdRp, did not show any effect in REMSA (Figure 4D). Taken together, our results suggest that PBTZs 1, 2, 4, and 5 may prevent the functionality of the virus RC that consists of viral and host proteins in intimate association with viral RNA, a macromolecular machine that still needs to be studied in detail.23
To pave the way toward future studies and in order to have an idea of the drug-like properties, we calculated in silico the physicochemical and pharmacokinetic properties of derivatives 1, 4, and 5 (Table 3).24 The compounds were predicted to have good solubility (Log S ∼ 2) and lipophilic/hydrophilic balance (Log D ∼ 2), with no significant inhibition of cytochrome (2C9), suggesting a low possibility for hepatotoxicity. Furthermore, the compounds are metabolically stable as revealed by RF_T_Half_Life descriptor and to potentially be absorbed after oral administration (HIA).
Table 3. In Silico Physicochemical and Pharmacokinetic Properties.
| Property | Desired values | 1 | 4 | 5 |
|---|---|---|---|---|
| log Sa | >1 | 2.1 | 2.1 | 2.0 |
| log Db | <5 | 2.3 | 2.1 | 2.3 |
| 2C9 pKic | <6 | 5.4 | 5.4 | 5.4 |
| BBB categoryd | – | “–” | “–” | “–” |
| HIA categorye | + | “+” | “+” | “+” |
| RF_T_Half_Lifef | stable | stable | stable | stable |
Intrinsic aqueous solubility. A Log S ≥ 1 corresponds to intrinsic aqueous solubility of greater than 10 μM.
Log D: logarithm of the octanol/water partition coefficient at pH = 7.4.
pKi values for CYP2C9 affinity. The defined threshold is to avoid drug–drug interactions due to inhibition of CYP2C9.
Predicts a classification of “+” for compounds which have a log([brain]:[blood]) ≥ −0.5 and “–” for compounds which have a ratio ←0.5.
Human Intestinal Absorption (HIA) Classification. Predicts a classification of “+” for compounds which are ≥30% absorbed and “–” for compounds which are <30% absorbed.
To conclude, the PBTZ derivatives we report here are potent antiflavivirus agents. In particular, C-4 aminoacyl PBTZs 1, 2, 4, and 5 show potent inhibition of both NS5 RdRp activity and its interaction with NS3 in biochemical assays, probably by targeting cavity B. These compounds represent the first-in-class NS5 RdRp and NS3–NS5 PPI dual inhibitors that are also able to exert potent and selective antiviral activity in cells against DENV1–4 serotypes and ZIKV. Mechanistic insights derived from resistant mutant selection studies on compound 4 identified a DENV2 quasispecies carrying mutations on the 3′-UTR, an important element that interacts with NS5 for the correct RC assembly and the RNA de novo synthesis. In addition, compound 4 was able to block NS5–3′UTR interaction as shown by REMSA. Therefore, all the results collectively indicate derivative 4 and other aminoacyl PBTZs as new interesting antiflavivirus agents that could act as RC disruptors by targeting NS5.
There is an urgent medical need to find new agents with broad-spectrum antiflavivirus activity, and our results strongly support future rounds of preclinical investigations for the best PBTZs.
Glossary
Abbreviations
- DENV
Dengue virus
- MoA
mode of action
- NS
nonstructural
- ORF
open reading frame
- PBTZ
pyridobenzothiazolone
- PPI
protein–protein interaction
- RC
replication complex
- RdRp
RNA-dependent RNA polymerase
- REMSA
RNA-Electrophoretic Mobility Shift Assay
- UTR
untranslated region
- WNV
West Nile virus
- YFV
Yellow Fever virus
- ZIKV
Zika virus.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00619.
Chemistry part, experimental and characterization data for all compounds, experimental procedures for the in silico studies, and all biological data (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Author Contributions
⊥ Rolando Cannalire and Kitti Wing Ki Chan equally contributed to the work.
This work has been realized in part thanks to (a) SILVER project of the European Commission within the seventh Framework Program Cooperation Project; grant agreement number: 2606444; (b) Scientific Independence of young Researchers (SIR) project from Italian Ministry of Education, University and Research; grant number: RBSI14C78S to G.M. and E.M.; and (c) National Medical Research Council grant NMRC/CBRG/0103/2016, National Research Foundation grant NRF2016NRF-CRP001-063 and a Ministry of Health grant (MOH-OFIRG18may-0006/2019) to S.G.V.
The authors declare no competing financial interest.
Supplementary Material
References
- Bhatt S.; Gething P. W.; Brady O. J.; Messina J. P.; Farlow A. W.; Moyes C. L.; Drake J. M.; Brownstein J. S.; Hoen A. G.; Sankoh O.; et al. The Global Distribution and Burden of Dengue. Nature 2013, 496, 504–507. 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . Ten threats to global health in 2019; https://www.who.int/emergencies/ten-threats-to-global-health-in-2019 (Accessed 2020-02-18). [Google Scholar]
- Wilder-Smith A.; Gubler D. J.; Weaver S. C.; Monath T. P.; Heymann D. L.; Scott T. W. Epidemic Arboviral Diseases: Priorities for Research and Public Health. Lancet Infect. Dis. 2017, 17 (3), e101–e106. 10.1016/S1473-3099(16)30518-7. [DOI] [PubMed] [Google Scholar]
- Wilder-Smith A.; Hombach J.; Ferguson N.; Selgelid M.; O’Brien K.; Vannice K.; Barrett A.; Ferdinand E.; Flasche S.; Mo; et al. Deliberations of the Strategic Advisory Group of Experts on Immunization on the Use of CYD-TDV Dengue Vaccine. The Lancet Infectious Diseases; Elsevier Ltd, 2019; pp e31–e38. [DOI] [PubMed] [Google Scholar]
- States M.; Strategic W. H. O.; Group A.; Grade T.; Sage T.; Surfhvv P.; Uh L. V.; Wkh H. L. Q.; Sxeolf Q.; Ri K.; et al. Weekly Epidemiological Record Relevé Épidémiologique Hebdomadaire. WHO 2018, 29, 389–396. [Google Scholar]
- Mackenzie J. Wrapping Things up about Virus RNA Replication. Traffic 2005, 6 (11), 967–977. 10.1111/j.1600-0854.2005.00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradrick S.; Ng W.; Soto-Acosta R.; Ooi E.; Garcia-Blanco M. The 5′ and 3′ Untranslated Regions of the Flaviviral Genome. Viruses 2017, 9 (6), 137. 10.3390/v9060137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollati M.; Alvarez K.; Assenberg R.; Baronti C.; Canard B.; Cook S.; Coutard B.; Decroly E.; de Lamballerie X.; Gould E. A.; et al. Structure and Functionality in Flavivirus NS-Proteins: Perspectives for Drug Design. Antiviral Res. 2010, 87, 125–148. 10.1016/j.antiviral.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y. P.; Zeng M.; Jiang B.; Zhang W.; Wang M.; Jia R.; Zhu D.; Liu M.; Zhao X.; Yang Q.; et al. Flavivirus RNA-Dependent RNA Polymerase Interacts with Genome UTRs and Viral Proteins to Facilitate Flavivirus RNA Replication. Viruses 2019, 11 ( (10), ), 929. 10.3390/v11100929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay M. Y. F.; Saw W. G.; Zhao Y.; Chan K. W. K.; Singh D.; Chong Y.; Forwood J. K.; Ooi E. E.; Grüber G.; Lescar J.; et al. The C-Terminal 50 Amino Acid Residues of Dengue NS3 Protein Are Important for NS3-NS5 Interaction and Viral Replication. J. Biol. Chem. 2015, 290 (4), 2379–2394. 10.1074/jbc.M114.607341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks A. J.; Johansson M.; John A. V.; Xu Y.; Jans D. A.; Vasudevan S. G. The Interdomain Region of Dengue NS5 Protein That Binds to the Viral Helicase NS3 Contains Independently Functional Importin Β1 and Importin α/β-Recognized Nuclear Localization Signals. J. Biol. Chem. 2002, 277 (39), 36399–36407. 10.1074/jbc.M204977200. [DOI] [PubMed] [Google Scholar]
- Lescar J.; Zou G.; Yap L. J.; Shochat S. G.; Shi P.-Y.; Yau Y. H.; Dong H.; Lim C. C.; Chen Y.-L. Functional Analysis of Two Cavities in Flavivirus NS5 Polymerase. J. Biol. Chem. 2011, 286 (16), 14362–14372. 10.1074/jbc.M110.214189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincetti P.; Caporuscio F.; Kaptein S.; Gioiello A.; Mancino V.; Suzuki Y.; Yamamoto N.; Crespan E.; Lossani A.; Maga G.; et al. Discovery of Multitarget Antivirals Acting on Both the Dengue Virus NS5-NS3 Interaction and the Host Src/Fyn Kinases. J. Med. Chem. 2015, 58 (12), 4964–4975. 10.1021/acs.jmedchem.5b00108. [DOI] [PubMed] [Google Scholar]
- Tarantino D.; Cannalire R.; Mastrangelo E.; Croci R.; Querat G.; Barreca M. L.; Bolognesi M.; Manfroni G.; Cecchetti V.; Milani M. Targeting Flavivirus RNA Dependent RNA Polymerase through a Pyridobenzothiazole Inhibitor. Antiviral Res. 2016, 134, 226–235. 10.1016/j.antiviral.2016.09.007. [DOI] [PubMed] [Google Scholar]
- Cannalire R.; Tarantino D.; Piorkowski G.; Carletti T.; Massari S.; Felicetti T.; Barreca M. L.; Sabatini S.; Tabarrini O.; Marcello A.; et al. Broad Spectrum Anti-Flavivirus Pyridobenzothiazolones Leading to Less Infective Virions. Antiviral Res. 2019, 167, 6–12. 10.1016/j.antiviral.2019.03.004. [DOI] [PubMed] [Google Scholar]
- Manfroni G.; Meschini F.; Barreca M. L.; Leyssen P.; Samuele A.; Iraci N.; Sabatini S.; Massari S.; Maga G.; Neyts J.; et al. Pyridobenzothiazole Derivatives as New Chemotype Targeting the HCV NS5B Polymerase. Bioorg. Med. Chem. 2012, 20 (2), 866–876. 10.1016/j.bmc.2011.11.061. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Soh T. S.; Zheng J.; Chan K. W. K.; Phoo W. W.; Lee C. C.; Tay M. Y. F.; Swaminathan K.; Cornvik T. C.; Lim S. P.; et al. A Crystal Structure of the Dengue Virus NS5 Protein Reveals a Novel Inter-Domain Interface Essential for Protein Flexibility and Virus Replication. PLoS Pathog. 2015, 11 (3), 1–27. 10.1371/journal.ppat.1004682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z.; Chen Y.; Schul W.; Wang Q.-Y.; Gu F.; Duraiswamy J.; Kondreddi R. R.; Niyomrattanakit P.; Lakshminarayana S. B.; Goh A.; et al. An Adenosine Nucleoside Inhibitor of Dengue Virus. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (48), 20435–20439. 10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voter A. F.; Keck J. L.. Development of Protein – Protein Interaction Inhibitors for the Treatment of Infectious Diseases. In Advances in Protein Chemistry and Structural Biology; Elsevier Inc., 2018; Vol. 111, pp 197–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filomatori C. V.; Iglesias N. G.; Villordo S. M.; Alvarez D. E.; Gamarnik A. V. RNA Sequences and Structures Required for the Recruitment and Activity of the Dengue Virus Polymerase. J. Biol. Chem. 2011, 286 (9), 6929–6939. 10.1074/jbc.M110.162289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Chan K. W. K.; Naripogu K. B.; Swarbrick C. M. D.; Aaskov J.; Vasudevan S. G. Subgenomic RNA from Dengue Virus Type 2 Suppresses Replication of Dengue Virus Genomes and Interacts with Virus-Encoded NS3 and NS5 Proteins. ACS Infect. Dis. 2020, 6, 436. 10.1021/acsinfecdis.9b00384. [DOI] [PubMed] [Google Scholar]
- Lescar J.; Soh S.; Lee L. T.; Vasudevan S. G.; Kang C.; Lim S. P. The Dengue Virus Replication Complex: From RNA Replication to Protein-Protein Interactions to Evasion of Innate Immunity. Adv. Exp. Med. Biol. 2018, 1062, 115–129. 10.1007/978-981-10-8727-1_9. [DOI] [PubMed] [Google Scholar]
- StarDrop. ADMEQSAR Models; https://www.optibrium.com/stardrop/stardrop-adme-qsar-models.php (Accessed 2020-02-18). [Google Scholar]
- BioSolveIT GmbH SeeSAR, Version 5.5; Sankt Augustin, Germany. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.