

Abstract
Breast cancer (BC) is the most diffused cancer type in women and the second leading cause of death among the female population. Effective strategies to fight estrogen responsive (ER+) BC, which represents 70% of all BC cases, rely on estrogen deprivation, via the inhibition of the aromatase enzyme, or the modulation of its cognate estrogen receptor. Current clinical therapies significantly increased patient survival time. Nevertheless, the onset of resistance in metastatic BC patients undergoing prolonged treatments is becoming a current clinical challenge, urgently demanding to devise innovative strategies. In this context, here we designed, synthesized, and performed in vitro inhibitory tests on the aromatase enzyme and distinct ER+/ER– BC cell line types of novel azole bridged xanthones. These compounds are active in the low μM range and behave as dual-mode inhibitors, targeting both the orthosteric and the allosteric sites of the enzyme placed along one access channel. Classical and quantum-classical molecular dynamics simulations of the new compounds, as compared with selected steroidal and nonsteroidal inhibitors, provide a rationale to the observed inhibitory potency and supply the guidelines to boost the activity of inhibitors able to exploit coordination to iron and occupation of the access channel to modulate estrogen production.
Keywords: Aromatase inhibitors, molecular dynamics, breast cancer, cytochromes P450, xanthone
Breast cancer (BC) is the most common malignancy in women and ranks the second place among cancer-related deaths in the female gender. The recent advances in genomics clarified that BC is a heterogeneous disease, characterized by different biological subtypes, leading to therapeutic choices based also on tumor genetic profiling.1,2 Approximately 75% of patients are hormone receptor-positive, requiring estrogens for tumor’s progression; thus, therapy focused on estrogen deprivation plays a pivotal role. Targeted therapy of BC is now based on two different approaches. Interference with binding of estrogens to their receptors (ERα) can be obtained via selective estrogen receptor modulators (SERMs), such as the prototype tamoxifen3 (Figure 1), and selective estrogen receptor degraders (SERDs), such as fulvestrant4 (Figure 1). Tamoxifen binds with high affinity to ERα and is used to significantly reduce the chance of BC in high-/average-risk women, although becoming ineffective in relapsing metastatic BC patients.5 Alternatively, more recently marketed drugs act by affecting the synthesis of estrogens via inhibition of the cytochrome P450 (CYP450) aromatase, the key enzyme for their biosynthetic process.6 Human aromatase (HA), produced by the CYP19A1 gene, catalyzes aromatization of androgens to estrogens, with a unique pathway in the steroidal hormone biosynthesis.7−9 HA is found in all estrogen producing tissues, such as ovaries and adrenal glands, but also in ER+ tumor cells. Third-generation steroidal and nonsteroidal aromatase inhibitors (AIs, exemestane, letrozole, and anastrozole, Figure 1) are now considered as first-line treatment for hormone-dependent BC.1,2
Figure 1.

Molecular structures of selective estrogen receptor modulators (SERMs, Tamoxifen and its metabolite Endoxifen) and degraders (SERD, Fulvestrant), along with the third-generation aromatase inhibitors (AIs, Exemestane, Letrozole and Anastrozole).
In spite of the unquestionable effectiveness of current therapies, hurdles regarding compliance with the diverse side effects of SERMs and AIs and the development of resistance still need to be solved, keeping ERα and HA as very attractive targets.2 Although a comprehensive picture of the molecular mechanisms involved in resistance onset is still missing, large-scale genomic investigations identified aggressive ERα somatic mutations, which make the receptor intrinsically active, even in the absence of estrogens.5 In this context, the strategy of completely abrogating estrogen production loses its efficacy. Remarkably, the release of HA crystal structure10 fostered intense investigations on its functional aspects11−13 and boosted the search for novel AIs.14−16
Nevertheless, the need of catching alternative approaches to counteract resistance onset to current therapies led to exploit alternative inhibitory/modulatorystrategies of estrogen biosynthesis. In this respect, allostery was praised as a possible viable route to fine-tune estrogen production, after some primary metabolites of tamoxifen, still endowed with significant ER modulation properties, were found to act even as AIs.17 This was supposed to contribute to the overall pharmacological effect of the drug. In particular, kinetic studies showed for endoxifen (Figure 1) a noncompetitive inhibition mechanism.17 A noncompetitive or mixed inhibition mechanism was also claimed for the marketed AI letrozole (LTZ), for other azole compounds used as pesticides.18 In a previous study, we identified allosteric binding pockets potentially responsible for this nonactive site-directed inhibition.19 Among these sites, one pocket overlapped with the heme proximal cavity, and a ligand binding at this site may prevent the coupling of HA with NADPH-cytochrome P450 reductase (CPR) and, thus, the electron flow necessary for catalysis; the second pocket was instead placed along one possible access/egress channel of the substrate/product to/from the active site, and a ligand binding at this site may result in blocking the substrate entrance to the active site. These pockets were recently targeted by distinct inhibitors to fine-tune HA activity for therapeutic benefit.20 Aiming at developing steroidal inhibitors’ derivatives via the functionalization of exemestane (EXE), it was noted that the addition of a hydrophobic tail tailored to occupy the allosteric cavity lying along one of the HA’s access channels resulted in nM inhibition, suggesting that the simultaneous occupation of the orthosteric and allosteric sites may be an effective inhibition strategy.21,22
As part of a project aimed at developing novel nonsteroidal AIs, some of us developed a series of imidazolylmethylxanthones (Figure 2), which resulted to be potent and selective inhibitors,23 ranging from μM to nM (potency ranking: position 4 > 1 > 3 > 2, Table 1) depending on the position of the chain carrying the heme-coordinating imidazole moiety on the central xanthone core.
Figure 2.

Design of potential dual-binding xanthone derivatives.
Table 1. IC50s Obtained on the Isolated Enzyme and GI50s on ER+ (MCF-7) and ER– (MDA-MB-231) Cell Lines, Distances between the Nitrogen of the Ligands and the Iron of the Heme, And Angle between the Planes of the Imidazole Ring and the Heme Moieties.
| Compound | IC50 (μM) | GI50 MCF-7 (μM) | GI50 MDA-MB-231 (μM) | Distance (Å) | Angle (deg) |
|---|---|---|---|---|---|
| LTZ | 0.0118 | 4.1 ± 1.120 | 34.0 ± 7.8 | 2.33 ± 0.15 | 91.8 ± 2.7 |
| 1 | 0.01723 | 69.1 ± 0.8 | 58.3 ± 11.7 | 2.30 ± 0.13 | 88.8 ± 2.0 |
| 2 | 0.15023 | 59.0 ± 2.6 | 44.3 ± 6.5 | 2.31 ± 0.15 | 89.0 ± 2.6 |
| 3 | 0.39023 | 19.9 ± 5.9 | 30.4 ± 4.6 | 2.23 ± 0.12 | 91.3 ± 2.1 |
| 1a | 5.55 ± 2.3 | 52.3 ± 16.3 | 21.1 ± 9.3 | 2.18 ± 0.10 | 92.3 ± 2.2 |
| 2a | 2.85 ± 0.1 | 6.3 ± 1.1 | 17.5 ± 7.9 | 2.15 ± 0.08 | 91.8 ± 2.4 |
| 3a | 0.77 ± 0.3 | >100 | >100 |
Concentration of drug required to inhibit cell growth by 50% as determined after 72 h continuous exposure to the tested compounds. Data represent the mean ± standard deviation of three independent experiments.
In this paper, we exploit the scaffold of previously reported xanthones to develop a dual-mode inhibition strategy (Figure 2) via the insertion of a pentynyloxy chain, which literature reported as the most suitable functional group for a favorable interaction with the hydrophobic residues lining the access channel when inserted on EXE.21 To this aim, docking calculations were initially performed following a protocol successfully used in previous studies.14 The calibration of the protocol and its accuracy were assessed by docking the most active imidazolylmethylxanthone compounds previously reported (1–3, Figure 3a) and the clinically used LTZ inhibitor.23 Indeed, the high potency of clinically employed nonsteroidal AIs (e.g., LTZ and anastrazole) is attributed to the formation of a coordination bond between the nitrogen of the azole ring and the heme iron atom, and the same mechanism is expected to be operative in previously reported imidazolylmethylxanthones.
Figure 3.

(a) Previously reported imidazolylmethylxanthones23 and (b) the newly synthesized compounds designed on the basis of docking simulations.
Consistently with previous findings, a standard docking protocol failed in finding binding poses with imidazole nitrogen coordinating the heme moiety.24,25 Since docking programs do not account for the formation of covalent/coordination bonds, especially with transition metal ions,26 these calculations were done by imposing a constraint between the Fe@heme-N@azole atoms. This approach generated binding poses with the imidazole ring in the proximity of the iron atom (Figure 4). In particular, the azole nitrogen of compound 1 laid at a distance of 2.47 Å from the iron and the imidazole ring adopted a suitable binding geometry (i.e., almost perpendicular to the heme plane). Furthermore, the aromatic moiety of the xanthone established π-stacking interactions with Trp224. As well, 2 nicely fitted in the binding pocket, although exhibiting a longer Fe–N distance (3 Å). The best binding pose of compound 3, which possessed the highest IC50 among the previously reported imidazolylmethylxanthones series, showed a considerable tilt of the azole ring with respect to the heme plane, possibly resulting in a lack of coordination. Nevertheless, the xanthone also established π-stacking interactions with Phe134. Thus, the observed binding poses provide a coarse rationale for the experimentally observed trend of IC50s (Figure 3a). However, even when introducing in the docking protocol a constraint to the Fe–N distance, we did not obtain any binding pose for LTZ. This is in line with the absence of any crystallographic structure capturing this inhibitor in complex with HA and suggests that considerable deformations of the active site is required to fit LTZ into the catalytic pocket. Hence, we adopted an induced fit protocol,27 in which the side chains of the binding site can be displaced to accommodate the ligand. This resulted in a LTZ’s binding pose with a Fe–N distance of 2.40 Å and in which one of its cyano groups is hydrogen (H)-bonded with the backbone of Met374 (Figure 4). The second cyano group, instead, laid at the entrance of the access channel.
Figure 4.

Best-ranked binding poses obtained for the AIs letrozole, exemestane functionalized with a pentynyloxy chain (EXEa),21 and compounds 1–3 and 1a–3a. The protein is shown in ribbons, the binding site is highlighted with a gray surface, while the inhibitors and the heme are shown in a balls and stick representation.
Taking advantage of the successful functionalization of steroidal inhibitors,21,22 and based on the docking poses of compounds 1–3 (Figure 4), we designed new derivatives (Figure 3b) by functionalizing the imidazolylmethylxanthones with a pentynyloxy chain. In order to fill the entrance of the access channel (Figure 4), functionalization in ortho or meta positions with respect to the imidazole ring appeared as the most suitable. Nevertheless, docking calculations, imposing a constraint on the Fe–N bond, identified binding poses only for compounds 1a and 2a bearing the hydrophobic tail in the meta position (Figures 3b and 4). These exhibited a Fe–N distance of 2.82 and 2.52 Å, respectively, with the pentynyloxy chain located within the access site, similarly to C6-substituted steroidal inhibitors.21 Moreover, compound 2a established π-stacking interaction with Trp224.
Considering the binding pose of compound 3, the entrance of the access channel could not be easily reached by the introduction of the rigid alkyloxy chain. Yet, from preliminary docking calculations without the Fe–N metal constraint, the resulting binding pose displayed the carboxy oxygen of the aromatic moiety located in the proximity of the iron. Thus, exploiting this possible interaction mode, we also designed a ligand (3a) with alkyloxy chain and the imidazolylmethyl moiety located at symmetric positions on different rings of the xanthone. In this case, an induced fit protocol identified a binding pose in which the oxygen of the xanthone was at 2.57 Å from the iron atom, while the pentynyloxy chain was located in a hydrophobic pocket lined by residues Leu301, Leu305, Leu25, Tyr220, Ile125, and Met127 (Figure 4).
For the synthesis of the designed alkynyloxy-substituted xanthones 1a and 2a (Scheme 1), 4-methoxy-2-methylphenol, obtained from the reduction of 2-hydroxy-5-methoxybenzaldehyde,28 or the commercially available 3-methoxy-5-methylphenol was reacted with 2-chlorobenzoic acid under Ullmann reaction conditions to give the corresponding diarylethers, which were then cyclized by treatment with polyphosphoric acid to obtain the xanthone cores (4, 7). Bromination of the methyl group with NBS and subsequent reaction with imidazole allowed obtaining the intermediates 5 and 8, which were then demethylated with AlCl3 to give the corresponding hydroxyl derivatives 6 and 9 and successively alkylated with 1-bromo-2-pentyne to get the final compounds.
Scheme 1.

Reagents and conditions: (i) K2CO3, Cu/CuI, pyridine, H2O, reflux, 2 h; (ii) H3PO4, P2O5, 120 °C, 7 h; (iii) NBS, BPO, CCl4, reflux, hν, 6 h; (iv) imidazole, CH3CN, reflux, N2, 6 h; (v) AlCl3, toluene, reflux, 3 h; vi) 1-bromo-2-pentyne, K2CO3, acetone, reflux, 18-26 h.
For the synthesis of xanthone 3a, 6-methoxy-3-imidazolylmethylxanthone 10(29) was subjected to the demethylation step followed by alkylation with 1-bromo-2-pentyne, as reported for xanthones 5 and 8, to obtain the desired compound (Scheme 2).
Scheme 2.

Reagents and conditions: (i) AlCl3, toluene, reflux, 3 h; (ii) 1-bromo-2-pentyne, K2CO3, acetone, reflux, 18–26 h.
The inhibitory potency of the newly synthesized compounds (1a–3a) against HA was tested at different concentrations (from 0.1 to 100 μM) to obtain IC50 values, which proved to be in the low μM range (from 0.77 to 5.55 μM). Thus, for derivatives 1a and 2a a marked reduction of the inhibitory potency, as compared to their parent compounds, was observed,23 whereas 3a retained a similar activity to 3 (Table 1). It seemed, thus, clear that the introduction of a long and rigid alkoxy chain close to the imidazole moiety (as in 1a and 2a) impaired an optimal positioning of the compounds in the HA’s active site, while the same chain located at the opposite side on the scaffold (3a) did not influence the interaction with the target. Moreover, the ability of compounds 1–3 and 1a–3a to inhibit the growth of ER+ (MCF-7) and ER– (MDA-MB-231) cell lines, expressed by the Growth Inhibition activity (GI50) values, was also evaluated (Figures S1 and S2). No appreciable differences were observed in the sensitivity of the analyzed cell lines, and this could possibly be related to the previously reported comparable expression of aromatase in MCF-7 and MDA-MB-231 cells30 and the specificity of the compounds toward the target. Among the six tested compounds, the previously reported 1–3 showed modest antiproliferative activities, with GI50 values >10 μM. Regarding the new substituted compounds, the same range of potency was seen with compound 1a, while 2a displayed a GI50 < 10 μM in MCF-7 (although not in MDA-MB-231) cell lines, comparable to LTZ.20 This observation suggests that the introduction of the alkoxy chain led to an increase in antiproliferative activity for compounds 1a (only on MDA-MB-231 cells) and particularly for 2a (on both cell lines) with respect to parent compounds 1 and 2.
In contrast, 3a was not able to inhibit cell growth at any concentration considered in both cell models, and the peculiar behavior of this compound may be ascribed to the different positioning of the long and rigid alkoxy chain as compared to 1a and 2a, for which the chain is placed on the same phenyl ring of the imidazolyl substituent. We remark that the MDA-MB-231, ER- cell lines, are not sensitive to the decreased level of circulating estrogens; therefore, LTZ or the other AIs investigated must be less effective in this type of cell lines.
To dissect the molecular facets underlying the observed inhibitory potency of the compounds synthesized, we applied a sophisticated computational workflow, able to accurately describe the formation of the coordination bond between imidazole rings of the inhibitors and the iron atom of the heme moiety: (i) first, classical molecular dynamics (MD) simulations constraining the Fe–N bond distance were performed to relax the protein, and next, (ii) quantum mechanics/molecular mechanics (QM/MM) MD simulations were done to account for the coordination bond and the binding of the inhibitors to HA.31 Due to the high computational costs of this latter type of calculations, if the formation of the Fe–N coordination bond had not occurred within 5 ps of QM/MM MD simulation, we performed metadynamics (MTD) simulations, using as collective variable (CV) the Fe–N distance to trigger the formation of the coordination bond (see Methods section). The binding pose obtained in this manner was then further relaxed by unbiased 10 ps long QM/MM MD. For all investigated compounds, we observed the formation of stable coordination bonds between the imidazole rings and the heme moiety (Figure 5) with the exception of compound 3a. Indeed, for the latter the potential coordination bond, observed in the docking pose, was lost during the QM/MM MD simulation. We, next, accurately monitored the structural parameters obtained from QM/MM MD and we compared them with the clinically used LTZ drug, in order to identify structural traits that may provide a rationale to the observed experimental data (Table 1). Despite the different potency, all compounds exhibit similar Fe–N bond lengths, with 1a and 2a being slightly shorter (2.18 and 2.15 Å, respectively). As well, a similar trend could be observed for the average angle between the planes composed by the imidazole ring of the inhibitors and the heme, which were almost perfectly perpendicular. This evidence suggests that the coordination bond length and orientation of the azole group is not critical for the reported IC50s.
Figure 5.
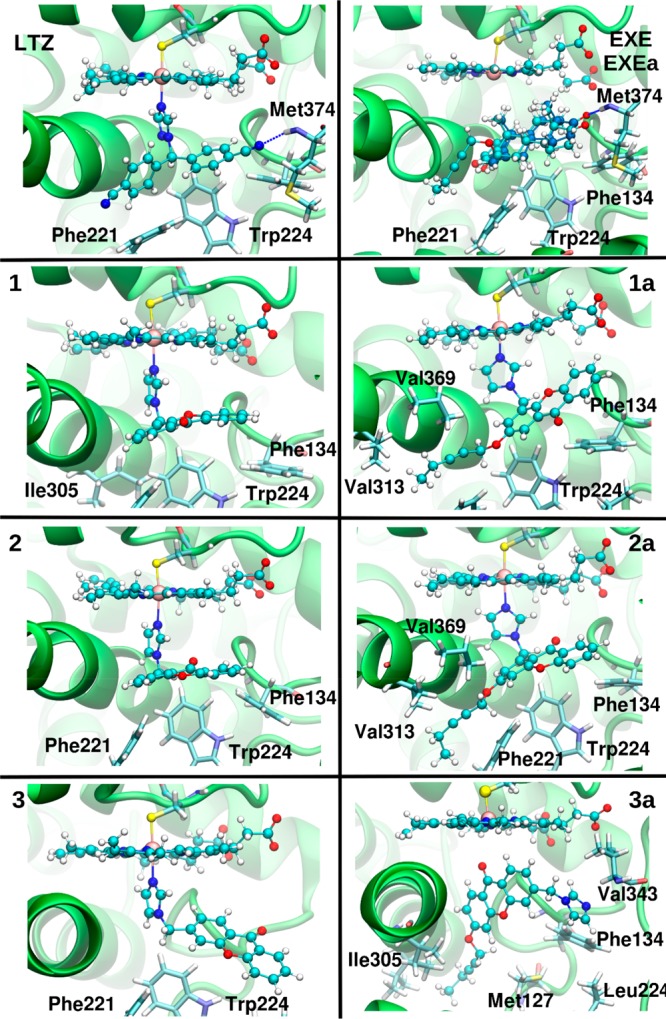
Representative structures as obtained from quantum-classical molecular dynamics simulations of letrozole (LTZ), exemestane (EXE) and its derivative (EXEa),21 and compounds 1–3 and 1a–3a. For comparison, the dual-mode steroidal inhibitor EXEa is also reported superimposed to EXE. The inhibitors and the heme moiety are shown in a ball and stick representation, while hydrophobic residues involved in stacking and hydrophobic interactions are highlighted as sticks. When present, hydrogen bonds are shown as dashed blue lines.
Surprisingly, the Fe–N distances calculated for 1a and 2a are similar to that of LTZ, suggesting that a perfect coordination geometry of the ligand to the heme iron is not the key element underlying its inhibitory activity. Nevertheless, LTZ, besides coordinating the heme iron, engages a persistent H-bond with Met374 and hydrophobic interactions with several residues located in the catalytic pocket (Phe221, Trp224, Phe132, Leu477, Val370, Leu372, and Ile133). These may both be critical elements to boost the LTZ potency. In spite of our refined computational protocol, we cannot exclude that the QM region, based on density functional theory, may experience some difficulties in describing the iron atom, thus not being able to fully account for the small differences observed between the low μM and nM range.25,31 Conversely, the steroidal inhibitor EXE lays in the active site showing a binding pose that perfectly resembles that of the endogenous substrate androstenedione. EXE H-bonds with Asp30932 and the backbone of Met374, similarly to LTZ (Figure 5). In this case, the addition of the hydrophobic tail does not alter the binding mode in the cavity and this protrusion snugly fits in the allosteric site (Figure 5).21 Aiming at rationalizing the differences in potency, we also monitored the effect of these inhibitors on the entry channels to the active site. While the apo enzyme is characterized by the presence of distinct ligand access routes (Figure S3),33,34 EXEa is able to perfectly fill ChII using its pentynyloxy side chain, triggering the closure of this channel. Apparently, for compound 1a, the hydrophobic tail does not completely fit into the channel and it only determines its rearrangement, leaving it partially open (Figure S3), possibly affecting the (residence-time) efficacy of the drug as well.
As a further element to explain the trend of potency, we also monitored the energetic costs of the active site rearrangements upon binding of the distinct drugs by considering the protein internal energies (Table 2) calculated via molecular mechanics generalized Born (MM-GBSA) on the QM/MM equilibrated trajectory. While EXE and its pentynyloxy-substituted derivative EXEa possess a similar deformation energy, compounds 1 and 3 show lower deformation values when compared to their derivatives 1a and 3a, suggesting that the presence of the tail imposes a strain in the orthosteric and allosteric sites.
Table 2. Energetic Costs of the Active Site Rearrangements upon Binding of the Distinct Drugs Considered as the Protein Internal Energies Obtained with the Molecular Generalized Born Surface Area (MM-GBSA) Method and Reported as the Sum of the Internal Bonds, Angles, and Dihedral Protein Contributions.
| Bond | Angles | Dihedrals | Total | |
|---|---|---|---|---|
| LTZ | 1740.3 ± 89.2 | 3166.5 ± 104.2 | 6802.4 ± 40.7 | 11709.2 ± 143.1 |
| EXE | 54.0 ± 8.4 | 3864.8 ± 43.3 | 5835.2 ± 27.3 | 9754.0 ± 51.9 |
| EXEa | 48.2 ± 0.8 | 3861.6 ± 43.3 | 5828.6 ± 29.6 | 9738.4 ± 52.5 |
| 1 | 2083.1 ± 47.2 | 3519.2 ± 50.7 | 6791.7 ± 36.9 | 12394.0 ± 78.5 |
| 1a | 2089.6 ± 39.6 | 3523.6 ± 35.6 | 6879.0 ± 37.9 | 12492.2 ± 65.4 |
| 2 | 1933.0 ± 30.2 | 3314.8 ± 52.8 | 6810.3 ± 40.9 | 12058.1 ± 73.3 |
| 2a | 1862.6 ± 32.3 | 3306.9 ± 41.8 | 6785.1 ± 34.3 | 11954.6 ± 63.0 |
| 3 | 1901.7 ± 33.1 | 3351.7 ± 46.3 | 6798.9 ± 31.6 | 12052.3 ± 65.1 |
| 3a | 1961.3 ± 31.7 | 3374.9 ± 41.2 | 6829.4 ± 26.3 | 12165.6 ± 58.3 |
Finally, to exclude that the increased size of the ligands may have hampered their binding to the active site and that their inhibitory activity may be ascribed to a nonactive site-directed inhibition, we did docking calculations, followed by classical MD simulations on the two allosteric pockets successfully exploited in our previous study.20 Only compound 3a was able to fit inside the HA access channel (Site 1, Figure S4), although suddenly dissociating after a few nanoseconds of MD simulation. In the case of the heme proximal cavity (Site 2), the putative binding site of CPR, compounds 1a–3a spontaneously dissociated after 50 ns of MD simulations (Figure S5).
Over the last four decades, aromatase enzyme has been a key target for the treatment and, more recently, prevention of ER+ BC. In this respect, although the release of the first HA crystal structure was a milestone for designing novel inhibitors, conventional docking methods experienced challenges to accurately describe the correct binding pose of AIs which form coordination bonds with the iron atom of the heme.2 This has heavily plagued research efforts in the rational design of a novel generation of AIs. This prompted the need of a higher level of theory to properly describe the coordination geometry, which however comes at high computational cost and long simulation time and can be applied to a limited number of molecules.26,35 We envision that in the future the use of the burgeoning deep learning techniques in drug design may readily overcome these challenges.
While awaiting this approach to revolutionize the field of metal-drug design, we have exploited here a complex computational protocol based on docking, classical and QM/MM biased and unbiased MD simulations to design and identify suitable binding poses of imidazolylmethylxanthones ligands’ derivatives able to inhibit HA targeting both the orthosteric and allosteric sites. In spite of the in silico prediction, the designed and synthesized compounds did not undergo a boost of potency, as revealed by in vitro enzymatic assays, being able to inhibit the enzyme in the low μM range. In contrast, the GI50 of 2a improved in cell-based studies suggesting the possible presence of an additional mechanism of action, involving possible off-target interactions, besides HA inhibition (Table S1).
A detailed structural analysis revealed that the addition of a pentynyloxy chain to imidazolylmethylxanthones, resulting in compound 1a and 2a, does not markedly affect the coordination geometry of the azole moiety to the iron, and the pentynyloxy chain fits within the access channel, prompting that the design of dual-mode nonsteroidal inhibitors coordinating the heme is very subtle. Stunningly, the coordination to heme iron is not pivotal to boost potency as also confirmed by compound 3a, which has an IC50 similar to 1a and 2a even though it is not coordinating the iron atom. By analyzing in detail the differences observed with more active compounds, we note that compounds 1a and 2a are unable to establish H-bond interaction with the active site, at variance of the third-generation AIs LTZ and EXE, and do not exploit at best the hydrophobic and π-stacking interactions. An energetic analysis also discloses that the pentynyloxy-substituted ligands 1a and 3a impose steric strain in the orthosteric and allosteric sites (Table 2) and that the designed compounds do not properly fit in the most favorable access channel to the active site.34 These data unequivocally unveil that the design of small-molecule HA inhibitors with potency in the nanomolar to low micromolar range depends on many different facts, among which the possibility of forming H-bonds and the deformation induced to the active site appear as the most relevant.
Besides rationalizing the critical structural elements underlying the activity of prototypical steroidal and nonsteroidal inhibitors, our outcomes provide critical guidelines for future development of next generation dual-mode inhibitors, maybe endowed with increased flexibility of the central core, that may become useful tools to exploit allosteric and orthosteric sites of HA and possibly of other CYP450s, involved in cancer progression, to modulate steroid metabolism.
Acknowledgments
AM thanks the Italian Association for Cancer Research (AIRC, MFAG Grant No 17134). AS was supported by a FIRC-AIRC “Mario e Valeria Rindi” fellowship for Italy. The authors gratefully acknowledge the computing time granted by the CINECA via the ISCRA B grant “TyrSwitch”.
Glossary
Abbreviations
- BC
breast cancer
- ER+
estrogen receptor positive
- ERα
estrogen receptor α
- SERM
selective estrogen receptor modulators
- SERD
selective estrogen receptor degraders
- CYP450
cytochrome P450
- HA
human aromatase
- AI
aromatase inhibitors
- LTZ
letrozole
- EXE
exemestane
- MD
molecular dynamics
- QM/MM
quantum mechanics/molecular mechanics
- MTD
metadynamics
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00591.
Experimental procedures for the syntheses and in vitro evaluation of compounds activities and simulation protocol and analysis; cellular proliferation assays, access/egress channels, binding pose, and representative structures; potential off-target interactions of the xanthone derivatives (PDF)
Author Contributions
⊥ JC and AS equally contributed to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- Feng Y.; Spezia M.; Huang S.; Yuan C.; Zeng Z.; Zhang L.; Ji X.; Liu W.; Huang B.; Luo W.; Liu B.; Lei Y.; Du S.; Vuppalapati A.; Luu H. H.; Haydon R. C.; He T. C.; Ren G. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. 10.1016/j.gendis.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinello A.; Ritacco I.; Magistrato A. Recent advances in computational design of potent aromatase inhibitors: open-eye on endocrine-resistant breast cancers. Expert Opin. Drug Discovery 2019, 14, 1065–1076. 10.1080/17460441.2019.1646245. [DOI] [PubMed] [Google Scholar]
- Lewis J. S.; Jordan V. C. Selective estrogen receptor modulators (SERMs): mechanisms of anticarcinogenesis and drug resistance. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2005, 591, 247–63. 10.1016/j.mrfmmm.2005.02.028. [DOI] [PubMed] [Google Scholar]
- McDonnell D. P.; Wardell S. E.; Norris J. D. Oral Selective Estrogen Receptor Downregulators (SERDs), a Breakthrough Endocrine Therapy for Breast Cancer. J. Med. Chem. 2015, 58, 4883–7. 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlin M.; Spinello A.; Pennati M.; Zaffaroni N.; Gobbi S.; Bisi A.; Colombo G.; Magistrato A. A Computational Assay of Estrogen Receptor alpha Antagonists Reveals the Key Common Structural Traits of Drugs Effectively Fighting Refractory Breast Cancers. Sci. Rep. 2018, 8, 649. 10.1038/s41598-017-17364-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D.; Lo J.; Egbuta C. Recent Progress in the Discovery of Next Generation Inhibitors of Aromatase from the Structure-Function Perspective. J. Med. Chem. 2016, 59, 5131–48. 10.1021/acs.jmedchem.5b01281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson E. R.; Mahendroo M. S.; Means G. D.; Kilgore M. W.; Hinshelwood M. M.; Graham-Lorence S.; Amarneh B.; Ito Y.; Fisher C. R.; Michael M. D.; et al. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr. Rev. 1994, 15, 342–55. 10.1210/edrv-15-3-342. [DOI] [PubMed] [Google Scholar]
- Spinello A.; Ritacco I.; Magistrato A. The Catalytic Mechanism of Steroidogenic Cytochromes P450 from All-Atom Simulations: Entwinement with Membrane Environment, Redox Partners, and Post-Transcriptional Regulation. Catalysts 2019, 9, 81. 10.3390/catal9010081. [DOI] [Google Scholar]
- Spinello A.; Pavlin M.; Casalino L.; Magistrato A. A Dehydrogenase Dual Hydrogen Abstraction Mechanism Promotes Estrogen Biosynthesis: Can We Expand the Functional Annotation of the Aromatase Enzyme?. Chem. - Eur. J. 2018, 24, 10840–10849. 10.1002/chem.201802025. [DOI] [PubMed] [Google Scholar]
- Ghosh D.; Griswold J.; Erman M.; Pangborn W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 2009, 457, 219–23. 10.1038/nature07614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgrignani J.; Magistrato A. Influence of the membrane lipophilic environment on the structure and on the substrate access/egress routes of the human aromatase enzyme. A computational study. J. Chem. Inf. Model. 2012, 52, 1595–606. 10.1021/ci300151h. [DOI] [PubMed] [Google Scholar]
- Sgrignani J.; Iannuzzi M.; Magistrato A. Role of Water in the Puzzling Mechanism of the Final Aromatization Step Promoted by the Human Aromatase Enzyme. Insights from QM/MM MD Simulations. J. Chem. Inf. Model. 2015, 55, 2218–2226. 10.1021/acs.jcim.5b00249. [DOI] [PubMed] [Google Scholar]
- Park J.; Czapla L.; Amaro R. E. Molecular simulations of aromatase reveal new insights into the mechanism of ligand binding. J. Chem. Inf. Model. 2013, 53, 2047–56. 10.1021/ci400225w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporuscio F.; Rastelli G.; Imbriano C.; Del Rio A. Structure-based design of potent aromatase inhibitors by high-throughput docking. J. Med. Chem. 2011, 54, 4006–17. 10.1021/jm2000689. [DOI] [PubMed] [Google Scholar]
- Favia A. D.; Nicolotti O.; Stefanachi A.; Leonetti F.; Carotti A. Computational methods for the design of potent aromatase inhibitors. Expert Opin. Drug Discovery 2013, 8, 395–409. 10.1517/17460441.2013.768983. [DOI] [PubMed] [Google Scholar]
- Sgrignani J.; Cavalli A.; Colombo G.; Magistrato A. Enzymatic and Inhibition Mechanism of Human Aromatase (CYP19A1) Enzyme. A Computational Perspective from QM/MM and Classical Molecular Dynamics Simulations. Mini-Rev. Med. Chem. 2016, 16, 1112–24. 10.2174/1389557516666160623101129. [DOI] [PubMed] [Google Scholar]
- Lu W. J.; Desta Z.; Flockhart D. A. Tamoxifen metabolites as active inhibitors of aromatase in the treatment of breast cancer. Breast Cancer Res. Treat. 2012, 131, 473–81. 10.1007/s10549-011-1428-z. [DOI] [PubMed] [Google Scholar]
- Egbuta C.; Lo J.; Ghosh D. Mechanism of inhibition of estrogen biosynthesis by azole fungicides. Endocrinology 2014, 155, 4622–8. 10.1210/en.2014-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgrignani J.; Bon M.; Colombo G.; Magistrato A. Computational approaches elucidate the allosteric mechanism of human aromatase inhibition: a novel possible route to Small-molecule regulation of CYP450s activities?. J. Chem. Inf. Model. 2014, 54, 2856–68. 10.1021/ci500425y. [DOI] [PubMed] [Google Scholar]
- Spinello A.; Martini S.; Berti F.; Pennati M.; Pavlin M.; Sgrignani J.; Grazioso G.; Colombo G.; Zaffaroni N.; Magistrato A. Rational design of allosteric modulators of the aromatase enzyme: An unprecedented therapeutic strategy to fight breast cancer. Eur. J. Med. Chem. 2019, 168, 253–262. 10.1016/j.ejmech.2019.02.045. [DOI] [PubMed] [Google Scholar]
- Ghosh D.; Lo J.; Morton D.; Valette D.; Xi J.; Griswold J.; Hubbell S.; Egbuta C.; Jiang W.; An J.; Davies H. M. Novel aromatase inhibitors by structure-guided design. J. Med. Chem. 2012, 55, 8464–76. 10.1021/jm300930n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roleira F. M. F.; Varela C.; Amaral C.; Costa S. C.; Correia-da-Silva G.; Moraca F.; Costa G.; Alcaro S.; Teixeira N. A. A.; Tavares da Silva E. J. C-6alpha- vs C-7alpha-Substituted Steroidal Aromatase Inhibitors: Which Is Better? Synthesis, Biochemical Evaluation, Docking Studies, and Structure-Activity Relationships. J. Med. Chem. 2019, 62, 3636–3657. 10.1021/acs.jmedchem.9b00157. [DOI] [PubMed] [Google Scholar]
- Gobbi S.; Zimmer C.; Belluti F.; Rampa A.; Hartmann R. W.; Recanatini M.; Bisi A. Novel highly potent and selective nonsteroidal aromatase inhibitors: synthesis, biological evaluation and structure-activity relationships investigation. J. Med. Chem. 2010, 53, 5347–51. 10.1021/jm100319h. [DOI] [PubMed] [Google Scholar]
- Verras A.; Kuntz I. D.; Ortiz de Montellano P. R. Computer-assisted design of selective imidazole inhibitors for cytochrome p450 enzymes. J. Med. Chem. 2004, 47, 3572–9. 10.1021/jm030608t. [DOI] [PubMed] [Google Scholar]
- Sgrignani J.; Casalino L.; Doro F.; Spinello A.; Magistrato A. Can multiscale simulations unravel the function of metallo-enzymes to improve knowledge-based drug discovery?. Future Med. Chem. 2019, 11, 771–791. 10.4155/fmc-2018-0495. [DOI] [PubMed] [Google Scholar]
- Spinello A.; Magistrato A. An omics perspective to the molecular mechanisms of anticancer metallo-drugs in the computational microscope era. Expert Opin. Drug Discovery 2017, 12, 813–825. 10.1080/17460441.2017.1340272. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–53. 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Minami N.; Kijima S. Reduction of Ortho-Hydroxyaromatic Carboxylic-Acids through Ethoxy-Carbonyl Derivatives with Sodium-Borohydride. Chem. Pharm. Bull. 1979, 27, 816–820. 10.1248/cpb.27.816. [DOI] [Google Scholar]
- Gobbi S.; Hu Q.; Zimmer C.; Belluti F.; Rampa A.; Hartmann R. W.; Bisi A. Drifting of heme-coordinating group in imidazolylmethylxanthones leading to improved selective inhibition of CYP11B1. Eur. J. Med. Chem. 2017, 139, 60–67. 10.1016/j.ejmech.2017.07.078. [DOI] [PubMed] [Google Scholar]
- Hevir N.; Trost N.; Debeljak N.; Rizner T. L. Expression of estrogen and progesterone receptors and estrogen metabolizing enzymes in different breast cancer cell lines. Chem.-Biol. Interact. 2011, 191, 206–16. 10.1016/j.cbi.2010.12.013. [DOI] [PubMed] [Google Scholar]
- Vidossich P.; Magistrato A. QM/MM molecular dynamics studies of metal binding proteins. Biomolecules 2014, 4, 616–45. 10.3390/biom4030616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nardo G.; Breitner M.; Bandino A.; Ghosh D.; Jennings G. K.; Hackett J. C.; Gilardi G. Evidence for an elevated aspartate pK(a) in the active site of human aromatase. J. Biol. Chem. 2015, 290, 1186–96. 10.1074/jbc.M114.595108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magistrato A.; Sgrignani J.; Krause R.; Cavalli A. Single or Multiple Access Channels to the CYP450s Active Site? An Answer from Free Energy Simulations of the Human Aromatase Enzyme. J. Phys. Chem. Lett. 2017, 8, 2036–2042. 10.1021/acs.jpclett.7b00697. [DOI] [PubMed] [Google Scholar]
- Ritacco I.; Saltalamacchia A.; Spinello A.; Ippoliti E.; Magistrato A. All-Atom Simulations Disclose How Cytochrome Reductase Reshapes the Substrate Access/Egress Routes of Its Partner CYP450s. J. Phys. Chem. Lett. 2020, 11, 1189–1193. 10.1021/acs.jpclett.9b03798. [DOI] [PubMed] [Google Scholar]
- Sgrignani J.; Magistrato A. First-principles modeling of biological systems and structure-based drug-design. Curr. Comput.-Aided Drug Des. 2013, 9, 15–34. 10.2174/1573409911309010003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.