

Abstract
Sphingosine 1-phosphate (S1P) is a bioactive lipid mediator associated with diverse homeostatic and signaling roles. Enhanced biosynthesis of S1P, mediated by the sphingosine kinase isozymes (SK1 and SK2), is implicated in several pathophysiological conditions and diseases, including skeletal muscle fibrosis, inflammation, multiple sclerosis, and cancer. Therefore, therapeutic approaches that control S1P production have focused on the development of SK1/2 inhibitors. In this framework, we designed a series of natural monosaccharide-based compounds to enhance anchoring of the known SK1 inhibitor PF-543 at the polar head of the J-shaped substrate-binding channel. Herein, we describe the structure-based design and synthesis of new glycan-containing PF-543 analogues and we demonstrate their efficiency in a TGFβ1-induced pro-fibrotic assay.
Keywords: Sphingosine 1-phosphate, skeletal muscle fibrosis, glycan derivatives, molecular modeling, sphingosine kinase 1
Sphingosine 1-phosphate (S1P, Figure 1) has been considered for a long time merely an ultimate metabolite of sphingolipid breakdown. However, the identification of the “inside-out signaling” mechanism associated with S1P production, release, and activity, allowed us to spotlight its key role as a bioactive signaling molecule which regulates many physiological processes as well as pathological ones.1,2 Intracellular S1P levels are tightly regulated by the crucial balance between its synthesis, mediated by two sphingosine kinase isozymes (SK1 and SK2), and reversible and irreversible degradation catalyzed by S1P phosphatases (SPP1 and SPP2) and S1P lyase, respectively.3 Specifically, SK1 and SK2 are expressed inside the cells where they show different subcellular localization that endows them with sometimes overlapping otherwise distinct biological effects. SKs catalyze ATP-dependent phosphorylation of sphingosine (Sph, Figure 1) inside the cells, and then S1P, besides acting as an intracellular mediator, can be exported in the extracellular milieu by means of unspecific ABC family members and specific spinster homologue 2 (SPNS2) transporters, where it interacts with its five specific GPCR receptors designated as S1PR1–5.4 Thus, SKs are key modulators of the dynamic S1P/sphingosine/ceramide ratio (sphingolipid rheostat model), which in turn affects cellular fate and the balance between cell survival and apoptosis.5
Figure 1.
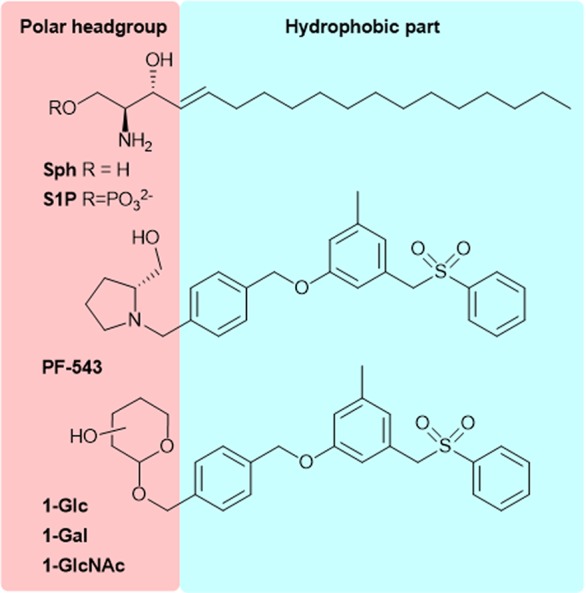
Structures of sphingosine (Sph), sphingosine 1-phosphate (S1P), PF-543, and the new glycoconjugates 1-Glc, 1-Gal, and 1-GlcNAc.
The discovery of a link between dysregulation of S1P levels and the onset and progression of several pathological settings (e.g., cancer, autoimmune diseases, atherosclerosis, fibrosis) spotlighted SKs as potential targets for drug discovery.6,7 In this scenario, we focused on the role of the SK1/S1P3 axis in the onset of skeletal muscle fibrosis, the hallmark of musculoskeletal myopathies. Experimental data showed that the TGFβ1 cytokine-mediated up-regulation of SK1 is responsible for the induction of the fibrotic program in myoblasts.8 In addition, the remodeling of the S1PR expression pattern occurs and it results in the increased expression of S1P3. Indeed, pharmacological as well as RNA interference approaches targeting SK1 or S1P3 dramatically altered the profibrotic effect induced by TGF1β.8 Moreover, S1P signaling has been recognized as a new drug target in relapsing-remitting multiple sclerosis. In this framework, a number of SK1 inhibitors have been developed to date.9−15 However, most of them are still limited by low potency and/or specificity, indicating the need for more performant SK1 inhibitors. Two possible strategies have recently emerged: (i) the targeting of the less conserved SK1 allosteric sites, in order to increase inhibitor affinity and selectivity;9 (ii) the integration of information from cocrystal structures of SK1 with known validated inhibitors14,15 into pharmacophoric models, with the aim to develop more potent inhibitors.
Conjugation of bioactive molecules with carbohydrates has emerged as a straightforward and promising approach to develop glycohybrids as a source of new drug candidates.16−26 The glycoconjugation approach has been exploited for a wide range of structurally diverse drugs ranging from small organic molecules up to, more recently, macromolecules (e.g., proteins and antibodies).16,17 This “sweet” functionalization endows researchers to exploit new opportunities to improve both the targeting21,23,26 and the pharmacokinetic properties of hydrophobic drugs.18,19 In particular, glycosylation has been exploited as a promising strategy to improve the half-life of therapeutic peptides and proteins (such as somatostatin and opioid drugs, to mention some of them)18−20 by inducing changes in their conformational structure and consequently in their physiochemical properties. Moreover, the introduction of a glycan moiety has been exploited both to enforce specific tumor-targeting of cytotoxic drugs21 and to improve the related aqueous solubility of validated proteinases inhibitors.24,25 The growing scientific interest in this field has inspired us to investigate the use of glycans as the polar headgroup of SK1 inhibitors. To this end, we selected one of the most potent and selective SK1 inhibitor reported so far,14PF-543, a nonlipid compound bearing 2-hydroxymethylpyrrolidine as a ring-constrained isostere of the aminopropanediol group of the natural substrate (Figure 1). Specifically, it was demonstrated that the pyrrolidine ring provides hydrogen-bonding interactions at the upper part of the cleft of the J-shaped pocket of the enzyme. In this work, we replaced the 2-hydroxymethylpyrrolidine moiety of PF-543 by selected monosaccharide units (i.e., d-glucose, N-acetyl-d-glucosamine, and d-galactose, Figure 1), with the aim to enhance the anchoring of the inhibitor at the J-shaped pocked of the enzyme by means of additional interactions provided by the sugar moiety. Herein, we describe the structure-based design and synthesis of monosaccharide-bearing PF-543 analogue SK1 inhibitors, and we provide evidence of their efficiency using a model of TGFβ1-induced skeletal muscle fibrosis.
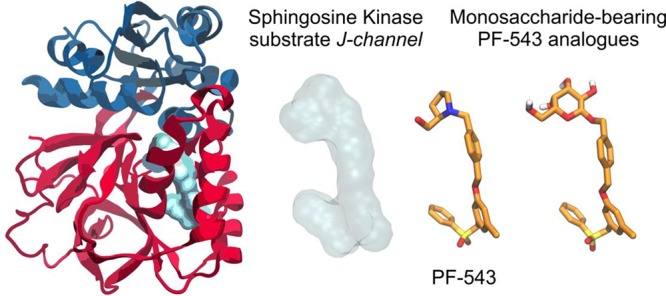
The design of the monosaccharide-bearing PF-543 analogues was based on the high-resolution crystal structure of SK1 in complex with the inhibitor PF-543.27 SK1 structure consists of an N-terminal domain that binds ATP and a C-terminal domain that harbors the substrate-binding site (Figure 2A). The inhibitor bound inside the highly hydrophobic Sph-binding cavity, termed the “J-channel” due to its shape, resembles the bent conformation of a lipid substrate (Figure 2B). The hydroxymethyl-pyrrolidine moiety of PF-543 occupies the position of a lipid’s headgroup next to the γ-phosphate of ATP,28 and it is anchored by two hydrogen bonds to the carboxylate group of Asp264. Remarkably, no other polar group of PF-543 was found to participate in hydrogen-bonding interactions within the J-channel.27 Still, the tight packing of the inhibitor at the hydrophobic pocket occupied by the terminal phenyl group (J-channel toe) and the placement of the sulfonylmethylene group at the heel of the J-channel (Figure 2B) has been proposed to confer the selectivity of PF-543 for SK1 (IC50 = 25 nM) versus SK2 (IC50 > 5,000 nM).11 Now, considering that a glycan moiety at the head of the J-channel would offer the potential for additional polar interactions, we retained the hydrophobic moiety of PF-543, which could be fine-tuned at a later stage to modulate the selectivity profile for SK1/SK2, as demonstrated recently.11
Figure 2.

(A) Structure of SK1 with bound ADP and the inhibitor PF-543 inside the lipid binding channel. SK1 and PF-543 are taken from the crystal structure with PDB ID 4V24(27) and the nucleotide by superimposing with PDB ID 3VZD.28 (B) Close-up view of the Sph-binding site, “J-channel”, illustrating the nonpolar residues (white stick-and-balls) that comprise the foot of the J-channel and the polar residues (green C, blue N, red O) around the polar head of the J-channel. PF-543 is shown with orange C sticks, and dashed lines indicate the hydrogen bonding interactions with Asp264 (SK1 isoform 2 numbering as in PDB 4V24). Isoform-specific residue changes at the “toe” and “heel” of the J-channel have been proposed to influence inhibitor selectivity for SK1 versus SK2. (C–E) Bound models of the designed analogues 1-Glc (C), 1-GlcNAc (D), and 1-Gal (E) inside the J-channel of SK1. The translucent surfaces indicate the space occupied by PF-543, whereas polar residues at the head of the J-channel are shown with ball-and-sticks. Hydrogen bonds are indicated by dashed lines with the heavy-atom distance in Å, whereas putative interactions within 3.5 Å are shown with dotted lines.
Indeed, our molecular docking calculations suggest that a β-d-glucopyranose moiety can be accommodated inside the Sph-binding site of SK1 without affecting the conformation of the hydrophobic moiety of PF-543 at the J-channel (Figure 2C). The selected bound conformation of 1-Glc exhibits two hydrogen bonds between 6-OH and the backbone NH groups of Asn200 and Ala201. In addition, 2-OH of 1-Glc is within hydrogen-bonding distance from the side-chain of Asp264, albeit the geometry is not ideal (Figure 2C). This is also the case for the 6-OH of 1-Glc and the distal Glu275, as well as the 4-OH of 1-Glc and the carboxylate group of Asp167, which is located at the N-terminal domain of SK1. The N-acetyl glucosamine derivative 1-GlcNAc is predicted to interact with SK1 via hydrogen bonds of its amide NH with the carboxylate group of Asp264 and its acetyl C=O with the hydroxyl group of Ser254 (Figure 2D). Notably, Ser254 has been shown to interact with the polar head of the substrate28 but not with PF-543.27 An additional H-bond could be formed by the 6-OH of 1-GlcNAc and the amide group of Ala201, including potential interactions with the side-chains of Asp167 and Glu275, similar to 1-Glc. With the aim to investigate the role of the sugar stereochemistry at position C-4, we also employed β-d-galactose as a polar head. Our calculations suggest that the 6-OH and 4-OH of 1-Gal could be engaged in hydrogen bonding interactions with the hydroxyl group of Ser254 and the amide NH group of Gly428 (Figure 2E). The 3-OH group of 1-Gal is also within hydrogen bonding distance from the side-chain of Asp167; however, at this bound pose there is no potential for direct interaction with Ala201, Asp264, or Glu275. Taken together, our analysis indicates that our design strategy is expected to yield analogues bearing the hydrophobic moiety of PF-543 that can be well accommodated within the J-channel of SK1. The introduction of a sugar moiety at the J-channel head endows the potential for additional hydrogen-bonding interactions with polar residues of SK1.
Compounds 1-Glc, 1-Gal, and 1-GlcNAc were synthesized following a convergent approach (Scheme 1). The hydrophobic scaffold 3 was prepared through an optimized procedure29 starting from commercially available 3-(bromomethyl)-5-methylphenyl acetate 2 (see SI). Compound 3 was then used as the acceptor in the glycosylation reactions to give the target compounds 1 (Scheme 1). In particular, the glucosyl 1-Glc and galactosyl 1-Gal derivatives were obtained through a glycosylation reaction between the per-pivaloylated glycosyl trichloroacetimidate donors (430 and 5,31 respectively) and acceptor 3, followed by acyl deprotection through Zèmplen transesterification. In both cases the glycosylation reactions proceed in high yield and complete stereoselectivity, and compounds 6a and 6b were recovered as pure β-glycosides (95% and 97% yield, respectively). A different strategy was used to prepare the N-acetyl d-glucosamine (GlcNAc) derivative 1-GlcNAc (Scheme 1). In this case, commercially available d-glucosamine penta-acetate 7 was reacted at high temperature with glycosyl acceptor 3 using ytterbium triflate as the catalyst to give 6c in satisfactory yield.32 Zemplén deacetylation of 6c with sodium methoxide in methanol gave efficiently (78%) the GlcNAc containing inhibitor 1-GlcNAc.
Scheme 1. Synthesis of 1-Glc, 1-Gal, 1-GlcNAc, and Ac-1-Glc.

TGF-β is involved in the regulation of skeletal muscle fibrosis as it was shown to be a potent inducer of myoblasts into myofibroblasts transdifferentiation,33 with a molecular mechanism relying on cytokine-mediated SK1 activation. On this basis, the ability of the new glycoconjugates to interfere with TGFβ1-induced fibrosis in C2C12 myoblasts was evaluated. PF-543 was used as reference control in this study at the concentration of 10 μM according with data in the literature.34−36 Cells were incubated with 10 μM of the inhibitors (e.g. PF-543, 1-Gal, 1-GlcNAc, and 1-Glc) for 30 min before the addition of the 5.0 ng/mL solution of TGFβ1. The expression of protein markers that correlate with muscle fibrosis was evaluated by Western blot analysis using specific anti-α-SMA and anti-SMα22. Total cell lysates were subjected to Western blot analysis to measure the relative content of fibrosis markers. In agreement with previous data,8 48 h TGFβ1 treatment induced the expression of fibrosis markers (Figure 3). Subsequently, treatment with PF-543 (10 μM) resulted in abrogation of the α-SMA expression induced by TGFβ1 whereas the expression of SMα22 was significantly affected (Figure 3). The three glycoconjugates (1-Glc, 1-Gal and 1-GlcNAc) displayed a variable effect according with the glycan residue (Figure 3). Both the α-SMA and SMα22 expression induced by TGFβ1 were not affected by 1-Gal, while 1-GlcNAc displayed a partial inhibition of α-SMA expression. Notably, 1-Glc significantly affected both the α-SMA and SMα22 expression induced by TGFβ1, an effect comparable to that displayed by PF-543 (Figure 3).
Figure 3.

Effect of glycoconjugates 1-Glc, 1-Gal, and 1-GlcNAc compared to PF-543 on TGFβ1-induced fibrosis marker expression in murine myoblasts. C2C12 myoblasts were seeded to 80% confluence and challenged with 5.0 ng/mL TGFβ1, PF-543, 1-Gal, 1-GalNAc, and 1-Glc (30 min, 10 μM). (A) The expression of protein markers which expression correlates with muscle fibrosis, has been evaluated by Western blot analysis, using specific anti-α-SMA and anti-SMα22. A representative blot is shown among three independent experiments with analogous results. (B and C) Densitometric analysis was performed of three independent experiments, and data, normalized to β-actin expression, were reported as mean ± SD of fold change above control untreated. The effect of TGFβ1 on fibrosis marker expression was significant by student t test. **P < 0.01. ***P < 0.001. The effect of pharmacological inhibition of SK1 on TGFβ1 induced fibrosis marker expression was significant by two-way ANOVA followed by Bonferroni posthoc test. #P < 0.05, ##P < 0.01, ###P < 0.001.
Considering that acetylation of the hydroxyl groups of glycans has been proved to be an efficient approach to promote transit of glycan containing molecules across the plasma membrane,37 we also sought to investigate the activity of the per-acetylated derivative of 1-Glc, Ac-1-Glc (Scheme 1). Therefore, a dose dependence of 1-Glc and Ac-1-Glc compared to the parental drug PF-543 on TGFβ1-induced fibrosis of murine myoblasts was performed in order to define the bioactive concentration. Western blot data (see SI, Figure S1) confirmed that 10 μM resulted in the most effective concentration on affecting the expression of both α-SMA and SMα22. Then, Western blot analysis was performed to detect the relative expression of fibrosis markers induced by TGFβ1 after 30 min and 2 h treatments with 1-Glc, Ac-1-Glc, and PF-543 as control (Figure 4). The TGFβ1-induced α-SMA expression was completely abrogated by Ac-1-Glc after 30 min, whereas the expression of SMα22 was significantly decreased after 2 h treatment (Figure 4). Notably, the effect of pharmacological inhibition of TGFβ1 induced α-SMA expression after 30 min Ac-1-Glc treatment was significantly higher compared to 1-Glc (two-way ANOVA followed by Bonferroni posthoc test, §§P < 0.01, Figure 4), supporting our rationale.
Figure 4.

Effect of 1-Glc and Ac-1-Glc compared to PF-543 in TGFβ1-induced fibrosis marker expression in murine myoblasts. C2C12 myoblasts were seeded to 80% confluence and treated with 5.0 ng/mL TGFβ1, PF-543, 1-Glc, and Ac-1-Glc (30 min, 120 min). (A) The expression of protein markers, which expression correlates with muscle fibrosis, has been evaluated by Western blot analysis, using specific anti-α-SMA and anti-SMα22. A representative blot is shown among three independent experiments with analogous results. (B and C) Densitometric analysis was performed of three independent experiments and data, normalized to β-actin expression, were reported as mean ± SD of fold change above control untreated. The effect of pharmacological inhibition of SK1 on TGFβ1 induced fibrosis marker expression was significant by two-way ANOVA followed by Bonferroni posthoc test. #P < 0.05, ##P < 0.01, ###P < 0.001. The effect of pharmacological inhibition of SK1 by 30 min Ac-1-Glc compared to 1-Glc on TGFβ1 induced αSMA expression was significant by two-way ANOVA followed by Bonferroni posthoc test. §§P < 0.01.
Laser scanning confocal microscopy was employed to outline the distribution of fibrosis markers that are functional cytoskeletal proteins, whose expression can be analyzed to follow the onset and progression of skeletal muscle fibrosis. Confocal immunofluorescence analysis revealed an enhanced expression of αSMA in C2C12 cells treated with 5.0 ng/mL TGFβ1 for 48 h, which resulted mainly localized within F-actin microfilaments (Figure 5). In agreement with previous results, the treatment with PF-543 as well as 1-Glc and Ac-1-Glc (10 μM) completely prevented the cytokine-induced expression of αSMA (Figure 5).
Figure 5.
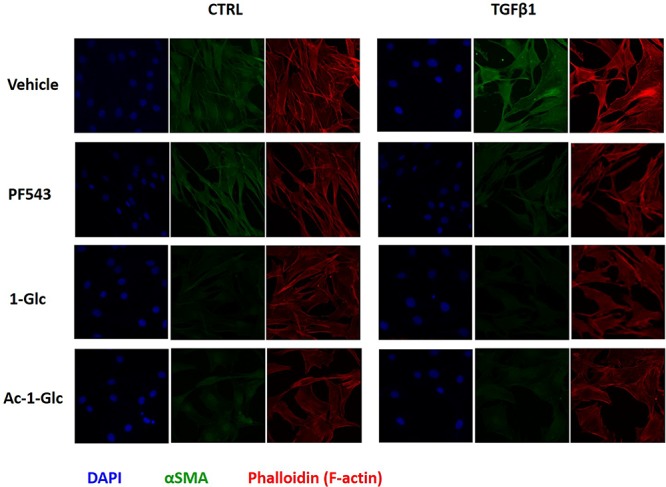
Confocal immunofluorescence analysis was performed in 24 h serum-starved C2C12 cells that were treated with 5.0 ng/mL TGFβ1, after a 2 h of incubation with PF-543, 1-Glc, and Ac-1-Glc. Anti-αSMA primary antibody and fluorescein-conjugated antimouse secondary antibody were used. DAPI was added to label nuclei and rhodamine-phalloidin as high-affinity F-actin probe in order to trace cytoskeletal microfilaments. The immunofluorescence images shown are representative of six fields for each condition.
It has been extensively demonstrated that TGF-β family members are implicated in the regulation of myogenic differentiation as they were shown to be potent inhibitors of myoblasts differentiation. Exogenous TGFβ1 was shown to prevent features of skeletal muscle differentiation, such as fusion and myotube formation, in different cultured cell lines in vitro,38,39 and the molecular mechanism at least in part relies on SK1 activation by the cytokine.8 For this reason, 1-Glc and Ac-1-Glc conjugates were assayed for the effect on TGFβ1-mediated myoblast differentiation compared to PF-543. For this purpose, Western blot analysis was performed to measure the relative expression of caveolin-3 (Cav-3) by TGFβ1 after 30 min, 1 h, and 2 h incubation with PF-543, 1-Glc, and Ac-1-Glc glycoconjugates. Data (see SI, Figure S2) showed that, as expected, TGFβ1 decreases skeletal muscle differentiation whereas this effect was significantly reverted by both 1-Glc and Ac-1-Glc and it was almost completely abolished at 2 h of incubation.
Taken together our findings suggest that the glucose-bearing derivative and in particular its acetylated form Ac-1-Glc is the most active compound, as it completely abrogates the TGFβ1 induced expression of a key fibrosis marker. Although additional structural and biochemical information is required, we can hypothesize that the lower potency of 1-Gal compared to 1-Glc and 1-GlcNAc is due to the lack of interaction with the side-chain of Asp264 (Figure 2C–E), a key residue that has been shown to provide the only hydrogen-bonding interactions of PF-543 within the J-channel of SK1. In conclusion, our work demonstrates that the use of natural monosaccharides as the polar headgroup of known SK inhibitors offers the potential to develop a new class of signaling molecules that regulate the expression of fibrosis markers. The biological effects of the new PF-543 analogues on TGFβ1-profibrotic effects in myoblasts suggest a possible interaction and inhibition of the catalytic site of SK1. However, further studies (e.g., binding data and further biochemical and pharmacological characterizations) are needed to corroborate this hypothesis. Finally, considering the sugar heterogeneity in terms of stereochemistry and availability of functional groups, our findings suggest that glycans can be employed as an additional tool to explore the pharmacological profile of modulators of the sphingolipid rheostat model.
Experimental Section
General
Standard laboratory procedures were followed to carry out the reactions and to prepare dry solvents. Optical rotations were measured with a PerkinElmer 241 polarimeter at 20 °C. 1H and 13C NMR spectra were recorded with a Bruker AVANCE-500 spectrometer at a sample temperature of 298 K. High-resolution mass spectra were collected by electrospray ionization (ESI) spectroscopy on a QTof SYNAPT G2Si Mass Spectrometer.
4-[(3-Methyl-5-(phenylsulfonylmethyl)-phenoxy)methyl]-phenylmethyl β-d-Glucopyranoside (1-Glc)
To a stirred solution of compound 6a (0.25 g, 0.028 mmol) in DCM (0.5 mL) was added sodium methoxide in methanol (0.1 M solution, 0.56 mL). The reaction was stirred at room temperature, and further portions of sodium methoxide (0.1 M solution, 0.30 mL) were added every 12 h. After 24 h the reaction mixture was neutralized with an ion-exchange resin (Dowex 50 × 8, H+ form), filtered, and concentrated. The crude product was subjected to flash chromatography (DCM/MeOH, 9:1) to give pure 1-Glc (0.012 g, 80%) as an amorphous white solid. [α]d20 = −32.0 (c = 0.5 in chloroform). 1H NMR (CDCl3/CD3OD, 2:1): δ = 7.68–7.62 (m, 3H, arom.), 7.53–7.47 (m, 2H, arom.), 7.44–7.39 (m, 2H, arom.), 7.37–7.33 (m, 2H, arom.), 6.74 (br s, 1H, arom.), 6.49 (br s, 1H, arom.), 6.48 (br s, 1H, arom.), 4.96–4.92 (m, 3H, OCHaHb and CH2SO2), 4.68 (d, 1H, J = 11.8 Hz, OCHaHb), 4.38 (d, 1H, J1,2 = 7.7 Hz, H-1), 4.28 (s, 2H, PhCH2O), 3.88 (dd, 1H, J5,6a = 2.7 Hz, J6a,6b = 12.0 Hz, H-6a), 3.74 (dd, 1H, J5,6b = 5.3 Hz, J6a,6b = 12.0 Hz, H-6b), 3.43–3.35 (m, 2H, H-3,4), 3.35–3.26 (m, 2H, H-2,5), 2.20 (s, 3H, CH3). 13C NMR (CDCl3/CD3OD, 2:1): δ = 158.6, 139.7, 137.6, 137.2, 136.4, 133.8, 128.9–127.4 (9C), 124.4, 116.4, 114.0, 102.0 (C-1), 76.6, 76.2 (C-5), 73.6 (C-2), 70.6 (OCH2), 70.3, 69.6 (CH2SO2), 62.6 (PhCH2O), 61.7 (C-6), 20.9 (CH3). HRMS (ESI): m/z calcd for C28H32O9NaS 567.1665 [M + Na]+, found 567.1665.
Acknowledgments
F.Co. and L.M. thank “Centro Interdipartimentale Grandi Apparecchiature (C.I.G.A.)” for HRMS analyses.
Glossary
Abbreviations
- Glc
d-glucose
- Gal
d-galactose
- GlcNAc
N-acetyl-d-glucosamine
- Sph
sphingosine
- S1P
sphingosine 1-phosphate
- SK
sphingosine kinase
- SPNS2
specific spinster homologue 2
- SPP
sphingosine 1-phosphate phosphatases
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00665.
Figures S1 and S2. Synthetic procedures for the synthesis of compounds 3, 6a–c, 1-Gal, 1-GlcNAc, and Ac-1-Glc. Computational and biological methods. 1H and 13C spectra of new compounds. (PDF)
Coordinate file of the SK1 bound with 1-Glc model (PDB)
Coordinate file of the SK1 bound with 1-GlcNAc model (PDB)
Coordinate file of the SK1 bound with 1-Gal model (PDB)
Author Contributions
⊥ A.P. and F.Ce. contributed equally to this work.
Author Contributions
# F.Co. and B.R. are colast and cocorresponding authors.
We thank Fondazione Cassa di Risparmio di Firenze (project #24081: Sviluppo di modulatori dei recettori per la sfingosina 1-fosfato: verso il controllo dell’atrofia muscolo scheletrica). B.R., F.C., J.T., and L.M. thank COST Action CA18013. B.R., J.T., F.Ce. and P.B. thank MIUR-Italy (“Progetto Dipartimenti di Eccellenza 2018–2022” allocated to Department of Chemistry “Ugo Schiff” and to the Department of Experimental and Clinical Biomedical Sciences “Mario Serio”).
The authors declare no competing financial interest.
Supplementary Material
References
- Takabe K.; Paugh S. W.; Milstien S.; Spiegel S. ″Inside-out″ signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol. Rev. 2008, 60 (2), 181–195. 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier A.; Hla T.. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366 ( (6463), ), eaar5551. 10.1126/science.aar5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiazek M.; Chacinska M.; Chabowski A.; Baranowski M. Sources, metabolism, and regulation of circulating sphingosine-1-phosphate. J. Lipid Res. 2015, 56 (7), 1271–1281. 10.1194/jlr.R059543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H.; Goetzl E. J. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat. Rev. Immunol. 2005, 5 (7), 560–570. 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- Newton J.; Lima S.; Maceyka M.; Spiegel S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333 (2), 195–200. 10.1016/j.yexcr.2015.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukocheva O. A.; Furuya H.; Ng M. L.; Friedemann M.; Menschikowski M.; Tarasov V. V.; Chubarev V. N.; Klochkov S. G.; Neganova M. E.; Mangoni A. A.; Aliev G.; Bishayee A. Sphingosine kinase and sphingosine-1-phosphate receptor signaling pathway in inflammatory gastrointestinal disease and cancers: A novel therapeutic target. Pharmacol. Ther. 2020, 207, 107464. 10.1016/j.pharmthera.2019.107464. [DOI] [PubMed] [Google Scholar]
- Khan F. I.; Lai D.; Anwer R.; Azim I.; Khan M. K. A. Identifying novel sphingosine kinase 1 inhibitors as therapeutics against breast cancer. J. Enzyme Inhib. Med. Chem. 2020, 35, 172–186. 10.1080/14756366.2019.1692828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cencetti F.; Bernacchioni C.; Nincheri P.; Donati C.; Bruni P. Transforming growth factor-beta1 induces transdifferentiation of myoblasts into myofibroblasts via up-regulation of sphingosine kinase-1/S1P3 axis. Mol. Biol. Cell 2010, 21 (6), 1111–1124. 10.1091/mbc.e09-09-0812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar O.; Ozkirimli E.; Ulgen K. Sphingosine kinase 1 (SK1) allosteric inhibitors that target the dimerization site. Comput. Biol. Chem. 2017, 69, 64–76. 10.1016/j.compbiolchem.2017.05.006. [DOI] [PubMed] [Google Scholar]
- Cao M.; Ji C.; Zhou Y.; Huang W.; Ni W.; Tong X.; Wei J. F. Sphingosine kinase inhibitors: A patent review. Int. J. Mol. Med. 2018, 41, 2450–2460. 10.3892/ijmm.2018.3505. [DOI] [PubMed] [Google Scholar]
- Adams D. R.; Tawati S.; Berretta G.; Rivas P. L.; Baiget J.; Jiang Z.; Alsfouk A.; Mackay S. P.; Pyne N. J.; Pyne S. Topographical Mapping of Isoform-Selectivity Determinants for J-Channel-Binding Inhibitors of Sphingosine Kinases 1 and 2. J. Med. Chem. 2019, 62, 3658–3676. 10.1021/acs.jmedchem.9b00162. [DOI] [PubMed] [Google Scholar]
- Magli E.; Corvino A.; Fiorino F.; Frecentese F.; Perissutti E.; Saccone I.; Santagada V.; Caliendo G.; Severino B. Design of Sphingosine Kinases Inhibitors: Challenges and Recent Developments. Curr. Pharm. Des. 2019, 25, 956–968. 10.2174/1381612825666190404115424. [DOI] [PubMed] [Google Scholar]
- Vettorazzi M.; Angelina E.; Lima S.; Gonec T.; Otevrel J.; Marvanova P.; Padrtova T.; Mokry P.; Bobal P.; Acosta L. M.; Palma A.; Cobo J.; Bobalova J.; Csollei J.; Malik I.; Alvarez S.; Spiegel S.; Jampilek J.; Enriz R. D. An integrative study to identify novel scaffolds for sphingosine kinase 1 inhibitors. Eur. J. Med. Chem. 2017, 139, 461–481. 10.1016/j.ejmech.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnute M. E.; McReynolds M. D.; Kasten T.; Yates M.; Jerome G.; Rains J. W.; Hall T.; Chrencik J.; Kraus M.; Cronin C. N.; Saabye M.; Highkin M. K.; Broadus R.; Ogawa S.; Cukyne K.; Zawadzke L. E.; Peterkin V.; Iyanar K.; Scholten J. A.; Wendling J.; Fujiwara H.; Nemirovskiy O.; Wittwer A. J.; Nagiec M. M. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444 (1), 79–88. 10.1042/BJ20111929. [DOI] [PubMed] [Google Scholar]
- Gustin D. J.; Li Y.; Brown M. L.; Min X.; Schmitt M. J.; Wanska M.; Wang X.; Connors R.; Johnstone S.; Cardozo M.; Cheng A. C.; Jeffries S.; Franks B.; Li S.; Shen S.; Wong M.; Wesche H.; Xu G.; Carlson T. J.; Plant M.; Morgenstern K.; Rex K.; Schmitt J.; Coxon A.; Walker N.; Kayser F.; Wang Z. Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg. Med. Chem. Lett. 2013, 23 (16), 4608–4616. 10.1016/j.bmcl.2013.06.030. [DOI] [PubMed] [Google Scholar]
- Solà R. J.; Griebenow K. Glycosylation of Therapeuic Proteins: an effective strategy to optimize efficacy. BioDrugs 2010, 24, 9–21. 10.2165/11530550-000000000-00000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi S. V.; Hussein W. M.; Varamini P.; Simerska P.; Toth I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem. Sci. 2016, 7 (4), 2492–2500. 10.1039/C5SC04392A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egleton R. D.; Mitchell S. A.; Huber J. D.; Janders J.; Stropova D.; Polt R.; Yamamura H. I.; Hruby V. J.; Davis T. P. Improved bioavailability to the brain of glycosylated Met-enkephalin analogs. Brain Res. 2000, 881 (1), 37–46. 10.1016/S0006-8993(00)02794-3. [DOI] [PubMed] [Google Scholar]
- Witt K. A.; Gillespie T. J.; Huber J. D.; Egleton R. D.; Davis T. P. Peptide drug modifications to enhance bioavailability and blood-brain barrier permeability. Peptides 2001, 22 (12), 2329–2343. 10.1016/S0196-9781(01)00537-X. [DOI] [PubMed] [Google Scholar]
- Albert R.; Marbach P.; Bauer W.; Briner U.; Fricker G.; Bruns C.; Pless J. SDZ CO 611: a highly potent glycated analog of somatostatin with improved oral activity. Life Sci. 1993, 53 (6), 517–25. 10.1016/0024-3205(93)90703-6. [DOI] [PubMed] [Google Scholar]
- Liu R.; Fu Z.; Zhao M.; Gao X.; Li H.; Mi Q.; Liu P.; Yang J.; Yao Z.; Gao Q. GLUT1-mediated selective tumor targeting with fluorine containing platinum(II) glycoconjugates. Oncotarget 2017, 8, 39476–39496. 10.18632/oncotarget.17073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher J. F.; Harrison A. W.; Bundy G. L.; Wilkinson K. F.; Rush B. D.; Ruwart M. J. Peptide to glycopeptide: glycosylated oligopeptide renin inhibitors with attenuated in vivo clearance properties. J. Med. Chem. 1991, 34 (10), 3140–3. 10.1021/jm00114a026. [DOI] [PubMed] [Google Scholar]
- Chen X.; Hui L.; Foster D. A.; Drain C. M. Efficient synthesis and photodynamic activity of porphyrin-saccharide conjugates: targeting and incapacitating cancer cells. Biochemistry 2004, 43 (34), 10918–10929. 10.1021/bi049272v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragai M.; Nativi C.; Richichi B.; Venturi C. Design in silico, synthesis and binding evaluation of a carbohydrate-based scaffold for structurally novel inhibitors of matrix metalloproteinases. ChemBioChem 2005, 6 (8), 1345–1349. 10.1002/cbic.200400456. [DOI] [PubMed] [Google Scholar]
- Nuti E.; Cuffaro D.; D’Andrea F.; Rosalia L.; Tepshi L.; Fabbi M.; Carbotti G.; Ferrini S.; Santamaria S.; Camodeca C.; Ciccone L.; Orlandini E.; Nencetti S.; Stura E. A.; Dive V.; Rossello A. Sugar-Based Arylsulfonamide Carboxylates as Selective and Water-Soluble Matrix Metalloproteinase-12 Inhibitors. ChemMedChem 2016, 11, 1626–1637. 10.1002/cmdc.201600235. [DOI] [PubMed] [Google Scholar]
- Christie M. P.; Simerska P.; Jen F. E.; Hussein W. M.; Rawi M. F.; Hartley-Tassell L. E.; Day C. J.; Jennings M. P.; Toth I. A drug delivery strategy: binding enkephalin to asialoglycoprotein receptor by enzymatic galactosylation. PLoS One 2014, 9 (4), e95024 10.1371/journal.pone.0095024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Knapp S.; Pyne N. J.; Pyne S.; Elkins J. M. Crystal Structure of Sphingosine Kinase 1 with PF-543. ACS Med. Chem. Lett. 2014, 5 (12), 1329–1333. 10.1021/ml5004074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Min X.; Xiao S. H.; Johnstone S.; Romanow W.; Meininger D.; Xu H.; Liu J.; Dai J.; An S.; Thibault S.; Walker N. Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure 2013, 21 (5), 798–809. 10.1016/j.str.2013.02.025. [DOI] [PubMed] [Google Scholar]
- Schnute M. E.; McReynolds M. D.; Carroll J.; Chrencik J.; Highkin M. K.; Iyanar K.; Jerome G.; Rains J. W.; Saabye M.; Scholten J. A.; Yates M.; Nagiec M. M. Discovery of a Potent and Selective Sphingosine Kinase 1 Inhibitor through the Molecular Combination of Chemotype-Distinct Screening Hits. J. Med. Chem. 2017, 60 (6), 2562–2572. 10.1021/acs.jmedchem.7b00070. [DOI] [PubMed] [Google Scholar]
- Brown P. D.; Willis A. C.; Sherburn M. S.; Lawrence A. L. Total synthesis and structural revision of the alkaloid incargranine B. Angew. Chem., Int. Ed. 2013, 52 (50), 13273–13275. 10.1002/anie.201307875. [DOI] [PubMed] [Google Scholar]
- Matto P.; Modica E.; Franchini L.; Facciotti F.; Mori L.; de Libero G.; Lombardi G.; Fallarini S.; Panza L.; Compostella F.; Ronchetti F. A general and stereoselective route to alpha- or beta-galactosphingolipids via a common four-carbon building block. J. Org. Chem. 2007, 72 (20), 7757–60. 10.1021/jo070849z. [DOI] [PubMed] [Google Scholar]
- Schultz V. L.; Zhang X.; Linkens K.; Rimel J.; Green D. E.; DeAngelis P. L.; Linhardt R. J. Chemoenzymatic Synthesis of 4-Fluoro-N-Acetylhexosamine Uridine Diphosphate Donors: Chain Terminators in Glycosaminoglycan Synthesis. J. Org. Chem. 2017, 82 (4), 2243–2248. 10.1021/acs.joc.6b02929. [DOI] [PubMed] [Google Scholar]
- Ismaeel A.; Kim J. S.; Kirk J. S.; Smith R. S.; Bohannon W. T.; Koutakis P. Role of transforming growth factor-β in skeletal muscle fibrosis: a review. Int. J. Mol. Sci. 2019, 20 (10), 2446. 10.3390/ijms20102446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Xia Y.; Yan W.; Zhang H.; Zhou F.; Zhao S.; Wang W.; Zhu D.; Xin C.; Lee Y.; Zhang L.; He Y.; Gao E.; Tao L. Sphingosine 1-phosphate signaling contributes to cardiac inflammation, dysfunction, and remodeling following myocardial infarction. Am. J. Physiol Heart Circ Physiol 2016, 310, H250–261. 10.1152/ajpheart.00372.2015. [DOI] [PubMed] [Google Scholar]
- González-Fernández B.; Sánchez D. I.; Crespo I.; San-Miguel B.; Álvarez M.; Tuñón M. J.; González-Gallego J. Inhibition of the SphK1/S1P signaling pathway by melatonin in mice with liver fibrosis and human hepatic stellate cells. Biofactors 2017, 43, 272–282. 10.1002/biof.1342. [DOI] [PubMed] [Google Scholar]
- Ju T.; Gao D.; Fang Z.-y. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem. Biophys. Res. Commun. 2016, 470, 728–734. 10.1016/j.bbrc.2016.01.053. [DOI] [PubMed] [Google Scholar]
- Bond M. R.; Zhang H.; Vu P. D.; Kohler J. J. Photocrosslinking of glycoconjugates using metabolically incorporated diazirine-containing sugars. Nat. Protoc. 2009, 4 (7), 1044–1063. 10.1038/nprot.2009.85. [DOI] [PubMed] [Google Scholar]
- Yablonka-Reuveni Z.; Rivera A. J. Proliferative dynamics and the role of FGF2 during myogenesis of rat satellite cells onisolatedfibers. Basic Appl. Myol. 1997, 7, 189–202. [PMC free article] [PubMed] [Google Scholar]
- Leask A.; Abraham D. J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18 (7), 816–27. 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.