

Abstract
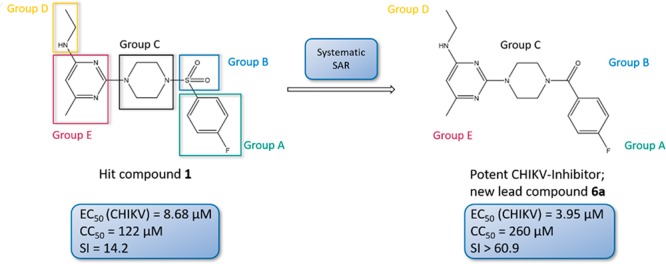
The chikungunya virus (CHIKV) is a mosquito-transmitted alphavirus, and it is the causative agent of chikungunya fever (CHIKF). Although it has re-emerged as an epidemic threat, so far there are neither vaccines nor pharmacotherapy available to prevent or treat an infection. Herein, we describe the synthesis and structure–activity relationship studies of a class of novel small molecule inhibitors against CHIKV and the discovery of a new potent inhibitor (compound 6a). The starting point of the optimization process was N-ethyl-6-methyl-2-(4-(4-fluorophenylsulfonyl)piperazine-1-yl)pyrimidine-4-amine (1) with an EC50 of 8.68 μM, a CC50 of 122 μM, and therefore a resulting selectivity index (SI) of 14.2. The optimized compound 6a, however, displays a much lower micromolar antiviral activity (EC50 value of 3.95 μM), considerably better cytotoxic liability (CC50 value of 260 μM) and consequently an improved SI of greater than 61. Therefore, we report the identification of a promising novel compound class that has the potential for further development of antiviral drugs against the CHIKV.
Keywords: Novel small-molecule antivirals, Chikungunya virus, Structure−activity relationship studies, Medicinal chemistry
Introduction
The chikungunya virus (CHIKV) is a member of the Togaviridae, genus alphavirus and was first described in East Africa (southern province of Tanzania) in 1952–1953.1,2 This mosquito-transmitted alphavirus (mainly by Aedes spp.) is the causative agent of chikungunya fever (CHIKF), a disease characterized by myalgia, polyarthralgia, fever, nausea, headaches, maculopapular rash, and complications including lymphopenia, lethal hepatitis, dermatologic lesions, and encephalitis.3−5 Although chikungunya fever is rarely fatal, symptoms such as myalgia and arthralgia have been reported to last for years after clearance of the infection, resulting in an impaired quality of life.6−8 Since the re-emergence of the virus in 2005, outbreaks have occurred in more than 40 countries throughout the world, not only in Southeast Asia and Africa but also in Europe (Italy and France) as well as America.9−11 Currently, neither vaccines nor pharmacotherapy are available to prevent or treat an infection.12−14 To date, the only available treatment is the alleviation of symptoms by using NSARs like ibuprofen, naproxen, or acetaminophen. Occasionally, corticosteroids are administered to patients to relieve symptoms like fever and pain.15−17 Up to now, the antiviral activity of small molecules, compounds from natural sources, and immune modulators on the replication of chikungunya virus have been investigated. Several compounds, ranging from antivirals with a broad spectrum like ribavirin to harringtonine (a cephalotaxine alkaloid) and IFN-α, were found to be active, targeting different stages in the alphavirus life cycle. Despite the demonstrated effectiveness and, in some cases, potent antiviral activity in vitro, none of these drugs passed the clinical trial phase.18−25 Also, the utilization of the already known antimalaria drug chloroquine as possible antiviral treatment is highly discussed regarding the potential risks and benefits for the patients.26−30 Therefore, the development of safe and effective new antivirals against CHIKV is highly desirable.31 This problem was addressed within the EU FP7 collaborative project SILVER. In this project, the small molecular hit CIM016321 (later referred as 1, Figure 1), which was identified previously by high-throughput screening by the Center for Innovation and Stimulation of Drug Discovery (CISTIM) and the University of Leuven (KU Leuven), was further investigated and a hit to lead program was initiated.
Figure 1.

For optimization, we divided the initial hit 1 into five parts. Compounds with modifications at the same part of the molecule are summarized to “Groups”.
To optimize 1, we divided the structure of this compound into five parts suitable for systematic variation (shown in Figure 1). The aim of the optimization was to achieve (i) an increase in antiviral activity and (ii) a decrease in cytotoxicity, therefore resulting in a compound exhibiting an improved selectivity index (SI). As guidance for systematic structural variations of the molecule, approaches like the concept of bioisosterism and the Topliss decision tree were applied.32−35
In this study, we have identified that analogues of 1 are potent and safe inhibitors of chikungunya virus in vitro. Moreover, structure–activity relationship studies unveiled the molecular requirements for highly active and safe antiviral compounds.
Results and Discussion
The initial hit 1 was prepared using a four-step-synthesis and was also established to give access to the entire compound series. The synthesis of these compounds is summarized in Scheme 1, starting from 2-chloro-N-ethyl-6-methylpyrimidin-4-amine (2a), which was synthesized following the procedures reported by Martyn et al.36 The reaction with tert-butyl piperazine-1-carboxylate, obtained according to the protocol from Moussa et al.,37 under microwave conditions gave a high yield of 85% of the desired Boc-protected piperazine–pyrimidine intermediate. The following deprotection and the N-sulfonamidation of piperazine were performed according to Martyn et al.,36 but with additional THF. The fourth step of the synthesis with p-fluorobenzenesulfonyl chloride afforded the desired target in a moderate yield of 32%.
Scheme 1. Synthesis of the Initial Hit 1.

Reagents and conditions: (a) EtOH, 24–48 h, rt; (b) EtOH, microwave, 3 min, 155 °C, 250 W, 12 bar, tert-butyl piperazine-1-carboxylate; (c) 4.5 M HCl, dioxane/THF, 24 h rt; (d) DCM, N(CH2CH3)3, 24 h rt, 4-fluorobenzenesulfonyl chloride.
The first alteration was performed using modifications on, or replacement of, the 4-fluorophenyl ring (Group A). All compounds from this group (5a–5s), except 5s, were synthesized following Scheme 2. Synthesis of 5s was achieved by reducing the nitro group using tin(II) chloride 5f to the corresponding amine. The first three steps of the synthesis remained the same as for 1, as well as the reaction conditions as described before. For the preparation of analogues, in the fourth step, instead of the 4-fluorobenzenesulfonyl chloride, a suitable sulfonyl chloride was used. The conditions for the reaction remained unchanged and provided analogues in 13–92% yield.
Scheme 2. Synthesis of the Compounds from Group A (5a–5r) and Group B (6a, 6b).

Reagents and condition: (a) DCM, N(CH2CH3)3, 24 h rt.
The synthesis of the analogues 6a and 6b from Group B, replacing the sulfonamide linker with an amide or a methyl group was carried out with the established synthesis route. The conditions and reagents remained the same as for 1 and Group A, however, with 4-fluorobenzoyl chloride or 4-fluorobenzyl chloride instead of the benzenesulfonyl chlorides (Scheme 2).
Substituents of the piperazine linker (Group C) where introduced to investigate the function of this group. The reaction for Group C (7a and 7b) followed the established synthesis route, with a small alteration for 7b: 2-chloro-N-ethyl-6-methylpyrimidin-4-amine (2a) was reacted directly with the tetrahydroquinoxaline (as modified linker) under microwave irradiation to access 4c. This reaction was performed without protecting one of the two amino groups before and gave still a high yield of 75%. The third step of the synthesis was identical to the fourth step of the previously used standard procedure–performed with 4-fluorobenzenesulfonyl chloride and triethylamine as a base in dichloromethane at room temperature for 24 h (7a: 55% yield, 7b: 28% yield; Scheme 3).
Scheme 3. Synthesis of the Compounds from Group C (7a, 7b).

Reagents and conditions: (a) EtOH, microwave, 3 min, 155 °C, 250 W, 12 bar; 3b, tert-butyl 1,4-diazepane-1-carboxylate; 4c, 1,2,3,4-tetrahydroquinoxaline; (b) 4b, 4.5 M HCl, dioxane/THF, 24 h rt; (c) DCM, N(CH2CH3)3, 24 h rt, 4-fluorobenzenesulfonyl chloride.
For Group D, the target compounds were accessed via the alteration of the first synthesis step. Instead of ethylamine as reactant, propane-2-amine, 2-methylpropan-2-amine, sodium ethoxide, or pyrrolidine were used to prepare the analogue compounds 8a–8d. The successive reaction between the substituted pyrimidine and the piperazine, following the sulfonamidation as the last step, was adopted according the established synthesis route (Scheme 4).
Scheme 4. Synthesis of the Compounds from Group D (8a–8d), from Group E (9a–9d), and 10a, 10b.

Reagents and conditions: (a) EtOH, 24–48 h rt; 2b/2j, propan-2-amine; 2c, 2-methylpropan-2-amin; 2d, sodium ethoxide; 2e, pyrrolidine; 2f–i, ethylamine; (b) EtOH, microwave, 3 min, 155 °C, 250 W, 12 bar, tert-butyl piperazine-1-carboxylate; (c) 4.5 M HCl, dioxane/THF, 24 h rt; (d) DCM, N(CH2CH3)3, 24 h rt, 4-fluorobenzenesulfonyl chloride.
Compounds of Group E (9a–9d) were synthesized following Scheme 4. The synthesis remained the same as for 1, as well as the reaction conditions. To prepare the analogues of this group, a differently substituted pyrimidine building block was used as starting reactant in the first step of the synthesis. The pyrimidine ring 2f (4-chloro-N-ethyl-6-methylpyrimidin-2-amine), used to prepare 9a, was obtained as a product out in the first step of the synthesis of 1 in a moderate yield of 17%.
Finally, the modifications with the most promising antiviral activities were combined, utilizing the established synthesis route described above (Scheme 4 and Scheme 5).
Scheme 5. Synthesis of 11a–11c.

Reagents and conditions: (a) DCM, N(CH2CH3)3, 24 h rt; 11a/11b, 4-chlorobenzoyl chloride; 11c, 4-fluorobenzoyl chloride.
All compounds were evaluated for their potential antiviral activity against CHIKV by determining the inhibition of CHIKV-induced cytopathogenic effect (CPE) in Vero cells (Table 1).
Table 1. Antiviral Evaluation of 1 and Analogues against CHIKV in Vero Cellsa.
| compound | EC50 (μM)b | EC90 (μM)c | CC50 (μM)d |
|---|---|---|---|
| 1 | 8.7 ± 1 | 14 ± 1 | 122 ± 24 |
| 5a | 14 ± 3 | 28 ± 6 | 156 ± 35 |
| 5b | 5 ± 0.4 | 7 ± 1 | 51 ± 19 |
| 5c | 7.1 ± 0.01 | 13 ± 2 | 44 ± 19 |
| 5d | 5.9 ± 0.2 | 8.7 ± 1 | 19 ± 2 |
| 5e | >233 | >233 | 45 ± 1 |
| 5f | 14 ± 3 | 24 ± 7 | 62 ± 1 |
| 5g | >259 | >250 | ND |
| 5h | 82 ± 11 | 125 ± 3 | 178 ± 31 |
| 5i | 15 ± 2 | 22 ± 2 | 95 ± 74 |
| 5j | 27 ± 9 | 52 ± 17 | 91 ± 7 |
| 5k | 20 ± 3 | 31 ± 2 | 131 ± 43 |
| 5l | 12 ± 5 | 19 ± 10 | 53 ± 7 |
| 5m | 16 ± 6 | 16 ± 4 | 67 ± 13 |
| 5n | 19 ± 5 | ND | 29 ± 3 |
| 5o | 10 ± 0.2 | 17 ± 5 | 181 ± 0.4 |
| 5p | 28 ± 5 | 36 ± 14 | 55 ± 3 |
| 5q | 75 ± 19 | >166 | 122 ± 23 |
| 5r | 10 ± 1 | 13 ± 3 | 15 ± 3 |
| 5s | 22 ± 3 | 51 ± 16 | 183 ± 17 |
| 6a | 4.0 ± 1 | 20 ± 8 | >260 |
| 6b | 2.5 ± 1 | 5 ± 1 | 69 ± 7 |
| 7a | 77 ± 5 | 123 ± 0.2 | 202 ± 18 |
| 7b | 16 ± 1 | 29 ± 8 | 106 ± 69 |
| 8a | 9.5 ± 2 | 17 ± 4 | 66 ± 6 |
| 8b | 12 ± 2 | ND | 19 ± 3 |
| 8c | >197 | >197 | ND |
| 8d | 33 ± 7 | 34 ± 1 | 50 ± 8 |
| 9a | 3.2 ± 0.2 | 14 ± 4 | 59 ± 18 |
| 9b | 48 ± 9 | 98 ± 34 | >329 |
| 9c | 14 ± 3 | 29 ± 2 | 91 ± 10 |
| 9d | 70 ± 0.2 | 124 ± 1 | 217 ± 3 |
| 10 | 37 ± 4 | 55 ± 8 | 76 ± 16 |
| 11a | 32 ± 2 | 108 ± 3 | 202 ± 27 |
| 11b | 51 ± 7 | 88 ± 17 | 204 ± 1 |
| 11c | 92 ± 9 | 102 ± 17 | 159 ± 31 |
EC50, EC90, and CC50 data represent median values ± standard deviation from at least three independent experiments. ND = not detectable.
The 50% effective concentration (EC50) is the concentration of compound that is required to inhibit virus-induced cell death by 50%.
The 90% effective concentration (EC90) is the concentration of compound that is required to inhibit virus-induced cell death by 90%.
The 50% cytostatic/cytotoxic concentration (CC50) is the concentration of compound that reduces the overall metabolic activity of the uninfected, compound-treated cells by 50% as compared to the untreated, uninfected cell.
The purpose of Group A was to investigate if a fluorine atom as substituent is essential, which turned out not to be the case: the analogue with the nonsubstituted phenyl ring (5a) also showed antiviral activity, although 5a was less active than 1 (EC50 = 14 ± 3 μM). In a next step, para fluorine was replaced by other halogens such as chlorine (5b), bromine (5c), and iodine (5d). All these compounds were more active than the initial hit: the chlorine-substituted 5b gave the best result with an EC50 of 5.3 μM, the iodine-substituted 5d showed similar antiviral activity with an EC50 of 5.9 μM, while the bromine-substituted 5c was less active (EC50 = 7.1 μM). The nitro-substituted 5f (EC50 = 14 ± 3 μM) and the methyl-substituted 5i (EC50 = 15 ± 2 μM) were also less active than 1. On the contrary, the trifluoromethyl substituted 5e was inactive, as well as 5g with CN substitution. The other para-substituted compounds (5h and 5s) showed less antiviral activity than the initial hit 1.
Shifting the para fluorine atom to other positions on the benzene ring resulted in less active compounds than the initial hit 1 (5j and 5k). Also, combining the ortho or meta to the para substitution did not result in an additive antiviral effect compared to 1: 5l with two fluorine atoms on the meta and para position and 5m with an ortho/para disubstitution pattern were less active than 1. Additionally, neither of the triple substituted compounds showed an improved additive antiviral effect (5n, 5o, and 5p). 5q, in which all hydrogen atoms on the phenyl ring were replaced by fluorine, was found to be poorly active. Conclusively, concerning the antiviral activity of the monosubstituted analogues, the 4-F substituted analogue is more active than the 3-F-substituted one, which is more active than the 2-F-substituted compound. Concerning the double- and triple-substituted compounds, the 3,4-F substituted 5l and the 2,3,4-F-substituted 5o are active; while the compounds with 2,4-F (5m) and 3,4,5-F substitution patterns (5n) are both only moderately active compared to 1.
To investigate if the double substitution on the aromatic ring with different halogens would also decrease the antiviral activity, 5r was prepared. It showed moderate activity against the virus, confirming our previous observation, that the chloride substitution enhances the antiviral activity. Nevertheless, the doubly substituted ring also showed a weaker antiviral effect using chlorides (5r) than the matching single substituted ring (5b).
Summarizing the results from Group A, 17 out of the 19 compounds were active against CHIKV, with three compounds being more potent than 1 (5b, 5c, and 5d). In those three compounds, other halogens replaced the fluorine at the para position. Therefore, replacing fluorine on the benzene ring by another halogen leads to an improvement in antiviral activity (chlorine > iodine > bromine > fluorine). 5b with the chlorine atom as substituent on the para position of the benzene ring showed the best test results; therefore, the fluorine/chlorine replacement should be considered for further optimization.
In Group B, both compounds showed impressive test results: 6a has an EC50 of 4.0 ± 1 μM, and 6b has an EC50 of 2.5 μM. Conclusively, both compounds are more than twice as active as 1. Although 6b is even more active than 6a, compound 6a should be considered for further optimization due to cytotoxicity reasons (1, CC50 = 122 μM, SI = 14.2; 6a, CC50 = 260 μM, SI = 60.9; 6b, CC50 = 69.3 μM, SI = 19.7). Therefore, compared to 1, the modification in 6a led to an improvement in activity, cytotoxicity, and selectivity, while the modification in 6b led to a higher cytotoxicity.
In Group C, the prepared analogues were less active than 1. Conclusively, 1,4-piperazine as a linker has proven to be the better choice to reach a potent antiviral activity.
The replacement of the ethylamine side chain with isopropylamine (8a) or tert-butylamine (8b) demonstrated good activity against the CHIKV, which unfortunately was accompanied by increased cytotoxicity (8a, CC50 = 66.4 μM, SI = 9.83; 8b, CC50 = 18.6 μM, SI = 1.17). In addition, it was investigated whether the nitrogen or the hydrogen atom of the ethylamine side chain is essential for activity and whether the antiviral activity of the compound would increase with higher molecular lipophilicity. Therefore, 8c was prepared in which an ethoxy side chain replaced the ethylamine side chain. However, 8c did not show any antiviral activity. To understand whether this loss of activity depends on the missing nitrogen atom or the absence of the hydrogen bond donor function, 8d was synthesized, in which a pyrrolidine ring replaced the ethylamine side chain. The result from 8d (EC50 of 33 ± 7 μM) indicated that the hydrogen bond donor is not essential for actively enhancing the antiviral activity, although its pure absence seemed to decrease the activity. Taken together with the results from Group D, the nitrogen atom of the ethylamine side chain seems to be essential for antiviral activity, while the hydrogen bond donor is not. Also, a bifurcation at the ethylamine side chain may be taken into consideration with caution.
In Group E, in a first step the optimal positions for the nitrogen atoms in the pyrimidine ring were investigated. In a second step, it was investigated whether the methyl group as a substituent on the pyrimidine ring is essential or not. The nitrogen at position R1 seemed to be necessary for good antiviral activity. Shifting the nitrogen from position R1/3 (as in the initial hit 1) to position R1/2 (9a) resulted in an improved antiviral activity.
Finally, the most interesting modifications were combined to either reach a higher antiviral activity (11a and 11c) or to deepen the knowledge of the chosen variation (10, 11b). The result of 10 and 11b underline the importance of the methyl group on the pyrimidine ring and the shifting of the nitrogen to position R1/2. Moreover, the decrease of activity with the cyclopropylamine side chain (10) , although its size is comparable to the original ethylamine, can possibly be explained by his impaired flexibility and different three-dimensional geometry.
Surprisingly, the combination of the most promising alterations (Group B:, amide; Group E, shifted N to position R1/2) in combination with either the best alteration of Group A (para chloride, 11a) or with the introduction of an isopropyl side chain (Group D, 11c) did not give the desired gain of antiviral activity.
Conclusion
In this study, we present several 2-(4-(phenylsulfonyl)piperazine-1-yl)pyrimidine and analogues as selective and potent inhibitors of CHIKV. All compounds described are easily accessible in a four-step synthesis route: starting from the required substituted pyrimidine reacting with the Boc-protected piperazine or other suitable nitrogen-containing functional groups under microwave irradiation, followed by deprotection and finally reaction with a benzenesulfonyl chloride. Additionally, the structural requirements for a significant anti-CHIKV activity have been determined on all five structural molecular subgroups. During this optimization process, 6a was identified as a potent and selective inhibitor of CHIKV, demonstrating a much better profile than the starting point, hit 1, and a selectivity index greater than 61. Furthermore, the optimized compound showed a wide-spectrum antiviral activity against all tested strains of CHIKV (data will be published elsewhere). Further structure–activity studies and optimization of the antiviral activity are ongoing and mechanism of action studies are being performed to determine the molecular target of this novel class of anti-CHIKV compounds. These studies point toward the viral capping machinery and more specifically to the viral protein nsP1 as the antiviral target of these compounds. Cross-resistance with another class of capping inhibitors, the MADTP series, was confirmed in antiviral assays (data not shown). As other classes of inhibitors that target the viral capping have been reported, the capping machinery of alphaviruses might be a hot spot for antiviral molecules.
Acknowledgments
We thank S. Delmotte, T. Bellon, and K. Donckers for their excellent technical assistance in the acquisition of the antiviral data. Furthermore, we are grateful to J. Wackerlig and D. Dobusch for the support with the chemical analysis of the compounds.
Glossary
ABBREVIATIONS
- THF
tetrahydrofuran
- EtOAc
ethyl acetate
- TEA
triethylamine
- DIPE
diisopropyl ether
- DMSO-d6
deuterated dimethyl sulfoxide
- dia
diazepane
- Ph
phenyl
- pyr
pyrimidine
- pip
piperazine
- ipr
isopropyl
- qu
quinoxaline
- TLC
thin layer chromatography
- ND
not detectable.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00662.
Cell lines and virus strain, instrumentation and experimental procedures, characterization and spectral data, HPLC-determined purity data, and NMR spectra for all the compounds (PDF)
Author Present Address
∥ CURA Marketing GmbH, Dr.-Franz-Werner-Straße 19, A-6020 Innsbruck, Austria.
Author Contributions
⊥ Moesslacher Julia and Battisti Verena have contributed equally to this manuscript. All authors have given approval to the final version of the manuscript.
This work was funded in parts (University of Innsbruck, KU Leuven) by the European Union Seventh Framework Program (FP7/2007-2013) under a SILVER grant (No. HEALTH-F3-2010-260644).
The authors declare no competing financial interest.
Dedication
This paper is dedicated to the memory of Prof. Maurizio Botta, who passed away much too early in August 2019.
Supplementary Material
References
- Robinson M. C. An Epidemic of Virus Disease in Southern Province, Tanganyika Territory, in 1952–1953. I. Clinical Features. Trans. R. Soc. Trop. Med. Hyg. 1955, 49 (1), 28–32. 10.1016/0035-9203(55)90080-8. [DOI] [PubMed] [Google Scholar]
- Thiberville S.-D.; Moyen N.; Dupuis-Maguiraga L.; Nougairede A.; Gould E. A.; Roques P.; De Lamballerie X. Chikungunya Fever: Epidemiology, Clinical Syndrome, Pathogenesis and Therapy. Antiviral Res. 2013, 99, 345–370. 10.1016/j.antiviral.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz O.; Albert M. L. Biology and Pathogenesis of Chikungunya Virus. Nat. Rev. Microbiol. 2010, 8 (7), 491–500. 10.1038/nrmicro2368. [DOI] [PubMed] [Google Scholar]
- Burt F. J.; Chen W.; Miner J. J.; Lenschow D. J.; Merits A.; Schnettler E.; Kohl A.; Rudd P. A.; Taylor A.; Herrero L. J.; et al. Chikungunya Virus: An Update on the Biology and Pathogenesis of This Emerging Pathogen. Lancet Infect. Dis. 2017, 17 (4), e107–e117. 10.1016/S1473-3099(16)30385-1. [DOI] [PubMed] [Google Scholar]
- Couderc T.; Lecuit M. Chikungunya Virus Pathogenesis: From Bedside to Bench. Antiviral Res. 2015, 121, 120–131. 10.1016/j.antiviral.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Moro M. L.; Grilli E.; Corvetta A.; Silvi G.; Angelini R.; Mascella F.; Miserocchi F.; Sambo P.; Finarelli A. C.; Sambri V.; et al. Long-Term Chikungunya Infection Clinical Manifestations after an Outbreak in Italy: A Prognostic Cohort Study. J. Infect. 2012, 65 (2), 165–172. 10.1016/j.jinf.2012.04.005. [DOI] [PubMed] [Google Scholar]
- Couturier E.; Guillemin F.; Mura M.; Leon L.; Virion J.-M.; Letort M.-J.; De Valk H.; Simon F.; Vaillant V. Impaired Quality of Life after Chikungunya Virus Infection: A 2-Year Follow-up Study. Rheumatology 2012, 51 (7), 1315–1322. 10.1093/rheumatology/kes015. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Morales A. J.; Restrepo-Posada V. M.; Acevedo-Escalante N.; Rodríguez-Muñoz E. D.; Valencia-Marín M.; Castrillón-Spitia J. D.; Londoño J. J.; Bedoya-Rendón H. D.; de Jesús Cárdenas-Pérez J.; Cardona-Ospina J. A.; et al. Impaired Quality of Life after Chikungunya Virus Infection: A 12-Month Follow-up Study of Its Chronic Inflammatory Rheumatism in La Virginia, Risaralda. Rheumatol. Int. 2017, 37 (10), 1757–1758. 10.1007/s00296-017-3795-1. [DOI] [PubMed] [Google Scholar]
- Rezza G.; Nicoletti L.; Angelini R.; Romi R.; Finarelli A. C.; Panning M.; Cordioli P.; Fortuna C.; Boros S.; Magurano F.; et al. Infection with Chikungunya Virus in Italy: An Outbreak in a Temperate Region. Lancet 2007, 370 (9602), 1840–1846. 10.1016/S0140-6736(07)61779-6. [DOI] [PubMed] [Google Scholar]
- Delisle E.; Rousseau C.; Broche B.; Leparc-Goffart I.; L’ambert G.; Cochet A.; Prat C.; Foulongne V.; Ferré J. B.; Catelinois O. Chikungunya Outbreak in Montpellier, France, September to October 2014. European Centre for Disease Prevention and Control 2015, 20, 21108. 10.2807/1560-7917.ES2015.20.17.21108. [DOI] [PubMed] [Google Scholar]
- Rashad A. A.; Mahalingam S.; Keller P. A. Chikungunya Virus: Emerging Targets and New Opportunities for Medicinal Chemistry. J. Med. Chem. 2014, 57 (4), 1147–1166. 10.1021/jm400460d. [DOI] [PubMed] [Google Scholar]
- Subudhi B.; Chattopadhyay S.; Mishra P.; Kumar A. Current Strategies for Inhibition of Chikungunya Infection. Viruses 2018, 10 (5), 235. 10.3390/v10050235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Sandoval A. 51 Years in of Chikungunya Clinical Vaccine Development: A Historical Perspective. Hum. Vaccines Immunother. 2019, 15 (10), 2351–2358. 10.1080/21645515.2019.1574149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J. V. J.; Lopes T. R. R.; De Oliveira-Filho E. F.; Oliveira R. A. S.; Durães-Carvalho R.; Gil L. H. V. G. Current Status, Challenges and Perspectives in the Development of Vaccines against Yellow Fever, Dengue. Acta Trop. 2018, 182, 257–263. 10.1016/j.actatropica.2018.03.009. [DOI] [PubMed] [Google Scholar]
- Caglioti C.; Lalle E.; Castilletti C.; Carletti F.; Capobianchi M. R.; Bordi L. Chikungunya Virus Infection: An Overview. New Microbiol. 2013, 36, 211–227. [PubMed] [Google Scholar]
- Kennedy Amaral Pereira J.; Schoen R. T. Management of Chikungunya Arthritis. Clin. Rheumatol. 2017, 36 (10), 2179–2186. 10.1007/s10067-017-3766-7. [DOI] [PubMed] [Google Scholar]
- World Health Organization . Guidelines on clinical management of Chikungunya Fever http://www.wpro.who.int/mvp/topics/ntd/Clinical_Mgnt_Chikungunya_WHO_SEARO.pdf (accessed Aug 2, 2019).
- da Silva-Júnior E. F.; Leoncini G. O.; Rodrigues É. E. S.; Aquino T. M.; Araújo-Júnior J. X. The Medicinal Chemistry of Chikungunya Virus. Bioorg. Med. Chem. 2017, 25 (16), 4219–4244. 10.1016/j.bmc.2017.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching K.-C.; Ng L. F. P.; Chai C. L. L. A Compendium of Small Molecule Direct-Acting and Host-Targeting Inhibitors as Therapies against Alphaviruses. J. Antimicrob. Chemother. 2017, 72 (11), 2973–2989. 10.1093/jac/dkx224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lani R.; Hassandarvish P.; Shu M.-H.; Hong Phoon W.; Jang Hann Chu J.; Higgs S.; Vanlandingham D.; Abu Bakar S.; Zandi K. Antiviral Activity of Selected Flavonoids against Chikungunya Virus. Antiviral Res. 2016, 133, 50–61. 10.1016/j.antiviral.2016.07.009. [DOI] [PubMed] [Google Scholar]
- Briolant S.; Garin D.; Scaramozzino N.; Jouan A.; Crance J. M. In Vitro Inhibition of Chikungunya and Semliki Forest Viruses Replication by Antiviral Compounds: Synergistic Effect of Interferon-Alpha and Ribavirin Combination. Antiviral Res. 2004, 61, 111–117. 10.1016/j.antiviral.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Kaur P.; Thiruchelvan M.; Lee R. C. H.; Chen H.; Chen K. C.; Ng M. L.; Chu J. J. H. Inhibition of Chikungunya Virus Replication by Harringtonine, a Novel Antiviral That Suppresses Viral Protein Expression. Antimicrob. Agents Chemother. 2013, 57 (1), 155–167. 10.1128/AAC.01467-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounce B. C.; Poirier E. Z.; Passoni G.; Simon-Loriere E.; Cesaro T.; Prot M.; Stapleford K. A.; Moratorio G.; Sakuntabhai A.; Levraud J.-P.; et al. Interferon-Induced Spermidine-Spermine Acetyltransferase and Polyamine Depletion Restrict Zika and Chikungunya Viruses. Cell Host Microbe 2016, 20 (2), 167–177. 10.1016/j.chom.2016.06.011. [DOI] [PubMed] [Google Scholar]
- Kaur P.; Chu J. J. H. Chikungunya Virus: An Update on Antiviral Development and Challenges. Drug Discovery Today 2013, 18 (19–20), 969–983. 10.1016/j.drudis.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelnabi R.; Neyts J.; Delang L.. Antiviral Strategies Against Chikungunya Virus. In Chikungunya Virus: Methods and Protocols; Chu J. J. H., Ang S. K., Eds.; Springer Science+Business Media: New York, 2016; Vol. 1426, pp 243–253. 10.1007/978-1-4939-3618-2_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamballerie X. D.; Boisson V.; Reynier J.-C.; Enault S.; Charrel R. N.; Flahault A.; Roques P.; Grand R. L. On Chikungunya Acute Infection and Chloroquine Treatment. Vector-Borne Zoonotic Dis. 2008, 8 (6), 837–840. 10.1089/vbz.2008.0049. [DOI] [PubMed] [Google Scholar]
- Roques P.; Thiberville S. D.; Dupuis-Maguiraga L.; Lum F. M.; Labadie K.; Martinon F.; Gras G.; Lebon P.; Ng L. F. P.; de Lamballerie X.; et al. Paradoxical Effect of Chloroquine Treatment in Enhancing Chikungunya Virus Infection. Viruses 2018, 10 (5), 268. 10.3390/v10050268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M.; Santhosh S. R.; Tiwari M.; Lakshmana Rao P. V.; Parida M. Assessment of in Vitro Prophylactic and Therapeutic Efficacy of Chloroquine against Chikungunya Virus in Vero Cells. J. Med. Virol. 2010, 82 (5), 817–824. 10.1002/jmv.21663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent M. J.; Bergeron E.; Benjannet S.; Erickson B. R.; Rollin P. E.; Ksiazek T. G.; Seidah N. G.; Nichol S. T. Chloroquine Is a Potent Inhibitor of SARS Coronavirus Infection and Spread. Virol. J. 2005, 2 (1), 69. 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarino A.; Di Trani L.; Donatelli I.; Cauda R.; Cassone A.; Damme V. New Insights into the Antiviral Effects of Chloroquine. Lancet Infect. Dis. 2006, 6 (2), 67–69. 10.1016/S1473-3099(06)70361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelnabi R.; Neyts J.; Delang L. Chikungunya Virus Infections: Time to Act, Time to Treat Transmission and Prevalence. Curr. Opin. Virol. 2017, 24, 25–30. 10.1016/j.coviro.2017.03.016. [DOI] [PubMed] [Google Scholar]
- Patani G. A.; LaVoie E. J. Bioisosterism: A Rational Approach in Drug Design. Chem. Rev. 1996, 96 (8), 3147–3176. 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- Brown N.Bioisosterism in Medicinal Chemistry. In Bioisosteres in Medicinal Chemistry; Brown N., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp 1–14. [Google Scholar]
- Topliss J. G. Utilization of Operational Schemes for Analog Synthesis in Drug Design. J. Med. Chem. 1972, 15 (10), 1006–1011. 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- Topliss J. G. A Manual Method for Applying the Hansch Approach to Drug Design. J. Med. Chem. 1977, 20 (4), 463–469. 10.1021/jm00214a001. [DOI] [PubMed] [Google Scholar]
- Martyn D. C.; Nijjar A.; Celatka C. A.; Mazitschek R.; Cortese J. F.; Tyndall E.; Liu H.; Fitzgerald M. M.; O’Shea T. J.; Danthi S.; et al. Synthesis and Antiplasmodial Activity of Novel 2,4-Diaminopyrimidines. Bioorg. Med. Chem. Lett. 2010, 20 (1), 228–231. 10.1016/j.bmcl.2009.10.133. [DOI] [PubMed] [Google Scholar]
- Moussa I. A.; Banister S. D.; Beinat C.; Giboureau N.; Reynolds A. J.; Kassiou M. Design, Synthesis, and Structure-Affinity Relationships of Regioisomeric N-Benzyl Alkyl Ether Piperazine Derivatives as σ-1 Receptor Ligands. J. Med. Chem. 2010, 53 (16), 6228–6239. 10.1021/jm100639f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.