

Abstract
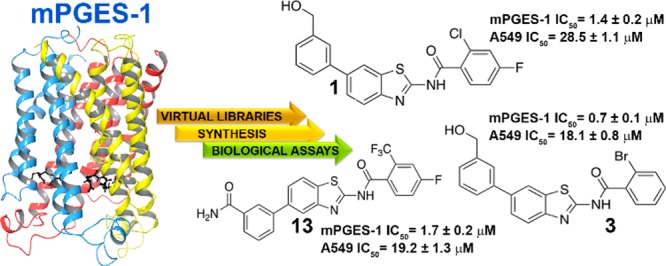
Microsomal prostaglandin E2 synthase-1 (mPGES-1), the terminal enzyme responsible for the production of inducible prostaglandin E2, has become an attractive target for the treatment of inflammation and cancer pathologies. Starting from an aminobenzothiazole scaffold, used as an unprecedented chemical core for mPGES-1 inhibition, a Combinatorial Virtual Screening campaign was conducted, using the X-ray crystal structure of human mPGES-1. Two combinatorial libraries (6 × 104) were obtained by decorating the aminobenzothiazole scaffold with all acyl chlorides and boronates available at the Merck database. The scientific multidisciplinary approach included virtual screening workflow, synthesis, and biological evaluation and led to the identification of three novel aminobenzothiazoles 1, 3, and 13 acting as mPGES-1 inhibitors. The three disclosed hits are able to inhibit mPGES-1 in a cell-free system (IC50 = 1.4 ± 0.2, 0.7 ± 0.1, and 1.7 ± 0.2 μM, respectively), and all are endowed with antitumoral properties against A549 human cancer cell lines at micromolar concentrations (28.5 ± 1.1, 18.1 ± 0.8, and 19.2 ± 1.3 μM, respectively).
Keywords: microsomal prostaglandin E2 synthase-1, prostaglandin, aminobenzothiazoles, virtual screening
Microsomal prostaglandin E2 synthase-1 (mPGES-1), the terminal enzyme responsible for the production of inducible prostaglandin (PG)E2, has become a well-established target for the treatment of cancer and inflammatory disorders, and its inhibition to control PGE2-related pathologies is an alternative option to targeting the upstream cyclooxygenases.1 Inhibition of mPGES-1 represents a promising strategy for the development of new anti-inflammatory and anticancer drugs with reduced side effects.2,3 Overexpression of mPGES-1 has been observed in inflammatory disorders as well as in many human tumors.4,5 Different clinical studies have shown increased levels of mPGES-1 in various human cancers, mainly colon,6 colorectal,7 prostate, stomach, pancreas, and cervix cancer.4 Also, the significant involvement of mPGES-1 in the genesis and progression of some tumor types has been recently recognized. Since the structural details of this glutathione-dependent enzyme have been progressively unravelled,8−13 it has become the focus of extensive studies that led to the discovery of several classes of inhibitors. However, despite the high number of inhibitors identified so far,14,15 only a few of them exhibit remarkable in vivo anticancer and anti-inflammatory activity, and to date, only GRC 27864 (Glenmark Pharmaceuticals Ltd.) and LY3023703 (Eli Lilly) have entered clinical development (phase I).16 In this context, the identification of novel mPGES-1 inhibitors represents an urgent need, especially for the treatment of colon and colorectal cancer (CRC) that are among the most widespread cancers associated with a high mortality rate.17 Continuing our research in this medicinal chemistry area, a new class of mPGES-1 inhibitors featuring an aminobenzothiazole scaffold was developed following a combinatorial approach (Figure 1). Considering the known inhibitory activity of PGE2 production by 2-aminothiazole derivatives18 and the significant biological properties of benzothiazoles,19−21 the aminobenzothiazole privileged scaffold22 was selected for our purpose. Hence, human mPGES-1 (PDB code: 4BPM)11 was used for the virtual screening study (see Supporting Information). The scientific multidisciplinary workflow (Figure 1) can be divided into the following steps: (a) creation of two combinatorial libraries; (b) molecular docking studies of the filtered combinatorial libraries and selection of the most promising hits (1–13, Figure 2); (c) synthesis of 1–13; (d) biological evaluation in cell-free and cell-based systems of the most promising compounds and identification of three novel hits (1, 3, and 13). The virtual libraries were built as illustrated in Figure 1, where all the acyl chlorides and boronates, available at the Merck database, were combined with 2-amino-6-bromobenzothiazole (scaffold A) and 2-amino-5-bromobenzothiazole (scaffold B).
Figure 1.

Scheme of the multidisciplinary approach used for the identification of novel mPGES-1 inhibitors.
Figure 2.

Molecular structure of compounds (1–13) selected from the two combinatorial libraries for the synthetic phase.
In more detail, the two scaffolds were combined with the 316 acyl chlorides at position 2 and 570 boronates at positions 5 and 6, respectively, obtaining 180,120 molecules for each library, using CombiGlide software23,24 (Figure 1).
After the generation of all tautomers and ionization states25 at a pH of 7.4 ± 1 and the filtering by their pharmacokinetic properties26 (see Supporting Information for further detail), ∼3 × 104 molecules for each scaffold (2-amino-6-bromobenzothiazoles and 2-amino-5-bromobenzothiazoles) were submitted to the successive Virtual Screening Workflow.27
Considering a docking score cutoff of −7.0 kcal/mol, 137 for the scaffold A and 190 for the scaffold B potential mPGES-1 inhibitors were then submitted to the successive detailed computational analysis. Thus, the most promising 13 molecules (1–13, Figure 2) were selected for synthesis, after excluding them to be “Pan-Assay Interference compounds” (SwissADME web tool;28 see Supporting Information). From a structural point of view, the mPGES-1 inhibitors reported to date8−11,13,29 have displayed two different binding modes (Figure 3) defining a total catalytic groove with a shape of “U” on the macromolecular surface (Figure 3). In more detail, these two models of interactions share a shallow groove located between the GSH binding site and the macromolecular surface, in the vicinity of the cytoplasmic part, mainly composed of aromatic (C:Phe44, C:His53) and polar (C:Arg52) residues (Figure 3). On the other hand, the first binding model type (PDB code: 4YK5, cocrystallized ligand depicted in purple)8 extends in the binding groove observable at the intersection between helix I of chain C and helix IV of chain A, characterized by polar (A:Gln134), aliphatic (C:Val24), and aromatic (C:Tyr28 and A:Tyr130) residues. Furthermore, the second binding model (PDB code: 4BPM, cocrystallized ligand depicted in green)11 extends its interaction in a pocket binding area detectable toward helix IV of chain A and away from the cytoplasmic part, formed by aliphatic (A:Pro124, A:Val128, A:Leu132) and polar (A:Ser127, A:Thr131) residues. On these premises, the combinatorial approach well matches with the possibility to explore the total binding groove with a U-shape (Figure 3), decorating the aminobenzothiazole scaffold (A and B) with residues of different size that could cover it.
Figure 3.
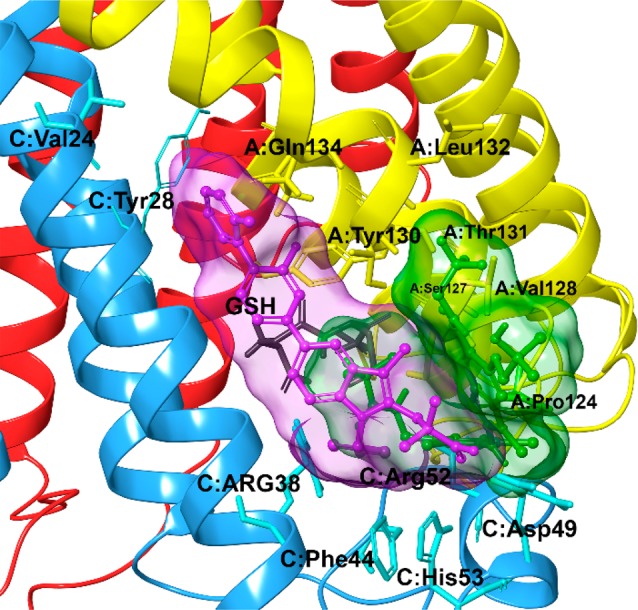
Two different models of interactions of cocrystallized mPGES-1 inhibitors on the receptor surface (pdb code: 4BPM).11 The receptor is represented by ribbons colored by chain (chain A is colored in yellow, chain B is colored in red, and chain C is colored in cyan). Ligand related to 4YK5(8) pdb code is depicted in purple; ligand related to 4BPM(11) pdb code is depicted in green.
After these steps, to retrospectively analyze the binding modes of novel binders and to filter molecular docking results, specific key interactions, reported by us12,30−34 and other research groups, were considered as essential contacts for mPGES-1 inhibition8−11,13,29 during the computational analysis. Hence, the optimal library members (1–13) were rationally selected for successive organic synthesis (Figure 2). The synthetic routes of the identified molecules are depicted in Figure 4. Pd-catalyzed Suzuki–Miyaura cross-coupling was performed between 3-(hydroxymethyl)phenylboronic acid I and 2-amino-6-bromobenzothiazole (scaffold A) or 2-amino-5-bromobenzothiazole (scaffold B), respectively. The Suzuki–Miyaura reaction was chosen for our purpose because it is an efficient and versatile method for the synthesis of highly functionalized biaryls in good yields under mild conditions. For the preparation of our compounds, the cross-coupling was performed under standard conditions using Pd(PPh3)4 as the catalyst and aqueous carbonate base in dioxane at 80 °C. Reactions occurred with 75–88% yields.
Figure 4.

Synthetic scheme of 5-substituted 2-aminobenzothiazole and 6-substituted 2-aminobenzothiazole. Reagents and conditions: (a) I, K2CO3, Pd(PPh3)4, 1,4-dioxane–H2O, 80 °C; (b) (CH3)3SiCl, pyridine, CH3CN, rt; (c) RCOCl; (d) I, K2CO3, Pd(PPh3)4, 1,4-dioxane–H2O, 80 °C; (e) (CH3)3SiCl, pyridine, CH3CN, rt; (f) RCOCl; (g) RCOCl, pyridine, CH3CN, rt; (h) II, K2CO3, Pd(PPh3)4, 1,4-dioxane–H2O, 80 °C.
Since the (hydroxymethyl)phenyl portion presented a primary alcohol, it was necessary to protect the hydroxyl group before acylation. We opted for transient protection by trimethylsilyl ether which was formed in situ in dry acetonitrile at room temperature with pyridine and removed during acidic workup. Acylation of the protected aminobenzothiazole nucleus was then accomplished by adding the proper acyl chloride to the reaction mixture. Compounds 1–10 were obtained with moderate to high yields (32–85%). For 11–13, a different synthetic approach was preferred because acylation of 2-amino-5-bromobenzothiazole gave less polar compounds with consequent more accessible purification procedures. Acylation reactions were conducted in dry acetonitrile with pyridine at room temperature. 16–18 were then subjected to Suzuki–Miyaura coupling with 3-aminocarbonylphenylboronic acid to give 11–13. All compounds were purified by reversed phase HPLC and obtained with >98% purity.
The effects of 1–13 against mPGES-1 activity were studied by monitoring the enzymatic conversion of the substrate PGH2 (20 μM) to PGE2 in a cell-free assay using microsomes of IL-1β-stimulated A549 cells as a source of mPGES-1 (Table S1 in Supporting Information). All compounds carrying the (hydroxymethyl)phenyl residue at position 5 of the benzothiazole scaffold (7–10) showed a residual mPGES-1 activity at 10 μM higher than 30%. The replacement of the (hydroxymethyl)phenyl residue at position 5 with a 3-aminocarbonylphenyl group in compounds 11 and 12 did not improve the potency against mPGES-1.
Indeed, compounds 11 and 12 with the same acyl group failed in mPGES-1 inhibition. However, when a 4-fluoro-2-(trifluoromethyl)phenyl moiety was introduced as an acyl moiety (13), mPGES-1 inhibitory activity was improved (Table S1 in Supporting Information). The introduction of the (hydroxymethyl)phenyl residue at position 6 of the benzothiazole further enhanced the potency. The presence of a pyridine ring in the amide group was detrimental, as shown for compounds 4 and 5. Replacing the pyridine with a 2,5-dimethylfuran (compound 6) yielded an IC50 value of 2.6 ± 0.1 μM. Contrariwise, the translation of the (hydroxymethyl)phenyl moiety from the 5- to 6-position increased the inhibitory potency in compounds 1 and 3, which are regioisomers of compounds 8 and 7, respectively (Table S1 in Supporting Information).
Based on the inhibitory activity against mPGES-1 (Table 1), 1, 3, 6, and 13 were selected as promising hits for further cell-based studies. Considering that mPGES-1 is found overexpressed in A549 human lung epithelial cancer cells,35 the effect of these compounds on PGE2 production in A549 cells was evaluated. In more detail, A549 cancer cells were incubated with IL-1β and test compounds (5 or 10 μM) for 24 h. All molecules tested reduced the cytokine-induced PGE2 production in a concentration-dependent manner (Figure 5) without significantly affecting cell viability at 24 h. The antiproliferative or cytotoxic effect of compounds 1, 3, 6, and 13 was evaluated in human A549 cells upon long-term incubation with increasing compound concentrations. The cancer cells were incubated for 48 and 72 h with 1, 3, 6, and 13 (5–100 μM) or the reference inhibitor CAY10526 (1–50 μM), and cell viability was monitored by MTT assay. The compound CAY10526 inhibits PGE2 production through the selective modulation of mPGES-1 expression but does not affect COX-2 expression,36 and it was used as a control in our experiments. All the compounds tested inhibited cancer cell viability (Table 2). Compounds 1, 3, and 13 were proven to be the most active and were used in further analysis.
Table 1. Effect of Tested Compounds on mPGES-1 Activitya.
| Compound | IC50 ± SD (μM) |
|---|---|
| 1 | 1.4 ± 0.2 |
| 3 | 0.7 ± 0.1 |
| 6 | 2.6 ± 0.1 |
| 13 | 1.7 ± 0.2 |
IC50 (μM) values are given as mean ± S.D. of single determinations obtained in three independent experiments.
Figure 5.

A549 cells were incubated for 24 h with 5 or 10 μM of compounds 1, 3, 6, and 13 in conditioned medium (1% FBS and 10 ng/mL IL-1β). PGE2 released into the medium was quantified by ELISA. The data are compared to vehicle (DMSO)-treated cells and expressed as mean ± SEM (pg/mL) of two independent experiments.
Table 2. Anti-proliferative Effect of mPGES-1 Inhibitors in the A549 Human Cancer Cell Line after 24, 48 or 72 h of Treatmenta.
| IC50 ± SD (μM) |
|||
|---|---|---|---|
| compd | 24 h | 48 h | 72 h |
| 1 | 98.2 ± 1.5 | 71.4 ± 1.6 | 28.5 ± 1.1 |
| 3 | 97.5 ± 2.0 | 57.5 ± 1.5 | 18.1 ± 0.8 |
| 6 | 95.0 ± 1.7 | 63.2 ± 2.1 | 55.2 ± 1.5 |
| 13 | 98.3 ± 1.9 | 50.6 ± 2.1 | 19.2 ± 1.3 |
| CAY10526 | 98.5 ± 1.6 | 12.5 ± 0.4 | 9.1 ± 0.5 |
IC50 (μM) values are given as mean ± S.D. of single determinations obtained in three independent experiments.
To explore the mechanisms underlying the repression of cancer cell viability, the effect of compounds 1, 3, and 13 was analyzed on cell cycle progression in A549 cancer cells by flow cytometry.
Compound concentrations were selected close or above the IC50 values for inhibition of PGE2 biosynthesis.
All compounds tested (1, 3, 13) induced a G0/G1 arrest in a time- and concentration-dependent manner (Figure 6A–C).
Figure 6.

A549 cancer cells were treated with compounds 1 (a), 3 (b), and 13 (c) at different concentrations for 48 or 72 h. The percentage of cell cycle stages was analyzed after DNA propidium iodide staining by flow cytometry. Results are expressed as means ± SEM of two independent experiments performed in triplicate (*P < 0.05, **P < 0.005,***P < 0.001).
For high concentrations of compound 3 (30 μM) and long-term incubation (72 h), the initial G0/G1 arrest was complemented by a G2/M arrest, which apparently prevents the transition of cells thereby again reducing the number of cells in G0/G1 phase.
Moreover, compounds 3 and 13 induced moderate programmed cell death at high concentrations (20–30 μM), as indicated by the increase of the subG0/G1 cell fraction (6 ± 0.5% after 48 h and 7 ± 0.7% after 72 h for 13; 5 ± 0.9% after 48 h and 18 ± 1.2 after 72 h for 3). Conversely, compound 1 did not show a cell-death-related hypodiploidy within 48 to 72 h (Figure 7A).
Figure 7.
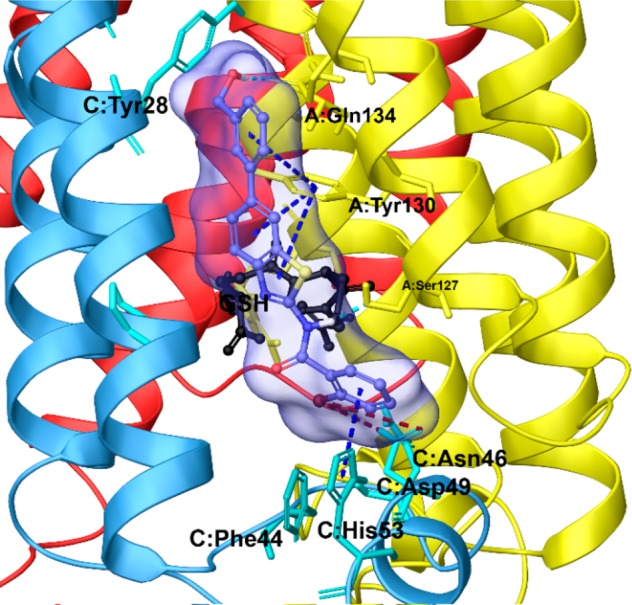
Three dimensional model of the pattern interaction between 3 (violet sticks) and mPGES-1 (colored by chain, namely chain A is depicted in yellow, chain B in red, and chain C in cyan ribbons).
The detailed description of the ligand/target interactions between mPGES-1 and 3, the most active compound of libraries (see above), was chosen as a representative case to elucidate at molecular level the structural basis responsible for enzyme inhibition (Figure 7). The benzothiazole scaffold assumes a binding mode similar to the cocrystallized ligand reported in purple in Figure 3. In more detail, 3 fits to the binding site (see above) defined by (a) the cytoplasmic part of the protein, featuring aromatic (C:Phe44, C:His53) and polar (C:Asp49) amino acids; (b) the cofactor (GSH) binding region in the deeper part of the binding pocket;37 (c) a binding groove between helix 1 of chain C and helix 4 of chain A, with polar (A:Gln134) and aromatic residues (C:Tyr28 and A:Tyr130). Furthermore, the substituents decorating the scaffold make specific contacts with the receptor counterpart: (a) the bromine atom forms halogen bonds with Asp49ChainC; (b) −OH and −NH groups establish hydrogen bonds with Gln134ChainA and Ser127ChainA; (c) benzothiazole and benzene rings are involved in two π–π stacking with Tyr130ChainA and His53ChainC, respectively.
In summary, the reported combinatorial approach, coupled with an efficient multistep virtual screening workflow, has accelerated and facilitated the identification of novel mPGES-1 inhibitors (1, 3, and 13), active at low micromolar concentrations against A549 human cancer cell lines. The discovered aminobenzothiazole scaffold can suppress PGE2 biosynthesis representing an interesting chemical core and a new promising template for developing anticancer agents that could be easily decorated according to convenient multicomponent organic reactions.
Furthermore, we have provided a suitable strategy based on a multistep virtual screening protocol of a combinatorial library for the identification of novel biologically active hits.
Glossary
Abbreviations
- CRC
colon and colorectal cancer
- IL
interleukin
- mPGES-1
microsomal prostaglandin E2 synthase
- LT
leukotriene
- PG
Prostaglandin
- SP
standard precision
- VSW
virtual screening workflow
- XP
extra precision
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.9b00618.
NMR spectra and synthetic procedures; experimental details of computational and biological studies (PDF)
Author Contributions
∇ M.G.C. and A.G. contributed equally to this work.
The research leading to these results has received funding from AIRC under IG 2018 - ID. 21397 project – P.I. Bifulco Giuseppe, and from the Deutsche Forschungsgemeinschaft, SFB1278 Polytarget, to O.W.
The authors declare no competing financial interest.
This paper was originally published ASAP on March 10, 2020. The surname of the fifth author was corrected, and the paper was reposted on March 31, 2020.
Supplementary Material
References
- Koeberle A.; Laufer S. A.; Werz O. Design and Development of Microsomal Prostaglandin E2 Synthase-1 Inhibitors: Challenges and Future Directions. J. Med. Chem. 2016, 59 (13), 5970–5986. 10.1021/acs.jmedchem.5b01750. [DOI] [PubMed] [Google Scholar]
- Elinav E.; Nowarski R.; Thaiss C. A.; Hu B.; Jin C.; Flavell R. A. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13 (11), 759–771. 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- Korniluk A.; Koper O.; Kemona H.; Dymicka-Piekarska V. From inflammation to cancer. Ir. J. Med. Sci. 2017, 186 (1), 57–62. 10.1007/s11845-016-1464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M.; Gokhale V.; Meuillet E. J.; Rosenberg D. W. mPGES-1 as a target for cancer suppression: A comprehensive invited review ″Phospholipase A2 and lipid mediators″. Biochimie 2010, 92 (6), 660–664. 10.1016/j.biochi.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen N. X.; Li D.; Stahle M. Transition from inflammation to proliferation: a critical step during wound healing. Cell. Mol. Life Sci. 2016, 73 (20), 3861–3885. 10.1007/s00018-016-2268-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y.; Nakatani Y.; Hara S. Role of microsomal prostaglandin E synthase-1 (mPGES-1)-derived prostaglandin E2 in colon carcinogenesis. Prostaglandins Other Lipid Mediators 2015, 121 (Pt A), 42–45. 10.1016/j.prostaglandins.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Loftus E. V. Jr. Epidemiology and risk factors for colorectal dysplasia and cancer in ulcerative colitis. Gastroenterol. Clin. North Am. 2006, 35 (3), 517–531. 10.1016/j.gtc.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Luz J. G.; Antonysamy S.; Kuklish S. L.; Condon B.; Lee M. R.; Allison D.; Yu X. P.; Chandrasekhar S.; Backer R.; Zhang A.; Russell M.; Chang S. S.; Harvey A.; Sloan A. V.; Fisher M. J. Crystal Structures of mPGES-1 Inhibitor Complexes Form a Basis for the Rational Design of Potent Analgesic and Anti-Inflammatory Therapeutics. J. Med. Chem. 2015, 58 (11), 4727–4737. 10.1021/acs.jmedchem.5b00330. [DOI] [PubMed] [Google Scholar]
- Schiffler M. A.; Antonysamy S.; Bhattachar S. N.; Campanale K. M.; Chandrasekhar S.; Condon B.; Desai P. V.; Fisher M. J.; Groshong C.; Harvey A.; Hickey M. J.; Hughes N. E.; Jones S. A.; Kim E. J.; Kuklish S. L.; Luz J. G.; Norman B. H.; Rathmell R. E.; Rizzo J. R.; Seng T. W.; Thibodeaux S. J.; Woods T. A.; York J. S.; Yu X. P. Discovery and Characterization of 2-Acylaminoimidazole Microsomal Prostaglandin E Synthase-1 Inhibitors. J. Med. Chem. 2016, 59 (1), 194–205. 10.1021/acs.jmedchem.5b01249. [DOI] [PubMed] [Google Scholar]
- Partridge K. M.; Antonysamy S.; Bhattachar S. N.; Chandrasekhar S.; Fisher M. J.; Fretland A.; Gooding K.; Harvey A.; Hughes N. E.; Kuklish S. L.; Luz J. G.; Manninen P. R.; McGee J. E.; Mudra D. R.; Navarro A.; Norman B. H.; Quimby S. J.; Schiffler M. A.; Sloan A. V.; Warshawsky A. M.; Weller J. M.; York J. S.; Yu X. P. Discovery and characterization of [(cyclopentyl)ethyl]benzoic acid inhibitors of microsomal prostaglandin E synthase-1. Bioorg. Med. Chem. Lett. 2017, 27 (6), 1478–1483. 10.1016/j.bmcl.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Li D.; Howe N.; Dukkipati A.; Shah S. T.; Bax B. D.; Edge C.; Bridges A.; Hardwicke P.; Singh O. M.; Giblin G.; Pautsch A.; Pfau R.; Schnapp G.; Wang M.; Olieric V.; Caffrey M. Crystallizing Membrane Proteins in the Lipidic Mesophase. Experience with Human Prostaglandin E2 Synthase 1 and an Evolving Strategy. Cryst. Growth Des. 2014, 14 (4), 2034–2047. 10.1021/cg500157x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren T.; Nord J.; Ek M.; Johansson P.; Liu G.; Geschwindner S. Crystal structure of microsomal prostaglandin E-2 synthase provides insight into diversity in the MAPEG superfamily. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (10), 3806–3811. 10.1073/pnas.1218504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuklish S. L.; Antonysamy S.; Bhattachar S. N.; Chandrasekhar S.; Fisher M. J.; Fretland A. J.; Gooding K.; Harvey A.; Hughes N. E.; Luz J. G.; Manninen P. R.; McGee J. E.; Navarro A.; Norman B. H.; Partridge K. M.; Quimby S. J.; Schiffler M. A.; Sloan A. V.; Warshawsky A. M.; York J. S.; Yu X. P. Characterization of 3,3-dimethyl substituted N-aryl piperidines as potent microsomal prostaglandin E synthase-1 inhibitors. Bioorg. Med. Chem. Lett. 2016, 26 (19), 4824–4828. 10.1016/j.bmcl.2016.08.023. [DOI] [PubMed] [Google Scholar]
- Wang D.; Dubois R. N. Prostaglandins and cancer. Gut 2006, 55 (1), 115–122. 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk C. D. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 2001, 294 (5548), 1871–1875. 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y.; Narumiya S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282 (16), 11613–11617. 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Ke J.; Yang Y.; Che Q.; Jiang F.; Wang H.; Chen Z.; Zhu M.; Tong H.; Zhang H.; Yan X.; Wang X.; Wang F.; Liu Y.; Dai C.; Wan X. Prostaglandin E2 (PGE2) promotes proliferation and invasion by enhancing SUMO-1 activity via EP4 receptor in endometrial cancer. Tumor Biol. 2016, 37 (9), 12203–12211. 10.1007/s13277-016-5087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B.; Chang H. H.; Medda F.; Gokhale V.; Dietrich J.; Davis A.; Meuillet E. J.; Hulme C. Synthesis and biological activity of 2-aminothiazoles as novel inhibitors of PGE2 production in cells. Bioorg. Med. Chem. Lett. 2012, 22 (10), 3567–3570. 10.1016/j.bmcl.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P. C.; Sinhmar A.; Sharma A.; Rajak H.; Pathak D. P. Medicinal significance of benzothiazole scaffold: an insight view. J. Enzyme Inhib. Med. Chem. 2013, 28 (2), 240–266. 10.3109/14756366.2012.720572. [DOI] [PubMed] [Google Scholar]
- Keri R. S.; Patil M. R.; Patil S. A.; Budagumpi S. A comprehensive review in current developments of benzothiazole-based molecules in medicinal chemistry. Eur. J. Med. Chem. 2015, 89, 207–251. 10.1016/j.ejmech.2014.10.059. [DOI] [PubMed] [Google Scholar]
- Baud M. G. J.; Bauer M. R.; Verduci L.; Dingler F. A.; Patel K. J.; Horil Roy D.; Joerger A. C.; Fersht A. R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018, 152, 101–114. 10.1016/j.ejmech.2018.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.; Dietrich J. Privileged scaffolds in lead generation. Expert Opin. Drug Discovery 2015, 10 (7), 781–790. 10.1517/17460441.2015.1041496. [DOI] [PubMed] [Google Scholar]
- Chen K.; Huang J.; Gong W.; Iribarren P.; Dunlop N. M.; Wang J. M. Toll-like receptors in inflammation, infection and cancer. Int. Immunopharmacol. 2007, 7 (10), 1271–1285. 10.1016/j.intimp.2007.05.016. [DOI] [PubMed] [Google Scholar]
- CombiGlide; Schrödinger, LLC: New York, NY, 2017.
- LigPrep; Schrödinger, LLC: New York, NY, 2017.
- QikProp; Schrödinger, LLC: New York, NY, 2017.
- Virtual Screening Workflow; Schrödinger, LLC: New York, NY, 2017.
- Daina A.; Michielin O.; Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjogren T.; Nord J.; Ek M.; Johansson P.; Liu G.; Geschwindner S. Crystal structure of microsomal prostaglandin E2 synthase provides insight into diversity in the MAPEG superfamily. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (10), 3806–3811. 10.1073/pnas.1218504110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iranshahi M.; Chini M. G.; Masullo M.; Sahebkar A.; Javidnia A.; Chitsazian Yazdi M.; Pergola C.; Koeberle A.; Werz O.; Pizza C.; Terracciano S.; Piacente S.; Bifulco G. Can Small Chemical Modifications of Natural Pan-inhibitors Modulate the Biological Selectivity? The Case of Curcumin Prenylated Derivatives Acting as HDAC or mPGES-1 Inhibitors. J. Nat. Prod. 2015, 78 (12), 2867–2879. 10.1021/acs.jnatprod.5b00700. [DOI] [PubMed] [Google Scholar]
- Chini M. G.; Ferroni C.; Cantone V.; Dambruoso P.; Varchi G.; Pepe A.; Fischer K.; Pergola C.; Werz O.; Bruno I.; Riccio R.; Bifulco G. Elucidating new structural features of the triazole scaffold for the development of mPGES-1 inhibitors. MedChemComm 2015, 6 (1), 75–79. 10.1039/C4MD00319E. [DOI] [Google Scholar]
- Lauro G.; Manfra M.; Pedatella S.; Fischer K.; Cantone V.; Terracciano S.; Bertamino A.; Ostacolo C.; Gomez-Monterrey I.; De Nisco M.; Riccio R.; Novellino E.; Werz O.; Campiglia P.; Bifulco G. Identification of novel microsomal prostaglandin E2 synthase-1 (mPGES-1) lead inhibitors from Fragment Virtual Screening. Eur. J. Med. Chem. 2017, 125, 278–287. 10.1016/j.ejmech.2016.09.042. [DOI] [PubMed] [Google Scholar]
- Di Micco S.; Spatafora C.; Cardullo N.; Riccio R.; Fischer K.; Pergola C.; Koeberle A.; Werz O.; Chalal M.; Vervandier-Fasseur D.; Tringali C.; Bifulco G. 2,3-Dihydrobenzofuran privileged structures as new bioinspired lead compounds for the design of mPGES-1 inhibitors. Bioorg. Med. Chem. 2016, 24 (4), 820–826. 10.1016/j.bmc.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Di Micco S.; Terracciano S.; Cantone V.; Fischer K.; Koeberle A.; Foglia A.; Riccio R.; Werz O.; Bruno I.; Bifulco G. Discovery of new potent molecular entities able to inhibit mPGES-1. Eur. J. Med. Chem. 2018, 143, 1419–1427. 10.1016/j.ejmech.2017.10.039. [DOI] [PubMed] [Google Scholar]
- Hanaka H.; Pawelzik S. C.; Johnsen J. I.; Rakonjac M.; Terawaki K.; Rasmuson A.; Sveinbjornsson B.; Schumacher M. C.; Hamberg M.; Samuelsson B.; Jakobsson P. J.; Kogner P.; Radmark O. Microsomal prostaglandin E synthase 1 determines tumor growth in vivo of prostate and lung cancer cells. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (44), 18757–18762. 10.1073/pnas.0910218106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero M. D.; Aquino M.; Bruno I.; Terencio M. C.; Paya M.; Riccio R.; Gomez-Paloma L. Synthesis and pharmacological evaluation of a selected library of new potential anti-inflammatory agents bearing the gamma-hydroxybutenolide scaffold: a new class of inhibitors of prostanoid production through the selective modulation of microsomal prostaglandin E synthase-1 expression. J. Med. Chem. 2007, 50 (9), 2176–2184. 10.1021/jm0700823. [DOI] [PubMed] [Google Scholar]
- Brock J. S.; Hamberg M.; Balagunaseelan N.; Goodman M.; Morgenstern R.; Strandback E.; Samuelsson B.; Rinaldo-Matthis A.; Haeggstrom J. Z. A dynamic Asp-Arg interaction is essential for catalysis in microsomal prostaglandin E2 synthase. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (4), 972–977. 10.1073/pnas.1522891113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.