

Abstract
Signaling bias is the propensity for some agonists to preferentially stimulate G protein-coupled receptor (GPCR) signaling through one intracellular pathway versus another. We previously identified a G protein-biased agonist of the D2 dopamine receptor (D2R) that results in impaired β-arrestin recruitment. This signaling bias was predicted to arise from unique interactions of the ligand with a hydrophobic pocket at the interface of the second extracellular loop and fifth transmembrane segment of the D2R. Here, we showed that residue Phe189 within this pocket (position 5.38 using Ballesteros-Weinstein numbering) functions as a micro-switch for regulating receptor interactions with β-arrestin. This residue is relatively conserved among class A GPCRs, and analogous mutations within other GPCRs similarly impaired β-arrestin recruitment while maintaining G protein signaling. To investigate the mechanism of this signaling bias, we used an active state structure of the β2-adrenergic receptor (β2R) to build β2R-WT and β2R-Y1995.38A models in complex with the full β2R agonist BI-167107 for molecular dynamics simulations. These analyses identified conformational rearrangements in β2R-Y1995.38A that propagated from the extracellular ligand binding site to the intracellular surface, resulting in a modified orientation of the second intracellular loop in β2R-Y1995.38A, which is predicted to affect its interactions with β-arrestin. Our findings provide a structural basis for how ligand binding site alterations can allosterically affect GPCR-transducer interactions and result in biased signaling.
Introduction
G protein-coupled receptors (GPCRs) represent the largest family of cellular receptors in mammals and are critical drug targets accounting for approximately one third of all FDA-approved drugs (1). These receptor proteins regulate multiple physiological processes by transducing extracellular stimuli, such as neurotransmitters, hormones, peptides, or light, into intracellular signals through activating both G protein-dependent and independent pathways, leading to second messenger generation and downstream signaling events. G protein-independent pathways are primarily mediated by β-arrestin proteins (2–4), which were originally identified as mediators of agonist-induced desensitization and receptor endocytosis, but were subsequently determined to also function as multi-valent scaffolding proteins that orchestrate various intracellular signaling pathways (5). While endogenous agonists promote GPCR signaling through the activation of both G proteins and β-arrestins, these events often occur in a temporally separate fashion (6–8). In contrast, some synthetic agonists have been described to preferentially activate discrete signaling pathways versus others, a phenomenon known as functional selectivity or biased signaling (9–13). The therapeutic potential of biased signaling is high because drugs that selectively modulate clinically relevant pathways, without affecting other signaling events, may exhibit fewer side effects (14, 15). Although the molecular mechanisms underlying biased signaling are not known with certainty, a leading hypothesis is that GPCRs can adopt distinct active conformational states that are selectively stabilized by different signaling biased ligands (16–20). A detailed understanding of the structural determinants underlying agonist-specific signaling states of GPCRs should allow for the rational design of novel functionally-selective agents (13).
Biased signaling can occur not only in response to ligands, but also from mutations in GPCRs resulting in restricted signaling to specific pathways. For instance, Caron and colleagues have used the evolutionary trace (ET) method (21) to identify D2 dopamine receptor (D2R) mutants that selectively signal through either G proteins or β-arrestins (22, 23). Similarly, Schönegge et al. (24) used the same ET method to identify signaling biased mutants of the β2-adrenergic receptor (β2R) that were selectively impaired in either Gi- or β-arrestin-, but not Gs-mediated signaling. Conversely, Donthamsetti et al. (25) reported a double mutant of the D2R that could robustly recruit β-arrestin but that was devoid of G protein-mediated signaling. β-arrestin-biased mutants of the M3 muscarinic receptor have also been developed for use in the “designer receptors exclusively activated by designer drugs” (DREADD) technology (26). None of the mutations described in these previous studies were within, or close to, the ligand binding sites, but rather were situated near the intracellular surface of the receptors. Further, the previously studied mutations were not investigated using related GPCRs and thus their generalizability is unclear.
We previously described a G protein-biased D2R agonist, MLS1547, that is efficacious for G protein-mediated signaling, but relatively ineffective in β-arrestin recruitment (27, 28). Structure-activity relationship analyses using MLS1547 and its analogs led to a pharmacophore model in the context of receptor structure to explain the biased signaling properties of this compound. This involved the interaction of the ligand with a hydrophobic pocket comprised of residues Ile184, Phe189 and Val190 within the fifth transmembrane region (TM5) and second extracellular loop (EL2) of the D2R. Here, we identify residue Phe189 in the D2R (position 5.38 using the Ballesteros-Weinstein numbering system (29)) as a micro-switch that regulates the active state for recruiting β-arrestin. Our findings showed that such a switch exists not only for the D2R, but also for several related GPCRs, including the β2R. Molecular dynamics simulations using an active state structure of the β2R (30) revealed that mutation of residue 5.38 resulted in conformational rearrangements that propagate from the extracellular ligand binding site to the intracellular surface, leading to an altered orientation of intracellular loop 2 (IL2), which is predicted to affect β-arrestin interactions, thus conferring biased signaling.
Results
Investigation of structural elements supporting signaling bias by the D2R agonist MLS1547
We previously suggested (27, 28) that the G protein-biased agonist MLS1547 uniquely interacts with a hydrophobic pocket of the D2R comprised of residues Ile184EL2, Phe1895.38, and Val1905.39 at the junction between the extracellular tip of TM5 and EL2 of the D2R (Fig. 1A) (31). Detailed structure-activity analyses showed that congeneric compounds of MLS1547 lacking a hydrophobic moiety oriented towards this pocket exhibit more balanced G protein- and β-arrestin-mediated signaling (27, 28), supporting the idea that ligand interactions with this pocket confer signaling bias. To further investigate the role of this binding pocket in D2R signaling, we created alanine mutations of the residues enclosing this pocket (I184EL2A, F1895.38A, and V1905.39A). We found that the singly mutated receptors were expressed at a comparable degree as the wild-type D2R (D2R-WT) in cells also expressing G protein (fig. S1A) and β-arrestin (fig. S1B) assay components. The I184EL2A or V1905.39A mutations decreased the potency and maximum response of MLS1547 for G protein activation, as measured using a bioluminesence resonance energy transfer (BRET)-based assay with biosensors fused to α and γ subunits of Go, an endogenous transducer of the D2R (32) (Fig. 1B, table S1). The effects of the F1895.38A mutation were more pronounced, resulting in a complete loss of MLS1547’s ability to activate Go (Fig. 1B, table S1). However, MLS1547 could still interact with the D2R F1895.38A, as demonstrated by its ability to functionally antagonize dopamine signaling (Fig. 1C) and compete for radioligand binding (table S2). These results suggest that the primary effect of the F1895.38A mutation is the elimination of MLS1547 efficacy for G protein activation. These results highlight the importance of this hydrophobic pocket and, in particular, identify Phe1895.38 in TM5 as a pivotal residue in regulating the biased signaling of MLS1547 through the D2R.
Figure 1. Investigation of structural elements supporting G protein-biased signaling by the D2R.
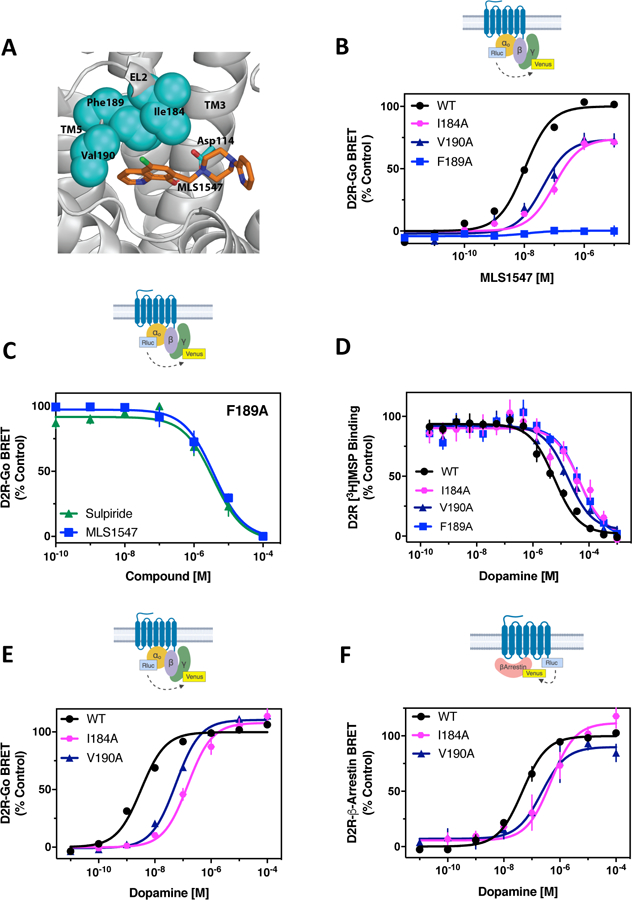
(A) Pharmacophore model for MLS1547 interactions with the D2R (modified from (27)). (B) The D2R-WT or the indicated D2R mutants were expressed in HEK293 cells with Goα1-Rluc8, β1, and γ2-mVenus. The cells were stimulated with MLS1547 and assayed for G protein activation by BRET. (C) HEK293 cells expressing D2R-F1895.38A, Goα1-Rluc8, β1, and γ2-mVenus were incubated with 13 µM (EC80) dopamine and the indicated concentrations of either sulpiride or MLS1547 and assayed for G protein activation by BRET. (D) Membrane preparations from HEK293 cells expressing either D2R-WT or D2R-I184EL2A, V1905.39A or F1895.38A were incubated with the indicated concentrations of dopamine and [3H]methylspiperone. Data are expressed as a percentage of the specific binding and fit using non-linear regression analyses (table S2). (E) HEK293 cells described in (B) were stimulated with dopamine and assayed for G protein activation. (F) The D2R-WT and indicated mutant receptors were fused to Rluc8 and expressed with β-arrestin2-mVenus and GRK2 in HEK293 cells. Dopamine-stimulated β-arrestin recruitment was assessed by BRET. Functional data are expressed as a percentage of the maximum dopamine or MLS1547 responses for D2R-WT (% control). Data in (B) to (F) represent the mean ± SEM values of 3–5 independent experiments performed in technical triplicate. Average EC50 and Emax values for functional assays are displayed in table S1.
Identification of a G protein signaling-biased mutant D2R
We next evaluated if perturbation of this hydrophobic pocket affected the signaling properties of dopamine through the D2R. Radioligand binding assays revealed that the I184EL2A, F1895.38A, and V1905.39A mutant receptors exhibited a 3–10-fold reduction in the affinity of dopamine (Fig. 1D, table S2). For the I184EL2A and V1905.39A mutants, the potency for dopamine activation of Go was reduced without changes in the maximum response (Fig. 1E, table S1). Similarly, BRET-based analysis of β-arrestin recruitment to the I184EL2A and V1905.39A mutants showed that the potency of dopamine was reduced without a change in Emax (Fig. 1F, table S1). For the D2R-F1895.38A mutant, the potency of dopamine was reduced for Go activation, similar to the I184EL2A and V1905.39A mutants, whereas the maximum response was comparable to that of the wild-type receptor (Fig. 2A, table S1). Similar results were observed using a different G protein-mediated assay measuring D2R-mediated inhibition of forskolin-stimulated cAMP accumulation (Fig. 2B, table S3). As with the Go activation assay, the D2R-F1895.38A mutant exhibited a decrease in dopamine potency for inhibiting cellular cAMP levels, although the maximal response for this response was comparable to that for D2R-WT (Fig. 2B, table S3). Thus, although the D2R-F1895.38A displayed reduced potency for dopamine stimulation of G protein-mediated signaling, the maximum response appeared to be unchanged. In contrast, dopamine was unable to stimulate β-arrestin recruitment for the D2R-F1895.38A, as assessed by either the β-arrestin BRET assay (Fig. 2C) or an enzyme complementation assay that measures the recruitment of β-arrestin to the receptor (Fig. 2D). Similar results were observed for other full agonists such that β-arrestin recruitment was either lost (for pramipexole and quinpirole) or greatly diminished (for rotigotine and apomorphine) with the D2R-F1895.38A mutant, whereas G protein activation was largely maintained with variable decreases in potency (fig. S2, A to H, and table S4). Calculation of bias factors that take into account effects on both EC50 and Emax (33) for rotigotine and apomorphine, which exhibit residual β-arrestin recruitment in the D2R-F1895.38A mutant, confirmed their G protein-mediated signaling bias (table S4).
Figure 2. The F1895.38A mutation confers G protein signaling bias in the D2R.
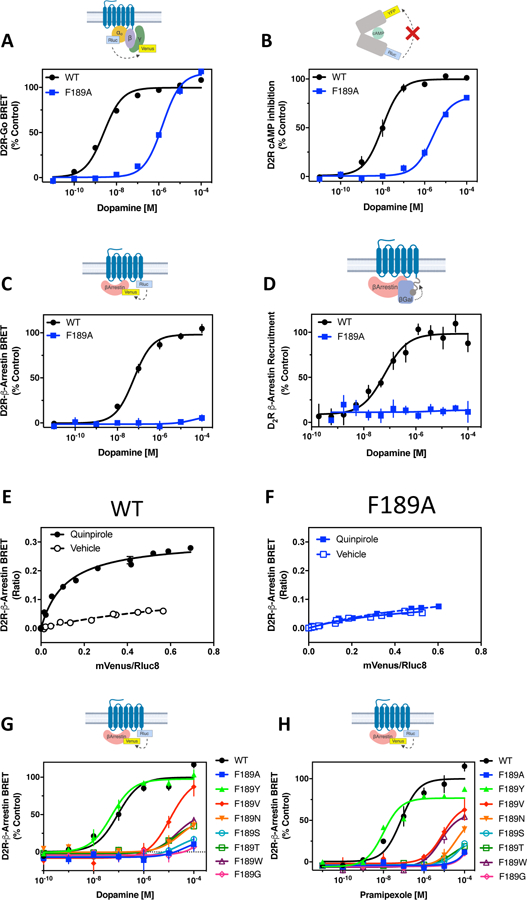
(A) HEK293 cells transiently expressing either D2R-WT or D2R-F1895.38A with Goα1-Rluc8, β1, and γ2-mVenus were stimulated with dopamine and assayed for G protein activation by BRET. Average EC50 and Emax values are displayed in table S1. (B) HEK293 cells transiently expressing D2R-WT or D2R-F1895.38A with the CAMYEL biosensor were assayed for inhibition of forskolin-stimulated cAMP production. Average EC50 and Emax values are displayed in table S3. (C) The D2R-WT and F1895.38A receptors were fused to Rluc8 and expressed in HEK293 cells with β-arrestin2-mVenus and GRK2. Dopamine-stimulated β-arrestin recruitment was assessed by BRET. Average EC50 and Emax values are displayed in table S1. (D) D2R-WT or D2R-F1895.38A were fused to a segment of β-galactosidase and expressed in CHO cells with β-arrestin2 fused to a complementing segment of β-galactosidase. Dopamine-stimulated complementation of β-galactosidase was measured. Average EC50 and Emax values are shown in table S3. (E to F) Molecular proximity between D2R-WT or D2R-F1895.38A and β-arrestin2 was detected with titration experiments performed in HEK293 cells. Cells expressing a fixed amount of D2R-WT-Rluc8 or D2R-F1895.38A-Rluc8 and increasing amounts of β-arrestin2-mVenus were incubated in the presence or absence of 10 μM quinpirole. β-arrestin recruitment was assessed by BRET. X-axes represent the ratio between the fluorescence emitted by β-arrestin2-mVenus and the luminescence emitted by D2R-WT or D2R-F1895.38A-Rluc8. Y-axes represent the BRET ratio. (G to H) The D2R-WT and indicated mutant receptors fused to Rluc8 were expressed in HEK293 cells with β-arrestin2-mVenus and GRK2. Dopamine- (G) or pramipexole (H)-stimulated β-arrestin recruitment was assessed by BRET. Average EC50 and Emax values are displayed in table S5. All functional data are expressed as percentage of the maximum response observed for D2R-WT. Data points in (A) to (H) represent mean ± SEM of 3–14 independent experiments performed in technical triplicate.
To further confirm the diminished ability of agonists to recruit β-arrestin to the D2R-F1895.38A mutant, we performed a BRET saturation assay (Fig. 2, E and F) in which the expression of the BRET donor (D2R-Rluc8) was held constant whereas that of the BRET acceptor (β-arrestin-mVenus) was increased, thus altering the donor/acceptor ratios. In the presence of the full D2R agonist quinpirole, the BRET signal saturated with increasing β-arrestin-mVenus when the D2R-WT was used (Fig. 2E). In contrast, using the D2R-F1895.38A mutant (Fig. 2F), quinpirole did not produce a saturable BRET signal, confirming the inability of this mutant to recruit β-arrestin in the presence of agonist.
One question concerning the differential effects of the D2R-F1895.38A mutation on G protein- and β-arrestin-mediated signaling was the degree of amplification in the G protein-mediated assays compared to the β-arrestin assays, which lack amplification. If the G protein-mediated assays were extremely amplified, then the F1895.38A mutation might negatively impact signaling efficacy without an observable effect on the maximum response in the assay. With respect to the D2R-WT, the potency of dopamine was 4–15-fold greater for stimulating G protein-mediated signaling compared to β-arrestin recruitment, suggesting some degree of amplication (tables S1 and S3). However, to assess this more directly, we compared the effects of a partial agonist of the D2R in the two assays. If the G protein-mediated assay was extremely amplified, then the relative Emax for the partial agonist should be much greater than that observed in the β-arrestin recruitment assay. The D2R partial agonist CAB02–110 (34) (compound 11)) was ~9 fold more potent in the G protein assay; however, its Emax (compared to dopamine) was only marginally higher compared to that in the β-arrestin assay (68% compared to 58%, respectively) (fig. S3). As observed previously (tables S1 and S3), dopamine was ~15-fold more potent in the G protein signaling assay (fig. S3). These results suggest that, although there was some degree of amplification in the G protein-mediated assay, it was not sufficiently high so as to obscure interpretation of the differential effects of the F1895.38A mutation on the two signaling arms of the D2R.
We further assessed D2R-mediated β-arrestin recruitment in response to either dopamine (Fig. 2G) or the D2R agonist pramipexole (Fig. 2H) in mutants in which Phe1895.38 was substituted using amino acid residues with different physicochemical properties. The only amino acid substitution that did not negatively impact agonist-stimulated β-arrestin recruitment was the replacement of phenylalanine with tyrosine, a structurally similar aromatic amino acid (Fig. 2, G and H, table S5). Together, these results indicate that the D2R-F1895.38A mutant is selectively biased towards G protein-mediated signaling and deficient with respect to β-arrestin recruitment.
Impairment of agonist-stimulated internalization of the D2R-F1895.38A mutant
A major function of β-arrestin recruitment is to initiate endocytosis of GPCRs into clathrin-coated pits, thereby removing them from the cell surface (3, 35, 36). Previously, we showed that β-arrestin2 mediates agonist-stimulated D2R internalization in neurons (37). To evaluate the internalization of the G protein-biased D2R-F1895.38A., we used [3H]sulpiride, a D2R antagonist that labels only cell surface receptors in intact cell binding assays due to its hydrophilicity and inability to cross the cell membrane (38, 39). Pretreatment with dopamine significantly decreased cell surface D2R-WT (Fig. 3A), as we have previously described (28, 38, 39). In contrast, dopamine pretreatment did not affect the cell surface binding of [3H]sulpiride in cells expressing the D2R-F1895.38A, indicating a lack of agonist-induced receptor internalization (Fig. 3B). We next measured constitutive BRET between the D2R and the plasma membrane-localized tyrosine kinase Lyn (40), which is decreased by agonist-induced internalization of the D2R. Treatment with dopamine dose-dependently reduced constitutive D2R-Lyn BRET in cells expressing the D2R-WT but not in those expressing the D2R-F1895.38A mutant. Together, these results suggest that impairment of β-arrestin recruitment in the D2R-F1895.38A functionally impacts β-arrestin-mediated downstream signaling processes as demonstrated by impaired receptor internalization.
Figure 3. The G protein-biased D2R-F1895.38A exhibits impaired agonist-induced internalization.
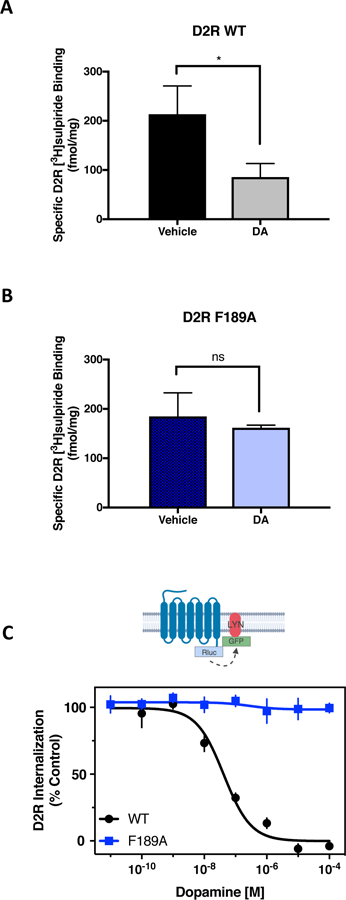
(A and B) HEK293 cells expressing either D2R-WT (A) or D2R-F1895.38A (B) were incubated for 1.5 hours with vehicle or 10 μM dopamine. Surface expression of the receptor was measured with an intact cell binding assay using [3H]sulpiride. Data are representative of three independent experiments. *p<0.05, unpaired Student’s t test. (C) HEK293 cells transiently expressing either D2R-WT-Rluc8 or D2R-F1895.38A-Rluc8 with LYN-rGFP were treated with increasing concentrations of dopamine for 10 min. The interaction between D2R and LYN was measured by BRET. In the graph, the constitutive basal BRET is defined as 100% control and maximum dopamine-induced decrease in BRET is defined as 0%. The EC50 for dopamine-induced internalization was 88 ± 19 nM. No measurable internalization was observed with the D2R-F1895.38A. Data in (A) to (C) are mean ± SEM of 4 independent experiments performed in technical triplicate.
Because the agonists rotigotine and apomorphine exhibit a very low, but measurable level of β-arrestin recruitment to the D2R-F1895.38A mutant (fig. S2E and S2G, and table S4), we wondered if these compounds would promote internalization of the mutant receptor to a corresponding low degree. Rotigotine and apomorphine stimulated maximal internalization of the D2R-WT, but did not promote substantial internalization of the D2R-F1895.38A (fig. S4A and S4B). These results suggest that, although β-arrestin is partially recruited to the mutant receptor in response to rotigotine and apomorphine, the resulting β-arrestin-D2R-F1895.38A interactions are no longer sufficient to promote receptor internalization.
Functional conservation of residue 5.38 in related GPCRs
Given the importance of Phe1895.38 in the D2R for determining signaling bias, we investigated the conservation of this and nearby residues among related GPCRs. An alignment of the residues surrounding the EL2-TM5 hydrophobic pocket revealed that residue 5.38 is relatively conserved such that either a phenylalanine or tyrosine is present in all of the catecholamine receptors and all but one of the serotoninergic receptors (Table 1). In fact, among all 286 human non-olfactory class A GPCRs, 88 (31%) possess Tyr whereas 42 (15%) contain Phe at position 5.38 (www.gpcrdb.org). Either Phe or Tyr at position at 5.38 enabled maximal agonist-stimulated β-arrestin recruitment to the D2R (Fig. 2G and 2H). An additional 27% of non-olfactory class A GPCRs possess Val at this position, which partially supports β-arrestin recruitment to the D2R (Fig. 2G and 2H). Given the high conservation of this residue, we wished to examine the closely related D3 and D4 dopamine receptors (D3R and D4R). Similar to the D2R, the D3R possesses a phenylalanine at position 5.38. Mutating this residue to alanine (D3R-F1885.38A) abolished dopamine-stimulated β-arrestin recruitment (Fig. 4A, table S6), whereas dopamine could still maximally activate G protein-mediated signaling, albeit with reduced potency (Fig. 4B, table S6). Nearly identical results were observed for the D4R, which contains a tyrosine at position 5.38 (Tyr1925.38). The D4R-Y1925.38A did not promote β-arrestin recruitment in response to dopamine stimulation (Fig. 4C, table S6), but fully activated Go when compared to the D4R-WT (Fig. 4D, table S6).
Table 1.
Alignment of hydrophobic pocket residues for select GPCRs.
| GPCR | ECL2-TM5 Sequence* | ||||||
|---|---|---|---|---|---|---|---|
| 45.52 | 5.35 | 5.36 | 5.37 | 5.38 | 5.39 | 5.40 | |
| D1R | Ser | Ser | Arg | Thr | Tyr | Ala | Ile |
| D2R | Ile | Asn | Pro | Ala | Phe | Val | Val |
| D3R | Ile | Asn | Pro | Asp | Phe | Val | Ile |
| D4R | Leu | Asp | Arg | Asp | Tyr | Val | Val |
| D5R | Ser | Asn | Arg | Thr | Tyr | Ala | Ile |
| 5-HT1AR | Ile | Asp | His | Gly | Tyr | Thr | Ile |
| 5-HT1BR | Val | His | Ile | Lys | Tyr | Thr | Val |
| 5-HT2AR | Leu | Asp | Asp | Asn | Phe | Val | Leu |
| 5-HT2BR | Leu | Phe | Gly | Asp | Phe | Met | Leu |
| 5-HT2CR | Leu | Asp | Pro | Asn | Phe | Val | Leu |
| ⍺1A | Ile | Glu | Pro | Gly | Tyr | Val | Leu |
| ⍺1B | Val | Glu | Pro | Phe | Tyr | Ala | Leu |
| ⍺1D | Ile | Glu | Ala | Gly | Tyr | Ala | Val |
| ⍺2A | Ile | Gln | Lys | Trp | Tyr | Val | Ile |
| ⍺2B | Leu | Glu | Ala | Trp | Tyr | Ile | Leu |
| ⍺2C | Leu | Glu | Thr | Trp | Tyr | Ile | Leu |
| β1R | Phe | Gln | Arg | Ala | Tyr | Ala | Ile |
| β2R | Phe | Gln | Gln | Ala | Tyr | Ala | Ile |
| β3R | Phe | Gln | Met | Pro | Tyr | Val | Leu |
| V2R | Ala | Arg | Arg | Thr | Tyr | Val | Thr |
Amino acid positions in transmembrane (TM) regions are delineated using Ballesteros and Weinstein nomenclature (29). The amino acid position in extracellular loop 2 (EL2) is delineated using the nomenclature developed by de Graaf et al. (77). Yellow indicates nonpolar amino acids with an aliphatic group (Ala, Val, Ile, Leu, Met, Gly); light green indicates hydrophobic amino acids with an aromatic ring (Phe, Tyr); dark green indicates Trp; purple indicates polar amino acids with an uncharged side chain (Ser, Thr, Asn, Gln); light grey indicates Pro; red indicates negatively charged amino acids (Glu, Asp); blue indicates positively charged amino acids (Arg, His, Lys).
Figure 4. Mutation of 5.38 confers G protein signaling bias in multiple GPCRs.
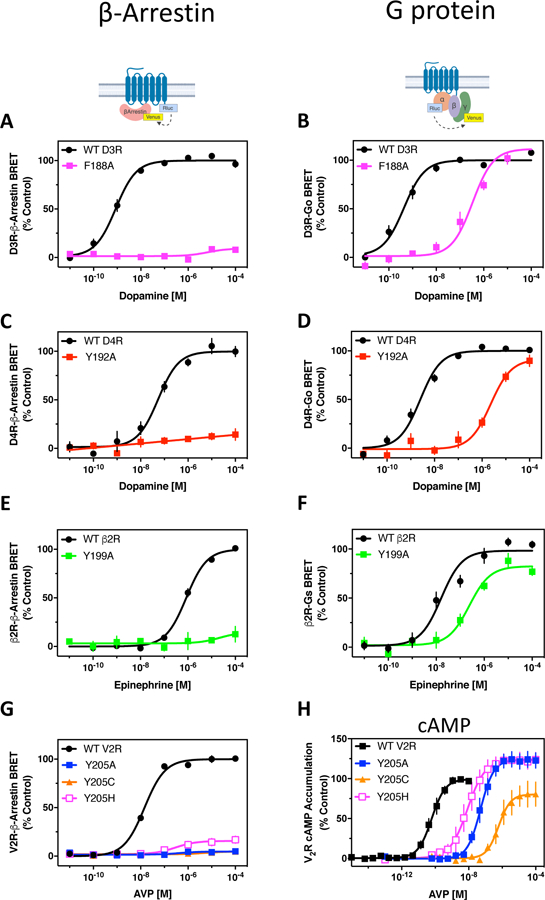
(A) The D3R-WT and D3R-F1885.38A were fused to Rluc8 and expressed in HEK293 cells with β-arrestin2-mVenus and GRK2. Dopamine-stimulated β-arrestin recruitment was assessed by BRET. (B) The D3R-WT and D3R-F1885.38A were expressed in HEK293 cells with Goα1-Rluc8, β1, and γ2-mVenus. The cells were stimulated with dopamine and assayed for G protein activation by BRET. (C) Dopamine-stimulated β-arrestin recruitment was assessed for the D4R-WT-Rluc-8 and D4R-Y1925.38A-Rluc8 as described in (A). (D) Dopamine-stimulated Go activation was assessed for the D4R-WT and D4R-Y1925.38A as described in (B). (E) The β2R-WT and β2R-Y1995.38A were fused to Rluc8 and expressed in HEK293 cells with β-arrestin2-mVenus. Epinephrine-stimulated β-arrestin recruitment was assessed by BRET. (F) The β2R-WT and β2R-Y1995.38A were expressed in HEK293 cells with Gαs-RLuc8, β1, and γ2-mVenus. The cells were stimulated with epinephrine and assayed for G protein activation by BRET. (G) V2R-WT or the indicated V2R mutant were fused to Rluc8 and expressed in HEK293 cells with β-arrestin2-mVenus and assayed for AVP-stimulated β-arrestin recruitment by BRET. (H) HEK293 cells expressing either V2R-WT or the indicated V2R mutant were assayed for AVP-stimulated cAMP accumulation using the TR-FRET-based Lance cAMP Detection kit. Data are expressed as a percentage of the maximum response for WT receptor. Average EC50 and Emax values are found in table S6. All data points represent the mean ± SEM of 3–6 independent experiments performed in technical triplicate.
This analysis was extended to the β2-adrenergic receptor (β2R), which, unlike the D2-like receptors, predominantly signals through Gs and activation of adenylyl cyclase. As observed with the dopamine receptors, mutation of residue 5.38 within the β2R (Y1995.38A) suppressed the ability of the endogenous agonist, epinephrine, to promote β-arrestin recruitment to the receptor (Fig. 4E, table S6), with minimal effects on epinephrine activation of Gs, as determined using a BRET-based assay (Fig. 4F, table S6). Similar results were observed using isoproterenol and BI-167107, which are β-adrenergic-selective full agonists (fig. S5A to S5D, table S6).
As for the D2R, we were interested in evaluating the degree of amplification in the β2R-mediated G protein signaling assay relative to that for β-arrestin recruitment. To this end, we compared the activities of two β2R partial agonists, formoterol and procaterol, with the the full agonist BI-167107 for Gs activation and β-arrestin recruitment following stimulation of the β2R-WT. As previously described (fig. S5C and S5D, table S6), the full agonist BI-167107 was slightly (5–7-fold) more potent for Gs activation compared to β-arrestin recruitment, suggesting some degree of amplification in the Gs assay (fig. S6A and S6B). When comparing the two functional responses, the EC50 values for the β2R partial agonists formoterol and procaterol were similar and, while the Emax value for formoterol was slighty higher in the Gs assay than for β-arrestin recruitment (90% compared to 70%, fig. S6A), the Emax values for procaterol in the two assays were comparable (fig. S6B). These results suggest that the Gs activation assay is only minimally amplified compared to the β-arrestin assay and imply a pivotal role for β2R residue 5.38 in the formation of an active state for recruiting β-arrestin.
We noted that there are two naturally occurring human polymorphisms at position 5.38 in the V2 vasopressin receptor (V2R), which appear to be associated with nephrogenic diabetes insipidus (41, 42). The V2R normally contains Tyr at position 5.38 (Y2055.38; Table 1); however, the disease-associated polymorphisms involve a change to either Cys or His, which result in decreased V2R-mediated cAMP accumulation (41–43). We examined the ability of the Y2055.38C and Y2055.38H polymorphisms, as well as a Y2055.38A construct, to recruit β-arrestin and increase cAMP levels. All of the V2R mutants were impaired in their ability to recruit β-arrestin when stimulated with the full agonist arginine vasopressin (AVP; Fig. 4G). Somewhat mixed results were obtained when intracellular cAMP levels were examined (Fig. 4H, table S6). The potency of AVP for stimulating cAMP accumulation was reduced for the three mutant receptors, which may relate to previous reports of reduced cAMP accumulation with the Y2055.38C and Y2055.38H mutants (41–43). In contrast, we observed a small, but statistically insignificant increase in the maximum AVP cAMP response with the Y2055.38A and Y2055.38H mutants (Fig. 4H, table S6) suggesting that perturbing Y2055.38 in the V2R may exert dose-dependent effects on agonist-stimulated G protein-mediated signaling.
Allosteric propagation of the β2R Y1995.38A perturbation to the intracellular surface
To investigate how alterations at position 5.38 of the receptor induce conformational rearrangements that propagate from the extracellular to the intracellular surface resulting in signaling bias, we used the β2R, for which high-resolution crystal structures of the active state are available, as a model system to study the underlying molecular mechanisms. Specifically, we used the crystal structure of β2R in an active conformation (PDB code 4LDE) (30), for building both β2R-WT and Y1995.38A models in complex with the high-affinity full agonist BI-167107 and carried out molecular dynamics (MD) simulations (table S7) to detect the perturbation of the Y1995.38A mutation on receptor conformation (Fig. 5, A to C). We compared the identities and interaction frequencies of the residues interacting with BI-167107 under both simulated conditions. We found that the shorter side chain in the Y1995.38A mutant resulted in 18.4% less frequent interactions with BI-167107 compared to Tyr1995.38 in the β2R-WT (table S8). Using this extent of difference as a heuristic threshold, we identified other residues that differentially interacted with BI-167107 in the β2R-WT compared to the β2R-Y1995.38A, meaning residues having >18.4% differences in interaction frequencies in the two conditions (table S8). In particular, Thr1644.56, which formed a hydrogen bond (H-bond) with the side chain -OH of Tyr1995.38 in the β2R-WT (fig. S7), did not form an H-bond in the Y1995.38A mutant and moved away from the ligand binding site (Fig. 5B and 5C). In addition, Tyr174EL2 in EL2 bent down to fill the space created by the loss of the bulky Tyr1995.38 side chain in the mutant construct (Fig. 5B).
Fig. 5. β2R-Y1995.38A changes the packing among TM4, TM5, and EL2.
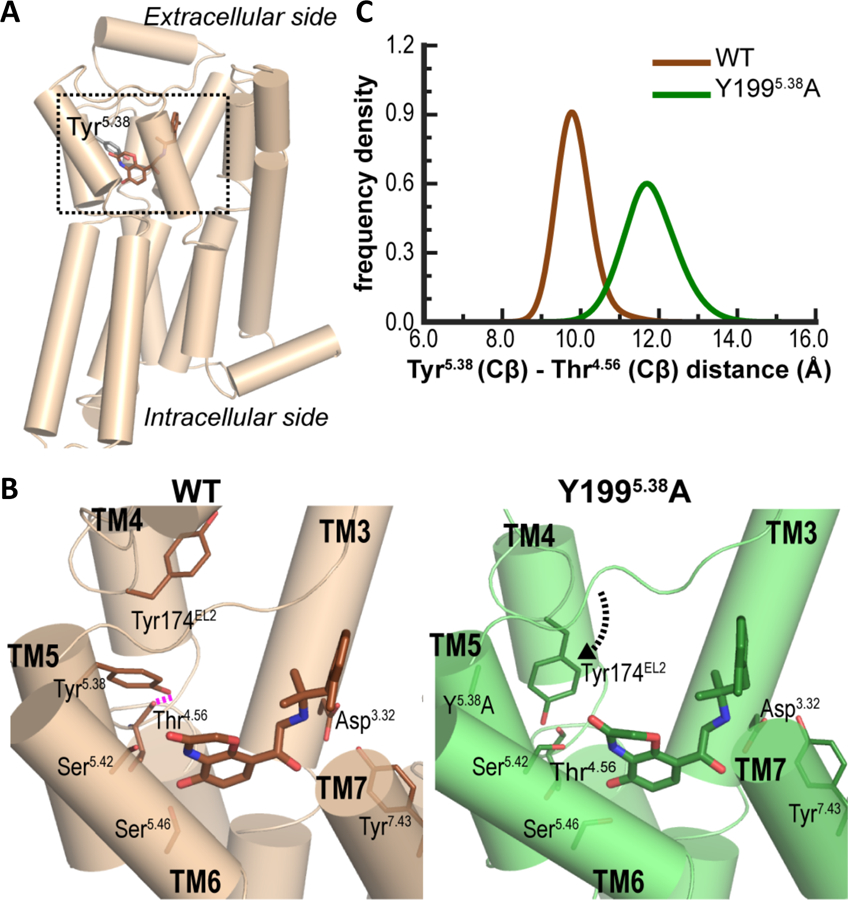
(A) The crystal structure of β2R-BI-167107 complex in active conformation (PDB ID 4LDE) (30). (B) Magnified view of the ligand binding site (the boxed region in (A)) in β2R-WT and β2R-Y1995.38A. In β2R-WT (brown structure), the sidechain -OH of Tyr5.38 forms a hydrogen bond (dashed magenta line) with the backbone oxygen of Thr4.56. In the absence of this H-bond, and the loss of the bulky sidechain of Tyr5.38 in β2R-Y1995.38A (green structure), Tyr174EL2 bends down to interact with BI-167107. (C) The distance between Tyr5.38 and Thr4.56 (Cβ-Cβ) is larger in β2R-Y1995.38A indicating a rearrangement of Thr4.56 away from the ligand binding site.
These rearrangements of local interactions near the extracellular position 5.38 propagated to the intracellular surface through the TM3-TM4-TM5 interface and resulted in changes in side chain positions such as Met1564.48, Ala2025.41, Ser2035.42, and Ser2075.46 at this interface (Fig. 6A). These coordinated changes consequently resulted in a different tilt of TM4, face shift of TM4 and TM5 on their extracellular sides, and an altered orientation of IL2 between TM3 and TM4 in the β2R-Y1995.38A compared to the β2R-WT (Figs. 6A, S8). Such coordinated changes between the extracellular and intracellular sides of TM4 are reminiscent of the differences between the cryogenic electron microscopy (cryo-EM) structure of the rhodopsin-Gi complex (PDB code 6CMO) (44) and the crystal structure of the rhodopsin-β-arrestin complex (PDB code 4ZWJ) (45) in the same region. Specifically, Trp1263.41, Cys1674.56, Phe1594.48, Trp175EL2, Phe2035.38, Tyr2065.41, Met2075.42, and His2115.46 in rhodopsin form a similar interaction network at the TM3-TM4-TM5 interface from the extracellular surface to the middle of the TM domain (Fig. 6B). The reconfiguration of these interactions in the two complexes affect TM4 and TM5 on their extracellular sides in a similar fashion as we observed when comparing the MD simulations of β2R-WT and β2R-Y1995.38A and appears to be associated with distinct IL2 conformations on the intracellular side: IL2 is helical in the rhodopsin-arrestin complex and extended in the rhodopsin-Gi complex. Thus, although the residue types are different between the β2AR and rhodopsin within this interface, it may serve as a common mechanistic pathway in propagating the impact of ligand binding from the extracellular to the intracellular side, and consequently be differentially affected by functionally-selective ligands resulting in biased signaling. The perturbation of this pathway, such as that by the Y1995.38A mutation in β2R (Figs. 6A, S8) or the F1895.38A mutation in D2R, even from an extracellular location, should have a similar impact.
Fig. 6. Local disruption near position 5.38 propagates to the intracellular loop 2 (IL2) through the TM3-TM4-TM5 interface.
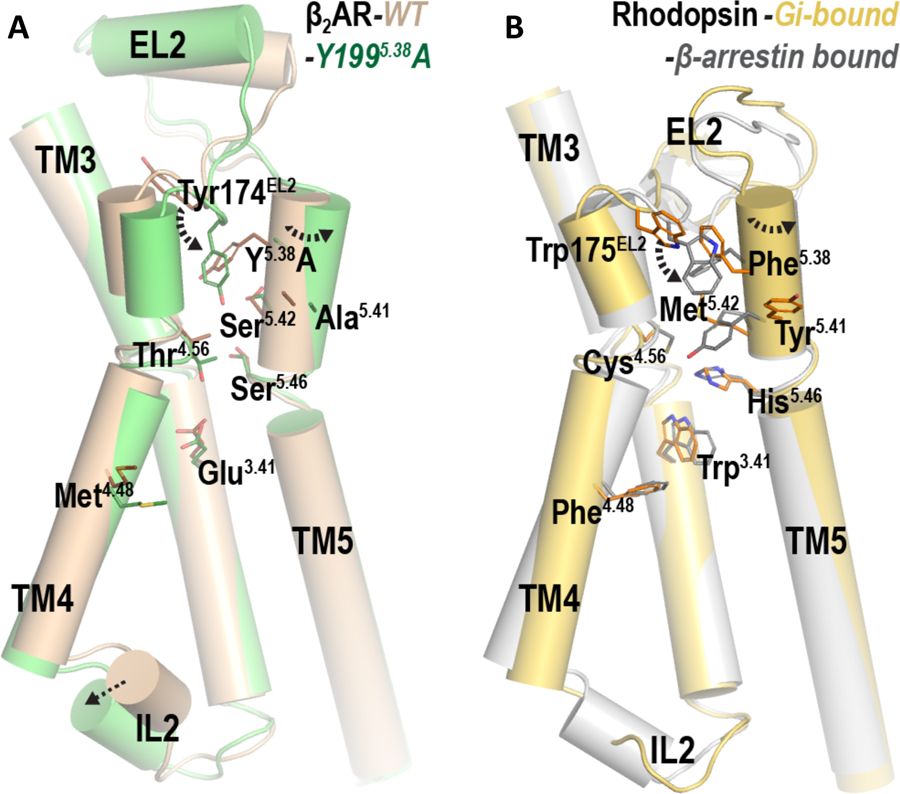
(A) The β2R-WT (brown) and β2R-Y1995.38A (green). (B) The rhodopsin-Gi complex (PDB code 6CMO) (44) (gold) and the rhodopsin-β-arrestin complex (PDB code 4ZWJ) (45) (silver).
DISCUSSION
It is widely appreciated that GPCRs typically signal through multiple pathways involving different transducers including both G proteins and β-arrestins. As for many GPCRs, biased agonists that selectively stimulate either G protein- (27, 28, 46–51) or β-arrestin-mediated pathways (13, 48, 52–55) have been discovered for the D2R, although the underlying molecular mechanisms are not well understood. Our previous studies (27, 28) with the G protein-biased agonist MLS1547 indicated that its diminished ability to recruit β-arrestin was correlated with its interaction with a hydrophobic pocket within the D2R consisting of residues Ile184EL2, Phe1895.38, and Val1905.39. These results suggest that structural features within this extracellular hydrophobic pocket may serve as a micro-switch to allosterically bias the intracellular signaling properties of the D2R. In support of this idea, McCorvy et al. (13) showed that modifying ligands based on the antipsychotic aripiprazole that result in strengthened interactions with D2R residue Ile184EL2 affect their ability to stimulate β-arrestin recruitment. To further test this hypothesis, we evaluated the impact of the single-point mutations I184EL2A, F1895.38A, and V1905.39A, on D2R signaling activity. Each of these alterations detrimentally affected the G protein-mediated signaling of MLS1547 with the F1895.38A mutant resulting in a complete loss of MLS1547 efficacy. These results further support the notion that this D2R pocket includes structural determinants that contribute to both ligand efficacy and signaling bias.
Somewhat different results were obtained with the mutant D2R constructs when we examined signaling in response to the endogenous neutral agonist dopamine. The I184EL2A and V1905.39A mutants displayed reduced dopamine-receptor binding affinity and potency for activating both G protein- and β-arrestin-mediated signaling without a loss of functional efficacy for either pathway. In contrast, whereas the F1895.38A mutant showed reduced dopamine binding affinity and potency for G protein-mediated signaling, as was observed with the other two D2R mutants, its efficacy for stimulating β-arrestin recruitment was completely eliminated. Further, the ability of dopamine to maximally activate G protein-mediated signaling was fully maintained. Because the BRET assays used in these experiments directly measure D2R-β-arrestin interactions, these results suggest that the D2R-F1895.38A was impaired in its ability to form an active conformation that recruits and activates β-arrestin. The observation that the D2R-F1895.38A failed to internalize in response to agonist stimulation, a process mediated by β-arrestin2 (37), further supports this conclusion. Together, these results provide evidence that the D2R-F1895.38A is biased for G protein-mediated signaling and that Phe1895.38 plays a pivotal role in regulating signaling bias.
A relatively high percentage of class A GPCRs, including catecholamine receptors, have the aromatic amino acids Phe or Tyr in the 5.38 position, suggesting conservation of function for these residues. Tyr was the only amino acid substitution for Phe1895.38 in the D2R that did not negatively impact agonist-stimulated β-arrestin recruitment. Further, substitution of the 5.38 residues in the closely related D3R (Phe1885.38) and D4R (Tyr1925.38) with alanine similarly eliminated agonist-stimulated β-arrestin recruitment while minimally affecting G protein-mediated signaling. Because D2-like receptors couple to the Gi/Go family, we extended our analyses to the β2R, which primarily activates Gs. An alanine mutation of the 5.38 residue (Tyr1995.38) in the β2R resulted in a G protein-signaling biased phenotype in which β-arrestin recruitment was negated while Gs activation was minimally affected. Thus, the retention of G protein signaling seen with the Phe/Tyr5.38A mutants for non-biased agonists appears to be independent of G protein coupling preference, whereas β-arrestin recruitment is consistently attenuated.
The nephrogenic diabetes insipidus-associated human polymorphisms at position 5.38 in the V2R result in Cys and His substitutions for Tyr at position 205 (Y2055.38C and Y2055.38H) (41, 42). We found that both of these alterations, as well as a V2055.38A substitution, were associated with a loss of V2R-β-arrestin interactions, in agreement with the results obtained with the D2-like receptors and β2R. The V2R mutants also exhibited a loss in potency for agonist-stimulated Gs protein-mediated signaling (cAMP accumulation), although maximum signaling activity was maintained. Human polymorphisms or genetic mutations that negatively affect V2R signaling result in nephrogenic diabetes insipidus (41–43); however, previous studies have emphasized diminished V2R-mediated cAMP accumulation or protein misfolding (41–43, 56, 57). Our current results now describe impaired β-arrestin recruitment associated with the V2R-Y205C/H5.38 polymorphisms, although further research is needed to determine how the loss of this pathway might be involved in nephrogenic diabetes.
Structural studies using crystallography and cryo-EM have provided new insights into the basis of GPCR activation and coupling with G proteins and β-arrestin signaling molecules. Different conformers of IL2 within the receptor appear to play a critical role in both of these interactions. Xu and colleagues have shown that in the rhodopsin-arrestin structure, the N- and C-domains of arrestin form a cleft between its middle and C-loops that the IL2 (in a helical conformation) of rhodopsin fits into (45). Notably, mutation of select middle or C-loop residues in arrestin, or IL2 residues in rhodopsin, weaken rhodopsin-arrestin interactions (45). Conversely, in the rhodopsin-Gi cryo-EM structure (44), the IL2 of rhodopsin is in an extended loop (see above) and exhibits less extensive interactions with Gi. In contrast, in the case of the β2R-Gs structure, the IL2 adopts a small two-turn helix that is important for Gs activation (58, 59). Dror and colleagues (60, 61) have provided evidence that arrestin activation is primarily achieved through interaction with the receptor core and intracellular loops of rhodopsin, specifically IL2 and to a lesser degree IL3.
Thus, the results of our MD simulations using the β2R as a model system, which predict an altered orientation of IL2 in response to the Y1995.38A mutation, are consistent with the observed loss or decrease in agonist-stimulated receptor-β-arrestin interactions. Further, our results with some agonists that exhibit limited β-arrestin recruitment to the D2R-F1895.38A, yet are incapable of promoting receptor internalization, suggest that this mutation cripples the receptor’s ability to activate β-arrestin. In contrast, altered IL2 conformations resulting from these mutations have a limited impact on G protein-mediated signaling, although agonist potencies for eliciting G protein activation were variably diminished. Based on the similar phenotypes of the aligned mutations in the highly homologous class A GPCRs investigated in this study, we propose that such a mechanistic pathway connecting an extracellular micro-switch in the ligand binding site to IL2, which directly couples to signaling proteins, is commonly involved in biased signaling. Indeed, different ligands have been previously shown to produce distinct conformations of IL2 in other GPCRs (20, 62).
Choi et al. have described a β2R mutation near the juncture of TM5 and IL3 that biases the receptor for G protein-mediated signaling due to defective GRK5-mediated receptor phosphorylation leading to diminished β-arrestin interactions (63). These investigators argued that the mutation did not affect intrinsic receptor-β-arrestin interactions because the fusion of a phosphorylated V2R peptide to the mutant β2R rescued its ability to undergo β-arrestin-mediated desensitization. Instead, they concluded that bias for or against β-arrestin-mediated signaling is mainly regulated through GRK-mediated receptor phosphorylation, which conceivably can be modulated by biased agonists. Although receptor phosphorylation by GRKs typically enhances β-arrestin association and its activation, this is not universal because GPCRs lacking C-termini or phosphorylation sites can still recruit and activate β-arrestin (61, 64). Indeed, we have previously shown that abrogation of GRK-mediated phosphorylation of the D1R (65) or D2R (38, 39) did not affect their ability to recruit and interact with β-arrestin. Thus, β-arrestin signaling bias can undoubtedly arise through different mechanisms that regulate β-arrestin interactions with the GPCR. For instance, Marureel et al. (66) have shown that a hydrogen-bond network between Ser2045.43 and Asn2936.55 in the β2R may underlie β-arrestin signaling bias for select β2R agonists.
In summary, our current study illustrates how structural pertubations in the extracellular ligand binding site can allosterically propagate to the intracellular surface of a GPCR and affect its ability to interact with signaling transducers, thus producing signaling bias. The further elucidation of structural determinants that underlie agonist-specific signaling states may assist in the rational design of novel functionally-selective agents that can serve as improved therapeutic agents.
MATERIALS and METHODS
Materials and Reagents
MLS1547 was originally obtained from the NIH Molecular Libraries Screening Center Network Library (27) and subsequently synthesized and verified for purity at the University of Kansas Specialized Chemistry Center by Dr. Kevin Frankowski (28). Gαo1-Rluc8, Gβ1, Gγ2-mVenus, Gαs-Rluc8, CAMYEL biosensor, β-Arrestin2-mVenus, human D2R-Rluc8, and human D3R-Rluc8 were kind gifts from Drs. Jonathan Javitch and Hideaki Yano at Columbia University. Additional human receptor cDNAs (D4R, β2R, V2R) were obtained at cDNA Resource Center (www.cdna.org). Further receptor constructs and mutants were prepared by Bioinnovatise (Rockville, MD). Constructs were prepared in pcDNA3.1 vectors and inserts were verified by sequencing. LYN-rGFP (40) was a kind gift from Dr. Michel Bouvier at the University of Montreal. All tissue culture media and supplies, were obtained from ThermoFisher Scientific (Carlsbad, CA). All other compounds and chemicals unless otherwise noted were obtained from Sigma Aldrich (St. Louis, MO).
Cell culture and transfection
HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin. CHO-K1-EA cells were cultured in Ham’s F12 media supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, and 300 µg/ml hygromycin. Cells were grown at 37˚C in 5% CO2 with 90% humidity. HEK293 cells were seeded in 100-mm or 35-mm plates and transfected overnight using a 1:3 ratio (1 µg DNA: 3 µl of polyethyleneimine (PEI)) diluted to 1 ml in non-supplemented DMEM and added (100 µl/ml) to the cells already in culture media. Media was replaced with complete media the following day. CHO-K1-EA cells seeded in 100- or 150-mm plates were transfected with TransIT-LT1 (Mirus Bio, Madison, WI) according to the manufacturer’s instructions. Media was replaced after 18 hours and cells were plated for experiments conducted the following day. Concentrations of DNA are indicated for each experiment type.
Bioluminescence resonance energy transfer (BRET) assays
HEK293 cells were transiently transfected with a BRET donor and corresponding BRET acceptor. Briefly, 4 x 106 cells/plate were seeded in 100-mm dishes and incubated overnight. BRET experiments were performed 48 hours after transfection. The amounts of cDNA used for each type of BRET assay varied. β-arrestin recruitment BRET assays used 1 μg of receptor-Rluc8 together with 5 μg of β-arrestin2-mVenus and 5 μg of GRK2 when indicated. G protein activation BRET assays used 1 μg of either Gαs-RLuc8 or 0.5 μg Gαo-RLuc8 together with Gγ2-mVenus, Gβ1, and the corresponding untagged receptor. Receptor internalization BRET assays used 1 μg of D2R-RLuc8 or D2R-F189A-Rluc8 and 5 μg of LYN-rGFP. On experiment day, cells were harvested, washed and resuspended in DPBS containing 200 μM sodium metabisulfite and 5.5 mM glucose. Cells were then plated in 96-well white, solid bottom plates (Greiner Bio-One) and incubated in the dark for 45 min. BRET signals were measured in the presence of 5 μM coelenterazine h (Nanolight Technology) for BRET1 (Rluc8-mVenus) or 2 μM Prolume Purple coelenterazine (Nanolight Technology) for BRET2 (Rluc8-rGFP). Dose-response curves were performed by adding coelenterazine h or Prolume Purple, as appropriate for the sensor, for 5 minutes followed by addition of the indicated concentrations of agonist for 5 minutes. BRET signals were determined by calculating the ratio of the light emitted by mVenus (535/30 nm) over that emitted by Rluc8 (475/30 nm) for BRET1, and the ratio of the light emitted by GFP2 or rGFP (515 nm) over that emitted by Rluc8 (410 nm) for BRET2 using a Pherastar plate reader (BMG Labtech, Cary, NC). Net BRET values were obtained by subtracting the background ratio from untreated cells. Agonist-promoted BRET changes were expressed as a percent of the maximum response of the wild type receptor for each ligand. For saturation BRET experiments, BRET donor concentrations (Rluc-tagged) were held constant and BRET acceptor amounts (mVenus-tagged) were increased. The net BRET values were obtained by subtracting the background ratio obtained from cells without BRET donor. Receptor expression levels were verified across experiments via measurement of Rluc8 for assays with a Rluc8-tagged receptor and found to be consistent from experiment to experiment. In addition, fluorescence levels were also monitored to control for expression across experiments by plating cells in 96-well black solid bottom plates (Greiner Bio-One) and measuring mVenus or GFP (480/530) emission.
CAMYEL biosensor assay for cAMP
Cyclic-AMP accumulation was measured by employing the CAMYEL (yellow fluorescence protein-Epac-Rluc) biosensor as previously described (67). Briefly, 4 x 106 HEK293 cells/plate were seeded on 100-mm dishes and incubated overnight. Cells were then transfected with 5 μg of untagged receptor and 5 μg of CAMYEL biosensor using the PEI method described above. BRET experiments were performed 48 hr after transfection. On experiment day, cells were harvested, washed and resuspended in DPBS containing 200 μM sodium metabisulfite and 5.5 mM glucose. Cells were plated in 96-well white, solid bottom-plates (Greiner Bio-One) and incubated in the dark for 45 min. For Gαo-mediated adenylyl cyclase inhibition, cells were pretreated for 5 min with 10 μM forskolin and 10 μM propranolol (to block endogenous β-adrenergic receptors). Cells were then stimulated for 5 min with agonist and BRET signal was determined by calculating the ratio of the light emitted by mVenus (535/30 nm) over that emitted by RLuc8 (475/30 nm) (BRET1) using a Pherastar plate reader (BMG Labtech, Cary, NC). The net BRET values were obtained by subtracting the background ratio from untreated cells. Agonist-promoted BRET changes were expressed as a percent of the maximum response of the wild type receptor for each ligand.
DiscoverX β-arrestin recruitment assay
The ability of the agonist-activated receptor to recruit β-arrestin2 was also determined using the DiscoverX PathHunter technology (DiscoverX, Fremont, CA). Assays were conducted, with minor modifications, as previously published by our laboratory (27). In brief, 1.5 million CHO-K1-EA cells stably expressing β-arrestin fused to an N-terminal deletion mutant of β-galactosidase were transfected 24 hours after seeding with 5 µg of either D2R-WT of D2R-F189A fused to a complementing N-terminal fragment of β-galactosidase using the TransIT-LT1 transfection reagent (Mirus Bio). 18 hr later, cells were detached and seeded at a density of 7000 cells/well in 384-well black bottom plates. After 24 hr of incubation, the cells were treated with multiple concentrations of compound in PBS and incubated at 37°C for 90 minutes. DiscoverX reagent was added to cells according to the manufacturer’s recommendations and incubated for 45 min at room temperature. Luminescence was measured on a Hamamatsu FDSS μCell reader. Data were collected as RLUs and subsequently normalized to a percentage of the control luminescence seen with a maximum concentration of dopamine using the D2R WT with 0% representing RLUs seen in the absence of any compound. The Hill coefficients of the concentration response curves did not differ from unity.
Lance assay for cAMP
Cyclic-AMP accumulation was measured by using the TR-FRET-based LANCE cAMP assay (Perkin Elmer). Briefly, 4 x 106 HEK293 cells/plate were seeded on 100-mm dishes and incubated overnight. Cells were then transfected with 5 μg of untagged V2R-WT, V2R-Y209A, V2R-Y209C or V2R-Y209H using the PEI method as described above. 16 h later the media was replaced with fresh media. 48 hours after transfection cells were harvested, washed and resuspended in HBSS containing 200 μM sodium metabisulfite and 20 μM HEPES, and were plated in 384-well white bottom plates at 1 x 106 cells/mL and 5 μL/well. Immediately after plating, cells were treated with 5 μl of varying concentrations of arginine vasopressin (AVP) and incubated for 30 min at room temperature. 5 μl of Tracer and 5 μl of a-cAMP were added to each well according to the manufacturer’s protocol and cells were incubated in the dark for 2 hours at room temperature. Plates were read on a PheraSTAR plate reader (BMG Labtech, Cary, NC) with excitation at 337 nm and emission at 620 nm and 665 nm. Data were obtained as the ratio between A (excitation at 337 nm/emission at 665 nm) and B (excitation at 337 nm/emission at 620 nm). Data are represented as a percentage of the maximum response of the wild type receptor.
Membrane [3H]methylspiperone binding assay
Radioligand competition and saturation binding assays were conducted with slight modifications as previously described by our laboratory (27, 28). For competition binding experiments, 1.5 x 106 CHO-K1-EA cells were seeded in 100-mm dishes and incubated overnight. The next day, cells were transfected with 10 μg of indicated non-tagged receptor construct using the TransIT-LT1 transfection reagent (Mirus Bio). For saturation binding experiments, 4 x 106 HEK293 cells were seeded in 100-mm dishes and incubated overnight. The next day, cells were transfected with 5 μg of indicated nontagged receptor along with 1 μg of Gαo-RLuc8, 5 μg of Gγ2-mVenus and 4 μg of Gβ1 or with 1 μg of indicated receptor tagged with Rluc8 along with 5 μg of β-arrestin2-mVenus and 5 μg of GRK2 using TransIT-LT1 (Mirus Bio). 48 hr after transfection, cells were dissociated from plates using EBSS-, and intact cells were collected by centrifugation at 900 g for 10 minutes. Cells were resuspended and lysed with 5 mM Tris-HCl and 5 mM MgCl2 at pH 7.4 at 4°C. Cell lysate was pelleted by centrifugation at 30,000g for 30 minutes and resuspended in EBSS + CaCl2 at pH 7.4. Cell lysates (100 μL, containing ∼10–20 μg of protein, quantified by the Bradford Assay) were incubated for 90 min at room temperature with the indicated concentrations of dopamine or MLS1547 and 0.2 nM [3H]methylspiperone (for competition binding assays) or the indicated concentrations of [3H]methylspiperone (for saturation binding assays) in a final reaction volume of 250 μL. Nonspecific binding was determined in the presence of 4 μM (+)-butaclamol. Bound ligand was separated from free by filtration through a PerkinElmer Unifilter-96 GF/C 96-well microplate using the PerkinElmer Unifilter-96 Harvester (PerkinElmer, Waltham, MA), washing three times with 1 ml/well ice-cold assay buffer. After drying, 50 μl of liquid scintillation cocktail (MicroScint PS; PerkinElmer) was added to each well, and plates were sealed and analyzed on a PerkinElmer Topcount NXT.
[3H]sulpiride binding assay in intact cells
Cell surface receptor expression was determined using the membrane impermeant radioligand [3H]sulpiride in intact cell binding assays (38, 39). 13 x 106 HEK293 cells were seeded in 150-mm dishes and incubated overnight. The next day, cells were transfected with 20 μg of non-tagged D2R-WT or D2R-F189A plus 20 μg of GRK2 using the PEI method described above. Cells were seeded into poly-D-lysine-coated 6-well plates 1 day before the assay at a density of 1 x 106 cells/well. 24 hr after plating, cells were incubated in the presence of either 0.2 mM sodium metabisulfite (control) or 0.2 mM sodium metabisulfite plus 30 μM dopamine in DMEM for 1.5 hours at 37°C. Stimulation was terminated by rapidly cooling the plates on ice and washing the cells three times with ice-cold EBSS. Cells were then incubated with 0.5 ml of [3H]sulpiride in EBSS (final concentration, 7.3 nM) at 4°C for 3.5 hours. Nonspecific binding was determined in the presence of 7.5 μM (+)-butaclamol. Cells were washed three times with ice-cold EBSS and removed from plates with 0.5 ml of 1% Triton X-100 and 5 mM EDTA in EBSS. Samples were mixed with 2 ml of liquid scintillation mixture and counted with a Beckman LS6500 scintillation counter. Cells used to measure protein concentration were incubated with EBSS without [3H]sulpiride, and a Bradford assay was used to determine total cellular protein concentration per well. Data are represented as specific binding in fmol/mg protein.
Bias factor calculation
Dose-response data for apomorphine and rotigotine were fitted to the following form of the operational model of agonism (68) to allow the quantification of biased agonism as described in (69):
Where Em is the maximal possible response of the system, Basal is the basal level of response, KA represents the equilibrium dissociation constant of the agonist (A) and τ is an index of the signalling efficacy of the agonist that is defined as RT/KE, where RT is the total number of receptors and KE is the coupling efficiency of each agonist-occupied receptor, and n is the slope of the transducer function that links occupancy to response. The analysis assumes that the transduction machinery used for a given cellular pathway are the same for all agonists, such that the Em and transducer slope (n) are shared between agonists. D2R WT and D2R F189A dose-response data for apomorphine and rotigotine were fit for each pathway (G protein and β-arrestin) to determine values of KA, τ and transduction coefficient (Log(τ/KA)). ΔTransduction coefficients (ΔLog (τ/KA) = Log (τ/KA)D2R WT - Log(τ/KA)D2R F189A) were calculated for the G protein and β-arrestin data for each compound. ΔΔLog (τ/KA) is obtained by substracting the Δtransduction coefficient for G protein from the Δtransduction coefficient for β-arrestin. The bias factors are the antilogs of ΔΔLog (τ/KA) and are shown in table S4.
Molecular dynamics (MD) simulation system setup and protocol
Initial coordinates of active state β2R bound to agonist BI-167107 were downloaded from (Protein Data Bank (PDB) entry 4LDE (30, 70). The BI-167107 bound β2R crystal structure was determined using a β2R-T4 lysozyme (β2R-T4L) fusion protein in which the T4L was fused to the N-terminus of the receptor in presence of camelid antibody fragment. We omitted T4L and camelid antibody fragment from all of our MD simulations. Additionally, unresolved parts of intracellular loop-3, N- and C-termini were omitted from the simulations. Four mutations (M96T, M98T, N187E, C265A according to UNIPROT numbering) that were introduced in the β2R crystal structure were mutated back to wild-type residues. Missing atoms of residues Lys60, Glu62, Lys149, Phe223, Gln224, Gln231, Lys263, Phe264, Lys270 were added using Maestro (Schrödinger, LLC). Asp792.50, Glu1223.41, and Asp1303.49 were protonated as described previously (71).
Prepared receptor–ligand complexes were inserted into explicit palmitoyl-2-oleoylphosphatidylcholine (POPC) lipid bilayer environment using Desmond MD System (version 4.5; D.E. Shaw Research, New York, NY). The system charges were neutralized, and 150 mM NaCl was added. Overall the simulation systems consisted of ~107889 atoms containing 297 lipid molecules, 58 sodium ions, 67 chloride ions, and 21092 explicit water molecules. To elucidate how Ala substitution for Tyr5.38, which lies towards the extracellular side in β2R, affects β-arrestin interactions with the intracellular side of the receptor, in silico β2R-Y1995.38A MD simulation systems were prepared from representative frames from equilibrated β2R-WT MD simulation trajectories.
MD simulation systems were simulated using Desmond MD System (version 4.5; D.E. Shaw Research, New York, NY) with the OPLS3 force field (72) and TIP3P water model. The protein-membrane relaxation was carried out with a protocol modified from that developed by Schrödinger, LLC. Briefly, the MD simulations were energy minimized and equilibrated for 1 ns with restraints on all protein and ligand heavy atoms, and then were equilibrated for 12 ns with restraints only on the protein backbone and ligand heavy atoms. For both the equilibrations and the following unrestrained production runs, we used Langevin constant pressure and temperature dynamical system (73) to maintain the pressure at 1 atm and the temperature at 310K, on an anisotropic flexible periodic cell with a constant-ratio constraint applied on the lipid bilayer in the X-Y plane. For both β2R-WT and β2R-Y1995.38A, we collected 14 trajectories with an aggregated simulation length of 21.0 μs (table S7).
Conformational analyses
To identify equilibrated portions of the MD trajectories that were used for the following conformational analysis, frames from β2R-WT and β2R-Y1995.38A MD trajectories were clustered using previously described Protein Interaction Analyzer (PIA) program (74). Briefly, the PIA clusters all frames from the MD trajectories based on a dissimilarity matrix of pairwise Cα-ifRMSDs (namely, the iterative fit RMSD of all the Cα atoms) (75) and ensures that the same clustering criteria are applied consistently for all of the trajectories across the simulated conditions. For this study, we utilized Cα atoms of previously identified 34 ligand binding residues (76) to perform clustering. These 34 residues include those at positions 2.61, 2.64, 2.65, EL1.50, 3.28, 3.29, 3.32, 3.33, 3.36, 3.37, 3.40, 4.57, EL2.52, 5.38, 5.39, 5.43, 5.46, 5.47, 6.44, 6.48, 6.51, 6.52, 6.55, 6.56, 6.58, 6.59, 7.32, 7.35, 7.36, 7.39, 7.42, and 7.43 according to Ballesteros-Weinstein numbering.
From the equilibrated portions of the MD trajectories, we computed the protein residues within 5 Å of the heavy atoms of BI-167107 and calculated the percentage time any protein residue heavy atom was within the distance cutoff (interactions frequencies). Residues having interactions frequencies >/= 25% in at least one of the conditions are given in Table S8.
Supplementary Material
Fig S1. D2R WT and D2R mutants express to a similar extent using transient transfection
Fig S2. Other D2R agonists exhibit G protein bias at the D2R-F1895.38A
Fig S3. Compound CAB02–110 is a partial agonist at both G protein activation and β-arrestin recruitment
Fig S4. Rotigotine- and apomorphine-stimulated internalization of the D2-WT and D2R-F1895.38A
Fig S5. The β2R-Y1995.38A exhibits G protein bias with different agonists
Fig S6. Partial agonist stimulation of β2R-mediated Gs activation and β-arrestin recruitment
Fig S7. Distribution of distance between the oxygen of Tyr5.38 sidechain hydroxyl and the backbone oxygen atom of Thr4.56 in the β2R-WT
Fig S8. IL2 moves downwards in the β2R-Y1995.38A mutant
Table S1. β-Arrestin-BRET and Go BRET signaling by D2R-WT and D2R mutants
Table S2. Affinity constants (Ki values) for MLS1547 and dopamine binding to D2R-WT and D2R mutants
Table S3. β-Arrestin recruitment and regulation of cAMP signaling by D2R-WT and D2R-F1895.38A
Table S4. β-Arrestin and Go BRET signaling for D2R-WT and D2R-F1895.38A in response to full D2R agonists
Table S5. β-Arrestin-BRET recruitment for D2R WT and various mutants at the Phe1895.38 position
Table S6. β-Arrestin- and G protein-mediated signaling by WT and 5.38 mutant D3R, D4R, β2R and V2R
Table S7. Summary of molecular dynamics simulations
Table S8. β2R-BI-167107 interacting residues.
Acknowledgments:
The authors wish to thank Megan Donegan for excellent technical assistance and Dr. J. Robert Lane for helpful discussions.
Funding: This study was supported by the Intramural Research Programs of the National Institute of Neurological Disorders and Stroke and the National Institute on Drug Abuse at the National Institutes of Health.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Hauser AS, Attwood MM, Rask-Andersen M, Schioth HB, Gloriam DE, Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov 16, 829–842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurevich VV, Gurevich EV, The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther 110, 465–502 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shenoy SK, Lefkowitz RJ, beta-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci 32, 521–533 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Q, Iverson TM, Gurevich VV, Structural Basis of Arrestin-Dependent Signal Transduction. Trends Biochem Sci 43, 412–423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peterson YK, Luttrell LM, The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol Rev 69, 256–297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ, Differential kinetic and spatial patterns of beta-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J Biol Chem 279, 35518–35525 (2004). [DOI] [PubMed] [Google Scholar]
- 7.L. M. J. Krasel C; Vilardaga M; Bunemann M, Kinetics of G-protein-coupled receptor signalling and desensitization. Biochem Soc Trans 32, 1029–31 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Nogueras-Ortiz C, Yudowski GA, The Multiple Waves of Cannabinoid 1 Receptor Signaling. Mol Pharmacol 90, 620–626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB, Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320, 1–13 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Violin JD, Lefkowitz RJ, Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci 28, 416–422 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Smith NJ, Bennett KA, Milligan G, When simple agonism is not enough: emerging modalities of GPCR ligands. Mol Cell Endocrinol 331, 241–247 (2011). [DOI] [PubMed] [Google Scholar]
- 12.McCorvy JD, Wacker D, Wang S, Agegnehu B, Liu J, Lansu K, Tribo AR, Olsen RHJ, Che T, Jin J, Roth BL, Structural determinants of 5-HT2B receptor activation and biased agonism. Nat Struct Mol Biol 25, 787–796 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCorvy JD, Butler KV, Kelly B, Rechsteiner K, Karpiak J, Betz RM, Kormos BL, Shoichet BK, Dror RO, Jin J, Roth BL, Structure-inspired design of beta-arrestin-biased ligands for aminergic GPCRs. Nat Chem Biol 14, 126–134 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soergel DG, Subach RA, Burnham N, Lark MW, James IE, Sadler BM, Skobieranda F, Violin JD, Webster LR, Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 155, 1829–1835 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Kingwell K, Pioneering biased ligand offers efficacy with reduced on-target toxicity. Nat Rev Drug Discov 14, 809–810 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K, Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, Baell JB, Sexton PM, Christopoulos A, Shaw DE, Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 503, 295–299 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK, Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504, 101–106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC, Structural features for functional selectivity at serotonin receptors. Science 340, 615–619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wingler LM, Elgeti M, Hilger D, Latorraca NR, Lerch MT, Staus DP, Dror RO, Kobilka BK, Hubbell WL, Lefkowitz RJ, Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 176, 468–478 e411 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.B. H. R. Lichtarge O, Cohen FE, An Evolutionary Trace Method Defines Binding Surfaces Common to Protein Families. J Mol Biol 257, 342–58 (1996). [DOI] [PubMed] [Google Scholar]
- 22.Peterson SM, Pack TF, Caron MG, Receptor, Ligand and Transducer Contributions to Dopamine D2 Receptor Functional Selectivity. PLoS One 10, e0141637 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peterson SM, Pack TF, Wilkins AD, Urs NM, Urban DJ, Bass CE, Lichtarge O, Caron MG, Elucidation of G-protein and beta-arrestin functional selectivity at the dopamine D2 receptor. Proc Natl Acad Sci U S A 112, 7097–7102 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schonegge AM, Gallion J, Picard LP, Wilkins AD, Le Gouill C, Audet M, Stallaert W, Lohse MJ, Kimmel M, Lichtarge O, Bouvier M, Evolutionary action and structural basis of the allosteric switch controlling beta2AR functional selectivity. Nat Commun 8, 2169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donthamsetti P, Gallo EF, Buck DC, Stahl EL, Zhu Y, Lane JR, Bohn LM, Neve KA, Kellendonk C, Javitch JA, Arrestin recruitment to dopamine D2 receptor mediates locomotion but not incentive motivation. Mol Psychiatry, (2018). [DOI] [PMC free article] [PubMed]
- 26.Nakajima K, Wess J, Design and functional characterization of a novel, arrestin-biased designer G protein-coupled receptor. Mol Pharmacol 82, 575–582 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Free RB, Chun LS, Moritz AE, Miller BN, Doyle TB, Conroy JL, Padron A, Meade JA, Xiao J, Hu X, Dulcey AE, Han Y, Duan L, Titus S, Bryant-Genevier M, Barnaeva E, Ferrer M, Javitch JA, Beuming T, Shi L, Southall NT, Marugan JJ, Sibley DR, Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Mol Pharmacol 86, 96–105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chun LS, Vekariya RH, Free RB, Li Y, Lin DT, Su P, Liu F, Namkung Y, Laporte SA, Moritz AE, Aube J, Frankowski KJ, Sibley DR, Structure-Activity Investigation of a G Protein-Biased Agonist Reveals Molecular Determinants for Biased Signaling of the D2 Dopamine Receptor. Front Synaptic Neurosci 10, 2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballesteros JA, Weinstein H, Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Met Neuro 25, 366–428 (1995). [Google Scholar]
- 30.Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK, Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang S, Che T, Levit A, Shoichet BK, Wacker D, Roth BL, Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 555, 269–273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang M, Spicher K, Boulay G, Wang Y, Birnbaumer L, Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc Natl Acad Sci U S A 98, 3577–3582 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenakin T, A Scale of Agonism and Allosteric Modulation for Assessment of Selectivity, Bias, and Receptor Mutation. Mol Pharmacol 92, 414–424 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Keck TM, Free RB, Day MM, Brown SL, Maddaluna MS, Fountain G, Cooper C, Fallon B, Holmes M, Stang CT, Burkhardt R, Bonifazi A, Ellenberger MP, Newman AH, Sibley DR, Wu C, Boateng CA, Dopamine D4 Receptor-Selective Compounds Reveal Structure-Activity Relationships that Engender Agonist Efficacy. J Med Chem 62, 3722–3740 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore CA, Milano SK, Benovic JL, Regulation of receptor trafficking by GRKs and arrestins. Annu Rev Physiol 69, 451–482 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Hanyaloglu AC, von Zastrow M, Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol 48, 537–568 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Skinbjerg M, Ariano MA, Thorsell A, Heilig M, Halldin C, Innis RB, Sibley DR, Arrestin3 mediates D(2) dopamine receptor internalization. Synapse 63, 621–624 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Namkung Y, Dipace C, Javitch JA, Sibley DR, G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J Biol Chem 284, 15038–15051 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Namkung Y, Dipace C, Urizar E, Javitch JA, Sibley DR, G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J Biol Chem 284, 34103–34115 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Namkung Y, Le Gouill C, Lukashova V, Kobayashi H, Hogue M, Khoury E, Song M, Bouvier M, Laporte SA, Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat Commun 7, 12178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yokoyama K, Yamauchi A, Izumi M, Itoh T, Ando A, Imai E, Kamada T, Ueda N, A low-affinity vasopressin V2-receptor gene in a kindred with X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol 7, 410–414 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Sangkuhl K, Rompler H, Busch W, Karges B, Schoneberg T, Nephrogenic diabetes insipidus caused by mutation of Tyr205: a key residue of V2 vasopressin receptor function. Hum Mutat 25, 505 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Postina R, Ufer E, Pfeiffer R, Knoers NV, Fahrenholz F, Misfolded vasopressin V2 receptors caused by extracellular point mutations entail congential nephrogenic diabetes insipidus. Mol Cell Endocrinol 164, 31–39 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Kang Y, Kuybeda O, de Waal PW, Mukherjee S, Van Eps N, Dutka P, Zhou XE, Bartesaghi A, Erramilli S, Morizumi T, Gu X, Yin Y, Liu P, Jiang Y, Meng X, Zhao G, Melcher K, Ernst OP, Kossiakoff AA, Subramaniam S, Xu HE, Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558, 553–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, Barty A, White TA, Yefanov O, Han GW, Xu Q, de Waal PW, Ke J, Tan MH, Zhang C, Moeller A, West GM, Pascal BD, Van Eps N, Caro LN, Vishnivetskiy SA, Lee RJ, Suino-Powell KM, Gu X, Pal K, Ma J, Zhi X, Boutet S, Williams GJ, Messerschmidt M, Gati C, Zatsepin NA, Wang D, James D, Basu S, Roy-Chowdhury S, Conrad CE, Coe J, Liu H, Lisova S, Kupitz C, Grotjohann I, Fromme R, Jiang Y, Tan M, Yang H, Li J, Wang M, Zheng Z, Li D, Howe N, Zhao Y, Standfuss J, Diederichs K, Dong Y, Potter CS, Carragher B, Caffrey M, Jiang H, Chapman HN, Spence JC, Fromme P, Weierstall U, Ernst OP, Katritch V, Gurevich VV, Griffin PR, Hubbell WL, Stevens RC, Cherezov V, Melcher K, Xu HE, Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523, 561–567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moller D, Banerjee A, Uzuneser TC, Skultety M, Huth T, Plouffe B, Hubner H, Alzheimer C, Friedland K, Muller CP, Bouvier M, Gmeiner P, Discovery of G Protein-Biased Dopaminergics with a Pyrazolo[1,5-a]pyridine Substructure. J Med Chem 60, 2908–2929 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Moller D, Kling RC, Skultety M, Leuner K, Hubner H, Gmeiner P, Functionally selective dopamine D(2), D(3) receptor partial agonists. J Med Chem 57, 4861–4875 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Weichert D, Banerjee A, Hiller C, Kling RC, Hubner H, Gmeiner P, Molecular determinants of biased agonism at the dopamine D(2) receptor. J Med Chem 58, 2703–2717 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Chen X, McCorvy JD, Fischer MG, Butler KV, Shen Y, Roth BL, Jin J, Discovery of G Protein-Biased D2 Dopamine Receptor Partial Agonists. J Med Chem 59, 10601–10618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonifazi A, Yano H, Ellenberger MP, Muller L, Kumar V, Zou MF, Cai NS, Guerrero AM, Woods AS, Shi L, Newman AH, Novel Bivalent Ligands Based on the Sumanirole Pharmacophore Reveal Dopamine D2 Receptor (D2R) Biased Agonism. J Med Chem, 60, 2890–2907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bonifazi A, Yano H, Guerrero AM, Kumar V, Hoffman AF, Lupica CR, Shi L, Newman AH, Novel and Potent Dopamine D2 Receptor Go-Protein Biased Agonists. ACS Pharmacol Transl Sci 2, 52–65 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J, Discovery of beta-arrestin-biased dopamine D2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc Natl Acad Sci U S A 108, 18488–18493 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hiller C, Kling RC, Heinemann FW, Meyer K, Hubner H, Gmeiner P, Functionally selective dopamine D2/D3 receptor agonists comprising an enyne moiety. J Med Chem 56, 5130–5141 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Mannel B, Dengler D, Shonberg J, Hubner H, Moller D, Gmeiner P, Hydroxy-Substituted Heteroarylpiperazines: Novel Scaffolds for beta-Arrestin-Biased D2R Agonists. J Med Chem 60, 4693–4713 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Mannel B, Hubner H, Moller D, Gmeiner P, beta-Arrestin biased dopamine D2 receptor partial agonists: Synthesis and pharmacological evaluation. Bioorg Med Chem 25, 5613–5628 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Armstrong SP, Seeber RM, Ayoub MA, Feldman BJ, Pfleger KD, Characterization of three vasopressin receptor 2 variants: an apparent polymorphism (V266A) and two loss-of-function mutations (R181C and M311V). PLoS One 8, e65885 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Erdelyi LS, Balla A, Patocs A, Toth M, Varnai P, Hunyady L, Altered agonist sensitivity of a mutant v2 receptor suggests a novel therapeutic strategy for nephrogenic diabetes insipidus. Mol Endocrinol 28, 634–643 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK, Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glukhova A, Draper-Joyce CJ, Sunahara RK, Christopoulos A, Wootten D, Sexton PM, Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci, 1, 73–83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Latorraca NR, Wang JK, Bauer B, Townshend RJL, Hollingsworth SA, Olivieri JE, Xu HE, Sommer ME, Dror RO, Molecular mechanism of GPCR-mediated arrestin activation. Nature 557, 452–456 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eichel K, Jullie D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita JB, Dror RO, von Zastrow M, Catalytic activation of beta-arrestin by GPCRs. Nature 557, 381–386 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez-Aguilar JM, Shan J, LeVine MV, Khelashvili G, Weinstein H, A functional selectivity mechanism at the serotonin-2A GPCR involves ligand-dependent conformations of intracellular loop 2. J Am Chem Soc 136, 16044–16054 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Choi M, Staus DP, Wingler LM, Ahn S, Pani B, Capel WD, Lefkowitz RJ, G protein-coupled receptor kinases (GRKs) orchestrate biased agonism at the beta2-adrenergic receptor. Sci Signal 11, (2018). [DOI] [PubMed] [Google Scholar]
- 64.Tobin AB, G-protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol 153 Suppl 1, S167–176 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim OJ, Gardner BR, Williams DB, Marinec PS, Cabrera DM, Peters JD, Mak CC, Kim KM, Sibley DR, The role of phosphorylation in D1 dopamine receptor desensitization: evidence for a novel mechanism of arrestin association. J Biol Chem 279, 7999–8010 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Masureel M, Zou Y, Picard LP, van der Westhuizen E, Mahoney JP, Rodrigues J, Mildorf TJ, Dror RO, Shaw DE, Bouvier M, Pardon E, Steyaert J, Sunahara RK, Weis WI, Zhang C, Kobilka BK, Structural insights into binding specificity, efficacy and bias of a beta2AR partial agonist. Nat Chem Biol 14, 1059–1066 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T, Hsueh R, Rebres RA, Ross EM, Taussig R, Fraser I, Sternweis PC, Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J Biol Chem 282, 10576–10584 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Black JW, Leff P, Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci 220, 141–162 (1983). [DOI] [PubMed] [Google Scholar]
- 69.Klein Herenbrink C, Verma R, Lim HD, Kopinathan A, Keen A, Shonberg J, Draper-Joyce CJ, Scammells PJ, Christopoulos A, Javitch JA, Capuano B, Shi L, Lane JR, Molecular determinants of the intrinsic efficacy of the antipsychotic aripiprazole. ACS Chem Biol, (2019). [DOI] [PMC free article] [PubMed]
- 70.Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK, Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, Shaw DE, Activation mechanism of the beta2-adrenergic receptor. Proc Natl Acad Sci U S A 108, 18684–18689 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, Kaus JW, Cerutti DS, Krilov G, Jorgensen WL, Abel R, Friesner RA, OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput 12, 281–296 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Feller SE, Zhang Y, Pastor RW, Brooks BR, Constant pressure molecular dynamics simulation: The Langevin piston method. The Journal of Chemical Physics 103, 4613–4621 (1995). [Google Scholar]
- 74.Stolzenberg S, Michino M, LeVine MV, Weinstein H, Shi L, Computational approaches to detect allosteric pathways in transmembrane molecular machines. Biochim Biophys Acta 1858, 1652–1662 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao C, Stolzenberg S, Gracia L, Weinstein H, Noskov S, Shi L, Ion-controlled conformational dynamics in the outward-open transition from an occluded state of LeuT. Biophys J 103, 878–888 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Michino M, Beuming T, Donthamsetti P, Newman AH, Javitch JA, Shi L, What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol Rev 67, 198–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Graaf C, Foata N, Engkvist O, Rognan D, Molecular modeling of the second extracellular loop of G-protein coupled receptors and its implication on structure-based virtual screening. Proteins 71, 599–620 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. D2R WT and D2R mutants express to a similar extent using transient transfection
Fig S2. Other D2R agonists exhibit G protein bias at the D2R-F1895.38A
Fig S3. Compound CAB02–110 is a partial agonist at both G protein activation and β-arrestin recruitment
Fig S4. Rotigotine- and apomorphine-stimulated internalization of the D2-WT and D2R-F1895.38A
Fig S5. The β2R-Y1995.38A exhibits G protein bias with different agonists
Fig S6. Partial agonist stimulation of β2R-mediated Gs activation and β-arrestin recruitment
Fig S7. Distribution of distance between the oxygen of Tyr5.38 sidechain hydroxyl and the backbone oxygen atom of Thr4.56 in the β2R-WT
Fig S8. IL2 moves downwards in the β2R-Y1995.38A mutant
Table S1. β-Arrestin-BRET and Go BRET signaling by D2R-WT and D2R mutants
Table S2. Affinity constants (Ki values) for MLS1547 and dopamine binding to D2R-WT and D2R mutants
Table S3. β-Arrestin recruitment and regulation of cAMP signaling by D2R-WT and D2R-F1895.38A
Table S4. β-Arrestin and Go BRET signaling for D2R-WT and D2R-F1895.38A in response to full D2R agonists
Table S5. β-Arrestin-BRET recruitment for D2R WT and various mutants at the Phe1895.38 position
Table S6. β-Arrestin- and G protein-mediated signaling by WT and 5.38 mutant D3R, D4R, β2R and V2R
Table S7. Summary of molecular dynamics simulations
Table S8. β2R-BI-167107 interacting residues.