

Abstract

Artesunic acid and artemisinin are natural substances with promiscuous anticancer activity against different types of cancer cell lines. The mechanism of action of these compounds is associated with the formation of reactive radical species by cleavage of the sesquiterpene pharmacophore endoperoxide bridge. Here we suggested topoisomerase 1 as a possible molecular target for the improvement of the anticancer activity of these compounds. In this context, we report that novel hybrid and dimer derivatives of artesunic acid and artemisinin, bearing camptothecin and SN38 as side-chain biological effectors, can inhibit growth of yeast cells overexpressing human topoisomerase 1 and its enzymatic activity in vitro. These derivatives showed also anticancer activity in melanoma cell lines higher than camptothecin and paclitaxel. In silico molecular docking calculations highlighted a common binding mode for the novel derivatives, with the sesquiterpene lactone scaffold being located near the traditional recognition site for camptothecin, while the bioactive side-chain effector laid in the camptothecin cleft.
Keywords: Artesunic acid, artemisinin, hybrid and dimer derivatives, topoisomerase 1 inhibitors, antimelanoma activity
Artesunic acid (1,Figure 1) is the hemisuccinate derivative of artemisinin (2), a natural sesquiterpene extracted from Artemisia annua (Figure 1). These compounds are antimalarial drugs1 able to produce reactive oxygen and carbon centered radicals by pH and metal mediated cleavage of the characteristic pharmacophore endoperoxide bridge.2 Artesunic acid and artemisinin derivatives also showed a promiscuous anticancer activity against different types of cancer cell lines,3−5 such as leukemia,6−8 melanoma, lung,9 colon, renal, ovarian, prostate, and breast,10,11 associated with the same type of radical mechanism12,13 as well as to the inhibition of both the mammalian sarco-endoplasmic calcium ATP-ase (SERCA) and the translationally controlled tumor protein (TCTP).14 Moreover, artemisinin derivatives target topoisomerase 115 and deregulate the c-Myc oncoprotein-topoisomerase cellular pathway, as in the case of artemisitene (4).16
Figure 1.

Chemical structures of artesunic acid (1), artemisinin (2), dihydroartemisinin (3), artemisitene (4), and artesunic acid and artemisinin hybrid and dimer derivatives 5–10.
Human DNA topoisomerase 1 (hTop1p) is a monomeric highly conserved enzyme composed by four domains: (a) the N-terminal domain containing the nuclear localization; (b) the core domain; (c) the linker domain; and (d) the C-terminal domain responsible for the cleavage of the DNA strand (Supporting Information SI#1).17,18 Inhibitors of hTop1p, including the alkaloid camptothecin (CPT), are currently used as anticancer drugs. CPT interacts reversibly with the enzyme blocking the DNA–protein complex after the cleavage and slowing the religation process. Single molecule experiments19 demonstrated that CPT derivatives significantly influence hTop1p-mediated DNA relaxation, with a more pronounced effect on the removal of positive (overwound) versus negative supercoils. Locking the DNA topoisomerase 1 protein clamp inhibits DNA rotation and induces cell lethality.20 The accumulation of positive supercoils ahead of the replication machinery induces potentially lethal DNA lesions. Furthermore, replication forks’ progression can generate double-strand breaks inducing apoptotic response.
Recently, with the aim to obtain a library of compounds endowed with antimelanoma activity, we synthesized a series of hybrids and dimers of artemisinin bearing some phytochemical products known for their anticancer activity.21 In particular, artemisinin and artesunic acid were combined with tyrosol, perillyl alcohol, and curcumin to give derivatives 5–10 (Figure 1). This library was screened against cervical cancer cell line (HeLa) and complementary metastatic melanoma cancer cell lines SK-MEL3, SK-MEL24, and RPMI7951, demonstrating an interesting biological activity. In this latter case, the occurrence of an alternative mechanism with respect to the cleavage of the endoperoxide bridge was hypothesized. Hence, we decided to evaluate the inhibitory activity of compounds 5–10 against hTop1p in Saccharomyces cerevisiae, which is a validated model system to test putative inhibitors and poisons of human enzymes in vivo.22−24
The experiments have been performed using plasmids expressing hTop1p under the control of Gal-1–10 promoter in a yeast strain EKY3 lacking the endogenous enzyme (SI#2).25,26 As shown in Figure 2, artemisinin (2) and compounds 9 and 10 did not show growth inhibitory activity. Artesunic acid (1) and dihydroartemisinin (3) showed a weak inhibition of the enzyme only at 30 μm (slightly improved in compounds 5 and 8), while derivatives 6 and 7 were the most active compounds showing an appreciable inhibitory effect at 10 μm. It is noteworthy that the growth inhibition was present only in cells overexpressing wild type human topoisomerase 1 gene, while overexpression of a CPT resistant mutant, where glycine 363 was changed into cysteine, did not show such an effect. This finding strongly suggested a similar mechanism of action between CPT and the artemisinin derivatives (SI#3). The most active compounds (including 1 as reference) were further evaluated, using the relaxing activity assay of hTop1p in vitro. As shown in Figure 3, compounds 6 and 7 were more effective than derivatives 1 and 8.
Figure 2.

Sensitivity of Saccharomyces cerevisiae EKY3 strain expressing human topoisomerase 1 was measured at different concentrations of artesunic acid (1), artemisinin (2), dihydroartemisinin (3), artemistene (4), and artesunic acid, as well as artemisinin hybrid and dimer derivatives 5–12. SN38 was used as reference compound (Ctrl: selective medium without compounds).
Figure 3.

Inhibition of the relaxing activity of hTop1p 1 in the presence of artesunic acid (1) and compounds 6–8, 11, and 12. SN38 was used as reference compound (pKS: negatively supercoiled pBlue-Script KSII(+) DNA; mix NT: not treated; topo: hTop1p; DMSO: dimethyl sulfoxide).
We further analyzed the cell phenotype, in order to better characterize the inhibitory effect of compounds 6 and 7. As shown in SI#4, cells treated with compound 6 exhibit a cell cycle defect, suggesting the activation of a DNA damage check point.
In silico molecular docking calculations (SI#6) were performed in order to rationalize the interaction between artemisnin derivatives and hTop1p. X-ray structure of this enzyme in complex with CPT was reported in the Protein Data Bank (PDB) with the code 1T8I.27 In this crystal structure CPT is stacked between DNA bases, making interactions with Arg364 and Asp533 by the aromatic nitrogen and the tertiary alcohol group, respectively. Considering that Camptotheca acuminata expresses topoisomerase with point mutations that confer resistance to CPT,28 we envisaged the possibility that Artemisina annua presents a mutated topoisomerase resistant to artemisinin. To confirm this idea, we aligned the primary sequences of hTop1p and topoisomerase from Artemisia annua (Uniprot sequence: A0A2U1QNR1|180-726, SI#5) obtaining a 50.19% of identity. Homology modeling showed that the only difference in the region of the active site was the substitution of Phe214 in Artemisia annua topoisomerase with Met428 in hTop1p (Figure 4A and SI#6). Starting from this result, we hypothesized the binding of the artemisinin-sesquiterpene moiety in the cavity generated by this substitution. In agreement with these findings, the sesquiterpene lactone portion occupied the pocket with Met428 mutation for all the compounds docked, while the phytochemical active moiety was located in the CPT recognition site.
Figure 4.
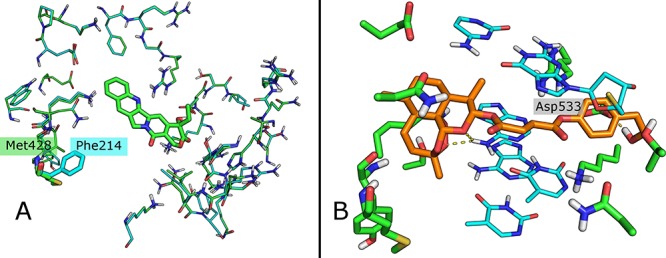
(A) Superposition of 1T8I crystal structure (green) and Artemisia annua topoisomerase homology model (Cyan); in light green CPT. The only residue difference Phe214/Met428 is reported in stick. (B) Binding mode of compound 7 (orange). Residues are reported in green and DNA in cyan.
In compound 7, for example, the artemisinin counterpart is positioned in the Met428 pocket forming two H bonds with the DNA, and the succinate linker is bounded in the CPT folder while the terminal OH is able to generate a further H bond with Asp533 like CPT (Figure 4B). In compound 6, the peroxide group of the sesquiterpene moiety makes two hydrogen bonds with an amine functionality of the DNA; the rest of the molecule interacts with Asn 720 and Arg 364 residues by a carbonyl of the linker and the phenolic group of the phytochemical counterpart, respectively (Figure 5B). Compound 5, whose aromatic phytochemical effector does not occupy well the recognition site, is the less active derivative of the three docked molecules (Figure 5A). Note that 5 has a shorter linker compared to derivatives 6 and 7 bearing the hemisuccinate moiety of artesunic acid. On the basis of these data, we synthesized hybrid derivatives 11 and 12 (Scheme 1) in which the artesunic acid scaffold is linked to CPT, and in alternative to SN38 (that is the active form of the anticancer drug irinotecan, Figure 6), in order to optimize the interaction with both hTop1p binding sites (SI#1).
Figure 5.

(A) Binding mode of compound 5 (salmon). Residues are reported in green and DNA in cyan. (B) Binding mode of compound 6 (light gray). Residues are reported in green and DNA in cyan.
Scheme 1. Synthesis of Artesunic Acid Hybrid Derivatives 11 and 12 Bearing CPT and SN 38 as Side-Chain Bioactive Effectors.

Figure 6.

Chemical structures of camptothecin, irinotecan, and SN 38.
Briefly, artesunic acid (1) was treated with CPT and SN 38 in dimethylformamide (DMF) in the presence of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP), to afford compounds 11 and 12 in 40% and 64% yields, respectively (Scheme 1 and SI#7). The stability of compound 12 was evaluated, as a selected example, by high performance liquid chromatography (HPLC) analyses solubilizing the compound in the cell culture medium. No significative amount of SN 38 was detected after 48 h treatment, confirming the stability of 12 in the analyzed conditions (see Supporting Information SI#8–11).
Derivatives 11 and 12 showed high inhibitory activity against hTop1p under both in vivo (Figure 2) and in vitro experiments (Figure 3). In particular, compound 12 was active at 3 μM in vivo and at 300 nM in vitro. On the contrary, compound 11 was more effective in vivo than in vitro. Moreover, compound 12 was not effective on CPT resistant mutant G363C confirming a mechanism of action similar to CPT (SI#3).
The anticancer activity of compounds 11 and 12 was evaluated by the cell survival 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay on the melanoma cancer cell lines RPMI7951 and SK-MEL24, and on the primary human fibroblast cell line C3PV, in the presence of CPT, SN38, and paclitaxel (Taxol), as references. It is noteworthy that compound 12 showed antimelanoma activity higher than CPT and paclitaxel, confirming the beneficial role of the artesunic acid pharmacophore in tuning of the biological activity (Table 1).
Table 1. Anticancer Activity of Compounds 11 and 12.a.
| IC50b |
||||
|---|---|---|---|---|
| Entry | Compound | C3PV | RPMI7951 | SK-MEL 24 |
| 1 | 11 | 0.3 ± 0.1 | 17.32 ± 3.4 | 0.85 ± 0.3 |
| 2 | 12 | 3.2 ± 2.4 | 0.05 ± 0.02 | 0.32 ± 0.03 |
| 3 | camptothecin | 0.45 ± 0.3 | 0.22 ± 0.09 | 0.46 ± 0.05 |
| 4 | SN 38 | 0.51 ± 0.26 | 0.26 ± 0.07 | 0.48 ± 0.07 |
| 5 | paclitaxel | 78.88 ± 0.79 | 0.013 ± 0.19 | 4.73 ± 0.83 |
Experiments were conducted in triplicate.
IC50 ± SD (half maximal inhibitory concentration ± standard deviation) values for all the compounds are expressed in μM unit.
The inhibitory activity of compounds 11 and 12 against hTop1p was rationalized by in silico molecular docking calculations using GLIDE and FRED software, respectively (SI#6). In the binding mode of compound 11, the interaction of the carbonyl oxygen of the lactam ring with the DNA and a new interaction (not reported before) with the Lys425 are evident (Figure 7A). Interestingly, the CPT portion of 11 is flipped compared to CPT alone, as shown by superposition of 11 with this natural product (SI#12). In compound 12, the SN38 counterpart is oriented like CPT in the recognition site of hTop1p (SI#13), with the tertiary alcohol interacting with the amino acid residue Asp533 (hid in Figure 7B). Probably, this interaction may be responsible for the higher antimelanoma activity of compound 12 with respect to 11.
Figure 7.
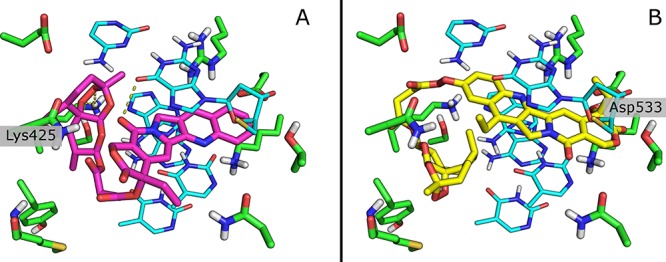
(A) Binding mode of compound 11 (magenta). Residues are reported in green and DNA in cyan. (B) Binding mode of compound 12 (yellow). Residues are reported in green and DNA in cyan.
In conclusion, the inhibition of yeast cells overexpressing hTop1p, associated with data on the relaxing activity in vitro, highlights the possibility that hTop1p might be a molecular target for the analyzed compounds. Moreover, growth inhibition studies on CPT resistant mutant of yeast EKY3 strain and cell phenotype analysis, strongly suggest that CPT and novel artemisinin derivatives share a similar mechanism of cell killing. In accordance with this hypothesis, the in silico molecular docking calculation used to depict the binding mode of compounds 5, 6, and 7 showed that the sesquiterpene lactone scaffold is positioned in a cavity near the traditional recognition site for CPT, while the bioactive side-chain effector laid in the CPT cleft. As a proof of concept compound 12, bearing SN38 as side-chain biological effector, was found to fit in the two binding pockets of topoisomerase 1, showing a mechanism of enzymatic and yeast cell growth inhibitions very similar to CPT.
Acknowledgments
This article is dedicated to Prof. Maurizio Botta for his contribution to the Medicinal Chemistry field. This work was supported by funds from MIUR Ministero dell’Istruzione, dell’Università della Ricerca Italiano, project PRIN 2017, ORIGINALE CHEMIAE in Antiviral Strategy - Origin and Modernization of Multi-Component Chemistry as a Source of Innovative Broad Spectrum Antiviral Strategy, cod. 2017BMK8JR (L.B. and R.S.) and AIRC - MFAG2017 (n. 20447) (A.P.). We are grateful to Agostino Bruno and Serena Rosignoli for their help in the computational analyses.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00131.
Structure of DNA topoisomerase 1B; cell culture condition; treatment protocol; statistical analysis; in vivo and in vitro assays; analysis of the EKY3 cell phenotype; sequence alignment of hTop1p and Artemisia annua topoisomerase; homology model, docking analyses; procedures for the synthesis of derivatives 11 and 12; HPLC analysis of compounds SN 38, 12, and 12 after treatment with the cell culture medium at room temperature for 48 h; superposition of CPT and compounds 11 and 12, respectively, in the active site of hTop1p in complex with DNA (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- White N. J. Qinghaosu (Artemisinin): The Price of Success. Science 2008, 320, 330–334. 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- Butler A. R.; Gilbert B. C.; Hulme P.; Irvine L. R.; Ren-Ton L.; Whitwood A. C. EPR Evidence for the Involvement of Free Radicals in the Iron-Catalysed Decomposi-tion of Qinghaosu (Artemisinin) and Some Derivatives; Antimalarial Action of Some Polycyclic Endoperoxides. Free Radical Res. 1998, 28, 471–476. 10.3109/10715769809066884. [DOI] [PubMed] [Google Scholar]
- Lai H. C.; Singh N. P.; Sasaki T. Development of artemisinin compounds for cancer treatment. Invest. New Drugs 2013, 31, 230–246. 10.1007/s10637-012-9873-z. [DOI] [PubMed] [Google Scholar]
- Fröhlich T.; Çapcñ Karagöz A.; Reiter C.; Tsogoeva S. B. Artemisinin-Derived Dimers: Potent Antimalarial and Anticancer Agents. J. Med. Chem. 2016, 59, 7360–7388. 10.1021/acs.jmedchem.5b01380. [DOI] [PubMed] [Google Scholar]
- Fröhlich T.; Reiter C.; Ibrahim M. M.; Beutel J.; Hutterer C.; Zeitträger I.; Bahsi H.; Leidenberger M.; Friedrich O.; Kappes B.; Efferth T.; Marschall M.; Tsogoeva S. B. Synthesis of Novel Hybrids of Quinazoline and Artemisinin with High Activities against Plasmodium falciparum, Human Cytomegalovirus and Leukemia Cells. ACS Omega 2017, 2, 2422–2431. 10.1021/acsomega.7b00310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter C.; Herrmann A.; Çapci A.; Efferth T.; Tsogoeva S. B. New artesunic acid homodimers: Potent reversal agents of multidrug resistance in leukemia cells. Bioorg. Med. Chem. 2012, 20, 5637–5641. 10.1016/j.bmc.2012.07.015. [DOI] [PubMed] [Google Scholar]
- Reiter C.; Capcñ Karagöz A.; Fröhlich T.; Klein V.; Zeino M.; Viertel K.; Held J.; Mordmüller B.; Emirdaǧ Öztürk S.; Anñl H.; Efferth T.; Tsogoeva S. B. Synthesis and study of cytotoxic activity of 1,2,4-trioxane- and egonol-derived hybrid molecules against Plasmodium falciparum and multidrug-resistant human leukemia cells. Eur. J. Med. Chem. 2014, 75, 403–412. 10.1016/j.ejmech.2014.01.043. [DOI] [PubMed] [Google Scholar]
- Capcı Karagoz A.; Reiter C.; Seo E.-J.; Gruber L.; Hahn F.; Leidenberger M.; Klein V.; Hampel F.; Friedrich O.; Marschall M.; Kappes B.; Efferth T.; Tsogoeva S. B. Access to new highly potent antileukemia, antiviral and antimalarial agents via hybridization of natural products (homo)egonol, thymoquinone and artemisinin. Bioorg. Med. Chem. 2018, 26, 3610–3618. 10.1016/j.bmc.2018.05.041. [DOI] [PubMed] [Google Scholar]
- Fröhlich T.; Ndreshkjana B.; Muenzner J. K.; Reiter C.; Hofmeister E.; Mederer S.; Fatfat M.; El-Baba C.; Gali-Muhtasib H.; Schneider-Stock R.; Tsogoeva S. B. Synthesis of Novel Hybrids of Thymoquinone and Artemisinin with High Activity and Selectivity Against Colon Cancer. ChemMedChem 2017, 12, 226–234. 10.1002/cmdc.201600594. [DOI] [PubMed] [Google Scholar]
- Fröhlich T.; Kiss A.; Wölfling J.; Mernyák E.; Kulmány A. E.; Minorics R.; Zupkó I.; Leidenberger M.; Friedrich O.; Kappes B.; Hahn F.; Marschall M.; Schneider G.; Tsogoeva S. B. Synthesis of Artemisinin-Estrogen Hybrids Highly Active against HCMV, P. falciparum, and Cervical and Breast Cancer. ACS Med. Chem. Lett. 2018, 9, 1128–1133. 10.1021/acsmedchemlett.8b00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efferth T.; Saverbrey A.; Olbrich A.; Gebhart E.; Rauch P.; Weber H. O.; Hengstler J. G.; Halatsch M. E.; Volm M.; Tew K. D.; Ross D. D.; Funk J. O. Molecular modes of action of artesunate in tumour cell lines. Mol. Pharmacol. 2003, 64, 382–394. 10.1124/mol.64.2.382. [DOI] [PubMed] [Google Scholar]
- Dhingra V.; Vishweshwar R. K.; Lakshmi N. M. Current status of artemisinin and its derivatives as antimalarial drugs. Life Sci. 1999, 66, 279–300. 10.1016/S0024-3205(99)00356-2. [DOI] [PubMed] [Google Scholar]
- Efferth T. Mechanistic perspectives for 1,2,4-trioxanes in anticancer therapy. Drug Resist. Updates 2005, 8, 85–97. 10.1016/j.drup.2005.04.003. [DOI] [PubMed] [Google Scholar]
- O’Neill P. M.; Barton V. E.; Ward S. A. The molecular mechanism of action of artemisinin - the debate continues. Molecules 2010, 15, 1705–1721. 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadioglu O.; Chan A.; Cong Ling Qiu A.; Wong V. K. M.; Colligs V.; Wecklein S.; Freund-Henni Rached H.; Efferth T.; Hsiao W. W. Artemisinin Derivatives Target Topoisomerase 1 and Cause DNA Damage in Silico and in Vitro. Front. Pharmacol. 2017, 8, 711. 10.3389/fphar.2017.00711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Li W.; Cui K.; Ji K.; Xu S.; Xu Y. Artemisitene suppresses tumorigenesis by inducing DNA damage through deregulating c-Myc-topoisomerase pathway. Oncogene 2018, 37, 5079–5087. 10.1038/s41388-018-0331-z. [DOI] [PubMed] [Google Scholar]
- Palle K.; Pattarello L.; van der Merwe M.; Losasso C.; Benedetti P.; Bjornsti M. A. Disulfide cross-links reveal conserved features of DNA topoisomerase I architecture and a role for the N terminus in clamp closure. J. Biol. Chem. 2008, 283, 27767–27775. 10.1074/jbc.M804826200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti P.; Benchokroun Y.; Houghton P. J.; Bjornsti M. A. Analysis of camptothecin resistance in yeast: relevance to cancer therapy. Drug Resist. Updates 1998, 1, 176–183. 10.1016/S1368-7646(98)80037-X. [DOI] [PubMed] [Google Scholar]
- Koster D. A.; Palle K.; Bot E. S.; Bjornsti M. A.; Dekker N. H. Antitumour drugs impede DNA uncoiling by topoisomerase I. Nature 2007, 448 (7150), 213–217. 10.1038/nature05938. [DOI] [PubMed] [Google Scholar]
- Woo M. H.; Losasso C.; Guo H.; Pattarello L.; Benedetti P.; Bjornsti M. A. Locking the DNA topoisomerase I protein clamp inhibits DNA rotation and induces cell lethality. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13767–13772. 10.1073/pnas.2235886100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botta L.; Filippi S.; Bizzarri B. M.; Zippilli C.; Meschini R.; Pogni R.; Baratto M. C.; Villanova L.; Saladino R.. Synthesis and Evaluation of Artemisinin-based Hybrid and Dimer Derivatives as Antimelanoma Agents. ACS Omega 2020, 5, 243. 10.1021/acsomega.9b02600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirigliano A.; Amelina A.; Biferali B.; Macone A.; Mozzetta C.; Bianchi M.; Mori M.; Botta B.; Pick H.; Negri R.; Rinaldi T. Statins interfere with the attachment of S. cerevisiae mtDNA to the inner mitochondrial membrane. J. Enzyme Inhib. Med. Chem. 2019, 35, 129–137. 10.1080/14756366.2019.1687461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirigliano A.; Stirpe A.; Menta S.; Mori M.; Dell’Edera D.; Pick E.; Negri R.; Botta B.; Rinaldi T. Yeast as a tool to select inhibitors of the cullin deneddylating enzyme Csn5. J. Enzyme Inhib. Med. Chem. 2016, 31, 1632–1637. 10.3109/14756366.2016.1160901. [DOI] [PubMed] [Google Scholar]
- Mannironi C.; Proietto M.; Bufalieri F.; Cundari E.; Alagia A.; Danovska S.; Rinaldi T.; Famiglini V.; Coluccia A.; La Regina G.; Silvestri R.; Negri R. An High-Throughput In Vivo Screening System to Select H3K4-Specific Histone Demethylase Inhibitors. PLoS One 2014, 9 (1), e86002 10.1371/journal.pone.0086002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsti M. A.; Benedetti P.; Viglianti G. A.; Wang J. C. Expression of human DNA topoisomerase I in yeast cells lacking yeast DNA topoisomerase I: restoration of sensitivity of the cells to the antitumor drug camptothecin. Cancer Res. 1989, 49, 6318–6323. [PubMed] [Google Scholar]
- Hann C.; Evans D. L.; Fertala J.; Benedetti P.; Bjornsti M. A.; Hall D. J. Increased camptothecin toxicity induced in mammalian cells expressing Saccharomyces cerevisiae DNA topoisomerase I. J. Biol. Chem. 1998, 273, 8425–8433. 10.1074/jbc.273.14.8425. [DOI] [PubMed] [Google Scholar]
- Staker B. L.; Feese M. D.; Cushman M.; Pommier Y.; Zembower D.; Stewart L.; Burgin A. B. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. 10.1021/jm049146p. [DOI] [PubMed] [Google Scholar]
- Sirikantaramas S.; Yamazaki M.; Saito K. Mutations in topoisomerase I as a self-resistance mechanism coevolved with the production of the anticancer alkaloid camptothecin in plants. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 6782–6786. 10.1073/pnas.0801038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.