

Abstract
TASIN (Truncated APC-Selective Inhibitors) compounds are selectively toxic to colorectal cancer cells with APC mutations, although their mechanism of action remains unknown. Here, we found that TASINs inhibit three enzymes in the post-squalene cholesterol biosynthetic pathway including EBP, DHCR7, and DHCR24. Even though all three of these enzymes are required for cholesterol biosynthesis, only inhibition of the most upstream enzyme, EBP, led to cancer cell death via depletion of downstream sterols, an observation that was confirmed by genetic silencing of EBP. Pharmacologic inhibition or genetic silencing of either DHCR7 or DHCR24 had no impact on cell viability. By using photo-affinity probes to generate a relationship between chemical structure and probe competition, we identified compounds that selectively inhibit either EBP or DHCR7. These studies identify EBP, but not downstream enzymes in the cholesterol biosynthetic pathway, as a target in APC mutant colorectal cancer and also have implications for the clinical development of highly selective EBP inhibitors.
Graphical Abstract
1. INTRODUCTION
APC is a tumor suppressor in colorectal cancer.1 Loss of function mutations in APC drive tumor growth by stabilizing the oncogenic transcription factor β-catenin.2 APC mutations are found in over 85% of familial adenomatous polyposis (FAP) patients,3 as well as over 80% of sporadic colorectal tumors;4 approximately 65% of these mutations cluster between amino acid 1,286 and 1,513.4
TASIN-1 (Truncated APC-Selective Inhibitor-1) was identified from a small molecule high-throughput screen for compounds that selectively target APC mutant colorectal cancer cells.5 TASIN-1 toxicity is only evident in lipoprotein deficient culture conditions, which implicates the sterol pathway as a potential target.5 A series of rescue experiments with different sterol intermediates predicted that TASIN-1 toxicity potentially depends on inhibiting the Emopamil binding protein (EBP).5 EBP is the 3-beta-hydroxysteroid-Delta(8),Delta(7)-isomerase in the post-squalene cholesterol biosynthetic pathway that can convert zymosterol to dehydrolathosterol, or zymostenol to lathosterol. The products of both reactions lead to cholesterol biosynthesis by one of two parallel pathways, known as the Bloch6 and Kandutsch/Russell7 pathway, respectively. Lathosterol but not zymosterol rescued TASIN-1 toxicity, supporting the hypothesis that TASIN-1 targets EBP. Furthermore, suppression of EBP by RNA interference reduced the proliferation of APC mutant colorectal cancer cells.5 Although this evidence is consistent with TASIN-1 targeting EBP, it remains unclear as to whether TASIN-1 directly binds to and inhibits EBP.
In an unbiased effort to identify the molecular target of TASIN, we used photochemical TASIN probes, which can be activated by ultraviolet (UV) light, to identify TASIN binding partners in live cells. Using this approach, we identified three TASIN targets in the cholesterol biosynthetic pathway: EBP, DHCR7, and DHCR24 (listed from upstream to downstream). Isotopic tracing of sterols in cells by mass spectrometry provides evidence that all three of these enzymes are inhibited by TASIN analogs at nanomolar concentrations. Even though inhibition of any enzyme blocks cholesterol biosynthesis, the toxic effects of TASIN are exclusively dependent on EBP inhibition. Using both pharmacologic and genetic systems, we found that EBP is essential in APC mutant cells because they are less efficient at scavenging cholesterol from LDL. We also identified TASIN–30 as a derivative that selectively inhibits EBP without affecting the activity of either DHCR7 or DHCR24.
2. RESULTS
2.1. Identification of the functional target of TASIN utilizing photoaffinity probes and medicinal chemistry.
To identify a photo-affinity TASIN probe, we synthesized derivatives in search of an active analog that contains a UV reactive group, such as a benzophenone or aryl azide, and an alkyne. Small molecules with these two chemical groups can be hypothetically used as chemical probes to identify proteins that bind TASIN. Upon exposure to ultraviolet treatment, benzophenone or aryl azide groups are expected to form a diradical or nitrene, respectively, which have the potential to covalently react with a proximal amino acid of a bound protein. The size of the resulting compound-protein complexes can be identified by performing SDS-PAGE on lysate that has been “clicked” with a fluorescent-azide. Click reactions efficiently and specifically conjugate a fluorescent azide to the alkyne group via a copper dependent azido-alkyne cycloaddition.
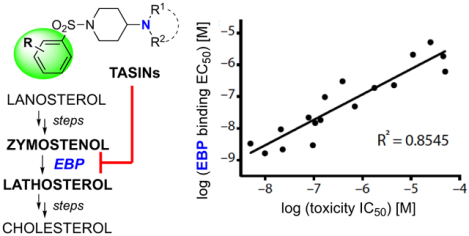
Based on an extensive medicinal chemistry program to establish structure-activity relationships within the TASIN series,8 we prepared several potential photoaffinity probes. One of these probes, TASIN-2 (Figure 1a), retained selective activity to DLD-1 cells. The viability of DLD-1 (APC mutant) cells treated with TASIN-2 was reduced by 50% (IC50) at 0.794 μM whereas HCT116 (wild-type APC) cells were insensitive at concentrations as high as 50 μM (Supplementary Fig. 1a). DLD-1 cells were treated with TASIN-2 in dose response and subjected to UV-B irradiation (310 nm). SDS-PAGE of clicked lysates revealed numerous dose-dependent, UV-dependent protein-compound conjugates, some of which were evident at concentrations as low as 0.003 μM (Figure 1b, Supplementary Figure 1b,c). To characterize which of these proteins might be related to TASIN activity, we tested whether other active TASIN analogs also bind these proteins by using a competition assay. For example, we measured the ability of TASIN-18 to interact with TASIN-2 bound proteins by repeating the UV crosslinking experiment with a fixed concentration of TASIN-2 and increasing concentrations of TASIN-18. The capacity of TASIN-18 to bind one of these proteins was measured by a reduction in TASIN-2 bound signal. We found that TASIN-18 reduced the fluorescent signal of four proteins in a dose-dependent fashion. We denoted these proteins by their estimated molecular weights as p18, p20, p27, and p70 (Figure 1c, Supplementary Fig. 1d). This suggested that one or more of these bands represented the target of TASIN compounds.
Figure 1.
TASIN binding to EBP leads to selective toxicity in APC mutant cells. (a) Chemical structure of TASIN-2, a photoaffinity probe. (b) SDS-PAGE analysis of DLD-1 cells incubated with different TASIN-2 concentrations in the presence or absence of UV light. (c) SDS-PAGE gel of DLD-1 cell lysates treated with TASIN-2 in the presence of different concentrations of TASIN-18, an active analog. (d) Correlation amongst 17 TASIN analogs for toxicity (IC50) and p20 binding (EC50). (e) Silver-stained SDS-PAGE gel of p20 purified from cell lysates of DLD-1 cells that were cross-linked with TASIN-2 in the presence or absence of an active competitor. (f) SDS-PAGE analysis of TASIN-2 binding to p20 in wild type and EBP null cells. (g) EBP is essential in DLD-1 cells grown in low serum conditions. Cell growth for DLD-1 cells expressing independent EBP sgRNAs in media conditions that include either 10% FBS (above) or 0.2% FBS (below).
We postulated that the concentration of TASIN-18 required to compete an individual protein band reflects its affinity for that protein. For example, 0.023 μM of TASIN-18 was sufficient to partially compete p20 but not p18 (Figure 1c) illustrating the ability to detect small differences in binding affinity between analogs. We reasoned that the concentration of an analog to reduce fluorescent signal by 50% (EC50) would correlate with its anti-proliferative potency (IC50) for on-target proteins. Thus, we repeated the aforementioned competition experiment with the TASIN-2 probe and 16 additional analogs, which represented a broad range of anti-proliferative potencies from 0.005 μM to 117 μM. (Supplementary Fig. 1e).
We plotted the competition IC50 for each analog versus its toxicity IC50 for each of the four bands (Figure 1d, Supplementary figure 1f). Among the four bands, p20 displayed the highest correlation of binding to toxicity with an R2 value of 0.8545, making this the most likely functional target of TASIN. Three other bands, p18, p27, and p70, displayed lower correlations between binding and toxicity, with R2 values of 0.738, 0.6121, and 0.2193, respectively.
To identify p20, we repeated the UV crosslinking experiments on a larger scale, and the click reaction was performed with a biotin-azide instead of a fluorescent-azide. By clicking biotin to compound bound proteins, we were able to use immobilized streptavidin to enrich and purify biotinylated proteins from the resulting lysate. The experiment was performed in the absence and presence of TASIN-18 as a competitor to help pinpoint p20. The eluate from streptavidin beads was analyzed by SDS-PAGE followed by silver staining, and as expected revealed a 20 kDa protein that was not present in the sample that included the competitor (Figure 1e, Supplementary Fig. 1g). We excised the p20 band and a gel slice of the corresponding molecular weight from the sample treated with the competitor. The gel slice was treated with trypsin, and the identity and abundance of the resulting peptides were determined by liquid chromatography and mass spectrometry (LC/MS). To identify proteins that were more abundant in the probe-only sample in comparison to the sample containing a competitor, we ranked all of the proteins identified in both samples by the ratios of their spectral index, which approximates abundance. The number one ranked protein with a ratio of 71.1 was Emopamil binding protein or EBP (Supplementary Tables 1, 2). To provide genetic evidence in support of our hypothesis that p20 is indeed EBP, we isolated two independent DLD-1 clones in which we silenced EBP using CRISPR/Cas9 technology (Supplementary Figure 1h). We observed no p20 crosslinking in DLD-1 EBP null cells providing genetic evidence that EBP encodes the protein at p20 (Figure 1f, Supplementary Fig. 1i).
EBP is an isomerase that converts zymostenol (or zymosterol) to lathosterol (or dehydrolathosterol), a required reaction in the post-squalene synthesis of cholesterol. TASIN toxicity is evident in DLD-1 cells cultured in 0.2% fetal bovine serum (FBS) supplemented with growth factors. Higher concentrations of FBS rescue the toxic effects of TASIN by providing a greater supply of cholesterol for lipoprotein uptake from the serum.5 Thus, the serum-rescue effect for TASIN is consistent with the hypothesis that TASIN inhibits the canonical activity of EBP. We thus tested whether EBP is essential to DLD-1 cells by reducing EBP expression using CRISPR/Cas9. When DLD-1 cells are grown in media containing 10% FBS, cells lacking EBP expression showed no growth deficit. By contrast, in low serum and hence low lipoprotein conditions, EBP silencing led to reduced viability (Figure 1g). Amongst the three different EBP single guide RNA’s, the reduction in viability correlated to the relative levels of EBP protein suppression (Figure 1h).
Next, we analyzed whether the selectivity observed with TASIN for APC mutant cells can be reproduced by EBP silencing. We compared the impact on cell growth between wild type cells and EBP null cells in DLD-1 and HCT116 cells, the latter of which is insensitive to TASIN. DLD-1 cells lacking EBP were unable to proliferate (Figure 1i). By contrast, HCT116 cells lacking EBP showed no growth deficit over 12 days (Figure 1i, Supplementary Figure 1h). Nonetheless, by day 20, EBP null HCT116 cells lost viability. Based on these observations, we concluded that in identical media conditions DLD-1 cells have a higher requirement for EBP activity than HCT116.
2.2. TASIN inhibits multiple enzymes in the cholesterol biosynthetic pathway.
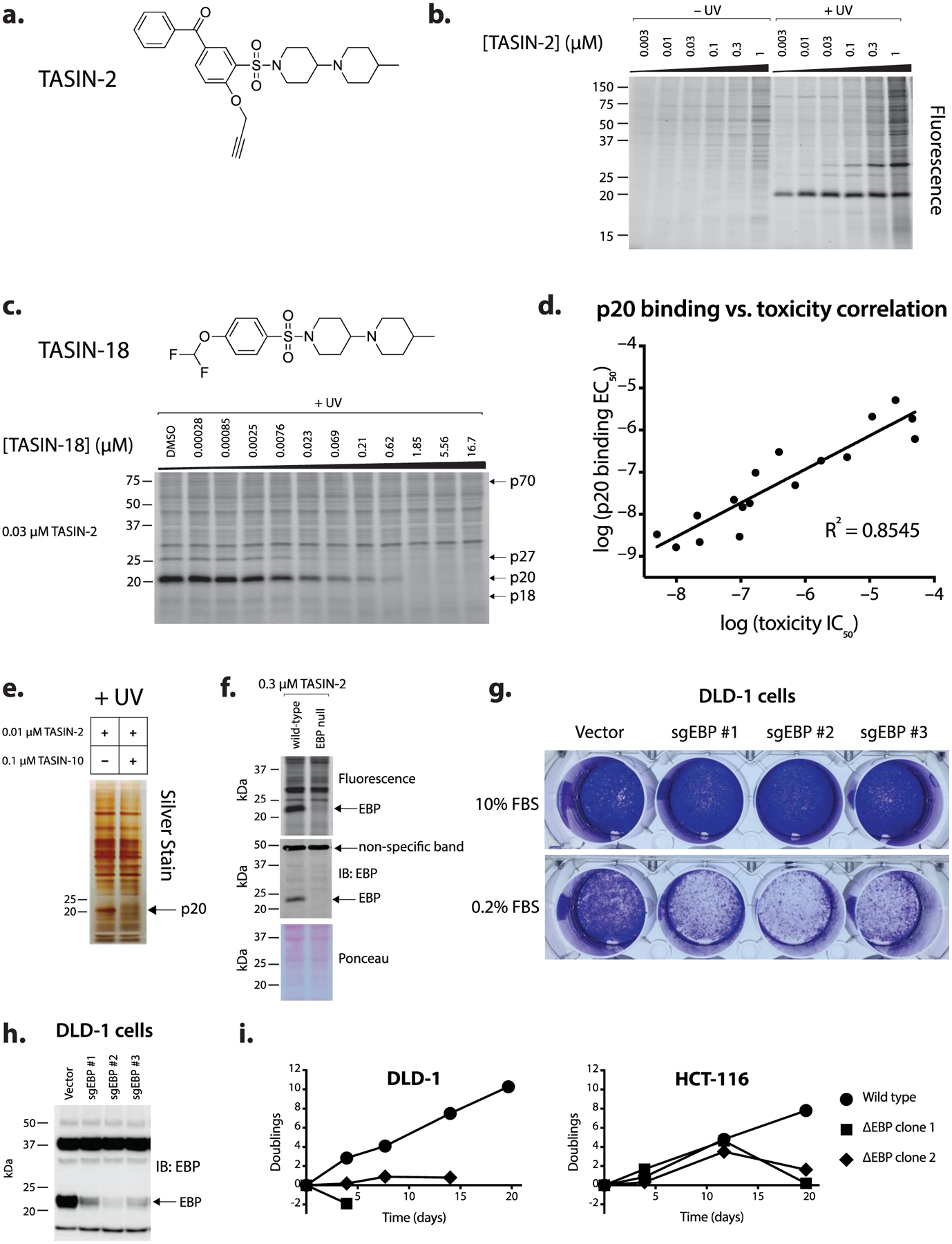
To test the hypothesis that TASIN inhibits EBP’s activity, we measured the levels of the following sterol intermediates upstream of EBP: lanosterol, ff-MAS (follicular fluid meiosis-activating sterol), zymosterol and dihydrolanosterol in cells treated between 0.03 and 10 μM of TASIN-18 (Figure 2a). TASIN-18 led to a dose-dependent increase in the levels of lanosterol, zymosterol, and ff-MAS, but had no impact on dihydrolanosterol levels. In comparison to DMSO treatment, 0.3 μM of TASIN-18 led to a 2.10, 5.97, and 108 fold increase in lanosterol, ff-MAS, zymosterol respectively (Figure 2b). The potency and magnitude of the increase is most evident for zymosterol, which is consistent with EBP inhibition leading to substrate accumulation for upstream enzymes.
Figure 2.
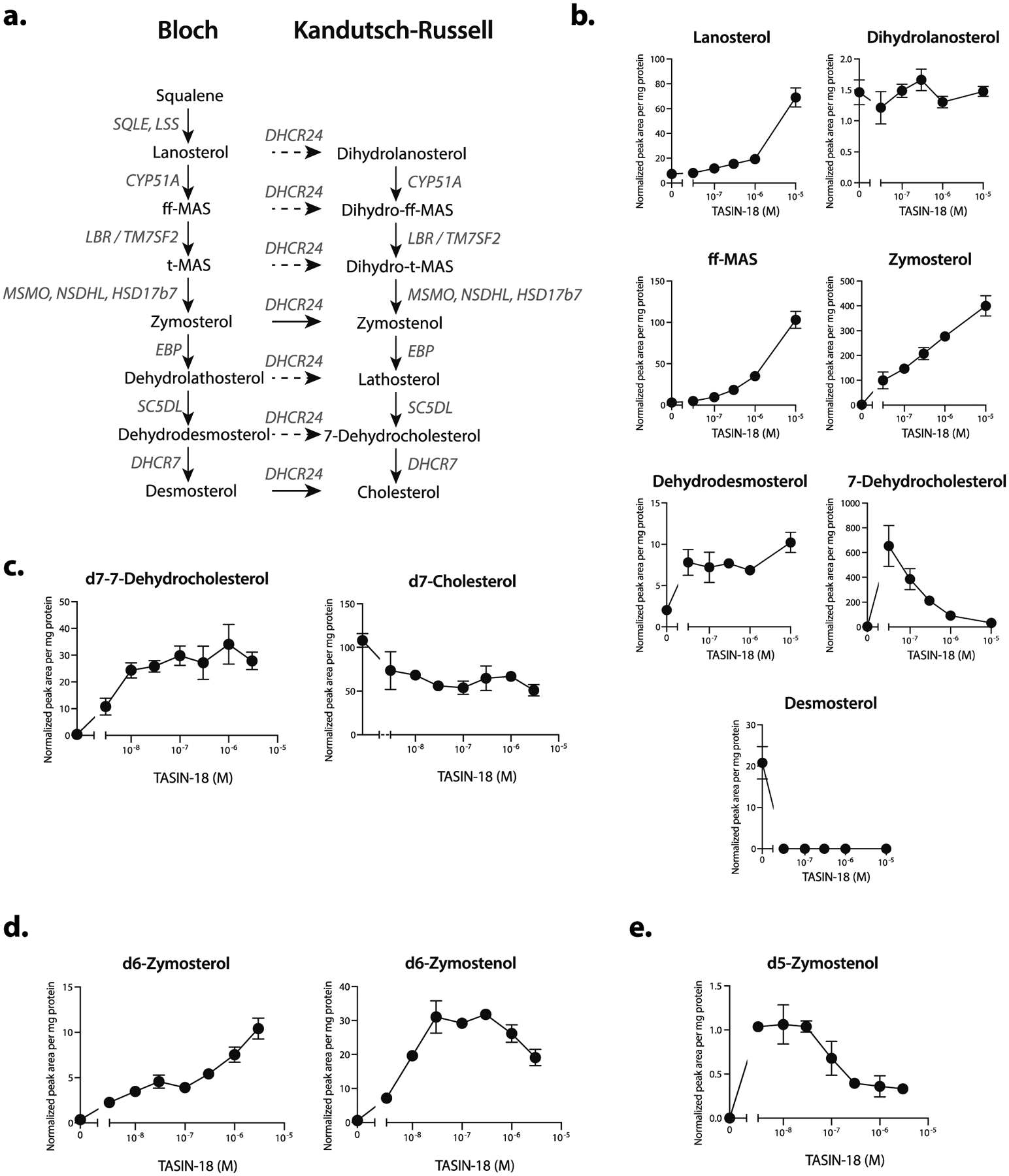
TASIN inhibits EBP, DHCR7 and DHCR24 in the cholesterol synthesis pathway. (a) Schematic of the post-squalene cholesterol synthesis pathway. The solid arrows represent zymosterol or desmosterol as the DHCR24 substrates for the Kandutsch-Russell (KR) or Bloch pathways, respectively.6,7 (b) Steady state sterol levels in DLD-1 cells following treatment with increasing concentrations of TASIN-18. (c) TASIN-18 inhibits DHCR7 based on isotopic tracing of d7-lathosterol. (d) TASIN-18 inhibits EBP and DHCR24 based on isotopic tracing with d6-lanosterol. (e) TASIN-18 inhibits DHCR24 based on d5-zymosterol isotopic tracing.
We also analyzed the following sterols downstream of EBP: dehydrodesmosterol, desmosterol, and 7-dehydrocholesterol. Our prediction was that TASIN-18 would inhibit EBP and lead to a decrease in these downstream sterol intermediates. Consistent with this hypothesis, desmosterol levels were undetectable in TASIN-18 treated cells, but surprisingly, we found that treatment with 0.03 μM of TASIN-18 caused a 170-fold increase in 7-dehydrocholesterol, the substrate for 7-dehydrocholesterol reductase (DHCR7) (Figure 2b). DHCR7 is an oxidoreductase that converts either 7-dehydrocholesterol or dehydrodesmosterol to cholesterol or desmosterol, respectively (Figure 2a). The observed increase in 7-dehydrocholesterol raised the possibility that TASIN-18 inhibits DHCR7 at even lower concentrations than it inhibits EBP. It is notable that the observed increase in 7-dehydrocholesterol was diminished with higher concentrations of TASIN-18. This effect could be explained by EBP inhibition at higher concentrations leading to a depletion of the DHCR7 substrate.
To directly test whether TASIN-18 inhibits DHCR7, we assayed DHCR7 activity independent of its effects on EBP. We pre-treated cells with lathosterol containing seven deuterium atoms in place of hydrogens complexed with beta-methyl cyclodextrin (d7-lathosterol-MCD), which is essential for aqueous solubility. DHCR7 activity following TASIN-18 treatment could thus be inferred by measuring the relative levels of its substrate, d7–7-dehydrocholesterol, and product, d7-cholesterol. Consistent with DHCR7 inhibition, we detected a dose-dependent increase in d7–7-dehydrocholesterol, which accumulated up to 90-fold compared to DMSO controls, while d7-cholesterol was reduced by as much as 50% (Figure 2c). Notably, 10 nM of TASIN-18 led to a 67-fold increase in d7–7-dehydrocholesterol, suggesting that TASIN-18 is a potent inhibitor of DHCR7. To infer the activity of EBP, we performed an analogous experiment using d6-lanosterol-MCD, which is expected to generate isotopically labeled d6-zymosterol. TASIN-18 treatment led to a dose dependent accumulation of both d6-zymosterol and d6-zymostenol, but the trend for each dose-response curve was different. An increase in zymosterol was apparent at 3 nM and continued to increase up to the highest dose we measured, 10 μM. By contrast, d6-zymostenol accumulated at dose below 1 μM, but unexpectedly declined at 1 and 3 μM. Zymosterol is converted to zymostenol by DHCR24, and the decrease in zymostenol levels raised the hypothesis that DHCR24 may also be inhibited by TASIN-18. To more directly test this hypothesis, we measured the impact of TASIN-18 on the production of d5-zymostenol in cells cultured with d5-zymosterol-MCD. D5-zymostenol accumulated at doses below 0.1 μM, consistent with EBP inhibition, but declined at higher doses, consistent with DHCR24 inhibition (Figure 2e). All together, we concluded that TASIN-18 inhibits the following three enzymes in the post-squalene cholesterol biosynthetic pathway albeit at different concentrations: DHCR7, EBP, and DHCR24 (in order of most to least potent).
2.3. DHCR7 is an off-target for TASIN.
Given that TASIN-18 inhibits DHCR7 at low nanomolar concentrations and even more potently than EBP, we set out to 1) determine if TASIN physically binds DHCR7 and 2) determine if inhibition of DHCR7 contributes to the cytotoxic activity of TASIN, which is also in the nanomolar range for DLD-1 cells. To address these questions, we utilized a photoaffinity probe with significant crosslinking for DHCR7. This molecule emerged from an independent and unrelated target identification project. We have an ongoing research effort to identify the target of 4C12, a small molecule that emerged from a screen for compounds with toxicity to mouse derived glioblastoma cell lines.9 In pursuit of this target, we found a photoaffinity probe, which is a modified analog of the 4C12 compound containing an aryl azide and alkyne (Figure 3a), crosslinks a 40 kDa protein in a UV dependent manner (Figure 3b, Supplementary Figure 3a). This band, which we refer to as p40, is diminished when the photo-affinity probe is co-incubated with excess 4C12 competitor (Figure 3c, Supplementary Figure 3b). Using the same strategy described for TASIN-2, we purified compound-bound proteins using streptavidin beads from lysates collected from crosslinked cells and clicked with a biotin azide. Purified proteins were identified and quantified using LC/MS from samples that were either treated with the probe alone or the probe plus 4C12. DHCR7 was enriched 21.4 fold in the probe only sample (Supplementary Table 4), raising the hypothesis that p40 is DHCR7. Although human DHCR7 is predicted to encode for a 54 kDa protein, prior reports show that it migrates as a 40 kDa protein on SDS-PAGE.10,11 We also show that C-terminally FLAG-tagged human DHCR7 overexpressed in DLD-1 cells migrates at approximately 43kDa (Supplementary Figure 3c).
Figure 3.
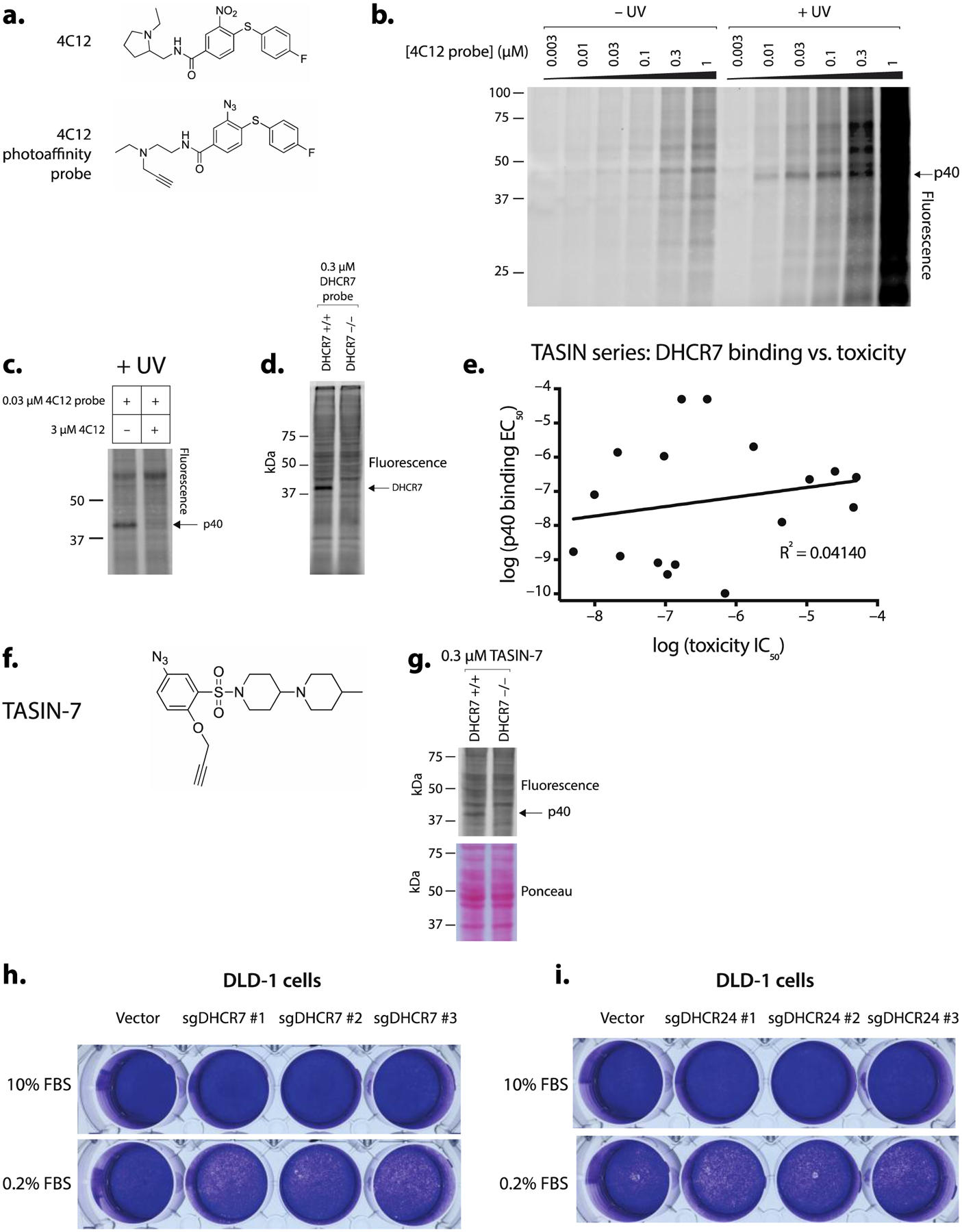
TASIN physically binds DHCR7, but loss of these two enzymes is of no functional consequence to the viability of DLD-1 cells. (a) Chemical structure of 4C12 (above) and a 4C12 photoaffinity probe (below). (b) SDS-PAGE analysis of lysate derived from DLD-1 cells incubated with 4C12 photoaffinity probes reveals a 40 kD protein (p40). (c) 4C12 competes for p40 crosslinking with the 4C12-probe. (d) p40 crosslinking is absent in DHCR7 −/− cells. (e) Competition assays for p40 with the TASIN series reveal no correlation between toxicity and DHCR7 binding. (f) Chemical structure of TASIN-7, a photoaffinity probe. (g) TASIN-7 crosslink p40 in wild type but not DHCR7 −/− cells. (h and i) DLD-1 cells expressing DHCR7 sgRNA (h) or DHCR24 (i) show no growth deficit in either media containing either 10% or 0.2% fetal bovine serum.
We sought genetic evidence implicating DHCR7 as p40 by evaluating p40 in DLD-1 cells that have a CRISPR/Cas9 generated frameshift mutation at Valine 126 DHCR7 (Supplementary Figure 3d) (hereafter referred to as DHCR7 −/−). The photo-affinity probe fails to crosslink p40 in DHCR7 −/− cells (Figure 3d, Supplementary Figure 3e) providing genetic evidence that p40 encodes DHCR7, and therefore hereafter, the probe is referred to as DTP (DHCR7 Targeting Probe).
We used this probe in competition assays with 18 TASIN analogs, similar to the experiment performed for TASIN-2. TASIN analogs exhibited different affinities for DHCR7, which we quantified with a binding EC50 (Supplementary Figure 3f). We found no correlation between the DHCR7 binding EC50 and the IC50 for toxicity (R2=0.0414) (Figure 3e). Based on these observations, we concluded that TASIN binding to DHCR7 has no consequence on cellular viability. An alternative explanation for our competition assays is that TASIN indirectly decreases expression of DHCR7 through a different binding event. To address this possibility, we sought to test whether a TASIN photo-affinity probe directly binds to DHCR7. TASIN-2 did not crosslink a 40 kDa protein, so we synthesized TASIN-7, a chemically distinct probe, which contains both an aryl azide and alkyne, with activity in DLD-1 cells (IC50 = 0.412 μM) (Figure 3f, Supplementary Figure 3g). TASIN-7 weakly crosslinked a 40 kDa protein that was present in the wild type cells but absent in DHCR7 −/− cells (Figure 3g, Supplementary Figure 3e) providing evidence that TASIN directly binds to DHCR7. Our pharmacology experiments suggested that TASIN analogs bind to and inhibit DHCR7, but also suggest that DHCR7 is not essential to DLD-1 cells. To genetically test the latter hypothesis, we stably co-expressed Cas9 and one of three single guide RNAs predicted to target DHCR7 in DLD-1 cells. Reduced DHCR7 expression was inferred from reduced p40 crosslinking in all three clones (Supplementary Figure 3h). When compared to the vector infected controls, no changes in viability were observed either in 10% FBS or 0.2% serum medium (Figure 3h), suggesting that the loss of DHCR7 in DLD-1 cells is of no functional consequence at least in these two media conditions.
In our initial survey of sterol levels using TASIN-18, we found evidence that TASIN-18 inhibited DHCR24 (Figure 2d,e). Thus far, we have not identified a TASIN probe for DHCR24. There are multiple explanations for this including that the effect of TASIN on DHCR24 is indirect or alternatively, TASIN binds to DHCR24 in a position such that photo-activation of our current probes do not covalently react. To genetically test whether TASIN’s activity could be explained by DHCR24 inhibition, we reduced DHCR24 expression by stably expressing Cas9 and sgRNA targeting DHCR24 (Supplementary Figure 3i). DLD-1 cells expressing DHCR24 sgRNA’s showed no growth deficit compared to mock treated cells in either 10% FBS (high sterol) or 0.2% FBS (low sterol) media (Figure 3i). Collectively, our experiments provide genetic and pharmacologic evidence that DHCR7 is a TASIN off-target, and genetic evidence that DHCR24 is off-target.
2.4. Identification of TASIN analogs selective for EBP.
To more precisely investigate TASIN’s mechanism of action, we sought to identify TASIN analogs that selectively target EBP. To identify such analogs, we determined each analog’s binding preference for EBP compared to DHCR7. The probe competition EC50 of each analog for p20 (EBP) or p40 (DHCR7) was quantified as previously described in Supplementary Figure 1h. Using 18 TASIN analogs, EBP selective compounds were ranked by the ratio of the EBP to DHCR7 binding EC50’s (Supplementary Figure 4a, 4b). TASIN-30 and TASIN-32, were found to have strong preferences for either EBP or DHCR7, respectively (Figure 4a). In the crosslinking assay, TASIN-32 competes with probe binding at DHCR7 with an EC50 = 0.1 nM and at EBP with EC50 = 49 nM), whereas TASIN-30 competes with EC50 > 50 μM (DHCR7) and 97 nM (EBP). Of note, compounds with selectivity for EBP all contained a trimethyl aryl group raising the hypothesis that this group reduces the affinity of TASIN for DHCR7 (Supplementary Figure 4a).
Figure 4.
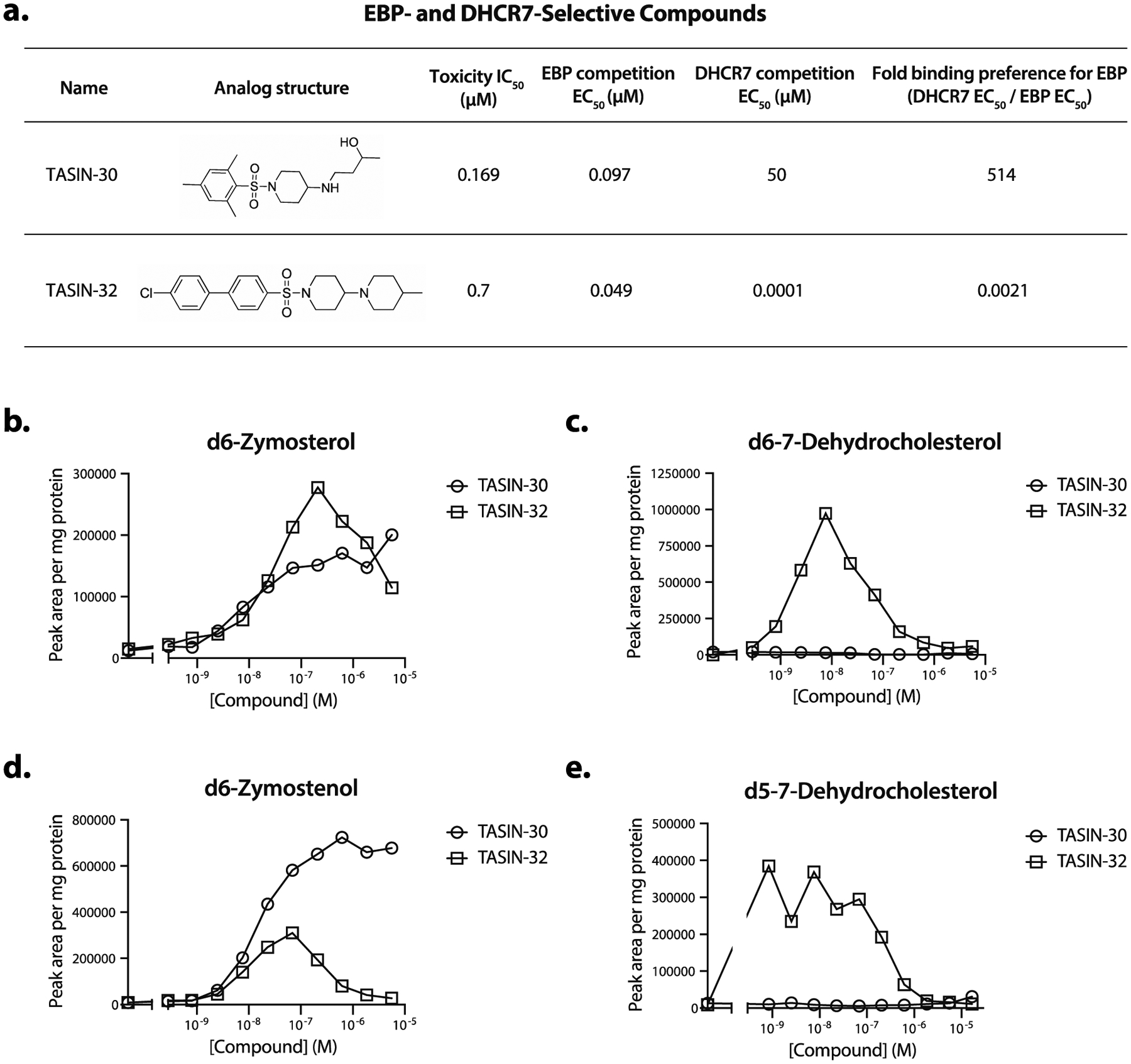
Identification of EBP-selective TASIN analogs. (a) TASIN-30 selectively binds EBP, whereas TASIN-32 is more selective for DHCR7. (b) Sterol tracing using an upstream sterol precursor demonstrates that TASIN-30 and TASIN-32 lead to accumulation of zymosterol, (c) but only TASIN-32 accumulates 7-dehydrocholesterol. (d) Sterol tracing with a downstream sterol precursor shows that TASIN-32 builds up 7-dehydrocholesterol but TASIN-30 does not. (e) TASIN-32 but not TASIN-30 inhibits DHCR24 based on reduced zymostenol levels.
We next used cellular activity assays for EBP or DHCR7 to determine whether competition EC50’s correlated with enzyme inhibition. DLD-1 cells incubated with d7-lanosterol were treated with varying doses of either TASIN-30 or TASIN-32 and sterol levels were then measured by LC/MS. Both TASIN-30 and TASIN-32 led to a dose dependent increase in d6-zymosterol, consistent with EBP inhibition (Figure 4b). TASIN-30 and TASIN-32 had different effects on the accumulation of d6–7-dehydrocholesterol. 7.62 nM of TASIN-32 led to accumulation of d6–7-dehydrocholesterol consistent with DHCR7 inhibition (Figure 4c). Notably, the potency of TASIN-32 is consistent with the DHCR7 binding EC50 we observed in competition assays. At doses above 7.62 nM, d6–7-dehydrocholesterol is gradually depleted (Figure 4c), which we attribute to the depletion of substrate by upstream inhibition of EBP. TASIN-30, on the other hand, showed no accumulation of d6–7-dehydrocholesterol at doses as high as 5.56 μM (Figure 4c), suggesting that it is a more potent inhibitor of EBP than DHCR7.
We also tested both TASIN-30 and TASIN-32 in a cellular assay that reports the activity of DHCR7 (independent of EBP) by using deuterium labeled lathosterol. TASIN-32 caused accumulation of d5–7-dehydrocholesterol at 1 nM (Figure 4d) while TASIN-30 caused no accumulation at a dose as high as 16.7 μM, confirming that it does not target DHCR7.
Our isotopic sterol tracing studies also show that TASIN-30 is selective for EBP over DHCR24. Like TASIN-18, TASIN-32 led to a decrease in d6-zymostenol levels at concentrations of 300 nM and higher (Figure 4e). By contrast, the increase in d6-zymostenol levels following TASIN-30 treatment saturated at 1 μM without any evidence of decrease. Taken together, these results suggest that TASIN-30 is selective for EBP over both DHCR7 and DHCR24.
2.5. Depletion of sterols due to loss of EBP, but not downstream enzymes, cause DLD-1 cell death.
After identifying EBP as the direct target of TASIN, we aimed to elucidate the mechanism by which loss of EBP causes cell death in DLD-1 cells. One possibility is that the accumulation of EBP substrates, either zymostenol or zymosterol, is toxic to cells. Alternatively, depletion of downstream sterol products may cause cell death. The fact that cholesterol rescues TASIN toxicity does not distinguish between these two possibilities because the addition of cholesterol blocks the synthesis of these substrates via negative feedback.12 We hypothesized that, if substrate accumulation is the proximate cause of death, we could prevent or delay cell death by co-incubating with compactin, an HMG-CoA reductase inhibitor, that stops the production of mevalonate, a required precursor for sterol synthesis. Contrary to the hypothesis, DLD-1 cells became increasingly sensitive to TASIN-30 when co-treated with increasing doses of compactin (Figure 5a). At the selected doses, treatment with compactin alone is non-toxic to DLD-1 cells (Supplemental Figure 5a) but significantly depletes zymosterol (Supplemental Figure 5b). Taken together, these experiments provide evidence that cell death from EBP inhibition is the consequence of depletion of downstream sterols, rather than build up of a toxic substrate. Consistent with this hypothesis, supplementation with lathosterol-MCD, but not zymosterol-MCD, was able to rescue DLD-1 cells from TASIN-30 cytotoxicity (Figure 5b). This model of toxicity predicts that DLD-1 cells are more dependent on sterol biosynthesis than the TASIN-insensitive cell line, HCT116. We tested this hypothesis by comparing the sensitivity of DLD-1 and HCT116 cells to compactin. Compactin had a more potent growth inhibitory effect on DLD-1 cells (IC50 = 3.2 μM) compared to HCT116 cells (IC50 ≈ 18.6 μM) (Figure 5c), supporting our hypothesis that the former is more dependent on sterol biosynthesis.
Figure 5.
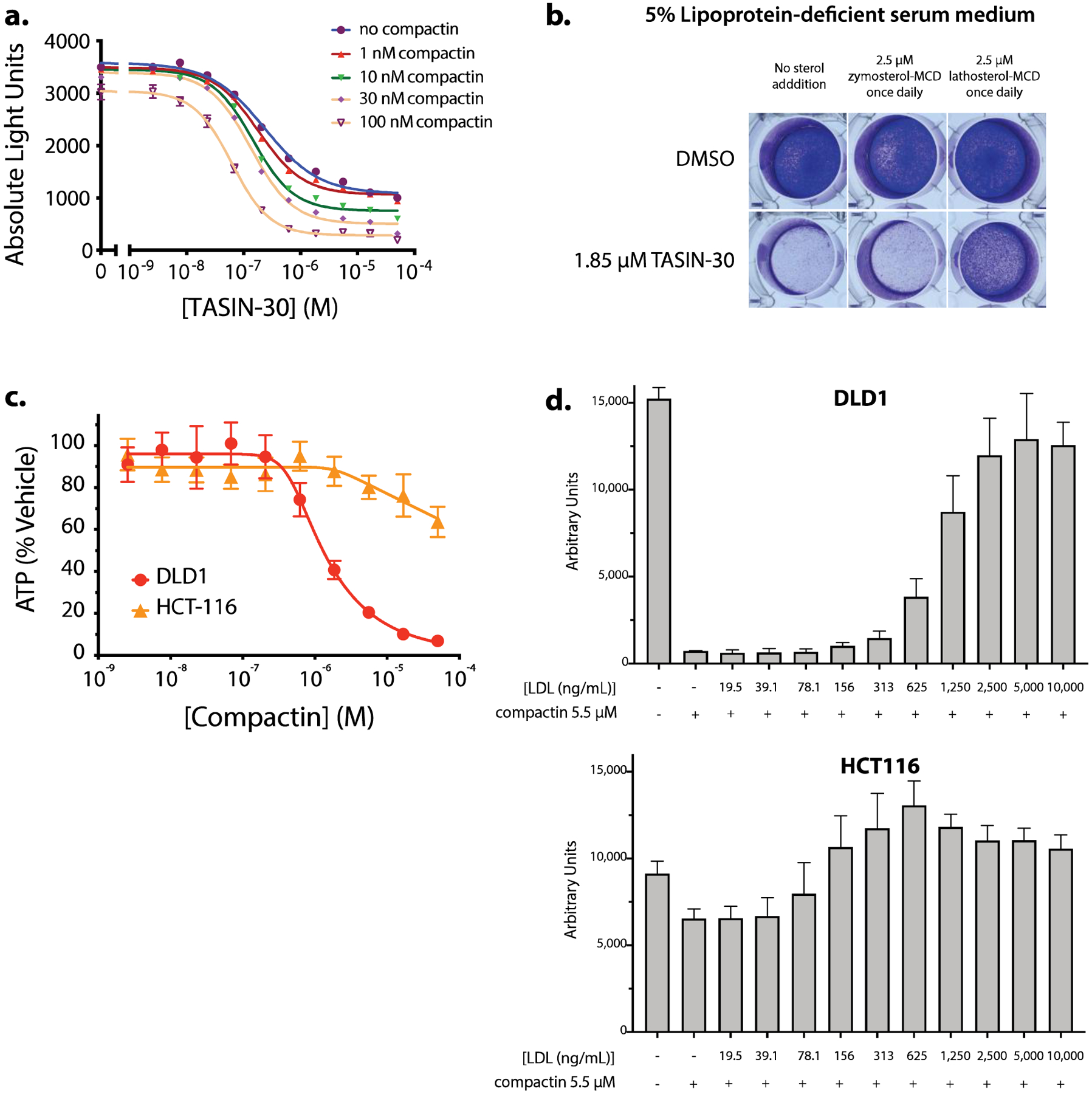
EBP inhibition kills cells by depletion of sterols and represents a transitional point in the sterol pathway of DLD-1 in terms of sterol essentiality. Compactin phenocopies TASIN selectivity suggesting different cholesterol requirements by the two cell lines. (a) Dose response of TASIN-30 with co-titration of compactin, an HMG-CoA Reductase inhibitor. Compactin sensitizes cells to TASIN. (b) Crystal Violet stain of DLD-1 cells treated with TASIN-30 in the presence or absence of zymosterol-MCD or lathosterol-MCD dosed once per day without media change. (c) Compactin is a more potent inhibitor of proliferation in DLD-1 (APC mutant) than HCT116 (APC wild type) cells. (d) Higher concentrations of LDL are required to rescue DLD1 (APC mutant) cells from compactin toxicity.
We postulated that DLD1 dependence on endogenous biosynthesis may be the consequence of reduced ability to scavenge cholesterol from the media. To test this hypothesis, we co-incubated either DLD1 or HCT116 cells with media containing increasing concentrations of LDL. We found that more than 10-fold higher concentrations of LDL was required to rescue DLD1 cells in comparison to HCT116 (Figure 5d). This difference suggests that APC mutant cells are less effective at scavenging cholesterol from LDL, and thus may have an increased dependence on endogenous cholesterol biosynthesis.
3. DISCUSSION
One strategy to develop new cancer therapies is to identify small molecules that are toxic to cells that harbor a cancer-causing mutation. In this regard, TASIN compounds were identified as selectively toxic to colorectal cancer cells that have mutations in the APC gene. Here, we discovered that TASIN inhibits EBP, DHCR7, and DHCR24 all of which are required enzymes for post-squalene cholesterol biosynthesis. Although inhibition of cholesterol biosynthesis is toxic to cells cultured in media that is low in cholesterol,5 not all steps of the pathway are essential in these conditions.13,14 As an extreme example, cholesterol-free mice have been generated by silencing DHCR24, demonstrating that desmosterol can substitute for cholesterol in vivo.15
Inhibition of different enzymes in the cholesterol biosynthetic pathway lead to disparate effects on cells. For example, patients with different loss of function mutations in sterol biosynthetic enzymes harbor different clinical phenotypes.16 Likewise, we found that in DLD-1 cells, loss of EBP but not DHCR7 or DHCR24, led to cell death. As expected, inhibition of EBP led to a rise in its substrates, zymosterol and zymostenol, raising the possibility of whether cells died due to depletion of downstream sterols or accumulation of upstream sterols. We used inhibitors of HMG-CoA reductase (HMGCR), an upstream enzyme in sterol synthesis, to reduce the levels of zymosterol and zymostenol in the setting of EBP inhibition. This did not rescue cells from TASIN toxicity but rather sensitized them to EBP inhibition, providing evidence that DLD-1 cells die as a result of sterol depletion. Therefore, differential sensitivity between loss of EBP and DHCR7 or DHCR24 suggest that 7-dehydrocholesterol or desmosterol might be able to substitute for cholesterol in a dividing cancer cell.
Toxicity as a consequence of sterol depletion in cultured cancer cells raises important questions concerning their selectivity to APC mutant cells. TASIN toxicity assays are not performed in fully defined media but rather in 0.2% FBS, which still has a low level of lipoproteins. Therefore, it is possible that APC wild type cells are able to more efficiently scavenge cholesterol through LDL receptor mediated uptake. Alternatively, APC mutant cells might have increased cholesterol “demand” in comparison to APC wild type cells, which leads to death when biosynthesis is blocked.
The non-cell autonomous mechanism of death also has implications for how we interpret cancer studies with TASIN in mice. TASIN led to the regression of xenograft tumors derived from human cancer cell lines with APC mutations but not those with wild type.5 In addition, genetically engineered models of APC mutant colorectal cancer had a lower tumor burden following TASIN treatment.5 Efficacy in the former model reflects the requirement of cholesterol biosynthesis in tumor maintenance. Although the latter model does not distinguish between tumor initiation and maintenance, it does suggest that cholesterol biosynthesis is important for tumor formation in the colon of immune-competent animals. Our findings suggest that increased dependence on cholesterol biosynthesis may be the result of a reduced efficiency to scavenge cholesterol from circulating LDL. These findings predict that TASINs may have synergistic activity with LDL lowering agents, which include HMG coA reductase inhibitors or antibodies that neutralize PCSK9.
Interestingly, EBP and DHCR7, two proteins that TASIN binds to and inhibits, have been purified from cells as members of a complex called the anti-estrogen binding site (AEBS). This complex was named for its ability to bind various non-steroidal anti-estrogens in vitro, such as tamoxifen.10,17 Like TASIN, tamoxifen and other AEBS binders lead to the inhibition of both EBP and DHCR7. In this report, we use specific photo-affinity probes for either EBP or DHCR7 to demonstrate that TASIN can bind to both proteins. Moreover, amongst a diverse set of analogues, we identified specific TASIN derivatives that are selective for one or the other protein. These probes can now be used to assess if compounds that bind the AEBS can independently bind EBP and DHCR7. In addition, we have generated cellular systems in which we have genetically silenced either EBP or DHCR7. These mutant cell lines can illuminate what if any role complex formation plays for AEBS compounds.
We identified several potential off-targets for TASIN that will be relevant to the development of a specific EBP targeting therapy for APC mutant colorectal cancer. Using a combination of isotopic sterol tracing and photo-affinity probes, we identified both DHCR7 and DHCR24 as off-targets. The probes allowed us to sort different TASIN derivatives by their ability to bind either DHCR7 or EBP, and revealed TASIN-30 as an analog that selectively binds EBP over DHCR7. Importantly, TASIN-30 also was selective for EBP over DHCR24. Sorting compounds by their affinity to EBP or DHCR7 revealed structural differences that may provide a rationale for enzyme-specific compound design. Of the 18 analogues tested, six were found to favor EBP binding, and all six had the same tri-methylated aromatic group. On the other hand, none of the DHCR7-selective compounds possessed this feature raising the hypothesis that this group prevents binding to DHCR7. Determining the structure of EBP and DHCR7 bound to TASIN could shed light on why this might be the case. Finally, TASIN probes bound to three other proteins denoted by their estimated molecular weight, p70, p27, and p18, which also qualify as off-targets. In particular, p18 and p27 binding to TASIN derivatives bound to p18 and p27 with a similar structure-activity relationship as binding to EBP suggesting a similar binding site on all three proteins. The identity of p18 and p27 are unknown. Recently, sequence alignment studies revealed a conserved domain between EBP, EBP-L, TM6SF2 and TMEM97 making the latter three proteins potential candidates for p18 or p27.18
Photo-crosslinking and competition assays like the ones described here provide a platform on which to develop specific inhibitors of enzymes in the cholesterol pathway. Such specific inhibitors of enzymes might help uncover the importance of these enzymes in diseases and may themselves have therapeutic value. For example, TASIN analogs have been shown to be efficacious in mouse models of multiple sclerosis through the accumulation of specific sterol intermediates,19 and it will be important to learn whether these effects are due to EBP or a yet unidentified target.
Supplementary Material
ACKNOWLEDGMENTS
J.K.D.B. acknowledges support of the Welch Foundation (Grant I-1422) and holds the Julie and Louis Beecherl, Jr., Chair in Medical Science. D. Nijhawan is supported by Welch Foundation I-1879, NIH R37CA226771, and NIH RO1CA217333. HRMS data were obtained from the Shimadzu Center for Advanced Analytical Chemistry (SCAAC) at UT Arlington. Andrew Lemoff and Hamid Mirzaei and colleagues at the U.T. Southwestern Medical Center proteomics core facility performed mass spectrometry analysis on TASIN bound proteins. R. Tomaino and colleagues at the Harvard Medical School Taplin Mass Spectrometry Facility performed mass spectrometry on 4C12-bound proteins. We would like to thank David G. McFadden and Michael S. Brown for critically evaluating the manuscript and helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Supplementary Tables 1–4 (xlsx)
Supplementary figures, experimental procedures and methods, and copies of 1H and 13C spectra (pdf)
J.K.D.B. and D.N. are co-founders of and have equity in Barricade Therapeutics, a company that aims to develop TASIN into therapeutic agents for the treatment of cancer and other diseases.
REFERENCES
- 1.Groden J; Thliveris A; Samowitz W; Carlson M; Gelbert L; Albertsen H; Joslyn G; Stevens J; Spirio L; Robertson M; Sargeant L; Krapcho K; Wolff E; Burt R; Hughes JP; Warrington J; McPherson J; Wasmuth J; Le Paslier D; Abderrahim H; Cohen D; Leppert M; White R, Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66 (3), 589–600. [DOI] [PubMed] [Google Scholar]
- 2.Munemitsu S; Albert I; Souza B; Rubinfeld B; Polakis P, Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci U S A 1995, 92 (7), 3046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powell SM; Petersen GM; Krush AJ; Booker S; Jen J; Giardiello FM; Hamilton SR; Vogelstein B; Kinzler KW, Molecular diagnosis of familial adenomatous polyposis. N Engl J Med 1993, 329 (27), 1982–7. [DOI] [PubMed] [Google Scholar]
- 4.Miyoshi Y; Nagase H; Ando H; Horii A; Ichii S; Nakatsuru S; Aoki T; Miki Y; Mori T; Nakamura Y, Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992, 1 (4), 229–33. [DOI] [PubMed] [Google Scholar]
- 5.Zhang L; Theodoropoulos PC; Eskiocak U; Wang W; Moon YA; Posner B; Williams NS; Wright WE; Kim SB; Nijhawan D; De Brabander JK; Shay JW, Selective targeting of mutant adenomatous polyposis coli (APC) in colorectal cancer. Sci Transl Med 2016, 8 (361), 361ra140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloch K, The biological synthesis of cholesterol. Science 1965, 150 (3692), 19–28. [DOI] [PubMed] [Google Scholar]
- 7.Kandutsch A; Russell A, Preputial gland tumor sterols III. A metabolic pathway from lanosterol to cholesterol. Journal of Biological Chemistry 1960, 235 (8), 2256–2261. [PubMed] [Google Scholar]
- 8.Wang W; Zhang L; Morlock L; Williams NS; Shay JW; De Brabander JK, Design and Synthesis of TASIN Analogues Specifically Targeting Colorectal Cancer Cell Lines with Mutant Adenomatous Polyposis Coli (APC). J Med Chem 2019, 62 (10), 5217–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y; Lim SK; Liang Q; Iyer SV; Wang HY; Wang Z; Xie X; Sun D; Chen YJ; Tabar V; Gutin P; Williams N; De Brabander JK; Parada LF, Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567 (7748), 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kedjouar B; de Medina P; Oulad-Abdelghani M; Payre B; Silvente-Poirot S; Favre G; Faye JC; Poirot M, Molecular characterization of the microsomal tamoxifen binding site. J Biol Chem 2004, 279 (32), 34048–61. [DOI] [PubMed] [Google Scholar]
- 11.Moebius FF; Fitzky BU; Lee JN; Paik YK; Glossmann H, Molecular cloning and expression of the human delta7-sterol reductase. Proc Natl Acad Sci U S A 1998, 95 (4), 1899–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown MS; Goldstein JL, Receptor-mediated endocytosis: insights from the lipoprotein receptor system. Proc Natl Acad Sci U S A 1979, 76 (7), 3330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez C; Martin M; Gomez-Coronado D; Lasuncion MA, Effects of distal cholesterol biosynthesis inhibitors on cell proliferation and cell cycle progression. Journal of lipid research 2005, 46 (5), 920–9. [DOI] [PubMed] [Google Scholar]
- 14.Xu F; Rychnovsky SD; Belani JD; Hobbs HH; Cohen JC; Rawson RB, Dual roles for cholesterol in mammalian cells. Proc Natl Acad Sci U S A 2005, 102 (41), 14551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wechsler A; Brafman A; Shafir M; Heverin M; Gottlieb H; Damari G; Gozlan-Kelner S; Spivak I; Moshkin O; Fridman E; Becker Y; Skaliter R; Einat P; Faerman A; Bjorkhem I; Feinstein E, Generation of viable cholesterol-free mice. Science 2003, 302 (5653), 2087. [DOI] [PubMed] [Google Scholar]
- 16.Herman GE; Kratz L, Disorders of sterol synthesis: beyond Smith-Lemli-Opitz syndrome. American journal of medical genetics. Part C, Seminars in medical genetics 2012, 160c (4), 301–21. [DOI] [PubMed] [Google Scholar]
- 17.Poirot M; Chailleux C; Fargin A; Bayard F; Faye JC, A potent and selective photoaffinity probe for the anti-estrogen binding site of rat liver. J Biol Chem 1990, 265 (28), 17039–43. [PubMed] [Google Scholar]
- 18.Sanchez-Pulido L; Ponting CP, TM6SF2 and MAC30, new enzyme homologs in sterol metabolism and common metabolic disease. Frontiers in genetics 2014, 5, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubler Z; Allimuthu D; Bederman I; Elitt MS; Madhavan M; Allan KC; Shick HE; Garrison E; M TK; Factor DC; Nevin ZS; Sax JL; Thompson MA; Fedorov Y; Jin J; Wilson WK; Giera M; Bracher F; Miller RH; Tesar PJ; Adams DJ, Accumulation of 8,9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 2018, 560 (7718), 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.