

Abstract
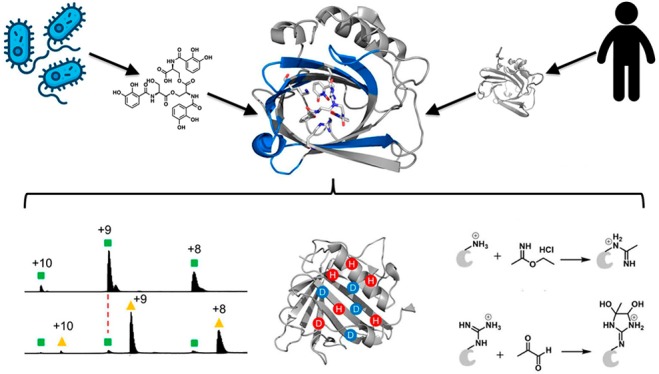
Both host and pathogen competitively manipulate coordination environments during bacterial infections. Human cells release the innate immune protein siderocalin (Scn, also known as lipocalin-2/Lcn2, neutrophil gelatinase-associated lipocalin/NGAL) that can inhibit bacterial growth by sequestering iron in a ferric complex with enterobactin (Ent), the ubiquitous Escherichia coli siderophore. Pathogenic E. coli use the virulence-associated esterase IroE to linearize the Ent cyclic trilactone to linear enterobactin (lin-Ent). We characterized lin-Ent interactions with Scn by using native mass spectrometry (MS) with hydrogen–deuterium exchange (HDX) and Lys/Arg specific covalent footprinting. These approaches support 1:1 binding of both Fe(III)-lin-Ent to Scn and iron-free lin-Ent to Scn. Both ferric and nonferric lin-Ent localize to all three pockets of the Scn calyx, consistent with Scn capture of lin-Ent both before and after Fe(III) chelation. These findings raise the possibility that Scn neutralizes both siderophores and siderophore-bound iron during infections. This integrated, MS-based approach circumvents the limitations that frustrate traditional structural approaches to examining Scn interactions with enterobactin-based ligands.
During infections, pathogenic bacteria must often overcome substantial thermodynamic barriers to obtain iron from host reservoirs. This competition between host and pathogen represents a robust tug-of-war over transition metal iron ions, which are growth-limiting nutrients for most bacteria. Many bacterial pathogens activate the biosynthesis of siderophores,1−3 low molecular-weight iron chelators that scavenge iron for bacterial use. In response to these bacteria, host cells secrete siderocalin (also known as lipocalin-2/Lcn2, neutrophil gelatinase-associated lipocalin/NGAL, or 24p3) to hinder iron acquisition by the pathogen.4−6 Unlike the canonical iron-binding proteins that chelate iron ions directly, Scn specifically binds ferric ions chelated with various catecholates that are either pathogen-derived siderophores1 or host-derived metabolites.4−7 The E. coli siderophore enterobactin (Ent) (Figure 1a) is the prototypical Scn ligand and was recently detected alongside Scn in the urine of patients with E. coli urinary tract infections (UTIs).7
Figure 1.

Chemical structure of enterobactin (Ent) (a) and linear enterobactin (lin-Ent) (b).
High quality X-ray crystal structure analyses of Scn reveal an eight-stranded antiparallel β-barrel core, defined as the calyx, that binds with ferric-catecholate complexes through positively charged side chains of three residues (R81, K125, and K134).2,8−11 These three residues form a distinctive hybrid electrostatic/cation-π interaction network with the delocalized π electrons of nearby ligand catechol functional groups. Additional interactions including van der Waals and hydrogen bonding are contributed by residues lining the calyx.1
Recent studies have used several biophysical tools to define structural interactions between Scn and potential ligands.1,5,8,11,12 Whereas optical1 and fluorescence1,5 spectroscopies can provide useful binding data, they provide only general structure information and report principally on transition metal complex binding that provides distinctive optical and fluorescence quenching signals. Although X-ray crystallography has provided detailed Scn structural data, the chemical instability of enterobactin during cocrystallization has been an irksome barrier to describing these complexes.4,8 The value of NMR is complicated for ferric ligands, where the presence of high-spin Fe(III) in the calyx would prevent useful spectroscopic analysis of Scn interactions.12 To circumvent these problems, we turned to fast, sensitive, mass spectrometry (MS)-based structural proteomics approaches that complement the available, high quality X-ray crystallographic structures. These MS techniques have been widely applied to protein–protein interactions,13−16 protein folding/unfolding,17,18 protein structure characterization,19,20 protein conformational dynamics,21−23 binding affinity,24 and binding stoichiometry determination.25
Here, we describe the application of MS-based structural proteomics to interrogate the interactions between Ent and Scn in intact, bound ferric and aferric complexes. We focused on Scn interactions with linear Ent (2,3-DHB-LSer)3 (lin-Ent) (Figure 1b), a major secreted form of enterobactin produced by extraintestinal pathogenic E. coli in culture26 and during human infections.7 Enterobacteria carrying the virulence-associated iroA locus encode IroE, an esterase that catalyzes monohydrolysis of the Ent trilactone to yield lin-Ent.7,27,28 Although fluorescence data support the binding of both ferric-Ent and ferric-lin-Ent to Scn,7,8 there is no structure for these complexes.
To characterize lin-Ent interactions with Scn, we combined native MS, hydrogen–deuterium exchange mass spectrometry (HDX-MS), and specific covalent footprinting. Native MS has few restrictions on mass range29,30 and establishes ligand binding and its stoichiometry. We observed the expected 1:1 binding between lin-Ent and Scn. HDX-MS31−33 provides localized (peptide-level) structural information on Scn upon ligand binding. We determined the regions of Scn that gain protection upon ligand binding. Specific covalent footprinting adds residue specificity to the HDX data and identifies the binding site.34−36 We used Lys and Arg-specific footprinting to follow conformational changes of the protein on the residue-level. The results indicate that ferric and aferric lin-Ent share a binding interface, with ferric lin-Ent-bound Scn showing higher stability. These observations support the intriguing possibility that Scn may capture Ent prior to iron-binding during infections.
The progressive design of our studies from “sketch” to “detail” provides holistic structural information on solution-state Scn and Scn complexes with both ferric and aferric ligands. The binding site we detected for ferric and aferric lin-Ent to Scn is consistent with crystallographic data, adding confidence to our methods. Moreover, we detected ligand-induced Scn conformational changes not seen in the X-ray crystal complex structure. This integrated platform also provides useful structural information from stoichiometry to conformational change and residue specific binding, and the approach can be applied to compare directly both ferric and aferric complexes. It will facilitate studies of Scn in complex with both bacterial and human derived ligands as we work toward therapeutic intervention.
Results and Discussion
Native MS
We used nondenaturing, native MS to (1) verify the molecular weight of Scn, (2) test whether both ferric and aferric lin-Ent bind to Scn, and (3) determine the binding stoichiometry of the complex.
Scn appeared at m/z 2068.3, 2298.0, and 2585.2 as [Scn +10H+]10+, [Scn +9H+]9+, and [Scn +8H+]8+ charge states, giving a MW of 20674.7 ± 3.2 Da (Figure 2). The peaks to the right of the main peaks indicate an impurity of Scn, likely Scn bearing a 115 Da modification. Following equilibration with Fe(III)-lin-Ent, we found a new peak shifted by 737.8 Da, corresponding to ferric lin-Ent bound to Scn in a 1:1 ratio. Although the m/z shifts to higher values, the charge state distribution does not change. Given that native protein charge states are mainly dependent on the folded protein’s exposed surface area,37,38 the unchanged Scn charge state of the lin-Ent-bound complex is attributable to a stable, high-order protein structure that changes little with binding. Its surface positive charge is unchanged where the −3 charge of the ferric lin-Ent ligand is neutralized by the three protonated amino acid side chains shielded within the calyx.
Figure 2.
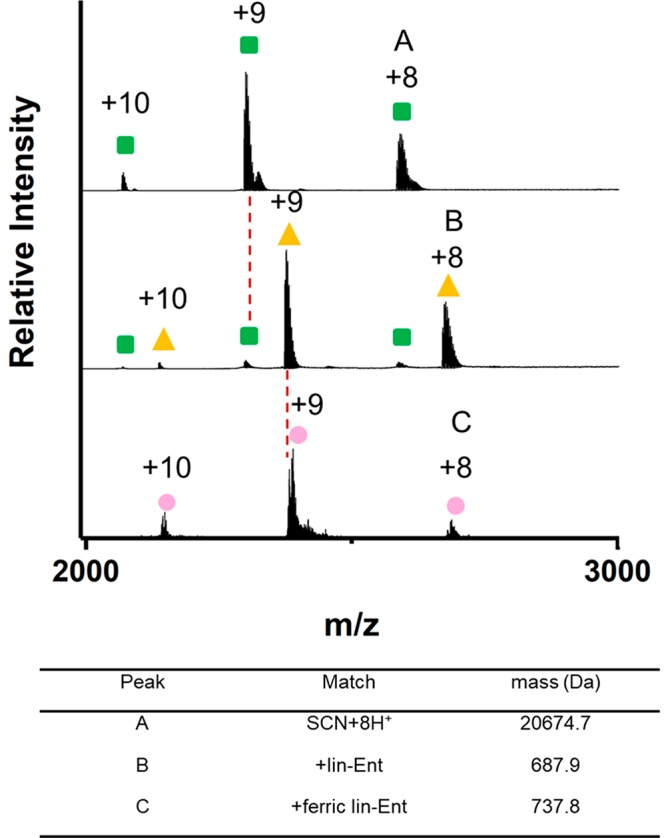
Native mass spectra of Scn (green square), the lin-Ent-bound Scn complex (yellow triangle), and the ferric lin-Ent-bound Scn complex (pink circle). (The concentrations of Scn, lin-Ent, and FeCl3 are 40 μM, 60 μM, and 72 μM, respectively.)
With lin-Ent binding, we saw an additional species shifted by 687.9 Da, allowing the conclusion that Scn can bind lin-Ent both in its iron-laden and iron-free state. Binding of aferric Ent to Scn was previously reported by cocrystallization during which the Ent was hydrolyzed.5 Fluorescence studies of Scn ligand binding rely on tryptophan quenching, which is likely due to a heavy-metal effect and therefore insensitive to aferric complexes. To our knowledge, this is the first convincing solution phase data of the aferric lin-Ent-Scn complex with intact lin-Ent. This result is further motivation to pursue MS-based studies of Scn complexes.
Peptide-Level HDX Analysis of Unbound-Scn
Because native spray MS provides limited structural detail on the protein–ligand interaction, we turned to HDX to obtain site-specific (peptide-level) information about likely binding sites. As a reversible footprinting approach with minimal perturbation of the protein, HDX should detect conformational changes of Scn at the peptide level. The normal quench for HDX is 4 M urea or guanidine at pH 2.5 to minimize HDX back exchange. Lack of significant change in the secondary structure of Scn from pH 7.4 to pH 2.01 indicates its high stability and forecasts obstacles in its digestion. We tried the traditional guanidine reagents, but this approach only achieved ∼87% coverage with 37 peptides in the digest (Figure S1). Traditional urea reagent similarly gave poor coverage of the protein (data not shown). In addition, there was a large amount of undigested protein as shown by the chromatogram (Figure S2). We assume that the 8-stranded antiparallel β-barrel fold makes Scn very resistant to denaturation under normal quench buffer conditions (pH = 2.5). Therefore, we sought an alternative way to denature Scn efficiently while minimizing HDX back exchange. Using trifluoroethanol as solvent to facilitate the denaturation of Scn, we obtained a coverage map of ∼93% of the full-length protein sequence with 92 digested peptides (Figure S3) and a negligible amount of undigested protein (Figure S4).
To obtain coarse-grained structural information, we measured the HDX for unbound Scn and compared it with that of aferric lin-Ent-bound and ferric lin-Ent-bound Scn at 37 °C within the same time range (10 s to 4 h) (Figures S5 and S6). (Note that there are two additional residues at the beginning of the expressed Scn sequence, so there is a two-residue shift of the residue numbers compared with those described previously (e.g., R81 shows up as R83, K125 shows up as K127, and K134 shows up as K136).)
Most of the N-terminal region (residues 1–33) is flexible or solvent-exposed, as shown by a more than 50% deuterium uptake within 10 s and an increasing extent of exchange to ∼70% at longer time. A four-turn α-helix (residues 147–162 within peptides 140–150 and 150–157) also shows solvent exposure or flexibility according to HDX. Peptide 158–176 in the C-terminal region maintains some protection, exhibiting a slow rate of HDX, which is attributed to the more ordered β-strand at the C terminus. With these exceptions, Scn is quite rigid and structured, as only a few peptides show 90% exchange within 15 min at 37 °C, in agreement with the X-ray crystal structure.8
Differential HDX Analyses of the lin-Ent-bound Scn and ferric lin-Ent-bound Scn Complexes
The highly sculpted Scn calyx lined with three positively charged residues (R81, K125, and K134) is thought to form hybrid electrostatic/cation-π interactions with catecholate ligands.1,4 To probe the siderophore binding induced conformational changes of Scn, we performed two sets of comprehensive differential HDX experiments of full length Scn: (1) in the presence and absence of aferric lin-Ent (Figures S5 and 3) and (2) in the presence and absence of ferric lin-Ent (Figures S6 and S7).
Figure 3.

Structure: Average percentage differences in deuterium uptake between apo-Scn and aferric lin-Ent-bound Scn as per color coding on an X-ray crystal structure of TrenCam-bound Scn (PDB 3HWG). Insets: HDX kinetic data for selected peptides in the bound and unbound state (Apo-Scn (black) and aferric lin-Ent-bound Scn (red)).
The first differential HDX analysis between apo-Scn and Scn bound with aferric lin-Ent should locate the siderophore binding site. To verify our conclusions that the differences in HDX kinetics between bound and unbound are significant, we compared the sum of all the differences in the kinetic plots to the propagation error (the square root of the sum of the squares of all standard deviations). If the accumulated differences are more than 3 times the propagation error, the probability that HDX occurring at that region is significantly different between bound and unbound is 99.7% (Figure S6a). Four regions of Scn exhibit significant deuterium uptake differences, including regions represented by peptides 34–42, 60–72, 94–109, and 124–139.
As a reference, we used a crystal structure of Scn bound with TrenCam, an Ent analog with a nonhydrolyzable, tertiary amine backbone (PDB 3HWG).5
Previous NMR and X-ray crystallography studies found R81, K125, and K134 (shown as R83, K127, and K136) to be important in binding.8,10 The decreased HDX seen in the proteolytic peptide 124–139 (containing K127 and K136) upon lin-Ent binding is consistent with the expected cation-π interactions (Figures 3 and S5). The peptide containing R83 does not show any noticeable deuterium uptake difference, possibly because the peptide covering R83 is long, and the deuterium uptake difference may be diluted by many invariant residues in the 14-residue peptide (Figure S5). In addition, binding sites are not always mapped by HDX.39 It is also possible that structural rigidity permits the ligand to interact with the R83 side chain without conformationally affecting the adjoining peptide backbone.
Peptides 34–42 and 60–72 do not contain residues known to bind the ligand directly but do exhibit protection upon lin-Ent binding. This suggests that conformational changes occur in these regions (Figures 3 and S5). According to the 3D structure, these two regions are part of the eight-stranded β barrel. Binding to lin-Ent may “squeeze” the core of this β barrel, inducing a tight, compact structure, thereby slowing HDX of some β-strands of the core structure. These peptides undergo slow HDX such that the difference in deuterium uptake does not become significant until 120 s. This divergence is consistent with protection in these two regions induced by ligand binding. Slow HDX and the delay before divergence emerges are consistent with rigidity in this region of the protein.
The region represented by peptide 94–109 contains part of two β-strands of the eight-stranded β barrel and a five-residue α-helix. This region shows ∼50% deuterium uptake within 10 s and ∼80% in 4 h (Figures 3 and S5). Its kinetic curves are divergent at the start of HDX, merge at later time points, and show high rates of exchange throughout. That this α-helix is more solvent exposed and likely more flexible than the core of the protein contributes to its fast HDX throughout. The β-strands are certainly more rigid, contributing to slower HDX for those regions.
We made a second HDX differential comparison of apo-Scn and ferric lin-Ent-bound Scn to understand the role of iron in the binding process (Figures S6b, S7, and S8). Four regions represented by peptic peptides 34–42, 60–72, 94–109, and 124–139 become protected by binding, nearly identical with the binding of aferric lin-Ent, and another region (81–94) now shows noticeable protection with ferric lin-Ent binding. Region 124–139, containing the two binding residues K127 and K136, shows more significant protection upon ferric lin-Ent binding compared to that of aferric (Figures 3 and S7). Compared with a small difference upon lin-Ent binding, the peptide 81–94 (containing binding residue R83) exhibits a greater difference in HDX upon ferric lin-Ent binding (Figures S5, S6b, S7). These findings suggest that the ferric ligand induces a more compact protein structure than the aferric ligand, potentially reflecting higher affinity binding.
Lys and Arg Footprinting
To pinpoint the residues that are involved in binding, we implemented specific and irreversible covalent footprinting. This approach should give higher spatial resolution than does HDX.
Because the binding residues are likely to be Lys and Arg, we chose two water-soluble reagents, ethyl acetimidate hydrochloride (ETAT) and methyl glyoxal (MG) (eqs 1 and 2), to modify Lys and Arg, respectively. Although these types of reagents have been used for protein modifications since the 1960s, their application in MS-based protein footprinting was more recent.13,40−102 These labeling reactions maintain the positive charge in the protein, and the irreversible binding permits a canonical bottom-up proteomics analysis. ETAT reacts with the amine group of Lys by an SN2 reaction giving a unique mass shift of +41.0265 Da (eq 1), whereas MG, a vicinal dicarbonyl, reacts with Arg to form a cyclic adduct that shifts the mass +72.0211 Da (eq 2). It is common for the MG-labeled product to lose one molecule of water.
 |
1 |
We evaluated the reactivity of ETAT and MG at different time points (Figure S9). The mass spectral peaks representing one ETAT and two ETAT modifications increase as the labeling time increases (e.g., ETAT modification extent of unbound Scn increases from 31% to 67% as time increases from 1 to 10 min) (Figure S9a and S9b). The difference between unbound and bound is readily apparent, becoming more so after 5 min (51% for Scn vs 30% for siderophore-bound Scn) (Figure S9c). The modification extent for MG footprinting also systematically increases with time (Figure S9d, 9e, 9f). We selected 5 min as the optimum labeling time to record differences between the three states.
We next asked whether ETAT and MG footprinting can identify lin-Ent and ferric lin-Ent binding sites of Scn with good spatial resolution. To answer this question, we digested the protein and analyzed the lysate by LC/MS/MS by using a typical proteomics protocol. The sequence coverage of Scn is ∼98%, ensuring a nearly complete picture of the protein. We integrated the extracted ion chromatogram (EIC) signal intensities for each labeled peptide and its unlabeled counterpart to give a modification ratio (Figures S10, S11, and S12). The labeled peptides are more hydrophobic, eluting later than their unmodified counterparts. MS/MS data confirm that K136 and K127 can become ETAT modified, and R83 can become MG modified. The most abundant charge states are the same before and after the labeling, consistent with ETAT and MG labeling maintaining a positive charge on Lys and Arg, respectively.
We can assign with high confidence that R83, K127, and K136 interact with the ligand, causing a decrease in their solvent accessible surface area upon binding (p value less than 0.01) (Figure 4). Indeed, these three positively charged residues were reported to intercalate with the catechol rings of the ligand and form a siderophore:Scn complex.1 The consistency between our results and those previously published confirms our understanding of Ent-Scn binding while validating the methods presented here as useful and accurate in Scn studies.
Figure 4.

Comparison of the modification extents of peptides produced by digestion after (a) 5 min of ETAT footprinting and after (b) 5 min of MG footprinting (Scn light gray, lin-Ent-bound Scn dark gray, and ferric lin-Ent-bound Scn blue or purple).
There are some residues that show less certain conformational change, including R74, R132, and R142. The p values for these are more than 0.01, decreasing confidence in their differences, but the modification extent difference is >50% (Figure 4b and Table S2). To check whether the decreased modification correlates with solvent accessible surface area (SASA), we calculated the SASA between Scn and ferric lin-Ent bound Scn (Figure S13). Upon ligand binding, there is an ∼60% MG modification decrease on R74, consistent with the decreased SASA (50.7 Å2 for Scn vs 36.1 Å2 for lin-Ent bound Scn). Although the HDX experiment did not provide information on peptide 73–80, the nearby region represented by peptide 58–72 exhibits pronounced protection following ligand binding. Thus, R74 may reorient as part of the conformational change in this region. R132 falls in the peptide region 124–139 where there is a significant deuterium uptake difference, suggesting this residue is associated indirectly with ligand binding. There is no significant difference in the SASA of R132 in bound and unbound states (Figure S13) so decreased footprinting upon ligand binding is likely explained by changes in chemical reactivity, steric hindrance of nearby amino acids, and the protein microenvironment. R142 undergoes a substantial decrease in SASA upon ligand binding (54 Å2 for Scn vs 8.0 Å2 for ferric lin-Ent-bound Scn). This is likely the cause for the difference in MG modification.
Residues K64, K100, and K126 show no difference in ETAT footprinting, indicating that they are not critical binding residues. Their corresponding peptides, 60–72 and 94–109, show measurable HDX differences. This discrepancy between ETAT footprinting and HDX results is consistent with remote conformational changes in this region, rather than direct ligand binding. This may occur when stabilization of Scn upon ligand binding limits peptide backbone flexibility and accessibility (monitored by HDX) more than the exposure of lysine residues to solvent (monitored by ETAT footprinting). This effect was also seen in other protein systems.41,42
Conclusions
Native MS, HDX, and chemical footprinting support a common Scn binding interface for lin-Ent and ferric-lin-Ent. The ferric lin-Ent interface is consistent with the model developed by the laboratory of Roland Strong.5 It is notable that the three pockets of the Arg and Lys-rich Scn calyx bind both ferric and aferric lin-Ent, because the latter molecule lacks the rigidity and anionic character of ferric Ent complexes. Ent fully occupies the first coordination sphere of Fe(III) with its six deprotonated catechol rings, making the Ent structure more rigid and sterically matched to the Scn calyx.2 Similar binding of ferric and aferric lin-Ent to the same Scn site suggests that Arg and Lys form similar cation-π interaction networks with ferric and aferric catechols. Minor differences in chemical footprinting may relate to differences in structure or stability between ferric and aferric lin-Ent.
These new data raise the possibility that Scn may counteract the bacterial enterobactin system not only by sequestering Ent-bound iron but also by sequestering aferric Ent. In this model, Fe(III) access to Ent is limited by sequestration of its ligands in the calyx, which is further shielded by the outward-facing Ent serine polyester backbone. Similar interactions are likely for cyclic Ent, which locks in the radial symmetry of catechols. In this context, Ent linearization confers no clear benefit to the pathogen. It is possible that linearization facilitates pathogenesis in other contexts28 or that its virulence-association reflects C-glycosylation of Ent catalyzed by the associated IroB. A better understanding of chemical microenvironments during infection may yield more definitive answers.
The analytical platform described here will be useful in studies of other ligands and mutant proteins, particularly in conjunction with crystallographic data. The ability to directly compare the ligand binding site and detect ligand-induced conformational changes of Scn for chemically labile ferric and aferric siderophores that defy stable crystallization is particularly useful. This ability arises because our approach does not rely on transition metal-dependent fluorescence quenching and avoids paramagnetic ferric ion interferences (e.g., as in NMR).
Finally, Lys and Arg specific footprinting, as a complement to HDX, pinpoints residue-level information about the protein structure and binding site. This can be used to ask detailed questions about the binding of other ligands to Scn.26 The overall approach can also be extended to other proteins with stable and rigid structures or proteins rich with Lys and Arg that are involved in interactions via salt bridges. The intrinsic protease resistance of Scn led us to introduce improvements in the digestion protocol for HDX by using trifluoroethanol to facilitate the denaturation of stable and rigid Scn. We boosted the coverage from ∼87% to ∼93% of the full-length protein sequence and increased peptic peptides from 37 to 92. Nonaqueous solvents may have broader advantages for other extracellular innate immune proteins and will be explored in future work.
Acknowledgments
We acknowledge National Institutes of Health grants RO1DK099534 and RO1DK111930 to J.P.H. and P41GM103422 and S10OD016298 to M.L.G. We also thank Protein Metrics and HDX Workbench for providing data processing software.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.9b00741.
Experimental methods, sequence coverage of HDX mapping (Figures S1, S3), chromatograms (Figures S2, S4, S10–S12), HDX kinetic plots (Figures S5, S7), cumulative changes in HDX rates (Figure S6), mapping of average percentage differences in deuterium uptake between two protein states (Figure S8), mass spectra of protein (Figure S9), solvent accessible surface area of Arg residues (Figure S13), and tables of modification ratios by ETAT and MG reagents (Tables S1, S2) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Abergel R. J.; Clifton M. C.; Pizarro J. C.; Warner J. A.; Shuh D. K.; Strong R. K.; Raymond K. N. (2008) The siderocalin/enterobactin interaction: A link between mammalian immunity and bacterial iron transport. J. Am. Chem. Soc. 130, 11524–11534. 10.1021/ja803524w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond K. N.; Dertz E. A.; Kim S. S. (2003) Enterobactin: An archetype for microbial iron transport. Proc. Natl. Acad. Sci. U. S. A. 100, 3584–3588. 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B. R.; Bogdan A. R.; Miyazawa M.; Hashimoto K.; Tsuji Y. (2016) Siderophores in iron metabolism: from mechanism to therapy potential. Trends Mol. Med. 22, 1077–1090. 10.1016/j.molmed.2016.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields-Cutler R. R.; Crowley J. R.; Hung C. S.; Stapleton A. E.; Aldrich C. C.; Marschall J.; Henderson J. P. (2015) Human urinary composition controls antibacterial activity of siderocalin. J. Biol. Chem. 290, 15949–15960. 10.1074/jbc.M115.645812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton M. C.; Rupert P. B.; Hoette T. M.; Raymondc K. N.; Abergel R. J.; Strong R. K. (2019) Parsing the functional specificity of Siderocalin/Lipocalin 2/NGAL for siderophores and related small-molecule ligands. J. Struct. Biol. 2, 100008. 10.1016/j.yjsbx.2019.100008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao G.; Clifton M. C.; Hoette T. M.; Mori K.; Deng S. X.; Qiu A.; Viltard M.; Williams D.; Paragas N.; Leete T.; Kulkarni R.; Li X.; Lee B.; Kalandadze A.; Ratner A. J.; Pizarro J. C.; Schmidt-Ott K. M.; Landry D. W.; Raymond K. N.; Strong R. K.; Barasch J. (2010) Iron traffics in circulation bound to a siderocalin (Ngal)-catechol complex. Nat. Chem. Biol. 6, 602–609. 10.1038/nchembio.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields-Cutler R. R.; Crowley J. R.; Miller C. D.; Stapleton A. E.; Cui W.; Henderson J. P. (2016) Human metabolome-derived cofactors are required for the antibacterial activity of siderocalin in urine. J. Biol. Chem. 291, 25901–25910. 10.1074/jbc.M116.759183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz D. H.; Holmes M. A.; Borregaard N.; Bluhm M. E.; Raymond K. N.; Strong R. K. (2002) The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 10, 1033–1043. 10.1016/S1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- Clifton M. C.; Corrent C.; Strong R. K. (2009) Siderocalins: siderophore-binding proteins of the innate immune system. BioMetals 22, 557–564. 10.1007/s10534-009-9207-6. [DOI] [PubMed] [Google Scholar]
- Goetz D. H.; Willie S. T.; Armen R. S.; Bratt T.; Borregaard N.; Strong R. K. (2000) Ligand preference inferred from the structure of neutrophil gelatinase associated lipocalin. Biochemistry 39, 1935–1941. 10.1021/bi992215v. [DOI] [PubMed] [Google Scholar]
- Miethke M.; Skerra A. (2010) Neutrophil gelatinase-associated lipocalin expresses antimicrobial activity by interfering with l-norepinephrine-mediated bacterial iron acquisition. Antimicrob. Agents Chemother. 54, 1580–1589. 10.1128/AAC.01158-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudevylle N.; Geist L.; Hötzinger M.; Hartl M.; Kontaxis G.; Bister K.; Konrat R. (2010) The v-myc-induced Q83 lipocalin is a Siderocalin. J. Biol. Chem. 285, 41646–41652. 10.1074/jbc.M110.123331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpikirati P.; Liu T.; Vachet R. W. (2018) Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods 144, 79–93. 10.1016/j.ymeth.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinz A. (2018) Cross-linking/Mass spectrometry for studying protein structures and protein-protein interactions: Where are we now and where should we go from here?. Angew. Chem., Int. Ed. 57, 6390–6396. 10.1002/anie.201709559. [DOI] [PubMed] [Google Scholar]
- Leitner A.; Faini M.; Stengel F.; Aebersold R. (2016) Crosslinking and mass spectrometry: An integrated technology to understand the structure and function of molecular machines. Trends Biochem. Sci. 41, 20–32. 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Xu G.; Takamoto K.; Chance M. R. (2003) Radiolytic modification of basic amino acid residues in peptides: Probes for examining protein-protein interactions. Anal. Chem. 75, 6995–7007. 10.1021/ac035104h. [DOI] [PubMed] [Google Scholar]
- Chen J.; Rempel D. L.; Gross M. L. (2010) Temperature Jump and Fast Photochemical Oxidation Probe Submillisecond Protein Folding. J. Am. Chem. Soc. 132, 15502–15504. 10.1021/ja106518d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogbein O. O.; Simmons D. A.; Konermann L. (2000) Effects of pH on the kinetic reaction mechanism of myoglobin unfolding studied by time-resolved electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 11, 312–319. 10.1016/S1044-0305(99)00149-X. [DOI] [PubMed] [Google Scholar]
- Lim J.; Vachet R. W. (2003) Development of a methodology based on metal-catalyzed oxidation reactions and mass spectrometry to determine the metal binding sites in copper metalloproteins. Anal. Chem. 75, 1164–1172. 10.1021/ac026206v. [DOI] [PubMed] [Google Scholar]
- Jumper C. C.; Schriemer D. C. (2011) Mass spectrometry of laser-initiated carbene reactions for protein topographic analysis. Anal. Chem. 83, 2913–2920. 10.1021/ac102655f. [DOI] [PubMed] [Google Scholar]
- Borotto N. B.; Zhang Z.; Dong J.; Burant B.; Vachet R. W. (2017) Increased β-Sheet dynamics and D-E Loop repositioning are Necessary for Cu(II)-induced amyloid formation by β-2-microglobulin. Biochemistry 56, 1095–1104. 10.1021/acs.biochem.6b01198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M.; Zhang B.; Cui W.; Gross M. L. (2017) Laser-initiated radical trifluoromethylation of peptides and proteins and its application to mass spectrometry-based protein footprinting. Angew. Chem., Int. Ed. 56, 14007–14010. 10.1002/anie.201706697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underbakke E. S.; Zhu Y.; Kiessling L. L. (2008) Isotope-coded affinity tags with tunable reactivities for protein footprinting. Angew. Chem., Int. Ed. 47, 9677–9680. 10.1002/anie.200803378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M. M.; Rempel D. L.; Du Z.; Gross M. L. (2003) Quantification of protein-ligand interactions by mass spectrometry, titration, and H/D exchange: PLIMSTEX. J. Am. Chem. Soc. 125, 5252–5253. 10.1021/ja029460d. [DOI] [PubMed] [Google Scholar]
- Jackson V. A.; Mehmood S.; Chavent M.; Roversi P.; Carrasquero M.; Toro D.; Seyit-Bremer G.; Ranaivoson F. M.; Comoletti D.; Sansom M. S.; Robinson C. V.; Klein R.; Seiradake E. (2016) Super-complexes of adhesion GPCRs and neural guidance receptors. Nat. Commun. 7, 11184. 10.1038/ncomms11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson J. P.; Crowley J. R.; Pinkner J. S.; Walker J. N.; Tsukayama P.; Stamm W. E.; Hooton T. M.; Hultgren S. J. (2009) Quantitative metabolomics reveals an epigenetic blueprint for iron acquisition in uropathogenic Escherichia coli. PLoS Pathog. 5, e1000305 10.1371/journal.ppat.1000305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H.; Fischbach M. A.; Liu D. R.; Walsh C. T. (2005) In vitro characterization of salmochelin and enterobactin trilactone hydrolases IroD, IroE, and Fes. J. Am. Chem. Soc. 127, 11075–11084. 10.1021/ja0522027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M.; Lin H.; Fischbach M. A.; Liu D. R.; Walsh C. T.; Groves J. T. (2006) Enzymatic tailoring of enterobactin alters membrane partitioning and iron acquisition. ACS Chem. Biol. 1, 29–32. 10.1021/cb0500034. [DOI] [PubMed] [Google Scholar]
- Heck A. J. (2008) Native mass spectrometry: a bridge between interactomics and structural biology. Nat. Methods 5, 927–933. 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- Politis A.; Schmidt C.; Tjioe E.; Sandercock A. M.; Lasker K.; Gordiyenko Y.; Russel D.; Sali A.; Robinson C. V. (2015) Topological models of heteromeric protein assemblies from mass spectrometry: Application to the yeast eIF3:eIF5 complex. Chem. Biol. 22, 117–128. 10.1016/j.chembiol.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fast C. S.; Vahidi S.; Konermann L. (2017) Changes in enzyme structural dynamics studied by hydrogen exchange-mass spectrometry: Ligand binding effects or catalytically relevant motions?. Anal. Chem. 89, 13326–13333. 10.1021/acs.analchem.7b03506. [DOI] [PubMed] [Google Scholar]
- Chung K. Y.; Rasmussen S. G.; Liu T.; Li S.; DeVree B. T.; Chae P. S.; Calinski D.; Kobilka B. K.; Woods V. L.; Sunahara R. K. (2011) Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature 477, 611–615. 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen J. R. (2009) Analysis of Protein Conformation and Dynamics by Hydrogen/Deuterium Exchange MS. Anal. Chem. 81, 7870–7875. 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borotto N. B.; Zhou Y.; Hollingsworth S. R.; Hale J. E.; Graban E. M.; Vaughan R. C.; Vachet R. W. (2015) Investigating therapeutic protein structure with diethylpyrocarbonate labeling and mass spectrometry. Anal. Chem. 87, 10627–10634. 10.1021/acs.analchem.5b03180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J.; Zhang H.; Gross M. L.; Blankenship R. E. (2009) Membrane orientation of the FMO antenna protein from Chlorobaculum tepidum as determined by mass spectrometry-based footprinting. Proc. Natl. Acad. Sci. U. S. A. 106, 6134–6139. 10.1073/pnas.0901691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.; Cheng M.; Gross M. L. (2019) Protein-metal-ion interactions studied by mass spectrometry-based footprinting with isotope-encoded benzhydrazide. Anal. Chem. 91, 1416–1423. 10.1021/acs.analchem.8b04088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebarle P.; Peschke M. (2000) Onthe mechanisms by which the charged droplets produced by electrospray lead to gas phase ions. Anal. Chim. Acta 406, 11–35. 10.1016/S0003-2670(99)00598-X. [DOI] [Google Scholar]
- Konijnenberg A.; Butterer A.; Sobott F. (2013) Native ion mobility mass spectrometry and related methods in structural biology. Biochim. Biophys. Acta, Proteins Proteomics 1834, 1239–1256. 10.1016/j.bbapap.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Konermann L.; Rodriguez A. D.; Sowole M. A. (2014) Type 1 and Type 2 scenarios in hydrogen exchange mass spectrometry studies on protein-ligand complexes. Analyst 139, 6078. 10.1039/C4AN01307G. [DOI] [PubMed] [Google Scholar]
- Chumsae C.; Gifford K.; Lian W.; Liu H.; Radziejewski C. H.; Zhou Z. S. (2013) Arginine modifications by methylglyoxal: Discovery in a recombinant monoclonal antibody and contribution to acidic species. Anal. Chem. 85, 11401–11409. 10.1021/ac402384y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krell T.; Pitt A. R.; Coggins J. R. (1995) The use of electrospray mass spectrometry to identify an essential arginine residue in type II dehydroquinases. FEBS Letters 360, 93–96. [DOI] [PubMed] [Google Scholar]
- Gao Y.; Wang Y. (2006) Site-selective modifications of arginine residues in human hemoglobin induced by methylglyoxal. Biochemistry 45, 15654–15660. [DOI] [PubMed] [Google Scholar]
- Janecki D. J.; Beardsley R. L.; Reilly J. P. (2005) Probing Protein tertiary structure with amidination. Anal. Chem. 77, 7274–7281. [DOI] [PubMed] [Google Scholar]
- Li K. S.; Chen G.; Mo J.; Huang R. Y.-C.; Deyanova E. G.; Beno B. R.; O’Neil S. R.; Tymiak A. A.; Gross M. L. (2017) Orthogonal mass spectrometry-based footprinting for epitope mapping and structural characterization: The IL-6 receptor upon binding of protein therapeutics. Anal. Chem. 89, 7742–7749. 10.1021/acs.analchem.7b01748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Limpikirati P.; Vachet R. W. (2019) Synergistic structural information from covalent labeling and hydrogen-deuterium exchange mass spectrometry for protein-ligand interactions. Anal. Chem. 91, 15248–15254. 10.1021/acs.analchem.9b04257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.