

Abstract
A number of structurally diverse D-mannose-derived lactols, including various deoxy-D-mannoses and conformationally restricted bicyclic D-mannoses, have been synthesized and investigated in mechanistic studies of β-mannosylation via Cs2CO3-mediated anomeric O-alkylation. It was found that deoxy mannoses or conformationally restricted bicyclic D-mannoses are not as reactive as their corresponding parent mannose. This type of β-mannosylation proceeds efficiently when the C2-OH is left free, and protection of that leads to inferior results. NMR studies of D-mannose-derived anomeric cesium alkoxides indicated the predominance of the equatorial β-anomer after deprotonation. Reaction progress kinetic analysis suggested that monomeric cesium alkoxides be the key reactive species for alkylation with electrophiles. DFT calculations supported that oxygen atoms at C2, C3, and C6 of mannose promote the deprotonation of the anomeric hydroxyl group by Cs2CO3 and chelating interactions between Cs and these oxygen atoms favour the formation of equatorial anomeric alkoxides, leading to the highly β-selective anomeric O-alkylation. Based on experimental data and computational results, a revised mechanism for this β-mannosylation is proposed. The utilization of this β-mannosylation was demonstrated by an efficient synthesis of the hexasaccharide core of complex fucosylated N-linked glycans.
Keywords: β-mannosylation, anomeric O-alkylation, carbohydrates, glycosylation, N-linked glycans
Graphical Abstract

Extensive mechanistic studies of β-mannosylation via Cs2CO3-mediated anomeric O-alkylation have been described. Experimental results from various NMR studies and reaction progress kinetic analysis as well as DFT calculations suggested that equatorial monomeric cesium alkoxides be the key reactive species for alkylation with electrophiles. A revised mechanism for this β-mannosylation is proposed. This β-mannosylation was utilized in the synthesis of the hexasaccharide core of complex fucosylated N-linked glycans.
Introduction
Tremendous glycobiological studies have demonstrated oligosaccharides and glycoconjugates play essential roles in numerous biological processes.1 In addition, carbohydrate molecules have been developed as effective therapeutic agents for treating various diseases. Sugar moieties, i.e. glycans, are also known to influence physical, chemical, and biological properties of their carrier molecules, including proteins, peptides, and small molecules.2 In order to study their biological functions, access to sufficient amounts of pure and structurally well-defined carbohydrate molecules need to be available to researchers. Isolation from natural sources sometimes has been successful, but the highly heterogeneous nature of glycoforms limit the practicality of this approach. In addition, the highly intrinsic complexity of the diverse carbohydrate structures poses a great challenge to the synthetic community, albeit great progress has been achieved in their chemical3 and chemo-enzymatic synthesis.4
Among various glycosidic linkages, β-mannopyranosides (oftentimes referred to as β-mannosides) exist in a wide range of biologically significant molecules including N-linked glycans, bacterial capsular polysaccharides, fungal metabolites, glycolipids, antimicrobial antibiotics, and lipopolysaccharides.5 As a class of 1,2-cis glycosides,6 β-mannopyranosides are exceptionally difficult to construct due to the steric effect of the axial C2-substituent as well as the absence of anomeric effect and neighboring group participation. Despite the fact that remarkable success has been achieved in stereoselective synthesis of β-mannosides,7 development of a mild and easily operable β-mannosylation method is desirable and of great interest to the carbohydrate community.
Initially developed by Schmidt8 and later by others,9 anomeric O-alkylation has been demonstrated as a viable alternative to traditional glycosylation for successful stereoselective synthesis of oligosaccharides and glycoconjugates. With respect to β-mannosides, Schmidt previously disclosed some studies on anomeric O-alkylation of partially or fully protected D-mannopyranoses with simple primary electrophiles in the presence of NaH or KOtBu as the base.10 Oftentimes, poor to moderate yields10c and moderate selectivity10d were observed. In addition, over-alkylation was found to be a problem when 3,4,6-tri-O-benzyl-D-mannopyranose (cf. 7, Table 1) was employed.10c Based on our success in stereoselective synthesis of 2-deoxy sugars,11,12 we recently reported a stereoselective synthesis of β-mannopyranosides by cesium carbonate (Cs2CO3)-mediated anomeric O-alkylation of D-mannose-derived 1,2-diols with primary or secondary electrophiles.13,14 It is worth noting that this mild and easily operable β-mannosylation method directly affords the desired β-mannosides with a free C2-OH at the mannose residue. The free C2-alcohol can be directly subjected to subsequent chemical transformations as demonstrated in a formal synthesis of potent calcium signal modulator acremomannolipin A15 and a concise synthesis of a trisaccharide oligomer of the Hyriopsis schlegelii glycosphingolipid, respectively.16
Table 1.
Anomeric O-alkylation of various deoxy-D-mannoses and bicyclic D-mannose-derived lactols with D-galactose-derived secondary triflate 6 in the presence of Cs2CO3.a,b

|
In our originally proposed hypothesis,13 after deprotonation of D-mannose 1 with excess amounts of base, a mixture of dianions 2 and 4 may be produced and interconvert into each other via open intermediate 3 (Scheme 1). Due to the chelation effect,10equatorial anomeric alkoxide 4 would be preferentially formed over the axial counterpart 2. In addition, equatorial anomeric alkoxide 4 was believed to be more nucleophilic than the axial counterpart 2 because of the double electron-electron repulsion (also known as kinetic anomeric effect, colored in purple).8,17 Subsequent SN2 reaction of equatorial anomeric alkoxide 4 with suitable electrophiles may afford desired β-mannopyranosides 5. It is believed that the synergy between kinetic anomeric effect and the chelation of cesium ion with oxygen atoms at C1 and C2 was the key for this β-selective anomeric O-alkylation to succeed (cf. Newman projection, Scheme 1).
Scheme 1. Initially proposed β-mannosylation via anomeric O-alkylation under dual control of kinetic anomeric effect and metal chelation.

Previously, in order to understand what structural features are critical for the β-mannosylation to be effective, 3,4,6-tri-O-benzyl-D-mannopyranose 7 and several D-mannose-derived lactol substrates including 3,4-di-O-benzyl-6-O-tert-butyldiphenylsilyl-D-mannose 8, 3,4,6-tri-O-benzyl-2-deoxy-D-mannose 9, 3,4-di-O-benzyl-6-deoxy-D-mannose (D-rhamnose) 11, 3,4-di-O-benzyl-2,6-dideoxy-D-mannose (D-olivose) 13, and a bicyclic 3-O-benzyl-4,6-O-benzylidene-D-mannose 16 were prepared and studied in the anomeric O-alkylation with D-galactose-derived secondary triflate 6 in the presence of cesium carbonate (Table 1).13 It was found that replacing the C6 benzyl ether of D-mannose 7 with a TBDPS ether led to lower yield of corresponding disaccharide 23 (40%, β only). Anomeric O-alkylation of 3,4,6-tri-O-benzyl-2-deoxy-D-mannose 9 with secondary triflate 6 (2.0 eq.) in the presence of Cs2CO3 (2.5 eq.) afforded the corresponding desired disaccharide 24 in 64% yield (β only), which was comparable to the reaction involving parent 3,4,6-tri-O-benzyl-D-mannose 7 (67% yield, β only). In addition, 3,4-di-O-benzyl-6-deoxy-D-mannose (D-rhamnose) 11 reacted with secondary triflate 6 to give corresponding desired disaccharide 26 in only 30% yield (β only). Furthermore, anomeric O-alkylation of D-olivose 13 and bicyclic D-mannose 16 with triflate 6 under the same conditions afforded desired disaccharides 28 and 31 in 15% and 0% yield, respectively.13 Those results suggested that our originally proposed working model for this β-mannosylation (cf. 4 in Scheme 1) may need to be revisited.
Results and Discussion
In order to gain more insight into this β-mannosylation, we prepared additional deoxy D-mannose-derived lactol substrates including 4,6-di-O-benzyl-3-deoxy-D-mannose 10, 4,6-di-O-benzyl-2,3-dideoxy-D-mannose 12,18 4-O-benzyl-3,6-dideoxy-D-mannose 14, and 4-O-benzyl-2,3,6-trideoxy-D-mannose 1519 (Table 1).20 It was found that Cs2CO3-mediated anomeric O-alkylation of 4,6-di-O-benzyl-3-deoxy-D-mannose 10, 4,6-di-O-benzyl-2,3-dideoxy-D-mannose 12, and 4-O-benzyl-3,6-dideoxy-D-mannose 14 with secondary triflate 6 (2.0 eq.) in the presence of Cs2CO3 (2.5 eq.) under the same conditions afforded 4,6-di-O-benzyl-3-deoxy-β-D-mannoside 25, 4,6-di-O-benzyl-2,3-dideoxy-β-D-mannoside 27, 4-O-benzyl-3,6-dideoxy-β-D-mannoside 29 in 40%, 26%, and 3% yields (β only), respectively. However, no 4-O-benzyl-2,3,6-trideoxy-β-D-mannoside 30 was detected when 4-O-benzyl-2,3,6-trideoxy-D-mannose 15 reacted with triflate 6 under the same conditions.
Next, direct competition experiments were carried out for comparative studies of the reactivity of the parent D-mannose 7 and its deoxy derivatives 9, 10, and 11. When a mixture of D-mannose 7 (1.0 eq.), 2-deoxy-D-mannose 9 (1.0 eq.), and triflate 6 (2.0 eq.) in 1,2-dichloroethane was warmed at 40 °C for 24 hours in the presence of 2.5 equivalents of Cs2CO3, β-mannoside 22 and 2-deoxy disaccharide 24 were obtained in 48% and 13%, respectively (β only) (a, Scheme 2). This interesting result demonstrates that D-mannose 7 is much more reactive than 2-deoxy-D-mannose 9 in this type of anomeric O-alkylation and suggests the important role of C2-OH of the D-mannose, despite the fact that previous experiments showed that anomeric O-alkylation of parent D-mannose 7 or 2-deoxy-D-mannose 9 with triflate 6 afforded 22 or 24 in similar yields and anomeric selectivity (Table 1, vide supra). Under exactly the same conditions, β-mannoside 22 and 3-deoxy disaccharide 25 were obtained in 30% and 15%, respectively (β only) (b, Scheme 2). Another competition experiment involving parent mannose 7 and 3,4-di-O-benzyl-6-deoxy-D-mannose 11 afforded β-mannoside 22 and 6-deoxy disaccharide 26 in 31% and 8%, respectively (β only) (c, Scheme 2). These results indicate that deoxy mannoses do not react as efficiently as their parent mannose in this type of Cs2CO3-mediated anomeric O-alkylation and suggest that all of the oxygen atoms at C2, C3 and C6 of mannose contribute to the reactivity of the mannose lactols.21
Scheme 2.

Competition anomeric O-alkylations between parent D-mannose and deoxy-D-mannoses.
Electronically, anomeric alkoxides derived from deoxy sugars should be more electron-rich than those derived from their corresponding parent sugars due to the absence of electron-withdrawing oxygen atom(s). In addition, deoxy sugars are less sterically hindered in comparison with their parent sugar molecules. Therefore, deoxy sugar-derived anomeric alkoxides should be more nucleophilic than those derived from their parent sugar molecules and afford the corresponding deoxy glycosides in higher yields via anomeric O-alkylation, which is consistent with our previous observations.11 However, our experimental results demonstrated that parent 3,4,6-tri-O-benzyl-D-mannose 7 was superior to those deoxy-D-mannoses (9-15) in this cesium carbonate-promoted anomeric O-alkylation (Table 1), which seemed somewhat contradictory to what we had observed previously for the stereoselective synthesis of 2-deoxy-β-glycosides.11 Such findings encouraged us to further investigate the mechanistic aspects of this β-selective mannosylation.
Previous studies also indicated that conformationally restricted bicyclic 3-O-benzyl-4,6-O-benzylidene-D-mannose 16 did not react with triflate 6 in the presence of cesium carbonate to afford corresponding disaccharide 31,13 which suggests that a conformationally flexible sugar ring may be critical for this type of anomeric O-alkylation. In order to further study the impact of the conformation and ring strain to the reactivity of the D-mannose-derived anomeric alkoxide in the β-mannosylation reaction, we prepared additional bicyclic D-mannose-derived lactol substrates. They include 3,4-O-bisketal-protected mannose 17, 4,6-di-O-benzyl-2,3-O-isopropylidene-D-mannose 18,22 3,6-anhydro-4-O-benzyl-D-mannose 19, 2,6-anhydro-3,4-di-O-benzyl-D-mannose 20, and 4-O-benzyl-3,6-O-(o-xylylene)-D-mannose 21.20,23 As shown in Table 1, Cs2CO3-mediated anomeric O-alkylation of 3,4-O-bisketal-protected mannose 17 and 4,6-di-O-benzyl-2,3-O-isopropylidene-D-mannose 18 bearing conformationally flexible C6-oxygen atom with secondary triflate 6 under standard conditions afforded 3,4-O-bisketal-protected mannoside 32 and 4,6-di-O-benzyl-2,3-O-isopropylidene-D-mannoside 33 in 23% and 34% yields (β only), respectively. However, none of 3,6-anhydro-4-O-benzyl-D-mannose 19, 2,6-anhydro-3,4-di-O-benzyl-D-mannose 20, and 4-O-benzyl-3,6-O-(o-xylylene)mannose 21 reacted with triflate 6 to afford detectable amounts of corresponding disaccharides 34, 35 or 36, respectively.24 These results suggested the relatively poor nucleophilicity of these bicyclic mannose-derived anomeric alkoxides. After careful analysis of the reaction mixtures, none of the bicyclic lactol donors (16, 17, 19-21) was detected except that 29% of the relatively more flexible 6,5-cis-fused lactol 18 was recovered. The decomposition of those strained bicyclic lactols may indicate their instabililty in addition to their low reactivity.25
Furthermore, we prepared 2,3,4,6-tetra-O-benzyl-D-mannopyranose 38 and studied this substrate in this Cs2CO3-mediated anomeric O-alkylation. As shown in Scheme 3, 2,3,4,6-tetra-O-benzyl-D-mannopyranose 38 was able to react with C4-triflate 6 (2.0 eq.) in the presence of cesium carbonate (2.5 eq.) to afford desired β-mannopyranoside 39β in 46% isolated yield and α-mannopyranoside 39α in 3% yield, respectively (β/α = 15.3/1). In addition, a small amount (5%) of β-glucoside 40 was also isolated from the reaction mixture. Presumably, deprotonation of the anomeric hydroxyl group of 38 followed by a sequential ring opening, epimerization of the C2 stereocenter through enolization of the aldehyde and re-protonation, and cyclization afforded a small amount of 2,3,4,6-tetra-O-benzyl-D-glucopyranose-dervived anomeric alkoxide 41. Anomeric O-alkylation of this alkoxide 41 gave rise to β-glucoside 40 under kinetic anomeric effect.8,17 This result indicated that 2,3,4,6-tetra-O-benzyl-D-mannopyranose 38 is less reactive than 3,4,6-tri-O-benzyl-D-mannopyranose 7, probably due to more severe steric effects. Despite that the presence of a free hydroxyl group at C2 of the mannose is not essential for this β-mannosylation, leaving this C2 hydroxyl group unprotected helps achieve superior yields and excellent β-selectivity.
Scheme 3.

Anomeric O-alkylation of 2,3,4,6-tetra-O-benzyl-D-mannopyranose.
Scheme 4.

Preparation of anomeric cesium alkoxides in CDCl3 and subsequent alkylation with allyl bromide.
Scheme 6.
Synthesis of the hexasaccharide core of fucosylated N-linked glycans.
We also carried out NMR studies for studying lactol 7 and its corresponding anomeric cesium alkoxide (42). In the event, to a 0.1 M solution of 3,4,6-tri-O-benzyl-D-mannose (7, α/β = 3.0/1) in CDCl326 was added 3 eq. of Cs2CO3 and the reaction mixture was stirred at 40 °C for 1 hour. The resulting mixture was allowed to stand for 0.5 hour and the supernatant, presumably containing cesium alkoxide (cf. 42, Scheme 4) in CDCl3, was taken out for NMR studies. It was found that cesium alkoxide 42 exists as a mixture of α and β anomers (α/β = 1/3.0).20 Next, 1.5 eq. of allyl bromide was added to this supernatant containing cesium alkoxide 42 in CDCl3 and the resulting mixture was stirred at 40 °C for 24 hours to afford desired allyl 3,4,6-tri-O-benzyl-β-D-mannoside (cf. 43,15 Scheme 4) in 23% yield. This result indicated that cesium alkoxide 42β is the reactive species in the anomeric O-alkylation.
We also prepared 3,4,6-tri-O-CD3-D-mannose (44) and its corresponding anomeric cesium alkoxide (45) for extensive NMR studies. The use of CD3 ether instead of benzyl ether protecting group (cf. 7) was to simplify the NMR spectra. As shown in Figure 1, 1H NMR spectrum of 3,4,6-tri-O-CD3-D-mannose (44) (0.1 M in CDCl3) showed that the more thermodynamically stable axial α-anomer was predominant before deprotonation due to the anomeric effect (α/β = 5.2/1).20 Next, 3 eq. of Cs2CO3 was added to this 0.1 M solution of 44 in CDCl3 and the reaction mixture was stirred at 40 °C for 1 hour. The resulting mixture was allowed to stand for 0.5 hour and the supernatant containing cesium alkoxide 45 was taken out and subjected to extensive NMR experiments (Scheme 5) at room temperature.20,27 The 1H NMR spectrum indicated that deprotonation of anomeric hydroxyl groups of 44 with Cs2CO3 cleanly afforded the corresponding anomeric cesium alkoxides (45) as a mixture of anomers (α/β = 1/5.0, Figure 1). Both signals of H-4 from cesium alkoxides 45β (major) and 45α (minor) were shown as triplets (J(H-3,H-4) = J(H-4,H-5) = 9.6 Hz) indicating a trans-diaxial relationship between H-3 and H-4 as well as H-4 and H-5.20 These data indicate that both anomeric cesium alkoxides (45α and 45β) mainly adopt a stable chair conformation at room temperature or 40 °C.27 In addition, proton signal broadening (except H4) may indicate the possible chelation of cesium with various oxygen atoms. The predominance of the equatorial β-anomer after deprotonation (α/β = 1/5.0) was probably due to the chelation of the cesium ion to C2-oxygen at room temperature, while in the axial α-anomer such a chelation would not be possible due to the 1,2-trans relationship. In addition, this solution of anomeric cesium alkoxide 45 in CDCl3 was treated with 1.5 eq. of allyl bromide and stirred at 40 °C for 24 hours to afford the desired allyl 3,4,6-tri-O-CD3-β-D-mannoside (46) in 22% yield (β only, Scheme 5). This result was comparable to that involving 3,4,6-tri-O-benzyl-D-mannose (7), which indicates that the benzyl protecting group is not required for this reaction. In addition, we also prepared 3,4,6-tri-O-(2-naphthylmethyl(NAP))-D-mannopyranose (47) and the corresponding cesium alkoxide 48 following the same procedure.20 Unfortunately, various attempts to crystallize D-mannose-derived anomeric alkoxides 42, 45, and 48 suitable for X-ray crystallographic analysis were unsuccessful.28
Figure 1. Comparison of.

1H NMR spectra of 3,4,6-tri-O-CD3-D-mannose (44, α/β = 5.2/1, colored in blue, bottom spectrum) and its corresponding cesium alkoxide 45 (α/β = 1/5.0, colored in red, top spectrum).
Scheme 5.

Revised mechanism for stereoselective β-mannosylation via Cs2CO3-mediated anomeric O-alkylation.
Previously, anomeric O-alkylation of 3,4,6-tri-O-benzyl-D-mannose 7 with allyl bromide in the presence of Cs2CO3 was found to afford allyl 3,4,6-tri-O-benzyl-β-D-mannoside 43 in 94% yield (β only).15 Based on that, we conducted reaction progress kinetic analysis involving two different initial concentrations of mannose 7, in order to establish whether monomeric cesium alkoxide 42β or its dimeric form is the key reactive intermediate for this anomeric O-alkylation (Figure 2).20 The measured concentration of mannose 7 and allyl β-D-mannoside 43 dependence on time is depicted in Figure 2 (a, 0.1 M of initial concentration of mannose 7), while kinetic data corresponding to 0.2 M of initial concentration of mannose 7 are provided in the Supporting Information.20 Next, kinetic analysis proved that this reaction exhibits a strict first-order dependence on mannose 7 at both 0.1 M and 0.2 M initial concentrations (b, Figure 2). As the deprotonation is complete in a relatively short time period (Scheme 4) and the subsequent SN2 reaction is believed to be the rate-determining step, our kinetic studies indicate that monomeric cesium alkoxide 42β should be the real reactive species for this anomeric O-alkylation.29
Figure 2.

Kinetic studies of anomeric O-alkylation of mannose 7 with allyl bromide (2.5 eq.) in the presence of Cs2CO3 (3.0 eq.) in 1,2-dichloroethane at 40 °C.
With all of the experimental results in hand, we next conducted computational studies to explore the role of Cs2CO3 in the anomeric O-alkylation and the origin of the decreased reactivities of deoxy-D-mannoses 9, 10, and 11.30 Based on the experimental studies discussed above, we surmised that the cesium salt may either promote the deprotonation of the anomeric OH group to form anomeric cesium alkoxides (e.g. 44) or enhance the nucleophilicity of the anomeric cesium alkoxides in the subsequent alkylation step. These two potential effects were computationally analyzed using density functional theory (DFT) calculations. All calculations were performed at the M06–2X/def2-QZVP/SMD(dichloroethane)//M06–2X/SDD-6–31G(d) level of theory (see SI for computational details).
We first investigated the relative acidities of the anomeric OH groups in tri-O-benzyl-D-mannose 7 and deoxy-D-mannoses 9, 10, and 11. We computed the reaction energies of the deprotonation of these substrates using anomeric cesium alkoxide 7-Cs as the base.31 All three deoxy-D-mannoses require much higher energy to deprotonate than D-mannose 7, indicating cesium alkoxide 7-Cs is much more stable than cesium alkoxides derived from the deoxy-D-mannoses. Examination of the lowest energy conformers of the deprotonated cesium alkoxides (Figure 3) indicated that the pyranose rings in 7-Cs adopts a 4C1 conformation. The 1C4 conformer of 7-Cs is 6.4 kcal/mol less stable (see SI for details). A strong chelation of the cesium ion with four oxygen atoms, O1, O2, O6, and O5 (the endocyclic oxygen), was observed.32 The lower stability of anomeric cesium alkoxides 9-Cs and 11-Cs is attributed to the lack of O2 and O6 substituents, respectively, which diminished the chelating Cs–O interactions. On the other hand, 10-Cs is stabilized by chelation with O1, O2, O6, and O5 atoms, which is similar to that in 7-Cs. Nonetheless, inductive effects of the C3-OBn group in 7-Cs provide additional stabilization of the negative charge in the alkoxide, and thus make the anomeric OH of 7 more acidic than that of 10.
Figure 3.

Relative acidity of anomeric OH of D-mannose 7 and deoxy-D-mannoses 9, 10, and 11. Cesium alkoxide 7-Cs is used as the base to calculate the reaction energies of all deprotonation reactions. All energies are in kcal/mol. Bond lengths are in angstrom.
Next, we investigated the relative nucleophilic reactivities of the anomeric cesium alkoxides using allyl bromide as the electrophile. The computed activation energies of the SN2 transition states with respect to each of the cesium alkoxide intermediates are shown in Figure 4. The allylation of all four cesium aloxides require comparable activation energies (ΔG‡ = 19.4 – 21.6 kcal/mol), suggesting once these alkoxides are formed, their reaction rates in the subsequent O-alkylation would be similar.
Figure 4.

Calculated activation free energies of the O-allylation of anomeric cesium alkoxides 7-Cs, 9-Cs, 10-Cs, and 11-Cs with allyl bromide. All energies are in kcal/mol. Bond lengths are in angstrom.
Taken together, the above computational analyses indicate the Cs ion forms strong chelating interactions with multiple oxygen atoms in anomeric alkoxides. The possible chelation of cesium ion with the oxygen atoms on the sugar is also supported by 133Cs NMR studies as well as 18-crown-6 titration studies.20 This stabilizing chelation effect promotes the deprotonation of D-mannose. The importance of the Cs–O chelation is evident in the experimentally observed low reactivities of deoxy-D-mannoses 9, 10, and 11. Removal of oxygen substituents at either C2, C3, or C6 positions destabilizes the chelated cesium alkoxides, and thus makes the deprotonation of anomeric hydroxyl group in deoxy-manoses more difficult.
Based on aforementioned discussions, we revised the originally proposed mechanistic hypothesis as shown in Scheme 5. After deprotonation of anomeric hydroxyl group of D-mannose 7 with cesium carbonate, a mixture of axial alkoxide 52 and equatorial alkoxide 54 may be produced and interconvert into each other via open aldehyde intermediate 53. Due to chelation, Equatorial anomeric cesium alkoxide 5433 (cf. 7-Cs, Figure 3), would be preferentially formed. In addition, the effective chelation may also alleviate the steric effect of the C2-hydroxyl group. Subsequent anomeric O-alkylation of the cesium alkoxide 54 with the assistance of kinetic anomeric effect, i.e. double electron-electron repulsion, with suitable electrophiles via SN2 substitution then affords the desired β-mannopyranosides 55 (Scheme 5).34
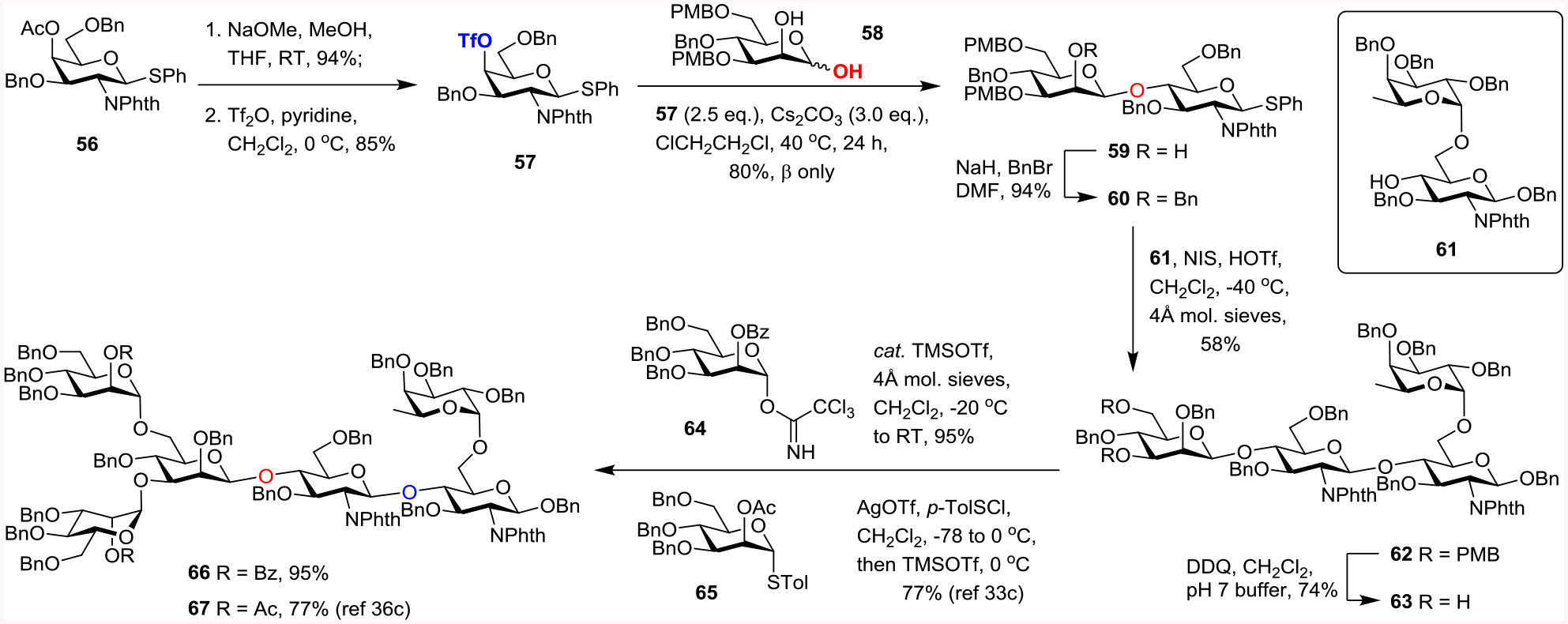
This β-mannosylation via anomeric O-alkylation was next utilized in the synthesis of the hexasaccharide core35 of complex fucosylated N-linked glycans.36 For instance, bi-antennary α-(2,3)-sialylated core-fucosylated N-glycan dodecasaccharide was found on alpha fetoprotein (AFP) isolated from patients with hepatocellular carcinoma. The structure of this N-glycan was believed to be a much more reliable marker than AFP to differentiate benign and malignant liver diseases.37 This dodecasaccharide was also detected on the surface of erythropoietin38 and is believed to be important for the in vivo activity of erythropoietin.39 As shown in Scheme 6, known protected β-D-galactosamine-derived thioglycoside 5640 underwent standard deacetylation (94%) followed by triflation of the resulting C4-alcohol to afford triflate 57 (85%). Next, cesium carbonate-mediated anomeric O-alkylation of known 3,6-di-O-(4-methoxybenzyl)-4-O-benzyl-D-mannopyranose 5813 with aforementioned triflate acceptor 57 (2.5 eq.) under the optimized conditions gave the desired β-disaccharide 59 in 80% yield (β only). Standard benzylation of the C2’-OH of disaccharide 59 afforded 60 which was subjected to the traditional glycosylation with known disaccharide acceptor 6136c (NIS, cat. TfOH) to produce the corresponding tetrasaccharide 62 in 58% yield. After DDQ-mediated deprotection of bis-PMB ethers of 62 under neutral conditions, 63 was obtained in 74% yield whose spectroscopic data were found to be identical to those previously reported.36c Previously, double α-mannosylation of tetrasaccharide 63 with thioglycoside donor 65 (AgOTf, p-toluenesulfenyl chloride; then catalytic TMSOTf) afforded hexasaccharide 67 in 77% yield.36c In our hands, double α-mannosylation of tetrasaccharide 63 with trichloroacetimidate donor 64 in the presence of catalytic amount of TMSOTf afforded complex hexasaccharide 66 in 95% yield.
Conclusions
In conclusion, various structurally diverse deoxy-D-mannoses and conformationally restricted bicyclic D-mannoses have been prepared and subjected to the mechanistic studies of this umpolung-type β-mannosylation via anomeric O-alkylation. Deoxy mannoses were found to be not as reactive as parent mannose, and conformationally restricted bicyclic D-mannoses afforded no product or lower yields than conformationally flexible D-mannoses. When the hydroxyl group at C2 was masked, this type of β-mannosylation did not proceed as efficiently, albeit the corresponding β-mannoside was still detected as the major product. Studies of various alkali metal bases indiated that cesium carbonate (Cs2CO3) was the optimal base for this β-selective anomeric O-alkylation.20 Extensive NMR studies demonstrated the formation of predominant equatorial anomeric cesium alkoxides after deprotonation, due to the chelation of the cesium ion to the oxygen atoms. In addition, the chelating interactions of cesium ion with the oxygen atoms on the sugar are also supported by 133Cs NMR experiments as well as 18-crown-6 titration studies.20 Kinetic studies supported that the anomeric O-alkylation involves monomeric cesium alkoxides as the key reactive species. DFT calculations suggested that that the oxygen atoms at C2, C3 and C6 of the mannose enhances the acidity of the anomeric hydroxyl group to facilitate the deprotonation by Cs2CO3. In particular, the C2-oxygen atom is believed to play a major role in the chelation with the cesium ion. Such chelation preferentially favors the formation of equatorial anomeric alkoxides, leading to the highly stereoselective alkylation to form the β-anomer of the products. Based on experimental data and computational results, a revised mechanism for this β-mannosylation is proposed. We also isolated and characterized a side product, 3,4,6-tri-O-benzyl-D-fructose, which was presumably formed via an α-keto rearrangement of the open aldehyde intermediate.20 This umpolung-type β-mannosylation has also been demonstrated in efficient synthesis of the hexasaccharide core of complex fucosylated N-linked glycans. Application of this method to the stereoselective construction of other types of challenging glycosidic linkages, such as β−2-amino-2-deoxy-D-mannosides, and synthesis of complex biologically significant carbohydrate molecules are currently underway.
Experimental Section
General procedure for Cs2CO3 mediated O-alkylation.
To a mixture of sugar lactol donor (0.1 mmol, 1.0 eq.), sugar derived triflate acceptor 6 (2.0 eq.), and Cs2CO3 (2.5 eq.) was added 1,2-dichloroethane (1.0 mL). The reaction mixture was stirred at 40 ℃ for 24 hours. The crude reaction mixture was purified by preparative TLC. The β-configuration of the new formed mannosidic linkage was unambiguously assigned by measuring the 1JC-H for the anomeric carbon.
Synthesis of the hexasaccharide core of the fucosylated N-linked glycans.
Phenyl-3,6-di-O-benzyl-4-O-trifluoromethylsulfonyl-2-deoxy-2-phthalimido-1-thio-β-D-galactopyranoside (57):
To a solution of known compound 56[40] (2.67 g, 4.6 mmol) in MeOH (25 mL) and THF (25 mL) was added 0.5 M NaOMe solution in MeOH (6.4 mL, 3.22 mmol). The mixture was stirred at room temperature for 7 hours. The reaction mixture was neutralized by the addition of Amberlyst IR-120 (H+), filtered, and concentrated. The residue was purified by silica gel column chromatography (Hexanes/EtOAc = 5/1 to 3/1) to give the corresponding alcohol (2.52 g, 94 %). To a solution of this alcohol (0.67 g, 1.2 mmol) and pyridine (1.0 mL, 12 mmol) in CH2Cl2 (3.0 mL) cooled at 0 ℃ was added Tf2O (0.35 mL, 2.0 mmol) dropwise. The resulting mixture was stirred at 0 ℃ for 2 h before being quenched with ice water. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (50 mL × 3). The combined organic layer was washed sequentially with saturated CuSO4 (50 mL × 3) and water (50 mL × 3), dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by silica gel column chromatography with CH2Cl2 to afford the sugar derived triflate 57 (725 mg, 85%). 1H NMR (600 MHz, CDCl3) δ 7.85 (m, 1H, HAr), 7.74 (m, 1H, HAr), 7.70 (m, 1H, HAr), 7.58 (dd, J = 7.3, 1.1 Hz, 1H, HAr), 7.42 – 7.32 (m, 7H, HAr), 7.25 – 7.18 (m, 3H, HAr), 6.98 – 6.93 (m, 3H, HAr), 6.90 – 6.83 (m, 2H, HAr), 5.54 (d, J = 2.9 Hz, 1H, H-4), 5.52 (d, J = 10.5 Hz, 1H, H-1), 4.70 (d, J = 12.6 Hz, 1H, -OCH2Ar), 4.65 (d, J = 11.2 Hz, 1H, -OCH2Ar), 4.52 – 4.45 (m, 2H, -OCH2Ar, H-2), 4.38 (dd, J = 10.5, 2.9 Hz, 1H, H-3), 4.21 (d, J = 12.6 Hz, 1H, -OCH2Ar), 4.01 (dd, J = 8.3, 5.6 Hz, 1H, H-5), 3.79 (dd, J = 9.2, 5.6 Hz, 1H, H-6a), 3.69 (dd, J = 9.2, 8.4 Hz, 1H, H-6b); 13C NMR (150 MHz, CDCl3) δ 168.05, 166.87, 137.36, 136.51, 134.26, 133.98, 132.81, 131.67, 131.62, 131.61, 129.01, 128.69, 128.44, 128.36, 128.34, 128.28, 128.24, 128.01, 123.80, 123.45, 118.68 (q, 1JC-F = 319.7 Hz), 84.21, 80.57, 75.14, 73.91, 72.97, 71.89, 67.20, 50.99.
Phenyl-O-2,4-di-O-benzyl-3,6-di-O-(para-methoxybenzyl)-β-D-mannopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-gluopyranoside (60):
To a mixture of mannopyranosyl donor 58[13] (1.02 g, 2.0 mmol), sugar-derived triflate acceptor 57 (3.7 g, 5.0 mmol), and Cs2CO3 (2.0 g, 6.0 mmol) was added 1,2-dichloroethane (20 mL). The reaction mixture was stirred at 40 ℃ for 24 hours. The crude reaction mixture was purified by silica gel column chromatography (Hexanes/EtOAc = 5/1 to 1/1) to give disaccharide 59 (1.73 g, 80%). To a solution of 59 (1.0 g, 0.93 mmol) in DMF (4.0 mL) cooled at 0 ℃ was added NaH (75 mg, 1.86 mmol, 60% in mineral oil) portion wise. The resulting mixture was stirred at 0 ℃ for 1 h before BnBr (0.17 mL, 1.4 mmol) was added. The reaction mixture was warmed up and stirred at ambient temperature for 3 h before being quenched with water. The resulting mixture was extracted with EtOAc three times, and combined organic extracts were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by silica gel column chromatography (Hexanes/EtOAc = 5/1 to 3/1) to give the title compound 60 (1.01 g, 94%). The 1JC-H of mannosidic anomeric carbon for 60 was determined to be 159.0 Hz. ; FT-IR (thin film): 3064, 3032, 2938, 2863, 1777, 1714, 1678, 1512, 1455, 1388, 1250, 1174, 1102, 1070, 917, 807, 701 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.80 (d, J = 7.3 Hz, 1H, HAr), 7.70 (m, 1H, HAr), 7.65 (m, 1H, HAr), 7.58 (d, J = 7.3 Hz, 1H, HAr), 7.50 – 7.44 (m, 2H, HAr), 7.42 – 7.38 (m, 2H, HAr), 7.37 – 7.27 (m, 11H, HAr), 7.24 – 7.13 (m, 9H, HAr), 6.89 – 6.81 (m, 4H, HAr), 6.78 – 6.69 (m, 5H, HAr), 5.52 (d, J = 9.9 Hz, 1H, H-1), 4.96 (d, J = 13.0 Hz, 1H, -OCH2Ar), 4.91 (s, 2H, -OCH2Ar), 4.86 (d, J = 10.9 Hz, 1H, -OCH2Ar), 4.60 (d, J = 12.0 Hz, 1H, -OCH2Ar), 4.56 (s, 1H, H-1′), 4.55 – 4.49 (m, 2H, -OCH2Ar), 4.49 – 4.41 (m, 4H, -OCH2Ar), 4.38 (d, J = 11.6 Hz, 1H, -OCH2Ar), 4.32 – 4.23 (m, 2H, H-3, H-2), 4.02 (dd, J = 9.8, 8.0 Hz, 1H, H-4), 3.90 (t, J = 9.6 Hz, 1H, H-4′), 3.81 – 3.78 (m, 4H, H-2′, -OCH3), 3.77 – 3.70 (m, 5H, -OCH3, H-6a, H-6′a), 3.66 – 3.59 (m, 3H, H-6b, H-6′b, H-5), 3.42 – 3.34 (m, 2H, H-3′, H-5′); 13C NMR (150 MHz, CDCl3) δ 168.08, 167.31, 159.25, 158.98, 139.03, 138.91, 138.63, 138.06, 133.86, 133.69, 132.82, 132.06, 131.80, 131.69, 130.74, 130.48, 129.33, 129.28, 128.87, 128.58, 128.38, 128.25, 128.06, 128.00, 127.94, 127.85, 127.78, 127.72, 127.66, 127.45, 126.82, 123.44, 123.33, 113.86, 113.73, 101.79, 83.37, 82.50, 79.26, 78.57, 76.04, 75.21, 75.11, 74.99, 74.81, 74.18, 73.59, 73.07, 71.61, 69.22, 68.91, 55.36, 55.31, 54.86; LRMS (ESI) calculated for C70H69NNaO13S [M+Na]+ 1187.44, found 1187.30.
Benzyl-O-2,4-di-O-benzyl-3,6-di-O-(para-methoxybenzyl)-β-D-mannopyranosyl-(1→4)-O-3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-gluopyranosyl-(1→4)-O-[2,3,4-tri-O-benzyl-α-L-fucopyranosyl-(1→6)]-3-O-benzyl-2-deoxy-2-phthalimido-β-D-gluopyranoside (62):
To a mixture of donor 60 (87 mg, 0.075 mmol), acceptor 61[36c] (45 mg, 0.05 mmol), activated 4 Å molecular sieves (300 mg), and NIS (84 mg) was added CH2Cl2 (2.5 mL). The solution was cooled to −40 ℃ and TfOH (2.0 μL) was added. The resulting mixture was stirred at this temperature overnight and then filtered through celite. The filtrate was quenched and washed with saturated Na2S2O3 aqueous solution. The organic layer was dried over Na2SO4, filtered, and concentrated. The crude reaction mixture was purified by preparative TLC (EtOAc/ CH2Cl2/Toluene = 1/5/5) to furnish the title tetra-saccharide 62 (57 mg, 58%). ; FT-IR (thin film): 3065, 3033, 2934, 2867, 1777, 1716, 1614, 1514, 1454, 1388, 1249, 1091, 1038, 749, 724, 700 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.88 (d, J = 7.3 Hz, 1H, HAr), 7.82 (d, J = 7.2 Hz, 1H, HAr), 7.76 – 7.56 (m, 7H, HAr), 7.50 (d, J = 7.3 Hz, 1H, HAr), 7.46 – 7.11 (m, 32H, HAr), 7.07 (m, 1H, HAr), 7.04 – 6.91 (m, 6H, HAr), 6.87 – 6.69 (m, 12H, HAr), 5.59 (d, J = 8.4 Hz, 1H, H-1), 5.01 – 4.91 (m, 5H), 4.91 – 4.81 (m, 4H), 4.80 – 4.72 (m, 2H), 4.66 – 4.47 (m, 9H), 4.45 – 4.26 (m, 8H), 4.21 – 4.16 (m, 2H), 4.12 (dd, J = 10.7, 8.5 Hz, 1H), 4.07 – 4.01 (m, 2H), 3.99 (dd, J = 10.2, 2.8 Hz, 1H), 3.90 – 3.85 (m, 2H), 3.83 – 3.77 (m, 4H), 3.77 – 3.72 (m, 3H), 3.68 (s, 3H), 3.65 – 3.57 (m, 3H), 3.41 (dd, J = 10.8, 3.0 Hz, 1H), 3.38 (dd, J = 9.4, 3.0 Hz, 1H), 3.35 (ddd, J = 9.8, 5.1, 1.7 Hz, 1H), 3.28 (ddd, J = 9.9, 3.0, 1.6 Hz, 1H), 1.01 (d, J = 6.5 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 168.27, 167.89, 167.78, 167.67, 159.19, 158.91, 139.21, 139.17, 139.05, 138.96, 138.86, 138.73, 138.07, 137.16, 133.91, 133.73, 133.67, 133.58, 132.01, 131.79, 131.66, 130.87, 130.59, 129.28, 129.19, 128.62, 128.60, 128.51, 128.42, 128.34, 128.23, 128.19, 128.10, 128.07, 128.05, 127.91, 127.89, 127.74, 127.72, 127.62, 127.60, 127.57, 127.55, 127.47, 127.44, 127.37, 127.08, 126.95, 126.72, 123.55, 123.31, 123.20, 113.81, 113.68, 101.45, 96.90, 96.86, 96.70, 82.46, 79.57, 79.24, 77.71, 77.33, 76.09, 76.05, 75.82, 75.30, 75.09, 75.06, 74.84, 74.82, 74.69, 74.63, 74.43, 74.36, 73.79, 73.39, 73.20, 73.09, 72.52, 71.30, 69.96, 69.27, 68.51, 66.07, 63.94, 56.68, 55.95, 55.35, 55.25, 16.51; LRMS (ESI) calculated for C119H118N2Na2O24 [M+2Na]2+ 1002.89, found 1002.80.
Benzyl-O-2-O-benzoyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1→3)-O-[2-O-benzoyl-3,4,6-tri-O-benzyl-α-D-mannopyranosyl-(1→6)]-O-2,4-di-O-benzyl-β-D-mannopyranosyl-(1→4)-O-3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-gluopyranosyl-(1→4)-O-[2,3,4-tri-O-benzyl-α-L-fucopyranosyl-(1→6)]-3-O-benzyl-2-deoxy-2-phthalimido-β-D-gluopyranoside (66):
To a solution of tetra-saccharide 62 (57 mg, 0.029 mmol) in CH2Cl2 (3.0 mL) was added a solution of pH 7 (1.0 mL) buffer and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (18 mg, 0.079 mmol). The resulting mixture was stirred at room temperature for 3 h before additional amount of DDQ (13 mg, 0.057 mmol) was added. The mixture was stirred for another 2 hours and another portion of DDQ (13 mg, 0.057 mmol) was added. After 2 hours, the mixture was quenched with saturated NaHCO3 solution and extracted with ethyl acetate (10 mL × 3). The organic layer was dried over Na2SO4, filtered, and concentrated. The crude reaction mixture was purified by preparative TLC (EtOAc/CH2Cl2 = 1/10) to furnish the corresponding diol 63 (37 mg, 74%). The spectroscopic data was consistent with the data reported in the literature.[36c] To a mixture of diol acceptor 63 (74 mg, 0.043 mmol), donor 64 (95 mg, 0.13 mmol), activated 4 Å molecular sieves (100 mg) was added CH2Cl2 (1.0 mL). The solution was cooled to −20 ℃ and TMSOTf (1.0 μL) was added. The resulting mixture was slowly warmed up to room temperature over 2 hours. The reaction was quenched with Et3N (200 μL) and filtered through celite. The filtrate was washed with saturated NaHCO3 aqueous solution and brine. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified by preparative TLC (EtOAc/Toluene = 1/10) to afford hexasaccharide 66 (115 mg, 95%). ; FT-IR (thin film): 3091, 3061, 3030, 2933, 2872, 1777, 1717, 1496, 1455, 1390, 1266, 1099, 1077, 737, 696 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.07 (dd, J = 8.2, 1.4 Hz, 2H), 7.83 – 7.78 (m, 2H), 7.76 (d, J = 7.4 Hz, 1H), 7.67 – 7.63 (m, 2H), 7.61 – 7.55 (m, 2H), 7.49 – 7.43 (m, 2H), 7.43 (m, 1H), 7.40 – 7.34 (m, 6H), 7.33 – 7.08 (m, 60H), 7.06 – 7.00 (m, 2H), 6.97 (t, J = 7.6 Hz, 2H), 6.95 – 6.92 (m, 2H), 6.90 – 6.87 (m, 2H), 6.76 – 6.68 (m, 4H), 6.63 – 6.55 (m, 1H), 6.50 (t, J = 7.5 Hz, 2H), 5.72 (t, J = 2.5 Hz, 1H), 5.54 (t, J = 2.3 Hz, 1H), 5.46 (d, J = 8.3 Hz, 1H), 5.20 (d, J = 2.0 Hz, 1H), 4.95 – 4.78 (m, 12H), 4.74 – 4.56 (m, 8H), 4.55 – 4.37 (m, 11H), 4.34 (d, J = 11.3 Hz, 1H), 4.29 (d, J = 12.3 Hz, 1H), 4.27 – 4.18 (m, 4H), 4.17 – 4.10 (m, 3H), 4.09 – 4.04 (m, 2H), 4.00 – 3.91 (m, 7H), 3.90 – 3.83 (m, 2H), 3.80 (m, 1H), 3.76 – 3.69 (m, 5H), 3.64 (dd, J = 10.8, 1.5 Hz, 1H), 3.61 – 3.53 (m, 5H), 3.29 (dd, J = 10.8, 3.4 Hz, 1H), 3.24 – 3.19 (m, 2H), 0.95 (d, J = 6.5 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 168.05, 167.87, 167.64, 165.56, 165.07, 139.01, 138.91, 138.89, 138.78, 138.76, 138.70, 138.41, 138.20, 138.04, 138.01, 137.94, 137.18, 133.75, 133.55, 133.24, 132.76, 131.95, 131.80, 131.63, 130.10, 130.00, 129.97, 129.95, 128.74, 128.58, 128.57, 128.54, 128.52, 128.49, 128.46, 128.37, 128.33, 128.30, 128.24, 128.21, 128.18, 128.02, 128.01, 127.98, 127.89, 127.87, 127.85, 127.77, 127.67, 127.63, 127.61, 127.59, 127.57, 127.53, 127.46, 127.43, 127.38, 127.15, 126.91, 126.84, 126.78, 123.52, 123.25, 102.06, 99.60, 98.34, 96.91, 96.69, 82.21, 79.77, 79.46, 78.35, 78.23, 77.80, 76.51, 76.31, 75.54, 75.24, 75.20, 75.17, 75.06, 74.81, 74.78, 74.58, 74.54, 74.47, 74.43, 74.16, 74.13, 73.85, 73.55, 73.41, 73.36, 72.66, 72.48, 72.10, 71.60, 71.03, 69.94, 69.08, 69.04, 69.00, 68.48, 67.99, 66.55, 66.04, 63.93, 56.64, 55.92, 16.51. LRMS (ESI) calculated for C171H166N2Na2O34 [M+2Na]2+ 1419.56, found 1419.80.
Supplementary Material
Acknowledgements
We are grateful to National Science Foundation (CHE-1464787 to X.L. and J.Z.), National Institutes of Health Common Fund Glycosciences Program (1U01GM125290 to P.L. and 1U01GM125289 to J.Z.), The University of Toledo, and University of Michigan‒Dearborn for supporting this research. Calculations were performed at the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF. We thank Dr. Yong Wah Kim for assistance in the 133Cs NMR studies, Mr. James K. Dunaway and Rodney A. Park from University of Toledo, and Mr. Luke Harding and Justin Woodward from University of University of Michigan‒Dearborn for experimental assistance.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].(a) Varki A, Glycobiology 1993, 3, 97–130. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bertozzi CR, Kiessling LL, Science 2001, 291, 2357–2364. [DOI] [PubMed] [Google Scholar]; (c) Varki A, Glycobiology 2017, 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].(a) Wyss DF, Choi JS, Li J, Knoppers MH, Willis KJ, Arulanandam ARN, Smolyar A, Reinherz EL, Wagner G, Science 1995, 269, 1273–1278. [DOI] [PubMed] [Google Scholar]; (b) Weymouth-Wilson AC, Nat. Prod. Rep 1997, 14, 99–110; [DOI] [PubMed] [Google Scholar]; (c) Kren V, Martinkova L, Curr. Med. Chem 2001, 8, 1303–1328. [DOI] [PubMed] [Google Scholar]
- [3]. For select reviews, see:; (a) Adero PO, Amarasekara H, Wen P, Bohé L, Crich D, Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bennett CS, Galan MC, Chem. Rev 2018, 118, 7931–7985. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zeng J, Xu Y, Wang H, Meng L, Wan Q, Sci. China Chem 2017, 60, 1162–1179. [Google Scholar]; (d) Li X, Zhu J, Eur. J. Org. Chem 2016, 4724–4767. [Google Scholar]; (e) Ranade SC, Demchenko AV, J. Carbohydr. Chem 2013, 32, 1–43. [Google Scholar]; (f) McKay MJ, Nguyen HM, ACS Catal. 2012, 2, 1563–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Li X, Zhu J, J. Carbohydr. Chem 2012, 31, 284–324. [Google Scholar]; (h) Zhu X, Schmidt RR, Angew. Chem. Int. Ed 2009, 48, 1900–1934; Angew. Chem. 2009, 121, 1932–1967. [DOI] [PubMed] [Google Scholar]; (i) Demchenko AV, Editor. Handbook of chemical glycosylation; Advances in stereoselectivity and therapeutic relevance. 2008, 501 pp; [Google Scholar]; (j) Fuegedi P, The Organic Chemistry of Sugars. Levy DE, Fuegedi P, Eds.; CRC Press, 2006, pp 89–179; [Google Scholar]; (k) Galonić DP, Gin DY, Nature 2007, 446, 1000–1007; [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Toshima K, Tatsuta K, Chem. Rev 1993, 93, 1503–1531. [Google Scholar]
- [4].(a) Li W, McArthur JB, Chen X, Carbohydr. Res 2019, 472, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang L-X, Amin MN, Chem. Biol 2014, 21, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gridley JJ, Osborn HMI, J. Chem. Soc., Perkin Trans 1, 2000, 1471–1491. [Google Scholar]
- [6].(a) Nigudkar SS, Demchenko AV, Chem. Sci 2015, 6, 2687–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Demchenko AV, Curr. Org. Chem 2003, 7, 35–79. [Google Scholar]; (c) Demchenko AV, Synlett 2003, 1225–1240. [Google Scholar]; (d) Tanaka M, Nakagawa A, Nishi N, Iijima K, Sawa R, Takahashi D, Toshima K, J. Am. Chem. Soc 2018, 140, 3644–3651. [DOI] [PubMed] [Google Scholar]
- [7].(a) Sasaki K, Tohda K, Tetrahedron Lett. 2018, 59, 496–503. [Google Scholar]; (b) Boltje TJ, Buskas T, Boons G-J, Nat. Chem 2009, 1, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ishiwata A, Lee YJ, Ito Y, Org. Biomol. Chem 2010, 8, 3596–3608. [DOI] [PubMed] [Google Scholar]; (d) Zeng Y, Kong F, Prog. Chem 2006, 18, 907–926. [Google Scholar]; (e) Barresi F, Hindsgaul O, Modern Methods in Carbohydrate Synthesis; Khan SH, A. R Eds. O’Neill, Harwood Academic Publishers: Amsterdam, 1996; 251–276. [Google Scholar]; (f) Toshima K, Tatsuta K, Chem. Rev 1993, 93, 1503–1531. [Google Scholar]; (g) Paulsen H, Angew. Chem. Int. Ed 1982, 21, 155–173; Angew. Chem. 1982, 94, 184–201. [Google Scholar]
- [8].(a) Schmidt RR, Michel J, Tetrahedron Lett. 1984, 25, 821–824. [Google Scholar]; (b) Schmidt RR, Angew. Chem. Int. Ed 1986, 25, 212–235; Angew. Chem. 1986, 98, 213–236. [Google Scholar]; (c) Schmidt RR, Pure Appl. Chem 1989, 61, 1257–1270. [Google Scholar]; (d) Schmidt RR, Klotz W, Synlett 1991, 168–170. [Google Scholar]; (e) Tsvetkov YE, Klotz W, Schmidt RR, Liebigs Ann. Chem 1992, 371–375. [Google Scholar]; (f) Schmidt RR, Front. Nat. Prod. Res 1996, 1, 20–54. [Google Scholar]
- [9].(a) Pertel SS, Gorkunenko OA, Kakayan ES, Chirva VJ, Carbohydr. Res 2011, 346, 685–688. [DOI] [PubMed] [Google Scholar]; (b) Ryan DA, Gin DY, J. Am. Chem. Soc 2008, 130, 15228–15229. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Morris WJ, Shair D. M, Org. Lett 2009, 11, 9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Trewartha G, Burrows JN, Barrett AGM, Tetrahedron Lett. 2005, 46, 3553–3556. [Google Scholar]; (e) Vauzeilles B, Dausse B, Palmier S, Beau J-M, Tetrahedron Lett. 2001, 42, 7567–7570. [Google Scholar]; (f) Izumi S, Kobayashi Y, Takemoto Y Org. Lett 2019, 21, 665–670. [DOI] [PubMed] [Google Scholar]
- [10].(a) Schmidt RR, Reichrath M, Angew. Chem., Int. Ed 1979, 18, 466–467. [Google Scholar]; (b) Schmidt RR, Reichrath M, Moering U, Tetrahedron Lett. 1980, 21, 3561–3564. [Google Scholar]; (c) Tamura J, Schmidt RR, J. Carbohydr. Chem 1995, 14, 895–911. [Google Scholar]; (d) Schmidt RR, Moering U, Reichrath M, Chem. Ber 1982, 115, 39–49. [Google Scholar]
- [11].Zhu D, Baryal KN, Adhikari S, Zhu J, J. Am. Chem. Soc 2014, 136, 3172–3175. [DOI] [PubMed] [Google Scholar]
- [12].Zhu D, Adhikari S, Baryal KN, Abdullah BN, Zhu J, J. Carbohydr. Chem 2014, 33, 438–451. [Google Scholar]
- [13].Nguyen H, Zhu D, Li X, Zhu J, Angew. Chem. Int. Ed 2016, 55, 4767–4771; Angew. Chem. 2016, 128, 4845–4849. [DOI] [PubMed] [Google Scholar]
- [14].Takahashi D, Trends. Glycosci. Glyc 2016, 28, E119–E120. [Google Scholar]
- [15].Li X, Berry N, Saybolt K, Ahmed U, Yuan Y, Tetrahedron Lett. 2017, 58, 2069–2072. [Google Scholar]
- [16].Bhetuwal BR, Woodward J, Li X, Zhu J, J. Carbohydr. Chem 2017, 36, 162–172. [Google Scholar]
- [17]. Previous studies also indicated that due to electron-electron repulsion anomeric C1-alkoxide is more nucleophilic than non-anomeric alkoxide, see: refs 9c, 11 and 12.
- [18].Yang MT, Woerpel KA, J. Org. Chem 2009, 74, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aidhen IS, Satyamurthi N, Indian J Chem., Sect B 2008, 47B, 1851–1857. [Google Scholar]
- [20]. See Supporting Information for details.
- [21]. See Supporting Information for additional direct competition experiments that produce similar results when allyl bromide is used as the electrophile.
- [22].Borodkin VS, Ferguson MAJ, V Nikolaev A, Tetrahedron Lett. 2001, 42, 5305–5308. [Google Scholar]
- [23]. For examples of using conformationally restricted o-xylylene protecting group in carbohydrate synthesis, see:; (a) Poss AJ, Smyth MS, Synth. Commun 1989, 19, 3363–3366. [Google Scholar]; (b) Balbuena P, Rubio EM, Mellet CO, Fernández JMG, Chem. Commun 2006, 2610–2612. [DOI] [PubMed] [Google Scholar]; (c) Imamura A, Lowary TL, Org. Lett 2010, 12, 3686–3689. [DOI] [PubMed] [Google Scholar]; (d) Okada Y, Asakura N, Bando M, Ashikaga Y, Yamada H, J. Am. Chem. Soc 2012, 134, 6940–6943. [DOI] [PubMed] [Google Scholar]; (e) Asakura N, Motoyama A, Uchino T, Tanigawa K, Yamada H, J. Org. Chem 2013, 78, 9482–9487. [DOI] [PubMed] [Google Scholar]; (f) Uchino T, Tomabechi Y, Fukumoto A, Yamada H, Carbohydr. Res 2015, 402, 118–123. [DOI] [PubMed] [Google Scholar]; (g) Zhang L, Shen K, Taha HA, Lowary TL, J. Org. Chem 2018, 83, 7659–7671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. When 2,6-anhydro-3,4-di-O-benzyl-D-mannose 20 was subjected to anomeric O-alkylation, the formation of corresponding disaccharide 35 was not observed, instead an enal (ref.S19, Supporting Information) was isolated in 50% yield. Presumably, enal was formed via deprotonation of anomeric hydroxyl of 20 followed by ring opening, enolization, and elimination of C3-benzyloxy group.
- [25]. A small amount of side product, 3,4,6-tri-O-benzyl-D-fructose 37 (Table 1), was always detected in all of the previous anomeric O-alkylation reactions involving 3,4,6-tri-O-benzyl-D-mannose 7 as the lactol donor. Presumably, 37 was obtained via an α-keto rearrangement of the open aldehyde intermediate derived from lactol 7.
- [26]. This cesium carbonate-mediated β-mannosylation via anomeric O-alkylation gives comparable results in 1,2-dichloroethane (DCE) and chloroform (CHCl3).
- [27]. Extensive NMR studies of anomeric cesium alkoxide (45) at room temperature or 40 ℃ did not show much difference.
- [28]. Attempted crystallization of anomeric alkoxides 42, 45, and 48 using various solvents or mixed solvents, e.g. dichloromethane, 1,2-dichloroethane, chloroform, and dichloromethane/n-pentane, was unsuccessful.
- [29]. Although the data prove that monomeric cesium alkoxide 42β should be the real reactive species for this anomeric O-alkylation at 0.1~0.2 M of initial concentration of mannose 7, other forms of cesium alkoxide 42β, e.g., dimeric form, can not be ruled out at higher concentration,
- [30].(a) Xu H, Muto K, Yamaguchi J, Zhao C, Itami K, Musaev DG, J. Am. Chem. Soc 2014, 136, 14834–14844. [DOI] [PubMed] [Google Scholar]; (b) Walden DM, Jaworski AA, Johnston RC, Hovey MT, Baker HV, Meyer MP, Scheidt KA, Cheong PHY, J. Org. Chem 2017, 82, 7183–7189. [DOI] [PubMed] [Google Scholar]; (c) Anand M, Sunoj RB, Schaefer HF III, J. Am. Chem. Soc 2014, 136, 5535–5538. [DOI] [PubMed] [Google Scholar]
- [31]. Computationally it is not feasible to accurately model the deprotonation using the experimentally used base Cs2CO3, which does not completely dissolve under the experimental conditions. Here, using 7-Cs as the model base will not affect the relative acidity trend.
- [32]. The chelating Cs–O distances in the optimized structures are typically shorter than 3 Å, which indicates relatively strong interactions, see:; Gagné OC, Hawthorne FC, Acta Crystallogr. B Struct. Sci 2018, 74, 63–78. [Google Scholar]
- [33]. Whether the C2-OH of intermediates 52 and 54 is deprotonated or not can not be concluded from the 1H NMR data; however, it is believed that it remains mainly as undeprotonated form in consideration of its pKa value. In addition, our experimental analysis of the weight of cesium alkoxide 42 is consistent with the presence of one cesium ion in the majority component.
- [34]. Deprotonation of C2-OH is not required for this anomeric O-alkylation to occur, as anomeric O-alkylation of 2,3,4,6-tetra-O-benzyl-D-mannopyranose also afford the desired β-mannoside in good yield and anomeric selectivity. However, we can not rule out the possibility that the C2-OH of mannose is deprotonated in a thermodynamically unfavorable, rate-limiting step to generate a bis-alkoxide which is rapidly alkylated at the anomeric position. We sincerely thank one of the reviewers for pointing this out.
- [35].Katoh T, Katayama T, Tomabechi Y, Nishikawa Y, Kumada J, Matsuzaki Y, Yamamoto K, J. Biol. Chem 2016, 291, 23305–23317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].(a) Wu B, Hua Z, Warren JD, Ranganathan K, Wan Q, Chen G, Tan Z, Chen J, E ndo A, Danishefsky SJ, Tetrahedron Lett. 2006, 47, 5577–5579. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu B, Tan Z, Chen G, Chen J, Hua Z, Wan Q, Ranganathan K, Danishefsky SJ, Tetrahedron Lett. 2006, 47, 8009–8011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sun B, Srinivasan B, Huang X, Chem. Eur. J 2008, 14, 7072–7081. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ, J. Am. Chem. Soc 2009, 131, 5792–5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].(a) Johnson PJ, Poon TCW, Hjelm NM, Ho CS, Ho SKW, Welby C, Stevenson D, Patel T, Parekh R, Townsend RR, Br. J. Can 1999, 81, 1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Taketa K, Electrophoresis 1998, 19, 2595–2602. [DOI] [PubMed] [Google Scholar]
- [38].Chen F-TA, Evangelista RA, Electrophoresis 1998, 19, 2639–2644. [DOI] [PubMed] [Google Scholar]
- [39].Watson E, Bhide A, van Halbeek H, Glycobiology 1994, 4, 227–237. [DOI] [PubMed] [Google Scholar]
- [40].Sawada T, Fujii S, Nakano H, Ohtake S, Kimata K, Habuchi O, Carbohydr. Res 2005, 340, 1983–1996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.