

Abstract
Similar to ischemic preconditioning, high-intensity exercise has been shown to decrease infarct size following myocardial infarction. In this article, we review the literature on beneficial effects of exercise, exercise requirements for cardioprotection, common methods utilized in laboratories to study this phenomenon, and discuss possible mechanisms for exercise-mediated cardioprotection.
Keywords: exercise, ischemic reperfusion, ischemic preconditioning, remote ischemic preconditioning
Beneficial Effects of Exercise
Nearly 60 years have passed since Morris and Crawford first published their groundbreaking findings in the British Medical Journal in 1958, proposing that an increased level of physical activity provides protection from cardiovascular events.1 In the following decades, exercise and an elevated level of physical fitness repeatedly displayed a strong correlation with a decreased risk of myocardial infarction (MI), all-cause mortality, and cardiovascular disease–related mortality.2–10 Based on these studies and more recent research,11–13 exercise appears to exert its beneficial effects by not only decreasing the likelihood of experiencing an MI but limiting the damage caused by ischemia and reperfusion if such an event does occur. Despite a common cause, these effects may exist due to vastly different mechanisms.
Exercise is generally considered to be protective against the risk of myocardial events by reducing several physiological risk factors such as elevated blood pressure, obesity, hyperlipidemia, and insulin resistance.14 Accompanying the reduction in risk factors, long-term exercise provides beneficial effects directly on the myocardium itself. Endurance exercise training has displayed the ability to positively impact the heart through beneficial hypertrophy, angiogenesis, and improved contractility.14,15 Additionally, individuals who exercise tend to experience relative bradycardia due to autonomic modulation, allowing for longer diastolic periods and elevated myocardial perfusion at submaximal workloads.14,16 Overall, these effects combine to improve vascular, endothelial, and myocardial health, contributing to the decreased event risk and all-cause mortality associated with exercise.
While these long-term adaptive changes in response to exercise have been associated with a decrease in the incidence of acute MI, exercise may also protect the heart in instances in which a cardiac event does take place. Although the mechanism is not fully understood, there is evidence to suggest that exercise may mimic the favorable, cardioprotective effects of ischemic preconditioning17 and attenuate cardiomyocyte death in the setting of myocardial ischemia–reperfusion (IR). In studies examining this effect, cardioprotection is determined by preservation of cardiac function via reduction or prevention of myocardial damage. Whether an intervention has resulted in cardioprotection can be established by demonstrating attenuation of cardiomyocyte death or reduction in infarct size. The surrogate markers for cardioprotection include a decreasing trend in cardiac enzymes (such as troponin T or I and creatine kinase-MB), preserved left ventricular (LV) ejection fraction (as assessed by echocardiography, myocardial nuclear ventriculography, or cardiac magnetic resonance imaging [MRI]), and reduced myocardial infarct size (which is also assessed with myocardial nuclear scanning, cardiac MRI, and in vivo studies by evaluation of cardiac tissues).
The protection provided by exercise appears to occur in a biphasic manner, with 2 separate “windows” of protection, beginning immediately after a single episode of exertion and continuing for multiple days.12,18–22 The first of these 2 windows begins immediately following the event and subsides within the first few hours following preconditioning. After approximately 24 hours, a second window of protection begins, extending for days to even weeks.22–24 The second window of protection has been reported to disappear after cessation of activity. For example, after 4 weeks of detraining, rats were found to have relatively less cardioprotection than those that continued to exercise. However, the rats that underwent detraining still maintained some form of cardioprotection compared to the control group.25,28 Similarly, exercise in humans has produced a cardioprotective state of increased nitric oxide and decreased endothelin 1 levels. The protective state lasted for 4 weeks after cessation of exercise, returning to baseline in 8 weeks.24 Despite similar protective effects during each window, the relationship between the mechanisms underlying these 2 time periods has yet to be determined.
Exercise Requirements for Cardioprotection
Duration
Beginning with the inaugural animal studies of exercise and protection from IR injury, conflicting opinions on the necessary intensity and duration to achieve cardioprotection have existed. An early study by Powers et al established that a rigorous 10-week training program at 75% to 80% of maximal oxygen consumption (VO2 max) was sufficient to improve myocardial contractile performance and reduce lipid peroxidation during IR in the rat in vivo.11 A further study established that exercise programs of 3 to 5 days at 60% to 70% VO2 max were found to be effective, improving contractile efficiency following IR in rats.13,26 Surpassing the 3-to 5-day success, researchers strive to see if any benefit of cardioprotection can be deemed from a single episode of exercise. Several subsequent studies effectively confirmed that cardioprotective benefits can be achieved in just 1 session.21,27–30 Despite this agreement with regard to duration, controversy continues regarding the intensity required in each individual session.
Intensity
In 2000, Lee et al utilized data gathered from the late 1980s to early 1990s to present the concept that short high-intensity bouts of exercise may be equally effective in protecting the cardiovascular system as longer, low-intensity workouts.31 To date, this has not been sufficiently proven in IR experiments. As mentioned previously, cardioprotective benefits have been observed from exercise at 60% to 70% VO2 max as well as 75% to 80% VO2 max, representing benefits from both moderate and vigorous intensities.11,13,26 Controversy exists in literature about the intensity of exercise and the level of cardioprotection. An earlier study comparing moderate-and high-intensity exercise in rats showed equivalent protection against IR injury.29 However, a study found that infarct size reduced proportionally to the intensity of the exercise in rats trained at 60% VO2 max versus rats trained at 80% VO2 max for 10 weeks.25 While the optimal intensity threshold has not been determined for IR protection, greater intensity overall does appear to be beneficial in long-term exercise regimens. In relation to event occurrence and long-term cardioprotection, Mittleman et al established a significantly decreased risk of MI following strenuous physical exertion in those who exercise regularly.30 Compared to individuals who exercise at an intensity of 6 metabolic equivalents of task or higher at least 5 per week, sedentary individuals have a nearly 50-fold increase in relative risk of MI following strenuous exertion. As reported by Swain et al, epidemiological studies comparing vigorous and moderate-intensity aerobic exercise suggest that vigorous exercise provides greater cardioprotective benefit due in part to risk factor modification.31 Although high-intensity exercise appears to be better in stimulating cardioprotection, it may not be well suited for elderly patients with comorbidities such as coronary artery disease or congestive heart failure. The American College of Sports Medicine recommends “30 minutes or more of moderate-intensity physical activity such as brisk walking on most, preferably all days of the week.”32 Moderate-intensity exercise is defined as exercise at an intensity of 40% to 60% of VO2 max.33 This recommendation seems reasonable in terms of inducing cardioprotection due to evidence of protection against IR injury in rats exposed to exercise at 55% VO2 max.29
Exercise: A Surrogate for Ischemic Preconditioning?
In 1986, Murry et al discovered the concept of “ischemic preconditioning,” the notion that a relatively brief cardiac ischemic event protects the heart against a subsequent more prolonged episode of ischemia. The degree of myocardial damage (i.e., infarct size) caused by prolonged ischemia and reperfusion was reduced in patients who were “preconditioned” by a preceding brief ischemic event compared to those who were not.17
Over time, it became well established that protection provided from preconditioning did not require application of brief ischemia directly to the heart itself. Remote ischemic preconditioning (RIPC) is the phenomenon whereby brief episodes of ischemia and reperfusion applied at distant tissues or organs render the myocardium resistant to a subsequent sustained episode of ischemia. This phenomenon was first described in 1993 by Przyklenk et al who demonstrated that brief ischemia in the circumflex coronary artery bed reduced subsequent infarction in the territory of the left anterior descending (LAD) artery.34 Interestingly, RIPC can provide a similar magnitude of protection as observed with local preconditioning.35 Despite promising findings in animal models, the results of human studies involving RIPC have been inconsistent. In 2007, Hausenloy et al reported decreased postoperative troponin leak in 27 patients who underwent RIPC prior to coronary artery bypass graft (CABG).36 A single-center, double-blind randomized study including 329 patients undergoing CABG also reported lower troponin leak as well as lower all-cause mortality in the RIPC group.37 In 2015, Meybohm et al conducted a prospective, double-blind, multicenter randomized controlled trial involving adults undergoing cardiac surgery. In contrast, the study did not observe any difference in composite primary end points or other outcomes between the RIPC and the control group.38 A recent Cochrane review including 29 studies involving 5392 patients undergoing CABG with or without valve surgery showed no evidence that RIPC has a treatment effect on clinical outcomes, such as mortality and post procedure MI. However, the Cochrane review did report moderate evidence that RIPC reduces the amount of troponin release post procedure.39 On review of the current evidence, it appears that RIPC does have a short-term effect on decreasing troponin release and protecting the myocardium during surgery. The effects of RIPC on other outcomes have been questionable. We feel that overall RIPC to an extent does exert cardioprotection in humans and the conflicting studies prove that we still do not have a strong understanding of the RIPC mechanism in order to exploit its full potential.
The fundamental difference between remote and local preconditioning is the fact that RIPC involves the communication or transfer of the protective trigger from a peripheral site to the heart. Both release of circulating humoral factors from the remote tissue during brief IR and neuronal transfer of the protective signal have been implicated to play a role. In 2009, Shimizu et al demonstrated in rats that transient limb ischemia released hydrophobic circulating factors of <150 kD which induced cardioprotection and were transferable across species.40 Lim et al suggested that RIPC included both a neural and a humoral component by demonstrating in mice that occlusion of either the femoral vein (humoral component) or a combined femoral and sciatic nerve resection (neural component) resulted in complete nullification of the cardioprotective effect of RIPC.41 In addition, Pickard et al suggested that release of humoral factors in RIPC required vagus nerve activation by showing that bilateral cervical vagotomy in rats abolished cardioprotection. Hexamethonium, a ganglionic blocker, was also shown to abrogate cardioprotection, suggesting that humoral factors may induce cardioprotection via recruitment of intrinsic cardiac ganglia.42
At present, the exact details of neuronal signal transfer in remote conditioning of the heart and their role in the release of humoral factors are not entirely clear. Despite this uncertainty, the following pathway has been suggested. Following brief cycles of ischemia and reperfusion of the upper limb, adenosine is believed to be released which not only stimulates the release of local humoral factors but also activates the sensory peripheral nerves. These nerves travel to the spinal cord and the brain stem, increasing parasympathetic activity, and possibly initiating cardioprotection via release of humoral factors from the pre-or postganglionic nerve endings of the heart.42 As this pathway suggests, rather than existing as mutually exclusive mechanisms, neural and humoral processes may act in concert to provide cardioprotection from remote conditioning.
A substantial body of evidence suggests that exercise is an effective surrogate for ischemic preconditioning with cellular mechanisms similar to both direct and RIPC.22 While exercise is not typically regarded as an ischemic event, physical activity stimulates the production of endogenous ligands (including, eg, adenosine) as in RIPC. One possible mechanism for this relationship is the elevated activity of anaerobic glycolysis in both strenuous exercise and RIPC. During RIPC, the remote ischemic tissue is forced to utilize anaerobic glycolysis to produce adenosine triphosphate (ATP) for cell survival. In exercise, although skeletal muscle cells preferably utilize glycogen and fatty acids for energy production, more intense activity requires cells to undergo some degree of anaerobic glycolysis.43 Thus, despite an approximate 5-fold increase in coronary flow during exercise, elevated myocardial oxygen demand and increased compressive forces on microvasculature may provide hypoxic conditions in the myocardium44 which could potentially act as stimuli for preconditioning. Whatever the mechanism of initiation, exercise has clearly displayed similarities to forms of ischemic preconditioning.
A common theme shared by RIPC (in particular, the humoral component of remote conditioning) and exercise is the release of circulating ligands that display the capability to interact with myocytes and trigger pathways that result in intracellular signaling associated with cardioprotection. These circulating ligands can be transmitted by the transfer of plasma or serum from a preconditioned or exercised donor to a naive acceptor participant, both within and among species.45,46 For example, postexercise plasma from humans has been shown to decrease cardiac infarct size by 40% in a rabbit Langendorff IR model.46 As discussed below, this property provides a significant opportunity to perform translational studies in which the mechanisms of RIPC and exercise, applied in human participants, can be investigated.
Mechanisms of Exercise Cardioprotection
Models and Methods
The established gold standard of cardioprotection by ischemic and remote preconditioning—and thus, by extrapolation, using exercise as the preconditioning stimulus—is reduction in myocardial infarct size. For preclinical models, in vivo experiments47 can be designed to both quantify infarct size and interrogate potential mechanisms that contribute to the infarct-sparing effect of a preceding period of exercise. For investigation of the mediators and mechanisms of exercise-induced cardioprotection in humans, alternative methods are clearly required. For global pathophysiologic assessment of parameters related to cardiac function and infarct size, the Langendorff isolated perfused heart has traditionally been used. In order to explore the phenomenon of exercise-induced cardioprotection on a more molecular level, various cell models have also been utilized. Although other methods have also been used, novel insights have been provided by testing of human-derived samples in Langendorff and cell culture studies.
Langendorff isolated buffer-perfused heart.
With the discovery of retrograde perfusion in the isolated heart model by Oskar Langendorff in 1895, the Langendorff isolated buffer-perfused heart method continues to provide a lasting contribution in the study of mammalian heart physiology due to the method’s ability to simulate heart function in an isolated organ system under physiological parameters.48 The Langendorff system’s ability to eliminate systemic influences, such as hormonal factors, autonomic regulation, and the inflammatory response, enables controlled variables to be applied to a cardiac model in order to assess cardiac function.44 Thus, the Langendorff model is extensively used in IR studies with the intent to gain insight into the mechanisms underlying IR injury.49
The model allows for cardiac variables, such as LV developed pressure, rate of change in LV developed pressure (dP/dt), coronary flow, and coronary pressure to be monitored during both ischemia and reperfusion. In addition, the measurement of infarct size using triphenyl tetrazolium chloride staining, along with cardiac markers of injury such as lactate dehydrogenase and troponin in coronary effluent, can be evaluated as end point indicators of IR injury.50 Collection of human serum or plasma following exercise (or samples obtained postexercise from preclinical models) and addition of these samples to the perfusate provide a robust translational and controlled method for examining the cardioprotective effects of exercise on IR injury within an intact heart.51
Cell models.
In addition to the Langendorff model, many previously conduced IR experiments have expanded their findings with results from cardiomyocyte cell culture studies. Early cardiomyocyte cell cultures involved isolation of cardiomyocytes from adult rats. In 1983, Nag et al developed a method to remove the hearts of adult rats, enzymatically dissociate the myocardium and maintain isolated myocyte suspensions in culture for 45 days.52 This established the feasibility of studying cardiac cells in a highly controlled environment. The basic concept of this cardiomyocyte isolation method has been expanded to include harvesting of cells from both neonatal and adult rats, with extension to multiple species including mice, guinea pig, and rabbits53–55 and most recently human cardiomyocytes.56–59 The model allows for the design of protocols measuring various aspects of cardiomyocyte function and integrity, including gene transcription and expression, protein content, cell signaling, growth, hypertrophy, and cell death.56,60–65 Researchers have also isolated and identified cardiomyocytes from the hearts of mice66 and rats exposed to exercise.67–70 In addition to the use of primary cultures of cardiomyocytes, researchers have utilized immortalized cell lines, including H9c2 rat myofibroblasts71–73 and murine-derived HL-1 cardiomyocytes, the only immortal cardiomyocyte cell line.74 The differences between primary culture and cell lines allow for varying factors to influence the environment of the IR cell model. For instance, primary cardiomyocytes begin to die within 20 to 30 minutes of ischemia; however, it takes several hours for HL-1 cells to die under ischemia.75 The immortalized cells are highly glycolytic compared to primary cultured adult cardiomyocytes. In recent years, human induced pluripotent stem cells-derived cardiomyocytes have been successfully cultured and provide a human cardiomyocytes model in which to research exercise effects.76–78
However, in relation to the ischemia and reperfusion process that occurs during acute MI, the cultured cardiomyocyte model provides a reductionist approach for examining the cellular mechanisms underlying cardioprotection. Exercise preconditioning through hypoxia reoxygenation is one of the mechanisms of exercise-induced cardioprotection. Reoxygenation causes reactive oxygen species (ROS), which may not only induce oxidative damage but also provide cardioprotection through activation of cell signalings.79 Many experiments have varied culture medium and examined the effects following exposure to hypoxia-reoxygenation.80–84 These in vitro conditions may not mimic the situation in vivo. For example, the death of cardiomyocytes following ischemia and reperfusion can continue up to 3 days post-reperfusion.85 However, the cell models are usually observed for less than 24 hours. The limitation of cell models restricts the ability to generalize research findings and the application of in vivo model can provide better comparison to human conditions.
LAD artery occlusion and IR model.
Myocardial injury through ligation of the LAD is an established in vivo model to mimic clinical MI. Measurement of the infarct size in LAD occlusion and IR is a validated method to evaluate the cardioprotective effect of exercise. In a study by de Waard et al, early exercise training by voluntary treadmill running started within 24 hours following a large MI had no effect on MI size but attenuated the MI-induced LV dysfunction without a detrimental effect on LV remodeling.86 Bito et al also reported that early exercise training after MI prevents cardiac contractiles dysfunction but no effect on cardiac remodeling.87 Exercise prior to a MI has the benefit of reducing infarct size and improving angiogenesis through activation of vascular endothelial growth factor.88 Continuous exercise training before MI and reinitiated early after a MI blunted the trends of infarct size-dependent systolic dysfunction and LV dilation.89 Interestingly, Vujic et al reported that exercise induces new cardiomyocyte generation in the adult mammalian heart by mouse MI model. Inhibition of miR-222, an miRNA-induced during exercise, blocked the cardiomyogenic response of exercise.90 Exercise training also protects from IR injury. Esposito et al elucidated the relationship of training intensity and cardioprotection after IR injury in rats.25
Mechanisms
The exact mechanism of exercise-induced cardioprotection against lethal IR injury is still not properly understood. Studies to date indicate that a number of factors and pathways are likely to be involved rather than a single mechanism. In the following paragraphs, we will highlight the factors that have thus far been suggested to play a role (Figure 1).
Figure 1. Proposed mechanisms of exercise cardioprotection.
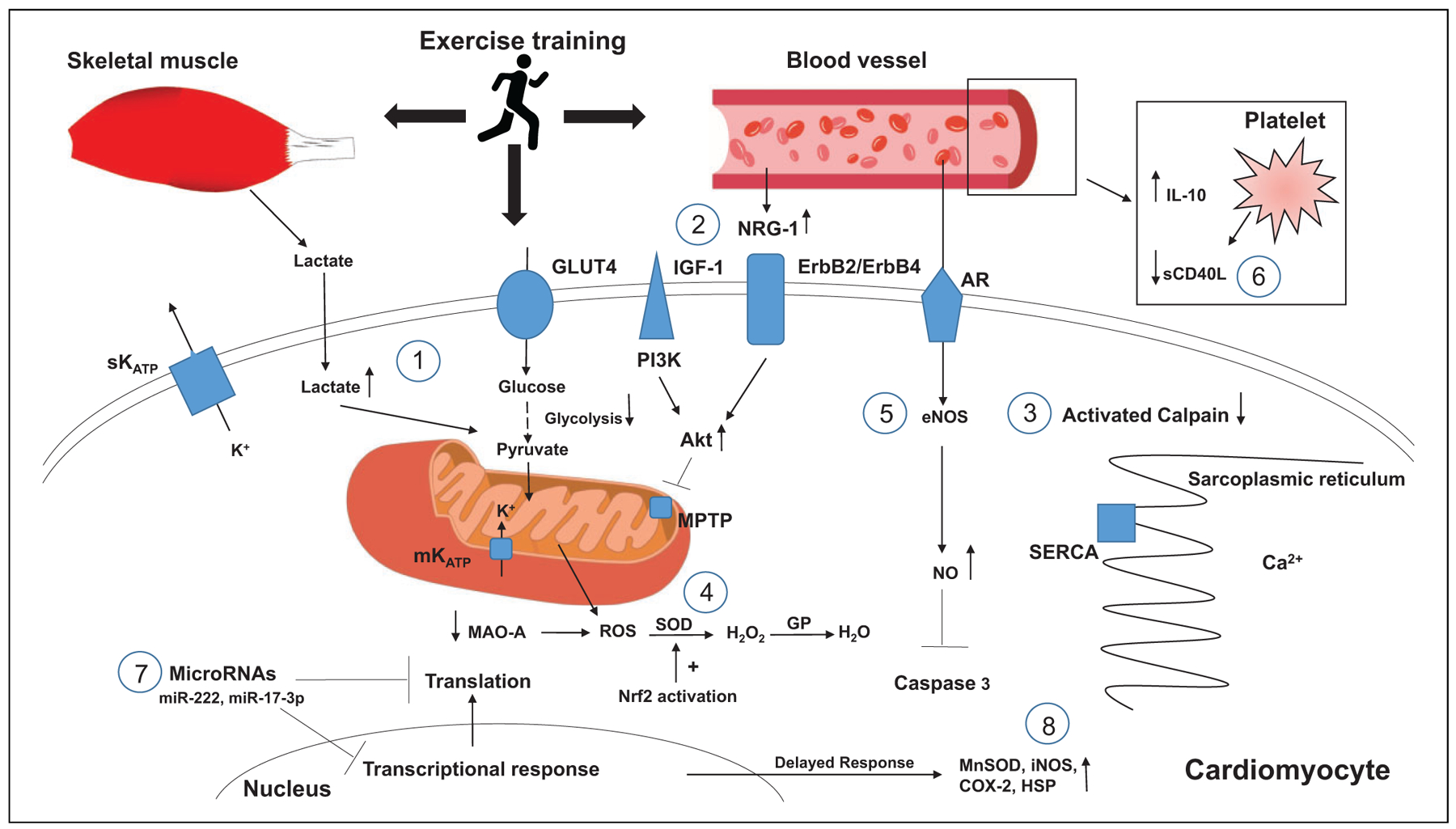
1. Lactate from skeletal muscle and intracellular cardiomyocytes provide substrate for ATP generation during anaerobic glycolysis.
2. NRG-1 levels increase binding to erbB2/erbB4, activating the PI3K/Akt pathway that inhibits MPTP opening during cellular hypoxia.
3. Intracellular calcium load is decreased, leading to decreased activation of calpain.
4. Decreased levels of MAO-A and increased SOD activity through activation of Nrf2 activation.
5. Endothelial eNOS and intracellular NO levels rise, inhibiting caspase 3 activity.
6. Decreased circulating sCD40L and increased production of IL-10.
7. Increased levels of miRNAs such as miR-222 and MiR-17–3p lead to inhibition of transcriptional and translation responses deleterious to cardiomyocyte function.
8. Genetic modification leads to de novo synthesis of effector proteins during second window of protection.
Alteration in cardiac metabolism during ischemia.
Under normal physiological conditions, fatty acid oxidation is essential for adult cardiomyocytes energy production. During an ischemic event, the decreased oxygen concentration inhibits oxidative phosphorylation in the mitochondria. To sustain ATP, cellular metabolism switches to utilizing glucose through anaerobic glycolysis, resulting in lactate formation and acidosis. The cumulative effect of decreased pH and ATP results in increased intracellular sodium and calcium concentrations, which eventually triggers the pathway of ischemic injury and, if sustained, leads to cell death.91 Depending on the conditions, cardiomyocytes are metabolically flexible92 and other substitute substrates, such as glucose, lactate, amino acid, and ketone bodies, can be oxidized by cardiomyocytes for generating ATP. Similarly, during exercise, the lactate produced by skeletal muscle is also oxidized by cardiomyocytes and contribute to ATP synthesis.93,94 Endurance exercise training has been shown to decrease the rate of glycolysis in rat heart during ischemia. Although the exact mechanism is unknown, exercise training could potentially improve the efficiency of cells in producing ATP during ischemia.95
Another potential mechanism of exercise-induced metabolic alteration is through the interaction between neuregulin (NRG) and the epidermal growth factor receptor/receptor tyrosine kinase (erbB) family of receptors. The NRGs are growth factors that are responsible for the growth and development of mammalian tissue, including skeletal and cardiac muscles. The NRG is believed to be involved in the regulation of glucose uptake in muscle cells, especially during the recruitment of glucose transporters to the cell surface and modulation of transporter expression.96 Thus, NRG may participate in improving the availability and the efficiency of glucose utilization during periods of prolonged ischemia. Jie et al demonstrated in an in vitro study that NRG may play an additional role in determining cell fate: Specifically, NRG-1 was associated with activation of the phosphatidylinositol 3-kinase (PI3K) pathway during oxidative stress in cardiac tissue, preventing the mitochondrial permeability transition pore (MPTP) from opening and limiting myocardial damage during the ischemic event.56 Additional studies have supported the role of NRG-1 activation of PI3K in cardiac protection and documented an elevation in plasma levels of NRG-1 in response to exercise.97,98 Furthermore, progressive resistance training has been shown to increase the expression of erbB2 receptors,99 and the presence of NRG β−1 in serum of humans has been established as a marker for cardiovascular fitness.100 Furthermore, progressive resistance training has been shown to increase the expression of erbB2 receptors,99 and the presence of NRG β −1 in human serum has been established as a marker for cardiovascular fitness.100
Intracellular calcium homeostasis.
Ischemia–reperfusion injury results in intracellular calcium overload which is responsible for a number of deleterious effects in cardiomyocytes, mainly mediated via the activation of calpain. Upregulation of this protease leads to degradation of structural proteins and activation of Bid, which causes mitochondrial dysfunction and proteolysis of sarco/endoplasmic reticulum calcium ATPase (SERCA) and ryanodine receptors, culminating in calcium dysregulation and hypercontracture of the cardiomyocytes.101 Exercised hearts have been shown to have better calcium handling through improved diastolic pressure during IR, decreased activation of calpain, and preserved SERCA when compared to nonexercised hearts.102–104
Redox homeostasis
Reactive oxygen species.
Another major player in IR injury is the generation of cytotoxic ROS due to depolarizations in mitochondrial membrane potential in cardiomyocytes at reperfusion105 and calcium-medicated damage in ischemia.106 The adverse consequences of ROS production include disruption of cell membranes via lipid peroxidation, causing damage and dysfunction of receptors and ionic channels. For example, ROS damage the sarcolemma, leading to an influx of calcium into the cytosol via sodium-calcium exchange and, as a result, intracellular calcium overload. The ROS also attracts neutrophils, contributing to further cell damage via increased inflammation. Reactive oxygen species, together with normalization of pH upon reperfusion, also contribute to the opening of the MPTP, which plays a role in triggering apoptosis via release of cytochrome C.107 As mentioned previously, opening of MPTP triggers apoptosis by disruption of the mitochondrial membrane potential, matrix swelling, rupture of mitochondrial membranes, and release of cytochrome C in the cytoplasm.22 Acidosis during ischemia maintains the MPTP in a closed state; however, upon reperfusion, lactate levels decrease and the pH rapidly normalizes, opening the MPTP. Exercise has been shown to delay the rapid normalization of the pH,108,109 therefore preventing MPTP opening and providing cardioprotection. Furthermore, exercise has been shown to sustain postischemic mitochrondrial bioenergetics and redox homeostasis, which is associated with preserved mitochrondrial membrane potential and protection against ventricular arrhythmias.69
Controlling ROS production and/or increasing the cellular concentration of antioxidant enzymes (including the superoxide dismutases [SODs], catalase, glutathione peroxidase, and glutathione reductase) have been long-standing, proposed strategies to decrease IR injury.110 However, the proposed strategies as of yet have not been proven to be “effective” in attenuating lethal IR injury and infarct size. Nonetheless, exercise has been shown increase the expression of endogenous antioxidants such as SOD1 and SOD2 in the mitochondria111,112 possibly via activation of the transcription factor nuclear factor erythroid-2 (Nrf2).113 In addition, exercise training has also been suggested to increase the activity of glutathione reductase in the heart via posttranslational modifications.114,115 Further studies have shown that exercise training decreases the production of ROS during IR from both subsarcolemmal and intermyofibrillar mitochondria and prevents the release of proapoptotic proteins from the cardiac mitochondria, hence providing cardioprotection.112 Finally, mitochondrial monoamine oxidase A (MAO-A) catalyzes the oxidative deamination of several monoamines, thus increasing ROS production.116 Exercise has been shown to decrease the expression of MAO-A in rat hearts.117 In summary, exercise training may provide cardioprotection by increasing antioxidant expression and attenuating ROS-mediated cell damage.
Nitric oxide.
In the initial minutes to hours following reperfusion, the endothelial cells become permeable, leading to edema, the expression of adhesion proteins, release of cytokines, and decrease in the production of nitric oxide (NO).118–120 Improvements in NO levels due to exercise can improve coronary hyperemia at the onset of reperfusion.119 However, the effect of NO-mediated reduction in reperfusion hyperemia is reduced with age.121 Nitric oxide as a mediator of exercise-induced cardioprotection also plays a role in upregulation of endothelial nitric oxide synthase (eNOS) protein and phosphorylation, inhibition of caspase 3 activity, and modification of complex I of the mitochondrial electron transport chain, resulting in decreased ROS production during IR.122
ATP-sensitive potassium channels.
Opening of ATP-sensitive potassium channels (KATP), present on both the sarcolemma (most likely in the intercalated disc in the cardiomyocytes) and the inner mitochondrial membranes, has been implicated to play a role in ischemic preconditioning cardioprotection. The sacrolemmal KATP channel is composed of 4 Kir6.x and 4 SURx subunits.123 However, there is a debate on mitochrondrial KATP channel.124 The proposed subunits of the mitochrondrial KATP channel are Kir1.1 (ROMK), poreforming subunits, and additional subunits for ATP inhibition of the channel complex. Unique short-form SUR2 splice variants have been proposed as a regulatory subunit of the mitochondrial KATP channel.123 Research has futher supported the role of these subunits in cardiac function, with Kir6.1, Kir6.2, and SUR2A found to be present in isolated ventricular myocytes.125 Cardiac KATP channels are favorably regulated by glycolytically derived ATP. Competition for local glycolytically derived ATP may also be responsible for the known functional interaction between the KATP channel and the Na+/K+ pump in the cardiac myocyte.123 This interaction has been suggested to be important in the protection provided by ischemic preconditioning. Transcription of KATP channel subunit mRNA, Kir6.1, SUR1, and SUR2A mRNA was upregulated in LAD rat hearts. These KATP channels open when ATP is depleted or in the setting of increased concentration of other endogenous factors such as NO, pH, fatty acids, adenosine, and acetylcholine.126,127 Possible mechanism by which sarcolemmal KATP offers cardioprotection is by shortening the cardiac action potential duration via acceleration of phase 3 repolarization that limits calcium entry into the cell via L-type calcium channels, preventing calcium overload within the cell.128 Sarcolemmal KATP can also trigger opening of mitochondrial KATP,129 which have been shown to attenuate IR-induced ventricular arrhythmias.130 Of potential interest, endurance exercise training has been shown to increase the expression of sarcolemmal KATP in cardiac myocytes.131–134 However, their opening appears to be more beneficial during ischemia not reperfusion.134
Inflammation.
During reperfusion, ROS, a neutrophil chemoattractant, has been implicated to contribute to further myocardial damage through increased inflammation. Moreover, the endothelial cells in the reperfused myocardium begin to express adhesion proteins, release cytokines, and reduce NO production, which promotes adhesion, accumulation, and activation of neutrophils. Thus, inflammation may exacerbate cardiomyocyte damage during IR, and exercise has been shown to play a role in decreasing inflammation135 and providing cardioprotection. However, inflammation is a complex and controversial topic in cardioprotection, and further studies are needed to validate its role.
sCD40L
Clinical studies consistently show that the interaction between damaged endothelium, platelets, and immune cells is responsible for the development and progression of atherosclerosis.136 Platelet activation leads to crucial integrin-mediated signaling cascades, which result in stable interactions between platelets and the endothelium as well as activation of glycoprotein IIb/IIIa receptors (primary aggregation receptors), release of α and dense granules, and circulating cytokines that activate the immune system and upregulate receptor expression on endothelial cells.136 CD40 receptors and their ligand CD40L are one such system that is rapidly inducible upon platelet activation and has been reported to play a role in atherosclerosis as well as in ischemic preconditioning.136 CD40 and CD40L are stored in the cytoplasm of platelets and are rapidly expressed on the platelet surface upon activation.137 CD40L is then cleaved into a biologically active circulating form, soluble CD40L (sCD40L), which binds to CD40 on endothelial cells. The binding of sCD40L evokes a subsequent increase in proinflammatory cytokine production and adhesion molecule expression on the endothelial cell surface such as E-selectin, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1.137 Ischemic preconditioning has been shown to prevent the increase in levels of circulating sCD40L during angioplasty, suggesting that preconditioning induces anti-inflammatory effects that may, potentially, contribute to cardioprotection.138 The association between exercise and reduced CD40 has been previously demonstrated. In mice, both moderate aerobic exercise and resistance training have been shown to decrease CD40 levels.139,140 While in humans, high-intensity exercise has been shown to decrease CD40L levels, decrease triglycerides, and increase high-density lipoproteins,141 whereas ultra-endurance exercise in athletes reduced sCD40L levels.142 In addition, exercise training has been shown to significantly reduce circulating sCD40L levels in patients with chronic heart failure.143 Therefore, exercise can contribute to cardioprotection by decreasing inflammatory mediators such as CD40 and reducing atherosclerosis, which is a major risk factor for cardiovascular disease.
Interleukins.
Interleukins (IL) are a subset of cytokines secreted by white blood cells that participate in the immune response. The IL-6 and IL-10 have been shown to increase with exercise and may have a role in cardioprotection. For example, evidence obtained in exercised mice has revealed an upregulation in serum levels of IL-6 and IL-6 receptors, which, in turn, are associated with an attenuation of IR-induced necrosis and arrhythmias. These changes in IL-6 were accompanied by increased expression of the phosphorylated forms of p44/42 MAPK (Thr202/Tyr204) and p38 MAPK (Thr180/Tyr182), processes implicated as possible mechanisms for cardioprotection.144 The IL-10 is an inhibitor of cytokine synthesis, and exogenous administration of this IL has been shown to reduce myocardial infarct size and inhibit neutrophil adhesion to vascular endothelium.145 Further studies have shown that exercise increases serum IL-10 levels and could prevent LV remodeling by inhibiting inflammatory cytokines.146,147
MicroRNAs.
An emerging trend in the debate of exercise-induced and ischemia-induced preconditioning is the study of microRNAs (miRNAs) and their role in the various pathways involved with cardioprotection. The miRNAs are small noncoding single-stranded RNA molecules that bind to 3’ untranslated region of their mRNA targets and downregulate gene expression via degradation or translational inhibition.148 Recent studies have suggested that families of miRNAs regulate signaling pathways involved in IR injury and may be a potential therapeutic target to reduce infarct size. Accordingly, identifying these miRNAs, their mechanism of action, and the phase of the IR injury cascade in which they are activated may provide novel insights. If miRNAs do contribute to cardioprotection, 1 potential strategy would be to create oligonucleotides with sequences similar to the identified miRNAs and use them to enhance prosurvival pathways in the setting of IR.149
To date, increased levels of miRNA-21, miRNA-92a, miRNA-126, miRNA-133, and miRNA-144 and decreased levels of miRNA-1, miRNA-29, miRNA-199a, and miRNA-320 along with others have been shown to be associated with cardioprotection.150 In addition, 4 microRNAs, miRNA-487b, miR-139–5p, miR-192, and miR-212, are reportedly modified by preconditioning.149 Exercise has been shown to increase the levels of miR-222, a miRNA necessary for cardiac growth and protection against pathological hypertrophy.66,151 Exercise also increases the levels of miR-21, miR-486, miR-378, and miR-940.152–155 This involvement of miRNAs and the documented ability for exercise to exert effects at an epigenetic level may provide clues that cardioprotection is regulated by more complex mechanisms than what could be explained by a simple humoral factor.
Transcription of de novo proteins.
The early phase of cardioprotection after exercise is mainly believed to be due to immediate release of activated transmitters. The mechanism of the second window of protection, which comes into effect after 24 hours, is relatively more complex and involves adjustment at the genetic level and de novo synthesis of effector proteins.156 Ischemic preconditioning is believed to produce trigger factors such as adenosine, opioids, and bradykinin. These factors activate intracellular signaling pathways that activate the transcription factors in the nucleus. Over the next 12 to 24 hours, the production of proteins (distal mediators) such as mitochondrial antioxidant manganese SOD, inducible NOS, cyclooxygenase 2, and heat shock protein ultimately activate the cardioprotective pathways. The protective pathways are usually activated within 24 to 48 hours following an ischemic insult and reperfusion injury.157 In addition, exercise preconditioning also stimulates the production of cytokines that activates the JAK/STAT pathway and results in the transcription of distal mediators.157
Future Direction and Clinical Implications
Despite the extensive history of research into exercise and cardioprotection, many questions remain unanswered. The possible ligands and cellular mechanisms underlying the benefits from exercise are still unclear, and further research is needed to understand what endogenous factors contribute to exercise-induced cardioprotection. Likewise, the duration and intensity of exercise that are required to induce cardioprotection, and the duration of such effects following physical activity, remain uncertain. Although long-term exercise training and single bouts have both displayed cardioprotective benefits, each may do so through different mechanisms and provide variable duration of protection—concepts that warrant future targeted investigation.
Pairing in vivo analysis with data obtained in the Langendorff isolated buffer-perfused heart and in cell culture models may contribute to the understanding of the mechanistic interactions contributing to exercise-induced cardioprotection. Translational studies with humans in moderate-intensity exercise regimens may also provide important mechanistic insights. Elucidation of these mechanisms may yield promising therapeutic targets for cardioprotection.
Acknowledgments
We thank Dr Karin Przyklenk of Cardiovascular Research Institute, Wayne State University School of Medicine, for sharing her valuable insight, knowledge, and suggestions. We also think Ms Hailan Hu for her excellent artistic work.
Funding
The author(s) received following financial support for the research, authorship, and/or publication of this article: This work was supported by University of Toledo, College of Medicine and Life Sciences, Dean’s fund.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Morris JN, Crawford MD. Coronary heart disease and physical activity of work: evidence of a national necropsy survey. Br Med J. 1958;2(5111):1485–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blair SN, Kohl HW 3rd, Paffenbarger RS Jr, Clark DG, Cooper KH, Gibbons LW. Physical fitness and all-cause mortality. A prospective study of healthy men and women. JAMA. 1989; 262(17):2395–2401. [DOI] [PubMed] [Google Scholar]
- 3.Paffenbarger RS Jr, Hyde RT, Wing AL, Hsieh CC. Physical activity, all-cause mortality, and longevity of college alumni. N Engl J Med. 1986;314(10):605–613. [DOI] [PubMed] [Google Scholar]
- 4.Paffenbarger RS Jr, Hyde RT, Wing AL, Lee IM, Jung DL, Kampert JB. The association of changes in physical-activity level and other lifestyle characteristics with mortality among men. N Engl J Med. 1993;328(8):538–545. [DOI] [PubMed] [Google Scholar]
- 5.Paffenbarger RS Jr, Wing AL, Hyde RT. Physical activity as an index of heart attack risk in college alumni. 1978. Am J Epidemiol. 1995;142(9):889–903; discussion 887–888. [DOI] [PubMed] [Google Scholar]
- 6.Sandvik L, Erikssen J, Thaulow E, Erikssen G, Mundal R, Rodahl K. Physical fitness as a predictor of mortality among healthy, middle-aged Norwegian men. N Engl J Med. 1993;328(8): 533–537. [DOI] [PubMed] [Google Scholar]
- 7.Berlin JA, Colditz GA. A meta-analysis of physical activity in the prevention of coronary heart disease. Am J Epidemiol. 1990; 132(4):612–628. [DOI] [PubMed] [Google Scholar]
- 8.Ekelund LG, Haskell WL, Johnson JL, Whaley FS, Criqui MH, Sheps DS. Physical fitness as a predictor of cardiovascular mortality in asymptomatic North American men. The Lipid Research Clinics Mortality Follow-up Study. N Engl J Med. 1988;319(21): 1379–1384. [DOI] [PubMed] [Google Scholar]
- 9.Leon AS, Myers MJ, Connett J. Leisure time physical activity and the 16-year risks of mortality from coronary heart disease and all-causes in the Multiple Risk Factor Intervention Trial (MRFIT). Int J Sports Med. 1997;18(suppl 3):S208–S215. [DOI] [PubMed] [Google Scholar]
- 10.Shortreed SM, Peeters A, Forbes AB. Estimating the effect of long-term physical activity on cardiovascular disease and mortality: evidence from the Framingham Heart Study. Heart. 2013; 99(9):649–654. [DOI] [PubMed] [Google Scholar]
- 11.Powers SK, Demirel HA, Vincent HK, et al. Exercise training improves myocardial tolerance to in vivo ischemia-reperfusion in the rat. Am J Physiol. 1998;275(5 pt 2):R1468–R1477. [DOI] [PubMed] [Google Scholar]
- 12.Brown DA, Jew KN, Sparagna GC, Musch TI, Moore RL. Exercise training preserves coronary flow and reduces infarct size after ischemia–reperfusion in rat heart. J Appl Physiol (1985). 2003; 95(6):2510–2518. [DOI] [PubMed] [Google Scholar]
- 13.Demirel HA, Powers SK, Zergeroglu MA, et al. Short-term exercise improves myocardial tolerance to in vivo ischemia–reperfusion in the rat. J Appl Physiol (1985). 2001;91(5):2205–2212. [DOI] [PubMed] [Google Scholar]
- 14.Shephard RJ, Balady GJ. Exercise as cardiovascular therapy. Circulation. 1999;99(7):963–972. [DOI] [PubMed] [Google Scholar]
- 15.Ellison GM, Waring CD, Vicinanza C, Torella D. Physiological cardiac remodelling in response to endurance exercise training: cellular and molecular mechanisms. Heart. 2012;98(1):5–10. [DOI] [PubMed] [Google Scholar]
- 16.Billman GE. Cardiac autonomic neural remodeling and susceptibility to sudden cardiac death: effect of endurance exercise training. Am J Physiol Heart Circ Physiol. 2009;297(4): H1171–H1193. [DOI] [PubMed] [Google Scholar]
- 17.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74(5):1124–1136. [DOI] [PubMed] [Google Scholar]
- 18.Tomai F, Perino M, Ghini AS, et al. Exercise-induced myocardial ischemia triggers the early phase of preconditioning but not the late phase. Am J Cardiol. 1999;83(4):586–588, A587-A588. [DOI] [PubMed] [Google Scholar]
- 19.Lambiase PD, Edwards RJ, Cusack MR, Bucknall CA, Redwood SR, Marber MS. Exercise-induced ischemia initiates the second window of protection in humans independent of collateral recruitment. J Am Coll Cardiol. 2003;41(7):1174–1182. [DOI] [PubMed] [Google Scholar]
- 20.Domenech R, Macho P, Schwarze H, Sanchez G. Exercise induces early and late myocardial preconditioning in dogs. Cardiovasc Res. 2002;55(3):561–566. [DOI] [PubMed] [Google Scholar]
- 21.Yamashita N, Baxter GF, Yellon DM. Exercise directly enhances myocardial tolerance to ischaemia–reperfusion injury in the rat through a protein kinase C mediated mechanism. Heart. 2001; 85(3):331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marongiu E, Crisafulli A. Cardioprotection acquired through exercise: the role of ischemic preconditioning. Curr Cardiol Rev. 2014;10(4):336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lennon SL, Quindry J, Hamilton KL, et al. Loss of exercise-induced cardioprotection after cessation of exercise. J Appl Physiol (1985). 2004;96(4):1299–1305. [DOI] [PubMed] [Google Scholar]
- 24.Maeda S, Miyauchi T, Kakiyama T, et al. Effects of exercise training of 8 weeks and detraining on plasma levels of endothelium-derived factors, endothelin-1 and nitric oxide, in healthy young humans. Life Sci. 2001;69(9):1005–1016. [DOI] [PubMed] [Google Scholar]
- 25.Esposito F, Ronchi R, Milano G, et al. Myocardial tolerance to ischemia–reperfusion injury, training intensity and cessation. Eur J Appl Physiol. 2011;111(5):859–868. [DOI] [PubMed] [Google Scholar]
- 26.French JP, Hamilton KL, Quindry JC, Lee Y, Upchurch PA, Powers SK. Exercise-induced protection against myocardial apoptosis and necrosis: MnSOD, calcium-handling proteins, and calpain. FASEB J. 2008;22(8):2862–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamashita N, Hoshida S, Otsu K, Asahi M, Kuzuya T, Hori M. Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J Exp Med. 1999;189(11): 1699–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alleman RJ, Stewart LM, Tsang AM, Brown DA. Why does exercise “trigger” adaptive protective responses in the heart? Dose Response. 2015;13(1): dose-response.14–023.Alleman. doi: 10.2203/dose-response.14-023.Alleman. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frasier CR, Moore RL, Brown DA. Exercise-induced cardiac preconditioning: how exercise protects your achy-breaky heart. J Appl Physiol (1985). 2011;111(3):905–915. [DOI] [PubMed] [Google Scholar]
- 30.Taylor RP, Harris MB, Starnes JW. Acute exercise can improve cardioprotection without increasing heat shock protein content. Am J Physiol. 1999;276(3 pt 2):H1098–H1102. [DOI] [PubMed] [Google Scholar]
- 31.Lee IM, Sesso HD, Paffenbarger RS Jr. Physical activity and coronary heart disease risk in men: does the duration of exercise episodes predict risk? Circulation. 2000;102(9):981–986. [DOI] [PubMed] [Google Scholar]
- 32.Pate RR, Pratt M, Blair SN, et al. Physical activity and public health. A recommendation from the Centers for Disease Control and Prevention and the American College of Sports Medicine. JAMA. 1995;273(5):402–407. [DOI] [PubMed] [Google Scholar]
- 33.Thompson PD, Buchner D, Pina IL, et al. Exercise and physical activity in the prevention and treatment of atherosclerotic cardiovascular disease: a statement from the Council on Clinical Cardiology (Subcommittee on Exercise, Rehabilitation, and Prevention) and the Council on Nutrition, Physical Activity, and Metabolism (Subcommittee on Physical Activity). Circulation. 2003;107(24):3109–3116. [DOI] [PubMed] [Google Scholar]
- 34.Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation. 1993;87(3):893–899. [DOI] [PubMed] [Google Scholar]
- 35.Liem DA, te Lintel Hekkert M, Manintveld OC, Boomsma F, Verdouw PD, Duncker DJ. Myocardium tolerant to an adenosine-dependent ischemic preconditioning stimulus can still be protected by stimuli that employ alternative signaling pathways. Am J Physiol Heart Circ Physiol. 2005;288(3): H1165–H1172. [DOI] [PubMed] [Google Scholar]
- 36.Hausenloy DJ, Mwamure PK, Venugopal V, et al. Effect of remote ischaemic preconditioning on myocardial injury in patients undergoing coronary artery bypass graft surgery: a randomised controlled trial. Lancet. 2007;370(9587):575–579. [DOI] [PubMed] [Google Scholar]
- 37.Thielmann M, Kottenberg E, Kleinbongard P, et al. Cardioprotective and prognostic effects of remote ischaemic preconditioning in patients undergoing coronary artery bypass surgery: a single-centre randomised, double-blind, controlled trial. Lancet. 2013;382(9892):597–604. [DOI] [PubMed] [Google Scholar]
- 38.Meybohm P, Bein B, Brosteanu O, et al. A multicenter trial of remote ischemic preconditioning for heart surgery. N Engl J Med. 2015;373(15):1397–1407. [DOI] [PubMed] [Google Scholar]
- 39.Benstoem C, Stoppe C, Liakopoulos OJ, et al. Remote ischaemic preconditioning for coronary artery bypass grafting (with or without valve surgery). Cochrane Database Syst Rev. 2017;(5): CD011719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shimizu M, Tropak M, Diaz RJ, et al. Transient limb ischaemia remotely preconditions through a humoral mechanism acting directly on the myocardium: evidence suggesting cross-species protection. Clin Sci (Lond). 2009;117(5):191–200. [DOI] [PubMed] [Google Scholar]
- 41.Lim SY, Yellon DM, Hausenloy DJ. The neural and humoral pathways in remote limb ischemic preconditioning. Basic Res Cardiol. 2010;105(5):651–655. [DOI] [PubMed] [Google Scholar]
- 42.Pickard JM, Davidson SM, Hausenloy DJ, Yellon DM. Codependence of the neural and humoral pathways in the mechanism of remote ischemic conditioning. Basic Res Cardiol. 2016; 111(4):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konstantinov IE, Li J, Cheung MM, et al. Remote ischemic preconditioning of the recipient reduces myocardial ischemia–reperfusion injury of the denervated donor heart via a KATP channel-dependent mechanism. Transplantation. 2005;79(12):1691–1695. [DOI] [PubMed] [Google Scholar]
- 44.Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88(3):1009–1086. [DOI] [PubMed] [Google Scholar]
- 45.Spurway NC. Aerobic exercise, anaerobic exercise and the lactate threshold. Br Med Bull. 1992;48(3):569–591. [DOI] [PubMed] [Google Scholar]
- 46.Ament W, Verkerke GJ. Exercise and fatigue. Sports Med. 2009; 39(5):389–422. [DOI] [PubMed] [Google Scholar]
- 47.Kaiser RA, Liang Q, Bueno O, et al. Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia– reperfusion-induced cell death in vivo. J Biol Chem. 2005; 280(38):32602–32608. [DOI] [PubMed] [Google Scholar]
- 48.Sahlin K Metabolic factors in fatigue. Sports Med. 1992;13(2): 99–107. [DOI] [PubMed] [Google Scholar]
- 49.Breivik L, Helgeland E, Aarnes EK, Mrdalj J, Jonassen AK. Remote postconditioning by humoral factors in effluent from ischemic preconditioned rat hearts is mediated via PI3K/Akt-dependent cell-survival signaling at reperfusion. Basic Res Cardiol. 2011;106(1):135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Michelsen MM, Stottrup NB, Schmidt MR, et al. Exercise-induced cardioprotection is mediated by a bloodborne, transferable factor. Basic Res Cardiol. 2012;107(3):260. [DOI] [PubMed] [Google Scholar]
- 51.Liao R, Podesser BK, Lim CC. The continuing evolution of the Langendorff and ejecting murine heart: new advances in cardiac phenotyping. Am J Physiol Heart Circ Physiol. 2012;303(2): H156–H167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olejnickova V, Novakova M, Provaznik I. Isolated heart models: cardiovascular system studies and technological advances. Med Biol Eng Comput. 2015;53(7):669–678. [DOI] [PubMed] [Google Scholar]
- 53.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97(24):2463–2469. [DOI] [PubMed] [Google Scholar]
- 54.Budas GR, Jovanovic S, Crawford RM, Jovanovic A. Hypoxia-induced preconditioning in adult stimulated cardiomyocytes is mediated by the opening and trafficking of sarcolemmal KATP channels. FASEB J. 2004;18(9):1046–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith RM, Suleman N, Lacerda L, et al. Genetic depletion of cardiac myocyte STAT-3 abolishes classical preconditioning. Cardiovasc Res. 2004;63(4):611–616. [DOI] [PubMed] [Google Scholar]
- 56.Jie B, Zhang X, Wu X, Xin Y, Liu Y, Guo Y. Neuregulin-1 suppresses cardiomyocyte apoptosis by activating PI3K/Akt and inhibiting mitochondrial permeability transition pore. Mol Cell Biochem. 2012;370(1–2):35–43. [DOI] [PubMed] [Google Scholar]
- 57.Baker JE, Su J, Koprowski S, Dhanasekaran A, Aufderheide TP, Gross GJ. Thrombopoietin receptor agonists protect human cardiac myocytes from injury by activation of cell survival pathways. J Pharmacol Exp Ther. 2015;352(3):429–437. [DOI] [PubMed] [Google Scholar]
- 58.Fang J, Fan L, Chen L, Chen X, Wu L. Coronary effluent from postconditioned hearts promotes survival of mesenchymal stem cells under hypoxia. Scand Cardiovasc J. 2014;48(2):120–127. [DOI] [PubMed] [Google Scholar]
- 59.Ladilov YV, Siegmund B, Piper HM. Protection of reoxygenated cardiomyocytes against hypercontracture by inhibition of Na+/H+ exchange. Am J Physiol. 1995;268(4 Pt 2): H1531–H1539. [DOI] [PubMed] [Google Scholar]
- 60.Skrzypiec-Spring M, Grotthus B, Szelag A, Schulz R. Isolated heart perfusion according to Langendorff – still viable in the new millennium. J Pharmacol Toxicol Methods. 2007;55(2):113–126. [DOI] [PubMed] [Google Scholar]
- 61.Bell RM, Mocanu MM, Yellon DM. Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J Mol Cell Cardiol. 2011;50(6):940–950. [DOI] [PubMed] [Google Scholar]
- 62.Hildebrandt H, Kahlert P, Heusch G, Kleinbongard P. Kinetics of cardioprotection by plasma dialysate from healthy volunteers undergoing remote ischemic preconditioning. Eur Heart J. 2015;36:580–580.25336228 [Google Scholar]
- 63.Nag AC, Cheng M, Fischman DA, Zak R. Long-term cell culture of adult mammalian cardiac myocytes: electron microscopic and immunofluorescent analyses of myofibrillar structure. J Mol Cell Cardiol. 1983;15(5):301–317. [DOI] [PubMed] [Google Scholar]
- 64.Springhorn JP, Ellingsen O, Berger HJ, Kelly RA, Smith TW. Transcriptional regulation in cardiac muscle. Coordinate expression of Id with a neonatal phenotype during development and following a hypertrophic stimulus in adult rat ventricular myocytes in vitro. J Biol Chem. 1992;267(20):14360–14365. [PubMed] [Google Scholar]
- 65.Autelitano DJ, Woodcock EA. Selective activation of alpha1A-adrenergic receptors in neonatal cardiac myocytes is sufficient to cause hypertrophy and differential regulation of alpha1-adrenergic receptor subtype mRNAs. J Mol Cell Cardiol. 1998; 30(8):1515–1523. [DOI] [PubMed] [Google Scholar]
- 66.Shi J, Bei Y, Kong X, et al. miR-17–3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia–reperfusion injury. Theranostics. 2017;7(3):664–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Farah C, Nascimento A, Bolea G, et al. Key role of endothelium in the eNOS-dependent cardioprotection with exercise training. J Mol Cell Cardiol. 2017;102:26–30. [DOI] [PubMed] [Google Scholar]
- 68.Bei Y, Xu T, Lv D, et al. Exercise-induced circulating extracellular vesicles protect against cardiac ischemia–reperfusion injury. Basic Res Cardiol. 2017;112(4):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alleman RJ, Tsang AM, Ryan TE, et al. Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics. Am J Physiol Heart Circ Physiol. 2016;310(10):H1360–H1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Feger BJ, Starnes JW. Exercise alters the regulation of myocardial Na(+)/H(+) exchanger-1 activity. Am J Physiol Regul Integr Comp Physiol. 2013;305(10):R1182–R1189. [DOI] [PubMed] [Google Scholar]
- 71.Zhao YY, Sawyer DR, Baliga RR, et al. Neuregulins promote survival and growth of cardiac myocytes. Persistence of ErbB2 and ErbB4 expression in neonatal and adult ventricular myocytes. J Biol Chem. 1998;273(17):10261–10269. [DOI] [PubMed] [Google Scholar]
- 72.Nesher M, Bai Y, Li D, Rosen H, Lichtstein D, Liu L. Interaction of atrial natriuretic peptide and ouabain in the myocardium. Can J Physiol Pharmacol. 2012;90(10):1386–1393. [DOI] [PubMed] [Google Scholar]
- 73.Obal D, Dai S, Keith R, et al. Cardiomyocyte-restricted overexpression of extracellular superoxide dismutase increases nitric oxide bioavailability and reduces infarct size after ischemia/reperfusion. Basic Res Cardiol. 2012;107(6):305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu X, Dai S, Wu WJ, et al. Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation. 2007;116(6):654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuznetsov AV, Javadov S, Sickinger S, Frotschnig S, Grimm M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxiareoxygenation. Biochim Biophys Acta. 2015;1853(2):276–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hwang HS, Kryshtal DO, Feaster TK, et al. Comparable calcium handling of human iPSC-derived cardiomyocytes generated by multiple laboratories. J Mol Cell Cardiol. 2015;85:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Casini S, Verkerk AO, Remme CA. Human iPSC-derived cardiomyocytes for investigation of disease mechanisms and therapeutic strategies in inherited arrhythmia syndromes: strengths and limitations. Cardiovasc Drugs Ther. 2017;31(3):325–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hidalgo A, Glass N, Ovchinnikov D, et al. Modelling ischemia– reperfusion injury (IRI) in vitro using metabolically matured induced pluripotent stem cell-derived cardiomyocytes. APL Bioengineering. 2018;2(2):026102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Samaja M, Milano G. Editorial – hypoxia and reoxygenation: from basic science to bedside. Front Pediatr. 2015;3:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Piao H, Takahashi K, Yamaguchi Y, Wang C, Liu K, Naruse K. Transient receptor potential melastatin-4 is involved in hypoxiareoxygenation injury in the cardiomyocytes. PLoS One. 2015; 10(4):e0121703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Du JK, Cong BH, Yu Q, et al. Upregulation of microRNA-22 contributes to myocardial ischemia–reperfusion injury by interfering with the mitochondrial function. Free Radic Biol Med. 2016;96:406–417. [DOI] [PubMed] [Google Scholar]
- 82.Panera N, Gnani D, Piermarini E, et al. High concentrations of H2O2 trigger hypertrophic cascade and phosphatase and tensin homologue (PTEN) glutathionylation in H9c2 cardiomyocytes. Exp Mol Pathol. 2016;100(1):199–206. [DOI] [PubMed] [Google Scholar]
- 83.Matsui T, Li L, del Monte F, et al. Adenoviral gene transfer of activated phosphatidylinositol 3’-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999; 100(23):2373–2379. [DOI] [PubMed] [Google Scholar]
- 84.Wu L, Tan JL, Wang ZH, et al. ROS generated during early reperfusion contribute to intermittent hypobaric hypoxiaafforded cardioprotection against postischemia-induced Ca(2+) overload and contractile dysfunction via the JAK2/STAT3 pathway. J Mol Cell Cardiol. 2015;81:150–161. [DOI] [PubMed] [Google Scholar]
- 85.Baines CP. How and when do myocytes die during ischemia and reperfusion: the late phase. J Cardiovasc Pharmacol Ther. 2011; 16(3–4):239–243. [DOI] [PubMed] [Google Scholar]
- 86.de Waard MC, van der Velden J, Bito V, et al. Early exercise training normalizes myofilament function and attenuates left ventricular pump dysfunction in mice with a large myocardial infarction. Circ Res. 2007;100(7):1079–1088. [DOI] [PubMed] [Google Scholar]
- 87.Bito V, de Waard MC, Biesmans L, et al. Early exercise training after myocardial infarction prevents contractile but not electrical remodelling or hypertrophy. Cardiovasc Res. 2010;86(1):72–81. [DOI] [PubMed] [Google Scholar]
- 88.Wu G, Rana JS, Wykrzykowska J, et al. Exercise-induced expression of VEGF and salvation of myocardium in the early stage of myocardial infarction. Am J Physiol Heart Circ Physiol. 2009; 296(2): H389–H395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Puhl SL, Muller A, Wagner M, et al. Exercise attenuates inflammation and limits scar thinning after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2015;309(2):H345–H359. [DOI] [PubMed] [Google Scholar]
- 90.Vujic A, Lerchenmuller C, Wu TD, et al. Exercise induces new cardiomyocyte generation in the adult mammalian heart. Nat Commun. 2018;9(1):1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wong R, Aponte AM, Steenbergen C, Murphy E. Cardioprotection leads to novel changes in the mitochondrial proteome. Am J Physiol Heart Circ Physiol. 2010;298(1):H75–H91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gaspar JA, Doss MX, Hengstler JG, Cadenas C, Hescheler J, Sachinidis A. Unique metabolic features of stem cells, cardiomyocytes, and their progenitors. Circ Res. 2014;114(8): 1346–1360. [DOI] [PubMed] [Google Scholar]
- 93.Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113(5):603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Goodwin GW, Taegtmeyer H. Improved energy homeostasis of the heart in the metabolic state of exercise. Am J Physiol Heart Circ Physiol. 2000;279(4):H1490–H1501. [DOI] [PubMed] [Google Scholar]
- 95.Burelle Y, Wambolt RB, Grist M, et al. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2004;287(3):H1055–H1063. [DOI] [PubMed] [Google Scholar]
- 96.Suarez E, Bach D, Cadefau J, Palacin M, Zorzano A, Guma A. A novel role of neuregulin in skeletal muscle. Neuregulin stimulates glucose uptake, glucose transporter translocation, and transporter expression in muscle cells. J Biol Chem. 2001;276(21): 18257–18264. [DOI] [PubMed] [Google Scholar]
- 97.Cai MX, Shi XC, Chen T, et al. Exercise training activates neuregulin 1/ErbB signaling and promotes cardiac repair in a rat myocardial infarction model. Life Sci. 2016;149:1–9. [DOI] [PubMed] [Google Scholar]
- 98.Fang SJ, Wu XS, Han ZH, et al. Neuregulin-1 preconditioning protects the heart against ischemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Chin Med J (Engl). 2010; 123(24):3597–3604. [PubMed] [Google Scholar]
- 99.LeBrasseur NK, Mizer KC, Parkington JD, Sawyer DB, Fielding RA. The expression of neuregulin and erbB receptors in human skeletal muscle: effects of progressive resistance training. Eur J Appl Physiol. 2005;94(4):371–375. [DOI] [PubMed] [Google Scholar]
- 100.Moondra V, Sarma S, Buxton T, et al. Serum neuregulin-1beta as a biomarker of cardiovascular fitness. Open Biomark J. 2009; 2:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Inserte J Calpains in the cardiovascular system. Cardiovasc Res. 2012;96(1):9–10. [DOI] [PubMed] [Google Scholar]
- 102.Bowles DK, Starnes JW. Exercise training improves metabolic response after ischemia in isolated working rat heart. J Appl Physiol (1985). 1994;76(4):1608–1614. [DOI] [PubMed] [Google Scholar]
- 103.French JP, Quindry JC, Falk DJ, et al. Ischemia–reperfusion-induced calpain activation and SERCA2a degradation are attenuated by exercise training and calpain inhibition. Am J Physiol Heart Circ Physiol. 2006;290(1):H128–H136. [DOI] [PubMed] [Google Scholar]
- 104.Sanchez G, Escobar M, Pedrozo Z, et al. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res. 2008;77(2): 380–386. [DOI] [PubMed] [Google Scholar]
- 105.Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010; 1797(6–7):865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Corbalan JJ, Kitsis RN. RCAN1-calcineurin axis and the setpoint for myocardial damage during ischemia–reperfusion. Circ Res. 2018;122(6):796–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hausenloy DJ, Yellon DM. Myocardial ischemia–reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013; 123(1):92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perrelli MG, Pagliaro P, Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World J Cardiol. 2011;3(6):186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Baines CP. The mitochondrial permeability transition pore and ischemia–reperfusion injury. Basic Res Cardiol. 2009;104(2): 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Braunersreuther V, Jaquet V. Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Curr Pharm Biotechnol. 2012;13(1):97–114. [DOI] [PubMed] [Google Scholar]
- 111.Powers SK, Quindry JC, Kavazis AN. Exercise-induced cardioprotection against myocardial ischemia–reperfusion injury. Free Radic Biol Med. 2008;44(2):193–201. [DOI] [PubMed] [Google Scholar]
- 112.Lee Y, Min K, Talbert EE, et al. Exercise protects cardiac mitochondria against ischemia–reperfusion injury. Med Sci Sports Exerc. 2012;44(3):397–405. [DOI] [PubMed] [Google Scholar]
- 113.Dinkova-Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52(suppl 1):S128–S138. [DOI] [PubMed] [Google Scholar]
- 114.Frasier CR, Moukdar F, Patel HD, et al. Redox-dependent increases in glutathione reductase and exercise preconditioning: role of NADPH oxidase and mitochondria. Cardiovasc Res. 2013;98(1):47–55. [DOI] [PubMed] [Google Scholar]
- 115.Frasier CR, Sloan RC, Bostian PA, et al. Short-term exercise preserves myocardial glutathione and decreases arrhythmias after thiol oxidation and ischemia in isolated rat hearts. J Appl Physiol (1985). 2011;111(6):1751–1759. [DOI] [PubMed] [Google Scholar]
- 116.Pchejetski D, Kunduzova O, Dayon A, et al. Oxidative stress-dependent sphingosine kinase-1 inhibition mediates monoamine oxidase A-associated cardiac cell apoptosis. Circ Res. 2007; 100(1):41–49. [DOI] [PubMed] [Google Scholar]
- 117.Kavazis AN, Alvarez S, Talbert E, Lee Y, Powers SK. Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am J Physiol Heart Circ Physiol. 2009;297(1): H144–H152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Piper HM, Meuter K, Schafer C. Cellular mechanisms of ischemia–reperfusion injury. Ann Thorac Surg. 2003;75(2): S644–S648. [DOI] [PubMed] [Google Scholar]
- 119.Loscalzo J, Vita JA. Ischemia, hyperemia, exercise, and nitric oxide. Complex physiology and complex molecular adaptations. Circulation. 1994;90(5):2556–2559. [DOI] [PubMed] [Google Scholar]
- 120.Joyner MJ, Wilkins BW. Exercise hyperaemia: is anything obligatory but the hyperaemia? J Physiol. 2007;583(pt 3):855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McDaniel J, Hayman MA, Ives S, et al. Attenuated exercise induced hyperaemia with age: mechanistic insight from passive limb movement. J Physiol. 2010;588(pt 22):4507–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Green DJ, Maiorana A, O’Driscoll G, Taylor R. Effect of exercise training on endothelium-derived nitric oxide function in humans. J Physiol. 2004;561(pt 1):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Foster MN, Coetzee WA. KATP channels in the cardiovascular system. Physiol Rev. 2016;96(1):177–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as a receptor for potassium channel openers. J Biol Chem. 1996;271(15):8796–8799. [DOI] [PubMed] [Google Scholar]
- 125.Singh H, Hudman D, Lawrence CL, Rainbow RD, Lodwick D, Norman RI. Distribution of Kir6.0 and SUR2 ATP-sensitive potassium channel subunits in isolated ventricular myocytes. J Mol Cell Cardiol. 2003;35(5):445–459. [DOI] [PubMed] [Google Scholar]
- 126.Hiraoka M, Furukawa T. Functional modulation of cardiac ATP-sensitive K(þ) channels. News Physiol Sci. 1998;13:131–137. [DOI] [PubMed] [Google Scholar]
- 127.Edwards G, Weston AH. KATP – fact or artefact? New thoughts on the mode of action of the potassium channel openers. Cardiovasc Res. 1994;28(6):735–737; discussion 741–735. [DOI] [PubMed] [Google Scholar]
- 128.Gross GJ, Peart JN. KATP channels and myocardial preconditioning: an update. Am J Physiol Heart Circ Physiol. 2003; 285(3):H921–H930. [DOI] [PubMed] [Google Scholar]
- 129.Patel HH, Gross ER, Peart JN, Hsu AK, Gross GJ. Sarcolemmal KATP channel triggers delayed ischemic preconditioning in rats. Am J Physiol Heart Circ Physiol. 2005;288(1):H445–H447. [DOI] [PubMed] [Google Scholar]
- 130.Quindry JC, Schreiber L, Hosick P, Wrieden J, Irwin JM, Hoyt E. Mitochondrial KATP channel inhibition blunts arrhythmia protection in ischemic exercised hearts. Am J Physiol Heart Circ Physiol. 2010;299(1):H175–H183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brown DA, Chicco AJ, Jew KN, et al. Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the KATP channel in the rat. J Physiol. 2005;569(pt 3):913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zingman LV, Zhu Z, Sierra A, et al. Exercise-induced expression of cardiac ATP-sensitive potassium channels promotes action potential shortening and energy conservation. J Mol Cell Cardiol. 2011;51(1):72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rottensteiner M, Leskinen T, Niskanen E, et al. Physical activity, fitness, glucose homeostasis, and brain morphology in twins. Med Sci Sports Exerc. 2015;47(3):509–518. [DOI] [PubMed] [Google Scholar]
- 134.Johnson MS, Moore RL, Brown DA. Sex differences in myocardial infarct size are abolished by sarcolemmal KATP channel blockade in rat. Am J Physiol Heart Circ Physiol. 2006; 290(6):H2644–H2647. [DOI] [PubMed] [Google Scholar]
- 135.Lakka TA, Lakka HM, Rankinen T, et al. Effect of exercise training on plasma levels of C-reactive protein in healthy adults: the HERITAGE Family Study. Eur Heart J. 2005;26(19): 2018–2025. [DOI] [PubMed] [Google Scholar]
- 136.Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357(24):2482–2494. [DOI] [PubMed] [Google Scholar]
- 137.Antoniades C, Bakogiannis C, Tousoulis D, Antonopoulos AS, Stefanadis C. The CD40/CD40 ligand system: linking inflammation with atherothrombosis. J Am Coll Cardiol. 2009;54(8): 669–677. [DOI] [PubMed] [Google Scholar]
- 138.Lee TM, Lin MS, Tsai CH, Chang NC. Effect of ischaemic preconditioning on regional release of inflammatory markers. Clin Sci (Lond). 2005;109(3):267–276. [DOI] [PubMed] [Google Scholar]
- 139.Cardinot TM, Lima TM, Moretti AI, et al. Preventive and therapeutic moderate aerobic exercise programs convert atherosclerotic plaques into a more stable phenotype. Life Sci. 2016; 153:163–170. [DOI] [PubMed] [Google Scholar]
- 140.Mardare C, Kruger K, Liebisch G, et al. Endurance and resistance training affect high fat diet-induced increase of ceramides, inflammasome expression, and systemic inflammation in mice. J Diabetes Res. 2016;2016:4536470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Santilli F, Vazzana N, Iodice P, et al. Effects of high-amount-high-intensity exercise on in vivo platelet activation: modulation by lipid peroxidation and AGE/RAGE axis. Thromb Haemost. 2013;110(6):1232–1240. [DOI] [PubMed] [Google Scholar]
- 142.Geertsema L, Lucas SJ, Cotter JD, Hock B, McKenzie J, Ferny-hough LJ. The cardiovascular risk factor, soluble CD40 ligand (CD154), but not soluble CD40 is lowered by ultra-endurance exercise in athletes. Br J Sports Med. 2011;45(1):42–45. [DOI] [PubMed] [Google Scholar]
- 143.Bjornstad HH, Bruvik J, Bjornstad AB, Hjellestad BL, Damas JK, Aukrust P. Exercise training decreases plasma levels of soluble CD40 ligand and P-selectin in patients with chronic heart failure. Eur J Cardiovasc Prev Rehabil. 2008;15(1):43–48. [DOI] [PubMed] [Google Scholar]
- 144.McGinnis GR, Ballmann C, Peters B, et al. Interleukin-6 mediates exercise preconditioning against myocardial ischemia reperfusion injury. Am J Physiol Heart Circ Physiol. 2015; 308(11):H1423–H1433. [DOI] [PubMed] [Google Scholar]
- 145.Hayward R, Nossuli TO, Scalia R, Lefer AM. Cardioprotective effect of interleukin-10 in murine myocardial ischemia–reperfusion. Eur J Pharmacol. 1997;334(2–3):157–163. [DOI] [PubMed] [Google Scholar]
- 146.Serra AJ, Santos MH, Bocalini DS, et al. Exercise training inhibits inflammatory cytokines and more than prevents myocardial dysfunction in rats with sustained beta-adrenergic hyperactivity. J Physiol. 2010;588(pt 13):2431–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kesherwani V, Chavali V, Hackfort BT, Tyagi SC, Mishra PK. Exercise ameliorates high fat diet induced cardiac dysfunction by increasing interleukin 10. Front Physiol. 2015;6:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ye Y, Perez-Polo JR, Qian J, Birnbaum Y. The role of micro-RNA in modulating myocardial ischemia–reperfusion injury. Physiol Genomics. 2011;43(10):534–542. [DOI] [PubMed] [Google Scholar]
- 149.Varga ZV, Zvara A, Farago N, et al. MicroRNAs associated with ischemia–reperfusion injury and cardioprotection by ischemic pre-and postconditioning: protectomiRs. Am J Physiol Heart Circ Physiol. 2014;307(2):H216–H227. [DOI] [PubMed] [Google Scholar]
- 150.Li J, Rohailla S, Gelber N, et al. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res Cardiol. 2014;109(5):423. [DOI] [PubMed] [Google Scholar]
- 151.Liu X, Xiao J, Zhu H, et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21(4):584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fernandes T, Barauna VG, Negrao CE, Phillips MI, Oliveira EM. Aerobic exercise training promotes physiological cardiac remodeling involving a set of microRNAs. Am J Physiol Heart Circ Physiol. 2015;309(4):H543–H552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xu T, Liu Q, Yao J, Dai Y, Wang H, Xiao J. Circulating microRNAs in response to exercise. Scand J Med Sci Sports. 2015; 25(2):e149–e154. [DOI] [PubMed] [Google Scholar]
- 154.Xu T, Zhou Q, Che L, et al. Circulating miR-21, miR-378, and miR-940 increase in response to an acute exhaustive exercise in chronic heart failure patients. Oncotarget. 2016;7(11): 12414–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ma Z, Qi J, Meng S, Wen B, Zhang J. Swimming exercise training-induced left ventricular hypertrophy involves microRNAs and synergistic regulation of the PI3K/AKT/mTOR signaling pathway. Eur J Appl Physiol. 2013;113(10):2473–2486. [DOI] [PubMed] [Google Scholar]
- 156.Pavo N, Lukovic D, Zlabinger K, et al. Sequential activation of different pathway networks in ischemia-affected and non-affected myocardium, inducing intrinsic remote conditioning to prevent left ventricular remodeling. Sci Rep. 2017;7:43958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hausenloy DJ, Yellon DM. The second window of preconditioning (SWOP) where are we now? Cardiovasc Drugs Ther. 2010; 24(3):235–254. [DOI] [PubMed] [Google Scholar]