

Abstract
Ultrafast magic angle spinning (MAS) technology and 1H detection have dramatically enhanced the sensitivity of solid-state NMR (ssNMR) spectroscopy of biopolymers. We previously showed that, when combined with polarization optimized experiments (POE), these advancements enable the simultaneous acquisition of multi-dimensional 1H- or 13C-detected experiments using a single receiver. Here, we propose a new sub-class within the POE family, namely HC-DUMAS, HC-MEIOSIS, and HC-MAeSTOSO that utilize dual receiver technology for the simultaneous detection of 1H and 13C nuclei. We also expand this approach to record 1H-, 13C-, and 15N-detected homonuclear 2D spectra simultaneously using three independent receivers. The combination of POE and multi-receiver technology will further shorten the total experimental times of ssNMR experiments for biological solids.
Keywords: Polarization Optimized Experiments (POE), Multi-receiver, Multi-acquisition, Solid-State NMR, Ultra-Fast Magic Angle Spinning, SIM-CP, HC-DUMAS, HC-MEIOSIS, HC-MAeS-TOSO
1. Introduction
Magic angle spinning solid-state NMR (MAS ssNMR) spectroscopy is maturing as a central technique for chemical, biochemical, and biophysical research, spurring its application to the characterization of the structure, dynamics, and ligand binding of protein fibrils, crystalline proteins, and membrane proteins at atomic resolution1–9. Higher magnetic field strengths and multidimensional experiments have provided powerful new tools for investigating larger biomacromolecules10,11. In the past few years, there has been a significant effort to accelerate MAS ssNMR experiments using dynamic nuclear polarization (DNP), paramagnetic relaxation enhancement (PRE), and ultra-fast spinning speeds12–15. Several technological developments have increased the scope of ssNMR applications to large biomolecular complexes. These advancements include low-E or E-free probes that increase the sensitivity of experiments, while avoiding heating caused by high-power RF pulses and enabling the analysis of temperature-sensitive biological samples under physiological conditions16,17. More recently, fast and ultrafast MAS probes as well as spectrometers equipped with field-gradient coils and multiple receivers enabled the development of cross-polarization (CP) and INEPT-based experiments for biosolids, with a dramatic increase in sensitivity and resolution18–21. During the past decade, our group has developed a general approach, namely, polarization optimized experiments (POE), which enables the concatenation of various pulse sequences into single experiments22,23. Using different strategies (i.e., DUMAS, MEIOSIS, and MAeSTOSO), we implemented the simultaneous acquisition of multiple 2D and 3D spectra at slow and fast spinning rates using either 1H- or 13C- detected single-receiver experiments22,24–27. Along with auxiliary developments made by other research groups, the POE family now includes 1H-detected experiments under fast MAS conditions, afterglow, dipolar-edited versions of DUMAS pulse sequences, and, more recently, hybrid pulse sequences combining CP and INEPT transfer periods27–34. These experiments reinforce the importance of developing multi-acquisition pulse sequences for ssNMR that utilize orphan spin operators (unused polarization) to enhance signal intensity and/or collect several datasets simultaneously23.
In ssNMR, 13C- and 15N-detected experiments have been paramount to the high-resolution spectral analysis of biomolecular systems1,2,35,36. While 1H-detected experiments using fast MAS have provided a significant boost in signal-to-noise, the resulting spectra are generally less resolved due to narrower chemical shift dispersion and line broadening from strong 1H-1H homonuclear dipolar couplings. In this work, we extend the POE methods using ultra-fast MAS and multi-receiver technologies to combine the benefits of 1H, 13C and 15N detection. Specifically, we present the multi-receiver implementation of three POE strategies, i.e., 1H- and 13C-detected DUMAS, MEIOSIS and MAeSTOSO (namely HC-DUMAS, HC-MEIOSIS, and HC-MAeSTOSO), and a triple-receiver (1H, 13C, and 15N) HCN-DUMAS. While specific combinations of POE are illustrated, the HC-DUMAS, HC-MEIOSIS, HC-MAeSTOSO, and HCN-DUMAS strategies can also be used to concatenate several other 1H, 13C, and 15N detected pulse sequences to speed up data acquisition. We demonstrate the performance of these new multi-acquisition POE methods, using a fully protonated uniformly U-13C,15N labeled microcrystalline GB1 protein and fMLF tripeptide samples.
2. Materials and methods
All the pulse sequences were implemented on a Bruker 600 MHz spectrometer equipped with a 1.3 mm fast MAS probe and Avance NEO® console with multi-receiver technology. The spectrometer was operated using TOPSPIN 4.0.2 software. U-13C and 15N labeled microcrystalline GB1 (β1 immunoglobulin binding domain of protein G) protein sample was prepared from the protocol (crystal form A) described previously by Rienstra and co-workers37,38. Approximately 2 to 3 mg of GB1 microcrystals, in residual precipitant solution, were packed into a 1.3 mm rotor. All the spectra were acquired with a MAS rate of 65 kHz. A sample temperature of 25 °C was maintained by setting the RF coil temperature to −20 °C to compensate for the heat induced by fast spinning as measured from the water resonance frequency.
All the experiments carried out on GB1 protein were acquired with a 2 s recycle delay, whereas a 3 s recycle delay was used for recording fMLF spectra. The 90° pulse length for 1H was set to 1.25 μs, whereas 90° pulses of 3 μs were used for 13C and 15N. For the GB1 sample, 1H solvent suppression was obtained by the MISSISSIPPI sequence (without gradients) with a 1H RF amplitude of 30 kHz applied for 200 ms (represented by tsupp in Figures 2A, 3A, 4A, and 5A)39. For HC-MAeSTOSO-4 and HC-MAeSTOSO-10 pulse sequences (Figures 4A and 5A), an additional water suppression (0.25*tsupp) period was used28. For the SIM-CP preparation period, simultaneous 1H-13C and 1H-15N Hartmann-Hahn matching conditions were obtained by using constant amplitude RF pulses on 13C and 15N, while the RF pulse on 1H was linearly ramped from 90 to 100%22,28,40. The optimized SIM-CP RF amplitudes for 1H, 13C, and 15N were 88.9, 23.7, and 23.6 kHz, respectively. For 13C-1H and 15N-1H back-CP periods, the optimal transfer was obtained using an 83.8 kHz RF amplitude on 1H with a 90 to 100% linear ramp, whereas RF amplitudes were set to 23.7 kHz for 13C and 15N. During NCA and NCO SPECIFIC-CP transfer, the 15N RF amplitude was set to 43 kHz, while the RF of 13C was linearly ramped from 85 to 100% with a maximum amplitude of 17 kHz41. Homonuclear DREAM polarization transfer was obtained with a linear RF ramp of 80 to 100% for CACO, and 85 to 100% for CACB using a 31.9 kHz 13C RF amplitude42. The 13C offset was set to 57 ppm during NCA and CACO, 175 ppm for NCO, and 45 ppm for CACB transfer periods. Mixing times for HH-RFDR sequence (Figures 5 and 7), were set from 0.5 to 2 ms43–45. During the t1 and t2 periods, a WALTZ16 sequence was used for heteronuclear decoupling with 10 kHz RF amplitude46. The RF carrier frequency for 1H, 13C and 15N were set to 4.7, 42, and 122 ppm, respectively. The contact times for CP and SIM-CP periods, were set to 1 ms, whereas 13C-1H and 15N-1H back-CP periods were set to 200 and 500 μs, respectively. The duration of NC SPECIFIC-CP and CACB DREAM transfer was set to 9.5 and 10 ms, respectively. The CXCX TOCSY mixing was obtained from a 12 ms WALTZ16 period with 13C RF amplitude set to 10 kHz. The t2 acquisition period for 1H (t2H) was set to 8.3 ms, whereas the 13C and 15N acquisition times were set to 25 ms (t2C and t2N). The total experimental time for the 2D HC-DUMAS, HC-MEIOSIS, and HC-MAeSTOSO-4 spectra of GB1 sample were 0.4, 3, and 3 h, respectively (Figures 2–4, and Table 1); whereas the HCN-DUMAS 2D spectra on fMLF sample was acquired in 1.4 h (Figure 7, and Table 2). For the HCN-DUMAS pulse sequence carried out on fMLF, suppression of the 1H background signal in the HH 2D experiment was achieved using a spin-echo sequence, τr-180°-τr before 1H acquisition, where τr corresponds to a rotor period of 15.38 μs.
Figure 2:
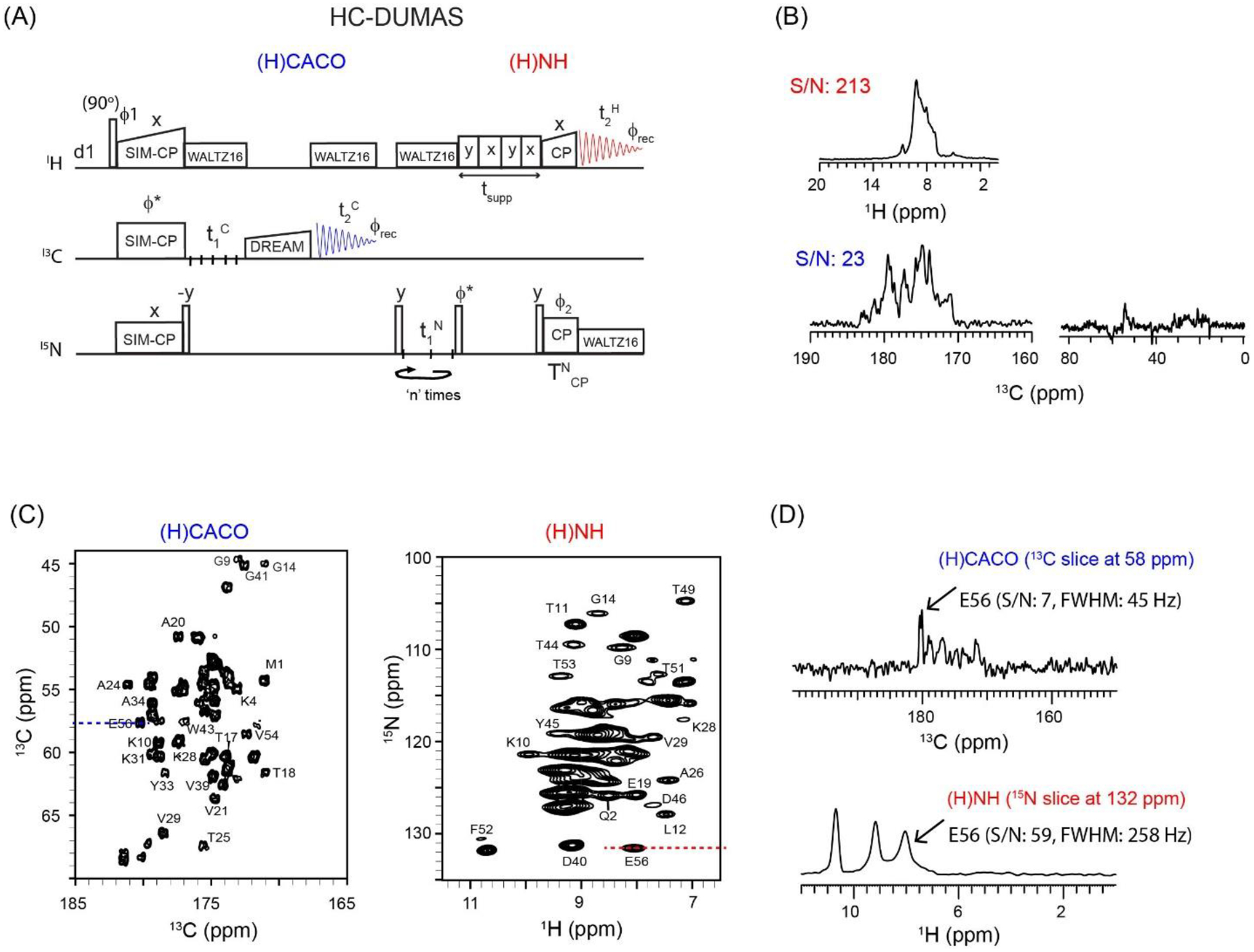
(A) 2D HC-DUMAS pulse sequence for simultaneous acquisition of (H)CACO and (H)NH experiments. The phase cycle is set to, ϕ1=y, −y; ϕ2=x, x, −x, −x; ϕrec=y, −y, −y, y. (B) One-dimensional (H)CACO and (H)NH spectra of GB1 protein acquired using the first increment of the pulse sequence setting t1C and t1N to zero, and n=2 (Table 1). (C) 2D (H)CACO and (H)NH spectra of GB1 acquired simultaneously using HC-DUMAS pulse sequence. (D) 1D cross sections for 1H and 13C dimensions extracted from the 2D (H)CACO and (H)NH spectra. The corresponding single receiver (1H or 13C) experiments were shown in references22,28.
Figure 3:
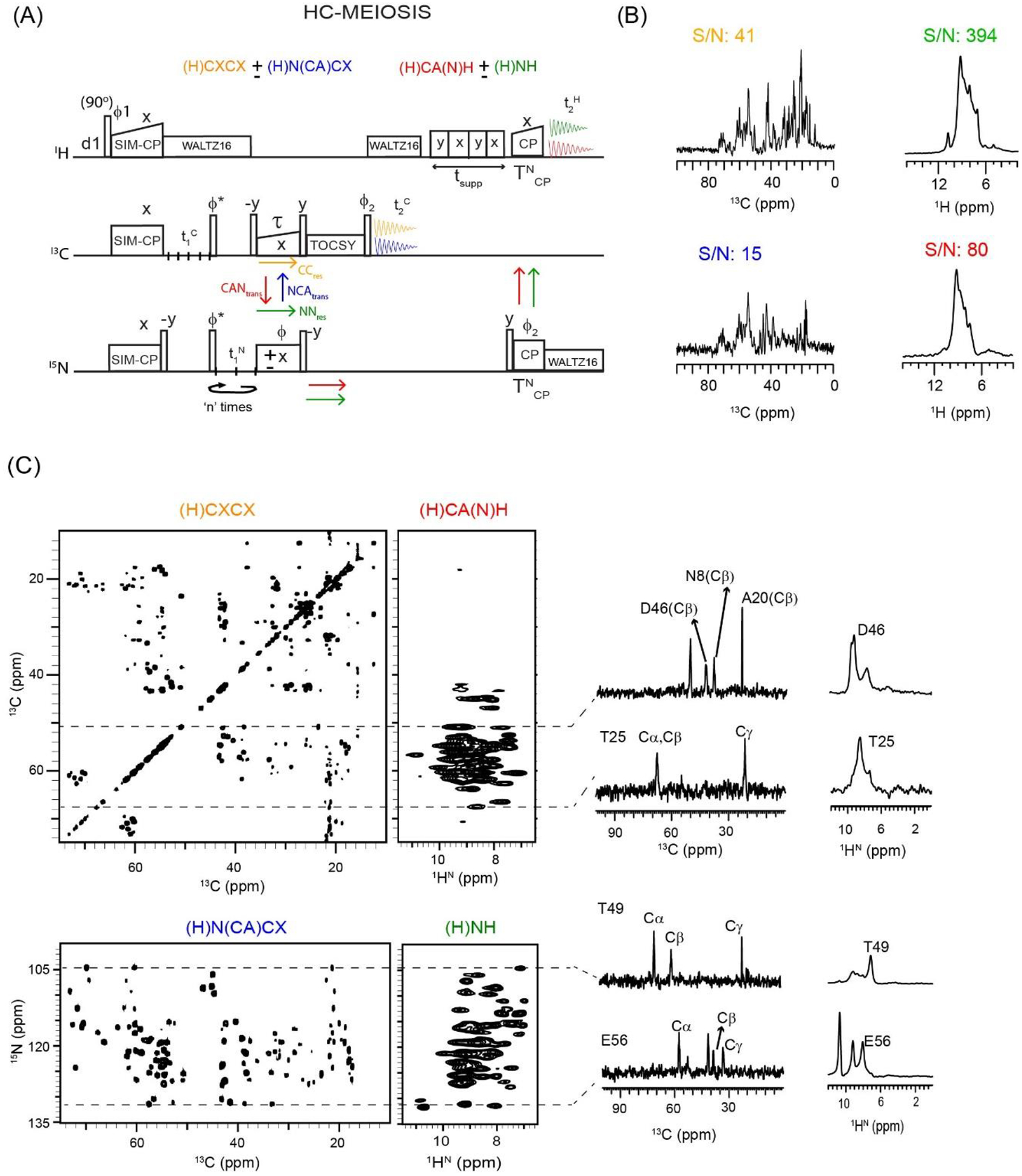
(A) HC-MEIOSIS pulse sequence for simultaneous acquisition of two pairs of 13C and 1H detected dual-receiver experiments. The phase cycle is set to, ϕ1=y, −y; ϕ2=x, x, −x, −x; ϕrec=y, −y, −y, y. (B) 1D HC-MEIOSIS spectra of GB1 protein obtained from the first increment of HC-MEIOSIS pulse sequence, and n=4 (Table 1). The corresponding integrated S/N values are indicated. (C) 2D HC-MEIOSIS spectra of GB1 protein, (H)CXCX, (H)N(CA)CX, (H)CA(N)H, and (H)NH acquired simultaneously using the pulse sequence in A. The 1D cross sections along the dotted lines are shown for 13C and 1H detected 2D spectra. The corresponding single receiver experiments were shown in references24,28.
Figure 4:
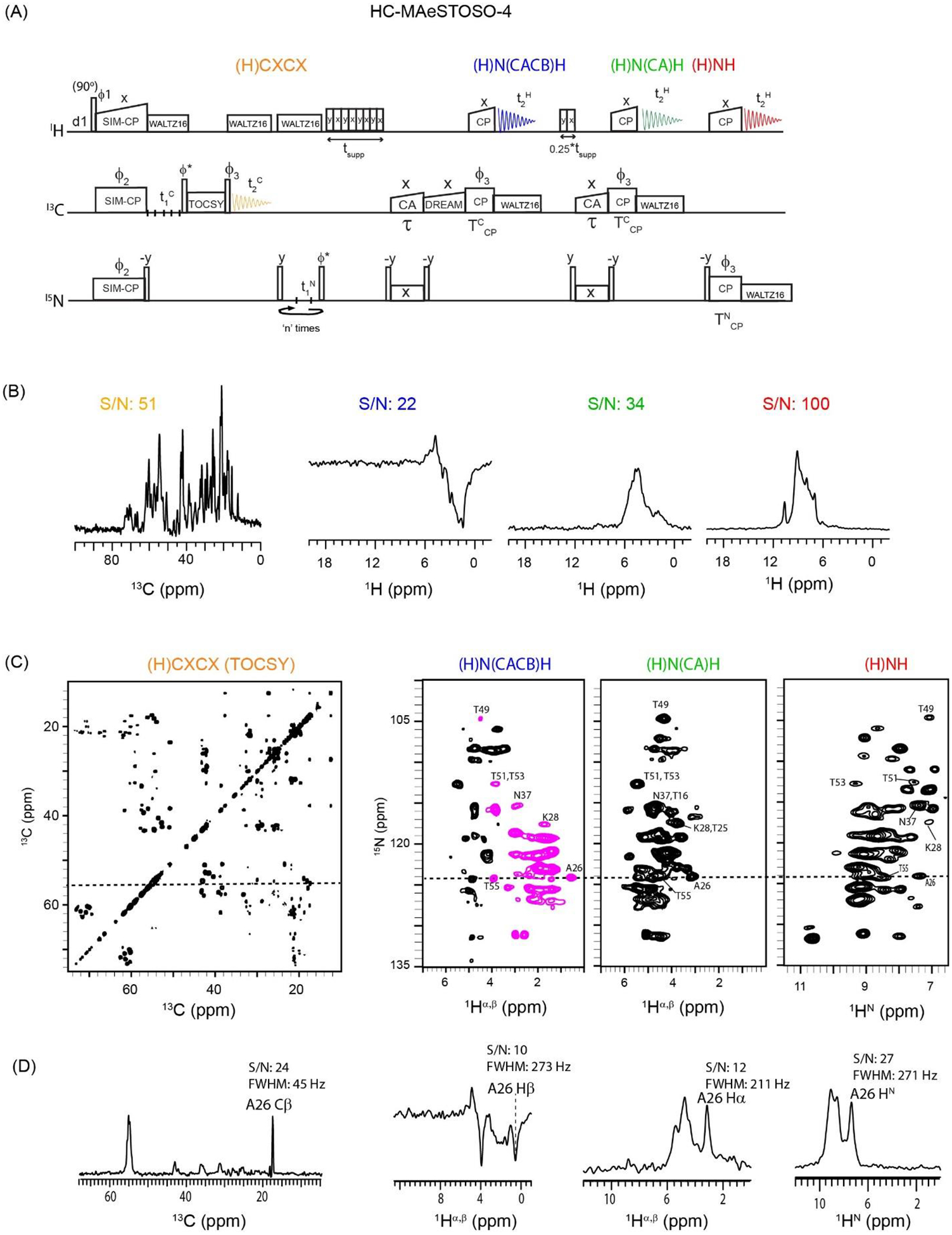
(A) Dual receiver HC-MAeSTOSO-4 pulse sequence for simultaneous acquisition of one 13C-detected and three 1H-detected experiments. The phase cycle is set to, ϕ1=y, -y; ϕ2=x, x, −x, −x; ϕ3=x, x, x, x, −y, −y, −y, −y; ϕrec=y, −y, −y, y, −y, y, y, −y. (B) 1D HC-MAeSTOSO-4 spectra of GB1 protein obtained by setting t1C and t1N to zero. (C) Simultaneous acquisition of 2D (H)CXCX, (H)N(CACB)H, (H)N(CA)H, and (H)NH spectra of GB1 using HC-MAeSTOSO-4 pulse sequence. (D) 1D cross sections along 13C (55.6 ppm) and 15N (124.2 ppm) dimensions and corresponding S/N and FWHM values. The corresponding single receiver experiments were shown in references25,28.
Figure 5:
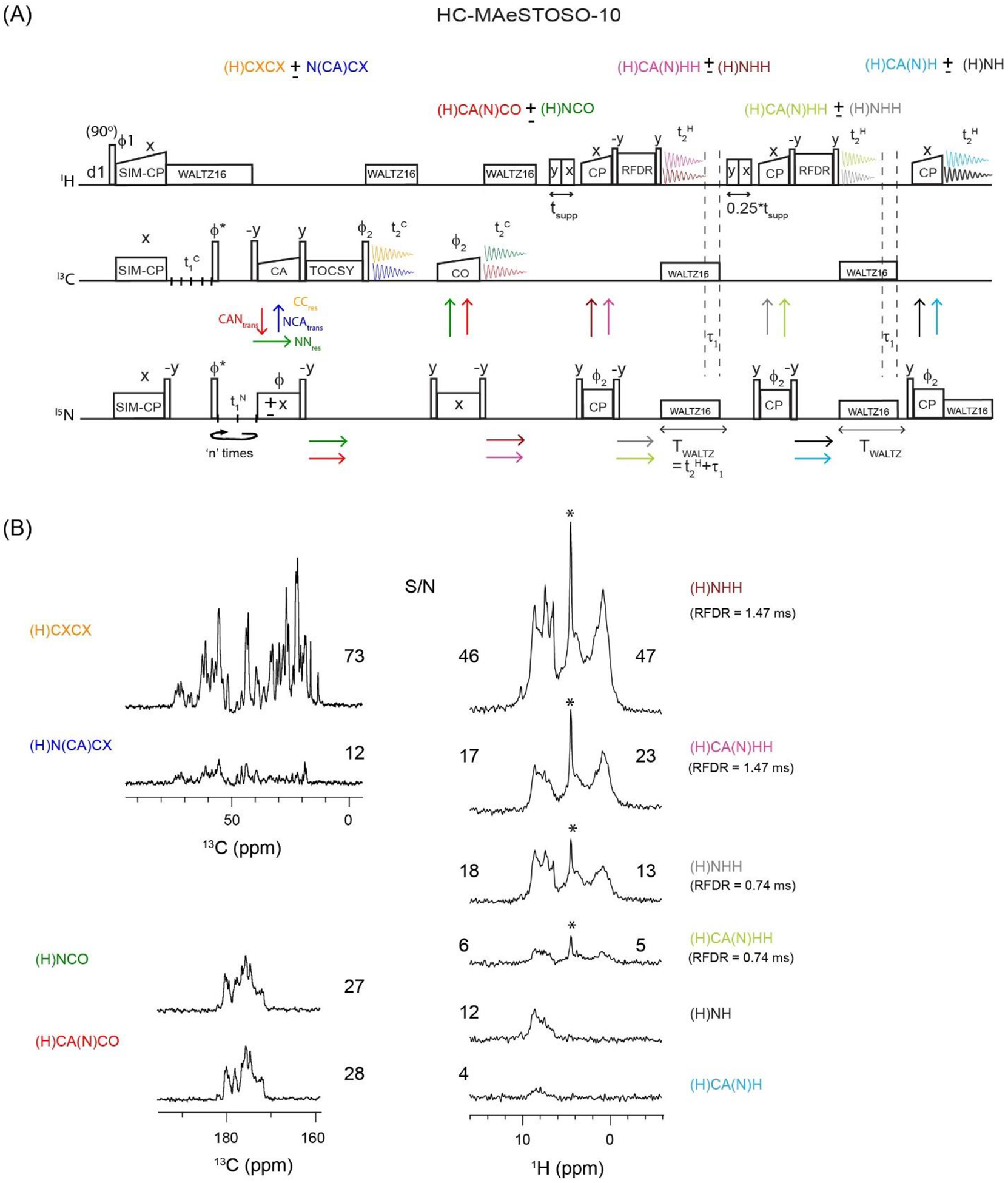
(A) Dual receiver HC-MAeSTOSO-10 pulse sequence for the simultaneous acquisition of four 13C detected, and six 1H detected experiments. The phase cycle is set to, ϕ1=y, −y; ϕ2=x, x, −x, −x; ϕrec=y, −y, −y, y. (B) HC-MAeSTOSO-10 1D spectra of GB1 protein obtained from the first increment (t1C, t1N=0). The integrated S/N values for each 1D spectra are shown in Figure 5B. For 1H spectra with HH RFDR mixing, the S/N values are calculated for HN (6 to 10 ppm) and HC (0 to 4 ppm) spectral regions, and respectively shown on the left and right sides of the 1D spectra. The 1H peak at 4.7 ppm, indicated by *, corresponds to water signal.
Figure 7:
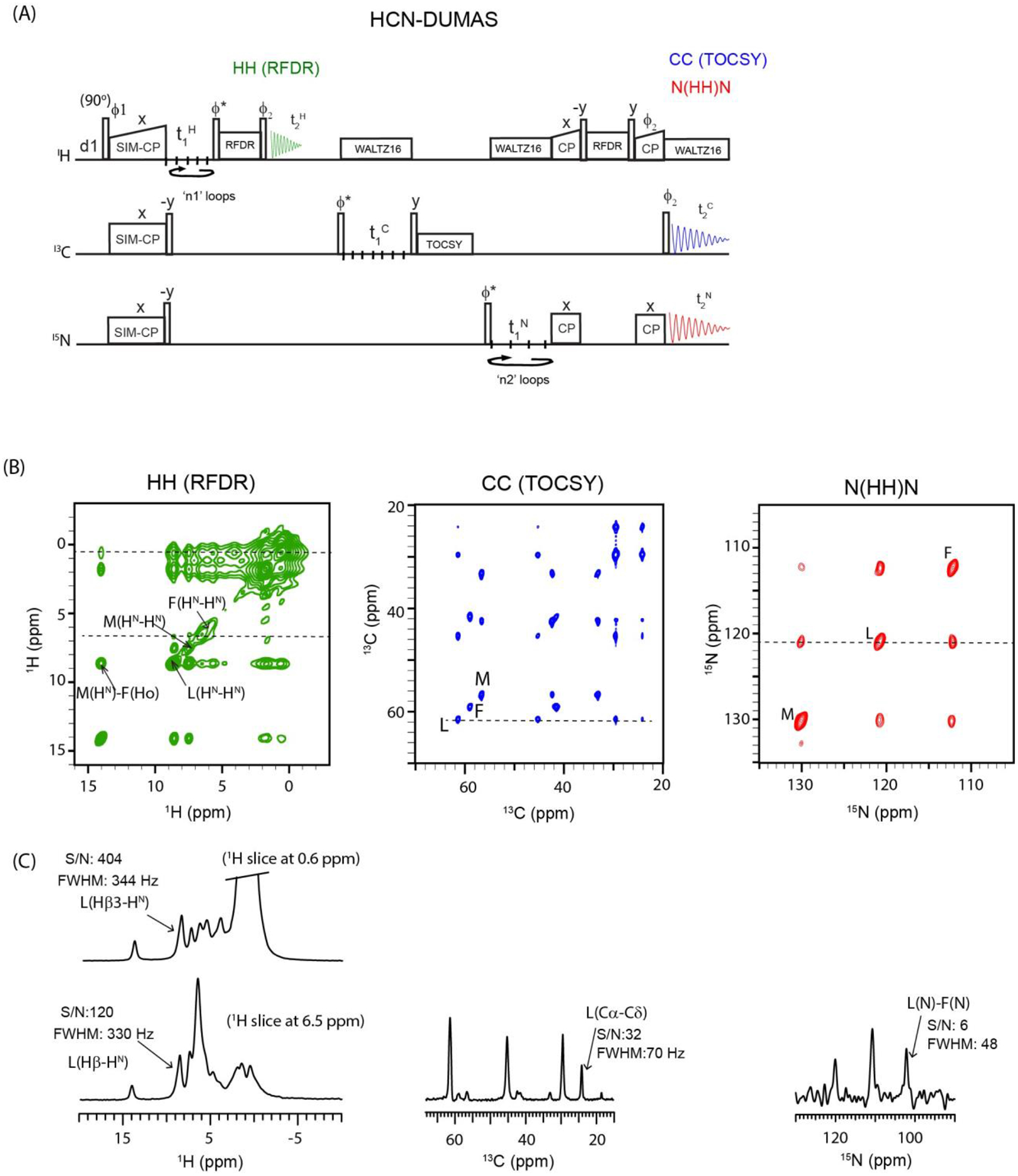
(A) Triple receiver HCN-DUMAS pulse sequence for simultaneous acquisition of 2D HH, CC, and N(HH)N homonuclear correlation experiments. The phase cycle is set to, ϕ1=y, −y; ϕ2=x, x, −x, −x; ϕrec=y, −y, −y, y. (B) 2D HH, CC, and N(HH)N spectra of fMLF tripeptide sample using HCN-DUMAS pulse sequence. (C) 1D cross-sections taken along 1H, 13C, and 15N dimensions, and corresponding S/N and FWHM values.
Table 1:
| POE Method | 13C detected experiments | 1H detected experiments | 13C t1 evolution (t1C) | 15N t1 evolutio n (t1N) | ns | nseff =n* ns | Texp | ~TCex | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dw (t1C) | ni (t1C) | T(t1C) | dw (t1N) | ni (t1N) | T(t1N) | n | |||||||
| HC-DUMAS | (H)CACO | (H)NH | 200 μs | 74 | 14.6 ms | 400 μs | 37 | 14.4 ms | 2 | 4 | 8 | 23 min | 42 min |
| HC-MEIOSIS = (ϕ = x, −x) | (H)CXCX (H)N(CA)CX |
(H)CA(N)H (H)NH |
100 μs | 148 | 14.7 ms | 400 μs | 37 | 14.4 ms | 4 | 16 | 64 | 3 h | 7.9h |
| HC-MAeSTOSO-4 | (H)CXCX | (H)N(CACB)H (H)N(CA)H (H)NH |
100 μs | 148 | 14.7 ms | 400 μs | 37 | 14.4 ms | 4 | 16 | 64 | 3 h | 6.3 h |
dw = dwell time; ni = no. of t1 increments; T (maximum t1 evolution time) = dw*(ni-1); n = no. of 15N t1 loops; ns = no. of scans; nseff = Effective number of scans for 15N edited spectrum; Texp = Total experimental time; TCexp= Estimated experimental time using conventional single acquisition pulse sequences; Condition for parallel 13C and 15N t1 evolutions: ni(t1C) = n*ni(t1N)22,26.
Table 2:
Experimental t1 evolution parameters for the HCN-DUMAS spectra reported in Figure 7.
| CXCX | HH | N(HH)N | Texp | ~TCexp | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dw (t1C) | ni (t1C) | T(t1C) | ns | ni (t1H) | T(t1H) | n1 | nseff =n1*ns | dw (t1N) | ni (t1N) | T(t1N) | n2 | nseff =n2*ns | |||
| 80 μs | 100 | 7.9 ms | 8 | 80 μs | 25 | 1.9 ms | 4 | 32 | 320 μs | 25 | 7.7 ms | 4 | 32 | 1.4 h | 3.2 h |
Condition for parallel 1H, 13C, and 15N t1 evolutions: ni(t1C) = n1*ni(t1H) = n2*ni(t1N).
All the 2D spectra were acquired with States mode during t1 evolution, by switching the phase ϕ* between y and −x47. Both t1 and t2 dimensions were processed with a 90° shifted sine bell window functions using NMRpipe scripts as previously described for multiple acquisition experiments26,48. The 2D data sets were analyzed using Sparky49. All multi-acquisition pulse sequences utilize multiple WALTZ16 decoupling periods on 1H, 13C, and 15N channels. As reported previously, we recommend to use different names for each WALTZ16 period in the programming code to avoid sharing of the pulse profile28. For reasons unknown at this point, we had to add three 13C (t2C) and one 1H (t2H) dummy acquisition periods at the end of the pulse program to successfully execute the HC-MAeSTOSO-4 experiment. Similarly, the HC-MAeSTOSO-10 pulse program required an additional two 13C and one 1H dummy t2 acquisition periods. Note that these dummy t2 acquisition periods contain no signal and are discarded during processing. We hope to rectify these issues with future software/hardware upgrades. The POE pulse programs will be available to users upon request.
3. Results
3.1. Polarization transfer efficiencies of CC and NC mixing periods
To design efficient and sensitive POE, it is important to optimize the transfer efficiencies for homo- and hetero-nuclear recoupling sequences. Figure 1 shows the comparison of optimized 13C spectral intensities obtained from the homo- and hetero-nuclear polarization transfer schemes, using the conventional 1D pulse sequences with 13C detection and WALTZ16 1H decoupling. Each of the 1D spectra was acquired using 512 scans. The complete experimental parameters are reported in section 2. To quantify the efficiencies of CC and NC transfer periods, the integrated intensities of the spectra in Figures 1B–F were normalized with respect to the Cα spectral region of 1H-13C CP spectrum (Figure 1A)50. In comparison to 1H-13C CP, the integrated intensities of SPECIFIC-CP NCA and NCO are 31 and 44%, respectively (Figures 1B and C). In general, the transfer efficiency of NCA is lower than NCO due to the high density of aliphatic protons and the CACB couplings. Note that typical NC transfer efficiencies range from 20–50%, depending on the RF parameters, MAS rate, spectral dispersion of CA and CO regions (i.e., B0 field) as well as sample conditions such as protein heterogeneity and extent of deuteration. The CACO and CACB DREAM transfer was optimized using the 15N CP-NCA-CACO (or CACB) pulse sequence, and the respective signal intensities are 19 and 10% (Figures 1D and E). The DREAM transfer is based on the double quantum recoupling Hamiltonian and inverts the phase of transferred polarization (CO or Cβ spectra of Figures 1D and E) with respect to the source polarization, Cα42. Figure 1F shows the 13C spectrum obtained with 15N-CP-NCA-CACX, where the CACX transfer is achieved using 12 ms TOCSY mixing period. Unlike CACB, the CACX TOCSY mixing period transfers the polarization from Cα to all intra-residue side chain carbons. The integrated intensities for CACB and CACX spectra were measured from 10 to 36 ppm, and 65 to 73 ppm, which correspond to Cβ resonances of Ser and Thr residues.
Figure 1:
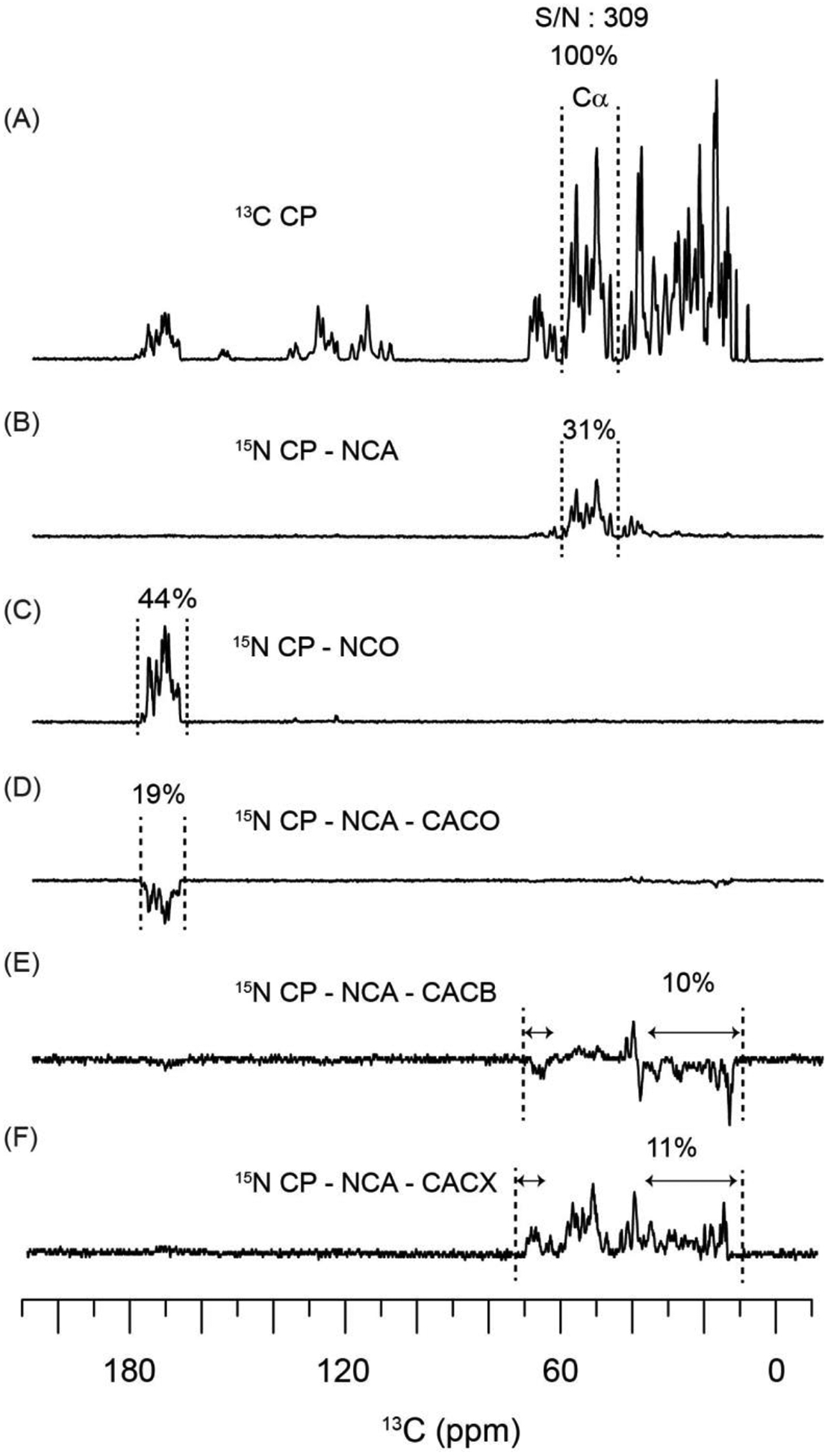
Comparison of 13C spectral intensities obtained from CC and NC polarization transfer periods, with respect to 13C CP. The spectra were recorded using conventional 13C detected 1D pulse sequences with WALTZ16 1H decoupling.
3.2. 1H- and 13C- detected DUMAS (HC-DUMAS) with dual receiver
DUMAS (DUal acquisition Magic Angle Spinning) pulse sequences begin with a SIM-CP preparation period that transfers the polarization from the 1H spin bath to 13C and 15N nuclei simultaneously22,51. The polarization transfer efficiency of CP and SIM-CP for GB1 protein is shown in Figure S1 (Supporting Information). The high efficiency of transfer for SIM-CP has been demonstrated previously for both microcrystalline and membrane proteins at slow and fast MAS rates22,28. The relative intensities of 13C spectra are almost identical for CP and SIM-CP schemes, while a minimal signal loss (5–8%) is observed for 15N. Using a single receiver, we previously showed that the 13C and 15N polarization generated by SIM-CP enables the simultaneous acquisition of pairs of 13C- or 1H-detected spectra with DUMAS and H-DUMAS strategies22,28. Analogously, the SIM-CP scheme can be used to develop HC-DUMAS sequences in which 13C- and 1H-detected experiments are acquired on two separate channels. Figure 2A shows the HC-DUMAS pulse sequence for the simultaneous acquisition of 2D 13C-detected (H)CACO and 1H-detected (H)NH spectra in 1st and 2nd acquisition periods, respectively. The SIM-CP preparation period is followed by 13C (t1C) and 15N (t1N) evolution periods. After t1 evolution, the 15N polarization is stored along the z-direction and a CACO DREAM mixing is applied on the 13C channel followed by the 1st acquisition period () that records a 2D (H)CACO spectrum. After the 1st acquisition, the solvent suppression is achieved by applying the MISSISSIPPI sequence for a time tsupp. The 15N z-polarization is then transferred to amide protons by applying a 90° pulse followed by a 15N-1H back-CP period, and subsequently used to obtain a 2D (H)NH spectrum in the 2nd acquisition period (), which is detected on the 1H channel. A key feature of POE is that the t1 evolution periods (t1C and t1N) used for 13C and 15N nuclei can be optimized separately as shown in Table 126. This enables the operator to optimize the t1 dwell-times and total evolution periods for each experiment. For the GB1 sample at 600 MHz 1H frequency, the indirect spectral widths for 13Cα and 15N were 5000 and 2500 Hz, respectively. Therefore, the 13Cα t1 dwell-time (t1C) for the (H)CACO experiment was set to 200 μs and 74 t1 increments were collected for a maximum 13C evolution time of 14.6 ms (Table 1). On the other hand, 15N dwell-time (t1N) was set to 400 μs, and two identical t1 evolution periods (n=2 in Figure 2A), each with 37 increments were recorded for a maximum t1 evolution period of 14.4 ms. As shown in Table 1, the t1 evolution parameters satisfy the condition, ni(t1C) = n * ni(t1N), where ‘ni’ represent number of t1 increments22. Therefore, rather than oversampling the 15N spectral width (t1), these cycles are instead used to acquire two data sets for the (H)NH experiment, which are added together during processing to essentially double the effective number of scans with respect to the (H)CACO experiment22,26.
Figure 2B shows the 1D CO and HN spectra of GB1 protein obtained from the first t1 increment (t1C and t1N = 0) of the HC-DUMAS pulse sequence using 4 scans. Note that the effective number of scans for HN spectrum correspond to 8 (nseff of Table 1). The integrated signal to noise ratios (S/N) are 23 and 213 for CO and HN, respectively. During the DREAM spin lock, the aliphatic 13C signals are dephased out and display weak intensities (Figure 2B). Figure 2C shows the 2D (H)CACO and (H)NH spectra acquired simultaneously using the HC-DUMAS pulse sequence with 4 scans per t1 increment and a total experimental time of 23 min. Several resolved peaks in the 2D (H)CACO and (H)NH spectra were assigned using the published data by Pintacuda and co-workers44. Figure 2D shows the cross-sections for E56 extracted from the 2D (H)CACO and (H)NH data sets with S/N values of 7 and 59, respectively. Overall, the 2D (H)CACO spectrum has lower S/N but has superior resolution in the direct dimension compared to the 2D (H)NH spectrum. The average CO and HN full-width-at-half-maximum (FWHM) line widths in the 2D (H)CACO and (H)NH spectra are 55 and 242 Hz, respectively. Note that the longer T2 relaxation times (or narrow line widths) of the CO nuclei resulted in the splitting of the resonances caused by CACO homonuclear J-couplings (Figures 2C and 2D). To further narrow the CO line widths, it is possible to use the BASHD acquisition method with 13C-13C homonuclear decoupling52. Note that for HC-DUMAS experiments, the high signal intensity of 1H can be exploited to record other 2D spectra, such as a (H)NHH experiment, by incorporating a HH mixing period prior to the 2nd acquisition. Similarly, the CACO DREAM mixing can be replaced by CACB or CXCX TOCSY sequences for 13C-detected experiments. Overall, the HC-DUMAS strategy is flexible enough to incorporate other mixing methods and optimize the sensitivity of 13C- and 1H-detected spectra53.
3.3. 1H- and 13C-detected MEIOSIS (HC-MEIOSIS) with dual receiver
In the MEIOSIS (Multiple ExperIments via Orphan SpIn operatorS) and H-MEIOSIS (1H-detected Multiple ExperIments via Orphan SpIn operatorS) methods two pairs of 13C- and 15N-edited spectra are recorded with either 13C- or 1H-detection using a single receiver24,28. A version of the HC-MEIOSIS (1H- and 13C- detected MEIOSIS) pulse sequence using two receivers is shown in Figure 3A. After SIM-CP, the 13C and 15N polarization pathways are evolved during the t1 period followed by bidirectional SPECIFIC-CP transfer from N to Cα and Cα to N (NCAtrans and CANtrans). Note that some residual polarization remains on both 13C and 15N after the SPECIFIC-CP sequence (CCres and NNres)24,29,31. The NCAtrans and CCres pathways are encoded within the 13C polarization, whereas the CANtrans and NNres pathways are encoded in the 15N polarization. To recover these pathways, both 15N and 13C spin operators are flipped to the z-direction by applying 90° pulses after SPECIFIC-CP. The CCres and NCAtrans pathways evolved under TOCSY mixing, followed by a 90° readout pulse that enables the acquisition of 2D (H)CXCX and (H)N(CA)CX spectra in the 1st acquisition period using the 13C receiver. The polarization from NNres and CANtran pathways is transferred to amide protons by applying a 90° pulse and NH back-CP period followed by detection on the 1H channel. This 2nd acquisition period gives 2D (H)NH and (H)CA(N)H spectra. As shown previously for the MEIOSIS and H-MEIOSIS experiments, the phase (ϕ) flip of the 15N SPECIFIC-CP pulse does not affect the residual polarization of the NNres and CCres pathways, but it does invert the phase of the CANtrans and NCAtrans pathways24. Therefore, the two polarization pathways in each of the 1st and 2nd acquisition periods are decoded by adding and subtracting the two data sets recorded with ϕ set to x and −x. Of course, the sensitivity of HC-MEIOSIS spectra depends on the intensities of the respective polarization pathways. Figure S2 shows the 1D MEIOSIS pulse sequence and corresponding intensities of the four polarization pathways (CCres, CANtrans, NNres, and NCAtrans) for GB1 using 13C detection. At the optimal SPECIFIC-CP transfer period (τ = 9.5 ms), approximately 30% of the 15N residual polarization (NNres) is observed; whereas the residual polarization for 13Cα and side chain atoms (Cβ, Cγ etc. or 13Cside chain) is 52 and 74%, respectively. Figure 3B shows the 1D spectra of 13C and 1H for GB1 obtained from the first increment (t1 = 0) of the HC-MEIOSIS experiment collected with 8 scans and a TOCSY mixing period of 12 ms. In the 1D (H)CXCX and (H)N(CA)CX spectra, the S/N of the 13C resonances (0–70 ppm) are 41 and 15, respectively. As expected, the 1D 13C spectrum from the (H)N(CA)CX experiment has the lowest S/N since it includes both heteronuclear (NCA) and homonuclear CACX TOCSY transfer periods. The integrated S/N of the 1H resonances between 6 and 12 ppm for the second set of 1D HC-MEIOSIS spectra, i.e., (H)CA(N)H and (H)NH, were 80 and 394, respectively. Using single receiver MEIOSIS or H-MEIOSIS experiments, the CCres pathway showed the highest S/N in the 2D experiments (13C-detected DARR or 1H-detected (H)CH)24,28. On the other hand, the CCres pathway in the HC-MEIOSIS experiment with dual receiver (i.e., 13C-detected (H)CXCX) shows a lower S/N compared to the NNres and CANtrans pathways that are recorded via 1H-detected experiments, i.e., (H)CA(N)H and (H)NH. Even though the (H)NH spectrum is recorded from the residual 15N polarization (~30%, Figure S2), it displays higher S/N with respect to the (H)CA(N)H spectrum, which requires an additional CAN transfer period. The 2D HC-MEIOSIS spectra of GB1 were recorded with 8 scans and consist of two data sets acquired in an interleaved manner with ϕ = x, −x (Figure 3C). For the 13C evolution (t1C), the dwell-time was set to 100 μs and 148 increments were collected, corresponding to a maximum evolution time of 14.7 ms (see Table 1). For the 15N indirect dimension, the t1 dwell-time was set to 400 μs with four identical t1 evolution periods (n=4 in Figure 3A) each with 37 increments and corresponds to a maximum evolution of 14.4 ms. The four 15N-edited 2D (H)N(CA)CX and (H)NH data sets were then added using post-acquisition processing scripts26. Essentially, the effective number of scans (nseff in Table 1) for the 15N-edited 2D (H)N(CA)CX and (H)NH spectra resulted to be four times higher than the 13C-edited 2D (H)CXCX and (H)CA(N)H spectra. Figure 3C shows the 1D cross-sections from the four 2D HC-MEIOSIS spectra. 1H-detected 2D (H)NH and (H)CA(N)H spectra have higher S/N, but their limited resolution can often prohibit residue-specific assignment. Conversely, the 13C-detected 2D (H)CXCX and (H)N(CA)CX spectra resolve many more peaks, but with lower S/N. The latter is evident from the NCA region of the N(CA)CX spectrum, which gives rise to 43 resolved peaks of GB1. In the (H)CXCX spectrum of Figure 3C, the intensities of the upper off-diagonal cross-peaks associated with residual polarization of 13Csidechain (72%) are relatively higher compared to the bottom off-diagonal cross-peaks originating from 13Cα residual polarization (52%). A similar observation was made for the 2D DARR spectrum obtained from the single receiver MEIOSIS pulse sequence24. The cross-peak intensities of the 2D (H)CXCX and (H)N(CA)CX spectra also depend on the efficiency of the homonuclear CC mixing period, which can be further improved using faster MAS rates (100–110 kHz)44. Similarly, the HC-MEIOSIS experiment can also be implemented with other CC mixing sequences such as CACO, CACB, PAR, and CHHC that can provide intra- and inter-residue CC correlations54–56. The four 2D HC-MEIOSIS spectra in Figure 3C were recorded in approximately 3 h (Texp). While the 2D (H)N(CA)CX and (H)CA(N)H spectra were recorded using transferred polarization, the 2D (H)CXCX and (H)NH spectra originate from the residual polarization, 72% for 13Cside chain and 30% for 15N (i.e., a factor of k = 0.72 and 0.3). To obtain these spectra at the same S/N using conventional single-acquisition sequences, the estimated time (k2*TExp) for each experiment is 1.6 h for the (H)CXCX experiment (~0.722*3 h), 0.3 h for the (H)NH experiment (~0.32*3 h) and 3 h for each of the (H)N(CA)CX and (H)CA(N)H experiments for a total of 7.9 h (3+3+1.6+0.3). Therefore, the HC-MEIOSIS multi-acquisition sequence, which requires just 3 h, provides a net savings of 62% in experimental time.
3.4. 1H- and 13C-detected MAeSTOSO (HC-MAeSTOSO) with dual receiver
Multiple acquisitions via sequential transfer of orphan spin polarization (or MAeSTOSO) strategy exploits 15N and 13C residual polarization originating from NC SPECIFIC-CP and CH or NH back-CP periods25,28. Figure 4A shows an example of the dual receiver HC-MAeSTOSO-4 pulse sequence. In this experiment, after SIM-CP, the 13C and 15N polarization is evolved during t1. Subsequently, the15N polarization is stored along the z-direction and the 13C polarization is used to acquire a 13C-detected (H)CXCX TOCSY experiment (1st acquisition period). After the 1st acquisition, a MISSISSIPPI sequence is used on the 1H channel to suppress the solvent signals. Then, the 15N polarization is transferred to Cβ using an NCA SPECIFIC-CP followed by a CACB DREAM transfer. A 13C-1H back-CP transfers the Cβ polarization to the covalently attached Hβ atoms, providing a 1H-detected 2D (H)N(CACB)H spectrum in the 2nd acquisition. At the end of the first SPECIFIC-CP period, ~30% of residual polarization remains on 15N and is stored along the z-direction by applying a 90° pulse (Figure S2). After the 2nd acquisition, the 15N residual polarization is transferred to Cα and then to Hα using another SPECIFIC-CP followed by a 13C-1H back-CP period. The Hα polarization is then detected in the 3rd acquisition to yield a 2D (H)N(CA)H spectrum. Note that the residual polarization at the end of the second SPECIFIC-CP period is approximately 9% (30% of the 30% remaining after the first SPECIFIC-CP), which is stored along the z-direction by a 90° pulse. After the 3rd acquisition, this 9% residual polarization is transferred to the amide protons using a 15N-1H back-CP and then detected in the 4th acquisition to give a 2D (H)NH spectrum. Both NCA and CACB transfer periods are applied prior to the 2nd acquisition since the intensity of the 15N polarization stored after the 1st acquisition is relatively high. On the other hand, the 3rd acquisition uses 30% residual 15N polarization, therefore only the NCA transfer period was incorporated prior to 13C-1H back-CP period. To recover the 9% 15N residual polarization a 15N-1H back-CP is used prior to the 4th acquisition period. A similar strategy was used for single receiver MAeSTOSO pulse sequences to optimize the sensitivity of multiple 2D spectra acquired simultaneously with NC and CC mixing periods25.
Figure 4B shows 13C and 1H 1D spectra of GB1 recorded from the first increment (t1 = 0) of the HC-MAeSTOSO-4 experiment using 16 scans. The 1D integrated S/N for 2D (H)CXCX, (H)N(CACB)H, (H)N(CA)H, and (H)NH are 51, 22, 34, and 100, respectively. In spite of the weak 15N residual polarization, the S/N of HN detected in the 4th acquisition is higher than the Hα and Hβ spectra recorded in the 2nd and 3rd acquisitions, respectively. This is because the Hβ spectrum requires NCA and CACB transfer steps, while the Hα spectrum only requires a NCA transfer step prior to 13C-1H back-CP period. In fact, these additional transfer periods significantly lower the signal intensities of Hα and Hβ resonances. The 2D (H)CXCX, (H)N(CACB)HB, (H)N(CA)HA, and (H)NH spectra of GB1 (Figure 4C) were acquired using 16 scans per t1 increment for a total of 3 h, as reported in Table 1. The DREAM CACB transfer inverts the phase of Cβ with respect to Cα peaks, therefore, the corresponding Hα and Hβ peaks in the 2D (H)N(CACB)HB spectrum have opposite signs42. Note that all three 1H-detected spectra of Figure 4C share the same 15N evolution period (t1N), and thus detect the same 15N chemical shift evolution for Hα, Hβ, and HN resonances. As an example, Figure 4C shows selected 15N intra-residue correlations with the corresponding Hα, Hβ, and HN atoms. The 1D cross-sections of the A26 resonances with respective S/N and FWHM values are shown in Figure 4D. The S/N for the Cβ, Hα, Hβ, and HN peaks of A26 are 24, 10, 12, and 27, respectively. In general, the required number of scans for recording multi-dimensional ssNMR experiments are estimated from the 1D integrated S/N from the 1st increment. However, the relative S/N of resulting 2D or 3D peaks may not follow the same proportionality. This is because the relative S/N of various residues depend on the efficiency of homo- and hetero-nuclear polarization transfer, which is variably affected by protein dynamics and spectral parameters such as the offset frequency and linewidths. As shown in Figures 3B and 3D, the 1D integrated S/N in the (H)NH spectrum is three times higher than the (H)N(CA)H, but the S/N of A26 peak in the 2D (H)NH spectrum is only 2.2 times higher compared to (H)N(CA)H. The linewidths of Hα, Hβ, and Hγ resonances are in the range of 200 to 270 Hz, while 13C linewidths in the 2D (H)CXCX spectrum were considerably narrower (~25–70 Hz). Also note that the (H)CXCX spectrum from the HC-MEIOSIS experiment (Figure 3A) uses ~50 to 70 % of the 13C residual polarization, whereas the (H)CXCX spectrum in the HC-MAESTOSO-4 (Figure 4A) is generated from the full 13C polarization. Therefore, the relative sensitivity of the (H)CXCX spectrum acquired from HC-MAeSTOSO-4 is higher than the one obtained from HC-MEIOSIS. The four 2D HC-MAeSTOSO-4 spectra of Figure 4C were recorded in only 3 h (Texp). To further summarize, while the (H)CXCX and (H)N(CACB)H spectra utilize the polarization transferred from the initial SIM-CP, the (H)N(CA)H and (H)NH spectra are recorded from 30 and 9% (or a k factor of 0.3 and 0.09) residual polarization originating from the SPECIFIC-CP periods. Therefore, using the single acquisition experiments, each of the (H)CXCX and (H)N(CACB)H spectra would require approximately 3 h, whereas the (H)N(CA)H with 30% (k = 0.3) and (H)NH with 9% (k = 0.09) polarization require 0.27 h (~0.32*Texp) and 0.02 h (~0.092*Texp)28. In other words, the total experimental time for all four spectra acquired separately would be 6.29 h (3.0+3.0+0.27+0.02), whereas using the HC-MAeSTOSO-4 strategy they were acquired in 3 h for a 53% reduction in acquisition time.
Recently, we demonstrated that it is possible to recover the residual polarization from both NC SPECIFIC-CP, and NH (or CH) back-CP periods28. This led to the simultaneous acquisition of ten 1H-detected experiments (H-MAeSTOSO-10). The dual receiver HC-MAeSTOSO-10 is shown in Figure 5A and acquires four 13C- and six 1H-detected experiments using five acquisition periods. In each acquisition period, a pair of 13C- and 15N-edited 2D spectra are recorded using two polarization pathways (represented by arrows in Figure 5A). Since these spectra are acquired with alternate phases (ϕ set to x and −x), the two polarization pathways in each acquisition period are decoded by adding and subtracting the data sets (similar to HC-MEIOSIS of Figure 3A). Note that the 13C detection periods (1st and 2nd acquisition) of the HC-MAeSTOSO-10 experiment resembles the MEIOSIS pulse sequence at slow spinning speeds24. In the HC-MAeS-TOSO-10 experiment, after first SPECIFIC-CP period, four MEIOSIS polarization pathways, CCres, CANtrans, NNres, and NCAtrans (color-coded in Figure 5A), are encoded in the 13C and 15N polarization. The CCres and NCAtrans pathways are subjected to a TOCSY mixing followed by the 1st acquisition period recording the 13C- and 15N-edited (H)CXCX and (H)N(CA)CX spectra. After the 1st acquisition period, the 15N polarization from NNres and CANtrans pathways is transferred to CO via SPECIFIC-CP followed by 2nd acquisition period that records the (H)CA(N)CO and (H)NCO spectra. At the end of second SPECIFIC-CP, the CANtrans and NNres pathways are encoded in the 15N residual polarization. Note that the application of two SPECIFIC-CP periods reduces the residual polarization to less than 10%. Nonetheless, this residual polarization is enough to acquire 1H-detected experiments. After the 2nd acquisition period, a MISSISSIPPI sequence is used for solvent suppression. Next, the two pathways encoded in the 15N polarization are transferred to amide protons by the NH back-CP scheme followed by HH RFDR mixing. The latter makes it possible to record the (H)NHH and (H)CA(N)HH spectra under 13C and 15N WALTZ16 decoupling applied during the 3rd acquisition. The 15N residual polarization after the NH back-CP period is stored along the z-direction by a 90° pulse on 15N. The residual 15N polarization from the NH back-CP is recovered after the first 1H acquisition by adding an additional WALTZ16 period (τ1), which keeps the 15N polarization in the z direction28. After the 3rd acquisition and the following τ1 period, the 15N residual polarization is exploited for two additional pairs of 13C and 15N edited (H)NHH and (H)CA(N)HH spectra in the 4th acquisition, and (H)NH and (H)CA(N)H spectra in the 5th acquisition. Compared to single acquisition experiments, the time saving of HC-MAeSTOSO-10 is similar to HC-MEIOSIS, i.e a time saving of up to 62%. In other words, additional acquisition periods (3rd, 4th and 5th) only contribute to marginal time savings because of weak residual polarization (less than 10%). Figure 5B shows ten 1D spectra of GB1 acquired using the dual-receiver HC-MAeSTOSO-10 pulse sequence with 32 scans and using the first increment (t1 = 0). The integrated S/N values of 13C- and 1H-detected spectra are shown in Figure 5B. In the 1H spectra, the water peak (indicated by an asterisk in Figure 5B) is not completely suppressed, which is probably due to the additional 1H pulses required for the RFDR mixing period. As shown in Figure 5B, in spite of the weak residual polarization, the S/N for the 1H-detected spectra in the 3rd and 4th acquisitions are comparable to those obtained with the 13C-detected experiments recorded in the 1st and 2nd acquisitions. Note that the application of (H)NHH and (H)CANHH 2D or 3D experiments requires higher proton resolution (~100 Hz 1H linewidth), which is achievable using combination high field NMR and fast MAS rates. In fact, 3D (H)NHH and (H)CHH experiments were utilized successfully to measure H-H distances for GB1 protein at 1 GHz using MAS rate of 100 kHz44.
3.5. 1H-, 13C-, and 15N-detected DUMAS (HCN-DUMAS) with triple receiver
Previous POE were designed using 13C and 15N polarization obtained from the 1H spin bath via a SIM-CP preparation period23. Using a triple receiver, we are able to assess both 13C and 15N transferred polarization as well as the residual 1H polarization that remains at the end of a SIM-CP period. This experiment is similar to the first multi-acquisition CP experiment on small organic molecules carried out with residual 1H polarization under CW decoupling57. In general, following a CP or SIM-CP period a residual polarization is left on 1H nuclei. This phenomenon can be understood by analyzing the CP dynamics in DQ (double quantum) and ZQ (zero quantum) sub-spaces58–60, where only a portion of the 1H spin polarization, namely the ZQ operator, is perpendicular to the ZQ dipolar Hamiltonian and evolves during CP. The oscillatory nature of the transferred 13C and 15N polarization is significantly damped by strong 1H-1H spin diffusion and T1ρ relaxation mechanisms, which leads to incomplete polarization transfer from 1H to 13C and 15N61. Even with fast MAS rates, strong HH dipolar couplings of fully protonated samples affect the CP transfer efficiency. In fact, these CP oscillations can be observed in samples where dipolar couplings are significantly scaled-down as for liquid crystals59,61.
Figure 6A shows the SIM-CP pulse sequence for simultaneous acquisition of residual 1H and transferred 13C and 15N polarization. After a SIM-CP period, the 1H receiver detects the residual polarization; whereas the 13C and 15N transferred polarization is stored along the z-direction by 90° pulses. After the 1H acquisition, another pair of 90° pulses on 13C and 15N creates transverse polarization that is simultaneously detected using 13C and 15N receivers, while decoupling the 1H channel with WALTZ16 sequence. Note that for protein samples, 1H detection requires solvent suppression using hard pulses (MISSISSIPPI), which dephase 1H residual polarization. For this reason, we chose a powdered sample of fMLF that does not require solvent suppression62. The pulse sequence of Figure 6A was carried out on the fMLF sample by varying the SIM-CP contact time (t). The integrated intensities for 1H, 13C, and 15N resonances were measured at various t and plotted in Figure 6B. The 1D spectra at t=0 and 1 ms are shown in Figure 6C. At t=0, maximum 1H intensity was observed, whereas 13C and 15N show no signal. The SIM-CP intensities of 13C and 15N gradually increases at higher t. For t=1 ms, both 13C and 15N intensities reached a plateau. Interestingly, the integrated 1H intensity from residual polarization drops down to 32% for side chain protons (−3 to 3 ppm), and to 18% for protons resonating between 3 to 10 ppm.
Figure 6:
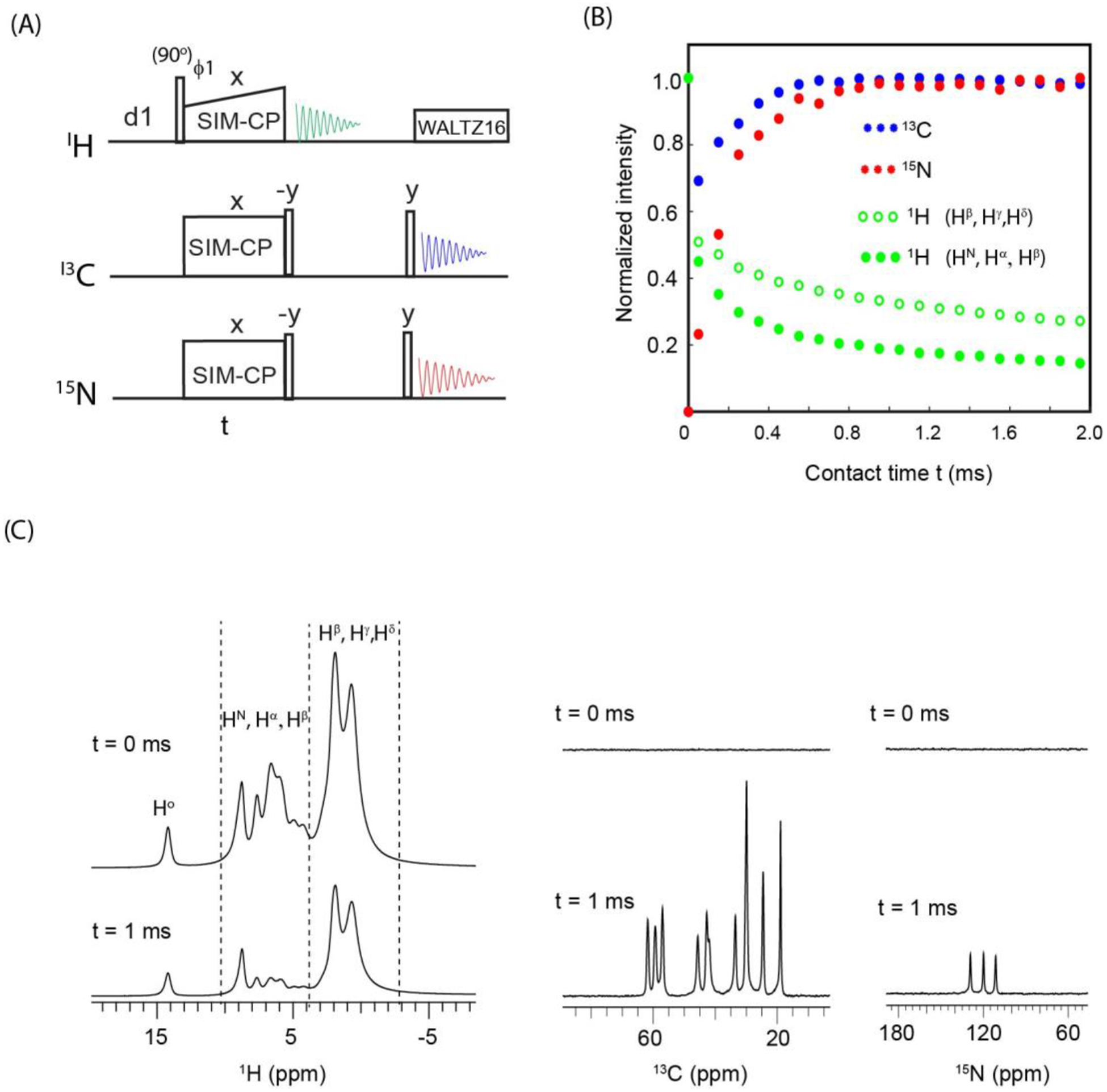
(A) 1D triple receiver SIM-CP experiment for monitoring residual 1H, and transferred 13C and 15N polarization. (B) Plot of the normalized intensities for SIM-CP polarization pathways obtained from the integrated intensities of 1H, 13C, and 15N 1D spectra of fMLF tripeptide sample, at contact times ranging from 0 to 2 ms. (C) 1H, 13C, and 15N 1D spectra at SIM-CP contact times 0 and 1 ms.
The SIM-CP sequence can be exploited to acquire three 2D homonuclear correlation experiments (Figure 7A) using two acquisition periods. In this pulse sequence, namely HCN-DUMAS, 13C and 15N polarization is stored along the z-direction after SIM-CP. The residual 1H polarization is evolved during a t1 period followed by a 90° pulse, a 1H-1H RFDR mixing, and another 90° pulse. Then, the 1H receiver records a 2D 1H-1H correlation experiment in the 1st acquisition period (t2H). After the 1H acquisition, the 13C polarization is evolved during t1 followed by a 90° pulse and TOCSY mixing. At the end of the TOCSY mixing period, the 13C polarization is kept along the z-direction by setting the duration of the TOCSY mixing to ‘n/4’ times of the WALTZ16 period, where ‘n’ is an integer28. After the 13C TOCSY mixing, the 15N z-polarization is flipped into the transverse plane by a 90° pulse and then evolved for a t1 period. Unlike 1H and 13C, the homonuclear dipolar- and J- couplings for 15N nuclei are very weak. Therefore, homonuclear 15N-15N transfer is achieved through protons using the NHHN sequence, which consists of a CP transfer from 15N to 1H followed by a HH RFDR mixing and another CP from 1H to 15N56,63,64. After the NHHN transfer steps, a 90° pulse on 13C flips the z-polarization to the transverse plane, and 15N and 13C polarization is then detected in the 2nd acquisition period by separate receivers under 1H decoupling, yielding 2D 13C-13C and 15N-15N homonuclear correlation spectra. Figure 7B shows the 2D HH, (H)CXCX, and N(HH)N homonuclear correlation spectra of the fMLF peptide, acquired simultaneously using the HCN-DUMAS pulse sequence. The successful implementation of double and triple receiver experiments relies on proper optimization of t1 evolution parameters for all nuclei. The t1 dwell-time is inversely proportional to the spectral width, whereas the number of t1 increments depends on T2 relaxation.
As for other POE, the HCN-DUMAS experiment can be implemented with separate t1 evolution parameters for 1H, 13C, and 15N nuclei (t1H, t1C, and t1N in Figure 7A). As shown in Table 2, the t1 evolution parameters satisfy the condition, ni(t1C) = n1 * ni(t1H) = n2 * ni(t1N). The spectra of Figure 7B were recorded using 80 μs t1 dwell time for 1H and 13C (Table 2). The 13C polarization was evolved for 7.9 ms with a maximum t1 evolution obtained with 100 t1 increments. For 1H evolution, four identical t1 periods (n1=4) were implemented using 25 t1 increments that corresponds to a maximum 1H t1 evolution of 1.92 ms. Similarly, for 15N t1 evolution, the dwell-time was set to 320 μs and four identical t1 periods (n2=4) were implemented using 24 increments with a maximum t1 evolution of 7.68 ms (see Table 2). Note that for fully protonated samples, 1H T2 relaxation is relatively short compared to 13C and 15N. Therefore, the HH 2D spectrum of Figure 7B was recorded with a shorter t1 evolution time (1.92 ms). The four identical 2D N(HH)N and HH data sets were added during the processing. The 1H-1H RFDR mixing times for HH and NHHN experiments were set to 0.96 and 1.92 ms, respectively. The 13C-13C TOCSY mixing was set to 12 ms, corresponding to five cycles of WALTZ16. Inter- and intra-residue correlations are shown in Figure 7C together with 1D cross-sections of the indirect dimensions for H, Cα, and N resonances of the Leu residues. The 2D N(HH)N spectrum of fMLF showed intense cross-peaks between the neighboring residues pairs, namely M-L and L-F, and relatively weaker cross-peaks between the 1st and 3rd residues (M and F). In the 2D CC spectrum, we observed total correlation of intra-residue 13C atoms for each of the three residues. In general, the peak intensities of homonuclear correlation spectra are similar for lower and upper off-diagonal cross-peaks. However, in the 2D HH spectrum obtained from HCN-DUMAS, the intensities of lower off-diagonal cross peaks (between 3 to 10 ppm along indirection 1H dimension) are much lower than the upper off-diagonal cross-peaks (0–3 ppm) that originated from high intensity sidechain protons (Figure 6C). Nevertheless, the high sensitivity of 1H detection leads to 1H cross peaks in both upper and lower off-diagonal regions of 2D spectra, as shown in the 1D cross sections of Figure 7C.
Using the HCN-DUMAS strategy, other combinations of 2D experiments can be designed. For example, the 1H residual polarization can be exploited to acquire a DQSQ experiment in the 1st acquisition65. Similarly, inter-residue CC correlations can be obtained in the 13C acquisition by replacing the TOCSY with a C(HH)C mixing period. The 2D HCN-DUMAS spectra of the fMLF sample (Figure 7B) were acquired using 8 scans for a total experimental time of 1.4 h. As shown in Figure 7C, in spite of using residual 1H polarization, the high sensitivity of 1H channel leads to higher S/N of HH cross peaks compared to the (H)CXCX and N(HH)N spectra. While the N(HH)N spectrum can be highly informative from having narrower linewidths, its feasibility for protein samples would be limited since longer experimental times are required to overcome the poor sensitivity associated with 15N-detection. The HCN-DUMAS can also be implemented with multiple 1H- and 13C-detected experiments, while acquiring the same N(HH)N experiment with 15N detection. In this experimental setup, HCN-DUMAS will have to be coded as two separate pulse sequences in two separate scripts, each with different HH and CC mixing times, but sharing the same NHHN mixing time. The two 2D HH and CC experiments would be processed as separate experiments, while the two resulting N(HH)N data sets can be added for signal enhancement. A similar experimental setup was previously described for a single receiver 13C-detected DUMAS experiment, which records 2D NCA and NCO spectra while acquiring a single DARR spectrum53.
4. Discussion
Multi-receiver technology represents a stepping-stone to expand the applications of POE methods to various multi-dimensional ssNMR experiments. To determine the efficiency of multi-receiver experiments, we used microcrystalline GB1 protein and fMLF tripeptide samples. The relative sensitivity of multi-receiver experiments depend on different parameters such as the gyromagnetic ratio, length of pulse sequences, as well as probe and RF console features such as quality factor (Q) and receiver digitization66–68. In general, the main drawback of multi-receiver experiments is the intrinsic low sensitivity of low γ nuclei such as 13C and 15N. Therefore, dual and multi-receiver experiments need to be acquired with a high number of scans to accumulate sufficient polarization for 13C and 15N-detected spectra, while highly sensitive 1H spectra would require far fewer scans68,69. Nevertheless, POE methods provide new ways to optimize the sensitivity, and therefore, make full use of multi-receiver technology.
As shown in Figure 2, the 2D (H)NH is a highly sensitive experiment, therefore, a 1H-detected (H)NH experiment is combined with another highly sensitive 13C-detected experiment such as CACO using HC-DUMAS strategy. On the other hand, the 13C-detected (H)CXCX and (H)N(CA)CX are low-sensitivity experiments due to NCA and CXCX transfer periods. Therefore, in the HC-MEIOSIS experiment (Figure 3), (H)CXCX and (H)N(CA)CX are combined with another pair of low-sensitivity 1H detected experiments (H)CA(N)H and (H)NH that utilize transferred and residual polarization from the SPECIFIC-CP period. Similarly, in the HC-MAeSTOSO-4 experiment (Figure 4), the low-sensitivity 13C-detected (H)CXCX experiment is combined with three other low-sensitivity 1H-detected experiments, (H)N(CACB)H, (H)N(CA)H, and (H)NH11. In comparison to single acquisition experiments, the time saving of HC-DUMAS is ~50%; whereas the simultaneous acquisition of four to ten spectra with HC-MEIOSIS and HC-MAeSTOSO lead to approximately 53 to 62% reduction in acquisition time. The application of triple receiver HCN-DUMAS experiment requires highly concentrated samples to achieve sufficient S/N for 15N-detected spectra. An alternative way to optimize the experimental time for HCN-DUMAS is to record multiple 2D HH and CC experiments, while acquiring a single N(HH)N experiment53. However, for fully protonated samples, broad 1H linewidths (200–400 Hz) represent a major drawback and lead to poorly resolved HH cross-peaks (Figure 7C). On the other hand, the 1H linewidths can be significantly narrowed for perdeuterated protein samples that give high resolution 1H spectra with 30 to 50 Hz 1H linewidths even at lower MAS rates of 30 to 40 kHz70–72. In comparison to fully protonated samples, the efficiency of SIM-CP is lower for perdeuterated samples due to the diluted 1H spin bath. As a future development of our approach, we will be testing the efficiency of SIM-CP and POE methods on fractionally perdeuterated protein samples.
High sensitivity and resolution are fundamental spectral requirements for the atomic resolution analysis of proteins by NMR spectroscopy. In spite of the low sensitivity, 13C-detected experiments are routinely used in solution- and ssNMR. In fact, 13C-detection offers unique advantages such as narrower linewidths and broad spectral dispersion. For example, in solution NMR, 13C-detected CON and CAN based experiments are used for analyzing protein samples (such as IDPs or intrinsically disordered proteins) that are poorly dispersed in the 1H dimension73,74. Similarly, 1H-detected fast MAS ssNMR spectra, such as CP-HSQC, suffer from broader 1H linewidths in fully protonated samples. For the GB1 spectra shown in Figures 2–4, the number of resolved peaks in a 1H-detected (H)NH spectrum is 19 (Figure 2C). Whereas for Cα and CO spectral regions in the 13C-detected (H)N(CA)CX (Figure 3C) and (H)CACO (Figure 2C) spectra, number of resolved peaks are 43 and 26, respectively. Note that the 1H line widths of microcrystalline GB1 are much narrower compared to more challenging heterogeneous systems such as fibrillar and membrane-bound proteins75,76. In other words, the 1H resolution can be even more compromised for such large systems. In practice, before proceeding to spectral analysis, it is necessary to test several 1H- and 13C-detected 2D experiments to optimize both the sensitivity and resolution. In addition, we recommend testing a range of protein sample conditions. For example, in the case of membrane proteins, it is important to optimize temperature, spinning speed, lipid composition, protein-to-lipid ratio as well as hydration levels. To this extent, the multi-receiver 2D experiments can provide multiple protein fingerprints, giving a comprehensive view of the 1H, 13C, and 15N spectral quality, which will be instrumental to optimize sample preparation/conditions as well as designing 3D experiments. Similarly, the 2D fingerprints are routinely used for screening protein samples with various mutations as well as by titrating with ligand or drug molecules. Such applications can be envisioned with POE to evaluate biological processes by recording multiple protein fingerprints simultaneously.
POE using fast MAS rates and multi-receiver technology can unleash the full potential of ssNMR applications to biomacromolecules. Several solution-NMR pulse sequences, such as PANSY and afterglow, have been developed for multi-receiver experiments66,69,77,78, while relatively few have been developed for ssNMR due to the limitations of 1H-detected experiments for solid samples. However, recent developments in fast MAS probes and perdeuteration have increased the sensitivity and resolution of 1H-detected experiments. In this work, we implemented the POE strategies to various multi-receiver experiments and evaluated relative S/N and FWHM of 1H, 13C and 15N detected spectra. To the best of our knowledge, this is the first experimental demonstration of DUMAS, MEIOSIS, and MAeSTOSO methods using multi-receiver technology and ultrafast magic angle spinning. Dual and multi-receiver experiments have also been demonstrated by other research groups. For example, the simultaneous acquisition of 13C- and 15N-detected experiments were shown for spectrometers equipped with two receivers at slow MAS rates68. Also, a recent study shown the application of triple receiver experiments (19F, 31P, and 27Al) to organometallic compounds79. Similarly, window 1H detection was used for dual receiver DUMAS and MEIOSIS experiments at slow MAS rates32. Dual receiver experiments were also implemented by concatenation of a 1H-detected 3D with 13C-detected 2D experiments at fast MAS rates using CP as preparation period80. More recently, we also demonstrated the application of POE methods using single receiver 1H detected ultrafast MAS experiments28. Multiple polarization pathways can also be detected in a single acquisition using Hadamard encoding and decoding of spin operators81. Such experiments have been reported for oriented (static) as well as for MAS pulse sequences24,30,60,82,83. On the other hand, the pulse sequences reported in this manuscript, are based on the multi-acquisition and enables the incorporation of various CC, NC, and HH mixing periods. The experiments reported in this work will help select and design novel ultrafast MAS POE methods using dual and triple receiver experiments. Although in principle it is possible to concatenate several different pulse sequences, it is important to select the POE method based on their performance. Therefore, the assessment of the relative S/N for 1H, 13C, and 15N detected experiments presented here will be instrumental to choose and tailor the experiments that fit the features of the protein sample under analysis.
Since HC-MEIOSIS and HC-MAeSTOSO utilize multiple transferred and weak residual polarization pathways, these methods are suited for combining two to five pairs of low and high sensitivity experiments. On the other hand, the HC-DUMAS method uses high intensity transferred polarization pathways, and therefore more suitable for combining a pair of high sensitivity experiments such as (H)CACO and (H)NH. The HC-MAeSTOSO methods incorporate the elements of HC-DUMAS and HC-MEIOSIS, but uses weak residual polarization for the 3rd through 5th acquisitions periods (Figures 4 and 5). For this reason, the 1st and 2nd acquisitions of HC-MAeSTOSO, are used for recording low sensitivity experiments that require a high number of scans, and enables the accumulation of enough residual polarization for the 3rd, 4th, and 5th acquisitions. Finally, the dual- or multi-receiver experiments reported here can be implemented on spectrometers equipped with a single receiver setup similar to solution NMR UTOPIA pulse sequences78 by switching the single receiver back and forth for 1H, 13C, and 15N acquisitions. Considering the advantages of 13C detection, we anticipate that the benefits of multi-receiver POE methods will be further enhanced when fast MAS probes are developed with improved 13C sensitivity.
5. Conclusions
POE methods allow the concatenation of almost all types of biomolecular ssNMR experiments, boosting the capability of ssNMR spectrometers at least two-fold. The combination of POE and multiple receivers opens up new horizons for pulse sequence development and propels the spectroscopic characterization of complex biomolecular systems. In this work, we have developed a new sub-class of POE (HC-DUMAS, HC-MEIOSIS, HC-MAeSTOSO, and HCN-DUMAS) with multiple receivers and ultrafast magic angle spinning to combine the benefits of 1H-, 13C-, and 15N-detected experiments. We anticipate that these experiments will be even more powerful when combined with other methods such as PRE, DNP, and non-uniform sampling.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institute of Health (GM 64742, HL 144130, 1S10OD021536 to G.V), the American Heart Association (19POST34420009 to D.W.), and the Minnesota NMR Center. We also thank Dr. J. Struppe and Dr. S. Pawsey from Bruker R&D for helpful discussions.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Andronesi OC et al. Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J Am Chem Soc 127, 12965–74 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Hu F, Luo W & Hong M Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science 330, 505–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma M et al. Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer. Science 330, 509–12 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SH et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 491, 779–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gustavsson M et al. Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban. Proc Natl Acad Sci U S A 110, 17338–43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wylie BJ et al. Ultrahigh resolution protein structures using NMR chemical shift tensors. Proc Natl Acad Sci U S A 108, 16974–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiang W, Yau WM, Lu JX, Collinge J & Tycko R Structural variation in amyloid-beta fibrils from Alzheimer’s disease clinical subtypes. Nature 541, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wasmer C et al. Amyloid fibrils of the HET-s(218–289) prion form a beta solenoid with a triangular hydrophobic core. Science 319, 1523–6 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Castellani F et al. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 420, 98–102 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Gan Z et al. NMR spectroscopy up to 35.2T using a series-connected hybrid magnet. J Magn Reson 284, 125–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbet-Massin E et al. Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J Am Chem Soc 136, 12489–97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maly T et al. Dynamic nuclear polarization at high magnetic fields. J Chem Phys 128, 052211 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wickramasinghe NP et al. Nanomole-scale protein solid-state NMR by breaking intrinsic 1HT1 boundaries. Nat Methods 6, 215–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penzel S et al. Spinning faster: protein NMR at MAS frequencies up to 126 kHz. J Biomol NMR 73, 19–29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Struppe J et al. Expanding the horizons for structural analysis of fully protonated protein assemblies by NMR spectroscopy at MAS frequencies above 100 kHz. Solid State Nucl Magn Reson 87, 117–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gor’kov PL et al. Using low-E resonators to reduce RF heating in biological samples for static solid-state NMR up to 900 MHz. J Magn Reson 185, 77–93 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Stringer JA et al. Reduction of RF-induced sample heating with a scroll coil resonator structure for solid-state NMR probes. J Magn Reson 173, 40–8 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Xue K et al. Limits of Resolution and Sensitivity of Proton Detected MAS Solid-State NMR Experiments at 111 kHz in Deuterated and Protonated Proteins. Sci Rep 7, 7444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbet-Massin E et al. Out-and-back 13C-13C scalar transfers in protein resonance assignment by proton-detected solid-state NMR under ultra-fast MAS. J Biomol NMR 56, 379–86 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Pinto C et al. Formation of the beta-barrel assembly machinery complex in lipid bilayers as seen by solid-state NMR. Nat Commun 9, 4135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gopinath T & Veglia G Probing membrane protein ground and conformationally excited states using dipolar- and J-coupling mediated MAS solid state NMR experiments. Methods 148, 115–122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gopinath T & Veglia G Dual acquisition magic-angle spinning solid-state NMR-spectroscopy: simultaneous acquisition of multidimensional spectra of biomacromolecules. Angew Chem Int Ed Engl 51, 2731–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gopinath T & Veglia G Orphan Spin Polarization: A Catalyst for High-Throughput Solid-State NMR Spectroscopy of Proteins. Annual Reports on NMR Spectroscopy 89, 103–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gopinath T & Veglia G Orphan spin operators enable the acquisition of multiple 2D and 3D magic angle spinning solid-state NMR spectra. J Chem Phys 138, 184201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gopinath T & Veglia G Multiple acquisitions via sequential transfer of orphan spin polarization (MAeSTOSO): How far can we push residual spin polarization in solid-state NMR? J Magn Reson 267, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gopinath T & Veglia G Experimental Aspects of Polarization Optimized Experiments (POE) for Magic Angle Spinning Solid-State NMR of Microcrystalline and Membrane-Bound Proteins. Methods Mol Biol 1688, 37–53 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Gopinath T, Wang S, Lee J, Aihara H & Veglia G Hybridization of TEDOR and NCX MAS solid-state NMR experiments for simultaneous acquisition of heteronuclear correlation spectra and distance measurements. J Biomol NMR 73, 141–153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gopinath T & Veglia G Proton-detected polarization optimized experiments (POE) using ultrafast magic angle spinning solid-state NMR: Multi-acquisition of membrane protein spectra. J Magn Reson 310, 106664 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banigan JR & Traaseth NJ Utilizing afterglow magnetization from cross-polarization magic-angle-spinning solid-state NMR spectroscopy to obtain simultaneous heteronuclear multidimensional spectra. J Phys Chem B 116, 7138–44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das BB & Opella SJ Simultaneous cross polarization to (13)C and (15)N with (1)H detection at 60kHz MAS solid-state NMR. J Magn Reson 262, 20–26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellstedt P et al. Solid state NMR of proteins at high MAS frequencies: symmetry-based mixing and simultaneous acquisition of chemical shift correlation spectra. J Biomol NMR 54, 325–35 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Sharma K, Madhu PK & Mote KR A suite of pulse sequences based on multiple sequential acquisitions at one and two radiofrequency channels for solid-state magic-angle spinning NMR studies of proteins. J Biomol NMR 65, 127–141 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Zhang R, Mroue KH & Ramamoorthy A Hybridizing cross-polarization with NOE or refocused-INEPT enhances the sensitivity of MAS NMR spectroscopy. J Magn Reson 266, 59–66 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gopinath T, Nelson SED & Veglia G (1)H-detected MAS solid-state NMR experiments enable the simultaneous mapping of rigid and dynamic domains of membrane proteins. J Magn Reson 285, 101–107 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fusco G et al. Direct observation of the three regions in alpha-synuclein that determine its membrane-bound behaviour. Nat Commun 5, 3827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gayen A, Leninger M & Traaseth NJ Protonation of a glutamate residue modulates the dynamics of the drug transporter EmrE. Nat Chem Biol 12, 141–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franks WT et al. Magic-angle spinning solid-state NMR spectroscopy of the beta1 immunoglobulin binding domain of protein G (GB1): 15N and 13C chemical shift assignments and conformational analysis. J Am Chem Soc 127, 12291–305 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Schmidt HL et al. Crystal polymorphism of protein GB1 examined by solid-state NMR spectroscopy and X-ray diffraction. J Phys Chem B 111, 14362–9 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou DH & Rienstra CM High-performance solvent suppression for proton detected solid-state NMR. J Magn Reson 192, 167–72 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hartmann SR & Hahn EL Nuclear Double Resonance in the Rotating Frame. Physical Review 128, 2042–2053 (1962). [Google Scholar]
- 41.Baldus M, Petkova AT, Herzfeld J & Griffin RG Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Molecular Physics 95, 1197–1207 (1998). [Google Scholar]
- 42.Verel R, Baldus M, Nijman M, van Os JWM & Meier BH Adiabatic homonuclear polarization transfer in magic-angle-spinning solid-state NMR. Chemical Physics Letters 280, 31–39 (1997). [Google Scholar]
- 43.Hu KN, Qiang W, Bermejo GA, Schwieters CD & Tycko R Restraints on backbone conformations in solid state NMR studies of uniformly labeled proteins from quantitative amide 15N-15N and carbonyl 13C-13C dipolar recoupling data. J Magn Reson 218, 115–27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andreas LB et al. Structure of fully protonated proteins by proton-detected magic-angle spinning NMR. Proc Natl Acad Sci U S A 113, 9187–92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nishiyama Y, Zhang R & Ramamoorthy A Finite-pulse radio frequency driven recoupling with phase cycling for 2D (1)H/(1)H correlation at ultrafast MAS frequencies. J Magn Reson 243, 25–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaka AJ, Keeler J, Frenkiel T & Freeman R An improved sequence for broadband decoupling: WALTZ-16. J Magn Reson 52, 335–38 (1983). [Google Scholar]
- 47.Cavanagh J, Fairbrother WJ, Palmer AG, Rance M & Skelton NJ Protein NMR Spectroscopy: Principles and Practice, 2nd Edition. Protein Nmr Spectroscopy: Principles and Practice, 2nd Edition, 1–888 (2007). [Google Scholar]
- 48.Delaglio FG, S; Vuister GW; Zhu G; Pfeifer J; and Bax A NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 49.Lee W, Tonelli M & Markley JL NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics 31, 1325–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Franks WT, Kloepper KD, Wylie BJ & Rienstra CM Four-dimensional heteronuclear correlation experiments for chemical shift assignment of solid proteins. J Biomol NMR 39, 107–31 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Nielsen AB et al. Simultaneous acquisition of PAR and PAIN spectra. J Biomol NMR 52, 283–8 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Struppe JO et al. Long-observation-window band-selective homonuclear decoupling: increased sensitivity and resolution in solid-state NMR spectroscopy of proteins. J Magn Reson 236, 89–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gopinath T & Veglia G Multiple acquisition of magic angle spinning solid-state NMR experiments using one receiver: application to microcrystalline and membrane protein preparations. J Magn Reson 253, 143–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lewandowski JR et al. Proton assisted recoupling at high spinning frequencies. J Phys Chem B 113, 9062–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fricke P et al. Backbone assignment of perdeuterated proteins by solid-state NMR using proton detection and ultrafast magic-angle spinning. Nat Protoc 12, 764–782 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Heise H, Seidel K, Etzkorn M, Becker S & Baldus M 3D NMR spectroscopy for resonance assignment and structure elucidation of proteins under MAS: novel pulse schemes and sensitivity considerations. J Magn Reson 173, 64–74 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Pines A, Gibby GM & Waugh JS Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys 59, 569–590 (1973). [Google Scholar]
- 58.Levitt MH, Suter D & Ernst RR Spin Dynamics and Thermodynamics in Solid-State Nmr Cross Polarization. Journal of Chemical Physics 84, 4243–4255 (1986). [Google Scholar]
- 59.Sinha N & Ramanathan KV Use of polarization inversion for resolution of small dipolar couplings in SLF-2D NMR experiments - an application to liquid crystals. Chemical Physics Letters 332, 125–130 (2000). [Google Scholar]
- 60.Gopinath T & Veglia G Sensitivity enhancement in static solid-state NMR experiments via single- and multiple-quantum dipolar coherences. J Am Chem Soc 131, 5754–6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muller L, Kumar A, Baumann T & Ernst RR Transient Oscillations in Nmr Cross-Polarization Experiments in Solids. Physical Review Letters 32, 1402–1406 (1974). [Google Scholar]
- 62.Rienstra CM et al. De novo determination of peptide structure with solid-state magic-angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A 99, 10260–5 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang R, Mroue KH & Ramamoorthy A Proton-Based Ultrafast Magic Angle Spinning Solid-State NMR Spectroscopy. Acc Chem Res 50, 1105–1113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wei YF & Ramamoorthy A 2D (15)N-(15)N isotropic chemical shift correlation established by (1)H-(1)H dipolar coherence transfer in biological solids. Chemical Physics Letters 342, 312–316 (2001). [Google Scholar]
- 65.Zhang R, Duong NT, Nishiyama Y & Ramamoorthy A 3D Double-Quantum/Double-Quantum Exchange Spectroscopy of Protons under 100 kHz Magic Angle Spinning. J Phys Chem B 121, 5944–5952 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Kupce E NMR with Multiple Receivers. Modern Nmr Methodology 335, 71–96 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Hong M & Yamaguchi S Sensitivity-enhanced static N-15 NMR of solids by H-1 indirect detection. Journal of Magnetic Resonance 150, 43–48 (2001). [DOI] [PubMed] [Google Scholar]
- 68.Herbst C et al. MAS solid state NMR of RNAs with multiple receivers. J Biomol NMR 41, 121–5 (2008). [DOI] [PubMed] [Google Scholar]
- 69.Kupce E, Kay LE & Freeman R Detecting the “Afterglow” of C-13 NMR in Proteins Using Multiple Receivers. Journal of the American Chemical Society 132, 18008–18011 (2010). [DOI] [PubMed] [Google Scholar]
- 70.Zhou DH, Graesser DT, Franks WT & Rienstra CM Sensitivity and resolution in proton solid-state NMR at intermediate deuteration levels: quantitative linewidth characterization and applications to correlation spectroscopy. J Magn Reson 178, 297–307 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Akbey U et al. Optimum levels of exchangeable protons in perdeuterated proteins for proton detection in MAS solid-state NMR spectroscopy. J Biomol NMR 46, 67–73 (2010). [DOI] [PubMed] [Google Scholar]
- 72.Chevelkov V, Rehbein K, Diehl A & Reif B Ultrahigh resolution in proton solid-state NMR spectroscopy at high levels of deuteration. Angewandte Chemie-International Edition 45, 3878–3881 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Chaves-Arquero B et al. A CON-based NMR assignment strategy for pro-rich intrinsically disordered proteins with low signal dispersion: the C-terminal domain of histone H1.0 as a case study. Journal of Biomolecular Nmr 72, 139–148 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Novacek J et al. 5D 13C-detected experiments for backbone assignment of unstructured proteins with a very low signal dispersion. J Biomol NMR 50, 1–11 (2011). [DOI] [PubMed] [Google Scholar]
- 75.Lalli D et al. Proton-Based Structural Analysis of a Heptahelical Transmembrane Protein in Lipid Bilayers. J Am Chem Soc 139, 13006–13012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loquet A et al. 3D structure determination of amyloid fibrils using solid-state NMR spectroscopy. Methods 138–139, 26–38 (2018). [DOI] [PubMed] [Google Scholar]
- 77.Kupce E & Freernan R Molecular structure from a single NMR experiment. Journal of the American Chemical Society 130, 10788–10792 (2008). [DOI] [PubMed] [Google Scholar]
- 78.Viegas A et al. UTOPIA NMR: activating unexploited magnetization using interleaved low-gamma detection. J Biomol NMR 64, 9–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martineau C, Decker F, Engelke F & Taulelle F Parallelizing acquisitions of solid-state NMR spectra with multi-channel probe and multi-receivers: Applications to nanoporous solids. Solid State Nuclear Magnetic Resonance 55–56, 48–53 (2013). [DOI] [PubMed] [Google Scholar]
- 80.Gallo A, Franks WT & Lewandowski JR A suite of solid-state NMR experiments to utilize orphaned magnetization for assignment of proteins using parallel high and low gamma detection. J Magn Reson 305, 219–231 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Gopinath T & Kumar A Hadamard NMR spectroscopy for two-dimensional quantum information processing and parallel search algorithms. J Magn Reson 183, 259–268 (2006). [DOI] [PubMed] [Google Scholar]
- 82.Gopinath T, Mote KR & Veglia G Proton evolved local field solid-state nuclear magnetic resonance using Hadamard encoding: theory and application to membrane proteins. J Chem Phys 135, 074503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sharma K, Madhu PK, Agarwal V & Mote KR Simultaneous recording of intra- and inter-residue linking experiments for backbone assignments in proteins at MAS frequencies higher than 60 kHz. J Biomol NMR (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.