

ABSTRACT
Background:
Sepsis is a life-threatening organ dysfunction initiated by a dysregulated response to infection, with imbalanced inflammation and immune homeostasis. Macrophages play a pivotal role in sepsis. N-[1-(1-oxopropyl)-4-piperidinyl]-N’-[4-(trifluoromethoxy)phenyl)-urea (TPPU) is an inhibitor of soluble epoxide hydrolase (sEH), which can rapidly hydrolyze epoxyeicosatrienoic acids (EETs) to the bio-inactive dihydroxyeicosatrienoic acids. TPPU was linked with the regulation of macrophages and inflammation. Here, we hypothesized that sEH inhibitor TPPU ameliorates cecal ligation and puncture (CLP)-induced sepsis by regulating macrophage functions.
Methods:
A polymicrobial sepsis model induced by CLP was used in our study. C57BL/6 mice were divided into four groups: sham+ phosphate buffer saline (PBS), sham+TPPU, CLP+PBS, CLP+TPPU. Mice were observed 48 h after surgery to assess the survival rate. For other histological examinations, mice were sacrificed 6 h after surgery. Macrophage cell line RAW264.7 was used for in vitro studies.
Results:
TPPU treatment, accompanied with increased EETs levels, markedly improved the survival of septic mice induced by CLP surgery, which was associated with alleviated organ damage and dysfunction triggered by systemic inflammatory response. Moreover, TPPU treatment significantly inhibited systemic inflammatory response via EETs-induced inactivation of mitogen-activated protein kinase signaling due to enhanced macrophage phagocytic ability and subsequently reduced bacterial proliferation and dissemination, and decreased inflammatory factors release.
Conclusion:
sEH inhibitor TPPU ameliorates cecal ligation and puncture-induced sepsis by regulating macrophage functions, including improved phagocytosis and reduced inflammatory response. Our data indicate that sEH inhibition has potential therapeutic effects on polymicrobial-induced sepsis.
Keywords: CLP, EETs, macrophages, sepsis, TPPU
Abbreviations: ALT, alanine aminotransferase, AST, aspartate aminotransferase, BUN, blood urea nitrogen, CLP, cecum ligation and puncture, CYP2J2, cytochrome P450 epoxygenase 2J2, DHETs, dihydroxyeicosatrienoic acids, EETs, epoxyeicosatrienoic acids, Scr, plasma creatinine, sEH, soluble epoxide hydrolase
INTRODUCTION
Sepsis is a life-threatening organ dysfunction initiated by a dysregulated response to infection, with an imbalance of inflammation and immune homeostasis (1), and is one of the leading causes of death in intensive care units (2, 3).
Historically, experts have argued that sepsis is a systemic inflammatory response elicited by excessive microorganism invasion; thus, many investigations have focused on the pro-inflammatory and anti-inflammatory systems. From this standpoint, investigators expected to improve the survival rate of sepsis by eliminating or blocking TNF-α and IL-1β signaling pathways, but made little progress (4, 5). Recent studies indicate that macrophages play a pivotal role in recognizing and eliminating pathogens through their diverse surface receptors and phagocytic function, thus preventing the occurrence of sepsis (6–8). Further, enhanced macrophage phagocytic and antimicrobial functions significantly lowered the mortality rate in sepsis, whereas blocking this process led to bacterial proliferation and spread, which may be regulated by surrounding molecules such as ATP, IL-4, or carbon monoxide (3, 9–11). Similarly, another study reported that disruption of macrophage phagocytic and migratory functions by destroying their actin cytoskeletal dynamics resulted in enhanced susceptibility to pathogen infection and increased mortality rate of sepsis (12). All these investigations indicate that improving macrophage function is a promising therapeutic strategy for sepsis, but its exact mechanisms remain elusive.
Epoxyeicosatrienoic acids (EETs), produced by cytochrome P450 epoxygenase that metabolizes arachidonic acid, can be rapidly hydrolyzed to bio-inactive dihydroxyeicosatrienoic acids (DHETs) by soluble epoxide hydrolase (sEH). TPPU, N-[1-(1-oxopropyl)-4-piperidinyl]-N’-[4-(trifluoromethoxy)phenyl)-urea, is an inhibitor of sEH that is capable of dampening EETs hydrolysis, thus improving its stabilization and levels (13–16). Previous studies showed that EETs and sEH inhibitors mitigated the progression of lipopolysaccharide (LPS)-induced sepsis, at least partly by regulating systemic inflammation and reducing organ dysfunction (17–19). Another investigation revealed that EETs modulated macrophage polarization and function, indicating that EETs also participated in regulating innate immune cells (20). However, whether EETs can alleviate polymicrobial infection-induced sepsis and its underlying mechanisms remains unknown. In this study, we hypothesized that sEH inhibitor, TPPU, ameliorates cecal ligation and puncture (CLP)-induced sepsis by regulating macrophage functions, including enhancing their phagocytosis function and subsequently reducing the inflammatory response.
MATERIALS AND METHODS
Animals
C57BL/6 male mice aged 8 to 12 weeks and weighing 23 g to 25 g were housed in a temperature-controlled room under a 12-h light–dark cycle with free access to drinking water and normal chow. All the care and experimental procedures of mice were approved by the Experimental Animal Research Committee of Tongji Medical College, Huazhong University of Science and Technology, and were performed in accordance with ARRIVE guidelines developed by the National Center for the Replacement, Refinement, and Reduction of Animals in Research. Mice were orally administered TPPU (Cayman Chemical, Ann Arbor, Mich) at a dose of 3 mg/kg/day or phosphate buffer saline (PBS) via gavage for 5 consecutive days (14). For survival rate estimation, 10 mice were used in each group whereas five mice per group were used for other experiments.
Establishment of sepsis model induced by cecal ligation and puncture
After oral administration of TPPU or PBS, cecal ligation and puncture (CLP) or sham surgery was performed as described previously (8, 10). Briefly, mice were anesthetized with pentobarbital (50 mg/kg i.p.). The abdominal skin was sterilized with iodine and a 1 cm laparotomy was performed on the medioventral line at about 1 cm under the xiphoid process to expose the cecum. About four-fifths of the cecum was ligated using 3-0 silk suture and was punctured thrice with a 20 1/2-gauge needle. Feces in the ligated cecum were extruded gently and the cecum was then returned to the peritoneal cavity. Finally, the abdomen was closed in two layers. For sham surgery, mice were subjected to the same procedures except for cecal ligation and puncture. For mortality rate estimation, mice were monitored every 3 h for the first 12 h and then every 6 h until 48 h. For further experiments, mice were anesthetized with pentobarbital (50 mg/kg i.p.) 6 h after surgery, and blood, peritoneal lavage fluids and organs were harvested.
Aerobic bacterial quantification
To determine bacterial growth and dissemination levels, the aerobic bacteria in blood, peritoneal lavage fluids, and spleens were assessed.
Six hours after CLP or sham surgery, blood was collected in sterile tubes with previously added EDTA for anticoagulation. Whole blood was diluted 10- to 104-fold with sterile PBS, and 20 μL was immediately plated on 5% solid agar medium for bacterial culture. The remaining portion was centrifuged at 3,000 rpm for 8 min, and the plasma was then isolated for further experiments.
Mice peritoneum lavage fluids were obtained in 5 mL of ice-cold PBS. Lavage fluids were collected and filtered through a 70-μm nylon filter (BD Biosciences) to remove debris. Peritoneal lavage fluids from mice that underwent sham surgery were diluted by 10-fold, whereas fluids from mice that underwent CLP surgery were diluted by 102–104-fold with sterile PBS and 20 μL of each were immediately plated on solid medium for bacterial culture. The remaining portions were stored at −80°C for future experiments (21).
To evaluate the bacterial seeding in organs, spleens were excised and homogenized in 1 mL sterile PBS. After vortexing, 20 μL of the fluids were immediately used for bacterial culture (21).
All solid media were incubated overnight at 37°C, and colony-forming units (CFUs) were calculated according to the dilution fold.
Liver and renal function assessment
Blood samples were collected in sterile tubes with previously added EDTA for anticoagulation, and then centrifuged at 3,000 rpm for 8 min to separate plasma. The levels of plasma alanine aminotransferase (ALT), aspartate aminotransferase (AST), creatinine, and blood urea nitrogen (BUN) were determined to assess the liver and renal functions using assay kits from Nanjing Jiancheng Bioengineering Institute according to the manufacturer's instructions.
Enzyme-linked immunosorbent assay
Plasma and peritoneal lavage fluids were prepared as described above. Inflammatory factors including TNF-a, IL-1β, IL-6, and IL-10 in mice plasma, peritoneal lavage fluids, and macrophage supernatants were determined using enzyme-linked immunosorbent assay (ELISA) kits (Ex-Cell Bio, Suzhou, China) according to the manufacturer's instructions.
Plasma EETs and DHETs determination
Plasma samples were collected as described above. Plasma 14,15-EET and 14,15-DHET were determined using an ELISA kit (Detroit R&D Inc, Detroit, Mich) according to the manufacturer's instruction.
Histological analysis
At the end of the study, mice were sacrificed 6 h after surgery; the lung, liver, and kidney were harvested and fixed in 4% paraformaldehyde, and then embedded in paraffin. Next, 5 μm sections from the lung, liver, and kidney were separately stained with hematoxylin–eosin (H&E). Morphologic alterations were examined by light microscope at ×400 magnification as previously described (22). Lung injury score was calculated by the following categories: neutrophil infiltration, pulmonary edema, hemorrhage, and disorganization of lung parenchyma. 0 = normal, 1 = light, 2 = moderate, 3 = severe, 4 = very severe (22). All histologic studies were performed in a blinded fashion.
Cell culture and treatment
The mouse macrophage cell line RAW264.7 was purchased from ATCC and incubated in RPMI 1,640 with 10% fetal bovine plasma in a 37°C sterile incubator with 5% CO2. Cells were seeded in 6- or 24 -well plates and exposed to TPPU (1 μM, Cayman Chemical, Ann Arbor, Mich) or 14,15-EET (1 μM, Cayman Chemical, Ann Arbor, Mich) for 4 h after they reached 80% to 90% confluence. LPS (1 μg/mL) was then added and co-incubated for 12 h. 14,15-EEZE (an inhibitor of 14,15-EET, 1 μM, Cayman Chemical, Ann Arbor, Mich) was added 1 h before LPS stimulation. Cell supernatants were collected for inflammatory cytokines detection (20).
Phagocytosis assay
To evaluate macrophage phagocytic activity, 1-μm fluorescein isothiocyanate (FITC) fluorescent microspheres (Invitrogen, Carlsbad, Calif) were used. Briefly, fluorescent microspheres were preconditioned with 3% bovine plasma albumin and 5% fetal bovine serum and added to the macrophages at a ratio of 100:1. After 30 min of co-incubation, the microspheres were removed from the cell plates, and the macrophages were washed three times with RPMI 1,640 medium to remove un-phagocytosed microspheres. Macrophages were observed and photographed at 488-nm excitation and 525-nm emission wave lengths under a fluorescence microscope and at ×200 magnification under a light microscope. At least five randomized fields of view were chosen.
Flow cytometry analysis
After various stimulations as described above, macrophages were harvested and incubated with 1 μm FITC fluorescent microspheres (Invitrogen, Carlsbad, Calif) at a ratio of 100:1 as described above, for 30 min in the cell incubator. Then phagocytosis was stopped by placing samples on ice and non-phagocyted microspheres were removed by centrifugation at 1,000 rpm for 5 min. Macrophages were washed thrice and resuspended in 0.2 mL PBS for flow cytometry analysis on a BD FACSort (BD Biosciences, San Jose, Calif) and the percentage of FITC+ cells was calculated.
Western blot
Total protein of macrophages was extracted using radio-immunoprecipitation assay lysis buffer according to the manufacturer's instruction (Beyotime, Shanghai, China). Proteins were separated by 10% SDS-PAGE, transferred onto polyvinylidene difluoride membranes, and incubated overnight with primary antibodies at 4°C. Rabbit polyclonal antibodies against phosphor-P38 (p-P38) (#4511T), P38 (t-P38) (#8690T), phosphor-JNK (p-JNK) (#9255S), phosphor-ERK (p-ERK) (#4370T), ERK (t-ERK) (#4695T), and glyceraldehyde-phosphate dehydrogenase (#5174T) were purchased from Cell Signaling Technology. Mouse polyclonal antibody against JNK (t-JNK) (#9252T) was also purchased from Cell Signaling Technology. Secondary antibodies were added and co-incubated for 2 h at room temperature. Protein bands were visualized using ECL detection reagent. The protein expression was quantified by densitometry and normalized to glyceraldehyde-phosphate dehydrogenase.
Statistical analysis
All data are expressed as means ± SEM and analyzed with SPSS 19.0 statistical software. Survival rates were determined by the Kaplan–Meier method and compared using the log-rank test, and other statistical differences were assessed by one-way ANOVA with post hoc analyses performed using the Student–Newman–Keuls method. P < 0.05 was considered statistically significant.
RESULTS
TPPU treatment decreased the mortality of septic mice induced by CLP
To assess the potential effects of TPPU treatment on sepsis, a polymicrobial sepsis model was generated by CLP surgery as described above. About 80% of the mice died in the first 24 h in the PBS-treated sepsis group and by 48 h, none of the mice remained alive. However, the survival rate of TPPU-treated septic mice was approximate 20% in the first 48 h after CLP surgery as shown in Figure 1. These data suggest that TPPU treatment improves the survival of septic mice induced by CLP surgery.
Fig. 1.
TPPU treatment decreased the mortality of septic mice induced by CLP.
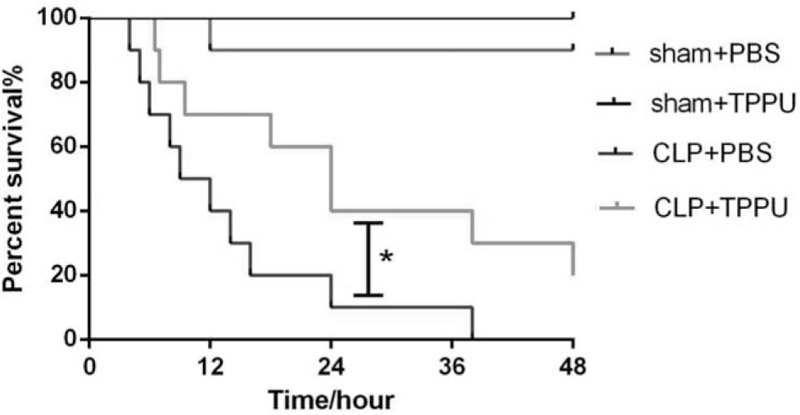
Survival curve of mice pretreated with TPPU or PBS following CLP or sham surgery. For the generation of the survival curve, 10 mice in each group were used. Data are expressed as means ± SEM. ∗P < 0.05 versus CLP+PBS group. CLP indicates cecal ligation and puncture; PBS, phosphate buffer saline.
TPPU treatment inhibited organ dysfunction in septic mice induced by CLP
The degree of organ dysfunction induced by sepsis is closely related to the mortality rate, and even a modest degree of organ dysfunction results in 10% mortality in patients hospitalized with infection (1, 4). In this study, organ function was assessed in septic mice. As shown in Figure 2A, histopathological features of organ damage were clearly observed in CLP-induced septic mice, compared with those in sham groups. In detail, less leukocyte infiltration (mainly neutrophils) in alveoli and lower alveolar wall thickness accompanied by a decreased lung injury score were observed in TPPU-treated septic mice, compared with those in PBS-treated septic mice, based on the results of lung sections stained with H&E (Fig. 2, A and B). Similarly, less peri-vascular leukocyte infiltration (mainly neutrophils) and edema were observed in liver sections from groups of TPPU-treated septic mice, compared with those in PBS-treated septic mice. In addition, less leukocyte infiltration (mainly neutrophils) was observed in glomerulus of kidney sections from groups of TPPU-treated septic mice, compared with those in PBS-treated septic mice. Moreover, liver and renal functions were also determined. As expected, increased ALT and AST levels were observed in the CLP+PBS group, compared with that in the sham+PBS group; interestingly, TPPU treatment markedly decreased the levels of ALT and AST in the CLP+TPPU group, compared with that in the CLP+PBS group as shown in Figure 2C and D. Similarly, increased plasma BUN and creatinine levels were observed in the CLP+PBS group, compared with that in the sham+PBS group; interestingly, TPPU treatment markedly decreased the levels of BUN and creatinine in the CLP+TPPU group, compared with that in the CLP+PBS group as shown in Figure 2E and F. Taken together, these data indicate that TPPU treatment alleviates organ damage and dysfunction in septic mice induced by CLP surgery.
Fig. 2.
TPPU treatment inhibited organ dysfunction in septic mice induced by CLP.
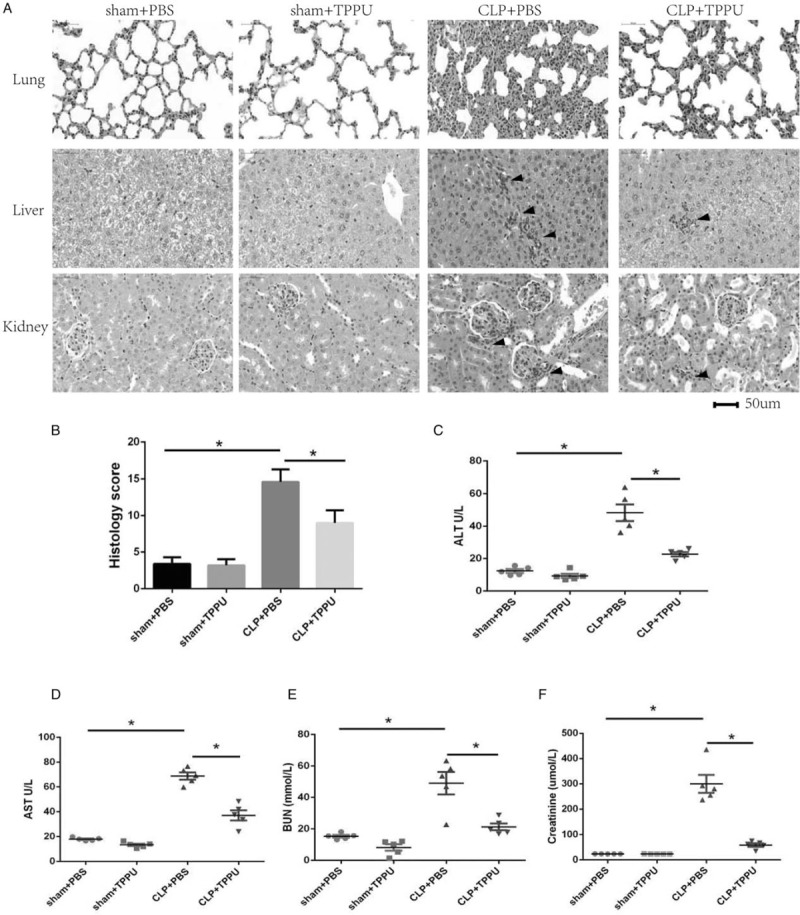
A, The representative sections (hematoxylin and eosin (H & E) staining) of mice lung, liver, and kidney. Histology score of lung injury (B). Levels of plasma ALT (C), AST (D), BUN (E), and creatinine (F). Data are expressed as means ± SEM, n = 5, ∗P < 0.05. CLP indicates cecal ligation and puncture; ALT, alanine aminotransferase; AST, aspartate aminotransferase; BUN, blood urea nitrogen.
TPPU treatment inhibited the systemic inflammatory response in septic mice induced by CLP
The occurrence and development of sepsis mainly result from overwhelming bacterial proliferation and dissemination, and excessive inflammatory cytokine release in an uncontrolled manner by immune cells (9). In this study, bacterial proliferation and related inflammatory responses were determined. CFUs from peritoneal cavity, whole blood, and spleens were calculated. Increased CFUs levels from peritoneal cavity, whole blood, and spleens were observed in the CLP+PBS group, compared with that in the sham+PBS group; interestingly, TPPU treatment markedly decreased the levels of CFUs from peritoneal cavity, whole blood, and spleens in the CLP+TPPU group, compared with that in the CLP+PBS group as shown in Figure 3A to C. These data indicate that TPPU treatment significantly prevents bacterial proliferation.
Fig. 3.
TPPU treatment inhibited systemic inflammatory response in septic mice induced by CLP.
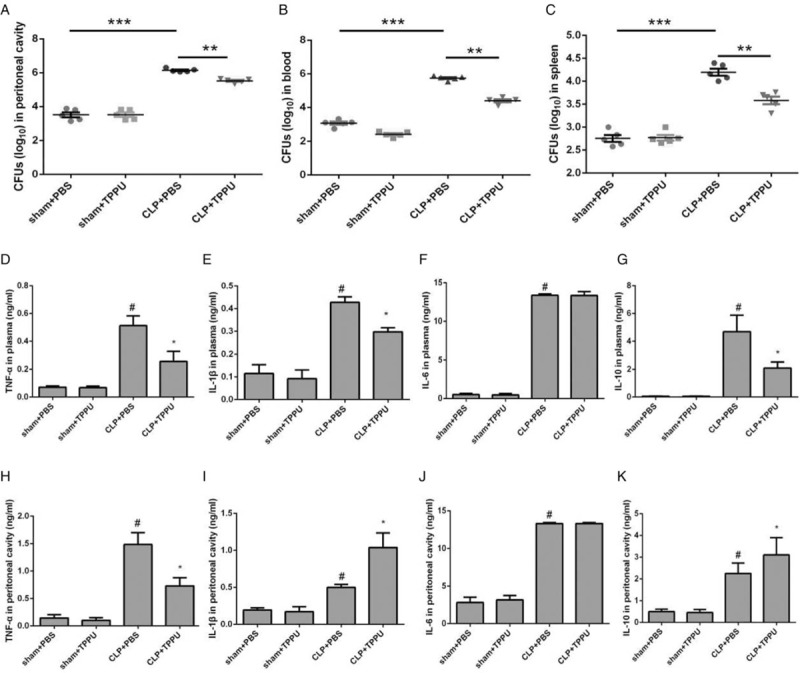
Bacterial burden in mouse peritoneal fluids (A), blood (B), and spleens (C). The cytokines levels of TNF-α (D), IL-1β (E), IL-6 (F), and IL-10 (G) in plasma. The cytokines levels of TNF-α (H), IL-1β (I), IL-6 (J), and IL-10 (K) in peritoneal lavage fluids. Data are expressed as means ± SEM, n = 5, ∗∗∗P < 0.001. ∗∗P < 0.01. #P < 0.05 versus Sham + PBS group. ∗P < 0.05 versus CLP+PBS group. CLP indicates cecal ligation and puncture; PBS, phosphate buffer saline.
Bacterial infection triggers inflammatory response that may progress to multiple organ dysfunction. In this study, the levels of inflammatory factors in the plasma and peritoneal cavity were determined. As shown in Figure 3D to K, the levels of TNF-α, IL-1β, IL-6, and IL-10 in both the plasma and peritoneal cavity were elevated intensively at 6 h after CLP surgery. Interestingly, the levels of TNF-α were decreased in both the plasma and peritoneal cavity in TPPU-treated CLP mice, compared with those in PBS-treated CLP mice (Fig. 3, D and H). The levels of IL-6 in both the plasma and peritoneal cavity remained almost consistent between TPPU- and PBS-treated CLP mice (Fig. 3, F and J). The levels of both IL-1β and IL-10 were decreased in the plasma of TPPU-treated CLP mice, compared with those in PBS-treated CLP mice (Fig. 3, E and G), but the levels of both were increased in peritoneal cavity of TPPU-treated CLP mice, compared with those in PBS-treated CLP mice (Fig. 3, I and K). Collectively, these data indicate that TPPU treatment inhibits the progression of the inflammatory response.
To further confirm the potential beneficial effects of TPPU treatment on the inflammatory response induced by CLP surgery, an in vitro study was carried out using RAW264.7 macrophages. RAW264.7 macrophages were pretreated with TPPU before LPS stimulation and cell supernatants were collected for ELISA. As shown in Figure 4, stimulation with LPS increased the concentrations of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 as well as the anti-inflammatory cytokine IL-10 (Fig. 4, A and B). As expected, pretreatment with TPPU significantly suppressed the increase in the levels of TNF-α, IL-1β, and IL-6 (Fig. 4A); in contrast, TPPU treatment further increased the level of IL-10, compared with LPS treatment alone as shown in Figure 4B.
Fig. 4.
TPPU treatment induced changes of cytokines release and MAPK signaling pathway in LPS-stimulated macrophages.
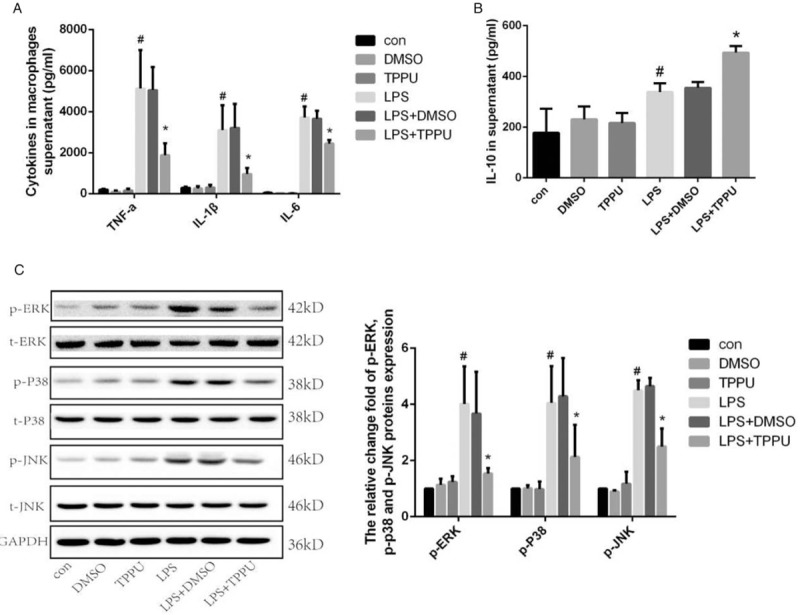
A, The concentrations of TNF-α, IL-1β, IL-6 in supernatants of macrophages. B, Concentration of IL-10 in supernatants of macrophages. C, Protein expression and phosphorylation of ERK1/2, p38MAPK (p38), and JNK in macrophages. Data are expressed as means ± SEM, #P < 0.05 versus Con group, ∗P < 0.05 versus LPS group. LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase.
The mitogen-activated protein kinase (MAPK) signaling pathway plays a key role in regulating the secretion of inflammatory cytokines in activated macrophages and is known for its involvement in inflammatory diseases such as sepsis. Here, we investigated whether the MAPK signaling pathway was involved in the anti-inflammatory effect of TPPU treatment on LPS-induced macrophages. As shown in Figure 4C, the phosphorylation levels of ERK1/2, p38 MAP kinase (p38), and JNK were higher in LPS-treated macrophages than those in the vehicle-treated group, whereas TPPU treatment prevented the increase in phosphor-ERK1/2, phosphor-p38, and phosphor-JNK levels (Fig. 4C). All these data demonstrate that TPPU treatment suppresses the inflammatory response in macrophages treated with LPS by inhibiting the MAPK signaling pathway in vitro.
Taken together, TPPU treatment inhibited the inflammatory response characterized by reduced bacterial proliferation and dissemination, and decreased inflammatory factors released in septic mice induced by CLP surgery.
TPPU treatment improved the phagocytic ability of LPS-stimulated macrophages
Macrophages play a key role in eliminating invasive bacteria through phagocytic and bactericidal functions in sepsis. In this study, the effects of TPPU treatment on macrophage phagocytic function were explored. As shown in Figure 5A, the number of fluorescent microspheres in RAW264.7 macrophages was increased in LPS-treated cells, compared with that in the control group; unexpectedly, TPPU treatment significantly increased the number of fluorescent microspheres in macrophages treated with LPS, suggesting that TPPU treatment improved the phagocytic function of macrophages. To further confirm the beneficial effects of TPPU treatment on phagocytic function of macrophages, flow cytometry analysis was performed. As expected, the percentage of FITC+ cells was significantly increased in TPPU-treated macrophages challenged with LPS, compared with that in the group of macrophages treated with LPS only as shown in Figure 5B. Collectively, these data indicate that TPPU treatment significantly strengthens the phagocytic function of LPS-treated macrophages.
Fig. 5.
TPPU treatment enhanced the phagocytosis of macrophages.
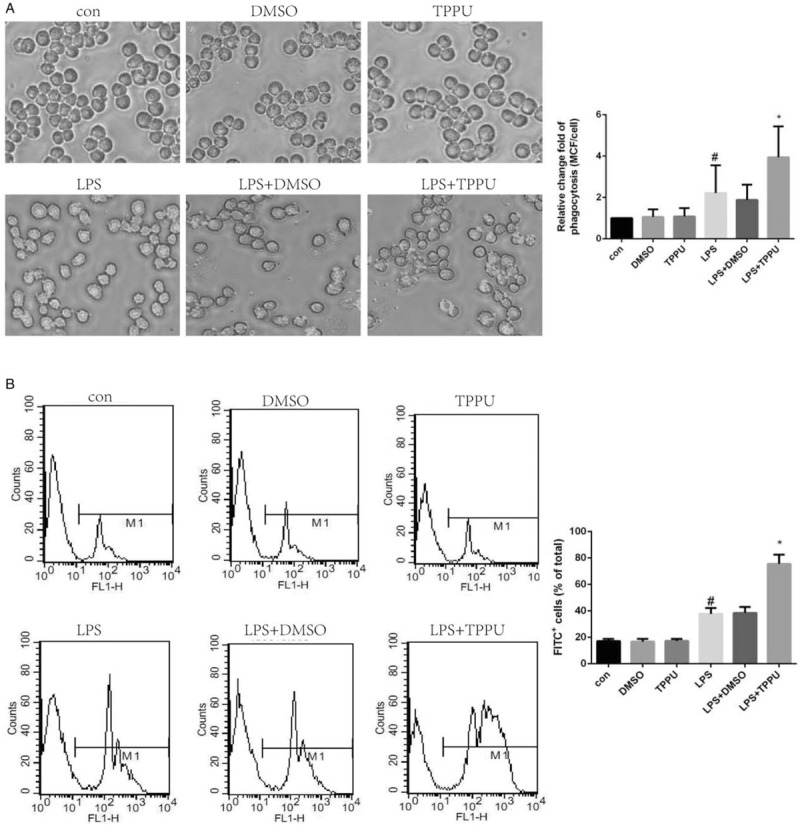
A, Macrophages pretreated with or without TPPU and LPS were co-incubated with fluorescent microspheres and observed and photographed under fluorescence microscope. Mean channel fluorescence (MCF) per cell was assessed. B, Macrophage phagocytic ability was assessed by flow cytometry. Data are expressed as means ± SEM, #P < 0.05 versus Con group, ∗P < 0.05 versus LPS group. LPS, lipopolysaccharide.
The effects of TPPU treatment on sepsis induced by CLP surgery are dependent on increased EETs level
TPPU, an inhibitor of sEH, is capable of increasing EETs levels indirectly. In this study, plasma 14,15-EET and 14,15-DHET levels were determined. As expected, increased plasma 14,15-EET level was observed in the sham+TPPU group compared with that in the sham+PBS group as shown in Figure 6A; similarly, increased plasma 14,15-EET level was observed in the CLP+TPPU group, compared with that in the CLP+PBS group (Fig. 6A). Interestingly, there was no significant difference in the level of plasma 14,15-DHET between the four groups. These data indicate that TPPU treatment in septic mice induced by CLP surgery is accompanied by increased plasma EETs level.
Fig. 6.
The effects of TPPU treatment on sepsis are dependent on increased EETs level.
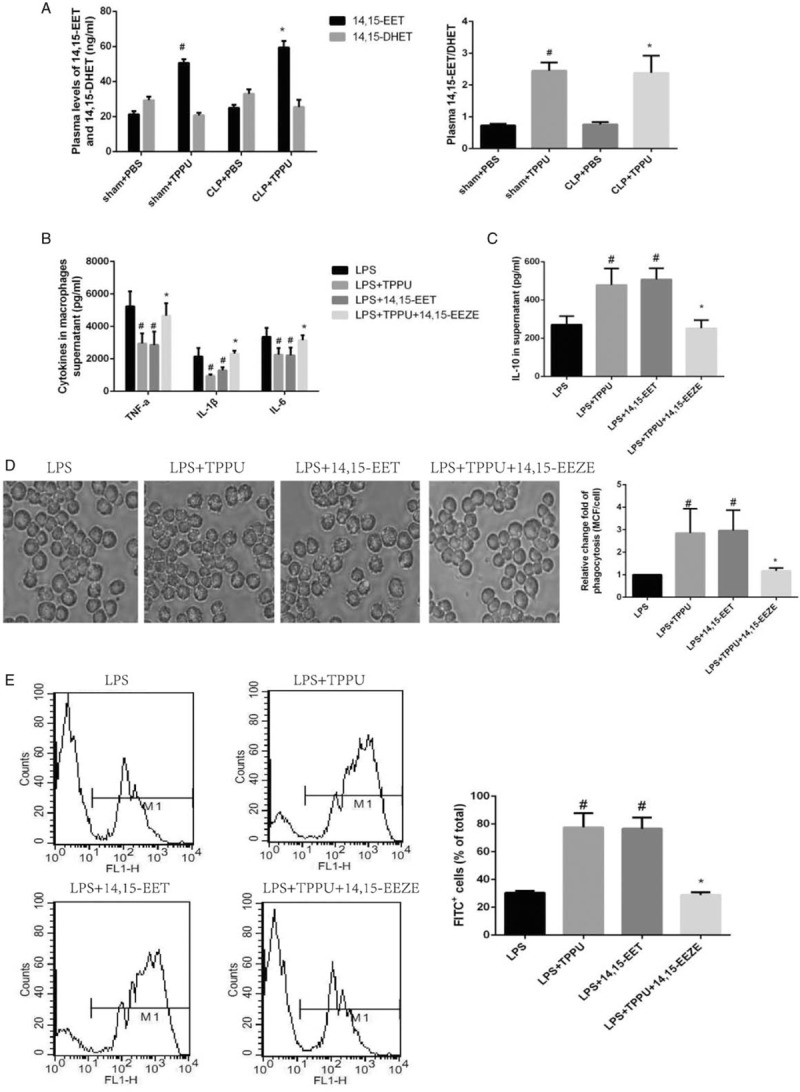
A, Concentrations of 14,15-EET and 14,15-DHET in mice plasma. Data are expressed as means ± SEM, #P < 0.05 versus Sham +PBS, ∗P < 0.05 versus CLP+PBS. B, The levels of TNF-α, IL-1β, and IL-6 in the supernatants of LPS-induced macrophages. Data are expressed as means ± SEM, #P < 0.05 versus LPS group, ∗P < 0.05 versus LPS+TPPU. C, Concentration of IL-10 in the supernatants of macrophages. Data are expressed as means ± SEM, #P < 0.05 versus LPS group, ∗P < 0.05 versus LPS+TPPU. D, Macrophages were co-incubated with fluorescent microspheres and photographed under fluorescence microscope. Macrophage phagocytosis was assessed by mean channel fluorescence (MCF) per cell. Data are expressed as means ± SEM, #P < 0.05 versus LPS group, ∗P < 0.05 versus LPS+TPPU. E, Macrophage phagocytosis was evaluated by flow cytometry. Data are expressed as means ± SEM, #P < 0.05 versus LPS group, ∗P < 0.05 versus LPS+TPPU. LPS, lipopolysaccharide.
Interestingly, a recent study indicated that sEH served as a key enzyme, which can metabolize docosahexaenoic acid into 19,20-dihydroxydocosapentaenoic acid that initiates pericyte loss and endothelial barrier breakdown (23). To identify whether EETs were involved in the beneficial effects of TPPU treatment on sepsis, 14,15-EET and its inhibitor 14,15-EEZE were used in this study. As shown in Figure 6B and C, TPPU and 14,15-EET treatment decreased the pro-inflammatory cytokines secretion, including TNF-α, IL-6, IL-1β, along with increased anti-inflammatory cytokine IL-10 secretion. Interestingly, 14,15-EEZE administration markedly abolished the effects of TPPU treatment on pro-inflammatory cytokines and IL-10 secretion as shown in Figure 6B and C. These data indicate that TPPU treatment inhibits the LPS-induced inflammatory response in macrophages via increased EETs level.
In addition, TPPU and 14,15-EET treatment markedly enhanced macrophage phagocytic ability respectively, compared with that in the LPS-treated control group, and interestingly, 14,15-EEZE administration markedly abolished the effects of TPPU treatment on macrophage phagocytic ability as shown in Figure 6D and E. These data indicate that TPPU treatment enhances the LPS-induced phagocytic ability in macrophages via increased EETs level. Taken together, these data indicate that TPPU treatment significantly improves the survival of septic mice induced by CLP surgery dependent on increased EETs level.
DISCUSSION
In the present study, our results demonstrated that TPPU treatment, accompanied by increased EETs level, markedly improved the survival of septic mice induced by CLP surgery, which was associated with alleviated organ damage and dysfunction triggered by systemic inflammatory response. Moreover, TPPU treatment significantly inhibited the systemic inflammatory response via EETs-induced inactivation of MAPK signaling pathway, due to enhanced macrophage phagocytic ability and subsequently reduced bacterial proliferation and dissemination with decreased inflammatory factors release.
LPS is a major component of gram-negative bacteria that activates the TLR4 signaling pathway, initiates inflammation in sepsis, and is widely used in sepsis studies (24, 25). Recent studies indicate that EETs prevent the progression of sepsis induced by LPS challenge in rodents. In addition to their effects on reducing inflammatory response and protecting against cardiac dysfunction during the acute phase of LPS-induced sepsis, EETs and sEH inhibition also increased the endothelial barrier function and protected against sepsis-induced lung injury in LPS-treated mice (19, 20). Furthermore, treatment with the sEH inhibitor TPPU alleviated LPS-induced pulmonary inflammation mechanistically via inhibiting macrophage pro-inflammatory cytokines release and the NF-κB signaling pathway (17, 18). In the present study, mice septic model was well established, and inhibition of sEH by TPPU treatment, accompanied by increased EETs level, markedly improved the survival rate in CLP-induced septic model. These data demonstrate that the sEH inhibitor TPPU not only inhibited the progression of sepsis induced by LPS stimulation, but also played a key role in polymicrobial-induced sepsis, which is more parallel to the clinical situation.
As professional phagocytic cells of the innate immune system, macrophages represent the first line of defense against bacterial infection (26). When pathogens invade, macrophages sense microbial products through their various surface receptors, resulting in macrophage activation and phagocytosis (27–30). On the other hand, microbial products bind to Toll-like receptors and activate several inflammatory signaling pathways including MAPK/NF-κB signaling pathway, which then contributes to the production of pro-inflammatory cytokines, e.g., TNF-α, IL-1β, and reactive oxygen species, to combat the invading organisms (8, 31–33). In addition to killing pathogens, pro-inflammatory cytokines are highly toxic to neighboring tissues and can cause dysregulated inflammation that results in severe septic shock and death (34). Various studies have revealed that controlling the inflammation produced by macrophages contributes to the survival of rodent models of sepsis (24, 25, 31, 32, 35, 36). Recent studies have identified the expression of CYP2J2 on human monocytes and macrophages (37, 38). Further observations revealed that CYP2J2 inhibited the activation of monocytes (38), and that CYP2J2 was upregulated during monocyte differentiation (37). sEH, which hydrolyzes EETs to the bio-inactive DHETs, is generally expressed on monocytes and macrophages, with its expression upregulated in the presence of LPS (39). Our previous study has demonstrated that increasing circulating EETs by CYP2J2 overexpression prevents LPS-induced cardiac dysfunction, which is associated with restrained inflammatory response (20). It is well established that LPS treatment activates the NF-κB signaling pathway, allowing NF-κB to translocate to the nucleus, leading to transcription of proinflammatory factors. Interestingly, maximal inhibition of LPS-induced NF-κB p65 activation was achieved by 11,12-EET, followed by 14,15-EET and 8,9-EET in macrophages (20). These data indicate that CYP epoxygenases/EETs have important regulatory roles in the functions of monocytes/macrophages. In the present study, TPPU treatment significantly strengthened the phagocytic function of macrophages. In addition, TPPU treatment inhibited the progression of systemic inflammatory response in vivo, as well as suppressed the inflammatory response in RAW 264.7 macrophages treated with LPS through the inhibition of MAPK signaling pathway.
Macrophages have various effects on sepsis including phagocytosis, bactericidal activity, inflammatory cytokines secretion, antigen presentation, and so on (40, 41). When microbes invade into organism, macrophages recognize and phagocytose the microbes, and then produce a series of pro-inflammatory cytokines to initiate the innate immune system. Moreover, peripheral neutrophils, lymphocytes, and monocytes were recruited to the inflammatory sites. During this process, excessive bacteria may induce exaggerated inflammatory response and cause organ damage. Bacterial load may be an important magnitude of inflammatory response whereas macrophage phagocytosis is the first step in eliminating bacteria (42). In our current study, TPPU treatment significantly reduced the bacterial load of septic mice with reduced inflammatory response and organ damage. In vitro, TPPU administration increased macrophage phagocytosis which may contribute to reduced bacterial load and inflammatory response. It is well known that macrophages play a regulatory role by releasing inflammatory mediators and recruiting PMNs. It is speculated that macrophages-initiated PMNs recruitment may contribute to reduced bacterial load, but still needs to be further explored.
In the acute phase of sepsis, macrophages are activated to release inflammatory cytokines and to perform phagocytosis or intracellular killing functions to limit bacterial dissemination and eliminate pathogens. In the late phase of sepsis, the immune system shifts to anti-inflammation and tissue repair. Any exaggerated responses in this process can cause dire outcomes. However, previous studies have revealed that these conditions can occur simultaneously, and that phagocytes from sepsis patients exhibit reduced inflammation and increased gene expression related to phagocytosis, pathogen killing, and tissue repair (11, 43). In the current study, we found that TPPU reduced the inflammatory response and bacterial growth in polymicrobial-induced sepsis. In vitro, we demonstrated that TPPU pretreatment decreased the inflammation in macrophages while upregulated phagocytosis function indicating that TPPU did not inhibit macrophage activation directly. Given the complex functions of macrophages in sepsis, we speculate that macrophages may undergo a functional reprogramming in sepsis; however, this still needs confirmation by other experiments.
It is well known that macrophages are hyperplastic, and can change their phenotype with micro-environment variation (44). Hypoxia-inducible factor 1α (HIF-1α) regulates various genes in macrophages and participates in regulating macrophage functions involved in inflammation, phagocytosis, and antimicrobial activity. A previous study revealed that HIF-1α overexpression inhibited macrophage inflammation while upregulated antimicrobial activity and tissue remodeling (11, 43). In addition, EETs played an important role in regulating HIF-1α activity (45, 46). Therefore, given the critical roles of HIF-1α on macrophages in sepsis, we speculate that TPPU treatment could regulate macrophage functions through HIF-1α, but this still needs to be further explored.
There are several limitations in this study. First, we observed only the acute phase of sepsis after surgery, whereas the mortality of sepsis exhibits three peaks in the clinic and a considerable number of patients die in the late phase. Second, the septic model was performed on young and healthy mice, whereas in the clinic, the number of patients with advancing age is increasing due to the exponential increase in the elderly population. Third, antibiotics were not used to prevent confusion between the effects of TPPU and antibiotics, while antibiotics are used as routine drugs in the clinic. Fourth, this is a preventive study, and so, TPPU treatment was started before the induction of sepsis, which is not consistent with the actual conditions in clinic. Fifth, different cytokines peak at different time points; thus, it is speculated that the similar plasma IL-6 levels between the TPPU group and vehicle group in septic mice could be due to the inappropriate time-point of plasma collection, which still needs to be explored further. Collectively, more studies are required to investigate the effects of TPPU on sepsis treatment.
In conclusion, TPPU treatment, accompanied by increased EETs level, markedly improved the survival of septic mice induced by CLP surgery, which was associated with alleviated organ damage and dysfunction triggered by systemic inflammatory response. Furthermore, TPPU treatment significantly inhibited the systemic inflammatory response, which was due to enhanced macrophage phagocytic ability and subsequently reduced bacterial proliferation and dissemination, and decreased inflammatory factors release. Our data provide evidences that sEH inhibition has potential effects on the treatment of polymicrobial-induced sepsis, but still warrants further clinical studies.
Footnotes
All the care and experimental procedures of mice were approved by the Experimental Animal Research Committee of Tongji Medical College, Huazhong University of Science and Technology, and were performed in accordance with ARRIVE guidelines developed by the National Center for the Replacement, Refinement, and Reduction of Animals in Research.
This work was partially supported by the funding from National Natural Science Foundation of China (No. 81471021) and Hu Bei Health and Family Planning Commission (WJ2015MB006).
ZC interpreted the data and wrote the manuscript. ZC and YT performed the in vivo experiments. ZC and JY performed the in vitro experiments. RD participated in the design of the study. YY and MF performed the histological examinations. JL and SH performed the flow cytometry analysis. DWW and LT contributed to the design of the study and revised the manuscript. XX designed the study and revised the manuscript. All authors read and approved the final manuscript.
The authors report no conflicts of interest.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The third international consensus definitions for sepsis and septic shock sepsis-3. JAMA 8 (315):801–810, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest 126 (1):23–31, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dahdah A, Gautier G, Attout T, Fiore F, Lebourdais E, Msallam R, Daëron M, Monteiro RC, Benhamou M, Charles N, et al. Mast cells aggravate sepsis by inhibiting peritoneal macrophage phagocytosis. J Clin Invest 124 (10):4577–4589, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delano MJ, Ward PA. The immune system's role in sepsis progression, resolution, and long-term outcome. Immunol Rev 274 (1):330–353, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med 348 (2):138–150, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Cavaillon J, Adib-Conquy M. Monocytes/macrophages and sepsis. Crit Care Med 33: suppl: S506–S509, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Serafini N, Dahdah A, Barbet G, Demion M, Attout T, Gautier G, Arcos-Fajardo M, Souchet H, Jouvin MH, Vrtovsnik F, et al. The TRPM4 channel controls monocyte and macrophage, but not neutrophil, function for survival in sepsis. J Immunol 189 (7):3689–3699, 2012. [DOI] [PubMed] [Google Scholar]
- 8.Liu W, Wu H, Chen L, Wen Y, Kong X, Gao WQ. Park7 interacts with p47phox to direct NADPH oxidase- dependent ROS production and protect against sepsis. Cell Res 25 (6):691–706, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Csoka B, Nemeth ZH, Toro G, Idzko M, Zech A, Koscso B, Spolarics Z, Antonioli L, Cseri K, Erdelyi K, et al. Extracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killing. FASEB J 29 (9):3626–3637, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, Kaczmarek E, Lee PJ, Zuckerbraun BS, Flavell R, et al. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest 124 (11):4926–4940, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng S, Scicluna BP, Arts RJW, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, Kox M, Manjeri GR, Wagenaars JAL, Cremer OL, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol 17 (4):406–413, 2016. [DOI] [PubMed] [Google Scholar]
- 12.Martins R, Maier J, Gorki AD, Huber KV, Sharif O, Starkl P, Saluzzo S, Quattrone F, Gawish R, Lakovits K, et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol 17 (12):1361–1372, 2016. [DOI] [PubMed] [Google Scholar]
- 13.Spector AA, Fang X, Snyder GD, Weintraub NL. Epoxyeicosatrienoic acids (EETs): metabolism and biochemical function. Prog Lipid Res 43 (1):55–90, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Ren Q, Ma M, Ishima T, Morisseau C, Yang J, Wagner KM, Zhang J, Yang C, Yao W, Dong C, et al. Gene deficiency and pharmacological inhibition of soluble epoxide hydrolase confers resilience to repeated social defeat stress. Proc Natl Acad Sci U S A 113 (13):E1944–E1952, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuloaga KL, Zhang W, Roese NE, Alkayed NJ. Soluble epoxide hydrolase gene deletion improves blood flow and reduces infarct size after cerebral ischemia in reproductively senescent female mice. Front Pharmacol 5:290, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sirish P, Li N, Liu JY, Lee KSS, Hwang SH, Qiu H, Zhao C, Ma SM, Lopez JE, Hammock BD, et al. Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc Natl Acad Sci U S A 110 (14):5618–5623, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong L, Zhou Y, Zhu Z, Liu T, Duan J, Zhang J, Li P, Hammcok BD, Guan C. Soluble epoxide hydrolase inhibitor suppresses the expression of triggering receptor expressed on myeloid cells-1 by inhibiting NF-kB activation in murine macrophage. Inflammation 40 (1):13–20, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Liu T, Duan J, Li P, Sun G, Liu Y, Zhang J, Dong L, Lee KSS, Hammock BD, et al. Soluble epoxide hydrolase inhibitor attenuates lipopolysaccharide-induced acute lung injury and improves survival in mice. Shock 47 (5):638–645, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong R, Hu D, Yang Y, Chen Z, Fu M, Wang DW, Xu X, Tu L. EETs reduces LPS-induced hyperpermeability by targeting GRP78 mediated Src activation and subsequent Rho/ROCK signaling pathway. Oncotarget 8 (31):50958–50971, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai M, Wu L, He Z, Zhang S, Chen C, Xu X, Wang P, Gruzdev A, Zeldin DC, Wang DW. Epoxyeicosatrienoic acids regulate macrophage polarization and prevent LPS-induced cardiac dysfunction. J Cell Physiol 230 (9):2108–2119, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belikoff B, Hatfield S, Sitkovsky M, Remick DG. Adenosine negative feedback on A2A adenosine receptors mediates hyporesponsiveness in chronically septic mice. Shock 35 (4):382–387, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nasseri S, Gurusamy M, Jung B, Lee D, Khang G, Doods H, Wu D. Kinin B1 receptor antagonist BI113823 reduces acute lung injury. Crit Care Med 43 (11):e499–e507, 2015. [DOI] [PubMed] [Google Scholar]
- 23.Hu J, Dziumbla S, Lin J, Bibli S, Zukunft S, de Mos J, Awwad K, Frömel T, Jungmann A, Devraj K, et al. Inhibition of soluble epoxide hydrolase prevents diabetic retinopathy. Nature 552 (7684):248–252, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielsen TB, Pantapalangkoor P, Luna BM, Bruhn KW, Yan J, Dekitani K, Hsieh S, Yeshoua B, Pascual B, Vinogradov E, et al. Monoclonal antibody protects against acinetobacter baumannii infection by enhancing bacterial clearance and evading Sepsis. J Infect Dis 216 (4):489–501, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin L, Zeng W, Zhang F, Zhang C, Liang W. Naringenin ameliorates acute inflammation by regulating intracellular cytokine degradation. J Immunol 199 (10):3466–3477, 2017. [DOI] [PubMed] [Google Scholar]
- 26.Lee W, Yan J, Kang J, Kim LK, Kim Y. Macrophage C-type lectin is essential for phagosome maturation and acidification during Escherichia coli-induced peritonitis. Biochem Biophys Res Commun 493 (4):1491–1497, 2017. [DOI] [PubMed] [Google Scholar]
- 27.Van Amersfoort ES, TJC Van Berkel, Kuiper J. Receptors, mediators, and mechanisms involved in bacterial sepsis and septic shock. Clin Microbiol Rev 16 (3):379–414, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding JL, Li P, Ho B. The Sushi peptides: structural characterization and mode of action against Gram-negative bacteria. Cell Mol Life Sci 65 (7–8):1202–1219, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaefer L. Complexity of danger: the diverse nature of damage-associated molecular patterns. J Biol Chem 289 (51):35237–35245, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robinson MJ, Sancho D, Slack EC, LeibundGut-Landmann S, Reis e Sousa C. Sousa CRE: Myeloid C-type lectins in innate immunity. Nat Immunol 7 (12):1258–1265, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Chang C, Hsu C, Lee A, Wang S, Lin K, Chang W, Peng H, Huang W, Chung C. 4-Acetylantroquinonol B inhibits lipopolysaccharide-induced cytokine release and alleviates sepsis through of MAPK and NFκB suppression. BMC Complement Altern Med 18 (1):1–11, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meira CS, Espírito Santo RFD, Dos Santos TB, Orge ID, Silva DKC, Guimarães ET, Aragão França LS, Barbosa-Filho JM, Moreira DRM, Soares MBP. Betulinic acid derivative BA5, a dual NF-kB/calcineurin inhibitor, alleviates experimental shock and delayed hypersensitivity. Eur J Pharmacol 815:156–165, 2017. [DOI] [PubMed] [Google Scholar]
- 33.Chen X, Cai X, Le R, Zhang M, Gu X, Shen F, Hong G, Chen Z. Isoliquiritigenin protects against sepsis-induced lung and liver injury by reducing inflammatory responses. Biochem Biophys Res Commun 496 (2):245–252, 2018. [DOI] [PubMed] [Google Scholar]
- 34.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11 (11):723–737, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiahou Z, Wang X, Shen J, Zhu X, Xu F, Hu R, Guo D, Li H, Tian Y, Liu Y, et al. NMI and IFP35 serve as proinflammatory DAMPs during cellular infection and injury. Nat Commun 8 (1):950, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Liu Z, Wu J, Bai B, Chen H, Xiao Z, Chen L, Zhao Y, Lum H, Wang Y, et al. New MD2 inhibitors derived from curcumin with improved anti-inflammatory activity. Eur J Med Chem 148:291–305, 2018. [DOI] [PubMed] [Google Scholar]
- 37.Nakayama K, Nitto T, Inoue T, Node K. Expression of the cytochrome P450 epoxygenase CYP2J2 in human monocytic leukocytes. Life Sci 83 (9–10):339–345, 2008. [DOI] [PubMed] [Google Scholar]
- 38.Bystrom J, Wray JA, Sugden MC, Holness MJ, Swales KE, Warner TD, Edin ML, Zeldin DC, Gilroy DW, Bishop-Bailey D. Endogenous epoxygenases are modulators of monocyte/macrophage activity. PLoS One 6 (10):e26591, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harris TR, Hammock BD. Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 526 (2):61–74, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili S, Mardani F, Seifi B, Mohammadi A, Afshari JT, Sahebkar A. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol 233 (9):6425–6440, 2018. [DOI] [PubMed] [Google Scholar]
- 41.Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity 41 (1):21–35, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stearns-Kurosawa DJ, Osuchowski MF, Valentine C, Kurosawa S, Remick DG. The pathogenesis of sepsis. Annu Rev Pathol 6:19–48, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shalova IN, Lim JY, Chittezhath M, Zinkernagel AS, Beasley F, Hernández-Jiménez E, Toledano V, Cubillos-Zapata C, Rapisarda A, Chen J, et al. Human monocytes undergo functional re-programming during sepsis mediated by hypoxia-inducible factor-1α. Immunity 42 (3):484–498, 2015. [DOI] [PubMed] [Google Scholar]
- 44.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41 (1):14–20, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suzuki S, Oguro A, Osada-Oka M, Funae Y, Imaoka S. Epoxyeicosatrienoic acids and/or their metabolites promote hypoxic response of cells. J Pharmacol Sci 108 (1):79–88, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Batchu SN, Lee SB, Samokhvalov V, Chaudhary KR, El-Sikhry H, Weldon SM, Seubert JM. Novel soluble epoxide hydrolase inhibitor protects mitochondrial function following stress. Can J Physiol Pharmacol 90 (6):811–823, 2012. [DOI] [PubMed] [Google Scholar]