

SUMMARY
Neurodegenerative diseases commonly involve the disruption of circadian rhythms. Studies indicate that mutant Huntingtin (mHtt), the cause of Huntington’s disease (HD), disrupts circadian rhythms often before motor symptoms are evident. Yet little is known about the molecular mechanisms by which mHtt impairs circadian rhythmicity and whether circadian clocks can modulate HD pathogenesis. To address this question, we used a Drosophila HD model. We found that both environmental and genetic perturbations of the circadian clock alter mHtt-mediated neurodegeneration. To identify potential genetic pathways that mediate these effects, we applied a behavioral platform to screen for clock-regulated HD suppressors, identifying a role for Heat Shock Protein 70/90 Organizing Protein (Hop). Hop knockdown paradoxically reduces mHtt aggregation and toxicity. These studies demonstrate a role for the circadian clock in a neurodegenerative disease model and reveal a clock-regulated molecular and cellular pathway that links clock function to neurodegenerative disease.
Graphical Abstract
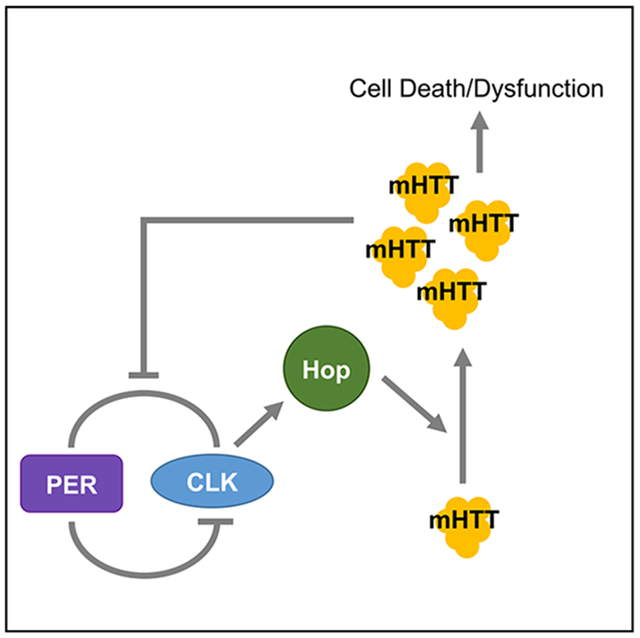
In Brief
Disruption of circadian rhythms is frequently observed across a range of neurodegenerative diseases. Here, Xu et al. demonstrate that perturbation of circadian clocks alters the toxicity of the mutant Huntingtin protein, the cause of Huntington’s disease (HD). Moreover, they reveal a key mechanistic link between the clock and HD.
INTRODUCTION
Considerable evidence suggests that disrupted clocks or sleep are associated with and potentially alter neurodegenerative disease processes (Musiek and Holtzman, 2016). In Alzheimer’s disease (Giubilei et al., 2001; Satlin et al., 1995; Stopa et al., 1999), Parkinson’s disease (Bordet et al., 2003; Kudo et al., 2011a), Huntington’s disease (Aziz et al., 2009; Kalliolia et al., 2014; Morton et al., 2005; Pallier et al., 2007; van Wamelen et al., 2013), chronic traumatic encephalopathy (CTE; Asken et al., 2016), traumatic brain injury (TBI; Mathias and Alvaro, 2012; Verma et al., 2007), and frontotemporal dementia (Anderson et al., 2009), circadian and/or sleep disruption has been consistently observed as evidenced by altered phase or amplitude of sleep-wake, activity, and/or temperature rhythms; melatonin and cortisol hormonal rhythms; and circadian pacemaker neuron and clock gene rhythms. In many cases, the circadian or sleep disruption precedes the appearance of the diagnostic symptoms of the disease, suggesting that they are impaired early in the disease process. For example, reduction in melatonin levels is observed in patients even in preclinical Alzheimer’s disease (AD) stages (Braak stages I–II; Wu et al., 2003). These studies suggest that the circadian clock is both a target of and may regulate neurodegenerative disease. Yet definitive evidence that the circadian clock can impact neurodegeneration in human disease models has been lacking.
To address the mechanistic relationship between circadian clocks and neurodegenerative diseases, we are using Huntington’s disease models. Polyglutamine (polyQ) expansion in Huntingtin (mHtt) leads to dysfunction and death of medium spiny neurons in the striatum and the characteristic Huntington’s chorea (Milnerwood and Raymond, 2010). Both human and animal studies indicate that mHtt also impairs circadian rhythms. Circadian behavioral rhythms are disrupted in Huntington’s disease (HD) patients and mouse models (Kudo et al., 2011b; Kuljis et al., 2012; Loh et al., 2013; Morton et al., 2005; Pallier et al., 2007). In HD patients, behavioral arrhythmicity is accompanied by disrupted melatonin rhythms (Aziz et al., 2009; Kalliolia et al., 2014) and reduced numbers of vasoactive intestinal peptide positive (VIP+) neurons in the master circadian pacemaker suprachiasmatic nucleus in HD patients (van Wamelen et al., 2013). In mouse models, suprachiasmatic nucleus (SCN) electrical activity can be disrupted in the presence of intact core molecular clock oscillations, indicating that clock output is disrupted (Kudo et al., 2011b). Molecular clocks are not only evident in the SCN but also in other brain regions, such as the striatum. In HD mouse models, disrupted molecular rhythms are observed in the SCN (Morton et al., 2005; Pallier et al., 2007) as well as in the striatum (Morton et al., 2005) and cortex (Fahrenkrug et al., 2007). In transgenic R6/2 mice, treatment with a sedative drug improved sleep-wake rhythms and reversed the dysregulated expression of Per2 and markedly improved their cognitive performance (Pallier et al., 2007; Pallier and Morton, 2009). Disrupted circadian rhythms and motor symptoms in mouse HD models were improved by time-restricted feeding (Wang et al., 2018; Whittaker et al., 2018). However, it was not known if these rhythm-improving interventions were solely symptomatic or if they altered the neurodegenerative process itself. Taken together, mHtt can disrupt the core clock and/or clock output of clock pacemaker neurons such as those in the SCN and may therefore contribute to behavioral rhythm disruption in HD. In addition, mHtt also targets clocks in the disease-relevant striatum. How mHtt and molecular clocks interact in clock-expressing neurons remains unclear.
To study how circadian disruption can modulate HD, we are using the fruit fly Drosophila, a well-established model organism in the study of circadian rhythms and neurodegenerative disease. The fruit fly is an evolutionarily conserved model system to study circadian rhythms (Allada and Chung, 2010; Tataroglu and Emery, 2015). In flies, the CLOCK (CLK)/CYCLE (CYC) transcription factor (reviewed in Mohawk et al., 2012; Zheng and Sehgal, 2008) activates period (per) and timeless (tim). PER and TIM proteins are in turn modified by protein kinases, such as DOUBLETIME (DBT), and inhibit CLK-CYC function (Kim et al., 2007; Kloss et al., 1998). The conservation extends to humans where mutations in the orthologs of fly per and Dbt are responsible for the circadian disorder advanced sleep phase syndrome (Toh et al., 2001; Xu et al., 2005), providing a compelling rationale for using the fly as a model for circadian rhythms and its interaction with Huntington’s pathology.
Valid Drosophila Huntington’s models have been generated by expression of human Htt with varying polyQ lengths. These fly models recapitulate many features of human HD. These include reduced lifespan (Lee et al., 2004; Wolfgang et al., 2005), motor deficits (Chongtham and Agrawal, 2016; Lee et al., 2004), neurodegeneration (Agrawal et al., 2005; Steffan et al., 2001), cytoplasmic followed by nuclear accumulation of mHtt (Jackson et al., 1998), and disrupted circadian rhythms (Sheeba et al., 2010). mHtt effects also depend on CAG repeat length (Jackson et al., 1998). In addition, the underlying molecular mechanisms of mHtt pathogenesis are conserved, including mTor (Ravikumar et al., 2004), histone acetylation (Ferrante et al., 2003; Hockly et al., 2003; Steffan et al., 2001), SUMOylation/ubiquitination (Steffan et al., 2004), and axonal transport (Gunawardena et al., 2003; Smith et al., 2014). The striking similarities between human, mammalian, and fly HD, including at the level of genes and therapeutics, indicate that the fly can make important contributions to understanding HD.
While circadian clock mutations can enhance neurodegeneration, especially under challenge, including in a disease model (Krishnan et al., 2012; Means et al., 2015; Musiek et al., 2013), whether these mutations exert their effects via their role in clocks versus pleiotropic effects remains unclear. Moreover, a role for the clock in mHtt toxicity has not been demonstrated. Here we provide evidence that circadian clocks can modulate neurotoxic effects of polyQ-containing proteins, such as those in HD, representing a potential therapeutic avenue to slow disease progression. In addition, we identify pathways that may mediate clock control of mHtt aggregation and toxicity, including a role for heat shock pathways.
RESULTS
Expression of mHtt in PDF Clock Neurons Suppresses Circadian Rhythmicity in a PolyQ-Length-Dependent Manner
To address the clocks-HD interaction, we did not attempt to fully model all aspects of HD in a fruit fly. Rather, we focused specifically on the role of the molecular clock in modulating mHtt in clock-containing neurons. This endophenotype is potentially highly relevant to HD as a subset of clock neurons in the suprachiasmatic nucleus are known to be lost in HD, suggesting that mHtt targets these neurons. In addition, striatal neurons also contain molecular clocks (see Morton et al., 2005), suggesting a potential cell autonomous mechanism by which clocks could impact striatal neurodegeneration in the pathognomonic target of HD.
We expressed human Htt genes in a subgroup of fly circadian neurons, PDF+ ventral lateral neurons (LNvs), using pdfGAL4. We mainly used two sets of human Htt transgenes: the first set, HttQ128 and HttQ0 (the non-pathogenic control for HttQ128 lacking polyQ repeats), contains exons 1, 2, and part of 3 of the Htt gene (Romero et al., 2008), and the second set, HttQ103-eGFP, HttQ72-eGFP, HttQ46-eGFP, and HttQ25-eGFP (the last is the non-pathogenic control for HttQ103/72/46) contains the first exon of the Htt gene with varying polyQ lengths (Zhang et al., 2010). The eGFP fusions facilitate tracking of polyQ aggregation (see below). PDF+ LNvs can be further divided into large LNvs (lLNvs) and small LNvs (sLNvs), the latter playing a crucial role in driving free-running rhythms under constant darkness conditions (Grima et al., 2004; Stoleru et al., 2005). Expression of HttQ128 in the PDF+ LNvs induces arrhythmicity and loss of the sLNv subset, comparable to the loss of subsets of SCN neurons observed in humans (Sheeba et al., 2008, 2010). Consistent with prior studies, we observed polyQ-length-dependent impairment of circadian rhythmicity (power-significance [P-S] for HttQ128 and HttQ103; Table 1). These effects are accompanied by age-dependent loss of a subset of sLNvs (Figures 1A and B). Not all toxic neurodegenerative genes induce these effects. Transgenic expression of Aβ42 induces retinal degeneration in flies (Burr et al., 2014). To determine if this transgene can alter rhythmicity, we drove expression in PDF neurons. However, it failed to reduce rhythmicity when expressed in PDF neurons, even after aging for several weeks (Figure 1C). Thus, we hypothesize that clock neurons (or at least a subset) are especially sensitive to mHtt consistent with HD neuropathology.
Table 1.
Polyglutamine-Length-Dependent Effects of Htt on Circadian Rhythmicity
| Period | P-S | N | R% | |
|---|---|---|---|---|
| Pdf-Gal4/U-HttQ0#10-2 | 24.5 ± 0.1 | 97 ± 10 | 16 | 94% |
| Pdf-Gal4/U-HttQ128#35 | – | 0 ± 0*** | 14 | 0% |
| Pdf-Gal4/+;U-HttQ25-eGFP/+ | 24.2 ± 0.1 | 93 ± 10 | 14 | 100% |
| Pdf-Gal4/+;U-HttQ46-eGFP/+ | 24.2 ± 0.1 | 99 ± 12 | 17 | 94% |
| Pdf-Gal4/+;U-HttQ72-eGFP/+ | 24.8 ± 0.1 | 84 ± 13 | 15 | 96% |
| Pdf-Gal4;U-HttQ103-eGFP/+ | 24.0 ± 0.1 | 42 ± 9*** | 16 | 75% |
p < 0.05,
p < 0.01,
p < 0.005 compared to non-pathogenic Htt control lines, respectively.
Figure 1. HttQ128 and HttQ103 Reduce sLNv Cell Number and Strongly Suppress PER Levels.

(A) sLNv cell number at different ages are shown for HttQ0 and HttQ128 expressing flies (n = 8–23).
(B) sLNv cell number at different ages are shown for HttQ103 expressing flies (n = 12–15). HttQ103 overexpression significantly decreases PDF+ sLNv number at D15 compared to HttQ25.
(C) Rhythmicity (P-S) is indicated for various genotypes, including flies with (Abeta42) and without (ctrl) Abeta42 in PDF neurons at either the age of day 3 (D3) or day 21 (D21) (n = 8–15).
(D and E) PER intensity at ZT0 in sLNvs and lLNvs in Pdf > HttQ0 and Pdf > HttQ128 flies (D; n = 6–23) and Pdf > HttQ25 and Pdf > HttQ103 flies (E; n = 7–28) at age day 15 is quantified and shown. (*p < 0.05, **p < 0.01, ***p < 0.005, two-tailed t test)
(F) Representative images of LNvs (sLNv, lLNv, and 5th sLNv) expressing HttQ0 or HttQ128 are shown. Staining of PER is indicated in green, while co-staining of PDF is indicated in red. Asterisks indicate sLNvs, while arrowheads indicate lLNvs. The PDF – 5th sLNv is labeled with “5th” if it is evident in the image. Scale bar indicates 10 μm.
In addition to cell loss, disrupted molecular clocks may also underlie poor behavioral rhythmicity in mHtt flies. It has been previously shown that the presence of even a single sLNv is sufficient for behavioral rhythms (Helfrich-Förster, 1998) and thus additional changes may be needed to suppress rhythms. Disruptions of circadian clock oscillations have been observed in mammalian HD models. Using both the HttQ128 and HttQ103 models, we observed strong reductions in PER levels at typical peak times of day in both the remaining sLNvs and the lLNvs, which do not undergo apparent cell loss (Figures 1D and 1F). These data also indicate that we are inducing functional levels of mHtt in both sets of neurons even though we do not observe lLNv cell loss. These potent clock-disrupting effects of mHtt on molecular rhythms are consistent with fly and mammalian studies (Khaskheli et al., 2017; Morton et al., 2005; Pallier et al., 2007). These data establish a molecular pathway, the core circadian clock, to assay mHtt effects on pre-degenerative neuronal function in a genetic model system.
Heterozygous ClkJrk Mutants Suppress mHtt Effects on sLNv Cell Number and mHtt Aggregate Formation
While effects of mHtt on the core clock have been previously observed, it is unclear if the clock can, in turn, impact HD pathogenesis. Genetic evidence suggests the clock may influence neuronal survival, but this has not been established in HD in flies or mammals. To test the role of the clock in HD pathogenesis, we tested the core clock mutant ClkJrk. ClkJrk contains a premature stop codon truncating the activation domain, but retaining its dimerization domain, resulting in a dominant-negative form of this transcriptional activator (Allada et al., 1998). Homozygotes are arrhythmic with low per RNA levels, while heterozygotes exhibit reduced levels with low-amplitude oscillations (Allada et al., 1998). As ClkJrk homozygotes apparently lack detectable sLNvs (Park and Hall, 1998), we tested heterozygotes. First, in flies expressing HttQ0, we did not observe any reduction in the number of sLNvs consistent with the idea that ClkJrk heterozygotes do not display developmental phenotypes evident in homozygotes (Figure 2A). Surprisingly, we found that more sLNvs were spared in HttQ128 expressing flies (Figure 2A). To further confirm that this is likely due to a circadian effect rather than a pleiotropic Clk function, we examined a second circadian clock mutant, per01, as well as per01; ClkJrk/+ double mutants. Interestingly, while per01 failed to suppress (or enhance) HttQ128 effects, per01 was able to suppress the neuroprotective effect of ClkJrk/+ (Figure 2A). The notion that per01 does not show the same phenotype as ClkJrk is not surprising as they “fix” the clock at opposite points in the cycle. The lack of a per01 phenotype could be explained by a ceiling to how much the clock can enhance mHtt effects. Nonetheless, we hypothesize that abrogation of PER repressor activity enhances CLK activated transcription suppressing the ClkJrk/+ effect. Importantly, it highlights the importance of the PER negative feedback loop, a cornerstone of the circadian clock, in HttQ128 effects. Moreover, it indicates that low per is not responsible for Clk effects as per01 does not have any apparent phenotype but instead likely reflects other CLK-activated genes.
Figure 2. mHtt-Induced sLNv Loss and Aggregate Formation in ClkJrk/+ Flies.

(A) The number of sLNvs present per brain hemisphere is indicated for various genotypes where HttQ0 and HttQ128 indicate fragments of Htt with (Q128) and without (Q0) polyglutamines. per01 and Jrk indicate per01 and ClkJrk mutants (n = 17~29; *p < 0.05, **p < 0.01, ***p < 0.005).
(B) Representative images of LNvs (sLNv and lLNv) expressing HttQ25-eGFP and HttQ46-eGFP (in green) are shown for wild-type (+/+) and ClkJrk/+ (Jrk/+). Co-staining for PDF is indicated in red. White arrowheads indicate lLNvs. White dot circles label sLNvs without aggregates. Yellow dash circles label sLNvs with aggregates. Example aggregates are pointed out by orange arrows.
Scale bar indicates 10 μm.
Misalignment between Endogenous Circadian Periodicity and Daily Light-Dark Cycles Can Suppress mHtt Effects on sLNv Cell Number and Aggregate Formation
One issue with the use of arrhythmic clock mutants is that one cannot easily distinguish between the role of a clock gene in the circadian clock from other pleiotropic effects of these genes. Circadian resonance experiments, where the timing of the endogenous clock is matched or mismatched with the cycles of daily environmental cycles (Ouyang et al., 1998), is the gold standard to definitively demonstrate functional roles of circadian timing. To test the role of circadian timing, we entrained pdfGAL4/UASHttQ128 flies under 12:12 and 10:10 light:dark (LD) cycles from the time of egg laying, i.e., throughout development. Thus, the only difference between the two groups is in the daily timing of the light:dark cycle to which the flies entrain. Flies are capable of entraining to these altered light:dark cycles as evidenced by the coherence (low SD) on the first day of constant darkness for the evening activity offset (Figure S1). Surprisingly, we found that more sLNvs were spared under 10:10 than under 12:12 conditions (Figure 3A). These effects were accompanied by a reduction in mHtt aggregates. Aggregates are quantified by identifying spots of high fluorescence intensity, reflecting the high concentrations of GFP-tagged proteins. Using the eGFP-tagged HttQ72, we found a reduction in the percentage of sLNvs that contain aggregates in 10:10 versus 12:12 conditions (Figures 3B and 3C).
Figure 3. Exposure to 10 h Light, 10 h Dark Cycles Reduces mHtt-Mediated sLNv Cell Loss and Aggregate Formation.
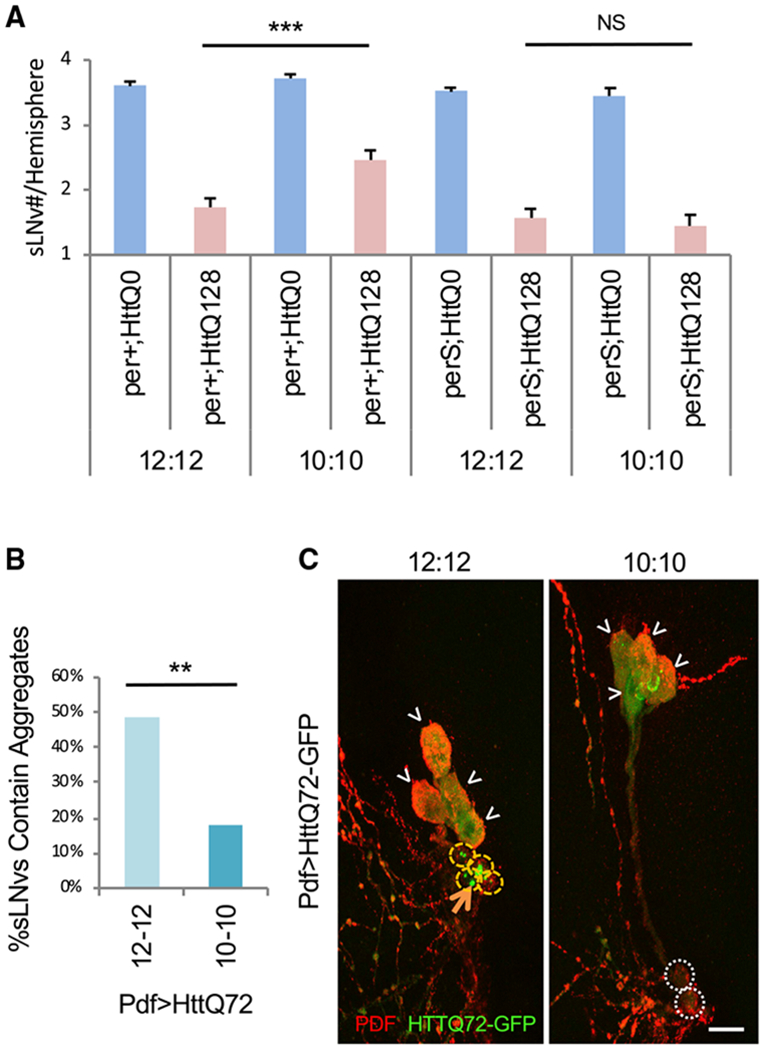
(A) The number of sLNvs present per brain hemisphere at age day 5 is indicated for various genotypes and environmental cycles, where HttQ0 and HttQ128 indicate fragments of Htt with (Q128) and without (Q0) polyglutamines. per+ and perS indicate wild-type and perS mutants (n = 36–62).
(B) Percentage of LNvs containing HttQ72-eGFP aggregates are indicated under 12 h light, 12 h dark conditions (12-12) and 10 h light, 10 h dark conditions (10-10; n = 37–39).
(C) Representative images of LNvs (sLNv and lLNv) expressing HttQ72-eGFP (in green) are shown under 12:12 and 10:10 conditions. Co-staining for PDF is indicated in red. White arrowheads indicate lLNvs. White dot circles label sLNvs without aggregates. Yellow dash circles label sLNvs with aggregates. Example aggregates are pointed out by orange arrows.
*p < 0.05, **p < 0.01, ***p < 0.005. Scale bar indicates 10 μm.
To determine if this effect was due to circadian clock timing, we examined perS flies with an endogenous period of 19 h that more closely aligns with the 10:10 cycle. We found that the neuroprotective effect of 10:10 is no longer evident, indicating the effect is related to circadian timing (Figure 3A). Thus, by first altering the timing of light:dark cycles, we can alter mHtt-induced cell loss, and then by altering the timing of the clock to more closely match these cycles, we can reverse this effect. These studies provide compelling evidence that altering clock timing in relation to environmental cycles can modify mHtt-induced effects.
As wild-type and perS flies are able to entrain to both 12:12 and 10:10 cycles, we more closely examined light:dark activity profiles in these flies, including in the HttQ128 models. Wild-type and HttQ128 flies display a robust evening anticipatory rise in locomotor activity in advance of lights-off under 12:12 conditions, consistent with the notion that non-PDF neurons are critical for evening anticipation (Figure S2). However, this peak is absent under 10:10 conditions, likely due to the delayed phase of entrainment. perS flies, on the other hand, show a phase advance of the evening peak in LD but, unlike wild-type, retain robust evening anticipation in 10:10 cycles (Figure S2). Thus, genetic and environmental conditions that retain evening anticipation exhibit normal mHtt effects, while in the case where evening anticipation is not evident during the light phase, we observe a reduction in those effects. We are not claiming a functional link between evening anticipation and mHtt effects. However, it is possible that clock-regulated genes may exhibit a similar pattern and the phase relationship between circadian gene expression and the timing of the light:dark cycle may be important for mHtt-induced neurodegeneration. Taken together, we have demonstrated through multiple lines of evidence that modulation of the circadian clock can alter the neurotoxic effects of mHtt, providing a molecular pathway impacting HD pathogenesis.
RNAi Screen of LNv Clock-Regulated Genes Identified Replicable Suppressors of mHtt-Induced Behavioral Arrhythmicity
As both environmental (10:10 LD) and genetic (ClkJrk/+) manipulations of the circadian clock reduced mHtt-induced neuronal loss, we hypothesized that manipulations of clock-controlled genes may mediate these effects. To identify these mediators, we performed RNA sequencing from fluorescence-activated cell sorting (FACS)-sorted PDF+ LNvs at 2 h time resolution across three LD cycles. We labeled PDF+ neurons with GFP, dissected and dissociated brains, and FACS-selected GFP+ neurons (Nagoshi et al., 2010). Libraries were generated (TruSeq Sample Preparation kit) and RNA sequencing (RNA-seq) was performed (HiSeq2000, Illumina). Sequence alignments and quantitation were performed using Kallisto (Bray et al., 2016). While these experiments were performed using two different temperatures and diets, we performed rhythmic gene detection across the 3 days of data using the boot empirical JTK_CYCLE with asymmetry search method with Benjamini-Hochberg correction for false discovery (boot eJTK; gammaBH < 0.05, fold change > 1.5) (Hutchison et al., 2018). We reasoned that robust clock-controlled genes should be cycling irrespective of diet and temperature. Using this approach, we identify 1,789 cycling genes, including the core clock genes, validating the overall approach (Figure S3). We will more fully describe this dataset elsewhere.
To identify clock-controlled genes (CCGs) that modulate mHtt toxicity, we performed an RNAi screen to knockdown CCGs and assayed HttQ128 effects on behavioral rhythmicity. We prioritized robustly cycling genes as well as those that have been previously implicated as mHtt modifiers in prior studies (Doumanis et al., 2009; Zhang et al., 2010). As behavioral rhythmicity is a functional readout of intact LNvs, it is possible that modifiers could improve rhythmicity by suppressing HttQ effects on neuron loss as well as pre-degenerative neuron function. To our knowledge, a genetic screen enabling discovery of modifiers of mHtt pre-degenerative function has not previously been performed in animals. We hypothesize that such “early” modifiers may highlight pathways with therapeutic potential before irreversible cell loss. We screened around 150 lines mainly from Bloomington TRiP collection and identified 16 primary hits that suppressed the effects of HttQ128 on behavioral rhythmicity (Figure 4A). Here we initially focus on Hsp70/90 Organizing Protein (Hop). Other modifiers will be described more fully elsewhere. Hop, also known as stress inducible protein STI1, functions as a co-chaperone for the major protein chaperones Hsp70 and 90 to facilitate protein folding (Baindur-Hudson et al., 2015). While the Hop transcript does not exhibit a rhythmic amplitude comparable to the core clock genes, it consistently peaks across all 3 days during the night-time (Figure 4B), a time around which CLK-activated cycling gene expression, such as per, is peaking (Figure S3). Using qPCR from FACS-sorted PDF+ LNvs, we found reductions in both Clk-activated tim transcript and Hop transcript in ClkJrk/+ mutants, consistent with the idea that Clk activates Hop (Figure 4C).
Figure 4. Identification of Hsp70/90 Organizing Protein (Hop) in a Screen for Cycling Genes That Suppress HttQ128-Induced Arrhythmicity.

(A) The x axis indicates ranking of screened RNAi lines based on their average rhythmicity (power-significance [P-S]) values in pdf > HttQ128 flies. The red line indicates the cut-off for RNAi to be considered modifiers, and the red circle (Ctrl) indicates the average P-S of the control. Two independent Hsp70/90 organizing protein (Hop) RNAi lines that are modifiers are indicated by black arrows.
(B) Transcript levels in transcripts per million (TPM) for Hop across three 24 h light:dark cycles. Light and dark periods are indicated in yellow and gray, respectively. Trend line indicates a 3 h window moving average. Each profile is in the following order: standard 1.5x sucrose-yeast (SY) food at 25°C, 1.5x SY at 18°C, and 0.5x SY at 25°C.
(C) LNv transcript levels at ZT14 for tim and Hop from ClkJrk/+ (ZT14 Jrk/+) are normalized to wild-type (ZT14 WT). Average levels for replicates are shown.
*p < 0.05 by t test.
Impairment of the Hsp70/90 Organization Protein Suppresses mHtt Toxicity
Several studies have shown that elevated expression or activity of heat shock proteins, including Hop itself, reduces mHtt or polyQ toxicity and/or aggregation formation (Chan et al., 2000; Hay et al., 2004; Kuo et al., 2013; Wolfe et al., 2013), while reduction of heat shock pathway components enhances mHtt or polyQ toxicity (Brehme et al., 2014; Wolfe et al., 2013). Surprisingly, Hop RNAi knockdown using two independent RNAi reagents partially suppressed arrhythmicity caused by HttQ128 (Figure 6A). On the other hand, Hop RNAi in otherwise wild-type flies did not further improve their rhythms, indicating the phenotype is mHtt dependent (Figure S4). To determine if these effects were specific to mHtt, we expressed a mutant form Tar Domain Protein 43 (A315T) that causes an inherited form of amyotrophic lateral sclerosis (ALS; Gitcho et al., 2008). Expression in PDF neurons caused suppression of free-running rhythmicity (Figure 5A). Previous studies had shown that overexpression of the fly homolog of glycogen synthase kinase 3, shaggy (sgg), could partially suppress TDP43A315T effects (Sreedharan et al., 2015), and in fact we observed similar results (Figure 5B). However, a Hop RNAi line that was able to suppress arrhythmicity due to mHtt was unable to do so with TDP43A315T (Figure 6B), revealing the polyQ specificity of Hop effects. In addition, knockdown of Hop also partially restored rhythmicity in flies expressing HttQ103-eGFP expression (Figure 5C). Rescue of behavioral rhythmicity was accompanied by a very modest (<1 neuron per hemisphere) increase in PDF+ sLNv cell number (Figure 5D).
Figure 6. Hop RNAi Suppresses mHtt Aggregate Formation and Increases pdfGAL4-Driven HttQ25-eGFP Levels.

(A) Percentage of LNvs (labeled with PDF in red; B) at age day 7 containing HttQ72-eGFP aggregates (in green) or in a TRiP RNAi library control background (TRiP Ctrl) and expressing Hop TRiP RNAi lines (RNAi TRiP #2; n = 26–29; *p < 0.05, **p < 0.01, ***p < 0.005). White arrowheads indicate lLNvs. White dot circles label sLNvs without aggregates. Yellow dash circles label sLNvs with aggregates. Example aggregates are pointed out by orange arrows. Scale bar indicates 10 μm.
(C) Representative images of LNvs (sLNv and lLNv) expressing HttQ25-eGFP are shown in TRiP RNAi library control background (TRiP Ctrl) and expressing Hop TRiP RNAi lines (Hop RNAi TRiP #2). Asterisks indicate sLNvs.
(D) GFP intensity in the sLNvs and lLNvs for TRiP Ctrl and Hop TRiP RNAi lines is quantified and shown (n = 8–20).
Scale bars indicates 10 μm. (n = 8–20; *p < 0.05, **p < 0.01, ***p < 0.005.
Figure 5. Hop RNAi Suppresses mHtt-Mediated Arrhythmicity, Aggregate Formation, and Cell Loss.

(A) Rhythmicity (P-S) is indicated for various genotypes, including flies expressing HttQ128 in PDF neurons in a TRiP RNAi library control background (HttQ128 TRiP Ctrl) and expressing two independent Hop TRiP RNAi lines (HttQ128 Hop RNAi TRiP #1 and #2; n = 17–19).
(B) Rhythmicity (P-S) is indicated for various genotypes, including flies expressing TDP43A315T in PDF neurons in a TRiP RNAi library control background (TDP43A315T TRiP Ctrl) and expressing Hop RNAi lines (TDP43A315T Hop TRiP #2; n = 11–22).
(C) The number of sLNvs present per brain hemisphere is indicated for various genotypes where either Hop RNAi (TRiP) or TRiP control and HttQ128 expression are shown (n = 18–19).
*p < 0.05, **p < 0.01, ***p < 0.005.
We then asked if Hop could affect mHtt levels or aggregation. In Hop RNAi flies, we examined HttQ72-eGFP and found a significantly reduced number of HttQ72-eGFP aggregates per sLNvs (Figures 6A and 6B). It is possible that Hop RNAi reduces the mHtt toxicity through diminishing the pdfGAL4 activity. As aggregation can independently affect mHtt levels, we examined the levels of non-pathogenic HttQ25-eGFP as a surrogate for assessing pdfGAL4 activity. We found that Hop knockdown actually increases HttQ25-eGFP levels (Figures 6C and 6D). Thus, our data suggest Hop suppression effects are not via Hop reduction of pdfGAL4 activity and are likely due to a direct effect on HttQ72 aggregation. Taken together, these data suggest that clock control of the Hop-driven protein chaperone cycle is a link between the clock and mHtt toxicity.
The finding that Hop reduction is neuroprotective could potentially be explained by proteotoxic stress induced by Hop loss resulting in induction of other chaperones. To test this hypothesis, we assessed the transcript levels of heat shock chaperones as well as heat shock factor (Hsf) after pan-cellular (Act5C-Gal4 mediated) Hop RNAi knockdown. Hop knockdown results in an upregulation of Hsp70 transcript but not Hsp40 or Hsf (Figure S5). Of note, Hsp70 can suppress mHtt-induced neurodegeneration (Jana et al., 2000; Warrick et al., 1999; Ormsby et al., 2013; Wacker et al., 2009). Thus, Hsp70 induction could provide a mechanism for Hop-knockdown-mediated neuroprotection.
DISCUSSION
Considerable evidence suggests that disrupted clocks or sleep are associated with and potentially alter neurodegenerative disease processes (Musiek and Holtzman, 2016). Yet it has been difficult to establish causal relationships between disrupted clocks and neurodegenerative disease. Here we provide evidence that altering clock function can impact the toxicity of mHtt in clock neurons. In addition, we performed a genetic screen of CCGs to identify candidate pathways that may mediate clock effects. These studies establish a functional role for the clock in a neurodegenerative disease model as well as identify putative molecular links between the clock and neurodegeneration.
This work demonstrated that altering clocks can modulate neurodegeneration in an animal model of disease, in this case, HD. We demonstrate that circadian clocks can modulate mHtt aggregation and cell death. First, partial abrogation of the core circadian transcription factor Clk is neuroprotective in mHtt models. Second, we show that that these effects are abrogated by loss of per, highlighting the important role of per negative feedback. Third, we found that alteration in the daily timing of light:dark cycles (10:10) is neuroprotective. Finally, speeding up the clock to closely match these cycles eliminates these effects. While clock-dependent neuropathological changes have been described in otherwise wild-type or neurodegeneration-prone animals (Kim et al., 2018; Krishnan et al., 2012; Means et al., 2015; Musiek et al., 2013), it remains unclear if these effects are via their functions in the clock or not. Moreover, we are unaware of clock-dependent changes in animal models of HD. Taken together, we have demonstrated that modulation of the circadian clock using multiple interventions can alter the neurotoxic effects of mHtt, providing a novel pathway impacting HD pathogenesis.
ClkJrk/+ effects are likely via their adult function in clocks, not via their potential pleiotropic non-circadian developmental role of Clk. While ClkJrk homozygotes exhibit selective developmental loss of the sLNvs, we actually observe an increase in sLNv cell number in mHtt-expressing ClkJrk/+ flies. Moreover, we find that ClkJrk/+ flies, unlike their homozygous mutant counterparts, exhibit a wild-type number of sLNvs and have a grossly normal morphology. The fact that the ClkJrk/+ effect can be partially rescued by per01, which itself does not display any apparent developmental abnormality, provides further support that ClkJrk effects on mHtt are not developmental but rather reflect the adult per feedback-loop-related function. As loss of per alone does not alter mHtt effects on cell number, our data are most consistent with the hypothesis that reduction in another Clk-activated gene could specifically mediate these effects. As a transcript that peaks in the night, at a time when many Clk target genes peak, and is reduced in ClkJrk/+ mutants, Hop represents a candidate direct mediator of Clk effects.
While our results provide abundant evidence for a clock-HD link, our data also indicate that not all circadian disruptions are created equal with respect to mHtt effects. In particular, the absence of an effect on cell loss in the arrhythmic per01 mutant does not preclude a role for the circadian clock in mHtt toxicity. It is known that neither clock gene knockouts nor ablation of the SCN alter learning and memory (Ruby et al., 2008). These results alone may suggest that the SCN has no role in learning and memory. Yet SCN-dependent dysrhythmia can disrupt memory processing (Ruby et al., 2008). Similarly, there are many examples of clock-regulated phenomena that impact a physiological or pathological output where genetic disruption of one limb of the clock has a robust phenotype but loss of the opposing limb has no detectable phenotype. For example, astrogliosis was also observed in other circadian clock mutants, namely double knockouts of Bmall partners Clock and Npas2 but not in Per1/Per2 double knockout (KO) mice (Musiek et al., 2013). Mutations in the positive or negative limb in the clock “fix” the clock at different points of a 24 h cycle. Thus, it remains possible that the clock may oscillate between a transcriptional state that modifies mHtt and one that does not. We hypothesize that loss of per fixes the clock at the point that it does not modify mHtt. Consistent this hypothesis, when moved from this point in the cycle in ClkJrk/+ mutants, we find that loss of per can enhance the toxicity of mHtt, providing compelling evidence for a functional role for the per feedback loop.
One surprise is that both our genetic and environmental perturbations to disrupt or alter clock function appear to be neuroprotective. This is evident both at the level of cell number and reduced number of aggregates. How could clock disruption be damaging in other contexts, even ones related to neuronal viability, but be protective in this case? First, our manipulations are comparatively subtle. ClkJrk heterozygotes retain some rhythmicity, albeit with reduced amplitude (Allada et al., 1998). Similarly, flies also are still able to entrain to 10:10 cycles although with an altered phase relative to the light:dark cycle. In the case of Clk/cyc homozygotes, it is not possible to assess cell loss as these flies do not have apparent sLNvs. One possibility is that our clock perturbations lead to Hop downregulation at the appropriate time of day leading to neuroprotection. Understanding the molecular mechanisms by which the clock can impact neurodegeneration under these conditions will help to test this model.
To address the molecular mechanisms by which the clock impacts HD, we applied a functional behavioral screen for modifiers of mHtt function that enabled the discovery of genes that are important for pre-degenerative mHtt-induced neuronal dysfunction. Unbiased genetic screens have been crucial in fly mHtt models of Huntington disease (Doumanis et al., 2009; Miller et al., 2010; Rincon-Limas et al., 2012; Zhang et al., 2010). Genetic screens in Drosophila have largely focused on those impacting the mHtt-dependent rough eye phenotype that may reflect developmental or cell death processes rather than earlier neural dysfunction. Retinal degeneration is not central for Htt-dependent motor dysfunction or HD progression. Thus, our model more closely approximates a known target of mHtt (clock-expressing neurons in the SCN and striatum) and can capture molecular pathways important for pre-cell death neural dysfunction. While we observed canonical cell loss, we also found robust reductions in the core clock protein PER, providing a robust marker for pre-cell death mHtt toxicity. We propose that the discovery of functional modifiers may provide effective therapeutic targets for intervention at an early stage before irreversible cell death has occurred.
One pathway through which mHtt can impair clock neuron function is via effects on the core clock. mHtt expression using the HttQ128 and HttQ103 models strongly suppresses PER levels in both the sLNvs and the lLNvs, consistent with effects observed in mammalian models. We do not yet know whether mHtt suppresses production of PER (e.g., its transcription) or enhances its degradation. Intriguingly, Clk contains a polyQ domain up to 33 amino acids long (Allada et al., 1998), suggesting that mHtt effects may act via sequestering this transcriptional activator. Given that low PER se does not lead to enhanced mHtt effects on cell death, the simplest interpretation is that mHtt effects on PER do not mediate mHtt effects on cell death but may instead contribute to disrupted behavioral rhythms.
Another paradoxical result is the finding that impairment of a heat shock chaperone pathway is neuroprotective with mHtt. In contrast to our results, overexpression of heat shock proteins (e.g., Hsp70/40/110) rescues polyQ-induced degeneration (Chan et al., 2000; Hay et al., 2004; Kuo et al., 2013). Reduction of Hop in C. elegans or yeast enhances toxicity from polyQ proteins (Brehme et al., 2014; Wolfe et al., 2013). Hop reduction can also enhance Tau-induced degeneration in the eye (Ambegaokar and Jackson, 2011). Here we find that Hop knockdown using two independent RNAi lines partially suppresses mHtt-induced arrhythmicity, aggregates, and cell loss. Interestingly, Hop knockdown also can suppress Tau-mediated neurodegeneration in flies (Butzlaff et al., 2015) and SCA3Q78-mediated neurodegeneration (VoSSfeldt et al., 2012). One possibility is that Hop knockdown may result in compensatory upregulation of other more efficacious chaperones with respect to mHtt. For example, inhibition of HSP90 leads to activation of Heat shock transcription factor 1 (HSF1) and thus results in induction of other heat shock (HS) proteins such as Hsp70 (Fujikake et al., 2008; Kudryavtsev et al., 2017). Consistent with this model, we observed induction of Hsp70 after Hop knockdown. Alternatively, Hop knockdown may impact other pathways that in turn lead to effects on mHtt. For example, Hop, as well as other heat shock components, are involved in the loading of small RNAs to the RNA-induced silencing complex (RISC) (Iwasaki et al., 2015).
Several features of our model suggest that the results described here will be broadly applicable. The underlying molecular mechanisms for both circadian clocks and HD are widely conserved between Drosophila and mammals. The expression of mHtt in clock-expressing neurons can model the established neurodegeneration of clock-expressing neurons in the striatum and suprachiasmatic nucleus seen in HD patients. As such, this central clock neuron model is likely more valid than eye models that have been prominently used for genetic screening.
The finding that the clock can modulate mHtt toxicity also has potential pathologic and therapeutic implications. One implication is that the clock could modulate various features of mHtt aggregate formation, dissociation, and/or degradation. For example, aggregation formation may be upregulated at night due to the nightly upregulation of Hop. Notably, clock control of heat shock pathway components has also been observed in mammals (Reinke et al., 2008). If a similar scenario is evident in human HDs, then timed therapeutic interventions, termed chronotherapy, could be efficacious. Similarly, we demonstrated that environmental manipulations can alter mHtt toxicity. Altering the phase of molecular oscillations relative to the environmental cycle may also provide a promising new therapeutic strategy for this fatal disease. Given the prevalence of circadian disruption across neurodegenerative disease, it will be of great interest to see how broad the role of the clock is in other neurodegenerative disease models.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ravi Allada (r-allada@northwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Adult male Drosophila melanogaster were used for most of the experiments (behavior, staining and qPCR) except for RNA-seq and the behavior for Abeta42 expressing flies. Fly stocks were maintained at room temperature. Crosses were set on the standard cornmeal-yeast-agar medium (81.4g cornmeal, 19.2g yeast, 11.1g soy flour, 6.42 g agar, 42.8ml corn syrup, 42.8ml molasses, 5.35ml propionic acid and 13.4ml ethanol and 1.43 g methylparaben per 1L of food), incubated under 12:12 or 10:10 (for the misalignment experiment) LD cycles at 25°C. Flies were collected within 24 hours after eclosing during the L phase and aged on the regular food under the LD condition their parents were entrained until they were used for behavior or staining experiments.
Adult female Pdf > Abeta42 flies were used for behavior assays due to the UAS-Abeta42 transgene inserted on the X chromosome. Crosses were set on regular food, and incubated under 12:12 LD cycles at 25°C. Females were collected within 24 hours after eclosing during the L phase and aged on the standard cornmeal-yeast-agar medium under 12:12 LD. Female control flies (Pdf-Gal4 X control strains) were acquired with the same procedure.
RNA-seq samples were prepared from adult female Pdf > mGFP flies. Flies were raised on standard cornmeal-yeast-agar medium at 25°C and collected within 5 days after eclosing. Flies were kept under 12:12LD on 1.5X Sucrose-Yeast (SY) fly food at 25°C (HC25), 0.5X SY fly food at 25°C (LC25) or 1.5X SY fly food at 18°C (HC18) for 5 days before sampled for dissection and FACS. Flies used for behavioral analysis were kept on tubes containing sucrose-agar food (5% sucrose and 2% agar) for 5 12:12 LD cycles (or 6 10:10 LD cycles) followed by 7 DD cycles.
METHOD DETAILS
Whole Mount Immunostaining
Fly crosses were set on the standard cornmeal-yeast-agar medium under 12:12 or 10:10 (for the misalignment experiment) LD cycles at 25°C. Flies eclosing within 24 hours were collected and kept under the conditions they were raised until the ages indicated in each experiment. Adult brains were dissected in PBS (137mM NaCl, 2.7mM KCl, 10mM Na2HPO4and 1.8mM KH2PO4) within 10 minutes. Then brains were fixed in 3.7% formalin solution (diluted from 37% formalin solution, Sigma-Aldrich) for 30 minutes. Brains were washed with 0.3% PBSTx (PBS with 0.3% Triton-X) for 4 times before primary antibody incubation. Primary antibodies were diluted in 0.3% PBSTx with 5% normal goat serum and incubation was done at 4°C overnight. Brains were washed for 4 times with 0.3% PBSTx after primary antibody incubation. Secondary antibodies were diluted in 0.3% PBSTx with 5% normal goat serum and incubation was done at 4°C overnight. Primary antibody dilutions were done as the followings: mouse anti-PDF (1:800, DSHB), rabbit anti-PER (1:8000, from Rosbash Lab). Secondary antibody dilutions were done as the followings: anti-mouse Alexa594 (1:800, invitrogen), anti-mouse Alexa488 (1:800, invitrogen), anti-rabbit Alexa594 (1:800, invitrogen), anti-rabbit Alexa488 (1:800, invitrogen), anti-rabbit Alexa647 (1:800, invitrogen).
Fly Stocks
RNAi lines used for screening and other overexpression lines were acquired from Bloomington Stock Center unless indicated separately. UAS-HttQ0/128 were kindly provided by Dr. Littleton. UAS-HttQ25/46/72/103-eGFP were kindly provided by Dr. Perrimon. UAS-TDP43-WT was kindly provided by Dr. Wu.
FACS, RNA Extraction from LNvs, RNA-seq and qPCR
LNvs were labeled with Pdf > mGFP. Fly brains were dissected at certain time points and processed as previously described (Kula-Eversole et al., 2010; Nagoshi et al., 2010). More specifically, aach brain was dissected in dissecting (pH7.4 9.9mM HEPES-KOH, 137mM NaCl, 5.4mM KCl, 0.17mM NaH2PO4, 0.22mM KH2PO4, 3.3mM glucose and 43.8mM sucrose) and kept in SMactive media (4.18mM KH2PO4, 1.05mM CaCl2, 0.7mM MgSO4•7H2O, 116mM NaCl, 8mM NaHCO3, 2mg/ml glucose, 2mg/ml trehalose 0.35mg/ml α-ketoglutaric acid, 0.06mg/ml fumaric acid, 0.6mg/ml malic acid, 0.06mg/ml succinic acid, 2mg/ml yeast extract with 20% non heat-inactivated FBS, 2 μg/ml insulin and 5mM pH6.8 Bis-Tris) on ice right after dissection. Both dissecting saline and SMactive media contain 20 μM DNQX, 50 μM APV and 0.1 μM TTX as final concentration. After finishing dissection of all the brains needed for one experiment, brains were washed with 500 μL dissecting saline twice (spin down at 2000rpm for 1min between washes). 100 μL Papain solution (50unit/ml) for each sample was activated at 37°C for 10min prior to use. Saline washed brains were incubated in at room for 30min in the Papain solution. Papain was deactivated by adding 500 μL SMactive media to the brain sample and the brains were washed with 500 μL SMactive media for 3 times. Cells were dissociated by triturating brains with self-made medium opening P1000 filter tip for around 30 times followed by triturating brains with self-made small opening P1000 filter tip for around another 30 times. Cells were filtered with nylon filters (SEFAR NITEX 03-100/32). GFP positive cells were isolated at Flow Cytometry Facility at Northwestern University. RNA from FACS sorted LNvs were extracted with PicoPure Knits. We synthesized 1st and 2nd strand cDNA from RNA first with Superscript III and DNA polymerase. Then we amplified the RNA by synthesizing more RNA from the cDNA template with T7 RNA polymerase. Amplified RNA was purified with RNeasy Mini Kit (QIAGEN). After the second round of cDNA synthesis from amplified RNA, the cDNA was submitted to HGAC at university of Chicago for library preparation and sequencing. Sequencing was done in HGAC at university of Chicago with Illumina HiSeq 2000. All samples are done with single end reads of 50 base pairs.
LNv cDNAs tested with qPCR were prepared in the same way described above. RNA from whole head samples was purified by TRIzol following the manufacturer protocol. Genomic DNA was digested by RQ1 DNase (Promega). First strand cDNA was synthesized using SuperScript III First-Strand Synthesis System (invitrogen) and applied for qPCR using SYBR Green supermix (BIO RAD).
QUANTIFICATION AND STATISTICAL ANALYSIS
Confocal Imaging and Data Quantification
Fly brains after immunostaining were imaged by Nikon C2 confocal. Data processing and quantification were done with Nikon NIS Elements. For sLNv number quantification, PDF+ sLNv number for each genotype undereach condition was counted blindly. Sample sizes indicated in the legends represent number of hemispheres used for each experiments combined from at least 2 replicates. Two tail t test was performed between two genotypes/conditions need to be compared.
For PER or GFP intensity measurements, the intermediate stack of each cell was chosen for measuring the mean intensity. Three areas for each hemisphere was randomly chosen and measured for background measurements. Cells in the same hemisphere used the same background mean intensity. The final mean intensity for PER or GFP signal from HttQ25-eGFP or HttQ46-eGFP was calculated by mean intensity measured from the middle stack of a cell minus the background mean intensity and then divided by the background mean intensity. Sample sizes indicated in the legends represent number of LNvs used for each experiments. Two tail t test was performed between two genotypes/conditions need to be compared.
For aggregation quantification, a threshold was applied to the channel used for imaging Htt aggregation (threshold was usually between 2500 to 3500, and the same threshold was used for control and experimental groups in a certain experiment). The percentage of cells containing aggregates passing the threshold was calculated. Sample sizes indicated in the legends represent number of LNvs used for each experiments. Z-statistic, and the corresponding p value, was determined for statistically comparing percentages.
Locomotor Activity Recording and Circadian Data Analysis
Behavior data recording, processing, plotting and analysis were done mainly as previously described (Pfeiffenberger et al., 2010a, 2010b). Fly locomotor activity was recorded from the Drosophila Activity Monitoring (DAM) data collection system and then extracted with DAM File Scan. Rhythmicity was measured by power - significance (P-S), parameters calculated by ClockLab. Activity actograms were plotted with either Counting Macro or ClockLab. Morning and evening Index were calculated with normalized activity given by output from Counting Macro. Morning Index = sum of normalized activity 3hr before light on / sum of normalized activity 6hr before light on. Evening Index = sum of normalized activity 3hr before light off / sum of normalized activity 6hr before light off (Seluzicki et al., 2014). The interval for summarizing the activities used for anticipation calculation is adjusted for perS under 12:12 (ZT6-8/ZT4-8) and 10:10 (ZT7.5-10/ZT5-10). The time of evening peak onset is calculated by finding the time when the largest increase in activity happens (Seluzicki et al., 2014). Data are represented as mean ± SD for onset time. Behavior tests for HttQ128 flies, with or without modifiers, were done with flies enclosed within 3 days by the 1st day of behavior run. Behavior test for HttQ103 flies, with or without modifiers, were done with day 4-6 old flies. All flies for behavior were entrained from the embryonic stage (after egglaying) under 12:12 LD cycles (or 10:10 LD as indicated in the text). Sample sizes indicated in the legends represent number of flies used for each experiments combined from at least 2 independent runs. Two tail t test was performed for P-S, anticipation index or activity peak onset time between two genotypes/conditions need to be compared.
RNA-seq and qPCR Data Analysis
Reads from each time point under each food/temperature condition were quantified against the Flybase transcript assembly, release 6.14, using Kallisto (Bray et al., 2016). Gene level quantification was obtained using tximport library, both for TPMs and counts data. Our LNv data comprise of three food/temperature combination conditions, with 12 time points per each condition: 1.5X Sucrose-Yeast (SY) fly food and 25°C),0.5X SY fly food and 25°C and 1.5X SY fly food and 18°C. Genes which do not pass the threshold of TPM >1 in at least 50% of samples were filtered out, leaving 7863 genes; conditions were concatenated to generate a 36 time points as an input data for Boot eJTK to determine cycling genes (Hutchison et al., 2018). We applied Benjamini-Hochberg (BH) correction method to Gamma p values calculated by Boot eJTK. BH corrected p value of less than 0.05 and fold change greater than 1.5 (between peak and trough) were used as a threshold for detection of cycling genes. For qPCR, relative RNA abundance was calculated based on delta CT values relative to the geometric average of CT values of Rp49 and Cam as internal controls (Vandesompele et al., 2002). The RNA abundance of a gene in the experimental sample is then normalized to the RNA abundance for the same gene in the corresponding control in each experiment for fold change calculations. Significance is determined by t test setting control to 1 in each experiment.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-PDF | Developmental Studies Hybridoma Bank (DSHB) | PDF C7 |
| Rabbit polyclonal anti-PER | Rosbash Lab | N/A |
| Goat anti-Mouse IgG Alexa Fluor 488 | Invitrogen | A-11001 |
| Goat anti-Rabbit IgG Alexa Fluor 488 | Invitrogen | A-11008 |
| Donkey anti-Mouse IgG Alexa Fluor 594 | Invitrogen | A-21203 |
| Donkey anti-Rabbit IgG Alexa Fluor 594 | Invitrogen | A-21207 |
| Donkey anti-Rabbit IgG Alexa Fluor 647 | Invitrogen | A-31573 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D(−)-APV | Sigma-Aldrich | A8054 |
| Tetrodotoxin | Sigma-Aldrich | T8024 |
| DNQX | Sigma-Aldrich | D0540 |
| Critical Commercial Assays | ||
| PicoPure RNA Isolation Kit | Applied Biosystems | KIT0204 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| SuperScript III First-Strand Synthesis System | Invitrogen | 18080051 |
| RQ1 RNase-Free DNase | Promega | M6101 |
| SsoAdvanced Universal SYBR® Green Supermix | BIO-RAD | 1725274 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: RNAi control for attP2 insertion lines: y[1] v[1]; P{y[+t7.7] = CaryP}attP2 | Bloomington Drosophila Stock Center | BDSC 36303 |
| D. melanogaster: RNAi of Hop (#1): y[1] sc[*] v[1]; P{y[+t7.7] v[+t1.8] = TRiP.HMS00779}attP2 | Bloomington Drosophila Stock Center | BDSC 32979 |
| D. melanogaster: RNAi of Hop (#2): y[1] sc[*] v[1]; P{y[+t7.7] v[+t1.8] = TRiP.HMS00965}attP2 | Bloomington Drosophila Stock Center | BDSC 34002 |
| D. melanogaster: overexpression of HttQ0 and HttQ128: UAS-HttQ0/128 | Lee et al., 2004 | N/A |
| D. melanogaster: overexpression of eGFP tagged HttQ25, HttQ46, HttQ72 and HttQ103: UAS-HttQ25/46/72/103-eGFP | Zhang et al., 2010 | N/A |
| D. melanogaster: overexpression of Abeta42: P{UAS-Abeta1-42.G}1 | Bloomington Drosophila Stock Center | BDSC 32038 |
| D. melanogaster: overexpression of TDP43A315T: UAS-TDP43-A315T-RFP | Guo et al., 2011 | N/A |
| w[1118]; P{w[+mC] = UAS-sgg.B}MB5 | Bloomington Drosophila Stock Center | BDSC 5361 |
| D. melanogaster: Gal4 transgene expressed by Pdf promoter: Pdf-Gal4 | Renn et al., 1999 | N/A |
| Oligonucleotides | ||
| Hop forward primer: AGCGTACAACGAGGGTCTCAAG | N/A | N/A |
| Hop reverse primer: TGCCCAGTTCCTTCTCCTTCC | N/A | N/A |
| Hsp70, Hsp40 and hsf forward and reverse primers | Gonsalves et al., 2011 | N/A |
| T7 oligo dT primer: GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGGTTTTTTTTTTTTTTTTTTTTTTTT | Nagoshi et al., 2010 | N/A |
| tim forward primer: GACTTGCCAAATCCCTCATC | N/A | N/A |
| tim reverse primer: GAAGCACTGCAACTCGATCA | N/A | N/A |
| Rp49 forward primer: CCGCTTCAAGGGACAGTATCTG | N/A | N/A |
| Rp49 reverse primer: ATCTCGCCGCAGTAAACGC | N/A | N/A |
| Cam forward primer: GGCACCATCACAACAAAGG | N/A | N/A |
| Cam reverse primer: CTTCTCGGATCTCCTCTTCG | N/A | N/A |
| Software and Algorithms | ||
| DAM Data Acquisition Software | Trikinetics | http://www.trikinetics.com |
| DAM File Scan Software | Trikinetics | http://www.trikinetics.com |
| ClockLab | Actimetrics | http://www.actimetrics.com/products/clocklab/ |
| Counting Macro | Pfeiffenberger et al., 2010b | N/A |
| Boot eJTK | Hutchison et al., 2018 | https://github.com/alanlhutchison/BooteJTK |
| kallisto | Bray et al., 2016 | https://github.com/pachterlab/kallisto |
| tximport | Soneson et al., 2015 | http://bioconductor.org/packages/release/bioc/html/tximport.html |
| Nikon NIS Elements | Nikon | https://www.nikon.com/products/microscope-solutions/lineup/img_soft/nis-elements/ |
Highlights.
Environmental and genetic clock perturbations alter mutant Huntingtin toxicity
Heat Shock Protein 70/90 Organizing Protein (Hop) is under circadian clock control
Knockdown of Hop reduces mutant Huntingtin toxicity
ACKNOWLEDGMENTS
We thank R. Morimoto, G. Beitel, and R. Cowell for helpful comments and advice. We thank J. Littleton, N. Perrimon, J. Wu, and the Bloomington Stock Center for fly strains. We thank M. Rosbash and M. Nitabach for antibodies. The research was supported by DARPA (D12AP00023) and the Brain Research Foundation. The content of the information does not necessarily reflect the position or the policy of the government, and no official endorsement should be inferred. F.X. was supported by a Northwestern University graduate fellowship in association with NIH T32HL007909.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.03.015.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agrawal N, Pallos J, Slepko N, Apostol BL, Bodai L, Chang LW, Chiang AS, Thompson LM, and Marsh JL (2005). Identification of combinatorial drug regimens for treatment of Huntington’s disease using Drosophila. Proc. Natl. Acad. Sci. USA 102, 3777–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allada R, and Chung BY (2010). Circadian organization of behavior and physiology in Drosophila. Annu. Rev. Physiol 72, 605–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allada R, White NE, So WV, Hall JC, and Rosbash M (1998). A mutant Drosophila homolog of mammalian Clock disrupts circadian rhythms and transcription of period and timeless. Cell 93, 791–804. [DOI] [PubMed] [Google Scholar]
- Ambegaokar SS, and Jackson GR (2011). Functional genomic screen and network analysis reveal novel modifiers of tauopathy dissociated from tau phosphorylation. Hum. Mol. Genet 20, 4947–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KN, Hatfield C, Kipps C, Hastings M, and Hodges JR (2009). Disrupted sleep and circadian patterns in frontotemporal dementia. Eur. J. Neurol 16,317–323. [DOI] [PubMed] [Google Scholar]
- Asken BM, Sullan MJ, Snyder AR, Houck ZM, Bryant VE, Hizel LP, McLaren ME, Dede DE, Jaffee MS, DeKosky ST, and Bauer RM (2016). Factors Influencing Clinical Correlates of Chronic Traumatic Encephalopathy (CTE): a Review. Neuropsychol. Rev 26, 340–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz NA, Pijl H, Frolich M, Schroder-van der Elst JP, van der Bent C, Roelfsema F, and Roos RAC (2009). Delayed onset of the diurnal melatonin rise in patients with Huntington’s disease. J. Neurol 256, 1961–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baindur-Hudson S, Edkins AL, and Blatch GL (2015). Hsp70/Hsp90 or-ganising protein (hop): beyond interactions with chaperones and prion proteins. Subcell. Biochem 78, 69–90. [DOI] [PubMed] [Google Scholar]
- Bordet R, Devos D, Brique S, Touitou Y, Guieu JD, Libersa C, and Destee A (2003). Study of circadian melatonin secretion pattern at different stages of Parkinson’s disease. Clin. Neuropharmacol 26, 65–72. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, et al. (2014). A chaperome subnetwork safe-guards proteostasis in aging and neurodegenerative disease. Cell Rep. 9, 1135–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr AA, Tsou WL, Ristic G, and Todi SV (2014). Using membrane-targeted green fluorescent protein to monitor neurotoxic protein-dependent degeneration of Drosophila eyes. J. Neurosci. Res 92, 1100–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butzlaff M, Hannan SB, Karsten P, Lenz S, Ng J, Voßfeldt H, Prüßing K, Pflanz R, Schulz JB, Rasse T, and Voigt A (2015). Impaired retrograde transport by the Dynein/Dynactin complex contributes to Tau-induced toxicity. Hum. Mol. Genet 24, 3623–3637. [DOI] [PubMed] [Google Scholar]
- Chan HYE, Warrick JM, Gray-Board GL, Paulson HL, and Bonini NM (2000). Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum. Mol. Genet 9,2811–2820. [DOI] [PubMed] [Google Scholar]
- Chongtham A, and Agrawal N (2016). Curcumin modulates cell death and is protective in Huntington’s disease model. Sci. Rep 6, 18736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doumanis J, Wada K, Kino Y, Moore AW, and Nukina N (2009). RNAi screening in Drosophila cells identifies new modifiers of mutant huntingtin aggregation. PLoS ONE 4, e7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenkrug J, Popovic N, Georg B, Brundin P, and Hannibal J (2007). Decreased VIP and VPAC2 receptor expression in the biological clock of the R6/2 Huntington’s disease mouse. J. Mol. Neurosci 31, 139–148. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, and Hersch SM (2003). Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci 23, 9418–9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, and Toda T (2008). Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem 283, 26188–26197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL 3rd, Bigio EH, Caselli R, et al. (2008). TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol 63, 535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giubilei F, Patacchioli FR, Antonini G, Sepe Monti M, Tisei P, Bastianello S, Monnazzi P, and Angelucci L (2001). Altered circadian cortisol secretion in Alzheimer’s disease: clinical and neuroradiological aspects. J. Neurosci. Res 66, 262–265. [DOI] [PubMed] [Google Scholar]
- Gonsalves SE, Moses AM, Razak Z, Robert F, and Westwood JT (2011). Whole-genome analysis reveals that active heat shock factor binding sites are mostly associated with non-heat shock genes in Drosophila melanogaster. PLoS ONE 6, e15934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grima B, Chelot E, Xia R, and Rouyer F (2004). Morning and evening peaks of activity rely on different clock neurons of the Drosophila brain. Nature 431, 869–873. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, and Goldstein LS (2003). Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40, 25–40. [DOI] [PubMed] [Google Scholar]
- Guo W, Chen Y, Zhou X, Kar A, Ray P, Chen X, Rao EJ, Yang M, Ye H, Zhu L, et al. (2011). An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol 18, 822–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, Mahal A, Smith DL, Woodman B, and Bates GP (2004). Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum. Mol. Genet 13, 1389–1405. [DOI] [PubMed] [Google Scholar]
- Helfrich-Förster C (1998). Robust circadian rhythmicity of Drosophila melanogaster requires the presence of lateral neurons: a brain-behavioral study of disconnected mutants. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol 182, 435–453. [DOI] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PAS, et al. (2003). Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 100, 2041–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison AL, Allada R, and Dinner AR (2018). Bootstrapping and Empirical Bayes Methods Improve Rhythm Detection in Sparsely Sampled Data. J. Biol. Rhythms 33, 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Sasaki HM, Sakaguchi Y, Suzuki T, Tadakuma H, and Tomari Y (2015). Defining fundamental steps in the assembly of the Drosophila RNAi enzyme complex. Nature 521, 533–536. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, Mac-Donald ME, and Zipursky SL (1998). Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 21, 633–642. [DOI] [PubMed] [Google Scholar]
- Jana NR, Tanaka M, Wang G.h., and Nukina N (2000). Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum. Mol. Genet 9, 2009–2018. [DOI] [PubMed] [Google Scholar]
- Kalliolia E, Silajdžić E, Nambron R, Hill NR, Doshi A, Frost C, Watt H, Hindmarsh P, Bjorkqvist M, and Warner TT (2014). Plasma melatonin is reduced in Huntington’s disease. Mov. Disord 29, 1511–1515. [DOI] [PubMed] [Google Scholar]
- Khaskheli MN, Baloch S, Sheeba A, Baloch S, Khan F, and Ansari MR (2017). Acute renal morbidities with obstetrical emergencies: An important women health issue. Pak. J. Med. Sci 33, 594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Ko HW, Yu W, Hardin PE, and Edery I (2007). A DOUBLETIME kinase binding domain on the Drosophila PERIOD protein isessential forits hyperphosphorylation, transcriptional repression, and circadian clock function. Mol. Cell. Biol 27, 5014–5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Subramanian M, Cho YH, Kim GH, Lee E, and Park JJ (2018). Short-term exposure to dim light at night disrupts rhythmic behaviors and causes neurodegeneration in fly models of tauopathy and Alzheimer’sdisease. Biochem. Biophys. Res. Commun 495, 1722–1729. [DOI] [PubMed] [Google Scholar]
- Kloss B, Price JL, Saez L, Blau J, Rothenfluh A, Wesley CS, and Young MW (1998). The Drosophila clock gene double-time encodes a protein closely related to human casein kinase Iepsilon. Cell 94, 97–107. [DOI] [PubMed] [Google Scholar]
- Krishnan N, Rakshit K, Chow ES,Wentzell JS, Kretzschmar D, and Giebultowicz JM (2012). Loss of circadian clock accelerates aging in neurode-generation-prone mutants. Neurobiol. Dis 45, 1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T, Loh DH, Truong D, Wu Y, and Colwell CS (2011a). Circadian dysfunction in a mouse model of Parkinson’s disease. Exp. Neurol 232, 66–75. [DOI] [PubMed] [Google Scholar]
- Kudo T, Schroeder A, Loh DH, Kuljis D, Jordan MC, Roos KP, and Colwell CS (2011b). Dysfunctions in circadian behavior and physiology in mouse models of Huntington’s disease. Exp. Neurol 228, 80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryavtsev VA, Khokhlova AV, Mosina VA, Selivanova EI, and Kabakov AE (2017). Induction of Hsp70 in tumor cellstreated with inhibitors of the Hsp90 activity: A predictive marker and promising target for radiosensitization. PLoS ONE 12, e0173640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kula-Eversole E, Nagoshi E, Shang Y, Rodriguez J, Allada R, and Rosbash M (2010). Surprising gene expression patterns within and between PDF-containing circadian neurons in Drosophila. Proc. Natl. Acad. Sci. USA 107, 13497–13502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuljis D, Schroeder AM, Kudo T, Loh DH, Willison DL, and Colwell CS (2012). Sleep and circadian dysfunction in neurodegenerative disorders: insights from a mouse model of Huntington’s disease. Minerva Pneumol. 51, 93–106. [PMC free article] [PubMed] [Google Scholar]
- Kuo Y, Ren S, Lao U, Edgar BA, and Wang T (2013). Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis. 4, e833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WCM, Yoshihara M, and Littleton JT (2004). Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 101, 3224–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh DH, Kudo T, Truong D, Wu Y, and Colwell CS (2013). The Q175 mouse model of Huntington’s disease shows gene dosage- and age-related decline in circadian rhythms of activity and sleep. PLoS ONE 8, e69993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias JL, and Alvaro PK (2012). Prevalence of sleep disturbances, disorders, and problems following traumatic brain injury: a meta-analysis. Sleep Med. 13, 898–905. [DOI] [PubMed] [Google Scholar]
- Means JC, Venkatesan A, Gerdes B, Fan JY, Bjes ES, and Price JL (2015). Drosophila spaghetti and doubletime link the circadian clock and light to caspases, apoptosis and tauopathy. PLoS Genet. 11, e1005171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J, Arrasate M, Shaby BA, Mitra S, Masliah E, and Finkbeiner S (2010). Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington’s disease molecular pathogenesis. J. Neurosci 30, 10541–10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, and Raymond LA (2010). Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci. 33, 513–523. [DOI] [PubMed] [Google Scholar]
- Mohawk JA, Green CB, and Takahashi JS (2012). Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci 35, 445–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, and Maywood ES (2005). Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J. Neurosci 25, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, and Holtzman DM (2016). Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, Roh JH, Ortiz-Gonzalez X, Dearborn JT, Culver JP, et al. (2013). Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Invest 123, 5389–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagoshi E, Sugino K, Kula E, Okazaki E, Tachibana T, Nelson S, and Rosbash M (2010). Dissecting differential gene expression within the circadian neuronal circuit of Drosophila. Nat. Neurosci 13, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormsby AR, Ramdzan YM, Mok YF, Jovanoski KD, and Hatters DM (2013). A platform to view huntingtin exon 1 aggregation flux in the cell reveals divergent influences from chaperones hsp40 and hsp70. J. Biol. Chem 288, 37192–37203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Y, Andersson CR, Kondo T, Golden SS, and Johnson CH (1998). Resonating circadian clocks enhance fitness in cyanobacteria. Proc. Natl. Acad. Sci. USA 95, 8660–8664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallier PN, and Morton AJ (2009). Management of sleep/wake cycles improves cognitive function in a transgenic mouse model of Huntington’s disease. Brain Res. 1279, 90–98. [DOI] [PubMed] [Google Scholar]
- Pallier PN, Maywood ES, Zheng Z, Chesham JE, Inyushkin AN, Dyball R, Hastings MH, and Morton AJ (2007). Pharmacological imposition of sleep slows cognitive decline and reverses dysregulation of circadian gene expression in a transgenic mouse model of Huntington’s disease. J. Neurosci 27, 7869–7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, and Hall JC (1998). Isolation and chronobiological analysis of a neuropeptide pigment-dispersing factor gene in Drosophila melanogaster. J. Biol. Rhythms 13, 219–228. [DOI] [PubMed] [Google Scholar]
- Pfeiffenberger C, Lear BC, Keegan KP, and Allada R (2010a). Locomotor activity level monitoring using the Drosophila Activity Monitoring (DAM) System. Cold Spring Harb. Protoc 2010, pdb.prot5518. [DOI] [PubMed] [Google Scholar]
- Pfeiffenberger C, Lear BC, Keegan KP, and Allada R (2010b). Processing circadian data collected from the Drosophila Activity Monitoring (DAM) System. Cold Spring Harb Protoc 2010, pdb.prot5519. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, and Rubinsztein DC (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet 36, 585–595. [DOI] [PubMed] [Google Scholar]
- Reinke H, Saini C, Fleury-Olela F, Dibner C, Benjamin IJ, and Schibler U (2008). Differential display of DNA-binding proteins reveals heat-shock factor 1 as a circadian transcription factor. Genes Dev. 22, 331–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renn SCP, Park JH, Rosbash M, Hall JC, and Taghert PH (1999). A pdf neuropeptide gene mutation and ablation of PDF neurons each cause severe abnormalities of behavioral circadian rhythms in Drosophila. Cell 99, 791–802. [DOI] [PubMed] [Google Scholar]
- Rincon-Limas DE, Jensen K, and Fernandez-Funez P (2012). Drosophila models of proteinopathies: the little fly that could. Curr. Pharm. Des 18, 1108–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E, Cha GH, Verstreken P, Ly CV, Hughes RE, Bellen HJ, and Botas J (2008). Suppression of neurodegeneration and increased neuro-transmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 57, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby NF, Hwang CE, Wessells C, Fernandez F, Zhang P, Sapolsky R, and Heller HC (2008). Hippocampal-dependent learning requires a functional circadian system. Proc. Natl. Acad. Sci. USA 105, 15593–15598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satlin A, Volicer L, Stopa EG, and Harper D (1995). Circadian locomotor activity and core-body temperature rhythms in Alzheimer’s disease. Neurobiol. Aging 16, 765–771. [DOI] [PubMed] [Google Scholar]
- Seluzicki A, Flourakis M, Kula-Eversole E, Zhang L, Kilman V, and Allada R (2014). Dual PDF signaling pathways reset clocks via TIMELESS and acutely excite target neurons to control circadian behavior. PLoS Biol. 12, e1001810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheeba V, Fogle KJ, Kaneko M, Rashid S, Chou YT, Sharma VK, and Holmes TC (2008). Large ventral lateral neurons modulate arousal and sleep in Drosophila. Curr. Biol 18, 1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheeba V, Fogle KJ, and Holmes TC (2010). Persistence of morning anticipation behavior and high amplitude morning startle response following functional loss of small ventral lateral neurons in Drosophila. PLoS ONE 5, e11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Rocha EM, McLean JR, Hayes MA, Izen SC, Isacson O, and Hallett PJ (2014). Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model of Huntington’s disease. Hum. Mol. Genet 23, 4510–527. [DOI] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Neukomm LJ, Brown RH Jr., and Freeman MR (2015). Age-Dependent TDP-43-Mediated Motor Neuron Degeneration Requires GSK3, hat-trick, and xmas-2. Curr. Biol 25, 2130–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, et al. (2001). Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 413, 739–743. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, et al. (2004). SUMO modification of Huntingtin and Huntington’s disease pathology. Science 304, 100–104. [DOI] [PubMed] [Google Scholar]
- Stoleru D, Peng Y, Nawathean P, and Rosbash M (2005). A resetting signal between Drosophila pacemakers synchronizes morning and evening activity. Nature 438, 238–242. [DOI] [PubMed] [Google Scholar]
- Stopa EG, Volicer L, Kuo-Leblanc V, Harper D, Lathi D, Tate B, and Satlin A (1999). Pathologic evaluation of the human suprachiasmatic nucleus in severe dementia. J. Neuropathol. Exp. Neurol 58, 29–39. [DOI] [PubMed] [Google Scholar]
- Tataroglu O, and Emery P (2015). The molecular ticks of the Drosophila circadian clock. Curr. Opin. Insect Sci 7, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptacek LJ, and Fu YH (2001). An hPer2 phosphorylation site mutation in familial Advanced Sleep-Phase Syndrome. Science 291, 1040–1043. [DOI] [PubMed] [Google Scholar]
- van Wamelen DJ, Aziz NA, Anink JJ, van Steenhoven R, Angeloni D, Fraschini F, Jockers R, Roos RA, and Swaab DF (2013). Suprachiasmatic nucleus neuropeptide expression in patients with Huntington’s Disease. Sleep (Basel) 36, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, and Speleman F (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, H0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Anand V, and Verma NP (2007). Sleep disorders in chronictraumatic brain injury. J. Clin. Sleep Med 3, 357–362. [PMC free article] [PubMed] [Google Scholar]
- VoSSfeldt H, Butzlaff M, PmSSing K, Ni Charthaigh RA, Karsten P, Lankes A, Hamm S, Simons M, Adryan B, Schulz JB, and Voigt A (2012). Large-scale screen for modifiers of ataxin-3-derived polyglutamine-induced toxicity in Drosophila. PLoS ONE 7, e47452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker JL, Huang SY, Steele AD, Aron R, Lotz GP, Nguyen Q, Giorgini F, Roberson ED, Lindquist S, Masliah E, and Muchowski PJ (2009). Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. J. Neurosci 29, 9104–9114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HB, Loh DH, Whittaker DS, Cutler T, Howland D, and Colwell CS (2018). Time-Restricted Feeding Improves Circadian Dysfunction as well as Motor Symptoms in the Q175 Mouse Model of Huntington’s Disease. eNeuro 5, ENEURO.0431-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JM, Chan HYE, Gray-Board GL, Chai Y, Paulson HL, and Bonini NM (1999). Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet 23, 425–428. [DOI] [PubMed] [Google Scholar]
- Whittaker DS, Loh DH, Wang HB, Tahara Y, Kuljis D, Cutler T, Ghiani CA, Shibata S, Block GD, and Colwell CS (2018). Circadian-based Treatment Strategy Effective in the BACHD Mouse Model of Huntington’s Disease. J. Biol. Rhythms 33, 535–554. [DOI] [PubMed] [Google Scholar]
- Wolfe KJ, Ren HY, Trepte P, and Cyr DM (2013). The Hsp70/90 cocha-perone, Sti1, suppresses proteotoxicity by regulating spatial quality control of amyloid-like proteins. Mol. Biol. Cell 24, 3588–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang WJ, Miller TW, Webster JM, Huston JS, Thompson LM, Marsh JL, and Messer A (2005). Suppression of Huntington’s disease pathology in Drosophila by human single-chain Fv antibodies. Proc. Natl. Acad. Sci. USA 102, 11563–11568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YH, Feenstra MGP, Zhou JN, Liu RY, Torano JS, Van Kan HJM, Fischer DF, Ravid R, and Swaab DF (2003). Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J. Clin. Endocrinol. Metab 88, 5898–5906. [DOI] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptacek LJ, and Fu YH (2005). Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature 434, 640–644. [DOI] [PubMed] [Google Scholar]
- Zhang S, Binari R, Zhou R, and Perrimon N (2010). A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics 184, 1165–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, and Sehgal A (2008). Probing the relative importance of molecular oscillations in the circadian clock. Genetics 178, 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.