

ABSTRACT
The TREK family of leak potassium channels has been found to play critical roles in nociception, sensitivity to general anaesthetics, neuroprotection, and memory. The three members of the family, TREK1, TREK2 and TRAAK establish the resting potential and modify the duration, frequency and amplitude of action potentials. Despite their apparent importance, the repertoire of regulatory interactions utilized by cells to control their expression is poorly understood. Herein, the contribution of miRNAs in the regulation of their post-transcriptional gene expression has been examined. Using different assays, miR-124 and to a lesser extent miR-128 and miR-183 were found to reduce TREK1 and TREK2 levels through specific binding to their 3ʹUTRs. In contrast, miR-9 which was predicted to bind to TRAAK 3ʹUTR, did not alter its expression. Expression of miR-124, miR-128 and miR-183 was found to mirror that of Trek1 and Trek2 mRNAs during brain development. Moreover, application of proinflammatory mediators in dorsal root ganglion (DRG) neurons revealed an inverse correlation between miR-124 and Trek1 and Trek2 mRNA expression. Voltage clamp recordings of TREK2-mediated currents showed that miR-124 reduced the sensitivity of TREK2-expressing cells to non-aversive warmth stimulation. Overall, these findings reveal a significant regulatory mechanism by which TREK1 and TREK2 expression and hence activity are controlled in neurons and uncover new druggable targets for analgesia and neuroprotection.
Abbreviations: microRNA: miRNA; UTR: untranslated region; K2p channels: two-pore domain K+channels; DRG: dorsal root ganglion; CNS: central nervous system; FBS: fetal bovine serum; TuD: Tough Decoy; TREK: tandem P-domain weak inward rectifying K+ (TWIK)-related K+ channel 1; TRAAK: TWIK-related arachidonic acid K+.
KEYWORDS: TREK1, TREK2, TRAAK, miR-124, miR-128, miR-183
Introduction
The two-pore domain K+ (K2p) channels stabilize the negative resting cell membrane potential, adjust the excitability and counteract the depolarization in mammalian cells. Although they are well known as ‘background/leak’ channels that remain active across the entire physiological voltage range, many of them are polymodal and respond to a wide range of diverse regulatory inputs such as temperature, hypoxia, hyperosmolarity, membrane stretch and swelling [1]. Among the K2p channels, the TREK subfamily members, TREK1/KCNK2, TREK2/KCNK10, and TRAAK/KCNK4 assemble as hetero/homo-dimers to produce outwardly rectifying currents with low basal activity [2]. TREK1 shares 65% and 45% amino acid identities with TREK2 and TRAAK, respectively, and less than 30% with other K2p four transmembrane domain channels [3,4]. They are expressed in both excitable and non-excitable cells and are thought to contribute significantly to pain perception [5–9], brain ischaemia [10], memory impairment [11], migraine [12], mental retardation [13], and morphine analgesia [14]. In addition, they have been found to respond positively to several neuroprotective agents, such as riluzole, nitric oxide, volatile anaesthetics, poly-unsaturated fatty acids, and lysophospholipids [15]. Despite progress in understanding their roles, it is still unclear how such diverse inputs modulate their expression or function. Currently, the only known mode of regulation is intracellular signalling via phosphorylation (by PKC and PKA) or dephosphorylation (by PLD2) of the channels that modulates their activity [4,16,17].
MicroRNAs (miRNAs) are a class of highly conserved, non-coding endogenous RNA molecules consisting of 17–24 nucleotides, that act to inhibit protein expression by partially hybridizing to complementary sequences in the 3ʹUTR of functionally-related target RNA transcripts [18]. They display a wide variety of expression patterns and many of them are differentially expressed during development or disease [19]. MiRNAs confer robustness to genetic programmes presumably by two opposing means: (a) miRNA and target mRNAs are either highly expressed in mutually exclusive tissues where the miRNA blocks translation of the unwanted mRNAs expressed from leaky promoters, (b) both the miRNA and target mRNAs are co-expressed in the same tissues where the miRNA acts as rheostat to dampen protein translation to optimal levels thus enabling customized expression [20,21]. Currently, some 2,700 mature miRNAs have been identified in humans (miRbase 22.1).
The growing body of evidence on the regulation of gene expression by miRNAs and the paucity of evidence on the regulation of TREK channels’ expression made us question whether miRNAs are involved in these channels’ expression. We tested a number of predicted conserved miRNAs and identified miR-124, miR-128 and miR-183 as new regulators of TREK channels’ expression. These miRNAs have overlapping expression patterns with Trek1 and Trek2 mRNAs and with the exception of miR-183, they are neuron-restricted, indicating that they refine endogenous TREK1 and TREK2 expression specifically in the nervous system.
Materials and methods
Ethics statement
All rodent tissues were obtained in accordance with the European Union (2003/65/CE) guidelines on the use of laboratory animals. Experimental protocols were approved by the Institutional Animal Care and Use Committee of BRFAA and the Veterinary Services of Attica prefecture (K/2134).
Generation of DNA constructs
The murine Trek1 (Kcnk2, 1879nt), Trek2 (Kcnk10, 814nt) and Traak (Kcnk4, 290nt) 3ʹUTRs were amplified from mouse brain genomic DNA using the proofreading Phusion polymerase (ThermoFisher, Massachusetts, USA) (for primer sequences and PCR conditions, see Supplementary Tables 1 and 2, respectively). The PCR products were cloned between the XhoI and NotI restriction sites of the psiCHECK-2 vector (Promega, Wisconsin, USA) downstream of the Renilla luciferase coding sequence.
To obtain murine Trek1 (3080nt) and Trek2 (2362nt) CDS plus 3ʹUTR, total RNA was extracted from CD1 mouse brains using the RNAzol RT reagent (Molecular Research Centre, Ohio, USA) according to manufacturer’s instructions. Afterwards, 1 μg of RNA was used as a template for cDNA synthesis using M-MLV Reverse Transcriptase (ThermoFisher) according to manufacturer’s protocol. The murine Trek1 (3080nt) and Trek2 (2362nt) CDS+3ʹUTR PCR products were then amplified using the Phusion polymerase (ThermoFisher) (for PCR conditions and primer sequences see Supplementary Tables 1 and 2) and cloned between the SalI and EcoRV restriction sites of the pENTR-GD expression vector (ThermoFisher).
Expression plasmids directing the synthesis of mmu-miR-9-2 (miRBase accession no. MI0000157), mmu-miR-124-1(miRBase accession no. MI0000716), mmu-miR-128-2 (miRBase accession no. MI0000726), and mmu-miR-183 (miRBase accession no. MI0000225) were prepared by cloning annealed oligonucleotides corresponding to the precursor miRNA sequences (pri-miRNA) into the pcDNA6.2-GW/EmGFP vector (ThermoFisher) as previously described [22]. Scramble 1, supplied by ThermoFisher, is predicted not to target any vertebrate gene. Scramble 2 was prepared by scrambling the mature sequence of mmu-miR-128-2 before inserting it in the pcDNA6.2-GW/EmGFP vector.
Anti-miRNA Tough Decoy (TuD) hairpin constructs were prepared by cloning annealed oligonucleotides corresponding to the reverse complement sequence of mature miR-124 or miR-128, as well as scramble, into the pENTR-U6 vector (ThermoFisher) as previously described (for primer sequences, see Supplementary Table 1) [23,24].
All constructs were Sanger-sequenced (Cemia Lab, Thessalia, Greece) before use (for primer sequences see Supplementary Table 1).
Site-directed mutagenesis
Point mutations for each miRNA binding site were inserted in Trek1 and Trek2 3ʹUTRs as previously described [25]. Briefly, proofreading PCR reactions with mutagenized primers were carried out using the psiCHECK-2 Trek1 and Trek2 3ʹUTR plasmids as templates. The reactions used the wild-type external primers and the mutagenized internal primers (for primer sequences see Supplementary Table 1). The PCR products were then gel-purified (GenElute™, Sigma-Aldrich, Missouri, USA) and equal amounts were PCR-assembled, two fragments at a time, using corresponding flanking primers. The PCR cycling conditions were as follows: five cycles with twice as long extension times and no primers (to allow assembly of full fragment), followed by 15 cycles of proofreading PCR to amplify the assembled fragment. The mutagenized final PCR products were then cloned between the XhoI and NotI restriction sites of the psiCHECK-2 vector and sequenced.
Cell-line culture and transfection
HEK293 and SK-N-SH cells were grown in high-glucose DMEM (Sigma-Aldrich) supplemented with 10% foetal bovine serum (FBS) (ThermoFisher) and 1% penicillin/streptomycin (Sigma-Aldrich). Cells were maintained at 37°C in a humidified 5% CO2 incubator (ThermoForma 3110, ThermoFisher). For luciferase reporter assays, HEK293 cells were transiently co-transfected, a day after plating in a 48-well plate (Greiner Bio-One diagnostics, Austria), with miRNAs and TREK reporter plasmids in a 3:1 ratio (0.3:0.1 μg) by using Lipofectamine 2000 according to the manufacturer’s protocol (ThermoFisher). For patch-clamp recordings, HEK293 cells were transiently co-transfected at plating with miR-124 and Trek2 CDS+3ʹUTR plasmids in a 3:1 ratio using Lipofectamine 2000. HEK293 cells were chosen for these experiments because they express low levels of neuronal miRNAs and are transfected with high efficiency (typically 70–90% at 48 hours verified by EmGFP expression). For RT-PCR and immunoblotting assays, SK-N-SH cells were co-transfected at plating with miRNAs and Trek1 or Trek2 expression plasmids in a 3:1 ratio (0.3:0.1 μg) by using Lipofectamine 2000 according to the manufacturer’s instructions (70–80% transfection efficiency verified by EmGFP expression). SK-N-SH cells were used in these experiments in order to examine whether these miRNAs regulate TREK levels in a neuronal-like context. To ensure that transfection efficiencies and TREK reporters’ expression were uniform across conditions, TREK plasmids and Lipofectamine 2000 reagent were prepared as master mix before aliquoting into tubes containing the different miRNA plasmids.
For co-transfection experiments using antagomiRs (80 nM), the 2ʹ-O-methyl anti-miRs sequences were: scramble, CAGUACUUUUGUGUAGUACAA, anti-miR-124, GGCAUUCACCGCGUGCCUUA; anti-miR-128, AAAGAGACCGGUUCACUGUGA and anti-miR-183, AGUGAAUUCUACCAGUGCCAUA (GenePharma, Shanghai, China).
The expression of the TREK family members is very low in both HEK293 and SK-N-SH cells (Supplementary Fig. 1A-C and data not shown), making these cell-lines suitable for regulation of expression studies. The expression levels of miR-9, miR-124, miR-128 and miR-183 in SK-N-SH cells before and after miR overexpression are shown in Supplementary Fig. 2A-E.
DRG neuron cultures
Dissociated, adult (2–3 month old) DRG neurons from male CD1 mice, were grown in Neurobasal medium with B-27 supplement (ThermoFisher), L-glutamine (ThermoFisher) and 2% heat-inactivated horse serum in poly-L-lysine (BD, New Jersey, USA) coated tissue culture dishes with (Luciferase experiments) or without (Gene expression studies) Nerve Growth Factor (NGF, 20 ng/ml) [26]. NGF enhances axonal regeneration but is not required for survival of adult sensory neurons [27,28].
For luciferase reporter analyses, freshly isolated DRG neurons were co-transfected with psiCHECK-2/Trek1 or Trek2 3ʹUTR plasmids and either TuD-scramble, TuD-124, or TuD-128 plasmids using Lipofectamine 3000 (ThermoFisher) in a 1:2 ratio (0.25:0.5 μg). Neurons were processed using the Dual-Luciferase reporter kit, 48 hours later. For mRNA and miRNA expression analysis, 24 hours after plating, neurons were treated with either Histamine (100 μM), Prostaglandin E2 (PGE2, 10 μM), Bradykinin (1 μM), Serotonin (10 μM) or NGF (50 ng/ml), and 24 hours later total RNA was isolated, cDNA was synthesized and real-time PCR (qPCR) was carried out as described below.
Luciferase reporter assay
Luciferase assays were performed 40–48 hours after transfection in HEK293 cells or DRG neurons with the Dual-Luciferase reporter kit (Promega) according to the manufacturer’s instructions, and measured with the Lumat LB9507 luminometer (Berthold Technologies, Bad Wildbad, Germany). Changes in the expression of Renilla luciferase (target) were calculated relative to firefly luciferase (internal reference) [22].
Total RNA isolation and cDNA synthesis
Total RNA was extracted from cell-lines and murine tissues using the RNAzol RT Reagent according to the manufacturer’s instructions (Molecular Research Centre). RNA quality was verified by measuring the absorbance at 260 and 280 nm (A260/280 of all samples >1.9). Human total RNA from different tissues was obtained from Zyagen (San Diego, CA, USA). First strand cDNA was synthesized from 0.1–0.6μg total RNA using random hexamer primers according to M-MLV reverse transcriptase protocol from ThermoFisher. For miRNA detection, a polyadenylation step before reverse transcription was included using Poly(A) polymerase (NEB, Massachusetts, USA), ATPs and a specific primer (panmiR) according to manufacturer’s instructions (see Supplementary Table 1 for sequence). The resulting cDNA was diluted with nuclease-free water (ThermoFisher) before use in quantitative and semi- quantitative PCR.
Semi-quantitative and quantitative PCR analysis
Semi-quantitative PCR was performed from cDNAs synthesized using whole brain extracts of different developmental stages (embryonic day 14 (E14) and 18 (E18), postnatal day 3 (P3), 13 (P13) and 70 (P70) dissected out from CD1 mice) using the Kapa Taq polymerase (Roche, Basel, Switzerland) and specific primers (Supplementary Tables 1 and 2). PCR products were then separated on 8% non-denaturing polyacrylamide gels that were subsequently stained with SyberGold (ThermoFisher). Images were captured with the Dolphin gel documentation system (Wealtech, Nevada, USA). The amplification bands corresponding to miRNAs, Trek1, Trek2, Gapdh and U6 mRNAs (the average of the latter two was used for normalization) were quantified by densitometric analysis using the image analysis software Image J (http://rsb.info.nih.gov/ij, NIH, USA).
cDNAs from tissues, adult DRG neurons or SK-N-SH cells transfected with the appropriate vectors, were used for qPCR. qPCR assay was based on SYBR green I binding dye and carried out in 96-well PCR white microplates (Roche) on a LightCycler 96 instrument (Roche). Fluorescence emission of the products and subsequent analysis were carried out with the LC96 software (Roche). The reaction mixture (20 μl total volume per well) included 9 μl of 10 x diluted cDNA, 200 nmol/l primers, 10 μl Kapa SYBR® Fast Universal 2 X qPCR Master Mix (Kapa Biosystems). Reactions were performed under the conditions shown in Supplementary Table 2. Dissociation (melting) curves were examined for each sample in order to assess the success of the amplification reactions. Each qPCR run included a no-cDNA template control and reverse transcription negative controls. All negative controls gave no or minimal detectable quantification cycle value, proving the lack of any contamination or non-specific signal. Each sample was tested in triplicate and data obtained from four independent experiments were used for analysis using the 2−ΔΔCt method [29] with Gapdh or U6 mRNAs serving as normalization controls.
Immunoblotting
Immunoblotting was carried out as previously described [30]. Briefly, equal amounts of whole cell extracts (20 μg) were analysed by 10% SDS-PAGE under reducing conditions. Total protein content was quantified by the Bradford Assay (BioRad Laboratories, Richmond, California, USA). The molecular weight of proteins was confirmed using a prestained protein ladder (PageRuler, ThermoFisher). The home-made rabbit anti-TREK1 antibody, kindly provided by Dr. Florian Lesage (Universite de Nice Sophia Antipolis, France) [31], the rabbit anti-TREK2 antibody (APC-055, Alomone) and the rabbit anti-TRAAK antibody (sc-50413, Santa Cruz Biotechnology, Santa Cruz, California, USA) were used in a 1:1000 dilution. The mouse anti-GAPDH antibody (sc-365062, Santa Cruz Biotechnology) was used in a 1:5000 dilution. The secondary HRP-conjugated anti-rabbit (CST#7074, Cell Signalling Technologies, Boston, Massachusetts, USA) and anti-mouse (CST#7076, Cell Signalling Technologies) antibodies were used in a 1:2000 dilution. The immunoreactive bands were revealed by using the enhanced chemiluminescence method (Clarity, Bio-Rad Laboratories, Richmond, California, USA). Each sample was tested in duplicate and samples obtained from four independent experiments were used for analysis. Densitometric analysis of immunoblots was performed using the image analysis software Image J (NIH, USA) and GAPDH was used for normalization. The expression levels of TREK1 and TREK2 proteins following plasmid overexpression in SK-N-SH or HEK293 cells are shown in Supplementary Fig. 1A-C.
Whole-cell patch clamp recordings
HEK293T cells were transiently co-transfected with miR-124 or scramble control and Trek2 CDS plus 3ʹUTR plasmids in a 3:1 ratio using Lipofectamine 2000. Electrophysiological recordings were performed 40–48 hours later using a WPC-100 (E.S.F. Electronic, Göttingen, Germany) patch clamp amplifier, in the voltage clamp mode. The extracellular solution composition was 140 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 4.54 mM NaOH at pH7.4. D-glucose, at 5 mM, was added right before the experiment. The intracellular (pipette) solution was 140 mM KCl, 1mM MgCl2, 10 mM HEPES, 5 mM EGTA/KOH, and 2 mM ATP-Na2- at pH 7.25 adjusted with NaOH. The bath electrode was placed in the same intracellular solution above an Agar bridge made with extracellular solution, to avoid the need to compensate the holding voltage for the liquid junction potential. Thick-walled borosilicate capillaries with filament (GC150F-10, Harvard Apparatus, Holliston, MA, USA) were pulled using a microprocessor-controlled vertical puller (PUL-100, World Precision Instruments, Sarasota, FL, USA) and exhibited a resistance between 2 and 4 MΩ. The EmGFP fluorescence was used to select transfected cells for patch clamping. After entering the whole-cell configuration, the capacitive currents were compensated using the R series, C slow and C fast adjustments of the amplifier until the shape of the test stimulus (20 mV square pulse) could be reproduced. The signal was filtered at 3 kHz and digitized at 5–25 kHz, using an Axon Instruments DigiData 1322A interface driven by pCLAMP 8.2 (Molecular Devices, Sunnyvale, CA, USA). To identify the current corresponding to TREK2, consecutive depolarizing voltage ramps from −100 to +50 mV, at 1 s interval were applied. Bath temperature was controlled with a custom-made system that was fast exchanging perfusate temperature using two Peltier elements. In order to measure and calibrate the bath temperature, a miniature T-type thermocouple (1T-1E, Physitemp, Clifton, NJ, USA) was placed close to the position where the recorded cell was found.
Statistical analysis
Data are presented as mean ± standard error of mean (SEM) from more than three independent experiments. Depending on the number of groups examined, comparisons were carried out using either Mann-Whitney U-test or Kruskal-Wallis test followed by Dunn’s post-hoc analysis (more than two groups). The threshold for significance was set to p-values less than 0.05. Statistical analysis was performed using the SPSS software (Release 10.0.1, IBM, IL, USA).
Results
Multiple miRNA target sites are predicted for Trek1, Trek2, and Traak 3’UTRs
To identify putative miRNA target sites located within the 3ʹUTR of TREK subfamily members predictions from three different algorithms were compiled and compared. These were the TargetScan (TargetScanMouse release 7.2) [32,33], DIANA-microT (DIANA-microT webserver v5.0) [34,35] and miRDB (accessed September 2019) [36] algorithms. miRNA-mRNA pairs eligible for experimental analysis were only those predicted by all three algorithms. Several putative target sites, which were conserved at orthologous positions across multiple vertebrate species, were identified by this analysis (Fig. 1A–C and Supplementary Fig. 3 [37]). They included sites for the highly-enriched neuronal miRNA miR-9, miR-124, miR-128, and the ubiquitous miR-183 [38,39,40 and Supplementary Table 3 for their expression analysis in 16 human tissues]. Interestingly, miR-124, miR-128 and miR-183 sites are predicted for both Trek1 and Trek2 3ʹUTRs reiterating the notion that Trek1 and Trek2 genes have arisen by gene duplication from a common ancestor [4]. The minimum free energy of the interaction between the miRNAs and the target sites is shown in Fig.1D. The lower the free energy, the higher the affinity that the miRNA has for the target mRNA. Further, the predicted target sites for miR-124 and miR-128 in Trek1 mRNA and miR-124, miR-128 and miR-183 in Trek2 mRNA are flanked by several AU nucleotides pointing to an improved targeting (Fig. 1A–D), Again, the lower the local AU score the better the accessibility [33].
Figure 1.

miRNA binding sites in Trek1, Trek2 and Traak 3ʹUTRs. Relative position and sequence conservation of miR-9, miR-124, miR-128, and miR-183 sites in (A) Trek1 3ʹUTR, (B) Trek2 3ʹUTR, and (C) Traak 3ʹUTR. D. Predicted hybridization and local AU scores using the RNAhybrid (webserver accessed December 2019) and TargetScan7.2 algorithms (webserver accessed September 2019), respectively [55]. The lower the scores, the higher the affinity of the miRNA for the target mRNA.
MiRNAs functionally interact with Trek1 and Trek2 3’UTRs, but not Traak 3’UTR
To test whether the predicted miRNA sites regulate the translation of the Trek mRNAs, two sets of expression plasmids were prepared: one set with the pri-miRNA sequences of the miRNAs inserted in the 3ʹUTR of Emerald GFP to overexpress the miRNAs via a RNA pol II promoter, and a set of reporter plasmids in which either the wild-type or the mutagenized (2 nucleotide substitutions in the seed sequence of predicted miRNA sites) 3ʹUTR of Treks was inserted downstream of a Renilla luciferase gene. The reporter plasmids, also, contained downstream a Firefly luciferase gene, that was used as an internal control to normalize for the differences in handling and transfection efficiencies (Fig. 2A). For the assay, the reporter was co-transfected with the miRNA expression plasmids into HEK293 cells and luciferase reporter expression was assessed 48 hours post-transfection.
Figure 2.
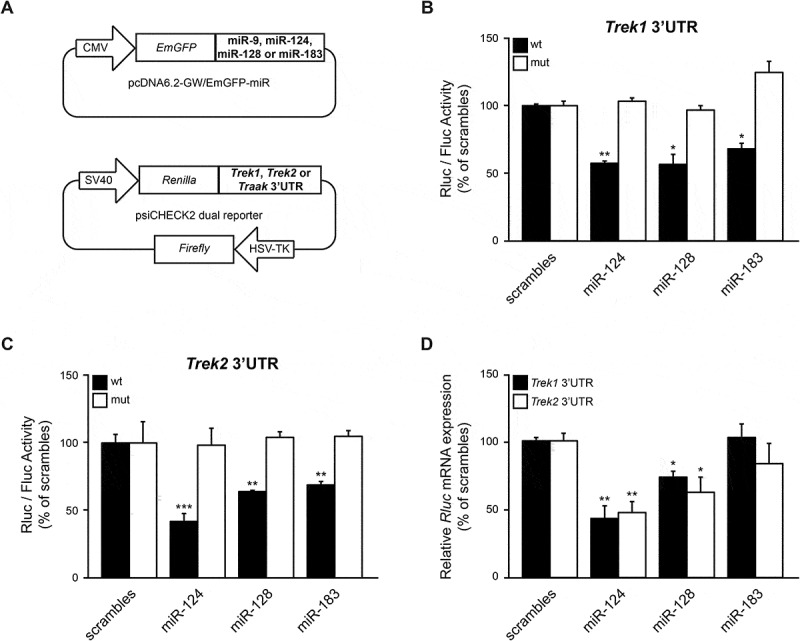
miR-124, miR-128, and miR-183 directly reduce TREK1 and TREK2 reporter protein expression. (A). Schematics of miRNA and TREK reporter constructs. (B, C). HEK293 cells were co-transfected with the TREK1 or TREK2 reporter and pri-miRNA expression vectors and luciferase activity was measured 48 hours later. These assays demonstrated that miR-124, miR-128, and miR-183 significantly reduced TREK1 and TREK2 reporter protein levels, compared to scramble controls. Subsequently, mutagenesis of the predicted miR-124, miR-128, and miR-183 binding sites reversed the inhibitory effect, indicating that these miRNAs directly suppress Trek1 and Trek2 mRNA expression by targeting the specified seed regions in the corresponding 3ʹUTR. (D). HEK293 cells were co-transfected with the Trek1 or Trek2 reporter genes and pri-miRNA expression vectors and Renilla mRNA levels were measured by RT-qPCR, 48 hours later. Firefly mRNA was used as an internal reference control. This analysis revealed that miR-124 and miR-128 overexpression significantly reduce Trek1 and Trek2 reporter mRNA levels, compared to scramble controls, indicating that these miRNAs drive Trek1 and Trek2 mRNA degradation. Data show the mean ± SEM from 6 independent transfections (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
These assays revealed that miR-124, miR-128, and miR-183 overexpression caused a 43.1% (p < 0.01), 43.3% (p < 0.05), and 31.7% (p < 0.05) decrease in the wild-type TREK1 reporter activity compared to control vectors, respectively (Fig. 2B, black bars). Mutagenesis of miR-124, miR-128, and miR-183 bindings sites on Trek1 3ʹUTR completely abolished the inhibitory effect mediated by these miRNAs (Fig. 2B, white bars). Furthermore, the miR-124, miR-128, and miR-183 overexpression caused a 58.9% (p < 0.001), 36.3% (p < 0.01), and 31.9% (p < 0.01) decrease in the wild-type TREK2 reporter activity compared to control vectors, respectively (Fig. 2C, black bars). Similarly to TREK1, mutations in the miR-124, miR-128, and miR-183 binding sites of Trek2 3ʹUTR completely abolished the inhibitory effect of these miRNAs (Fig. 2C, white bars). In contrast to the above results, miR-9 overexpression did not reduce TRAAK reporter expression (Supplementary Fig. 4A). Overall, these findings indicate that miR-124, miR-128, and miR-183 directly interact with the identified regions in Trek1 and Trek2 3ʹUTRs to inhibit translation from the chimeric transcripts.
To test whether miR-124, miR-128, and miR-183 drive mRNA degradation of Renilla hybrid mRNAs, HEK293 cells were co-transfected with Trek1 or Trek2 reporter plasmids and miRNA expression plasmids. 48 hours later, total RNA was reverse transcribed and cDNA products were analysed by qPCR, using primers for Renilla mRNA. Firefly mRNA was used as an internal reference control. The qPCR analysis revealed that miR-124 and miR-128, but not miR-183, overexpression significantly decreased Renilla mRNA levels of both TREK1 and TREK2 reporters, indicating that these two miRNAs lead to Trek1 and Trek2 mRNA degradation (Fig. 2D).
MiRNAs reduce Trek1 and Trek2 mRNA and protein levels in SK-N-SH neuroblastoma cells
To examine whether these miRNAs regulate TREK levels in a neuronal-like context, SK-N-SH neuroblastoma cells were also employed. Initially, the levels of endogenous miR-9, miR-124, miR-128, and miR-183 were assessed by qPCR. The values of qPCR analyses were normalized by the delta threshold cycle values (ΔCT) method using Gapdh mRNA as internal reference control. Specifically, miRNA endogenous expression was quantified as ΔCT values, defined as CT of the target miRNA minus the CT of Gapdh mRNA. These assays revealed that endogenous miR-128 appeared at mean ΔCT −1.18 ± 0.47, more than five PCR cycles earlier than miR-9 (mean ΔCT 8.21 ± 0.6), miR-124 (mean ΔCT 4.93 ± 0.54) or miR-183 (mean ΔCT 5.31 ± 0.44) indicating that miR-128 is the most abundant among the four miRNAs (Supplementary Fig. 2A).
The same assays were, then, repeated after transfection with miRNA plasmids. qPCR assays followed by the 2-ΔΔCt analysis in SK-N-SH cells transfected with the miRNA plasmids showed that miR-9 levels increased 68-fold, miR-124 levels increased 20-fold, miR-128 levels 2-fold and miR-183 levels 18.5-fold compared to controls (Supplementary Fig. 2 B-E).
Having evaluated miRNA expression in SK-N-SH cells, TREK mRNA and protein levels were assessed following co-transfection of these cells with plasmids expressing TREK channels (Supplementary Fig. 1A, B) and either miRNA plasmids or 2ʹ-O-methyl miRNA inhibitors (antagomiRs). Results of RT-qPCR from RNA extracts using Trek1 specific primers revealed that Trek1 mRNA levels were decreased 19.5% (p < 0.05) by miR-124, but were not affected by miR-128 or miR-183 overexpression (Fig. 3A, black bars). Similarly, Trek2 mRNA levels were decreased 36% (p < 0.05) following miR-124 overexpression, but were not affected by either miR-128 or miR-183 overexpression (Fig. 3A, white bars). Sequestration of the highly abundant miR-128 with anti-miR-128 oligos in SK-N-SH cells resulted in a 10-fold increase (p < 0.01) in Trek1 mRNA levels (Fig. 3B, black bars). Further, Trek1 mRNA levels were not affected by anti-miR-124 and anti-miR-183 treatments. In contrast, the application of anti-miR-124, anti-miR-128, and anti-miR-183 significantly increased Trek2 mRNA levels by 45.2% (p < 0.05), 195.3% (p < 0.05) and 139% (p < 0.01), respectively (Fig. 3B, white bars).
Figure 3.
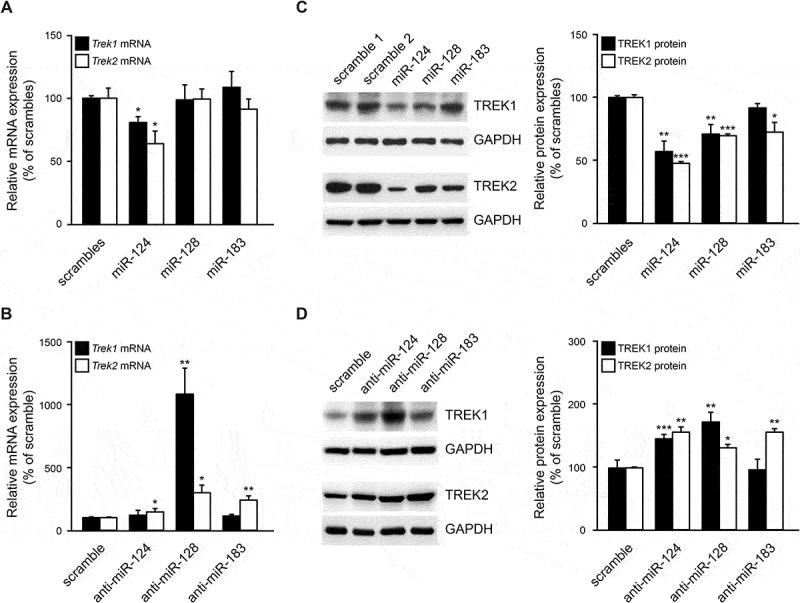
miR-124 and miR-128 regulate TREK1 and TREK2 expression. (A, B). SK-N-SH cells were co-transfected with Trek1 or Trek2 CDS plus 3ʹUTR plasmids and either pri-miRNA expression plasmids or 2ʹ-O-methyl miRNA inhibitors (antagomiRs). 48 hours later, Trek1 and Trek2 mRNA levels were measured by RT-qPCR. Gapdh mRNA was used as an internal reference control. These assays demonstrated that miR-124 overexpression significantly reduced Trek1 and Trek2 mRNA levels, while anti-miR-128 significantly increased Trek1 and Trek2 mRNA levels, compared to scramble controls. Trek2 mRNA levels were also increased by anti-miR-124 and anti-miR-183. (C, D). SK-N-SH cells were co-transfected with Trek1 or Trek2 CDS+3ʹUTR plasmids and either pri-miRNA expression plasmids or 2ʹ-O-methyl miRNA inhibitors. 48 hours later, TREK1 and TREK2 protein levels were measured by immunoblotting. GAPDH was used as an internal control for loading. These assays demonstrated that miR-124 and miR-128 overexpression significantly reduced TREK1 and TREK2 levels, while TREK2 protein levels were also reduced by miR-183 overexpression. Anti-miR −124 and −128 oligos significantly increased TREK1 levels, compared to scramble controls, while TREK2 levels were increased by all three anti-miR oligos. Data show the mean ± SEM from 4 RT-PCR and 5 immunoblotting independent experiments (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Immunoblots from whole protein extracts revealed a significant reduction in TREK1 levels by miR-124 and miR-128, but not miR-183 overexpression; in particular, miR-124, miR-128, and miR-183 caused a 42.9% (p < 0.01), 29% (p < 0.01) and 8.1% (p = 0.06) reduction in TREK1 protein levels, respectively (Fig. 3C, black bars). Immunoblots for TREK2 revealed a significant reduction in its protein levels after overexpression of all three miRNAs. In particular, miR-124, miR-128, and miR-183 caused a 52.3% (p < 0.001), 30.2% (p < 0.001) and 27.6%, (p < 0.05) reduction in TREK2 protein levels, respectively (Fig. 3C, white bars). Application of antagomiRs revealed that TREK1 protein levels were significantly increased by anti-miR-124 (46.4%, p < 0.001) and anti-miR-128 (73.4%, p < 0.01), but not anti-miR-183 (Fig. 3D, black bars). Finally, anti-miR-124, anti-miR-128, and anti-miR-183 application increased TREK2 protein levels 58.2% (p < 0.001), 32.7% (p < 0.001), and 58.8% (p < 0.05), respectively (Fig. 3D, white bars).
Traak mRNA and protein expression following miR-9 modulation were also investigated but no significant change was observed, in accordance with the luciferase findings (Supplementary Fig. 4B, C).
Overall, the data from this set of experiments indicate that neuronal miR-124 is the most potent miRNA at regulating TREK1 and TREK2 channel expression. Neuronal miR-128 is the second most effective miRNA at regulating both channels’ expression. In addition, ubiquitous miR-183 regulates TREK2, but not significantly TREK1 levels. Altogether, Trek2 mRNA is more amendable to miRNA regulation.
miR-124 expression inversely correlates with Trek1 and Trek2 mRNA expression in DRG neurons
To place our findings within a physiological context, we assessed the expression of miR-124, miR-128, miR-183, Trek1 and Trek2 mRNAs, in the central nervous system of mice throughout development. Fig. 4A shows that the levels of miR-124, miR-128, and miR-183 broadly correlate with the expression of Trek1 and Trek2 mRNAs, with lower levels early in embryonic development that reach a peak late in adulthood. These data support the notion that these miRNAs regulate Trek1 and Trek2 mRNA expression primarily postnatally and that they act to tune, rather than to eliminate, Trek1 and Trek2 expression in the nervous system.
Figure 4.
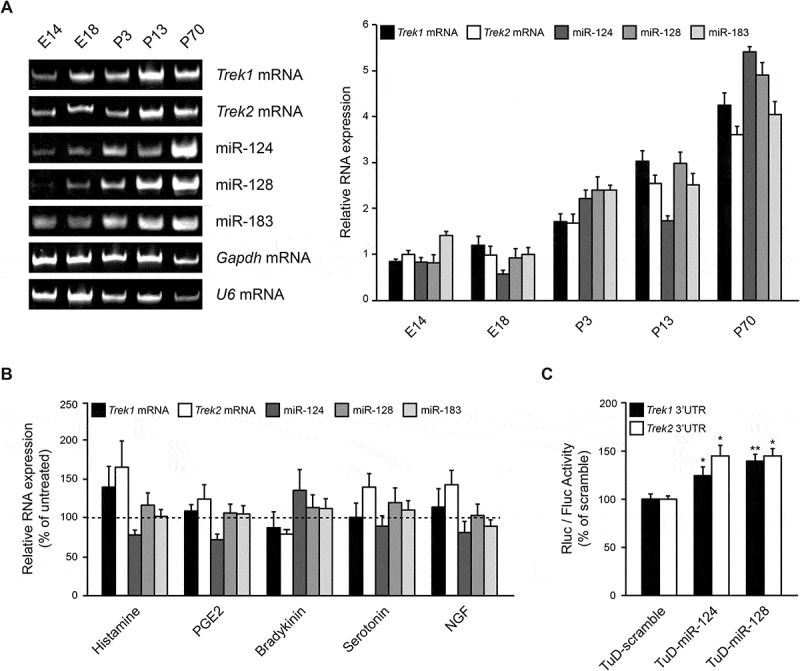
miR-124 expression inversely correlates with Trek1 and Trek2 mRNA expression. (A). Representative gel and graph of the RT-PCR amplification products of whole brains from different developmental stages (embryonic day 14 (E14) and 18 (E18), postnatal day 3 (P3), 13 (P13) and 70 (P70)) extracted from mice showing the expression of miR-124, miR-128, miR-183, Trek1 and Trek2 mRNAs throughout development in the brain. Cycles were falling within the linear range of amplification and the number of PCR cycles for each primer pair was different. The amount of starting template for each condition was normalized to Gapdh mRNA. Data show the mean ± SEM from 3 independent experiments. These data demonstrated that miR-124, miR-128, miR-183, Trek1 and Trek2 mRNAs are co-expressed in the nervous system and levels increase with ageing. (B). Primary cultures of adult DRG neurons were incubated with the pro-inflammatory mediators Histamine (100 μM), Prostaglandin E2 (PGE2, 10 μM), Bradykinin (1 μM), Serotonin (10 μM) and Nerve Growth Factor (NGF, 50 ng/ml) and 24 hours later, total RNA was extracted and analysed by RT-qPCR. U6 mRNA was used as an internal reference control. These assays demonstrated that miR-124 expression inversely correlates with Trek1 and Trek2 mRNAs expression, compared to untreated cells. Data show the mean ± SEM from 4 independent experiments. C. miR-124 and miR-128 regulate TREK1 and TREK2 expression in DRG neurons. Primary cultures of adult DRG neurons were co-transfected with psiCHECK-2/Trek1 or Trek2 3ʹUTR plasmids and either of TuD-scramble control, TuD-124, or TuD-128 plasmids and luciferase activity was measured 48 hours later. These assays demonstrated that endogenous neuronal miR-124 and miR-128 bind to luciferase mRNAs bearing Trek1 or Trek2 3ʹUTR and block their translation. Data show the mean ± SEM from 5 independent experiments (*, p < 0.05; **, p < 0.01).
Considering the established role of TREKs in pain perception, the effects of different noxious signals in the expression of miR-124, miR-128, and miR-183 and Trek1 and Trek2 mRNAs were also investigated. For this series of experiments, primary cultures of adult dorsal root ganglia (DRG) neurons were incubated with the pro-inflammatory mediators Histamine, PGE2, Bradykinin, Serotonin and NGF. Some of these agents have been previously shown to induce changes in the biophysical properties and expression levels of several ion channels and receptors involved in pain signalling, including the two pore domain K channels [5,41]. Following 24 hours incubation with these agents, total RNA was extracted and analysed by RT-qPCR. Results from these neuronal extracts revealed that the levels of Trek1, Trek2 mRNAs and miR-124, but not miR-128 and miR-183 were affected by these pro-inflammatory mediators, although not to a significant extend (Fig. 4B). Nevertheless, there was a clear inverse correlation between miR-124 expression, which is the most abundant miRNA in neurons [42,43], and Trek1 and Trek2 mRNA expression. Due to the limited number of DRG neurons obtained at this developmental stage, not enough protein could be extracted to detect TREK channels by immunoblotting.
To further confirm miRNA regulatory function in the context of DRG neurons, the ability of endogenously-expressed neuronal miR-124 and miR-128 to regulate the levels of the highly responsive TREK1 and TREK2 reporters was also evaluated. For these experiments, highly efficient TuD sponge plasmids [24] for miR-124 and miR-128 were constructed and transfected together with the Trek1 or Trek2 reporter plasmid in freshly dissociated DRG neurons. Luciferase activity, assessed 48 hours later, revealed a 25% (p < 0.05) and 42% (p < 0.01) increase in chimeric TREK1 protein levels following TuD-124 and TuD-128 plasmid overexpression, respectively (Fig. 4C, black bars). Similarly, chimeric TREK2 protein levels were increased 41% and 49% (both at p < 0.05) following TuD-124 and TuD-128 plasmid overexpression, respectively (Fig. 4C, white bars). These data indicate that endogenous neuronal miR-124 and miR-128 bind to mRNAs bearing Trek1 or Trek2 3ʹUTRs and block their translation.
miR-124 controls TREK2 current density in a heterologous expression system
To assess whether miRNA regulation impacts on the current density of TREK channels in the plasma membrane, TREK2-generated currents were recorded after transient co-overexpression of Trek2 and miR-124 plasmids in HEK293 cells. The voltage ramps applied to identify TREK2 currents were of 1 second duration, ranging from −100 to +50 mV, at 1 second intervals (Fig. 5A). The TREK2 current observed at 25°C was strongly outward rectifying in physiological K+ concentrations (4 mM extracellular, 140 mM intracellular) as previously reported [4]. Upon thermal activation at 32°C [44], however, the average amplitude of the TREK2 current measured at 50 mV was significantly increased (Fig. 5B). Fig. 5 C, D show that, at the baseline current, no significant differences between scramble control and miR-124 transfected cells were observed. At 32°C, however, the average amplitude of the TREK2 current was significantly impaired in miR-124 expressing cells (p < 0.01). To calculate the actual decrease in the current upon thermal activation the baseline values (25°C) were subtracted from the values recorded at 32°C and a 2-fold decrease was confirmed (p < 0.05) (Fig. 5E). Hence, miR-124, by modulating the expression of TREK2 at the post transcriptional level, interferes with the current elicited by TREK2 channel upon thermal activation.
Figure 5.
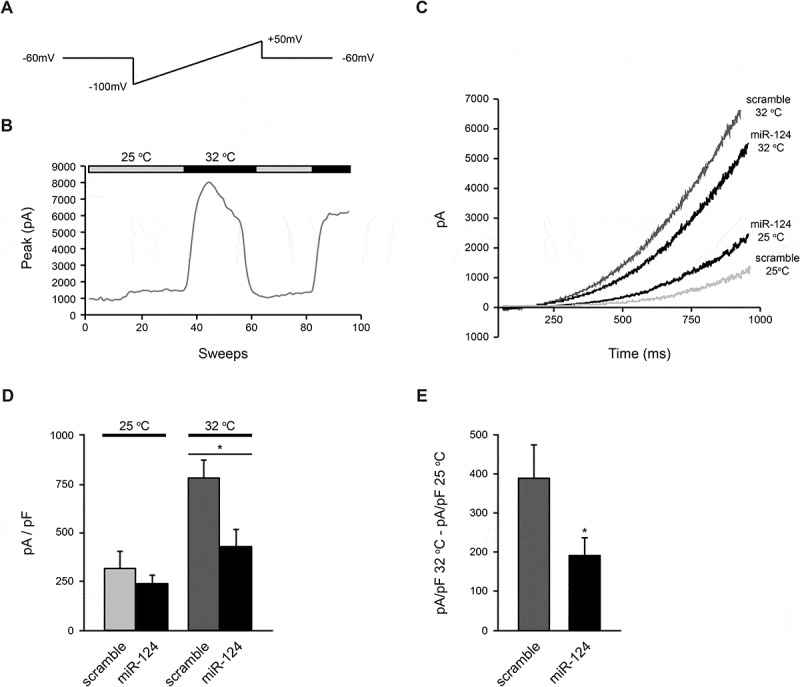
miR-124 interferes with the current elicited by TREK2 upon its thermal activation via direct regulation of TREK2 protein expression. (A). Voltage ramps applied to identify TREK2 currents. They were of 1 second duration, ranging from −100 mV to +50 mV, at 1 second intervals. (B). Time-course of the heating effect on the current mediated by the TREK2 channel measured at 50 mV. (C). Representative TREK2-mediated currents elicited by depolarizing voltage ramps at 25°C and 32 °C. (D). The amplitude values of the average current mediated by TREK2 in cells transfected with either scramble control (n = 19 at 25°C and n = 14 at 32°C) or miR-124 (n = 14 at 25°C and n = 14 at 32°C). These assays demonstrated that the expression of miR-124 significantly reduced the amplitude of the currents at 32°C. (E). The increase in the current upon thermal activation was calculated by subtracting the values at 25°C from the values recorded at 32°C. Analysis revealed that the expression of miR-124 significantly reduced TREK2 currents. Data show the mean ± SEM from 3 independent experiments (*, p < 0.05).
Discussion
This study has revealed, for the first time, a previously unknown mechanism by which TREK1 and TREK2 expression levels are regulated in the nervous system. Multiple evidence presented herein suggest that three miRNAs, miR-124, miR-128 and miR-183 bind directly to the 3ʹUTRs of Trek1 and Trek2 mRNAs and result in the reduction of their protein levels. Sequence analysis of the miRNA recognition elements in the 3ʹUTR of Trek1 and Trek2 mRNAs showed that they are conserved among vertebrate species. In vitro experiments, in which the miRNA ‘seed’ regions have been mutagenized, resulted in an unhindered protein expression of the chimeric luciferase reporter bearing the Trek1 and Trek2 3ʹUTRs following miRNA overexpression. Gain-of-function experiments revealed that overexpression of miR-124, miR-128 or miR-183 results in down-regulation of TREK1 and TREK2 protein expression levels, whereas loss-of-function experiments showed that inhibition of endogenous miR-124, miR-128 or miR-183 by antisense oligonucleotides or TuD constructs promote the expression of TREK1 and TREK2 proteins or chimeric reporters.
The possibility that TREK1 and TREK2 expression is also regulated indirectly by these miRNAs cannot be excluded. miR-124 is an inducer of autophagy [45,46] and it is likely that TREK1 and TREK2 protein levels, unlike mRNA levels, are modulated to some extent by this function.
Examination of miR-124, miR-128, miR-183, Trek1 and Trek2 mRNA expression patterns during CNS development showed that all three miRNAs as well as Trek1 and Trek2 mRNAs are upregulated from embryonic day 14 to peak levels at postnatal day 70, the later age analysed. This finding suggests that miR-124, miR-128, and miR-183 play a role in modulating Trek1 and Trek2 mRNA translation throughout development proportionally, rather than abruptly inhibiting their expression in the CNS.
Different noxious signals have also been examined for their role on miRNA and Trek1 and Trek2 mRNA expression in DRGs. While miR-128 and miR-183 levels were not affected, there was an inverse correlation recorded between Trek1 and Trek2 mRNA levels and miR-124. Knowing that TREK channels modulate, among others, thermal sensitivity in cells, clamp recordings of action potential threshold in scramble control vs miR-124 transfected cells at baseline and after heat stimulation were measured. As would have been expected from its direct interaction with the Trek2 mRNA, miR-124 overexpression reduced the current elicited upon thermal activation of TREK2. This finding is important as it indicates that other miRNA functions do not negate direct effects on TREK activity. Previously, miR-124 and to lesser extend miR-183 have been investigated with respect to neuropathic pain. miR-124 was found to be downregulated in rats after chronic sciatic nerve injury and its overexpression repressed mechanical allodynia and heat hyperalgesia via decrease in cytokine production [47]. Similar conclusions were obtained using the GRK2 model of chronic pain [48]. In a model of bone cancer pain, miR-124 levels were also downregulated and intrathecal injection of miR-124 mimics alleviated cancer pain [49]. Animals with conditional deletion of the miR-183 cluster (miR-183/96/182) in neurons were found to respond normally to light touch, cold, heat, or pinprick but were hypersensitive to mechanical stimuli [50]. Further, in the chronic compress injury model, miR-183 was shown to target mTOR and relieve neuropathic pain [51]. Hence, considering that TREK channels contribute significantly to pain perception [5–9], it is likely that part of the miR-124 and miR-183 effects observed in these studies are mediated via these channels.
TRAAK regulation by miR-9 was also investigated but no effect was observed. One possible reason is that the predicted miR-9 target site is AU-rich and such regions decrease the stability of seed pairing interactions, hence lowering the chances of successful inhibition [52,53]. Further, miRNAs with AU-rich seed regions, such as miR-9, have more 3′UTR binding sites, a consequence of the AU-rich nucleotide composition of 3′UTRs, which dilutes their inhibitory effect on each of their targets (‘off targeting’) [52]. Last, there is a high number of GC bases immediately flanking the right-hand side of the target site on Traak mRNA which is not favouring miRNA accessibility [33].
In conclusion, this study provides strong evidence that miR-124, miR-128 and miR-183 regulate TREK1 and TREK2 levels and widens our knowledge on their role in the pathophysiology of peripheral neuropathies. Additional studies will reveal whether these miRNAs could serve as novel analgesics as well as anaesthetics and neuroprotectors. Along these lines, miRNAs are easily druggable and several have already been approved for therapy [54], thus they can quickly be tested for the benefit of patient community.
Funding Statement
This work was supported by a bilateral Greek-Romanian grant from Greek General Secretariat for Research and Technology (GSRT) to ED (Grant ID 11ROM12_4_ET30).
Acknowledgments
We thank the BRFAA animal facility employees and particularly Paulos Alexakos for excellent veterinary assistance. We are grateful to Florian Lesage (Universite de Nice Sophia Antipolis, France) for the TREK1 antibody.
Disclosure statement
No potential conflict of interest was reported by the authors.
Authors’ Contributions
Conceived the study: ED. Designed and optimized protocols: ED, AB. Prepared plasmid constructs: ED, MP. Performed tissue culture and molecular biology experiments: MP, ED. Performed electrophysiology experiments: LM, TS. Analyzed data: MP, ED, LM, TS, AB, SGD, PP. Wrote manuscript: ED. All authors read, edited and approved the final manuscript.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Feliciangeli S, Chatelain FC, Bichet D, et al. The family of K2P channels: salient structural and functional properties. J Physiol. 2015;593:2587–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mathie A, Veale EL.. Two-pore domain potassium channels: potential therapeutic targets for the treatment of pain. Eur J Physiol. 2015;467:931–943. [DOI] [PubMed] [Google Scholar]
- [3].Bang H, Kim Y, Kim D. TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem. 2000;275:17412–17419. [DOI] [PubMed] [Google Scholar]
- [4].Lesage F, Terrenoire C, Romey G, et al. Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J Biol Chem. 2000;275:28398–28405. [DOI] [PubMed] [Google Scholar]
- [5].Alloui A, Zimmermann K, Mamet J, et al. TREK-1, a K+ channel involved in polymodal pain perception. Embo J. 2006;25:2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Acosta C, Djouhri L, Watkins R, et al. TREK2 expressed selectively in IB4-binding C-fiber nociceptors hyperpolarizes their membrane potentials and limits spontaneous pain. J Neurosci. 2014;34:1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dadi PK, Vierra NC, Days E, et al. Selective small molecule activators of TREK-2 channels stimulate dorsal root ganglion c-fiber nociceptor two-pore-domain potassium channel currents and limit calcium influx. ACS Chem Neurosci. 2017;8:558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pereira V, Busserolles J, Christin M, et al. Role of the TREK2 potassium channel in cold and warm thermosensation and in pain perception. Pain. 2014;155:2534–2544. [DOI] [PubMed] [Google Scholar]
- [9].Poupon L, Lamoine S, Pereira V, et al. Targeting the TREK-1 potassium channel via riluzole to eliminate the neuropathic and depressive-like effects of oxaliplatin. Neuropharmacology. 2018;140:43–61. [DOI] [PubMed] [Google Scholar]
- [10].Li ZB, Zhang HX, Li LL, et al. Enhanced expressions of arachidonic acid-sensitive tandem-pore domain potassium channels in rat experimental acute cerebral ischemia. Biochem Biophys Res Commun. 2005;327:1163–1169. [DOI] [PubMed] [Google Scholar]
- [11].Pan YP, Xu XH, Wang XL. mRNA expression alteration of two-pore potassium channels in the brain of beta-amyloid peptide25-35-induced memory impaired rats. Acta pharmaceutica Sinica. 2003;38:721–724. [PubMed] [Google Scholar]
- [12].Lafreniere RG, Cader MZ, Poulin JF, et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med. 2010;16:1157–1160. [DOI] [PubMed] [Google Scholar]
- [13].Barel O, Shalev SA, Ofir R, et al. Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am J Hum Genet. 2008;83:193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Devilliers M, Busserolles J, Lolignier S, et al. Activation of TREK-1 by morphine results in analgesia without adverse side effects. Nat Commun. 2013;4:2941. [DOI] [PubMed] [Google Scholar]
- [15].Lamas JA, Fernandez-Fernandez D. Tandem pore TWIK-related potassium channels and neuroprotection. Neural Regen Res. 2019;14:1293–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chemin J, Girard C, Duprat F, et al. Mechanisms underlying excitatory effects of group I metabotropic glutamate receptors via inhibition of 2P domain K+ channels. Embo J. 2003;22:5403–5411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Comoglio Y, Levitz J, Kienzler MA, et al. Phospholipase D2 specifically regulates TREK potassium channels via direct interaction and local production of phosphatidic acid. Proc Natl Acad Sci U S A. 2014;111:13547–13552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Doxakis E. Principles of miRNA-target regulation in metazoan models. Int J Mol Sci. 2013;14:16280–16302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wienholds E, Plasterk RH. MicroRNA function in animal development. FEBS Lett. 2005;579:5911–5922. [DOI] [PubMed] [Google Scholar]
- [20].Hornstein E, Shomron N. Canalization of development by microRNAs. Nat Genet. 2006;38(Suppl):S20–4. [DOI] [PubMed] [Google Scholar]
- [21].Peterson KJ, Dietrich MR, McPeek MA. MicroRNAs and metazoan macroevolution: insights into canalization, complexity, and the Cambrian explosion. BioEssays. 2009;31:736–747. [DOI] [PubMed] [Google Scholar]
- [22].Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285:12726–12734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bak RO, Hollensen AK, Primo MN, et al. Potent microRNA suppression by RNA Pol II-transcribed ‘Tough Decoy’ inhibitors. Rna. 2013;19:280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Haraguchi T, Ozaki Y, Iba H. Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res. 2009;37:e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Paschou M, Doxakis E. Neurofibromin 1 is a miRNA target in neurons. PloS One. 2012;7:e46773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Doxakis E, Huang EJ, Davies AM. Homeodomain-interacting protein kinase-2 regulates apoptosis in developing sensory and sympathetic neurons. Curr Biol. 2004;14:1761–1765. [DOI] [PubMed] [Google Scholar]
- [27].Lindsay RM. Nerve growth factors (NGF, BDNF) enhance axonal regeneration but are not required for survival of adult sensory neurons. J Neurosci. 1988;8:2394–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tong JX, Eichler ME, Rich KM. Intracellular calcium levels influence apoptosis in mature sensory neurons after trophic factor deprivation. Exp Neurol. 1996;138:45–52. [DOI] [PubMed] [Google Scholar]
- [29].Koukouraki P, Doxakis E. Constitutive translation of human alpha-synuclein is mediated by the 5ʹ-untranslated region. Open Biol. 2016;6:160022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fragkouli A, Koukouraki P, Vlachos IS, et al. Neuronal ELAVL proteins utilize AUF-1 as a co-partner to induce neuron-specific alternative splicing of APP. Sci Rep. 2017;7:44507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Maingret F, Lauritzen I, Patel AJ, et al. TREK-1 is a heat-activated background K(+) channel. Embo J. 2000;19:2483–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Agarwal V, Bell GW, Nam JW, et al. Predicting effective microRNA target sites in mammalian mRNAs. eLife. 2015;4:e05005. DOI: 10.7554/ELIFE.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Grimson A, Farh KK, Johnston WK, et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kiriakidou M, Nelson PT, Kouranov A, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Paraskevopoulou MD, Georgakilas G, Kostoulas N, et al. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013;41:W169–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43:D146–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fehlmann T, Ludwig N, Backes C, et al. Distribution of microRNA biomarker candidates in solid tissues and body fluids. RNA Biol. 2016;13:1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Guo Z, Maki M, Ding R, et al. Genome-wide survey of tissue-specific microRNA and transcription factor regulatory networks in 12 tissues. Sci Rep. 2014;4:5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ludwig N, Leidinger P, Becker K, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cohen A, Sagron R, Somech E, et al. Pain-associated signals, acidosis and lysophosphatidic acid, modulate the neuronal K(2P)2.1 channel. Mol Cell Neurosci. 2009;40:382–389. [DOI] [PubMed] [Google Scholar]
- [42].Bak M, Silahtaroglu A, Moller M, et al. MicroRNA expression in the adult mouse central nervous system. Rna. 2008;14:432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kang D, Choe C, Kim D. Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J Physiol. 2005;564:103–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gong X, Wang H, Ye Y, et al. miR-124 regulates cell apoptosis and autophagy in dopaminergic neurons and protects them by regulating AMPK/mTOR pathway in Parkinson’s disease. Am J Transl Res. 2016;8:2127–2137. [PMC free article] [PubMed] [Google Scholar]
- [46].Yao L, Zhu Z, Wu J, et al. MicroRNA-124 regulates the expression of p62/p38 and promotes autophagy in the inflammatory pathogenesis of Parkinson’s disease. Faseb J. 2019;33:8648–8665. [DOI] [PubMed] [Google Scholar]
- [47].Zhang Y, Liu HL, An LJ, et al. miR-124-3p attenuates neuropathic pain induced by chronic sciatic nerve injury in rats via targeting EZH2. J Cell Biochem. 2019;120:5747–5755. [DOI] [PubMed] [Google Scholar]
- [48].Willemen HL, Huo XJ, Mao-Ying QL, et al. MicroRNA-124 as a novel treatment for persistent hyperalgesia. J Neuroinflammation. 2012;9:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Elramah S, Lopez-Gonzalez MJ, Bastide M, et al. Spinal miRNA-124 regulates synaptopodin and nociception in an animal model of bone cancer pain. Sci Rep. 2017;7:10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Peng C, Li L, Zhang MD, et al. miR-183 cluster scales mechanical pain sensitivity by regulating basal and neuropathic pain genes. Science. 2017;356:1168–1171. [DOI] [PubMed] [Google Scholar]
- [51].Xie X, Ma L, Xi K, et al. MicroRNA-183 suppresses neuropathic pain and expression of AMPA receptors by targeting mTOR/VEGF signaling pathway. Cell Physiol Biochem. 2017;41:181–192. [DOI] [PubMed] [Google Scholar]
- [52].Garcia DM, Baek D, Shin C, et al. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nat Struct Mol Biol. 2011;18:1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ui-Tei K. Optimal choice of functional and off-target effect-reduced siRNAs for RNAi therapeutics. Front Genet. 2013;4:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chakraborty C, Sharma AR, Sharma G, et al. Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol Ther Nucleic Acids. 2017;8:132–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kruger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006;34:W451–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.