

ABSTRACT
Marek’s disease (MD), induced by Marek’s disease virus (MDV), is a lymphotropic neoplastic disease and causes huge economic losses to the poultry industry. Non-coding RNAs (ncRNAs) play important regulatory roles in disease pathogenesis. To investigate host circular RNA (circRNA) and microRNA (miRNA) expression profile, RNA sequencing was performed in tumourous spleens (TS), spleens from the survivors (SS) without any lesion after MDV infection, and non-infected chicken spleens (NS). A total of 2,169 circRNAs were identified and more than 80% of circRNAs were derived from exon. The flanking introns of 1,744 exonic circRNAs possessed 579 reverse complementary matches (RCMs), which mainly overlapped with chicken repeat 1 family (CR1F). It suggested that CR1F mediated the cyclization of exons by intron pairing. Out of 2,169 circRNAs, 113 were differentially expressed circRNAs (DECs). The Q-PCR and Rnase R digestion experiments showed circRNA possessed high stability compared with their linear RNAs. Integrated with previous transcriptome data, we profiled regulatory networks of circRNA/long non-coding RNA (lncRNA)-miRNA-mRNA. Extensive competing endogenous RNA (ceRNA) networks were predicted to be involved in MD tumourigenesis. Interestingly, circZMYM3, an intronic circRNA, interacted with seven miRNAs which targeted some immune genes, such as SWAP70 and CCL4. Gga-miR-155 not only interacted with circGTDC1 and circMYO1B, but also targeted immune-related genes, such as GATA4, which indicated the roles of non-coding RNAs played to mediate immune responsive genes. Collectively, this is the first study that integrated RNA expression profiles in MD model. Our results provided comprehensive interactions of ncRNAs and mRNA in MD tumourigenesis.
KEYWORDS: Chicken, Marek’s disease, circRNA, miRNA, lncRNA, mRNA, regulatory network
Introduction
Marek’s disease (MD), a T-cell lymphoma disease, caused by Marek’s disease virus (MDV), can induce T-cell lymphoma, immunosuppression and neurological disorders, and bring huge losses to the global poultry industry [1,2]. The mortality of susceptible chicken infected with MDV reached up to 100%. MD in chicken is also a good biomedical research model for virus-induced lymphoma disease [3,4]. Up to date, many researchers reported that non-coding RNAs (ncRNAs), including microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs), play important role in the occurrence of diseases [5–9]. Some lncRNAs and miRNAs involved in MD have been reported [10], but circRNA profile in MD is not yet investigated. CircRNA, was a type of endogenous non-coding RNA, is the product of back-splicing. Unlike linear RNA, circRNA, without cap and poly A tails, is stable and not easily degraded by Rnase [11,12]. CircRNAs are related to cell development [13] and various diseases, such as cardiovascular disease, severe acne, traumatic spinal cord injury and oral squamous cell carcinoma [14–17]. CircRNA possesses many important functions, including acting as microRNA (miRNA) sponges [18,19], interacting with proteins to regulate gene expression [20], regulating transcription and splicing [21,22] and translation into a protein or polypeptide [23]. It is reported that exon-derived circRNAs acted as miRNAs sponges and involved in many diseases such as glioblastoma [18], hepatocellular [24], gastric cancer [25]. The circRNA profiles have been studied in chicken muscle [13] and follicular [26]. Zhang, et al. (2017) profiled the circRNA expression of livers in ALV-J-resistant and ALV-J-susceptible chickens and concluded that circRNAs were related to chicken resistance to ALV-J-induced tumour formation [27]. Qiu, et al. (2018) investigated the circRNA expression profiles of the spleen tissues of normal chickens and ALV-J-infected chickens and declared that circRNA was involved in the tumourigenesis [28].
In this study, we comprehensively analyzed transcriptomes and features of circRNA and miRNA in MDV-infected and non-infected spleen and constructed ceRNA network including circRNA, lncRNA, miRNA and mRNA to identify epigenetic factors involved in MD tumourigenesis.
Results
Profiling of circRNAs in chicken spleens
To profile circRNA landscape in chicken spleens during MDV-induced transformation stage, we characterized circRNA transcripts in 14 samples using rRNA-depleted RNA-Seq, sequencing data have been submitted to Gene Expression Omnibus (GSE138600). A total of 161.35 gigabytes data, and 157.99 gigabytes were remained after filtering the adapters and low-quality reads. Clean reads were mapped against the reference genome and unmapped reads were saved followed by anchors splitting and re-alignment.
We identified 2,169 circRNAs with at least two independent reads spanning over back-splice junction sites via two software of find_circ and CIRI2 (Supplementary Table 1). Among 2,169 circRNAs, 1,744 were produced from exons of 1,172 genes, out of which, 855 parental genes generated one circRNA per gene, while, the other genes could produce two or more circular informs (Fig. 1(a)). In total of 286 were generated from intergenic region and the remaining were intronic circRNAs (Fig. 1(b)). The length of circRNAs ranged from 185 nt to 91,159 nt and most of them located in chromosome 1 (Fig. 1(c)). The counts of the reads that spanned over back-splice junction sites were normalized as the number of transcripts per million (TPM) (Supplementary Fig. 1). The circRNAs originated from exon, intron and intergenic region showed no significant changes in length and expression abundance (TPM) (Fig. 1(d)). The back-splicing events in chicken showed that circRNA preferred to be generated from exon regions. The length of flanking introns of circRNAs’ parental exon was of median 7,208 bp and ranged from 12 bp to 193 data, 653 bp, which was significantly longer than that of the linear exons (Fig. 1(e)), which indicated that exons with long flanking intron sequences tended to generate circRNAs. We also observed that introns flanking circRNAs’ parental exons contained many repetitive elements. Of note, simple repeats and interspersed repeats extensively existed in introns bracketing exonic circRNAs, including long interspersed nuclear elements (LINEs, especially chicken repeat 1 family), short interspersed nuclear elements (SINEs), low complexity sequences, satellites and microsatellites (Fig. 1(f)).
Figure 1.
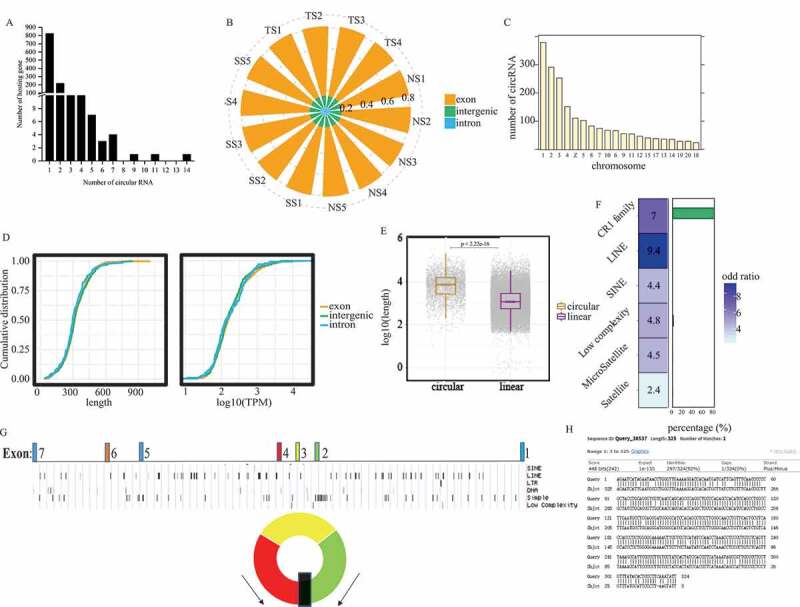
Identification of circRNA in chicken spleens. (a) The distribution of host genes encoding different number of novel circRNAs in chicken spleens. (b) The proportion of different categories of circRNAs in chicken spleens. Most of circRNAs were derived from exon regions of their host genes while a few circRNAs were formed by circularization of intronic or intergenic sequences. (c) The distribution of novel circRNAs in different chromosomes. Huge chromosomes produced more circRNAs in chicken genome. (d) Cumulative distribution of circRNA length (left) and expression (right). Three different types of circRNAs showed no significant changes in length and expression abundance (TPM). (e) Box plot showing the length of flanking introns of circRNAs’ host exon and the other exons. Introns surrounding circRNAs’ host exon significantly larger than that of other spliced exons. (f) Over-representation test showed that simple repeats and interspersed repeats were significantly enriched in circRNA flanking introns (p < 0.01, fisher exact test). Moreover, by employing alignment in each circRNA flanking intron pair, over 80% of RCMs contained chicken repeat 1 family (left). (g) The genomic regions of RUNX2. (h) The RCM in the long intron flanking sequences of the upstream of exon 2 and the downstream of exon 4.
We aligned the flanking intron pairs of exons encoding circRNA to identify reverse complementary matches (RCMs) using Basic Local Alignment Search Tool (BLAST). Total of 579 RCMs were observed. Among them, 576 were shared in the three groups, one was only shared in SS and NS, and two were only shared in TS and SS. Out of 579 RCMs, 284 were overlapped with CR1 family. Fisher exact test showed RCMs were highly enriched in CR1 family (Supplementary Table 2; Fig. 1(f)). It suggested that CR1 family within RCMs facilitated the circularization of their embedded exon. For example, the genomic structure of the circRUNX2.1 (Table 1, gga_circ_0009435) showed that the RCM existed in the long flanking intron of exon (Fig. 1(g,h)). We further analyzed sequence feature of RCMs in chickens, and three most prevalent sequence motifs were identified (Supplementary Fig. 2). However, only 23% of 579 RCMs possessed at least one of these motif sites.
Table 1.
The specific information of 18 circRNAs.
| Circ_ID | Chr | Parental gene | Length (nt) | Junction | Name |
|---|---|---|---|---|---|
| gga_circ_0011261 | 4 | ZMYM3 | 272 | Intron 10pa | circZMYM3 |
| ggs_circ_0014428 | 7 | GTDC1 | 282 | Intron 3p-exon 4a | circGTDC1 |
| gga_circ_0014634 | 7 | MYO1B | 1251 | Exon 11–13 | circMYO1B |
| gga_circ_0009435 | 3 | RUNX2 | 546 | Exon 2–4 | circRUNX2.12 |
| gga_circ_0009437 | 3 | RUNX2 | 162 | Exon 6 | circRUNX2.2b |
| gga_circ_0015132 | 8 | AMY2A | 605 | Exon 5–9 | circAMY2A |
| gga_circ_0003618 | 1 | na | 305 | Intergenic_region | circNA1 |
| gga_circ_0008071 | 2 | UBE2E2 | 281 | Exon 4–5 | circUBE2E2 |
| gga_circ_0002209 | 16 | TAP1 | 315 | Exon 6–7 | circTAP1 |
| gga_circ_0000062 | 10 | na | 181 | Intergenic_region | circNA2 |
| gga_circ_0000179 | 10 | na | 308 | Intergenic_region | circNA3 |
| gga_circ_0003057 | 1 | RUNX1 | 411 | Exon 2–3 | circRUNX1 |
| gga_circ_0000542 | 11 | WWOX | 357 | Exon 3–4+ intron 3p1 | circWWOX |
| gga_circ_0003849 | 1 | DACH1 | 278 | Exon 2–3 | circDACH1 |
| gga_circ_0007434 | 2 | na | 507 | Intergenic_region | circNA4 |
| gga_circ_0016716 | Z | NTRK2 | 718 | Exon 2–3+ exon 10–11 | circNTRK2 |
| gga_circ_0016532 | Z | SMARCA2 | 385 | Exon 3–4 | circSMARCA2 |
| gga_circ_0005414 | 1 | LRP6 | 329 | Exon 8–9 | circLRP6 |
ap means the partial sequence of the region
bcircRUNX2.1 and circRUNX2.2 were originated from the same parental gene but different exons.
The profile of circRNAs in three groups
Combining with our previous data [29], we conducted a comparison between the linear splice junction sites of parental genes and the back-splice junction sites of circRNAs. We measured the correlation of expression between circRNAs and their host genes in non-infected chicken to investigate their relationship. CircRNAs exhibited an extremely low degree of correlation with their host genes (Figure 2(a)). A weak Pearson correlation coefficient of 0.058 suggested that the frequency of head-to-tail splicing events was largely independent of its parental gene expression. We then evaluated the expression variation of circRNAs and their parental genes in three groups. Coefficient of variance of each RNA was computed as the measurement of expression level variance. CircRNAs tended to have higher expression variance relative to their parental genes, which indicated that the biogenesis of circRNAs is prone to be regulated by multiple factors in different individuals (Fig. 2(b)).
Figure 2.
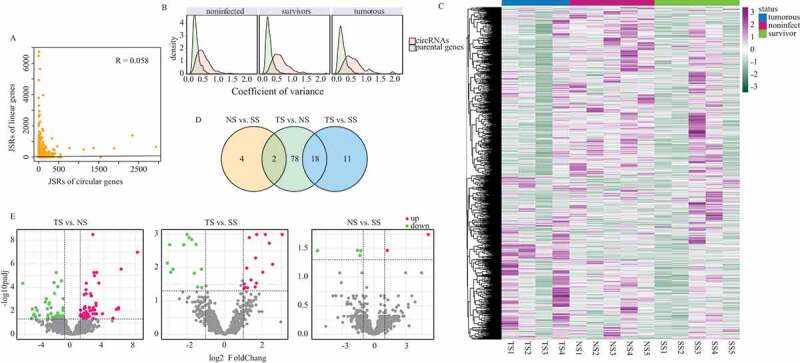
Expressional profile of circRNAs. (a) Expression correlation between circRNAs and their respective parental genes. Reads that spanned junction site reads (JSRs) were compared between circular and linear RNAs. (b) Expression variance of circRNAs and their parental genes in three groups. (c) Heatmap showing the expression of all circRNAs identified in this research. Read counts of each circRNAs were scaled into Z score. (d) Number of differentially expressed circRNAs in three different contrasts. (e) Volcano plots depicting differentially expressed circRNAs in three contrasts. The horizontal dashed lines represent the adjusted p value cut-off of 0.01 and the perpendicular dashed lines represent |LFC| cut-off of 1.
Next, differentially expressed circRNAs (DECs) were identified by DEGseq2 (Supplementary Table 3). CircRNAs shared the similar expression profiles were clustered, and the results showed some circRNAs predominately expressed in tumourous group and some mainly expressed in non-infected group (Fig. 2(c)). In total of 98, 29 and 6 differentially expressed (DE) circRNAs in TS vs. NS, TS vs. SS and NS vs. SS, respectively (Fig. 2(d)). In TS vs. NS, 58 circRNAs up-regulated while 40 circRNAs down-regulated in TS group; 13 circRNAs were higher expressed in TS group relative to SS group, whereas 16 circRNAs had lower expression levels in TS group than that in SS group. Only six DE circRNAs were found in NS vs. SS contrast, of which two circRNAs up-regulated in NS group and four circRNAs down-regulated (Fig. 2(e)).
Out of 579 RCM circRNAs (with RCM in their flanking regions), 30 circRNAs were DE with 28 in TS vs. NS, 6 in TS vs. SS and 1 in NS vs. SS. In further analysis, we found that RCM circRNAs were not over-represented in DE circRNA category compared with other non-RCM circRNAs (p value = 0.91, fisher exact test), that suggested there was no relationship between circRNA bearing RCM and its differential expression. Additionally, 284 out of 579 RCM circRNAs were overlapped with chicken repeat family (CR1F circRNA), 14 CR1F circRNAs DE in TS vs. NS, 1 in TS vs. SS and 0 in SS vs. NS (Supplementary Table 2). To further investigate the relationship between chicken repeat 1 family and MD, we used Chi-squared test to analyze whether CR1F circRNAs were prone to enrich in any comparison group. The results showed that CR1F circRNAs were not significantly over-represented in any comparison compared with RCM circRNAs that had no CR1F overlaps (p value = 0.18, chi-squared test).
The gene ontology analysis showed that parental genes of DE circRNAs in TS vs. NS preferentially enriched in apoptotic process and regulatory region nucleic acid binding (Supplementary Table 4; Fig. 3(a)). Parental genes of DE circRNAs in TS vs. SS were mainly enriched in DNA repair and immune response (Fig. 3(b)). But no significant hits of parental genes-associated pathways were discovered in three comparisons.
Figure 3.
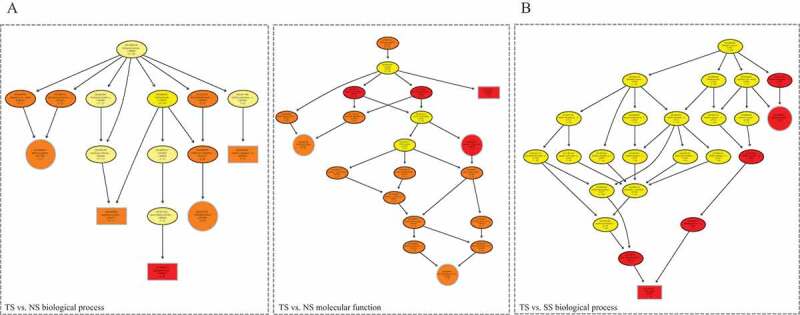
GO enrichment of host genes of differentially expressed circRNAs. Directed acyclic graph (DAG) described the significant biological processes and molecular functions of DE circRNAs in TS vs. NS contrast (a) and TS vs. SS contrast (b).
To analyze the effects influencing differential expression aside from clinical status, principal components analysis was performed on three groups based on expression levels of DE circRNAs, which displayed no significant sex or other effects (Supplementary Fig. 3).
Experimental validation of circRNAs
To verify the accuracy of transcriptome sequencing, we selected four DE circRNAs randomly, and divergent primer and convergent primer were designed to identify the junction sequence of circRNA. As shown in agarose gel electrophoresis (Fig. 4), the products could be amplified by convergent primers with either DNA or cDNA used as template; however, divergent primer could amplify product only using cDNA as template. Sanger sequencing of products amplified from cDNA showed junction sequences (Fig. 4). Q-PCR results showed that the expression of these circRNAs was corresponding with that in RNA-Seq, which illustrated that data from RNA-Seq were accurate and credible (Fig. 5). Four circRNAs, as well as their parental genes, were digested by Rnase R to investigate the stability of circRNA. Q-PCR results showed that circRNAs parental genes were DE (p < 0.01, Fig. 6(a)), while, the expression of circRNAs had no significant difference between Rnase R treatment and non-treatment group (p > 0.05, Fig. 6(b)).
Figure 4.
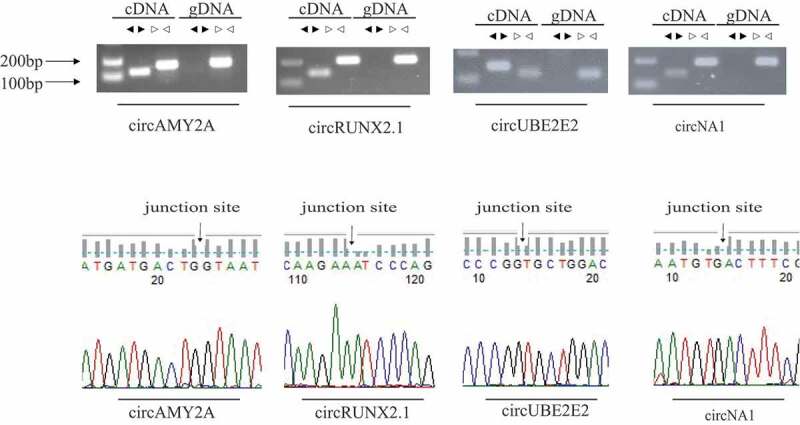
Experimental validation of circular RNAs. (a) Divergent primers and convergent primers were used to amplify circRNAs in cDNA and genomic DNA (gDNA). Black triangles represent divergent primers white triangles represent convergent primers. (b) Sanger sequencing confirmed the back-splicing junction sequence of circRNAs.
Figure 5.
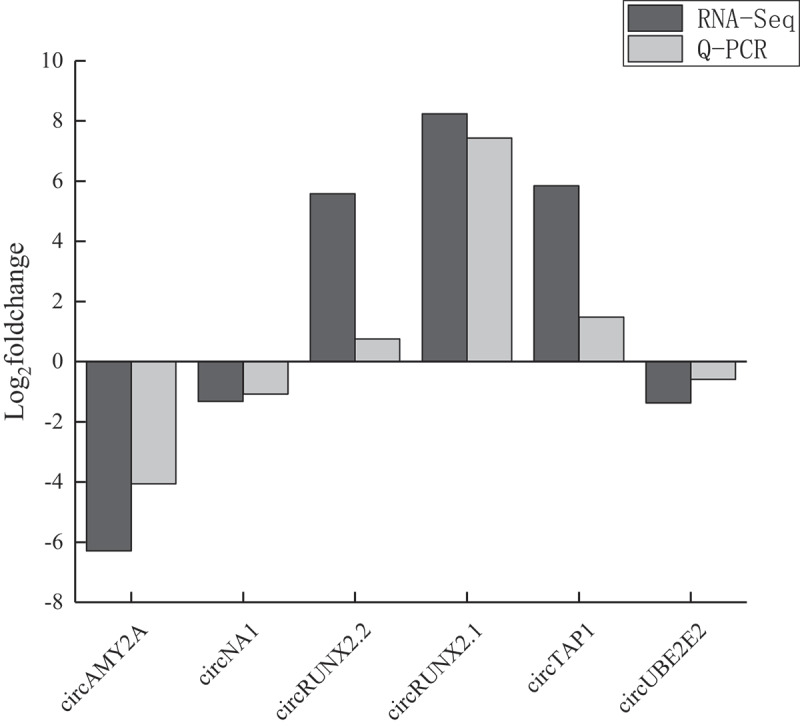
Validation of the gene expression profile by real-time PCR. Six circRNAs were detected by Q-PCR. X-axis represents gene name, y-axis represents log2foldchange, black column represents expression data from RNA-Seq, light grey column means expression data from Q-PCR.
Figure 6.
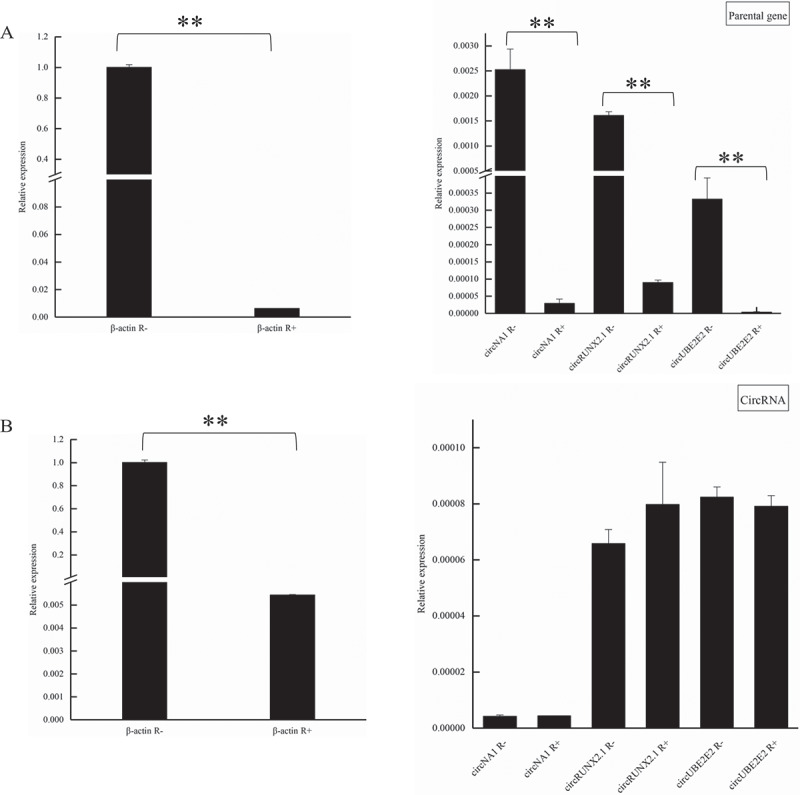
The resistance of circRNAs to Rnase R digestion. (a) The expression of β-actin and parental genes of circRNAs with or without Rnase R treatment. (b) The expression of β-actin and circRNAs with or without Rnase R treatment. R+ represents that the RNA was digested by Rnase R, R- represents that RNA without digested by Rnase R. **means p < 0.01.
miRNA transcriptome analysis
In order to investigate the interaction of circRNAs and miRNAs in MD tumourigenesis, miRNA sequencing for tumourous and non-infected spleens was performed, sequencing data had been submitted to Gene Expression Omnibus (GSE138601). Reads shorter than 17 bp and longer than 35 bp were discarded and remaining reads were aligned onto the chicken genome. Uniquely mapped reads were passed into the downstream analyses. Detail sample information, quality control and mapping statistics were listed in Supplementary Table 5. DE miRNAs (DE miRNAs) were identified by DESeq2 (Supplementary Fig. 4A). We obtained 33 and 64 DE miRNAs in TS vs. NS contrast and TS vs. SS contrast, respectively, of which 29 miRNAs deregulated in both contrasts (Supplementary Table 6; Supplementary Fig. 4(b,c)). Specifically, no DE miRNAs were detected in NS vs. SS contrast.
Delineation of the circRNA/lncRNA-miRNA-mRNA interplays network
To elucidate the miRNA regulatory function of DE circRNAs in our study, we used miRanda to predict potential miRNA seed sites. We combined previous lncRNA and mRNA data, and investigated the interactions of DE lncRNAs and the circRNA-miRNA-mRNA (Fig. 7(a)), 107 DE lncRNAs were participated in this network, and 29 DE lncRNAs were implicated in the network with the gga-miR-155 acting as a hub gene (Supplementary Table 7). Most of circRNAs were shared by at most three miRNAs. However, circZMYM3 (Table 1, gga_circ_0011261) contained the greatest number of seven miRNA binding sites.
Figure 7.
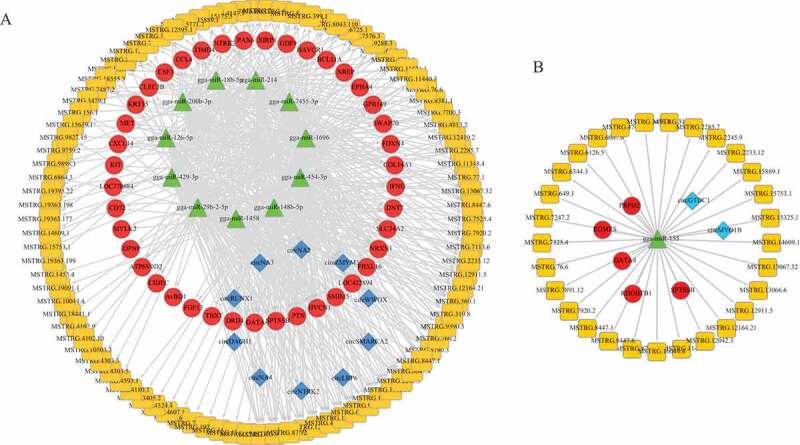
Regulatory network of circRNA/lncRNA-miRNA-mRNA pairs in chicken spleens with Marek’s disease virus infection. (a) Interaction network of DE lncRNAs, circRNAs and mRNA. (b) Interaction network of miR-155 and its potential target lncRNAs, circRNAs and mRNAs.
We used Cytoscape to visualize this network wherein circRNAs, lncRNAs and mRNAs were connected by the same target miRNAs (Fig. 7(a)) [30]. In the networks, gga-miR-214 had a putative binding seed targeting SWAP switching B-cell complex 70 kDa subunit (SWAP70), an immune response-related gene, which was predicted to interact with circZMYM3 and 55 lncRNAs. C-C Motif Chemokine Ligand 4 (CCL4), a chemokine, involving in diverse immune responses, was predicted to interact with gga-miR-429-3p and gga-miR-200b-3p that were decoyed by circZMYM3. gga-miR-155 was predicted to interact with 29 lncRNAs, two circRNAs, circGTDC1 and circMYO1B (Table 1, gga_circ_0014428 and gga_circ_0014634), and five mRNA (Fig. 7(b)).
Potential coding ability analysis
Recent studies have found that circRNA has the potential to encode proteins by IRES, we performed IRES prediction of circRNA sequences and junction sequences (Supplementary Table 8). According to prediction results, there were 2,474 IRESs and the prediction score of some IRESs was up to 0.9974 which indicated an extremely high potential of coding protein.
Discussion
In human genome, less than 2% DNA is translated into protein, and a large amount of DNA sequence is transcripted into RNA without translation activity, which is called non-coding RNA (ncRNA). ncRNA was originally regarded as junk of gene transcription [31]. With the development of high-throughput sequencing technologies, more and more ncRNAs were identified and manifested to play key roles in biological processes, cell development and various diseases [32–34]. CircRNA is single-stranded RNA with a covalently closed loop structure [16]. Fischer and Leung (2017) revealed that circRNAs are stable, dynamically, tissue-specific manner, and involved in apoptosis, oxidative stress, protection against microbe infections and the mediation of cellular homoeostasis [35]. Thus, we investigated the circRNA expression profiles of MDV-infected chicken spleen and to analyze the function of circRNAs in tumour formation process. In this study, we identified 2,169 circRNAs in 14 spleens tissues, which was lower than other researches [13,26]. This might be explained by dynamic expression of circRNA in a tissue-specific manner [35].
Previous study reported that MDV replicated in B cell and T cells resulting in clinical signs such as paralysis and immune suppression. MDV-associated immunosuppression mainly reflected in virus-induced changes in immune regulation and tumour cell-induced immunosuppression [36]. Our previous study investigated the lncRNA-mRNA expression profiles of NS, TS, SS and found that several DE genes were related to B-cell activation and Wnt signalling pathway. Furthermore, some MD-resistant candidate genes, such as IGF-1, CTLA4, HDAC9, SWAP70, CD72, JCHAIN, CXCL12 and CD8B were also identified [29].
CircRNAs and lncRNAs can serve as miRNA sponge to regulate the expression of mRNA by competing endogenous RNAs (ceRNAs) mechanism. Combining previous lncRNA and mRNA data, we found that 2 circRNAs and 29 lncRNAs could bind with miR-155 (Fig. 7(b)). Yao, et al. (2009) also found that miRNA-155, an MDV-1-encoded functional orthologue of MDV-miR-M4, specifically down-regulated in MDV-transformed tumour cells [37]. Study revealed that miR-155 facilitated the suppression of host innate immunity to latent viral and the immortalization of Epstein-Barr virus [38]. Besides that, miR-155 was associated with various diseases, such as, Hodgkin’s lymphoma, primary mediastinal B-cell lymphoma, diffuse large B-cell lymphoma [39], chronic lymphocytic leukaemia [40], pancreatic tumour [41] and breast cancer [42]. Notably, deleting two copy of mdv1-miR-M4 of the infectious BAC clone pRB-1B5 resulted in significant decline in MD incidence. However, after supplementing one of gga-miR-155, which was functional homolog of mdv1-miR-M4, the decrease of incidence was rescued [43]. Therefore, miR-155 played important roles in multiple diseases, including MD. In this study, miR-155 in ceRNA network acted as hub gene, and five DE mRNA targets were identified, such as GATA4, which participated in the regulation of disease occurrence [44,45].
The generation mechanisms of circRNA include lariat-driven circularization, intron-pairing-driven circularization and RNA-binding-proteins-driven circularization [46]. There is emerging evidence that exonic circRNAs in human and other model organisms are bracketed by long introns which contain reverse complementary matches (RCMs) that promote circularization of circRNAs [47]. Human ALU repeats [48] and mammalian-wide interspersed repeats (MIRs) [49] could mediate circRNAs’ formation. In order to investigate circRNA generation mechanism in chickens, we analyzed RCMs in flanking intron sequence of parental exon of circRNAs. We found that there were 579 RCMs, and out of which CR1 family occupied a large proportion. We speculated that CR1 family might facilitate the circulation of exon. However, circRNAs that possessed RCMs were not associated with a particular MD status.
Intron circRNA (ciRNA) generally located in nucleus and regulated its parental gene transcription [21,50,51]. Zhang et al. (2014) revealed that the intronic circRNA boasted few miRNA binding sites [52]. Chen, et al. (2019) revealed that circAGO2 which generated from the intron region of AGO2, mainly located in the cytoplasm, and circAGO2 mediated the translocation of HuR from nuclear to cytoplasm, which can inhibit the gene silence induced by AGO2/miRNA network and promoted the tumourigenesis and aggressiveness [53]. However, in our study, circZMYM3, intron-derived circRNA, was predicted to possess seven miRNA binding sites and those miRNAs targeted genes that involved in immune response. It indicated that some ciRNA might regulate miRNA and mRNA through ceRNA mechanism other than only modulating their host gene.
Recently, studies reported that some circRNAs showed coding ability, and their peptides or proteins products exhibited important regulatory roles in cell development, especially in diseases [54,55]. Gu, et al. (2018) reported that circgprc5a translated into polypeptide to promote bladder cancer growth and metastasis [56]. It is reported that circRNA with a ribosome entry site (IRES) can be translated, and have translation ability [57]. However, Guo, et al. (2014) found that the translation efficiency of circular RNA was lower than that of linear RNA in human U2OS cells and they also found that there was no evidence showed that ribosome protected fragment (RPF) was related to the translation of circRNA [58]. In this study, IRESfinder software was used to identify the IRES of circRNAs, we found 2,474 IRESs in all circRNAs, out of which, 218 circRNAs possessed high score (score > 0.90). Maybe some of these circRNAs could be translated, which will make ceRNA network complicated. However, translation capability of circRNAs with high score needs further verification.
Materials and methods
Biological samples
The information of experimental samples was described in previous study [59]. Briefly, 150 1-d-old specific-pathogen-free White Leghorn (BWEL) chicks were divided into two groups. One hundred chicks were infected intraperitoneally with 2,000 plaque-forming units (PFUs) of the MDV-GA, and the remaining 50 birds were injected with the same volume of diluent (0.2 mL) as non-infected controls. Two groups were housed in different isolators. After 31 days of infection, the chickens were continuously observed, and the individuals with severe clinical conditions were euthanized, four tumourous spleens (TS) of which were sampled. Meanwhile, five non-infected spleens (NS) of chickens in the control group were collected. The trial period lasted to 56 days post infection, and all remaining birds were euthanized. Five MDV-infected chicken without any clinical signs were regarded as survivors and their spleens were sampled (SS). All tissues were preserved in RNA fixer at 4°C overnight and transferred to −80°C for further study. All animal handling procedures were conducted according to regulations and guidelines established by Animal Care and Use Committee of China Agricultural University (Approval ID: XXCB-20,090,209).
RNA isolation and quality assessment
The total RNA was extracted from spleen tissues by Trizol (Invitrogen, CA) according to the manufacturer’s instruction. The NanoPhotometer® spectrophotometer (IMPLEN, USA) was adopted to monitor RNA purity. RNA concentration was measured via Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, USA).
Library construction and RNA sequencing
CircRNA sequencing: A total of 5.0 μg per sample RNA was used. Firstly, ribosomal RNA was removed by Epicentre Ribo-zero™ rRNA Removal Kit (Epicentre, USA) and ethanol precipitation was used to clean up residual rRNA. Secondly, 3 U/μg of Rnase R (Epicentre, USA) was used to digest the linear RNA. The sequencing libraries were generated by NEBNext® Ultra™ Directional RNA Library Prep Kit for Illumina® (NEB, USA) following manufacturer’s recommendations and library quality was assessed by the Agilent Bioanalyzer 2100 system. The libraries were sequenced on an Illumina Hiseq 4000 platform and 150 bp paired-end reads were generated.
Small RNA sequencing: A total of 3 μg total RNA per sample was used as input material for the small RNA library. Sequencing libraries were generated using NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB, USA) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. NEB 3ʹ SR Adaptor was directly and specifically ligated to 3ʹ end of miRNA. After the 3ʹ ligation reaction, the single-stranded DNA adaptor was transformed into a double-stranded DNA molecule. Then 5ʹ ends adapter was ligated to 5ʹ ends of miRNAs and the first strand cDNA was synthesized. PCR amplification was performed using LongAmp Taq 2X Master Mix, SR Primer for Illumina and index (X) primer. Library quality was assessed on the Agilent Bioanalyzer 2100 system using DNA High Sensitivity Chips.
Quality control and mapping to the reference genome
Raw reads in fastq format were firstly processed through quality control. In this step, reads containing adapter, poly-N and low-quality bases at high proportion were removed to obtain clean reads. All the downstream analyses were based on the clean reads with high quality. Reference genome and annotation files were downloaded from genome website (version: Gallus_gallus-5.0; GCA_000002315.3).
For circRNAs, index of the reference genome was built using bowtie2 v2.2.8 and paired-end clean reads were aligned to the reference genome using Bowtie with default arguments [60]. Regards to miRNAs, short reads were aligned to reference genome using Burrows-Wheeler Aligner (BWA) with ‘aln’ and ‘samse’ options and default parameters [61]. Annotated chicken miRNAs file was downloaded from miRbase (http://www.mirbase.org/index.shtml) [62]. Reads counts that assigned to genome were computed by FeatureCounts (0.18.1) [63].
CircRNA identification and sequence feature analysis of circRNAs’ parental sequences
The circRNAs were detected by two softwares of find_circ (https://github.com/marvin-jens/find_circ) [64] and CIRI2 [65]. The intersecting results from two tools were considered as candidate circRNAs. For downstream quantitative analyses, candidate circRNAs with at least two back-splicing junction reads in each sample were remained. The intron sequences of exonic circRNAs’ parental genes and repetitive sequences in chicken were downloaded from UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables). For the identification of reverse complementary matches (RCMs), we aligned two introns’ sequences flanking the same exonic circRNA using Basic Local Alignment Search Tool (BLAST) with ‘blastn’ task, ‘-word_size 7’ and ‘-evalue 20’. The upstream and downstream introns of an exonic circRNA were input as query sequence and subjected sequences, respectively. For each intron pair, several alignments were obtained, and the alignment with lowest e-value that passed the threshold was regarded as reverse complementary match (RCM). Then we applied software Bedtools to compare the RCM genomic coordinates with those of chicken repeat elements and over-presentation test was conducted by fisher exact test to discover significant overlaps of RCMs and repeat elements. We further used MEME suite to identify novel motifs within RCMs with arguments ‘-mod zoops -minw 6 -maxw 50’ [66].
Differential expression and functional enrichment analysis of circRNAs
Differential expression analysis of NS, TS and SS groups was performed using the DESeq2 R package (1.22.1) based on the negative binomial distribution [67]. Variables derived from sex and days post infection (DPI) were added in design formula thus the effects of sex and DPI were corrected by generalized linear model. The p values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. CircRNAs and miRNAs subject to criteria of q value < 0.05 and log2foldchange (LFC) > 1 were assigned as DE circRNAs and miRNAs between every comparison among tumourous spleen, non-infected spleen and survivor spleen.
Gene ontology (GO) enrichment and KEGG pathway (http://www.genome.jp/kegg/) analysis for host genes of DE circRNAs were conducted by the TopGO R package, and KOBAS software, respectively [68,69]. GO terms and KEGG pathway with corrected p < 0.05 were considered to be significantly enriched.
CircRNA/lncRNA-miRNA-mRNA network analysis
Combined with our previous lncRNA and mRNA data (GSE124133), circRNA/lncRNA-miRNA-mRNA network was constructed to reveal their potential interactions in MDV-infected spleens. miRNA binding sites of all circRNAs and lncRNAs were predicted using miRanda (paring score ≥ 150; free energy ≤ −7). All circRNA/lncRNA-miRNA-mRNA pairs were subjected to hypergeometric test and p values were adjusted by the Benjamini and Hochberg’s approach. circRNA/lncRNA-miRNA-mRNA pairs with q value < 0.05 were regarded as candidate miRNA regulatory networks. Cytoscape software was applied to construct the circRNA/lncRNA-miRNA-mRNA regulatory networks based on predicted miRNA binding sites [30]. Internal ribosome entry sites (IRES) of circRNA were predicted by IRESfinder software (http://iresite.org/IRESite_web.php?page=introduction) to investigate translation potential of circRNAs without 5ʹ cap structure.
Validation of circRNAs
Divergent and convergent primers were designed to amplify PCR product using candidate circRNAs sequences as template. Divergent primers were designed in the region about 100 ~ 200 bp surrounding junction site, and convergent primers were designed in one exon of host gene. Primer information was shown in Supplementary Table 9. PCR products amplified by divergent primers were sequenced to confirm the junction site of circRNAs.
The stability of circRNA was detected by the treatment of Rnase R kit (Geneseed, China) following the manufacturer’s instructions. Briefly, a total of 2 μg RNA was digested in 6 U Rnase R for 8 min at 37°C, then, incubated in 70°C for 10 min. The digested product was used to detect the expression of circRNA and linear genes.
cDNA synthesis and quantitative real-time PCR (Q-PCR)
Total of 1 μg RNA was used for reverse transcription using Circular RNA Fluorescence Quantitative PCR Reverse Transcription Kit (Geneseed, China) following the manufacturer’s instructions. Q-PCR was conducted using Circular RNA Fluorescence Quantitative PCR kit (Geneseed, China) according to the manufacturer’s instructions. The β-actin gene was used as reference gene for circRNAs [13]. The Q-PCR was programed in a ABI PRISM ®7500 Sequence Dection System as follows: 95°C for 5 min; 40 cycles of 95°C for 15 s, 60°C for 12 s, 72°C for 34 s and 72°C for 5 min. The relative expression of circRNAs was calculated using 2−∆∆Ct method. The detail of primer sequences was showed in Supplementary Table 9.
Conclusion
We investigated circRNA profile and comprehensively analyzed circRNA/lncRNA-miRNA-mRNA regulatory networks and found extensive interactions of these RNAs involved in MD tumourigenesis. In addition, we found that long flanking intron of exonic circRNAs possessed repeats and might induced the circulation of exon by RCMs. RCMs were mainly overlapped with chicken repeat 1 family, indicating CR1’s important role in the formation of circRNA in chickens.
Funding Statement
This work was financially supported by The National Natural Science Foundation of China [31301957 and 31320103905], The Programs for Chang jiang Scholars and Innovative Research Team in University [IRT_15R62], Young Scientist Supporting Project, Beijing Key Laboratory for Animal Genetic Improvement and Farm Animals Germplasm Resource Platform.
Data availability
All data of this study are included in this published article and its supplementary information files. RNA-seq data are available at NCBI Gene Expression Omnibus (GEO, GSE138601 and GSE138600).
CircRNA data (GSE138600) are available at: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138600), miRNA data (GSE138601) are available at: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138601).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Biggs PM, Nair V.. The long view: 40 years of Marek’s disease research and avian pathology. Avian Pathol. 2012;41:3–9. [DOI] [PubMed] [Google Scholar]
- [2].Jarosinski KW, Tischer BK, Trapp S, et al. Marek’s disease virus: lytic replication, oncogenesis and control. Expert Rev Vaccines. 2006;5:761–772. [DOI] [PubMed] [Google Scholar]
- [3].Calnek BW. Pathogenesis of Marek’s disease virus infection. Curr Top Microbiol Immunol. 2001;255:25–55. [DOI] [PubMed] [Google Scholar]
- [4].Burgess SC, Young JR, Baaten BJG, et al. Marek’s disease is a natural model for lymphomas overexpressing hodgkin’s disease antigen (CD30). Proc National Acad Sci. 2004;101:13879–13884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sabin LR, Delas MJ, Hannon GJ. Dogma derailed: the many influences of RNA on the genome. Mol Cell. 2013;49:783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18:5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Parnas O, Corcoran DL, Cullen BR. Analysis of the mRNA targetome of microRNAs expressed by Marek’s disease virus. mbio. 2014;5:e01060–01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hentze MW, Preiss T. Circular RNAs: splicing’s enigma variations. Embo J. 2013;32:923–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Suzuki H, Zuo Y, Wang J, et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006;34:e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ouyang H, Chen X, Wang Z, et al. Circular RNAs are abundant and dynamically expressed during embryonic muscle development in chickens. DNA Res. 2018;25(1):71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Qin C, Liu CB, Yang DG, et al. Circular RNA expression alteration and bioinformatics analysis in rats after traumatic spinal cord Injury. Front Mol Neurosci. 2018;11:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wang YF, Li BW, Sun S, et al. Circular RNA expression in oral squamous cell carcinoma. Front Oncol. 2018;8:398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Altesha MA, Ni T, Khan A, et al. Circular RNA in cardiovascular disease. J Cell Physiol. 2019;234:5588–5600. [DOI] [PubMed] [Google Scholar]
- [17].Liang J, Wu X, Sun S, et al. Circular RNA expression profile analysis of severe acne by RNA-Seq and bioinformatics. J Eur Acad Dermatol Venereol. 2018;32:1986–1992. [DOI] [PubMed] [Google Scholar]
- [18].Wang R, Zhang S, Chen X, et al. CircNT5E acts as a sponge of miR-422a to promote glioblastoma tumorigenesis. Cancer Res. 2018;78:4812–4825. [DOI] [PubMed] [Google Scholar]
- [19].Kabir TD, Ganda C, Brown RM, et al. A microRNA-7/growth arrest specific 6/TYRO3 axis regulates the growth and invasiveness of sorafenib-resistant cells in human hepatocellular carcinoma. Hepatology. 2018;67:216–231. [DOI] [PubMed] [Google Scholar]
- [20].Du WW, Yang W, Liu E, et al. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44:2846–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li Z, Huang C, Bao C, et al. Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22:256–264. [DOI] [PubMed] [Google Scholar]
- [22].Ashwal-Fluss R, Meyer M, Pamudurti NR, et al. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56:55–66. [DOI] [PubMed] [Google Scholar]
- [23].Fan X, Yang Y, Wang Z. Pervasive translation of circular RNAs driven by short IRES-like elements. BioRxiv. 2018;473207. DOI: 10.1101/473207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Han D, Li J, Wang H, et al. Circular RNA circMTO1 acts as the sponge of microRNA-9 to suppress hepatocellular carcinoma progression. Hepatology. 2017;66:1151–1164. [DOI] [PubMed] [Google Scholar]
- [25].Cheng J, Zhuo H, Xu M, et al. Regulatory network of circRNA-miRNA-mRNA contributes to the histological classification and disease progression in gastric cancer. J Transl Med. 2018;16:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shen M, Li T, Zhang G, et al. Dynamic expression and functional analysis of circRNA in granulosa cells during follicular development in chicken. BMC Genomics. 2019;20:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang XH, Yan YM, Lei XY, et al. Circular RNA alterations are involved in resistance to avian leukosis virus subgroup-J-induced tumor formation in chickens. Oncotarget. 2017;8:34961–34970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Qiu L, Chang G, Bi Y, et al. Circular RNA and mRNA profiling reveal competing endogenous RNA networks during avian leukosis virus, subgroup J-induced tumorigenesis in chickens. PLoS One. 2018;13:e0204931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].You Z, Zhang Q, Liu C, et al. Integrated analysis of lncRNA and mRNA repertoires in Marek’s disease infected spleens identifies genes relevant to resistance. BMC Genomics. 2019;20:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kumar L, Shamsuzzama, Haque R,et al. Circular RNAs: the emerging class of non-coding RNAs and their potential role in human neurodegenerative diseases. Mol Neurobiol. 2017;54:7224–7234. [DOI] [PubMed] [Google Scholar]
- [32].Elling R, Chan J, Fitzgerald KA. Emerging role of long noncoding RNAs as regulators of innate immune cell development and inflammatory gene expression. Eur J Immunol. 2016;46:504–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li M, Liu Y, Zhang X, et al. Transcriptomic analysis of high-throughput sequencing about circRNA, lncRNA and mRNA in bladder cancer. Gene. 2018;677:189–197. [DOI] [PubMed] [Google Scholar]
- [34].Meng S, Zhou H, Feng Z, et al. CircRNA: functions and properties of a novel potential biomarker for cancer. Mol Cancer. 2017;16:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Fischer JW, Leung AK. CircRNAs: a regulator of cellular stress. Crit Rev Biochem Mol Biol. 2017;52:220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schat KA. Marek’s disease immunosuppression. In: Marek's disease an evolving problem. Davison TF and Nair VK, eds. London: Elsevier Academic Press; 2004;142–155. [Google Scholar]
- [37].Yao Y, Zhao Y, Smith LP, et al. Differential expression of microRNAs in Marek’s disease virus-transformed T-lymphoma cell lines. J Gen Virol. 2009;90:1551–1559. [DOI] [PubMed] [Google Scholar]
- [38].Lu F, Weidmer A, Liu CG, et al. Epstein-Barr virus-induced miR-155 attenuates NF-kappaB signaling and stabilizes latent virus persistence. J Virol. 2008;82:10436–10443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kluiver J, Poppema S, De Jong D, et al. BIC and miR-155 are highly expressed in hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 2005;207:243–249. [DOI] [PubMed] [Google Scholar]
- [40].Fulci V, Chiaretti S, Goldoni M, et al. Quantitative technologies establish a novel microRNA profile of chronic lymphocytic leukemia. Blood. 2007;109:4944–4951. [DOI] [PubMed] [Google Scholar]
- [41].Gironella M, Seux M, Xie MJ, et al. Tumor protein 53-induced nuclear protein 1 expression is repressed by miR-155, and its restoration inhibits pancreatic tumor development. Proc Natl Acad Sci. 2007;104:16170–16175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Iorio MV, Ferracin M, Liu CG, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070. [DOI] [PubMed] [Google Scholar]
- [43].Zhao Y, Xu H, Yao Y, et al. Critical role of the virus-encoded microRNA-155 ortholog in the induction of Marek’s disease lymphomas. PLoS Pathog. 2011;7:e1001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Takagi K, Moriguchi T, Miki Y, et al. GATA4 immunolocalization in breast carcinoma as a potent prognostic predictor. Cancer Sci. 2014;105:600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Agnihotri S, Wolf A, Munoz DM, et al. A GATA4-regulated tumor suppressor network represses formation of malignant human astrocytomas. J Exp Med. 2011;208:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fan X, Weng X, Zhao Y, et al. Circular RNAs in cardiovascular disease: an overview. Biomed Res Int. 2017;2017:5135781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ivanov A, Memczak S, Wyler E, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10:170–177. [DOI] [PubMed] [Google Scholar]
- [49].Yoshimoto R, Rahimi K, Hansen T, et al. Biosynthesis of circular RNA ciRS-7/CDR1as is mediated by Mammalian-Wide Interspersed Repeats (MIRs). biorxiv. 2019. DOI: 10.1101/411231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205–211. [DOI] [PubMed] [Google Scholar]
- [51].Zhang Y, Zhang XO, Chen T, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51:792–806. [DOI] [PubMed] [Google Scholar]
- [52].Zhang Y, Yang L, Chen LL. Life without A tail: new formats of long noncoding RNAs. Int J Biochem Cell Biol. 2014;54:338–349. [DOI] [PubMed] [Google Scholar]
- [53].Chen Y, Yang F, Fang E, et al. Circular RNA circAGO2 drives cancer progression through facilitating HuR-repressed functions of AGO2-miRNA complexes. Cell Death Differ. 2019;26:1346–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Legnini I, Di Timoteo G, Rossi F, et al. Circ-ZNF609 is a circular rna that can be translated and functions in myogenesis. Mol Cell. 2017;66:22–37.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yang Y, Gao X, Zhang M, et al. Novel role of FBXW7 circular RNA in repressing glioma tumorigenesis. JNCI: Journal of the National Cancer Institute. 2018;110:304–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gu C, Zhou N, Wang Z, et al. circGprc5a promoted bladder oncogenesis and metastasis through gprc5a-targeting peptide. Mol Ther Nucleic Acids. 2018;13:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chen CY, Sarnow P. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. science. 1995;268:415–417. [DOI] [PubMed] [Google Scholar]
- [58].Guo JU, Agarwal V, Guo H, et al. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15:409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lian L, Qu LJ, Sun HY, et al. Gene expression analysis of host spleen responses to Marek’s disease virus infection at late tumor transformation phase. Poult Sci. 2012;91:2130–2138. [DOI] [PubMed] [Google Scholar]
- [60].Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Griffiths-Jones S, Grocock RJ, Van Dongen S, et al. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Liao Y, Smyth GK, Shi W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- [64].Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. [DOI] [PubMed] [Google Scholar]
- [65].Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform. 2018;19:803–810. [DOI] [PubMed] [Google Scholar]
- [66].Bailey TL, Boden M, Buske FA, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Xie C, Mao X, Huang J, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39:W316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Alexa A, Rahnenfuhrer J. topGO: enrichment analysis for gene ontology. R Package Version. 2018;2:34. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data of this study are included in this published article and its supplementary information files. RNA-seq data are available at NCBI Gene Expression Omnibus (GEO, GSE138601 and GSE138600).
CircRNA data (GSE138600) are available at: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138600), miRNA data (GSE138601) are available at: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE138601).