

ABSTRACT
MicroRNAs (miRNAs) are small RNAs that regulate mRNA expression and have been targeted as biomarkers of organ damage and disease. To explore the utility of miRNAs to assess injury to specific tissues, a tissue atlas of miRNA abundance was constructed. The Rat Atlas of Tissue-specific and Enriched miRNAs (RATEmiRs) catalogues miRNA sequencing data from 21 and 23 tissues in male and female Sprague-Dawley rats, respectively. RATEmiRs identifies tissue-enriched (TE), tissue-specific (TS), or organ-specific (OS) miRNAs via comparisons of one or more tissue or organ vs others. We provide a brief overview of RATEmiRs and present how to use it to detect miRNA expression abundance of candidate biomarkers as well as to compare the expression of miRNAs between rat and human. The database is available at https://www.niehs.nih.gov/ratemirs/
KEYWORDS: RATEmiRs, microRNA, biomarker, RNA-Seq, miRNA expression, tissue-specific, tissue-enriched, organ-specific
Introduction
MicroRNAs (miRNAs) are short, non-coding RNAs of approximately 22 nucleotides in length that regulate gene expression by selectively binding to the 3ʹ untranslated regions of messenger RNAs (mRNAs), thereby inhibiting translation or degrading the transcript [1,2]. Recently there has been great enthusiasm for using miRNAs as biofluid-based biomarkers of diseases and toxicity [3–9]. miRNAs are highly sequence conserved across mammalian species and some are stable in extracellular environments upon release from cells, either by active transport or passively through membrane leakage [10–14]. An example is miR-122, which has been shown to be specifically expressed in the liver and which has been investigated as a potential blood-based biomarker of various types of liver disease or dysfunction in both experimental animals and in humans [9,15–17]. The Human miRNA Tissue Atlas [18] and mimiRNA [19] are resources that catalogues miRNA expression profiles from multiple organs, cell types, whole blood, blood components (serum and plasma) and cell isolates, or body fluids such as urine and saliva to assess miRNA abundance. Rodent model systems have been used to identify miRNAs as biomarkers of adverse toxicity to drugs for safety assessment, and there has been great interest in translating rodent miRNA biomarkers to clinical practice [9,20–22]. Access to a resource containing the baseline level of miRNA expression in a wide variety of rodent animal tissues will facilitate the evaluation of miRNAs as potential modern-day therapeutic targets and biomarkers of tissue injury from environmental stressors, disease, or toxic exposure.
To meet this translational need, the Rat Atlas of Tissue-specific and Enriched miRNAs (RATEmiRs) database was developed to permit user-friendly querying of next-generation sequencing miRNA expression data (The Rat microRNA Body Atlas) [23] from 21 and 23 tissues (14 organs) of toxicologic interest in male and female Sprague-Dawley rats, respectively, to identify tissue-enriched (TE), tissue-specific (TS) or organ-specific (OS) miRNAs [24]. Currently, there is no other publicly available rat miRNA expression database of whole-body organs and tissues. A TE miRNA is one which is abundantly expressed in a tissue vs the other tissues given the parameter setting for analysis. A TS miRNA is one which is abundantly expressed in only a given tissue. An OS miRNA is one which is abundant in only all the tissues that are part of an organ. The RATEmiRs database is freely available and can be found at https://www.niehs.nih.gov/ratemirs/. To construct the database, RNA sequencing (RNA-Seq) was used because it was recently shown to outperform microarray in terms of differential gene expression, edging it mainly in the accuracy of low-abundance transcripts [25]. The RNA-Seq platform is therefore optimal for low expression miRNA profiling when sequenced at sufficient depth; via this technology, novel transcripts can be discovered [26,27].
The Sprague-Dawley rat was utilized to generate the rat miRNA atlas as this model is widely used to evaluate pharmaceutical safety, cancer risks in the 2-year bioassay, and for basic biomedical research. Expression profiling of naive rats provides a baseline assessment of potential miRNA biomarkers exclusivity, enrichment in a subset of tissues, or ubiquitous expression across multiple tissues [23]. However, there is no current standard method for analysing RNA-Seq data and as such, different bioinformatic pipelines can yield varying results [25,28]. To address the variability, three bioinformatics pipelines are implemented in RATEmiRs to report tissue abundance of the rat miRNA body atlas data. miRNAs that are detected in consensus across all three analytical pipelines are more likely to be enriched in a given tissue or organ as compared to those miRNAs that may be implicated by only one or two pipelines.
Ideally, a candidate miRNA biomarker should be TS, detectable in biofluids acquired in a non-invasive manner, and should be translatable across species of interest (in this case, rodent and human) [29,30]. Using case examples with RATEmiRs we can show how the system can be utilized to 1) assess baseline expression exclusivity of potential biomarkers in rat tissues and 2) compare the normalized expression of miRNAs between rat and human tissues [19] for comparative transcriptomic purposes.
Results
RATEmiRs functionality
RATEmiRs contains three bioinformatics pipelines to detect abundant miRNAs. The Eli Lilly pipeline uses non-negative matrix factorization (NMF) as the core analytic method [31]. NMF applies multivariate analysis and linear algebra on the miRNA data to determine expression abundance within tissues/organs. The National Institutes of Environmental Health Sciences (NIEHS) pipeline uses quasi-Poisson modelling of the data [32]. This statistical method accounts for the dispersion (variation) in the count data when assessing the miRNA abundance expression. The Maastricht University pipeline uses percentage of mapped reads to assess miRNA abundance. Based on each pipeline’s data preprocessing criteria [23,24], the number of replicate biological samples for each tissue may differ (Table 1). Inclusion of a larger number of biological replicates increases the power of the analysis and captures individual variation in miRNA transcript abundance.
Table 1.
Number of TE miRNAs per tissue using pipeline default settings.
| # of TE miRNAs (Pipeline sample size) |
||||
|---|---|---|---|---|
| Tissues | Lilly | NIEHS | Maastricht | Intersection |
| Adrenal | 27 (10) | 46 (10) | 2 (10) | 1 |
| Muscle biceps‡ | 57 (10) | 20 (10) | 6 (10) | 3 |
| Brainstem† | 98 (10) | 186 (10) | 5 (10) | 2 |
| Cerebellum† | 16 (10) | 116 (10) | 4 (10) | 1 |
| Cerebrum† | 69 (10) | 128 (10) | 4 (10) | 4 |
| Cortex# | 4 (10) | 10 (10) | 0 (10) | 0 |
| Dorsal root ganglion (DRG/Uk) | 33 (10) | 231 (4) | 3 (10) | 2 |
| Duodenum+ | 75 (10) | 14 (9) | 0 (10) | 0 |
| Stomach glandular (Gln)* | 38 (10) | 30 (10) | 0 (10) | 0 |
| Heart | 31 (10) | 20 (10) | 5 (10) | 2 |
| Hippocampus† | 75 (10) | 30 (10) | 0 (10) | 0 |
| Ileum+ | 43 (10) | 19 (8) | 9 (10) | 2 |
| Jejunum+ | 26 (10) | 25 (10) | 0 (10) | 0 |
| Kidney# | 2 (10) | 10 (10) | 0 (10) | 0 |
| Liver | 49 (10) | 22 (10) | 5 (10) | 2 |
| Medulla# | 2 (10) | 92 (10) | 0 (10) | 0 |
| Stomach non-glandular (NGln)* | 6 (10) | 92 (10) | 3 (10) | 0 |
| Ovary | 19 (5) | 3 (5) | 0 (5) | 0 |
| Pancreas | 33 (10) | 42 (10) | 10 (10) | 7 |
| Muscle soleus‡ | 48 (10) | 21 (10) | 4 (10) | 3 |
| Testicle | 202 (5) | NA (1) | 53 (5) | 42ⱡ |
| Uterus | 9 (5) | 27 (5) | 0 (5) | 0 |
| Whole Blood | 25 (10) | 191 (6) | 19 (10) | 4 |
Denotation of tissues that are part of an organ: #: Kidney; *: Stomach; +: Intestine; †: Brain; ‡: Muscle.
Intersection of the three pipelines. ⱡ Indicates that the tissue-enriched (TE) miRNAs are from the Lilly and Maastricht pipelines only.
NA: Not applicable due to sample size of 1.
RATEmiRs use case #1: discovery of TE, TS, or OS miRNA
A common need for biomarker developers and gene expression analysts is to identify miRNAs that are highly specific to a tissue or organ type. RATEmiRs provides a handy platform for generating hypotheses that may lead to 1) highly specific candidate miRNA biomarkers in bio-fluids, and 2) identification of tissue-specific regulatory functions governed by miRNAs that are uniquely expressed in a given tissue. Queries can be performed using individual data analysis pipelines or combined to obtain consensus miRNAs that are robustly identified as enriched, independently of the data analysis pipeline used. This analysis can be found under the tabs ‘Data Driven TE & TS’ or “Data Driven ‘OS’ (the data-driven nomenclature refers to the fact that the query is relying on the pipeline analysis). The Lilly and Maastricht pipelines mapped the miRNA sequencing data to mature miRNAs using miRBase [33–36] v20 and iso-miRNAs. NIEHS mapped to mature miRNAs using miRBase v19 and Maastricht also mapped the data to identify novel miRNAs (those not found in miRBase). Data from miRBase is populated into RATEmiRs in order to update the annotation of the miRNAs according to the current version available (v22). Therefore, when all three pipelines are used, the intersect of the Venn diagram will be of any mature miRNAs found in common to be TE, TS or OS. If users desire to identify abundantly expressed iso-miRNAs in a tissue or organ, then selecting the Lilly and Maastricht pipelines simultaneously will uncover any such miRNAs. Lastly, if users need to identify novel miRNAs that are abundantly expressed in a tissue or organ, then selecting the Maastricht pipeline is the option of choice.
The default web page for RATEmiRs is the Data Driven TE and TS tab (Fig. 1A). The interface involves five steps (numbered in circles) to perform an analysis for identification of TE or TS miRNAs. Step 1 is to select which type of miRNA is desired: TE or TS. TE miRNAs are largely abundant in one or more tissues. TS miRNAs are exclusively expressed in a single tissue. Step 2 is to select the data analysis pipeline(s) to investigate and set parameters or one may use the defaults. For TE analysis, Lilly and Maastricht use a percentage of the total expression of each miRNA in one tissue vs all tissues whereas NIEHS uses an F-like test statistic for the comparison based on a quasi-likelihood ratio test to detect differential expression and obtain a p-value of the significance [32]. Step 3 is to select one tissue from the left drop down box to compare to other tissues listed in the right drop down box. Step 4 is to consider adjusting the filtering of miRNAs based on a number of mapped read counts to reduce background noise. Step 5 is to click the GO button to run the analysis and launch the results page.
Figure 1.
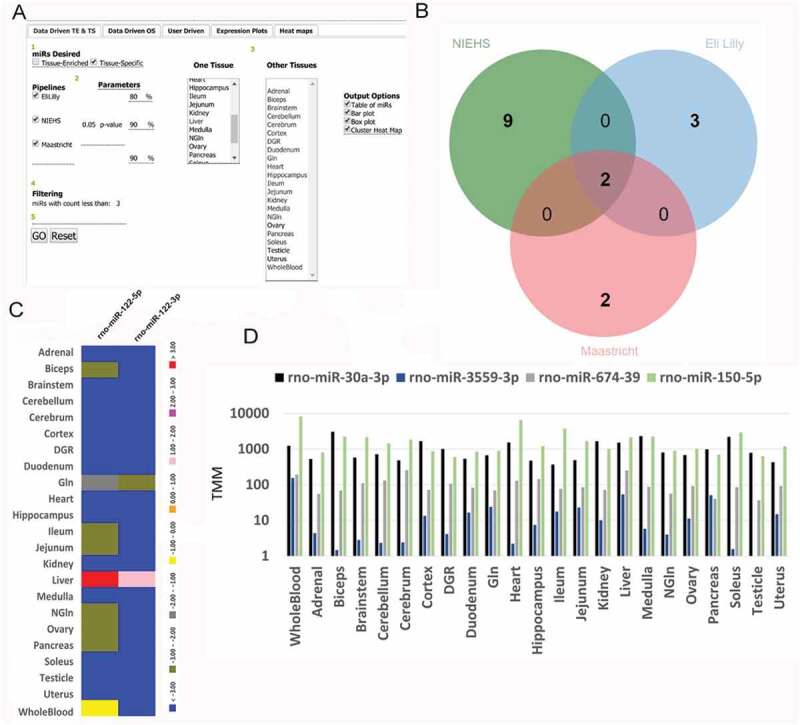
RATEmiRs interface and output results. A) Data Driven TE and TS analysis tab. Other tabs are for Data Driven OS, User Driven, viewing expression plots and viewing heat maps. Mouseover of a bolded title or parameter setting describes the function or parameter setting value used in the analysis. B) Overlap of liver TS miRNAs. When two or more pipelines are selected, a Venn diagram is generated depicting the overlap of the miRNAs. C) Heat map of the miRNA expression for rno-miR-122-3p and rno-miR-122-5p. The y-axis is the tissues and the x-axis is the miRNAs. The colour for the heat map expression is the NIEHS TMM data scaled to values between − 4 and + 4. D) Bar plot expression of NIEHS TMM expression for rno-miR-30a-3p, rno-miR-3559-3p, rno-miR-674-3p and rno-miR-150-5p. The colour in the legend represents a particular miRNA. The y-axis is TMM in log base 10 scale and the x-axis is the tissues.
As shown in Table 1, each pipeline reports TE miRNAs from one tissue vs selected other tissues comparisons. TE miRNAs identified across all selected pipelines suggest higher confidence whereas nonoverlapping miRNAs may represent pipeline-specific results as stated above or previously published [37]. For example, querying RATEmiRs with the default parameters for TE miRNAs in the muscle bicep yielded 57, 20 and 6 miRNAs from the Lilly, NIEHS and Maastricht pipelines, respectively. Only three miRNAs are in the intersection (rno-miR-483-3p, rno-miR-483-5p and rno-miR-675-3p) and only four miRNAs in the overlap between Lilly and NIEHS (rno-miR-1-3p, rno-miR-133a-3p, rno-miR-133a-5p and rno-miR-675-5p). Keep in mind that overlaps are constrained to mature miRNAs. Many of the 50 miRNAs unique to the Lilly pipeline are iso-miRs.
When two or more pipelines are selected for analysis, a link is provided to display a Venn diagram of the abundant miRNAs identified by each pipeline (Fig. 1B). Clicking on a section of the diagram will display the overlapping miRNAs and a table of the overlaps is downloadable. Although individual pipelines have their specific naming conventions of the miRNAs, when the query for abundant miRNAs compares multiple pipelines, the annotation of the miRNAs is reconciled by lookup tables in RATEmiRs and then presented in the Venn diagram overlap with common miRBase identifiers. The analysis output is provided as downloadable tables of the abundant miRNAs, bar graphs, box plots and clustergrams. Interactive profile plots of the abundant miRNAs and a heat map (Fig. 1C) of the scaled (to values between − 4 and + 4) expression data are accessible through the interface.
A similar tab for identifying OS miRNAs is available. The steps are the same as the TE and TS analysis, except that only one organ is selected. The expression of the miRNAs in the tissue(s) of the organ is compared to the tissue(s) from the other organs. Finally, a User Driven tab is available as a portal to supply miRBase miRNA IDs to RATEmiRs to display the expression data.
RATEmiRs use case #2: verifying tissue expression of candidate miRNA biomarkers
Another common need for biomarker development is to identify the tissue of origin of a miRNA that is leaked into a biofluid (such as blood or urine). Because biofluids such as blood have access to a variety of organs via normal circulation and perfusion processes, it can be difficult to ascertain which tissue the biofluid-based biomarker arose. Complicating this assessment is that many diseases or injury processes affect more than one organ or propagate endocrine miRNA signalling, further complicating tissue-of-origin assessments. Alternatively, it may be desirable to determine cross-species relevance and utility of miRNA biomarkers identified in another species (such as human to rat or vice versa). To facilitate this, especially if a user is not sure they need miRNA localization queries, the ‘User Driven’ tab is available to assess tissue expression abundance of specific miRNAs input by the user. To take advantage of this function, users may select the desired analysis pipeline(s) and then enter either a single miRNA or list of mature miRNA names, using standard miRNA nomenclature (e.g. rno-miR-122-3p is an acceptable input, but miR-122 or 122 is not at this time). Users may select the desired pipeline(s) and the tissues in which to perform the comparison. For biomarker discovery, it may be desirable to compare expression across all tissues. However, users can compare a subset of two or more tissues depending on their needs. After making these selections, users should click the ‘GO’ button at the bottom of the page. A new page will open showing normalized expression values for the selected tissues and selected pipeline(s). Users can also access data visualizations for the output results by clicking either the ‘Expression Plots’ or ‘Heat maps’ tabs.
miR-122 is perhaps one of the most extensively studied miRNAs in terms of a translational biomarker due to its high tissue specificity and role in maintaining liver function. It is highly expressed in the liver [23], making up the majority of the miRNA milieu in liver tissue [38]. Recent studies have shown that miR-122 is a potential biomarker for liver injury and disease in rodents [39], canines [17], and in humans [40]. Querying of miRNAs in RATEmiRs in a liver vs all other tissues comparison or a liver vs all other organs comparison identifies rno-miR-122-3p and rno-miR-122-5p as TS and OS. As shown in Fig. 1C, both of the miRNAs are not highly detected in any other tissue. They are essentially exclusively expressed in the liver although the expression of rno-miR-122-3p is more repressed than rno-miR-122-5p in the whole blood.
Another example of using RATEmiRs to detect a potential candidate biomarker is rno-miR-132 which is OS in the rat cerebrum tissue in all 3 pipelines using default parameters except for Maastricht University where the total expression for the tissue was set to 70%. Recently, miR-132 has been purported to be important for regulating neuronal function in humans and its two homologous miRNAs (hsa-miR-132-5p and hsa-miR-132-3p) has been followed up as potential clinical biomarkers for Alzheimer’s disease, Parkinson’s disease and other neurodegenerative diseases or disorders [41].
Recently several rat miRNAs were classified as potential biomarkers of early, mid and late stages of drug-induced liver injury (DILI) [22]. miRNA rno-miR-122-5p was among them. Four of the miRNAs (rno-miR-30a-3p, rno-miR-3559-3p, rno-miR-674-3p and rno-miR-150-5p) are detected in the RATEmiRs whole blood data/NIEHS pipeline as TE with a p-value < 0.05 (Fig. 1D). In fact, whole blood, heart and ileum tissues show increased expression of rno-miR-150-5p than the liver, and other tissues have relatively similar expressions of rno-miR-30a-3p and rno-miR-674-39 as in the liver. Furthermore, the whole blood shows more expression of rno-miR-3559-3p than the liver.
miRNAs can be released from cells through at least three mechanisms: active ATP dependent release involving secretion of miRNAs from cells in the form of microvesicles, including exosomes and shedding vesicles, and passive release from cells including necrosis, apoptosis and drug-induced membrane perturbations. Additionally, miRNAs can be released from cells through protein–RNA interactions as high-density lipoprotein can bind miRNAs and deliver them to cells and miRNAs in the plasma can be found bound to Argonaute proteins [42–44]. The contribution of miRNAs in the blood may be a function of the level of a miRNA in a given tissue, the size of the tissue and the blood flow received by that tissue as well as half-lives of various cell types in the tissue and state of health of the tissue. In order for a miRNA to be a TS marker of organ injury, it should be specifically expressed in a single organ. If a miRNA is expressed in multiple tissues, drug-induced toxicity may cause membrane perturbations in specific organs which may lead to miRNA changes in the blood without injury to other organs. Thus, in the case of miR-150-5p, drug-induced membrane perturbations could cause release of the miRNA form cardiac tissue while the liver may remain unaffected, in which case miR-150-5p would not be indicative of liver injury, but a cardiac effect. Given these assessments, it is difficult to classify the aforementioned DILI miRNA biomarkers as being exclusively released from the liver and circulating in the blood. Thus, it might be worthwhile for investigators to use RATEmiRs or similar databases when interpreting miRNA expression data in studies designed to identify miRNAs as biomarkers.
Comparing similarity of miRNA expression between rat and human
Rodent model systems are practical to use for assessing toxicity of chemicals and for prioritizing biomarkers of diseases and then extrapolating to humans. TE and TS miRNAs identified by querying the RATEmiRs database may be of interest to compare the abundance in human tissues. mimiRNA [19] is a publicly available resource of human tissue miRNA expression data collected from various sources and platforms (sequencing, microarray and quantitative real-time PCR). We identified seven miRNAs (miR-142-3p, miR-142-5p, miR-324-3p, miR-324-5p, miR-335, miR-429 and miR-431) that have matching miRBase IDs and expression data in the same five tissues (cerebellum, duodenum, hippocampus, ileum and jejunum) between our rat data and the human data (Table 2). The seven miRNAs were the ones which matched IDs exactly between the rat and human and were expressed in at least one of the tissues that are in common between the two species. The miRNA expression data from each tissue and source were scaled between 0 and 1,000, and then correlated using a similarity metric that ranges between 0 and 1 (see the Materials and methods section). None of the seven miRNAs have a high similarity in the cerebellum. However, all of them have high similarity (> ~0.9) in the intestinal tissues (duodenum, ileum and jejunum) and three miRNAs (miR-142-3p, miR-142-5p and miR-335) have high similarity (> ~0.98) in the hippocampus.
Table 2.
Similarity measurements of miRNAs expression between rat and human.
| rat miR | human miR | Cerebellum | Duodenum | Hippocampus | Ileum | Jejunum |
|---|---|---|---|---|---|---|
| rno-miR-142-3p | hsa-miR-142-3p | 0.51 | 0.98 | 0.98 | 0.86 | 0.98 |
| rno-miR-142-5p | hsa-miR-142-5p | 0.51 | 0.96 | 0.98 | 0.96 | 0.96 |
| rno-miR-324-3p | hsa-miR-324-3p | 0.51 | 0.96 | 0.51 | 0.96 | 0.97 |
| rno-miR-324-5p | hsa-miR-324-5p | 0.51 | 0.96 | 0.51 | 0.96 | 0.96 |
| rno-miR-335 | hsa-miR-335 | 0.51 | 0.96 | 0.98 | 0.96 | 0.96 |
| rno-miR-429 | hsa-miR-429 | 0.51 | 0.97 | 0.51 | 0.96 | 0.96 |
| rno-miR-431 | hsa-miR-431 | 0.51 | 0.97 | 0.51 | 0.96 | 0.96 |
The miRNA similarity measurements were computed for each tissue by first normalizing the miRNA TMM data from the NIEHS pipeline to scale the data between 0 and 1,000 in the same fashion as the Human data from the mimiRNA expression profiler. Then the absolute difference between the normalized data for each miRNA in the tissues was used as a similarity measurement ranging between 0 and 1.
Summary
The RATEmiRs database provides easy access to miRNA-seq baseline expression data in an assortment of rat tissues and organs of biologic interest. The system has three bioinformatics pipelines to permit detection of TE, TS, or OS miRNA abundance in tissue or organ vs other tissues or organs comparisons. The query interface provides a user-friendly portal on top of the analytic framework of the pipelines. Candidate miRNA biomarkers can be verified for exclusivity in a particular tissue or organ. In addition, using a simple normalization of miRNA expression data from RATEmiRs, miRNAs from the human tissue atlas can be correlated with the rat to assess concordance.
Materials and methods
Tissue sample collection, RNA preparation and processing
Details of organ and tissue acquisition and RNA extraction from the rats are as previously described [23]. Briefly, five male and five female Sprague-Dawley rats 12–13 weeks in age with average weights between 250 and 300g, were obtained from Charles Rivers Laboratories, maintained in rooms on a 12-h light/12-h dark cycle with temperature between 68° and 79° F, relative humidity between 30% and 70% and then euthanized by CO2 asphyxiation at 10:00 am on 4 November 2012. The rats were necropsied and the organs were processed in batches according to approximate tissue size then placed into RNAlater® at 4°C for 24 h. In batch #1, greater than 150 mg of liver, stomach, ileum, jejunum and duodenum were placed into RNAlater® for subsequent total RNA isolation. The following portions of the intestine corresponding to the segment that is routinely collected for histology were collected: the stomach with approximately 5 cm of the duodenum attached, a 10 cm section from the midpoint of the jejunum and the distal 5 cm of ileum. A syringe containing RNAlater® was used to flush the contents from the lumen of the collected segments prior to placing them into the RNAlater® collection tube. In batch #2, ~150 mg of kidney, pancreas, brain, testis, biceps and soleus were placed into RNAlater® for subsequent total RNA isolation. In batch #3, 30–50 mg of adrenal, heart, ovary and uterus were placed into RNAlater® for subsequent total RNA isolation. The samples were then transferred to-20o C the following day. All procedures in this protocol are in compliance with the U.S. Department of Agriculture’s (USDA) Animal Welfare Act (9 CFR Parts 1, 2, and 3); the Guide for the Care and Use of Laboratory Animals: Eighth Edition, (Institute for Laboratory Animal Research, The National Academies Press, Washington, D.C.); and the National Institutes of Health, Office of Laboratory Animal Welfare. Whenever possible, procedures in this study were - designed to avoid or minimize discomfort, distress, and pain to animals. The protocols were reviewed and approved by the Covance Institutional Animal Care and Use Committee.
Sequencing data and analysis
miRNA libraries were constructed according to Illumina’s TruSeq small RNA sample prep protocol (Illumina, San Diego CA), 50bp single-end fragments were generated and sequenced on Illumina HiSeq 2000 sequencers resulting in about 4–5 million reads per sample. The raw data that support the findings of this study are openly available in the Gene Expression Omnibus (GEO) [45,46] at https://www.ncbi.nlm.nih.gov/geo/, series accession reference number GSE78031. The bioinformatics pipelines for analysis [23,24] entail non-negative matrix factorization (NMF) [31], quasi-Poisson modelling[32] and percentage of total mapped reads by Eli Lilly, the National Institute of Environmental Health Sciences (NIEHS), and Maastricht University, respectively.
Overview of the RATEmiRs system
The RATEmiRs web application database system is comprised of three components [24]:
A relational database that stores the next-generation RNA sequencing (RNA-Seq) of miRNAs from 21 and 23 tissues in male and female Sprague-Dawley rats, respectively. The tissues make up 14 organs (Table 1): adrenal, brain, dorsal root ganglion (DRG), heart, intestine, kidney, liver, muscle, ovary, pancreas, stomach, testicle, uterus, whole blood. The mapped read count data for each miRNA was normalized by the trimmed mean of M-values (TMM) method [47] in order to harmonize (i.e. scale) the transcript measurements for display purposes. Database tables are included to convey each miRNA according to its tissue/organ of origin, with updated annotations of the miRNAs according to the current version of miRBase.
An analysis server that processes the miRNA expression data through three bioinformatics pipelines for on-the-fly comparisons of one tissue/organ vs other tissues/organs.
A web application server that accepts and executes query and analysis requests from a client web browser to and from the database and the analytic server. Results are reported in the web browser in table format displaying the TMM values for each miRNA in the selected tissues with a column denoting the Tissue Specificity Index (TSI) [48] for TE and TS miRNA or Organ Specificity Index (OSI) in the case of OS miRNA searches.
RATEmiRs is available at https://www.niehs.nih.gov/ratemirs/using any web browser. Google Chrome and Mozilla Firefox are supported and highly recommended for optimal user experience.
Tissue/organ specificity index
RATEmiRs computes the TSI as reported by Ludwig et al. (2016) or OSI in the case of OS to assess the abundance of miRNA expression in a set of tissues or organs [48]. TSI/OSI ranges between 0 and 1 where values near 0 represent a miRNA expressed in many or possibly all the tissues/organs, and measurements close to 1 indicate a miRNA expressed exclusively in one tissue/organ. For the jth miRNA
where N is the number of tissues or organs and is the average expression of the jth miRNA in the ith tissue or organ in the case of OSI, normalized by the maximal average expression of the jth miRNA in any of the tissues or organs.
Correlating miRNA expression between rat and human
To normalize the data for comparison, the data were scaled using the following score as previously described [19].
where is the ith miRNA average expression measurement in the jth tissue, is the average expression of all miRNAs in the jth tissue and SDj the standard deviation of the miRNAs in the jth tissue. This normalization scales the data between 0 and 1,000. To measure the correlation of the scaled miRNA data between human (hs) and rat (rn), the following similarity metric was used:
The similarity metric D ranges between 0 and 1.
Acknowledgments
The rat atlas miRNA study was supported by Eli Lilly. The Health and Environmental Sciences Institute (HESI) scientific initiative is primarily supported by in-kind contributions (from public and private sector participants) of time, expertise, and experimental efforts. These contributions are supplemented by direct funding (that largely supports program infrastructure and management) that was provided by HESI’s corporate sponsors. A list of supporting organizations (public and private) is available at http://www.hesiglobal.org. The authors thank Drs. Scott Auerbach and Arun Pandiri for their comments to improve the manuscript. This research was supported, in part, by the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Environmental Health Sciences (NIEHS). The authors greatly appreciate the NIEHS Office of Scientific Computing and the NIEHS Computer Technology Branch for computational, web and application development resources to support RATEmiRs.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. [DOI] [PubMed] [Google Scholar]
- [2].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. [DOI] [PubMed] [Google Scholar]
- [3].Harrill AH, McCullough SD, Wood CE, et al. MicroRNA biomarkers of toxicity in biological matrices. Toxicol Sci. 2016;152:264–272. [DOI] [PubMed] [Google Scholar]
- [4].Sheinerman KS, Toledo JB, Tsivinsky VG, et al. Circulating brain-enriched microRNAs as novel biomarkers for detection and differentiation of neurodegenerative diseases. Alzheimers Res Ther. 2017;9:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kumar P, Dezso Z, MacKenzie C, et al. Circulating miRNA biomarkers for Alzheimer’s disease. PLoS One. 2013;8:e69807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Salloum-Asfar S, Satheesh NJ, Abdulla SA. Circulating miRNAs, small but promising biomarkers for autism spectrum disorder. Front Mol Neurosci. 2019;12:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hwang SR, Tham NTT, Lee SH, et al. Comparison of microRNA expressions for the identification of chemical hazards in in vivo and in vitro hepatic injury models. J Appl Toxicol. 2019;39:333–342. [DOI] [PubMed] [Google Scholar]
- [8].Howell LS, Ireland L, Park BK, et al. MiR-122 and other microRNAs as potential circulating biomarkers of drug-induced liver injury. Expert Rev Mol Diagn. 2018;18:47–54. [DOI] [PubMed] [Google Scholar]
- [9].Wang K, Zhang S, Marzolf B, et al. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proc Natl Acad Sci U S A. 2009;106:4402–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vrijens K, Bollati V, Nawrot TS. MicroRNAs as potential signatures of environmental exposure or effect: a systematic review. Environ Health Perspect. 2015;123:399–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kondkar AA, Abu-Amero KK. Utility of circulating microRNAs as clinical biomarkers for cardiovascular diseases. Biomed Res Int. 2015;2015:821823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Marrone AK, Beland FA, Pogribny IP. The role for microRNAs in drug toxicity and in safety assessment. Expert Opin Drug Metab Toxicol. 2015;11:601–611. [DOI] [PubMed] [Google Scholar]
- [13].Coenen-Stass AML, Pauwels MJ, Hanson B, et al. Extracellular microRNAs exhibit sequence-dependent stability and cellular release kinetics. RNA Biol. 2019;16:696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhao C, Sun X, Li L. Biogenesis and function of extracellular miRNAs. ExRNA. 2019;1. [Google Scholar]
- [15].Laterza OF, Scott MG, Garrett-Engele PW, et al. Circulating miR-122 as a potential biomarker of liver disease. Biomark Med. 2013;7:205–210. [DOI] [PubMed] [Google Scholar]
- [16].Shifeng H, Danni W, Pu C, et al. Circulating liver-specific miR-122 as a novel potential biomarker for diagnosis of cholestatic liver injury. PLoS One. 2013;8:e73133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Harrill AH, Eaddy JS, Rose K, et al. Liver biomarker and in vitro assessment confirm the hepatic origin of aminotransferase elevations lacking histopathological correlate in beagle dogs treated with GABAA receptor antagonist NP260. Toxicol Appl Pharmacol. 2014;277:131–137. [DOI] [PubMed] [Google Scholar]
- [18].Fehlmann T, Ludwig N, Backes C, et al. Distribution of microRNA biomarker candidates in solid tissues and body fluids. RNA Biol. 2016;13:1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ritchie W, Flamant S, Rasko JE. mimiRNA: a microRNA expression profiler and classification resource designed to identify functional correlations between microRNAs and their targets. Bioinformatics. 2010;26:223–227. [DOI] [PubMed] [Google Scholar]
- [20].Wolenski FS, Shah P, Sano T, et al. Identification of microRNA biomarker candidates in urine and plasma from rats with kidney or liver damage. J Appl Toxicol. 2017;37:278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nassirpour R, Homer BL, Mathur S, et al. identification of promising urinary MicroRNA biomarkers in two rat models of glomerular injury. Toxicol Sci. 2015;148:35–47. [DOI] [PubMed] [Google Scholar]
- [22].Kagawa T, Shirai Y, Oda S, et al. Identification of specific MicroRNA biomarkers in early stages of hepatocellular injury, cholestasis, and steatosis in rats. Toxicol Sci. 2018;166:228–239. [DOI] [PubMed] [Google Scholar]
- [23].Smith A, Calley J, Mathur S, et al. The rat microRNA body atlas; evaluation of the microRNA content of rat organs through deep sequencing and characterization of pancreas enriched miRNAs as biomarkers of pancreatic toxicity in the rat and dog. BMC Genomics. 2016;17:694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bushel PR, Caiment F, Wu H, et al. RATEmiRs: the rat atlas of tissue-specific and enriched miRNAs database. BMC Genomics. 2018;19:825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang C, Gong B, Bushel PR, et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat Biotechnol. 2014;32:926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Campbell JD, Liu G, Luo L, et al. Assessment of microRNA differential expression and detection in multiplexed small RNA sequencing data. RNA. 2015;21:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Creighton CJ, Reid JG, Gunaratne PH. Expression profiling of microRNAs by deep sequencing. Brief Bioinform. 2009;10:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wu PY, Wang MD. The selection of quantification pipelines for illumina RNA-seq data using a subsampling approach. IEEE EMBS Int Conf Biomed Health Inform. 2016;2016:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Robinson S, Pool R, Giffin RB, Institute of Medicine (U.S.) . Forum on drug discovery development and translation. Emerging safety science: workshop summary. Washington, DC: National Academies Press; 2008. [PubMed] [Google Scholar]
- [30].Diamandis EP. Cancer biomarkers: can we turn recent failures into success? J Natl Cancer Inst. 2010;102:1462–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Brunet JP, Tamayo P, Golub TR, et al. Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A. 2004;101:4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lund SP, Nettleton D, McCarthy DJ, et al. Detecting differential expression in RNA-sequence data using quasi-likelihood with shrunken dispersion estimates. Stat Appl Genet Mol Biol. 2012;11. [DOI] [PubMed] [Google Scholar]
- [33].Griffiths-Jones S, Grocock RJ, van Dongen S, et al. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Griffiths-Jones S, Saini HK, van Dongen S, et al. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bisgin H, Gong B, Wang Y, et al. Evaluation of bioinformatics approaches for next-generation sequencing analysis of microRNAs with a toxicogenomics study design. Front Genet. 2018;9:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chang J, Nicolas E, Marks D, et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004;1:106–113. [DOI] [PubMed] [Google Scholar]
- [39].Madboly AG, Alhusseini NF, Abd El Rahman SM, et al. Serum miR-122 and miR-192 as biomarkers of intrinsic and idiosyncratic acute hepatotoxicity: A quantitative real-time polymerase chain reaction study in adult albino rats. J Biochem Mol Toxicol. 2019;33:e22321. [DOI] [PubMed] [Google Scholar]
- [40].Rissin DM, Lopez-Longarela B, Pernagallo S, et al. Polymerase-free measurement of microRNA-122 with single base specificity using single molecule arrays: detection of drug-induced liver injury. PLoS One. 2017;12:e0179669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Qian Y, Song J, Ouyang Y, et al. Advances in roles of miR-132 in the nervous system. Front Pharmacol. 2017;8:770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Turchinovich A, Weiz L, Langheinz A, et al. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011;39:7223–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Vickers KC, Palmisano BT, Shoucri BM, et al. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011;13:423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen X, Liang H, Zhang J, et al. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012;22:125–132. [DOI] [PubMed] [Google Scholar]
- [45].Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ludwig N, Leidinger P, Becker K, et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016;44:3865–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]