

ABSTRACT
Mutation of the essential yeast protein Ipa1 has previously been demonstrated to cause defects in pre-mRNA 3ʹ end processing and growth, but the mechanism underlying these defects was not clear. In this study, we show that the ipa1-1 mutation causes a striking depletion of Ysh1, the evolutionarily conserved endonuclease subunit of the 19-subunit mRNA Cleavage/Polyadenylation (C/P) complex, but does not decrease other C/P subunits. YSH1 overexpression rescues both the growth and 3ʹ end processing defects of the ipa1-1 mutant. YSH1 mRNA level is unchanged in ipa1-1 cells, and proteasome inactivation prevents Ysh1 loss and causes accumulation of ubiquitinated Ysh1. Ysh1 ubiquitination is mediated by the Ubc4 ubiquitin-conjugating enzyme and Mpe1, which in addition to its function in C/P, is also a RING ubiquitin ligase. In summary, Ipa1 affects mRNA processing by controlling the availability of the C/P endonuclease and may represent a regulatory mechanism that could be rapidly deployed to facilitate reprogramming of cellular responses.
KEYWORDS: Polyadenylation, ubiquitination, alternative mRNA processing
Introduction
Tight regulation of gene expression is critical for proper cellular function. A step of mRNA synthesis that can influence gene expression is mRNA polyadenylation, an essential maturation step in which mRNA precursor is trimmed by cleavage at its 3ʹ end and a poly(A) tail added. Regulating the overall efficiency at which this processing occurs is a mechanism by which cells could globally alter the amount of mRNAs that reach the cytoplasm for translation. In a different regulatory strategy, a process known as alternative polyadenylation results in a change in the cleavage site position to yield specific mRNA isoforms with different lengths of 3ʹ untranslated (3ʹ UTRs) or coding regions. Shortening of the 3ʹ UTR removes binding sites for factors that influence mRNA stability, translation, subcellular localization, and partner protein interaction, while coding region shortening alters protein function [1,2]. Alternative polyadenylation plays an important and increasingly appreciated role in regulation of gene expression, and the choice of poly(A) site has been shown to vary with changes in cell state during development, tissue differentiation, tumorigenesis, senescence, and as part of the response to stress or infection [3–17].
Regulation of polyadenylation can be elicited by changing the relative concentration of the core mRNA 3ʹ end processing factors or their recruitment to RNA Polymerase II (Pol II) elongation complexes, by affecting how quickly Pol II reaches downstream poly(A) sites, and in some cases, by the expression of positive or negative regulators [7,10,12,18]. Post-translational modification of processing factors is another mechanism for regulation of mRNA 3ʹ end formation. Modifications such as phosphorylation [19–26], acetylation [27], and sumoylation [28] have all been reported to affect the activity of the Cleavage/Polyadenylation (C/P) complex or usage of alternative poly(A) sites. Many studies have also shown that experimental depletion of subunits of the C/P complex can lead to changes in the choice of poly(A) sites (reviewed in [7] and [12]).
In yeast, the C/P complex is composed of CPF, CF IA, and CF IB. CPF contains 14 subunits and includes the enzymes of the complex, which are the Ysh1 endonuclease, the Pap1 poly(A) polymerase, and two phosphatases, Ssu72 and Glc7, which can act on RNA Polymerase II (Pol II). CF IA, which has four subunits (Clp1, Pcf11, Rna14 and Rna15), provides RNA recognition and scaffolding functions to bring together CPF, CF IB, and Pol II. CF IB is the RNA-binding protein Hrp1. The C/P machinery is highly conserved from yeast to mammals [10,29,30], and proteins with sequence, functional, and/or structural homology to almost all of the yeast subunits can be found in the mammalian C/P complex.
While much progress has been made recently to elucidate the composition of the C/P complex and its structural organization [29,30], regulatory mechanisms that lead to changes in levels of C/P subunits are not so well defined. One mechanism that has been described is regulation of the amount of the CF IA subunits Pcf11 and Rna14 and their metazoan homologs PCF11 and CSTF3 by premature transcription termination upstream of the gene’s stop codon [31]. In addition, alterations in the expression of mRNAs encoding C/P subunits have been reported upon cell differentiation, increased proliferation, or in response to external stimuli [32], but these changes do not always correlate with changes in protein levels [5,33].
The levels of C/P proteins could also be regulated by changes in translation or protein stability. We have previously shown that ubiquitination, a modification that can lead to protein degradation, can modulate C/P efficiency, but molecular targets were not identified [34]. An attractive target for this type of regulation would be the endonuclease (Ysh1 in yeast and CPSF73 in mammals) [35]. The activity of this enzyme initiates mRNA 3ʹ end processing, and if it is depleted, 3ʹ ends are not generated for poly(A) addition, and Pol II transcription does not terminate efficiently [36–38].
In this study, we describe how the level of Ysh1 is regulated by ubiquitin-mediated proteasomal degradation. This targeting of Ysh1 is modulated by the presence of Ipa1, an essential protein that was initially discovered as a factor whose genetic interaction profile was very similar to those of mRNA 3ʹ end processing factors [39]. Ipa1 forms a stable complex with only the Ysh1 and Mpe1 subunits of the C/P complex [39,40]. We have previously shown that the ipa1-1 mutant allele causes a defect in both the cleavage and poly(A) addition steps of mRNA 3ʹ end processing in vitro, lengthening of mRNAs in vivo, inefficient transcription termination downstream of poly(A) sites and at snoRNA genes, and poor recruitment of the Pta1 subunit of the C/P complex to the 3ʹ end of an mRNA gene [39,41]. We now show that Ipa1 exerts its effects on mRNA 3ʹ end processing by blocking the ubiquitination of Ysh1, a regulatory mechanism that has not previously been reported.
Results
Ysh1 protein level is decreased by Ipa1 mutation
Because the ipa1-1 phenotypes were reminiscent of those of 3ʹ-end processing mutants, we investigated whether the levels of subunits of the C/P complex were altered in ipa1-1. Western blot analysis of whole-cell extract of ipa1-1 showed most subunits were not affected. However, some subunits (Rna14, Clp1, Cft1, and Swd2) were increased in level (Fig. 1A), while Ysh1 was the only subunit that showed a striking decrease. Thus, the ipa1-1 mutation alters the stoichiometry of C/P complex subunits in ways that could explain the processing defects.
Figure 1.
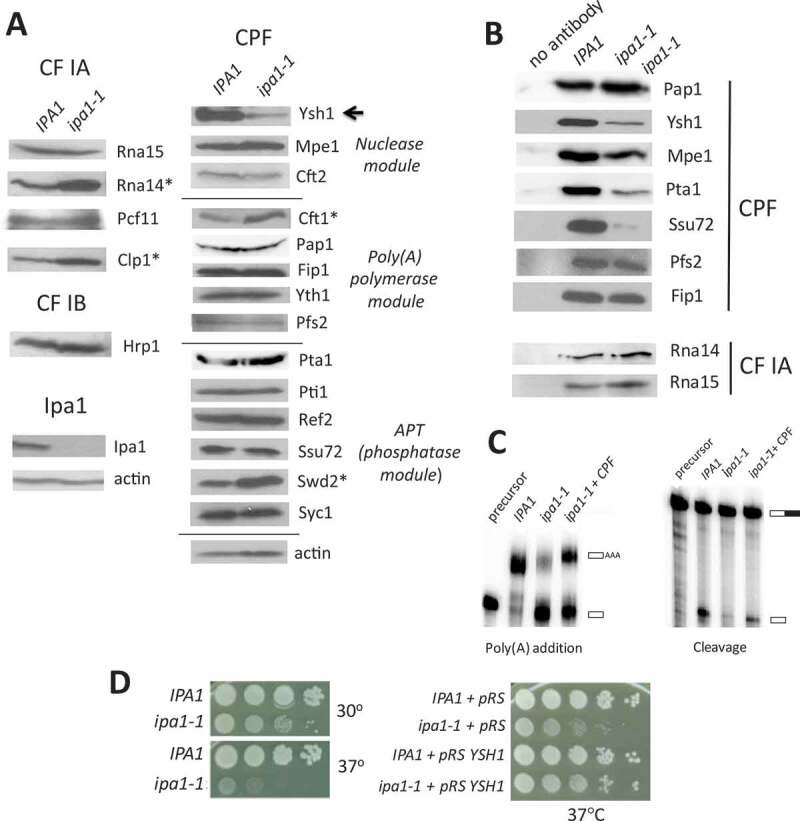
The ipa1-1 mutant negatively affects CPF stoichiometry, cell growth and mRNA 3ʹ end processing in vitro.
(A) Expression of subunits of the C/P complex in IPA1 and ipa1-1 cells. Cells expressing Myc13 tagged Ipa1 were grown at 30°C and shifted to 37°C for 1 hour. Cell extracts were analysed by Western blotting using antibodies against the Myc epitope, C/P subunits, Syc1 (an APT-specific subunit) [43] and actin as a loading control. The Ysh1 subunit (arrow) is decreased in ipa1-1 cells, and Rna14, Clp1, Cft1 and Swd2 are increased in level (asterisks). (B) Interaction of C/P subunits with the poly(A) polymerase Pap1. Co-immunoprecipitation was performed with Pap1 antibody and extract from IPA1 or ipa1-1 cells. The eluates were analysed by Western blotting with antibodies against C/P subunits. Pull-down from IPA1 extract using protein A beads without Pap1 antibody is shown in the left column. (C) Cleavage and poly(A) addition activities are rescued by addition of purified CPF to ipa1-1 extract. Processing extracts were prepared from IPA1 and ipa1-1 cells grown at 30°C and then shifted to 37°C for 1 hour. For poly(A) addition, extracts were incubated with ATP and P32-labelled RNA that ends at the GAL7 poly(A) site. For cleavage-only assays, extracts were incubated with 3ʹ-dATP (a polyadenylation terminator) and P32-labelled RNA containing sequence upstream and downstream of the GAL7 poly(A) site. RNA products were resolved on a denaturing polyacrylamide gel and visualized with a phosphorimager. Positions of substrate and products are indicated on the right (open rectangles indicate sequence upstream of the cleavage site, the filled rectangle is sequence downstream, and the open rectangle labelled AAA is polyadenylated product). The lane marked ‘precursor’ indicates the unreacted substrate. (D) Relative growth of IPA1 (wild-type) and ipa1-1 strains at 30°C and 37°C. For the left panel, cells were grown in liquid YPD, 10-fold serially diluted, spotted onto YPD plates, and incubated for 3 days at the indicated temperatures. For the right panel, cells were transformed with the pRS315 plasmid with or without the YSH1 gene and spotted on CM-leu plates to maintain selection of the plasmid.
Recent findings indicate that CPF can be sub-divided into three enzymatic modules: a nuclease module, a phosphatase module (also called APT for Associated with Pta1), and a polymerase module [40,42,43]. To test whether the CPF organization is compromised in ipa1-1, we performed a co-immunoprecipitation experiment using wild-type or ipa1-1 extracts and antibody against Pap1. The composition of immunoprecipitated proteins was assessed by Western blotting with antibodies against subunits of CPF and CF IA. Consistent with the low level of Ysh1 in ipa1-1 extract, less Ysh1 was incorporated into CPF (Fig. 1B). Mpe1, Pta1, and Ssu72 also showed decreased incorporation into CPF despite normal levels in the ipa1-1 total extract (Fig. 1B). The loss of these subunits is likely due to low Ysh1 incorporation into CPF as Ysh1 directly interacts with Mpe1 and Pta1, and Pta1 in turn recruits Ssu72 to the processing machinery [42,44,45]. This result supports the proposal of Casanal, et al. [42] that the nuclease module functions as a bridge to connect the polymerase and phosphatase modules. The interactions of Pap1 with Pfs2 and Fip1 were not affected, as would be expected given that Fip1 directly contacts both Pap1 and Pfs2 [46,47]. Furthermore, the cross-factor interaction between CPF and the CF IA subunits Rna14 and Rna15 was not impaired by the ipa1-1 mutation. Consistent with defects in CPF composition in ipa1-1, defects in cleavage and poly(A) addition seen with in vitro reactions using the GAL7 poly(A) site were rescued by addition of TAP-purified CPF to ipa1-1 extract (Fig. 1C).
The ipa1-1 mutant bears four point mutations (I109M, K121I, G226E, and D270V). These mutations result in a loss of Ipa1 protein (Fig. 1A), and the I109M and K121I mutations are in a region identified as interacting with Ysh1 by crosslinking mass-spectrometry [40]. The mutant is slow growing compared to wild-type cells at 30°C, and this growth defect is worsened at 37°C (Fig. 1D, left panel). Our previous report had found that the termination defect and the decrease in the amount of Pta1 recruited to the 3ʹ end of a transcribed gene in vivo in the ipa1-1 mutant can be abrogated by Ysh1 overexpression [41]. Consistent with this result, supplementing the ipa1-1 mutant with additional copies of the YSH1 gene on a low copy plasmid was able to rescue the growth defect observed in ipa1-1 (Fig. 1D, right panel). These results indicate that the growth defect conferred by ipa1-1 is in large part due to the low level of Ysh1.
mRNA expression changes in ipa1-1 support a processing defect due to Ysh1 and Ipa1 deficiency
We next tested whether the ipa1-1 mutation altered the efficiency of 3ʹ-end processing in vivo by looking at the level of endogenous transcripts that contain sequence upstream and downstream of the poly(A) sites of PDC1 and RPP1B, which are genes with single poly(A) sites. These transcripts represent RNA that has not been processed and can be detected by RT-qPCR with a primer pair that spans the poly(A) site. In agreement with the defect caused by ipa1-1 in the in vitro cleavage/polyadenylation reaction, mutation of IPA1 greatly increased the amount of unprocessed pre-mRNA at the PDC1 and RPP1B poly(A) sites in vivo (Fig. 2A). Overexpression of YSH1 restored much of the processing at these poly(A) sites, indicating that the depletion of Ysh1 is a major contributor to the ipa1-1 3ʹ end processing defect.
Figure 2.
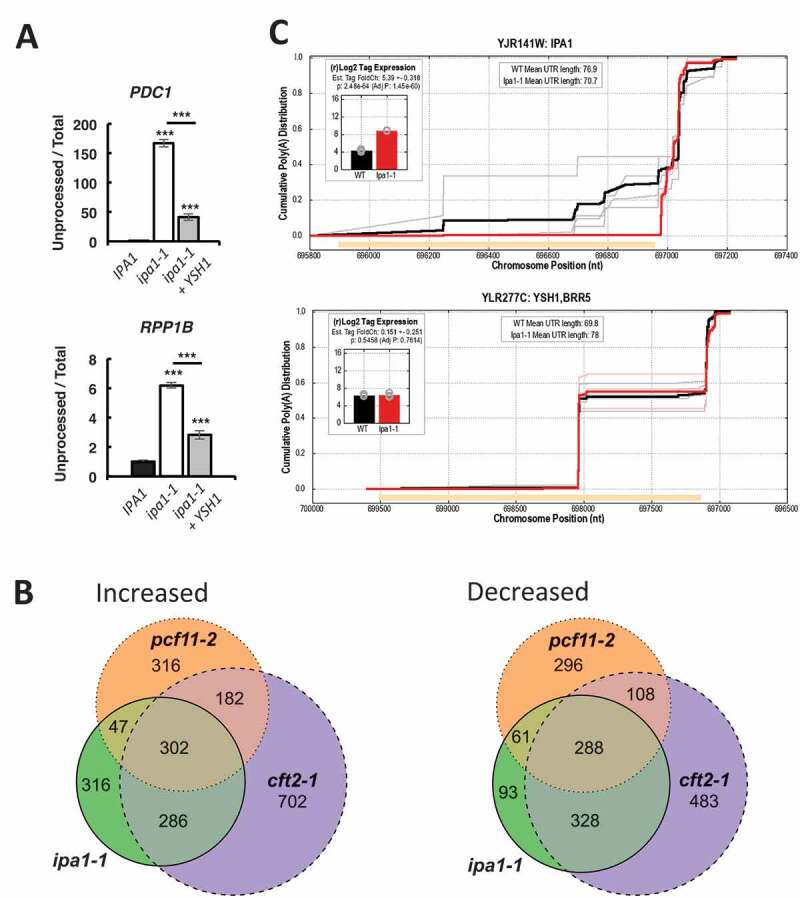
mRNA expression changes support a processing defect due to Ysh1 and Ipa1 deficiency in the ipa1-1 mutant.
(A) Determination of the level of unprocessed transcripts from the PDC1 and RPP1B genes, as determined by RT-qPCR using primers on either side of the poly(A) site (unprocessed) and a primer pair near the 3ʹ end of the ORF (total). (B) The intersection of genes with increased (left panel, log2 fold-change > 0.5, p-value < 0.2) or decreased (right panel, log2 fold-change < 0.5, p-value < 0.2) expression in each of three mutant strains – ipa1-1 (green), pcf11-2 (orange), and cft2-1 (purple). Expression data were obtained from Costanzo et al. [39] and processed as described in Pearson et al. [41]. (C) Cumulative polyadenylation distribution (CPD) plots for the IPA1 and YSH1 genes for wild-type (black) and ipa1-1 (red), averaged from three biological replicates each, with traces for individual replicates in grey or pink. The CPD plot [5] shows the empirical likelihood of polyadenylation at or before each position in the gene. Orange bars show coding sequence, with plots oriented such that the direction of transcription is left to right. The inserts show the log2 tag counts corresponding to mRNAs that are polyadenylated in the 3ʹ UTR of IPA1 or YSH1 for wild-type (black) and ipa1-1 (red). The log2 tag counts correspond to reads from the RNA seq data of Costanzo et al. [39], which uses a method that only returns reads from the sequence immediately upstream of poly(A) sites. The sequencing data of Costanzo et al. [39] were also used to obtain the CPD plots.
To further explore the link between the ipa1-1 mutation and a defect in mRNA 3ʹ end processing, we examined the RNA-seq data sets of Costanzo et al. [39] to determine changes in the mRNA expression profiles of the ipa1-1 mutant compared to that of two mutants with strong mRNA 3ʹ end processing defects, cft2-1 and pcf11-2 [39]. Cft2 directly contacts Ysh1 in the CPF complex [40], an interaction conserved with the mammalian homologs, CPSF100 and CPSF73 [48], and Pcf11 is a subunit of a different factor, CF IA. For the genes with increased expression in ipa1-1, 73% were shared with those up in cft2-1 and 43% with pcf11-2 (Fig. 2B). Of the genes in ipa1-1 that are decreased in expression, 80% are shared with those decreased in cft2-1 and 45% with pcf11-2. The finding that the majority of the ipa1-1-sensitive genes are shared with cft2-1 supports the idea that a major defect in ipa1-1 is loss of CPF function, and in particular, a defect in the nuclease module of which Ysh1 and Cft2 are part.
Interestingly, when we analysed the sequencing data of Costanzo et al. [39], the IPA1 mRNA in the ipa1-1 mutant showed significantly increased expression (log2 fold-change over wild-type of 5.4) and a suppression of poly(A) sites within the IPA1 open reading frame (Fig. 2C, top panel). In wild-type cells, ~30% of the IPA1 transcripts end within the ORF, but in ipa1-1, these are completely suppressed, leading to more full-length mRNA isoforms. This observation suggests that the mutant cells are using this mechanism to try to compensate for the loss of the Ipa1 protein (Fig. 1A) and that Ipa1 auto-regulates its own expression.
Ysh1 is degraded via the ubiquitin-proteasome system in ipa1-1
We next investigated the reason for the strong reduction in Ysh1 protein in ipa1-1. RNA sequencing analysis of mRNAs in the ipa1-1 mutant and the isogenic wild-type strain showed no change in YSH1 mRNA level or shifts in poly(A) site usage (Fig. 2C, bottom panel). Approximately half of the YSH1 transcripts use a poly(A) site within the coding sequence, but usage of this site does not change in ipa1-1 (Fig. 2C). We thus considered that the decrease in Ysh1 protein in ipa1-1 could be attributed to degradation by the ubiquitin-proteasome system. Inactivating the proteasome in the ipa1-1 strain by deleting the UMP1 gene, which is needed for efficient proteasome assembly [49], restored the level of Ysh1 to one comparable to that observed in wild-type cells (Fig. 3A). This result suggests that Ysh1 in ipa1-1 might be post-translationally modified by ubiquitin and targeted for proteasomal degradation. To test this, extracts from strains expressing Myc-tagged Ysh1 were prepared using 2% SDS to disrupt protein–protein interactions, following the protocol described by Laney and Hochstrasser [50] for analysis of ubiquitinated proteins. High molecular weight Ysh1 species were observed in the ipa1-1 ump1▵ cells (Fig. 3B). Similar results were observed if extracts were prepared in 8M urea (Fig. S1A). To further characterize the larger species, Ysh1 was immunoprecipitated from diluted SDS extracts, and immunoprecipitates probed with antibody against ubiquitin or against the Myc epitope on Ysh1 (Fig. 3C). Some of the larger Ysh1 species were lost during the immunoprecipitation, perhaps due to the presence of deubiquitinases in the extract. However, high molecular weight species containing ubiquitin were detected in the precipitate from ipa1-1 ump1▵ cells expressing Myc-Ysh1, but not from ipa1-1 ump1▵ cells lacking the Myc tag or from cells with the unmutated IPA1 gene (Fig. 3C, lower panel), confirming the accumulation of ubiquitinated Ysh1 when Ipa1 is defective.
Figure 3.
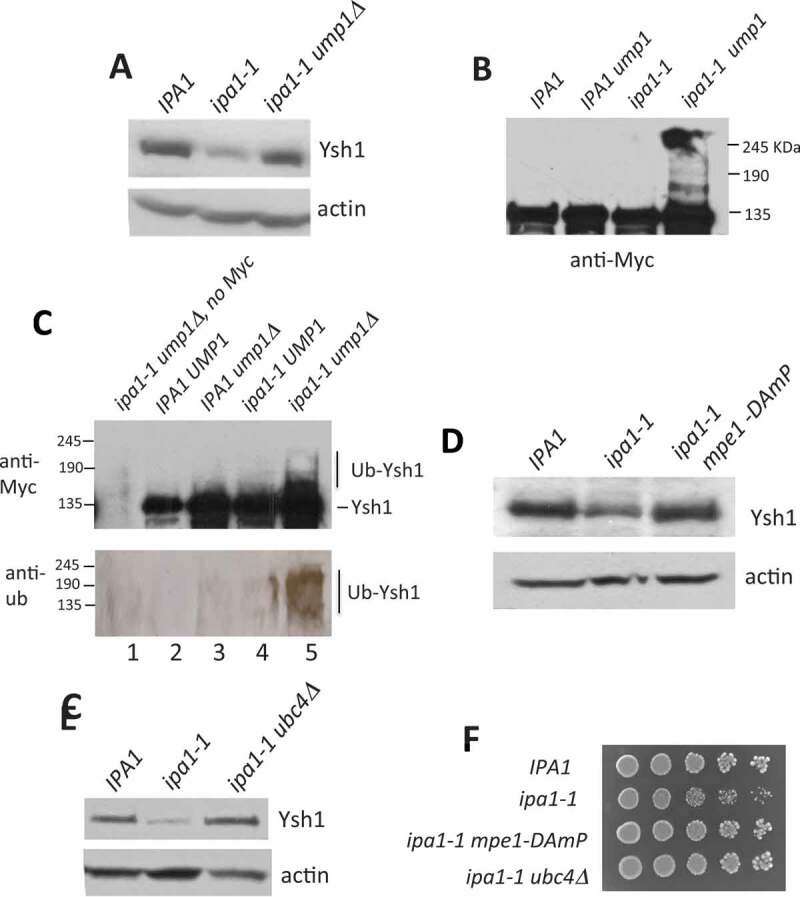
Ysh1 is degraded via the ubiquitin-proteasome system in ipa1-1.
(A) In vivo expression of Ysh1 in ipa1-1 and ipa1-1 ump1▵. Cells were grown at 30°C and then shifted to 37°C for 1 hr. Extracts from these cells were analysed by Western blotting using an antibody against Ysh1 or actin. (B) Cell lysate prepared from the ipa1-1 ump1▵ cells under denaturing conditions shows higher molecular weight Ysh1 species. Immunoblots were probed with antibody against a Myc tag on Ysh1. (C) Ysh1 is ubiquitinated in vivo in ipa1-1. Ysh1 was immunoprecipitated from the indicated cell extracts. The immunoprecipitate in lane 1 was obtained from ipa1-1 ump1▵ cells with untagged Ysh1. The immunoprecipitates were analysed by Western blotting using antibody against Myc to detect Ysh1 or against ubiquitin. To concentrate the Western signal, a high-percentage acrylamide gel was used for immunoblotting with anti-ubiquitin. (D, E) Expression of Ysh1 in the ipa1-1 mutant or the ipa1-1 mutant which also contained either a deletion of the UBC4 gene (ubc4▵) or a mutation of the MPE1 gene (mpe1-DAmP) [39], in which the MPE1 3ʹ UTR is disrupted by insertion of the ampicillin gene, which destabilizes the mRNA, leading to reduced protein expression. Experiments were performed as described in (A). (F) Growth of ipa1-1 relative to ipa1-1 paired with mutation of MPE1 or UBC4. Cells were grown on YPD plates at 30°C.
Ubiquitination requires the sequential actions of three proteins: E1, the ubiquitin-activating enzyme; E2, a ubiquitin-conjugating enzyme; and E3, a ubiquitin ligase [51,52]. Yeast has only one E1, but multiple E2 and E3 enzymes. To identify E2 and E3 enzymes responsible for Ysh1 ubiquitination in ipa1-1, we mutated candidate E2s or E3s in the ipa1-1 strain and observed Ysh1 protein level. We reasoned that if an enzyme responsible for Ysh1 ubiquitination in ipa1-1 was not functional, Ysh1 would not be targeted for degradation and would remain at a level comparable to that of wild-type. Mpe1, which is a CPF subunit in the nuclease module, interacts directly with Ysh1 [40] and contains a RING finger-like domain found in one type of E3 ubiquitin ligases [53]. RBBP6, the mammalian homolog of Mpe1, has ubiquitin ligase activity [54,55]. Thus, Mpe1 is a natural candidate for the E3 responsible for Ysh1 ubiquitination. Indeed, when MPE1 was mutated in ipa1-1, Ysh1 protein level was restored to that seen in wild-type cells (Fig. 3D) and growth of ipa1-1 improved (Fig. 3F), suggesting that this particular E3 is the primary regulator of Ysh1 levels when Ipa1 is defective.
For a candidate E2 enzyme, we focused on Ubc4 because of reported genetic interactions linking Ubc4 with Ipa1 and the C/P machinery. Of the eleven E2 enzymes in S. cerevisiae [56], only Ubc4 showed a significant positive genetic interaction with Ipa1 [39], as determined by improved growth when mutations in both IPA1 and UBC4 are paired in the same cell. This genetic interaction suggests that Ipa1 action is antagonistic to that of Ubc4. In addition, we had earlier found that the growth defect of the mpe1-C182G,C185G mutant was suppressed by overexpression of HEL2, encoding a Ring Finger ubiquitin ligase that physically interacts with Ubc4 [34,57,58]. Consistent with these interactions, mutation of UBC4 restored Ysh1 protein level to a normal level in ipa1-1 (Fig. 3E) and improved growth of ipa1-1 (Fig. 3F). Thus, even though Ipa1 has been shown to physically interact with other E2 proteins [59], including Ubc1, Ubc5, Ubc7, Ubc8, Ubc11, Ubc13, and Pex4, the presence of wild-type genes encoding these enzymes or that of the close Ubc4 paralog, Ubc5, is not sufficient to cover for loss of Ubc4 in the ipa1-1 mutant.
UBC4 and MPE1 are required for in vitro ubiquitination of Ysh1
As described above, mutating UBC4 or MPE1 in the ipa1-1 strain restored the Ysh1 level in vivo. However, restoration of Ysh1 protein level could be caused by indirect effects of these mutations. Ubc4 functions in various cellular pathways, and because Mpe1 is involved in 3ʹ-end processing, its mutation could affect gene expression of relevant factors. Thus, we developed an in vitro ubiquitination assay to investigate whether Ubc4 and Mpe1 are directly involved in Ysh1 ubiquitination. Extracts were made from either wild type or mutant cells and incubated with ubiquitin and CPF purified from yeast expressing 3xHA-TAP-tagged YSH1 in the mpe1-1 mutation, which causes Mpe1 to dissociate from CPF [53]. We were unable to purify Ysh1 alone as we have found that it is prone to aggregation.
Using anti-HA antibody, we observed the appearance of high molecular weight Ysh1 species upon addition of recombinant ubiquitin and CPF (Fig. 4A, lane 3) to wild-type extract. These species did not appear when ATP was omitted (Fig. 4A, lane 2) or when methylated ubiquitin (me-ub) was used in the reactions (Fig. 4A, lane 4), or when a reaction was performed without adding Ysh1 (Fig. S1E). ATP is required for the addition of ubiquitin to an active-site cysteine residue on the ubiquitin-activating enzyme [52], and methylation of the ϵ-amino group of lysines blocks ubiquitin from forming polyubiquitin chains [60]. These results indicate that the larger forms of Ysh1 corresponded to polyubiquitinated Ysh1. When extracts made from ubc4▵ or mpe1-1 strains were used, the larger species did not form efficiently (Fig. 4B and 4C, lanes 3). However, when recombinant Ubc4 or Mpe1 was added to the reactions, the larger Ysh1 species accumulated, showing that the missing factor needed for Ysh1 polyubiquitination in the ubc4▵ or mpe1-1 extracts was Ubc4 or Mpe1, respectively (Fig. 4B and 4C, lanes 4). Polyubiquitination of Ysh1 was abrogated when me-ub was used instead of ubiquitin (Fig. 4B and 4C, lanes 5).
Figure 4.
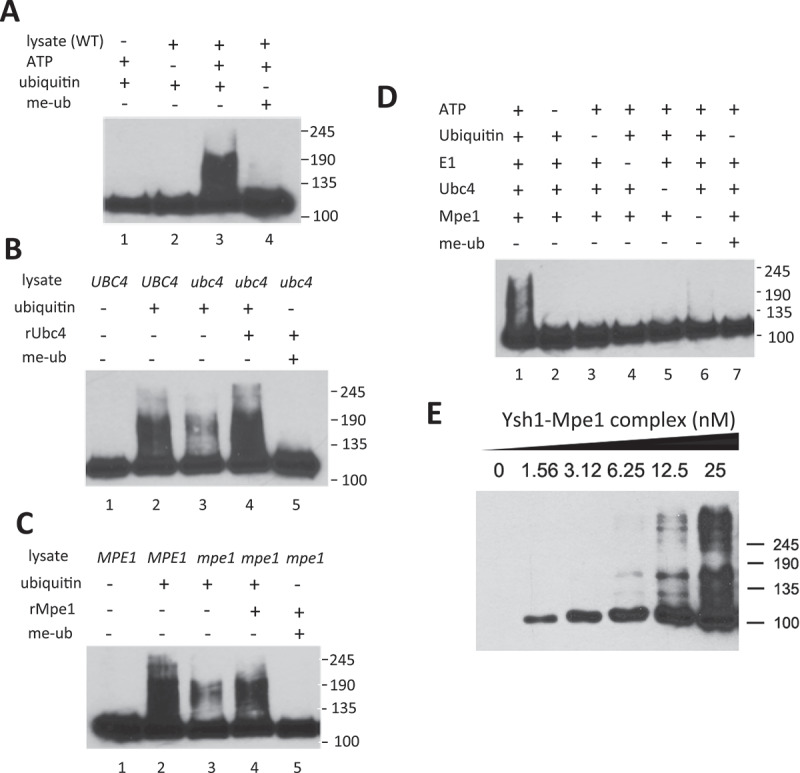
Ubc4 and Mpe1 are required for in vitro ubiquitination of Ysh1.
(A) In vitro ubiquitination of Ysh1 in cell extract. Extract from wild-type cells was incubated with ubiquitin and CPF purified from yeast expressing 3xHA-TAP-tagged YSH1. Components of the in vitro reaction were omitted as indicated, and methylated ubiquitin (me-ub) replaced ubiquitin in lane 4. Ysh1 was detected with HA antibody. (B) Lysate prepared from ubc4Δ cells does not efficiently ubiquitinate Ysh1 unless recombinant Ubc4 is added. Reactions in lanes 1 and 2 use wild-type extract (UBC4). Reactions were performed as described in Panel A, and Ysh1 was detected with HA antibody. (C) Lysate prepared from mpe1-1 cells does not efficiently ubiquitinate Ysh1 unless recombinant Mpe1 is added. Reactions were performed as described in Panel A, and Ysh1 was detected with HA antibody. (D) In vitro ubiquitination assay with purified proteins. The reaction in lane 1 contains E1 (Uba1), Ubc4, Mpe1, ATP, ubiquitin, and CPF containing Ysh1-HA3. For lanes 2–7, each component is omitted as indicated, or ubiquitin is replaced with methylated ubiquitin (me-ub). Ysh1 was detected with HA antibody. (E) A Ysh1-Mpe1 heterodimer is ubiquitinated in the reconstituted assay. Increasing amounts of the dimer were added as indicated and reactions performed as described in Panel D. In all of the experiments in this figure, marker positions are shown on the right of each panel.
A purified in vitro system ubiquitinates Ysh1
The data presented above confirm a requirement of Ubc4 and Mpe1 for Ysh1 ubiquitination. We next tested whether Ubc4 and Mpe1 were sufficient to direct Ysh1 ubiquitination by establishing an in vitro ubiquitination assay with purified proteins. His6-tagged Ubc4 and Mpe1 were expressed in E. coli and purified by Ni-chelating affinity chromatography. CPF containing Ysh1-HA3 was purified from yeast as described above, and recombinant Uba1 (the E1 ubiquitin-activating enzyme) was obtained commercially. When a mixture of Ysh1, Ubc4, Mpe1, Uba1 and ubiquitin was incubated with ATP, Ysh1 was ubiquitinated as shown by the appearance of high molecular weight species (Fig. 4D, lane 1). When any component was omitted, or when ubiquitin was substituted with me-ubiquitin, these species disappeared, indicating that they correspond to polyubiquitinated Ysh1 (Fig. 4D and Fig. S1F). These data indicate that Ubc4 as a ubiquitin conjugase and Mpe1 as a ubiquitin ligase, along with Uba1 and ubiquitin, are sufficient to mediate Ysh1 ubiquitination.
We also tested whether a Ysh1–Mpe1 complex expressed and purified from insect cells (Fig. S1C) was a substrate for ubiquitination in the reconstituted assay. Ysh1 was readily ubiquitinated when provided in complex with Mpe1, with the ubiquitinated species reaching a larger size than in the previous assay (Fig. 4E), indicating that Ysh1 that is only in complex with Mpe1 is also substrate for ubiquitination.
Our data showing that the Ysh1 level decreased when Ipa1 was depleted (Fig. 1A) suggested that Ipa1 might somehow block Ysh1 ubiquitination and degradation. However, addition of recombinant Ipa1 to the in vitro ubiquitination reaction using cell extract showed no inhibition of ubiquitination in comparison to bovine serum albumin (BSA) added in the same amounts (Fig. S1G). This finding suggests that Ipa1 is not acting directly to prevent Ubc4 and Mpe1 from modifying Ysh1.
Discussion
Regulating mRNA polyadenylation can help a cell achieve optimal protein production for a given condition or cell state. Because of this potential, a greater knowledge of the fundamental regulatory strategies governing polyadenylation can contribute to understanding of normal cell physiology and dysregulation in the disease state [1]. One mechanism to regulate mRNA polyadenylation is to limit the amount of the core processing machinery, which can contribute to a general decrease in mRNA synthesis when less synthetic capacity is needed. This mechanism can also force favouring of poly(A) sites with better matches to conserved sequence elements on the precursor mRNA, leading to mRNA isoforms with different properties. In this study, we show that the budding yeast protein Ipa1 controls the level of Ysh1, the essential endonuclease that cuts mRNA precursor at the poly(A) site in preparation for polyadenylation and mRNA nuclear export. Loss of Ipa1 leads to depletion of the Ysh1 protein without affecting YSH1 mRNA level. Other subunits of the C/P complex are not depleted, indicating selective targeting of Ysh1. We discuss below possible mechanisms by which Ipa1 could act to regulate Ysh1 levels.
Our findings show that increasing the copy number of the YSH1 gene suppressed the temperature-sensitive growth and the in vivo processing defect of ipa1-1, indicating that these defects are largely due to loss of Ysh1. The Ysh1 depletion in ipa1-1 cells is a consequence of ubiquitin-mediated proteasomal degradation that is facilitated by Mpe1, which, like Ysh1, is an integral subunit of the nuclease module of CPF [40]. Mpe1 possesses a ubiquitin-like domain, a zinc knuckle, and a RING finger domain [34]. Thus, by sequence, Mpe1 is also a member of the RING family of ubiquitin ligases [61], and our results confirm this predicted activity. We have also identified Ubc4 as a ubiquitin conjugase that participates in Ysh1 modification. The sequence of Ipa1 indicates that like Mpe1, it is a ubiquitin ligase, but of the HECT family [41,59]. While Ipa1 possesses homology to the E2 interaction domain of this group of proteins, it does not have an active site cysteine [59], suggesting it does not function like a conventional HECT-type E3, which covalently receives ubiquitin from a partner E2 and transfers it to the target protein.
The molecular mechanism by which Ipa1 prevents Ysh1 degradation is not clear. Biochemical fractionation indicates that while Ipa1 exists in a complex with only the Ysh1 and Mpe1 subunits of CPF, it is not a component of CPF itself [39,40,42]. The direct interaction of Ipa1 with Ysh1 [40] and intracellular presence of the Ipa1/Ysh1/Mpe1 trimer support a role involving physical contact between the two proteins. It is possible that Ipa1, through its interaction with the intrinsically disordered C-terminal domain of Ysh1 [40], protects Ysh1 from degradation until it is incorporated into CPF. However, addition of Ipa1 to an in vitro assay did not block Ysh1 ubiquitination, suggesting Ipa1 is not preventing productive interaction of a ubiquitin conjugase like Ubc4 with the Mpe1/Ysh1 dimer.
Since Ipa1 is found in both nucleus and cytoplasm [59], another role for Ipa1 could be as a co-chaperone to help Ysh1 fold correctly during or after translation (Fig. 5). The Hsp70 and Hsp90 chaperones fold substrate proteins, and accessory proteins called co-chaperones modulate chaperone activity and deliver client proteins [62,63,64]. Ysh1 physically interacts with Hsp82 [65], a member of the Hsp90 family, and several other proteins which assist in protein folding were associated with Ipa1, including Cpr6 and Aha1 (two co-chaperones of Hsp82), the Cct4, Cct6, and Cct8 subunits of the cytosolic Chaperonin Containing TCP1 complex, Hsp42 (a member of the small heat shock chaperone family), Ssa3 (one of the yeast HSP70 ATPases), and Ydj1 (an Ssa3 co-chaperone and Hsp40 family member) [39]. Cells mutated in both IPA1 and HSC82, encoding a paralog of Hsp82, grow more poorly than cells defective for only IPA1 or HSC82 [39], suggesting that the two genes lie in compensatory pathways.
Figure 5.
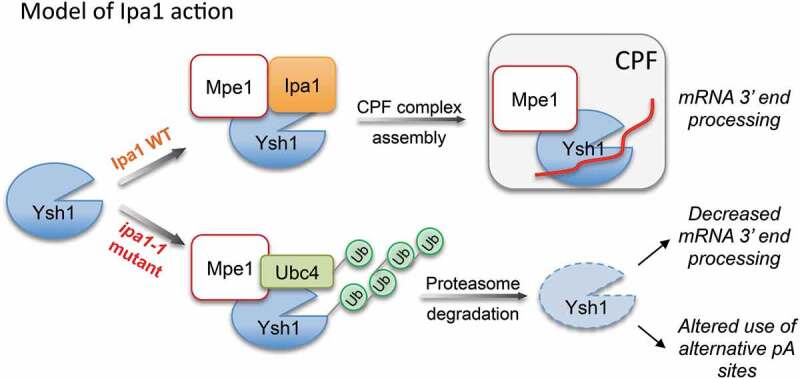
Model for the action of Ipa1 in maintaining the level of Ysh1. In the presence of Ipa1, Ysh1 is correctly folded, and the Ysh1/Mpe1 complex can be incorporated into CPF, leading to efficient mRNA 3ʹ end processing. In the absence of Ipa1, Ysh1 would not fold properly, leading to ubiquitination and destruction of Ysh1, inefficient mRNA 3ʹ end processing, and alterations in the choice of the cleavage site.
Some co-chaperones direct Hsp70/Hsp90 chaperones to participate in either protein degradation or folding [62,63]. Co-chaperones with this activity can be RING-type ubiquitin ligases such as CHIP, and this function has been proposed for the Mpe1 human homolog RBBP6 [66]. Thus, it is possible that Ipa1 acts by recruiting a chaperone complex to Ysh1 or otherwise promoting the folding activity of the chaperones. Once folded, the Ysh1/Mpe1 complex could be incorporated into CPF (Fig. 5), through a switch in which Ipa1 interacting at the Ysh1 C-terminal domain is replaced with the CPF Cft2 subunit [40]. In the absence of Ipa1, Ysh1 would not fold properly, leading to ubiquitination and destruction of Ysh1, inefficient mRNA 3ʹ end processing, and as we described previously, alterations in the choice of poly(A) site position [41]. Our model for Ipa1 action predicts that the Ipa1/Ysh1/Mpe1 complex might be exquisitely tuned for folding or instead, rapid degradation of Ysh1, depending on the needs of the cell. However, we cannot exclude other possible models. For example, Ipa1 could recruit or regulate the abundance of a deubiquitinase that counteracts the activity of Ubc4 and Mpe1 on Ysh1, although there is no evidence for this from reported physical interactions. Ubiquitination does not necessarily lead to degradation [67], and Ipa1 could promote ubiquitination of a factor in a way that regulates its activity towards Ysh1.
Our study suggests that yeast has evolved an easily tunable mechanism that ensures the correct amount of Ysh1 when cells are growing in optimal conditions. It is possible that this mechanism could also allow rapid removal of Ysh1 and a general down-regulation of the polyadenylation capacity of the cell in less favourable growth conditions. Having to modulate only one subunit of the C/P complex (i.e. Ysh1) would provide a means to quickly match this aspect of mRNA synthesis to changing growth conditions. Interestingly, the Ipa1 homolog, UBE3D/UBE2CBP, physically interacts in quantitative proteomics screens with CPSF-73 [68,69], the counterpart of Ysh1. While UBE3D has been implicated in cell cycle regulation [70], age-related macular degeneration [71], the inflammatory condition of aggressive juvenile periodontitis [72], and alterations in the fatty acid profile of muscle [73], its functional role in mammalian cells have not been determined. Future experiments may not only elucidate the precise mechanism by which Ipa1 modulates Ysh1 degradation but also explore whether this regulatory relationship is conserved across eukaryotes.
Materials and methods
Yeast strains and plasmids construction
The S. cerevisiae strains used in this study are given in Table 1. SDL98, SDL99, SDL107, and SDL116 were constructed by inserting a Myc13 tag at the C terminus of the endogenous YSH1 gene as described [74].
Table 1.
Yeast strains used in this study.
| Strain | Genotype |
|---|---|
| W303a | ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 |
| BY4741a | his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 |
| SDL164 | As BY4741a but ipa1-1::KanR [39] |
| SDL101 | As BY4741a but ipa1-1::KanR, ump1Δ::LEU2 (This study) |
| SDL89 | As BY4741a but ipa1-1::KanR,ubc4Δ::natR [39] |
| SDL90 | As BY4741a but ipa1-1::KanR,mpe1-DAmP::natR [39] |
| SDL106 | As BY4741a but ipa1-1::KanR,ubc7Δ::LEU2 [39] |
| SDL98 | As BY4741a but YSH1-Myc13::HisMX6 (This study) |
| SDL99 | As BY4741a but ipa1-1::KanR,YSH1-Myc13::HisMX6 (This study) |
| SDL107 | As BY4741a but ump1Δ::LEU2,YSH1-Myc13::HisMX6 (This study) |
| SDL116 | As BY4741a but ipa1-1::KanR, ump1Δ::LEU2, YSH1-Myc13::HisMX6 (This study) |
| SDL57 | BY4742a; his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 ubc4Δ::LEU2(This study) |
| SDL110 | As W303a but IPA1-Myc13::HisMX6 (This study) |
| SDL103 | As BY4741a but IPA1-Myc13::HisMX6 (This study) |
| SDL104 | As BY4741a but ipa1-1-Myc13::HisMX6 (This study) |
| mpe1-1 | W303a; ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 mpe1-1 [53] |
For Ysh1 purification in yeast, theYSH1-3xHA-TAP (SDL184) strain was constructed using mpe1-1 (W303a; ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100 mpe1-1) [53], as the host (described below). SDL110 (W303a; ade2-1 ura3-1 his3-11,15 trp1-1 leu2-3,112 can1-100)
For pRS315-YSH1, the YSH1 is under the control of the MPE1 promoter and terminator sequences. The coding sequence of YSH1 was amplified from genomic DNA by PCR using the upstream primer NdeI-BRR5-fwd (5ʹ -CTCTACATATGGAGCGAACAAATACAACAACATTC-3ʹ) and the downstream primer NotI-BRR5-rev (5ʹ-tacagGCGGCCGCTTAACATAGCGGTGTGACCAAATTACC-3ʹ). The PCR product was digested with NdeI and NotI and was cloned into the pRS315-MPE1 vector [34], which was digested with the same restriction enzymes.
pET21b-MPE1 construction was described in Lee and Moore [34]. For pTD68-Flag-UBC4 construction, DNA sequence containing BamHI-Flag-UBC4-XhoI was ordered as gBlocks from IDT (sequence shown below), digested with BamHI and XhoI, and cloned into pTD68 digested with the same restriction enzymes. The construction of pTD68 was described in Uehara et al. [75]. The UBC4 sequence (underlined below) was E.coli codon optimized.
5ʹACTCATGGATCCGACTATAAAGATGACGACGACAAGGGTGGATCGGGCTCGTCTTCTAAGCGCATTGCAAAAGAACTGTCTGATCTTGAGCGTGATCCGCCAACGTCGTGCTCTGCTGGGCCTGTAGGGGATGATTTGTACCATTGGCAAGCGTCCATCATGGGACCTGCTGACAGTCCGTATGCTGGGGGCGTATTTTTTCTTTCCATCCATTTTCCGACAGATTACCCTTTTAAGCCCCCTAAGATCTCCTTCACCACGAAAATTTACCATCCTAACATCAACGCAAATGGAAACATTTGCCTTGATATCTTAAAGGACCAGTGGAGCCCGGCACTTACACTTAGTAAAGTATTGCTTTCTATTTGTTCGTTGCTGACGGACGCCAATCCAGACGATCCATTGGTCCCAGAGATCGCTCATATTTACAAGACTGACCGCCCTAAGTACGAAGCGACCGCACGCGAATGGACAAAAAAATATGCCGTCTAACTCGAGTGTCTG3ʹ
For pTD68-V5-IPA1 construction, the same scheme was used as described for pTD68-Flag-UBC4. The DNA sequence containing BamHI-V5-IPA1-XhoI was ordered as gBlocks from IDT (sequence shown below). The IPA1 sequence (underlined below) was E.coli codon optimized.
ACTCATGGATCCGGTAAACCAATCCCAAATCCGTTACTTGGGCTTGATTCTACTGGGATGGTACAATACGTAGTGGAATGGTTACCACGCATCCAGAGTATCAGCGTTGTAGTAGAGGGGTGGAAGCAGGTGGAAATCAAAAATTTGAAAGACACGCTGATGTCCATCTCTGGTGACGAGGAGCAGGTAGAAGACATCCTTTTACCCGTTGAAGTAGAGGAAAAAGTAGATGCTTCCTATAAATTCAAGAACCGCGGTAAAGACTTGGAGTGGATGACCAAGCTGCGTTCCAAAAGCTCGAAGATCTATGATAGCTCGATCATGTCACTGCCGGACGGTCGTTGGACAAAGGAGGAGTTGCGCTCCGACTCGGACTTCTCTATCGAATGCCTTAATTGTAAGCAGAAGATCATTAGCAAAGACAACTGTCAAGTTTTGAACGATATGCCGAGCGAATTCTGGTTCGAACTTATGGATTATTGGCACTGCCACAAACCGGATGTAAAAGAGGATAAATCTTCATACACGCGCTTTGAGACCCTTAAACCGTCCAAAAATGAAATTCTTATTGGGTCTTCCTATTTCCAAGGCACTCCTGCGACGTTTGAAAACGTAGCAACGACGAAAGAAAATGACAATGTTCTGTGCATCAAATGTTCAGCTGTGCTTGGACAAGTTACTGCAGGTAGCTTATACAAGCTTCACAAGTGGAAACTGCAATTAATCCGTTCAGGAAACACATACAAGTTCCCTCCCGAATGTGATATTACCATCTCATTAATTAATGTAGTTAAAGCGAATTCCTGCCGCTATGTTCTGGTGAAATGCAAAACTGAAAGTTTACTGGTCTGGATCTTTAGTGTGGACATCGGAGTTACTTTGACGGGAAACAAATCGTTCAAACGTGCGATGAAGCTGCTTTACACTAATTCTGTCACTACAATCAACCGCTGCCTTAATCGCCAAGTGGTAGAGGAGTTGGACTTCCAGGAAACCTCGTTTAACGCATTCTATTCCGCCTTGCAACATACAAATGCTTTGCTTCCATCGAGTATGAAAAAAATTGGGGAGTGGACAATTTCATATACCTCGCTTATCTAACTCGAGTGTCTG
The YSH1-3xHA-TAP in mpe1-1 background was generated by inserting the 3xHA-TAP sequence immediately upstream of the stop codon of YSH1 as previously described with some modifications [76]. In brief, 3xHA-TAP-TRP1 bracketed by sequences flanking the YSH1 stop codon was amplified by PCR with pBS1479 as a template by using Q5 High-Fidelity DNA polymerase (NEB) and primers BRR5-3HA-TAP-fwd (5ʹ-AGGGTGGAAAGCCTCTTAAATATTGGTGGTAATTTGGTCACACCGCTATGTGGATCCGGTTCTTTAATTAACATCTTTTACCCATACGATGTTCCTGACTATGCGGGCTATCCGTATGACGTCCCGGACTATGCAGGATCCTATCCATATGACGTTCCAGATTACGCTGCTCAGTGCGGAGGATCCATGGAAAAGAGAAG-3ʹ) and BRR5-3HA-TAP-rev (5ʹ-CGTAAAGAGTGATTAAGGTAAACAATAAAAGGCAGTTCTAACCAGATTTTGGTTTGGTATTACTTCTATAAAGTAGTCTACTTAGTATGCGTAACTGTTTtacgactcactataggg-3ʹ). Each primer has a pBS1479 specific sequence (underlined) and a 50-base (fwd primer) and 100-base (reverse primer) YSH1-specific sequence. mpe1-1 cells were transformed with the YSH1-3xHA-TAP-TRP1 PCR product, followed by selection for the TRP1 marker. Transformants harbouring YSH1-3xHA-TAP-TRP1 were verified by PCR and the expression of YSH1-3xHA-TAP was confirmed by Western blot.
Yeast spot assay and cultures
Growth properties were analysed by growing strains in liquid yeast extract-peptone-dextrose (YPD) at room temperature to an optical density at 600 nm (OD600) of 1.0, spotting 5 of 10-fold serial dilutions onto YPD plates, and incubating the plates for 3 days at 30°C or 37°C.
Affinity purification of Ysh1-3HA-TAP and PTA1-TAP
Yeast expressing 3xHA-TAP-tagged YSH1 in the mpe1-1 background [53] were grown in 8 L of YPD at 25°C to an OD600 = 0.7 and shifted to 37°C for 1.5 h. For purification of CPF, FY23 cells carrying PTA1-TAP [22] were grown in 8 L of YPD at 30°C to an OD600 = 1.0. Extracts were prepared as described previously [77] except omitting the ammonium sulphate precipitation step. CPF was purified according to the standard TAP procedure [76] except that the IgG beads were washed with high salt buffer (1.5 M NaCl) for CPF used in the in vitro ubiquitination assays. The eluates were separated on an SDS-8% polyacrylamide gel and detected by silver staining (Fig. S1B). Ysh1 was confirmed by Western blot analysis using anti-HA antibody.
Preparation of extracts and in vitro mRNA 3ʹ end processing assays
Preparation of yeast processing extracts and processing assays were done as described previously [77]. For the rescue experiment with TAP-purified CPF, 10–30 ng of CPF was mixed with yeast extracts and incubated for 5 min at 25°C before RNA substrate was added. Western blotting to determine levels of proteins in the processing extracts was performed according to standard procedures. The monoclonal antibodies against Pta1 and Pap1 have been described previously [77]. Polyclonal antibodies against Rna14 and Rna15, and Ysh1 were a gift from H. Domdey. Polyclonal antibody against Mpe1 was a gift from F. Wyers. The antibodies against Ssu72, Pfs2, Fip1, and Yth1 have been described previously [5].
Co-immunoprecipitation assays
Yeast processing extracts (1 mg) prepared as described previously [77] were incubated with anti-Pap1 antibody overnight at 4°C in 500 μl of buffer IP-150 (10 mM Tris-HCl at pH 7.9, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 μM pepstatin A, 0.6 μM leupeptin, 0.05% NP-40). IP samples were then incubated with protein G beads for 1 hr at 4°C, washed 5X with 1 ml of buffer IP-150, and eluted with 70 μl of SDS sample buffer (without reducing reagent) for 15 min at 25°C. The eluates were resuspended with SDS sample buffer, boiled for 3 min, and analysed in an SDS-10% polyacrylamide gel.
In vivo detection of Ysh1 ubiquitination
The method for purifying ubiquitinated proteins under denaturing conditions was described previously by Laney and Hochstrasser [78]. For our experiments, cells were grown in 20 ml of minimal medium at 30°C to an OD600 = 0.5 and shifted to 37°C for 1 hour. Cells were resuspended in 200 μl SDS buffer (2% SDS and 45 mM HEPES, pH 7.5), boiled for 15 min, and centrifuged at 14,000 x g for 5 min. The supernatant was diluted with 4 ml ice-cold Triton lysis buffer (1% Triton X-100, 150 mM NaCl, 50 mM HEPES, pH 7.5, 5 mM EDTA, 20 mM NEM, 1 mM 1,10-phenanthroline monohydrate, 1 mM PMSF, 2 μM pepstatin A, and 0.6 μM leupeptin) and incubated with anti-Myc antibody for 30 min at 4°C. Protein G beads (Santa Cruz) were then added and rotated for another 30 min. Beads were washed three times with 1 ml Triton lysis buffer containing 0.05% SDS, resuspended in SDS sample buffer, boiled for 5 min, and analysed in an SDS-PAGE. Western blotting was performed according to standard procedures except that after transfer, the nitrocellulose membrane was submerged in water and autoclaved for 40 min to improve immuno-detection of ubiquitin, blocked with 5% bovine serum albumin, and then probed with anti-ubiquitin from Santa Cruz (sc-8017). For detection of Ysh1, anti-Myc antibody (9E10) was used.
Recombinant protein expression and purification
Mpe1 was expressed and purified from E. coli Rosetta (DE3) cells according to Lee and Moore [34]. Recombinant Ubc4 and Ipa1 were expressed and purified from E. coli Rosetta cells as in Cho et al. [79] with the following exceptions. Buffer A was composed of 50 mM sodium phosphate buffer, pH 8.0, 300 mM NaCl, 10 mM Imidazole, 5% glycerol, 5 mM β-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 μM pepstatin A, and 0.6 μM leupeptin. Buffer B was composed of 50 mM sodium phosphate buffer, pH 8.0, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 μM pepstatin A, and 0.6 μM leupeptin. Ipa1-His6 was expressed from a pET21a plasmid, and protein purified by nickel affinity chromatography. Ubc4 was expressed from the pTD68 plasmid [75], which adds a His6-SUMO-FLAG tag to the N-terminus of Ubc4, and purified as described in Uehara, et al. [75]. After purification of the Ubc4 fusion protein by nickel‐affinity chromatography, the His6‐SUMO tag was released using His6‐tagged SUMO protease. Cleavage reactions were passed through Ni‐NTA resin to remove free H‐SUMO and the protease. The purified proteins (Fig. S1D) were dialysed against buffer D (50 mM Tris, pH 7.9, 150 mM NaCl, 0.5 mM DTT, 20% glycerol, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 μM pepstatin A, and 0.6 μM leupeptin).
For expression of the Ysh1–Mpe1 complex in Sf9 insect cells, we used Bac-to-Bac technology (Thermo Fisher) as described by Cooper et al. [80]. The Mpe1 ORF tagged with Myc (5ʹ) and His6 (3ʹ) was inserted into pFastBac-Dual after the pPolh promoter using the SacI and NotI sites, and the Ysh1 ORF tagged with His6 (5ʹ) and 3HA (3ʹ) was inserted into pFastBac-Dual after the p10 promoter using the XhoI and NheI sites. The pFASTbac-Dual-Ysh1-Mpe1 recombinant plasmid was validated by Sanger sequencing. The construct was transformed into DH10Bac cells and positive colonies were selected by Bluo-Gal. Bacmid DNA was prepared by using the P1, P2 and N3 buffer from Qiagen miniprep kit (Cat. No. 27106) followed by isopropanol precipitation. The purified bacmid DNA was used for transfection in Sf9 insect cell to make baculovirus.
For expression and purification of the Ysh1–Mpe1 complex, Sf9 cells were grown in Sf900-SFM medium (Thermo Fisher) to a density of 2 × 106 cells/ml and infected with recombinant baculovirus by adding 7.5 ml of the viral stock from the third passage (P3) to 1.5 litres of cells in a 5-litre incubation bottle. Seventy-two hours after infection, cells were harvested and resuspended in solubilization buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM imidazole, 10% glycerol, 10 uM pepstatin A, 100 uM leupeptin and 1 mM PMSF). The suspension was lysed by passing it three times through an M-110S microfluidizer according to the manufacturer’s instructions. Supernatant was collected from lysate by centrifugation at 4,000 g for 25 min. Ysh1 and Mpe1 in 45 mL of supernatant were isolated by adding 1 mL Ni Superflow resin (GE Healthcare). After incubation for 1 h, the resin was washed three times with 15 column volumes of 50 mM Tris pH 8.0, 150 mM NaCl and 10 mM imidazole. Ysh1 and Mpe1 were eluted with 50 mM Tris pH 8.0, 150 mM NaCl, and 300 mM imidazole and concentrated in a centrifuge filter (molecular weight cut-off of 30 kDa, Millipore EMD). Ysh1–Mpe1 complex was further purified by anti-HA beads (Thermo Fisher, #26181) according to the manufacture instructions and analysed by coomassie blue staining and Western Blot using anti-HA antibody (sc-7392) and Anti-MYC antibody (sc-40) from Santa Cruz (Fig. S1C). The concentrations of purified proteins were determined by BCA protein assay (Thermo Fisher).
In vitro ubiquitination assays
Yeast crude extracts were prepared as described in Zhao et al. [77]. The extracts are diluted to 1 μg/μl in buffer D (50 mM Tris, pH 7.9, 150 mM NaCl, 0.5 mM DTT, 20% glycerol, 1 mM phenylmethylsulfonyl fluoride [PMSF], 2 μM pepstatin A, and 0.6 μM leupeptin). The reaction includes 2 μg of extract, 2 μl of 5X reaction buffer (100 mM Tris pH 7.5, 100 mM MgCl2, 10 mM ATP, and 5 mM DTT), 15 μM ubiquitin aldehyde (Boston Biochem U-201), 0.2 mM ubiquitin (Boston Biochem U-100sc) or 0.2 mM me-ubiquitin (Boston Biochem U-501), and the CPF-high-salt fraction in a final volume of 10 μl. For recombinant Ubc4 and Mpe1 add-back experiments, protein was added to 1.5 μM in a final volume of 10 μl. These reactions were carried out at 37°C for 10 min. The reactions with purified components shown in Fig. 3D include 10 nM Uba1 (Boston Biochem E-300), 200 nM Ubc4, 200 nM Mpe1, 0.2 mM ubiquitin or 0.2 mM me-ubiquitin and ~200 nM Ysh1-high-salt fraction and 2 μl of 5X reaction buffer (same as above) in a final volume of 10 μl. These reactions were carried out at 30°C for 20 min. The in vitro ubiquitination assays (total reaction volume of 20 μL) shown in Fig. 3E were performed using Uba1 (125 nM), Ubc4 (500 nM), 1 μg/μL ubiquitin, the Ysh1–Mpe1 complex expressed in insect cells at the indicated concentrations in 100 mM Tris, pH 7.5, 100 mM MgCl2, 10 mM ATP, 4 mM MgCl2, and 5 mM DTT for 30 min at 30°C. Reactions were stopped by addition of SDS sample buffer containing 4% β-mercaptoethanol, heated at 95°C for 5 min, and resolved on 10% Bis-Tris gels or by SDS-PAGE followed by immunoblotting with the indicated antibodies.
qRT-PCR analysis
Total RNA from wild-type or mutant cells was isolated using the Hot Phenol Method [81] and Heavy Phase Lock Gel tubes (Quantabio), and treated with RQ1 DNase (Promega). DNA-free RNA was subjected to reverse transcription using random hexamers. The resulting cDNA samples were analysed using real-time PCR analysis performed in a 12 µl reaction with 0.5 µl of 10 µM forward and reverse primers, 5 µl of SYBR Green Supermix (BIO-RAD), 1 µl cDNA, and 5 µl of distilled water. The primer sequences are listed in Table 2. The expression of the unprocessed transcript of a given gene was normalized to the expression of total mRNA of that gene.
Table 2.
Primers used for RT-qPCR.
| Primer Name | Primer Sequence 5ʹ-3’ |
|---|---|
| PDC1 Forward | GCCAGTCTTCGATGCTCCAC |
| PDC1 Total Reverse | ATCGCTTATTGCTTAGCGTTGG |
| PDC1 pA Span Reverse | ACTGTCGGCAACTTCTTGTCTGG |
| RPP1B Total Forward | ACGCTAAGGCTTTGGAAGGTAAGGA |
| RPP1B Total Reverse | AACCGAAACCCATGTCGTCGTCAGA |
| RPP1B pA Span Forward | GACGACGACATGGGTTTCGGT |
| RPP1B pA Span Reverse | TCGTAGCCCTTTCGTATGGACA |
Global gene expression analysis
RNA sequencing data sets for the ipa1-1, pcf11-2 and cft2-1 were obtained from Costanzo et al. [39] and processed and analysed as described previously for the ipa1-1 data set [41]. Differential expression assessment was made with DESeq2 [80], using a threshold of log2 fold-change > 0.5, p-value < 0.2 for increased genes, or log2 fold-change < 0.5, p-value < 0.2 for decreased genes, and based only upon poly(A) site tags that supported a full-length mRNA transcript. Venn diagrams were generated with eulerr [50]. Cumulative polyadenylation distribution (CPD) plots were determined as described in Graber et al. [5].
Supplementary Material
Funding Statement
This work was supported by the National Institutes of Health [P20GM103423 and P20GM104318];National Science Foundation [MCB‐1244043];
Acknowledgments
We thank members of the laboratories of C. Moore and A. Bohm for critical input regarding our experiments and manuscript and to Ellen White and Katya Heldwein for help with protein expression in insect cells. We are grateful to A. Greenleaf, D. Bentley, H. Domdey, M. Hampsey, M. Henry, and F. Lacroute for the generous gifts of antibodies used in this study. This work was supported by NSF grant MCB‐1244043 to C. Moore. J. Graber was partially supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant numbers P20GM103423 and P20GM104318.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Gruber AJ, Zavolan M.. Alternative cleavage and polyadenylation in health and disease. Nat Rev Genet. 2019;20:599–614. [DOI] [PubMed] [Google Scholar]
- [2].Mayr C. Regulation by 3ʹ-Untranslated Regions. Annu Rev Genet. 2017;51:171–194. [DOI] [PubMed] [Google Scholar]
- [3].Chen M, Lyu G, Han M, et al. 3ʹ UTR lengthening as a novel mechanism in regulating cellular senescence. Genome Res. 2018;28:285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].de Lorenzo L, Sorenson R, Bailey-Serres J, et al. Noncanonical alternative polyadenylation contributes to gene regulation in response to hypoxia. Plant Cell. 2017;29:1262–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Graber JH, Nazeer FI, Yeh PC, et al. DNA damage induces targeted, genome-wide variation of poly(A) sites in budding yeast. Genome Res. 2013;23:1690–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jia X, Yuan S, Wang Y, et al. The role of alternative polyadenylation in the antiviral innate immune response. Nat Commun. 2017;8:14605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].MacDonald CC. Tissue-specific mechanisms of alternative polyadenylation: testis, brain, and beyond (2018 update). Wiley Interdiscip Rev RNA. 2019;10:e1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Neve J, Patel R, Wang Z, et al. Cleavage and polyadenylation: ending the message expands gene regulation.s. RNA Biology. 2017;14:865–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pai AA, Baharian G, Page Sabourin A, et al. Widespread shortening of 3ʹ untranslated regions and increased exon inclusion are evolutionarily conserved features of innate immune responses to infection. PLoS Genet. 2016;12:e1006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shi Y, Manley JL. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015;29:889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Tang S, Patel A, Krause PR. Herpes simplex virus ICP27 regulates alternative pre-mRNA polyadenylation and splicing in a sequence-dependent manner. Proc Natl Acad Sci U S A. 2016;113:12256–12261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18:18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yoon OK, Brem RB. Noncanonical transcript forms in yeast and their regulation during environmental stress. RNA. 2010;16:1256–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zheng D, Wang R, Ding Q, et al. Cellular stress alters 3ʹUTR landscape through alternative polyadenylation and isoform-specific degradation. Nat Commun. 2018;9:2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Masamha CP, Wagner EJ. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis. 2018;39:2–10. [DOI] [PubMed] [Google Scholar]
- [16].Park HJ, Ji P, Kim S, et al. 3ʹ UTR shortening represses tumor-suppressor genes in trans by disrupting ceRNA crosstalk. Nat Genet. 2018;50:783–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sadek J, Omer A, Hall D, et al. Alternative polyadenylation and the stress response. Wiley Interdiscip Rev RNA. 2019;10:e1540. [DOI] [PubMed] [Google Scholar]
- [18].Elkon R, Ugalde AP, Agami R. Alternative cleavage and polyadenylation: extent, regulation and function. Nat Rev Genet. 2013;14:496–506. [DOI] [PubMed] [Google Scholar]
- [19].Mizrahi N, Moore C. Posttranslational phosphorylation and ubiquitination of the Saccharomyces cerevisiae Poly(A) polymerase at the S/G(2) stage of the cell cycle. Mol Cell Biol. 2000;20:2794–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Colgan DF, Murthy KG, Zhao W, et al. Inhibition of poly(A) polymerase requires p34cdc2/cyclin B phosphorylation of multiple consensus and non-consensus sites. Embo J. 1998;17:1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim H, Lee JH, Lee Y. Regulation of poly(A) polymerase by 14-3-3epsilon. Embo J. 2003;22:5208–5219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].He X, Moore C. Regulation of yeast mRNA 3ʹ end processing by phosphorylation. Mol Cell. 2005;19:619–629. [DOI] [PubMed] [Google Scholar]
- [23].Ryan K. Pre-mRNA 3ʹ cleavage is reversibly inhibited in vitro by cleavage factor dephosphorylation. RNA Biol. 2007;4:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Tang HW, Hu Y, Chen CL, et al. The TORC1-regulated CPA complex rewires an RNA processing network to drive autophagy and metabolic reprogramming. Cell Metab. 2018;27:1040–54 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kajitani N, Glahder J, Wu C, et al. hnRNP L controls HPV16 RNA polyadenylation and splicing in an Akt kinase-dependent manner. Nucleic Acids Res. 2017;45:9654–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dermody JL, Dreyfuss JM, Villen J, et al. Unphosphorylated SR-like protein Npl3 stimulates RNA polymerase II elongation. PloS One. 2008;3:e3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shimazu T, Horinouchi S, Yoshida M. Multiple histone deacetylases and the CREB-binding protein regulate pre-mRNA 3ʹ-end processing. J Biol Chem. 2007;282:4470–4478. [DOI] [PubMed] [Google Scholar]
- [28].Richard P, Vethantham V, Manley JL. Roles of sumoylation in mRNA processing and metabolism. Adv Exp Med Biol. 2017;963:15–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xiang K, Tong L, Manley JL. Delineating the structural blueprint of the pre-mRNA 3ʹ-end processing machinery. Mol Cell Biol. 2014;34:1894–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kumar A, Clerici M, Muckenfuss LM, et al. Mechanistic insights into mRNA 3ʹ-end processing. Curr Opin Struct Biol. 2019;59:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kamieniarz-Gdula K, Gdula MR, Panser K, et al. Selective roles of vertebrate PCF11 in premature and full-length transcript termination. Mol Cell. 2019;74:158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rehfeld A, Plass M, Krogh A, et al. Alterations in polyadenylation and its implications for endocrine disease. Front Endocrinol (Lausanne). 2013;4:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shell SA, Hesse C, Morris SM Jr., et al. Elevated levels of the 64-kDa cleavage stimulatory factor (CstF-64) in lipopolysaccharide-stimulated macrophages influence gene expression and induce alternative poly(A) site selection. J Biol Chem. 2005;280:39950–39961. [DOI] [PubMed] [Google Scholar]
- [34].Lee SD, Moore CL. Efficient mRNA polyadenylation requires a ubiquitin-like domain, a zinc knuckle, and a RING finger domain, all contained in the Mpe1 protein. Mol Cell Biol. 2014;34:3955–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mandel CR, Kaneko S, Zhang H, et al. Polyadenylation factor CPSF-73 is the pre-mRNA 3ʹ-end-processing endonuclease. Nature. 2006;444:953–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Eaton JD, Davidson L, Bauer DLV, et al. Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Genes Dev. 2018;32:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Baejen C, Andreani J, Torkler P, et al. Genome-wide analysis of RNA Polymerase II termination at protein-coding genes. Mol Cell. 2017;66:38–49. [DOI] [PubMed] [Google Scholar]
- [38].Schaughency P, Merran J, Corden JL. Genome-wide mapping of yeast RNA polymerase II termination. PLoS Genet. 2014;10:e1004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Costanzo M, VanderSluis B, Koch EN, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353(6306). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hill CH, Boreikaite V, Kumar A, et al. Activation of the endonuclease that defines mRNA 3ʹ ends requires incorporation into an 8-subunit core cleavage and polyadenylation factor complex. Mol Cell. 2019;73:1217–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pearson EL, Graber JH, Lee SD, et al. Ipa1 is an RNA polymerase II elongation factor that facilitates termination by maintaining levels of the poly(A) site endonuclease Ysh1. Cell Rep. 2019;26:1919–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Casanal A, Kumar A, Hill CH, et al. Architecture of eukaryotic mRNA 3ʹ-end processing machinery. Science. 2017;358:1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lidschreiber M, Easter AD, Battaglia S, et al. The APT complex is involved in non-coding RNA transcription and is distinct from CPF. Nucleic Acids Res. 2018;46:11528–11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].He X, Khan AU, Cheng H, et al. Functional interactions between the transcription and mRNA 3ʹ end processing machineries mediated by Ssu72 and Sub1. Genes Dev. 2003;17:1030–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ghazy MA, He X, Singh BN, et al. The essential N terminus of the Pta1 scaffold protein is required for snoRNA transcription termination and Ssu72 function but is dispensable for pre-mRNA 3ʹ-end processing. Mol Cell Biol. 2009;29:2296–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Preker PJ, Lingner J, Minvielle-Sebastia L, et al. The FIP1 gene encodes a component of a yeast pre-mRNA polyadenylation factor that directly interacts with poly(A) polymerase. Cell. 1995;81:379–389. [DOI] [PubMed] [Google Scholar]
- [47].Ohnacker M, Barabino SM, Preker PJ, et al. The WD-repeat protein pfs2p bridges two essential factors within the yeast pre-mRNA 3ʹ-end-processing complex. Embo J. 2000;19:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Dominski Z, Yang XC, Purdy M, et al. A CPSF-73 homologue is required for cell cycle progression but not cell growth and interacts with a protein having features of CPSF-100. Mol Cell Biol. 2005;25:1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Ramos PC, Hockendorff J, Johnson ES, et al. Ump1p is required for proper maturation of the 20S proteasome and becomes its substrate upon completion of the assembly. Cell. 1998;92:489–499. [DOI] [PubMed] [Google Scholar]
- [50].Oliveros JC An interactive tool for comparing lists with Venn’s diagrams. 2007-2015. http://bioinfogp.cnb.csic.es/tools/venny/index.html
- [51].Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. 2017;86:129–157. [DOI] [PubMed] [Google Scholar]
- [52].Varshavsky A. The ubiquitin system, an immense realm. Annu Rev Biochem. 2012;81:167–176. [DOI] [PubMed] [Google Scholar]
- [53].Vo LT, Minet M, Schmitter JM, et al. Mpe1, a zinc knuckle protein, is an essential component of yeast cleavage and polyadenylation factor required for the cleavage and polyadenylation of mRNA. Mol Cell Biol. 2001;21:8346–8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chibi M, Meyer M, Skepu A, et al. RBBP6 interacts with multifunctional protein YB-1 through its RING finger domain, leading to ubiquitination and proteosomal degradation of YB-1. J Mol Biol. 2008;384:908–916. [DOI] [PubMed] [Google Scholar]
- [55].Di Giammartino DC, Li W, Ogami K, et al. RBBP6 isoforms regulate the human polyadenylation machinery and modulate expression of mRNAs with AU-rich 3ʹ UTRs. Genes Dev. 2014;28:2248–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Finley D, Ulrich HD, Sommer T, et al. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics. 2012;192:319–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Matsuo Y, Ikeuchi K, Saeki Y, et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat Commun. 2017;8:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Singh RK, Gonzalez M, Kabbaj MH, et al. Novel E3 ubiquitin ligases that regulate histone protein levels in the budding yeast Saccharomyces cerevisiae. PloS One. 2012;7:e36295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lutz AP, Schladebeck S, Renicke C, et al. Proteasome activity is influenced by the HECT_2 protein Ipa1 in budding yeast. Genetics. 2018;209:157–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hershko A, Heller H. Occurrence of a polyubiquitin structure in ubiquitin-protein conjugates. Biochem Biophys Res Commun. 1985;128:1079–1086. [DOI] [PubMed] [Google Scholar]
- [61].Metzger MB, Pruneda JN, Klevit RE, et al. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta. 2014;1843:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Edkins AL. CHIP: a co-chaperone for degradation by the proteasome. Subcell Biochem. 2015;78:219–242. [DOI] [PubMed] [Google Scholar]
- [63].Rosenzweig R, Nillegoda NB, Mayer MP, et al. The Hsp70 chaperone network. Nat Rev Mol Cell Biol. 2019;20:665–680. [DOI] [PubMed] [Google Scholar]
- [64].Nathan DF, Vos MH, Lindquist S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc Natl Acad Sci U S A. 1997;94:12949–12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Zhao R, Davey M, Hsu YC, et al. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell. 2005;120:715–727. [DOI] [PubMed] [Google Scholar]
- [66].Kappo MA, Ab E, Hassem F, et al. Solution structure of RING finger-like domain of retinoblastoma-binding protein-6 (RBBP6) suggests it functions as a U-box. J Biol Chem. 2012;287:7146–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18:579–586. [DOI] [PubMed] [Google Scholar]
- [68].Hein MY, Hubner NC, Poser I, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–723. [DOI] [PubMed] [Google Scholar]
- [69].Huttlin EL, Bruckner RJ, Paulo JA, et al. Architecture of the human interactome defines protein communities and disease networks. Nature. 2017;545:505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kobirumaki F, Miyauchi Y, Fukami K, et al. A novel UbcH10-binding protein facilitates the ubiquitinylation of cyclin B in vitro. J Biochem. 2005;137:133–139. [DOI] [PubMed] [Google Scholar]
- [71].Huang LZ, Li YJ, Xie XF, et al. Whole-exome sequencing implicates UBE3D in age-related macular degeneration in East Asian populations. Nat Commun. 2015;6:6687. [DOI] [PubMed] [Google Scholar]
- [72].Offenbacher S, Divaris K, Barros SP, et al. Genome-wide association study of biologically informed periodontal complex traits offers novel insights into the genetic basis of periodontal disease. Hum Mol Genet. 2016;25:2113–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rovadoscki GA, Pertile SFN, Alvarenga AB, et al. Estimates of genomic heritability and genome-wide association study for fatty acids profile in santa ines sheep. BMC Genomics. 2018;19:375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Longtine MS, McKenzie A 3rd, Demarini DJ, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in saccharomyces cerevisiae. Yeast. 1998;14:953–961. [DOI] [PubMed] [Google Scholar]
- [75].Uehara T, Parzych KR, Dinh T, et al. Daughter cell separation is controlled by cytokinetic ring-activated cell wall hydrolysis. Embo J. 2010;29:1412–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Puig O, Caspary F, Rigaut G, et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. [DOI] [PubMed] [Google Scholar]
- [77].Zhao J, Kessler M, Helmling S, et al. Pta1, a component of yeast CF II, is required for both cleavage and poly(A) addition of mRNA precursor. Mol Cell Biol. 1999;19:7733–7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Laney JD, Hochstrasser M. Analysis of protein ubiquitination. Curr Protoc Protein Sci. 2011;66. Chapter 14:Unit145:14.5.1–14.5.13. [DOI] [PubMed] [Google Scholar]
- [79].Cho H, McManus HR, Dove SL, et al. Nucleoid occlusion factor SlmA is a DNA-activated FtsZ polymerization antagonist. Proc Natl Acad Sci U S A. 2011;108:3773–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Cooper RS, Georgieva ER, Borbat PP, et al. Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat Struct Mol Biol. 2018;25:416–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ares M. Isolation of total RNA from yeast cell cultures. Cold Spring Harb Protoc. 2012;2012:1082–1086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Oliveros JC An interactive tool for comparing lists with Venn’s diagrams. 2007-2015. http://bioinfogp.cnb.csic.es/tools/venny/index.html