

ABSTRACT
Members of the conserved Argonaute (Ago) protein family provide defence against invading nucleic acids in eukaryotes in the process of RNA interference. Many prokaryotes also contain Ago proteins that are predicted to be active nucleases; however, their functional activities in host cells remain poorly understood. Here, we characterize the in vitro and in vivo properties of the SeAgo protein from the mesophilic cyanobacterium Synechococcus elongatus. We show that SeAgo is a DNA-guided nuclease preferentially acting on single-stranded DNA targets, with non-specific guide-independent activity observed for double-stranded substrates. The SeAgo gene is steadily expressed in S. elongatus; however, its deletion or overexpression does not change the kinetics of cell growth. When purified from its host cells or from heterologous E. coli, SeAgo is loaded with small guide DNAs whose formation depends on the endonuclease activity of the argonaute protein. SeAgo co-purifies with SSB proteins suggesting that they may also be involved in DNA processing. The SeAgo-associated small DNAs are derived from diverse genomic locations, with certain enrichment for the proposed sites of chromosomal replication initiation and termination, but show no preference for an endogenous plasmid. Therefore, promiscuous genome sampling by SeAgo does not have great effects on cell physiology and plasmid maintenance.
KEYWORDS: Argonaute, programmable DNA nuclease, SeAgo, Synechococcus elongatus, ori and ter sites
Introduction
Conserved Argonaute (Ago) proteins, present in all domains of life, use nucleic acid guides to recognize complementary genetic targets. Eukaryotic RNA interference systems rely on this activity for suppression of invading genetic elements, regulation of gene expression and epigenetic silencing [1–3]. Diverse Ago variants are also encoded in the genomes of many bacterial and archaeal species but their in vivo functions remain poorly understood. Many prokaryotic Agos (pAgos) contain predicted catalytic sites in the PIWI domain that likely enable them to process nucleic acid targets, but their in vivo activities and natural substrates remain unknown in most cases [3–6].
In vitro analysis of several pAgos demonstrated that they can indeed cleave their nucleic acid targets in a guide-dependent manner at precisely defined positions [7–16]. Interestingly, unlike their eukaryotic counterparts, studied pAgos prefer DNA substrates both in vitro and in vivo; several proteins were also shown to cleave RNA, albeit with a lower efficiency [7–10,12,13,15–17]. Since the active site of pAgos can catalyse endonucleolytic cleavage of a single nucleic acid strand, pAgos preferentially act on single-stranded substrates, while their ability to target double-stranded DNA strongly depends on the AT-content of the target, the presence of single-stranded regions, and the reaction temperature [15–17]. Some pAgos were shown to process double-stranded DNA in a guide-independent manner, resulting in its gradual fragmentation. Such ‘chopping’ activity was proposed to generate guide molecules that might be then loaded into pAgo [13,16,18].
Most analysed pAgo proteins were isolated from thermophilic prokaryotes and showed low levels of activity at moderate temperatures. Recent analysis of two proteins from mesophilic bacteria, CbAgo (from the Gram-positive Clostridium butyricum) and LrAgo (from the cyanobacterium Limnothrix rosea), revealed that they are able to cleave single-stranded DNA at much lower temperatures than their thermophilic counterparts [15,16]. However, the ability of CbAgo and LrAgo to process double-stranded DNA in a guide-dependent manner also greatly depends on the temperature. Previous in vivo studies of catalytically active pAgos were performed exclusively with thermophilic proteins. It was shown that TtAgo (from Thermus thermophilus), PfAgo (Pyrococcus furiosus) and MjAgo (Methanocaldococcus jannaschii) preferably attack plasmid substrates and decrease plasmid content and/or transformation efficiencies in their native hosts, suggesting their role in cell defence against foreign DNA [9,13,17]. Analysis of the specificity of DNA targeting in vivo was performed only for TtAgo in the heterologous E. coli system, at a much lower temperature than is optimal for TtAgo. It was shown that TtAgo is loaded with plasmid-derived but not chromosomal small DNA species, enriched for a cytidine residue in their 5ʹ-end [17]. However, due to the suboptimal temperature of TtAgo expression and inability to purify small nucleic acids from its native host, T. thermophilus, it remains unclear whether such preferential targeting of plasmid DNA reflects a natural resistance of the genome against pAgo action. It is also unknown whether pAgos from mesophilic bacteria can process genomic DNA or invading nucleic acids in vivo under physiological conditions.
Here, we characterize the SeAgo protein from the model mesophilic cyanobacterium Synechococcus elongatus. The closest homolog of SeAgo among the previously studied pAgos is TtAgo [3,6]. Structural modelling of SeAgo, based on the published structure of TtAgo, reveals a characteristic bilobal structure, with the nucleic acid binding cleft located between the N-PAZ and MID-PIWI lobes and the catalytic tetrad of negatively charged residues located in the PIWI domain (Fig. 1A, B). We show here that SeAgo acts as a DNA-guided DNA endonuclease in vitro and is autonomously loaded with small guide DNAs when expressed in either S. elongatus or E. coli. The regions preferentially targeted by SeAgo in the chromosomal DNA include putative sites of replication initiation and termination. Despite its constant low expression level during cell growth, SeAgo deletion or overexpression does not affect cell viability. Therefore, SeAgo can promiscuously probe various chromosomal locations without disturbing cell growth or genome integrity.
Figure 1.

Site-specific ssDNA cleavage by SeAgo. (A) Structural model of SeAgo. The N-, PAZ, MID, and PIWI domains are shown in yellow, red, green and blue, respectively. The modelling was performed using the Phyre2 web tool [19], based on the structure of TtAgo from T. thermophilus (PDB 2ETN [11]) as the template. (B) Structure of TtAgo (4NCB [8]). (C) Scheme of the guide and target DNAs used in the cleavage assay (see Table S1 for complete oligonucleotide sequences). (D) Kinetics of ssDNA cleavage by SeAgo in the presence of Mg2+ and Mn2+. The reaction products were analysed by denaturing urea PAGE followed by SYBR Gold staining. The cleavage site is located in the middle of the 50 nt target oligonucleotide, which also contains a 3ʹ-Cy3 fluorophore (T-tDNA_Cy3, Table S1), so that the resulting 25 nt 3ʹ-fragment has lower mobility in the gel than the 5ʹ-fragment (see Fig. S3). (E) Effects of mismatches in different guide parts on target DNA cleavage by SeAgo. The positions and the identities of the mismatches are indicated above the gel. (F) Quantification of the cleavage efficiencies for guide DNAs containing mismatches at various positions relative to the control fully complementary guide oligonucleotide (fc, fold-change). Means and standard deviations from three independent experiments are shown (* – p < 0.05; ** – p < 0.01). (G) Effect of SeSSB on ssDNA cleavage by SeAgo. The experiments were performed with 500 nM SeAgo, 500 nM guide DNA, 100 nM target DNA and 400 nM SeSSB in Mn2+-containing buffer. Positions of the target (T), guide (G) and product (P) DNAs are indicated.
Materials and methods
S. elongatus growth and transformation
Wild-type strain of S. elongatus PCC7942 was obtained from Invitrogen. Cyanobacterial strains were maintained in liquid BG11 or on solid BG11 plates [20] under continuous light conditions (~250 μE m−2 s−1) at 30°C with appropriate antibiotics, liquid medium was bubbled with air containing 1.0% (v/v) CO2.
The SeAgo gene (WP_011378069.1) was amplified by PCR from genomic DNA of S. elongatus PCC7942, fused to the N-His6-tag sequence and cloned downstream of the PSBAII promoter into the pSyn6 vector containing the SpR cassette (GeneArtTM kit, Invitrogen) for further integration into the neutral site (NS1) of the chromosome (↑AgoWT strain). Mutant variant of the SeAgo gene with alanine substitutions of three aspartic acid residues in the catalytic tetrad (D516A, D584A and D709A; ↑AgoCD strain) was generated by site-directed PCR mutagenesis and cloned into the same vector. An empty pSyn6 vector was used to obtain a control strain with the SpR cassette in the NS1 site of the chromosome. To obtain the ∆Ago (luciferase) strain, the firefly luciferase gene [21] was cloned with site-specific flanks into the pSyn6 vector for further integration instead of the SeAgo gene in the chromosome.
Cell transformation was performed as described in the GeneArt™ kit manual for S. elongatus (Invitrogen). Briefly, S. elongatus cells were grown to OD750 = 1.0, 1 mL of the culture was placed in a culture tube, the cells were washed three times with 1 ml of BG11 medium and resuspended in 100 µl of the medium. 0.1 µg of plasmid DNA in ultrapure water was added, the culture was incubated in the dark for 4 h at 34°C and spread on BG11 agar plates with 10 µg/ml spectinomycin. Positive transformants were selected, checked by PCR screening and used for further experiments.
RT-qPCR
To measure the levels of SeAgo expression at different growth stages, 50 ml samples were taken from cell cultures at 4, 6, 8 and 10 days after inoculation. The cells were collected by centrifugation at 4500 g for 7 min at 4°C. The supernatant was decanted and total RNA was extracted by Trizol. Any remaining DNA was removed using the gDNA Removal Kit (Jena Bioscience, GmbH). RNA concentration was measured by a NanoDrop 3300 fluorospectrometer. cDNA was synthesized from ~0.5 µg of RNA with a RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) using gene-specific primers. The cDNA was diluted 10-fold and used as a template with primers to amplify a 190 bp fragment of the SeAgo gene by qPCRmix-HS SYBR Master Mix (Evrogen) in a Roche LightCycler 96 System. The rnpB gene was used as a reference with a 162 bp amplicon. Three biological and three technical replicates were performed for each sample along with no template and no reverse transcriptase controls. The resulting CT values were averaged, and the 2− ΔΔCT method was used for quantification of relative gene expression levels.
Luciferase assay
For luciferase assays, a Promega luciferase assay system was used. Cells were gently pelleted by centrifugation at 13000 g for 15 s and lysed with 150 µl of the Promega CCLR buffer. Twenty microliters of the lysate was added to 45 µl of the Promega assay buffer containing luciferin at 25°C, and luminescence was measured with a Modulus Microplate Reader (Turner BioSystems, Inc.) with default parameters. Luminescence was normalized to the total protein amount, experiments were independently repeated three times and the normalized luminescence values were averaged.
SeAgo expression and purification in S. elongatus
The ↑AgoWT strain was used for SeAgo purification from S. elongatus. Cells were grown in the BG11 medium at 30°C under continuous illumination and bubbled with air containing 1.0% (v/v) CO2 until OD750 = 0.6. The cells were collected by centrifugation and stored at – 80°C. Cell pellet was resuspended in Co2+-chromatography buffer A (40 mM potassium-phosphate buffer pH 7.4, 0.2 M NaCl, 0.05 M KCl) supplemented with EDTA-free protease inhibitor cocktail (Roche) and disrupted by a high-pressure homogenizer at 25000 psi. The lysate was clarified by centrifugation at 32000 g for 30 min and the supernatant was loaded onto Co2+-charged TALON Metal Affinity Resin (Clontech) for 2 h. The beads were washed twice with Co2+-buffer A, then with the same buffer containing 10 mM and 20 mM of imidazole, and eluted with buffer containing 100 mM imidazole. The resulting SeAgo sample was used for the preparation of associated nucleic acids and proteins. In control experiments, the S. elongatus cell culture was mixed with an equal amount of Rhodobacter sphaeroides cell culture prior to protein purification and all subsequent procedures were performed in the same way.
Small nucleic acid purification, sequencing, and analysis
Nucleic acids were extracted from purified SeAgo using Proteinase K treatment followed by extraction with neutral phenol/chloroform and ethanol precipitation. The samples were treated by alkaline phosphatase (part of the samples was left untreated) and labelled with γ-[32P]-ATP using T4 polynucleotide kinase for further visualization. RNase and DNase treatment were performed by incubation with 25 units of RNase A (Thermo Scientific) or 2 units of DNase I (Thermo Scientific) at 37°C for 30 min. The resulting nucleic acids were resolved by 15% urea-PAGE and eluted from the gel by 0.4 M NaCl with further ethanol precipitation. Small DNA libraries for high throughput sequencing were prepared using a bridged ligation approach [22]. Simultaneous ligation of 5ʹ- and 3ʹ-linkers was performed in 20 µl volume containing 100 pmol 50 and 30 linker and bridge oligonucleotides, 5% PEG8000, 1× T4 DNA ligase buffer, and 1 µl of T4 DNA ligase (New England Biolabs, 400,000 units/ml). The samples were incubated for 4 h at 25°C, the products were resolved by 15% urea-PAGE, eluted by 0.4 M NaCl and ethanol precipitated. Standard Illumina primers were used to create indexed libraries. Small DNA sequencing was performed in the rapid run mode (single-end, 50 bp reads) on the Illumina Hiseq 2500 platform (Millard and Muriel Jacobs Genetics and Genomics Laboratory).
For small DNA analysis, adaptor sequences were trimmed using the cutadapt utility (v 3.6) and processed reads were mapped to the S. elongatus PCC7942 genome and pANL plasmid sequences (GenBank: CP000100.1) or Rhodobacter sphaeroides ATCC17025 genome (GenBank: CP000143.2) using bowtie aligner (v. 1.2.3) allowing 0 mismatches. Data analysis was done using Perl and R scripts. The small DNA coverage was calculated using command line utilities (Samtools v.1.9, Bedtools v.2.28.0). Data visualization and statistical analysis were done using R, including the ggplot2 package [23]. Presumable dif-site in the S. elongatus PCC7942 genome was identified using a Python script and bacterial dif consensus as the query. The full sequencing data have been deposited to the 4TU.Centre repository and are available via https://doi.org/10.4121/uuid:6388f6cd-5516-407e-8964-b451b603c79.
Protein mass spectrometry
For identification of partner proteins of SeAgo, it was purified from the ↑AgoWT strain as described above (‘SeAgo expression and purification in S. elongatus’); the empty strain was used as a control. The SeAgo samples eluted from TALON Metal Affinity Resin were used for Liquid chromatography-tandem mass spectrometry analysis. The samples were analysed by Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer coupled with EASY-nLC 1000 Liquid Chromatograph (Thermo Fisher Scientific) in The Proteomics Core Facility of the University of Southern California. Data analysis and spectra matching were performed using Skyline [24]. The mass spectrometry data have been deposited to the 4TU. Centre repository and are available via https://doi.org/10.4121/uuid:6388f6cd-5516-407e-8964-b451b603c79a
SeAgo expression and purification in E. coli
The wild-type SeAgo gene was cloned into the pLATE52 expression vector in frame with the N-terminal His6 tag. A codon-optimized version of the gene (see the sequence in Supplementary Material online) was obtained using the online IDT Codon Optimization Tool for E. coli, the gene was synthesized by IDT and cloned into the pET28 expression vector in frame with the N-terminal His6 tag. E. coli BL21(DE3) cells carrying expression plasmids with either wild-type or optimized SeAgo were cultivated in the LB medium at 37°C until OD600 = 0.5. Cold shock was performed by incubation of cell cultures in an ice bath for 30 min, the cultures were induced with 0.2 mM IPTG and aerated for 12 h at 16°C. The cells were collected by centrifugation and stored at – 80°C. Cell pellet was resuspended in the Co2+-chromatography buffer (30 mM Tris-HCl pH 7.9, 0.5 M NaCl, 5% glycerol) supplemented with EDTA-free protease inhibitor cocktail (Roche) and disrupted by a high-pressure homogenizer at 18,000 psi. The lysate was clarified by centrifugation at 32,000 g for 30 min and the supernatant was loaded onto a HiTrap TALON crude column charged with Co2+ (GE Healthcare). The column was washed extensively with the Co2+-buffer, then with the buffer containing 10 and 20 mM imidazole, and the proteins were eluted with the buffer containing 100 mM imidazole. Fractions containing SeAgo were concentrated by ultrafiltration using an Amicon 50K filter unit (Millipore) and diluted with 20 mM Tris-HCl (pH 7.9) to 50 mM NaCl concentration. The sample was loaded onto a Heparin FF column (GE Healthcare) equilibrated with 20 mM Tris-HCl containing 50 mM NaCl, washed with 10 volumes of the same buffer and eluted with a linear NaCl gradient (0.1–1.0 M). The samples containing SeAgo (eluted at 300–350 mM NaCl) were concentrated using Amicon 50K and purified on a Superose 6 10/300 GL column (GE Healthcare) equilibrated with buffer GF (10 mM Tris-HCl pH 7.9, 0.5 M NaCl, 5% glycerol, 1 mM DTT, 0.5 mM EDTA). Fractions containing SeAgo were concentrated using Amicon 50K, aliquoted, and flash-frozen in liquid nitrogen. The heparin chromatography step was omitted during purification of the wild-type, non-codon-optimized version of SeAgo. The purity of the final protein samples was assessed by denaturing SDS-PAGE followed by Coomassie staining. The protein concentration was determined by the Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad). The mutant variant of SeAgo with alanine substitutions of the catalytic tetrad residues was obtained by site-directed mutagenesis of the SeAgo gene, expressed and purified in the same way. Small DNAs were extracted from SeAgo after the first purification step (Co2+-chromatography) as described above.
SeSSB cloning, expression, and purification
Nucleotide sequence of the SeAgo SSB gene (WP_011377525.1; S. elongatus strain PCC7942) was obtained by PCR from genomic DNA of S. elongatus and cloned into the pET28 vector in frame with the N-terminal His6 tag. E. coli BL21(DE3) cells carrying the expression vector were cultivated in the LB medium for 18 h at 16°C with the addition of 0.2 mM IPTG at the beginning of growth. The cells were collected by centrifugation and stored at – 80°C. SeSSB was purified by Co2+-NTA-affinity chromatography in the same way as SeAgo, except for the final elution step that was performed with buffer containing 200 mM imidazole. Fractions containing SeSSB were loaded onto a HiTrap Q HP anion exchange chromatography column (GE Healthcare) equilibrated with buffer Q-start (Tris-HCl pH 7.9 30 mM, DTT 1 mM, EDTA 1 mM), washed with at least 10 column volumes of the same buffer and eluted with a linear NaCl gradient (0.5–1 M). Samples containing SeSSB (eluted at 300 mM NaCl) were aliquoted and flash-frozen in liquid nitrogen.
Nucleic acid cleavage assays
The sequences of all guide and target oligonucleotides are shown in Supplementary Table S1. The cleavage assays were performed at 37°C in reaction buffer containing 10 mM Tris-HCl pH 7.9, 100 mM KCl, 1 mM DDT and 2 mM MnCl2 (or MgCl2 when indicated). 500 nM SeAgo was mixed with 500 nM guide DNA and incubated for 10 min for guide loading. Target DNA was added to 100 nM. In reactions containing SeSSB, 400 nM SSB was added to target DNA and incubated for 10 min at 37°C prior to the addition of SeAgo. The reaction was stopped after indicated time intervals by mixing the samples with equal volumes of stop solution containing 8 M urea and 20 mM EDTA. The cleavage products were resolved by 15-19% denaturing PAGE, stained with SYBR Gold (Invitrogen), visualized with Typhoon FLA 9500 or ChemiDoc XP, and analysed by the ImageQuant (GE Healthcare) software.
For analysis of plasmid DNA cleavage, the final SeAgo concentration was 500 nM, guide DNAs were added to 500 nM when indicated. When using two guide molecules, they were independently loaded into SeAgo at equimolar ratios and combined together to the final concentration of 500 nM. The pJET_target plasmid was added to the reaction mixtures at 2 nM final concentration, followed by incubation for indicated time intervals at 37°C. If SeSSB was used, 400 nM SSB was incubated with plasmid DNA for 10 min prior to the addition of SeAgo. For analysis of the effect of transcription on SeAgo activity, E. coli core RNA polymerase (150 nM) and the σ70 subunit (450 nM) were preincubated with plasmid DNA for 10 min at 37° and mixed with SeAgo preloaded with guide DNAs, followed by addition of NTPs (100 µM each, final concentration). The reactions were stopped by treatment with Proteinase K for 20 min at 37°C (and RNAse A in the case of transcription), the samples were mixed with 6×Purple Loading Dye (New England Biolabs) and the cleavage products were resolved by native 1% agarose gel electrophoresis.
Results
Endonuclease activity of SeAgo in vitro
To analyse SeAgo activities in vitro, we cloned the SeAgo gene, expressed it in E. coli and purified the protein by Co2+-affinity chromatography and gel filtration. The level of heterologous expression and the yield of SeAgo in E. coli was low (0.015 mg per gram of cell pellet after two steps of purification). To improve protein purification, we also obtained a synthetic codon-optimized version of the SeAgo gene (see Materials and methods for details). The optimized SeAgo was purified in three steps, including Co2+-affinity, heparin-affinity chromatography and gel filtration (Fig. S1A), with the final yield of 0.011 mg/g. Both protein preparations were analysed in parallel, described in this and subsequent sections.
To study the endonuclease activity of SeAgo, we performed an in vitro cleavage assay using single-stranded DNA or RNA guide and target oligonucleotides (Fig. 1C, see Table S1 for guide and target sequences). SeAgo was first loaded with a guide, followed by the addition of target DNA or RNA. SeAgo loaded with guide DNA cleaved complementary single-stranded DNA (ssDNA) target, resulting in the appearance of two-DNA fragments, corresponding to the 5ʹ- and 3ʹ-parts of the target molecule (Fig. 1D). No cleavage was observed for other combinations of guide and target molecules (guide DNA/target RNA; guide RNA/target DNA; guide RNA/target RNA), or in the absence of guides (Fig. S2A). DNA cleavage depended on the catalytic activity of SeAgo since no cleavage was observed in the case of a mutant variant of SeAgo with substitutions of three of the catalytic tetrad residues (catalytically dead protein, SeAgo CD; Fig. S2B). The efficiency of cleavage was higher in the presence of Mn2+ ions, while only weak cleavage was detected in the presence of Mg2+ (Fig. 1B).
Similarly to previously studied pAgo proteins, SeAgo cleaves target DNA between the 10th and 11th guide positions (Fig. 1D and Fig. S2). The cleavage position was verified by direct comparison of the lengths of resulting DNA fragments with synthetic oligonucleotide markers corresponding to the expected reaction products (Fig. S2D). Furthermore, to identify the 5ʹ- and 3ʹ-cleavage products, we used a DNA target that contained 32P and Cy3 labels at its 5ʹ- and 3ʹ-ends, respectively (Fig. S3A). While the expected cleavage site was located in the middle of the 50 nt target oligonucleotide (Table S1), the resulting 3ʹ-fragment should have lower mobility in the gel due to the presence of the 3ʹ-Cy3 fluorophore. As expected, the lower mobility reaction product contained the Cy3 label, while the higher mobility band contained the 32P-label and therefore corresponded to the 5ʹ-target fragment (Fig. S3A).
We further tested whether the efficiency of endonuclease cleavage by SeAgo depends on the structure of the 5ʹ-end of guide DNA. It was found that SeAgo can cleave the DNA target with similar efficiencies when loaded with guides containing four different 5ʹ-nucleotides (Fig. S2C; Table S1). Interestingly, SeAgo could also cleave the ssDNA target using both 5ʹ-phosphorylated and 5ʹ-OH guide molecules. In this case, an additional cleavage site was detected one nucleotide closer to the 5ʹ-end of the target DNA (between the 11th and 12th guide positions), suggesting that interactions of SeAgo with the 5ʹ-guide phosphate may help to define the precise position of the target cleavage (Fig. S3B).
Mismatches between the guide and target nucleic acids can have adverse effects on the activity of both eukaryotic and prokaryotic Ago proteins [16,25–29]. To study the effect of mismatches on target processing, we loaded SeAgo with a series of DNA guides each containing a single mismatch at every position, and analysed cleavage of the same target DNA. We found that mismatches in the seed region (positions 2–8) did not inhibit and could even stimulate target cleavage by SeAgo, as observed for guides with nucleotide substitutions at the 5th and 6th positions (Fig. 1E, F). In contrast, target cleavage was strongly inhibited by mismatches in the 3ʹ-part of guide DNA relative to the site of cleavage (positions 12–15).
Recent analysis of TtAgo showed that its activity can be stimulated by the single-stranded DNA binding protein (SSB) [30], which might expose the ssDNA substrate in a more accessible conformation and facilitate its recognition. To test if the activity of SeAgo is increased in the presence of SSB, we expressed the SeSSB protein of S. elongatus and purified it from E. coli. The addition of SeSSB had no stimulatory effect on ssDNA cleavage by SeAgo (Fig. 1E).
In addition to the site-specific cleavage of the target DNA, we observed slow non-specific DNA degradation with our SeAgo preparations. The non-specific cleavage products could be seen as higher mobility bands below guide DNA and below the 5ʹ-cleavage product that appeared after prolonged incubation with SeAgo (Fig. 1D, Fig. S2A, C). This activity was comparable for Mn2+ and Mg2+-containing reactions (Fig. 1D), was independent of mutations in the active site of SeAgo (Fig. S2B), and was observed for SeAgo fractions after all three purification steps (Fig. S1C). The full-length target DNA and the 3ʹ-cleavage product that both contained the Cy3 fluorophore at their 3ʹ-ends were not degraded (Fig. 1D, Fig. S3A), and the shortened guide DNAs retained the 32P-label at their 5ʹ-end (Fig. S3B). Sequence analysis and structural modelling of SeAgo did not reveal any additional nuclease domains within the protein (Fig. 1A). Therefore, we conclude that this activity likely results from an admixture of a 3ʹ-exonuclease that could not be completely removed during the purification procedure. At the same time, it does not significantly affect the pattern of DNA cleavage by SeAgo, since no additional specific cleavage products were observed with the shorter guide DNA accumulated during the reaction (Fig. 1, Figs. S2 & S3).
Processing of double-stranded DNA by SeAgo
Previously studied pAgo proteins, including TtAgo, MjAgo, CbAgo and LrAgo, were shown to cleave double-stranded DNA substrates at specific sites defined by guide DNAs, or perform guide-independent ‘chopping’ of dsDNA, depending on the reaction conditions [13,15–18].
To study the ability of SeAgo to process dsDNA, we used two guide molecules targeting two different strands in a plasmid substrate, with the predicted cleavage sites separated by two nucleotides (Fig. 2A). In the absence of guides, SeAgo could slowly linearize the supercoiled plasmid (Fig. 2B, lanes 4–5). The linearization of the plasmid depended on the presence of the intact active site in SeAgo and was not observed in the case of the mutant SeAgo with substitutions of the catalytic tetrad residues (SeAgo CD; Fig. S1D). After prolonged incubation (120 min, lane 5) with empty SeAgo a light smear of shorter DNA fragments appeared, possibly as a result of guide-independent plasmid chopping. Surprisingly, the addition of either one or two guide molecules (Fig. 2B, lanes 6–11) had little effect on plasmid cleavage. Similar results were obtained with another plasmid, which was also processed by SeAgo independently of the addition of guide DNAs (Fig. S4B). Analysis of the presence of small DNAs in SeAgo preparations demonstrated that most nucleic acids are removed during the purification procedure (Fig. S1B, also see below), indicating that SeAgo primarily processes dsDNA substrates in a guide-independent manner.
Figure 2.
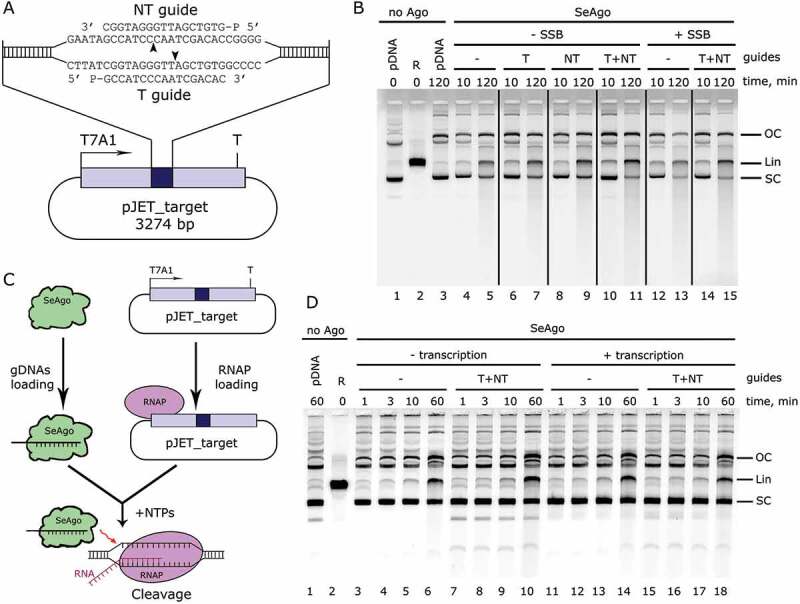
Plasmid DNA processing by SeAgo. (A) Scheme of the target plasmid (pJET_target). Positions of guide DNAs, promoter (T7A1) and terminator (T) regions are indicated. The predicted cleavage sites are shown with arrowheads. (B) Analysis of plasmid processing by nondenaturing agarose gel-electrophoresis. The plasmid was incubated with empty SeAgo or guide-loaded SeAgo in the absence or in the presence of SeSSB. (C) Scheme of an experiment aimed at the analysis of a possible role of transcription in DNA processing by SeAgo. (D) Analysis of the effect of transcription on plasmid processing. The reactions were performed either in the absence or in the presence of RNA polymerase and NTPs. SeAgo, guide DNA and plasmid DNA were present at 500 nM, 500 nM and 2 nM, respectively. T, guide DNA corresponding to the transcribed DNA strand in the target region; NT, guide DNA corresponding to the nontranscribed DNA strand; SC, supercoiled plasmid; OC, open-circle relaxed plasmid; Lin, linearized plasmid.
Guide-dependent dsDNA cleavage by SeAgo likely requires prior unwinding of two strands of DNA. Therefore, we tested whether the efficiency of dsDNA cleavage could be increased by factors that stimulate DNA unwinding: at increased temperature, in the presence of SSB or during ongoing transcription of the target region, which would transiently melt dsDNA. Similar patterns of plasmid DNA cleavage were observed at 37°C, 45°C, and 53°C (Fig. S4B). SeSSB also did not affect DNA processing, either in the absence or in the presence of guide DNAs (Fig. 2B, lanes 12–15). We then performed the cleavage reaction in the presence of RNA polymerase and NTPs, resulting in transcription initiation from a promoter located upstream of the plasmid region complementary to the guide DNA molecules (Fig. 2C). The efficiency of plasmid linearization by SeAgo was not affected by transcription, independently of the presence of guide DNAs (Fig. 2D, compare lanes 3–10 with 11–18). Therefore, transcription has little effect on dsDNA processing by SeAgo, at least under particular in vitro conditions.
Analysis of SeAgo expression in S. elongatus
To get insight into functions of SeAgo in its host cells, we generated a series of mutant strains of S. elongatus PCC 7942. In a control strain, we introduced a spectinomycin resistance (SpR) cassette into the NS1 region of the genome (‘empty’ strain, Fig. 3A). In the ΔAgo strain, the SeAgo gene was replaced with a luciferase gene (firefly luciferase) placed under control of the SeAgo promoter. In the ↑AgoWT strain, an additional copy of the SeAgo gene, containing an N-terminal His6-tag, was placed under control of the strong promoter of photosystem II (PSBA2), thus giving an increased level of SeAgo expression. In the ↑AgoCD strain, the catalytically dead variant of the SeAgo gene with substitutions of catalytic residues was placed in the same context.
Figure 3.

Expression of SeAgo in S. elongatus. (A) Schematics of the S. elongatus strains used for in vivo analysis. Positions of the chromosomal pAgo gene (replaced with luciferase in the ΔAgo strain), spectinomycin resistance gene, His6-tagged wild-type or catalytically inactive SeAgo, and corresponding promoter regions are indicated. (B) Comparison of the growth curves for the wild-type and ΔAgo (luciferase) strains, with an indication of time points used for the analysis of SeAgo expression. (C) Measurements of the SeAgo expression in the wild-type strain of S. elongatus by RT-qPCR. The mRNA levels are normalized to the rnpB housekeeping gene and are shown relative to the expression level at the 4th day of cell growth. (D) Analysis of the activity of the luciferase gene placed under control of the SeAgo promoter in the ΔAgo strain. The luminescence levels were normalized by the amounts of total protein in the samples after cell lysis. (E) Comparison of the growth curves for various strains of S. elongatus analysed in this study.
We first studied SeAgo expression in the wild-type strain of S. elongatus, and also analysed the activity of the SeAgo promoter at different stages of cell growth (Fig. 3B). The levels of SeAgo mRNA were measured at 4, 6, 8 and 10 days of cell growth by real-time quantitative RT-PCR (Fig. 3C). The level of endogenous SeAgo expression was low relative to the control housekeeping gene rnpB, encoding for the catalytic RNA component of ribonuclease P (~1%, Fig. S5). At the same time, its expression was stable, with no more than two-fold changes over the measurement period (Fig. 3C and Fig. S5). In the case of the overexpression strain (↑AgoWT), the levels of SeAgo expression were increased about 200-fold in comparison with wild-type cells, and became comparable to the reference housekeeping gene; these levels were also stable during the measurements (Fig. S5). Furthermore, we analysed the activity of the luciferase reporter inserted in the SeAgo genomic locus and expressed under control of the SeAgo promoter (in the ΔAgo strain). The levels of luciferase expression did not change more than two-three fold during the analysed time period (Fig. 3D). Thus, we conclude that the SeAgo promoter is active in the S. elongatus cells and SeAgo is steadily expressed during cell growth, although at a low level.
To reveal the possible effects of changes in the SeAgo expression on cell growth, we compared the growth kinetics for various strains of S. elongatus. All the strains were cultivated for a month under identical conditions with constant illumination, as described in Materials and Methods. Analysis of the optical densities did not reveal strong differences in the growth rates between different strains over this period (Fig. 3E), suggesting that neither deletion nor increased expression of wild-type or mutant SeAgo significantly affected cell physiology.
Analysis of SeAgo-bound small DNA guides
To study whether SeAgo associates with guide molecules in bacterial cells, we analysed nucleic acids co-purified with SeAgo expressed in native (S. elongatus) or heterologous (E. coli) cells (Fig. 4A). One-step purification of His6-tagged chromosomally encoded SeAgo from S. elongatus showed that it was associated with 14–20 nt molecules that were sensitive to DNase and resistant to RNase treatment (Fig. 4A, left panel, lanes 1–3). SeAgo-bound nucleic acids could not be efficiently labelled with polynucleotide kinase without prior treatment with phosphatase, resulting in the removal of pre-existing 5ʹ-phosphate (compare lanes 1 and 4). Thus, in S. elongatus cells SeAgo is bound to 5ʹ-phosphorylated small DNAs.
Figure 4.

Analysis of small DNAs associated with SeAgo in vivo. (A) Isolation of small nucleic acids bound to SeAgo during its expression in S. elongatus (left) or E. coli (right). The experiments were performed with His6-tagged chromosomally encoded SeAgo in S. elongatus (↑AgoWT strain) or plasmid-encoded SeAgo in E. coli. After cell lysis, SeAgo was purified by Co2+-affinity chromatography, associated nucleic acids were (or were not) treated with alkaline phosphatase, labelled using γ-32P-ATP and polynucleotide kinase (PNK, radioactive labelling is indicated with asterisks), and treated with DNase I or RNaseA, as indicated on the figure, followed by denaturing PAGE. DNA markers are shown on the right of each gel. (B) Length distribution of SeAgo-associated small DNAs in sequenced libraries. (C) Sequence logos for small DNAs aligned by their 5ʹ-ends; only reads ≥16 nt were used for this analysis. (D) Distribution of small DNAs along with the endogenous pANL plasmid. The small DNA coverage is plotted as rpkm (reads per kilobase per million reads in the small DNA library; open orange circles) over the plasmid coordinate. The blue line shows local polynomial regression fitting (LOESS smoothing). The predicted origin of replication is shown with a two-way red arrow [31]. (E) Distribution of small DNAs along the S. elongatus genome. Genomic positions are indicated on the x-axis. The rpkm values were averaged over 5 kb windows. The three major peaks of small DNAs and predicted positions of the replication origin and termination regions are indicated.
SeAgo was also associated with 5ʹ-phosphorylated small DNAs when expressed in heterologous E. coli; however, in this case, they had a broader length distribution and included 14–24 nt long species (Fig. 4A, right panel, lanes 6–9; also see Fig. S1B). Importantly, no small DNAs were found in association with the catalytically dead version of SeAgo, demonstrating that its catalytic activity is essential for small DNA biogenesis (lane 11). At the same time, the different length distributions of small DNAs in S. elongatus and E. coli suggest that additional cellular nucleases may participate in their processing, in particular, resulting in their shortening in S. elongatus.
To get insight into the sequence composition of guide DNAs associated with SeAgo in S. elongatus, we performed high-throughput sequencing of small DNAs co-purified with SeAgo (in four independent biological replicates, see Materials and Methods for details). Analysis of the length distribution of the resulting DNA sequences revealed that the major fraction is 17–18 nt long, corresponding to the lengths of starting small DNAs associated with SeAgo (Fig. 4B). SeAgo-bound small DNAs have very diverse sequences with no significant nucleotide bias at any position, except for a slight enrichment for guanine at the first position (Fig. 4C).
Since Ago proteins can be loaded with nucleic acid guides in solution, it was possible that some small DNAs co-purified with SeAgo might have been formed and associated with it during cell lysis and protein purification, thus distorting the profile of genuine guides bound to SeAgo in the cells. To test this possibility, we mixed the culture of S. elongatus with a distant bacterial species, Rhodobacter sphaeroides, prior to cell lysis followed by purification and sequencing of SeAgo-bound nucleic acids. Mapping of the small DNA reads to the S. elongatus and R. sphaeroides genomes revealed that the overwhelming majority of sequences corresponded to the genome of S. elongatus, with only ~1-2% mapping to the heterologous genome. The average GC-content of small DNAs mapped to each genome corresponded to the average GC-content of the respective genomic DNA (56.5% vs. 51.3% for small DNAs and genomic DNA from S. elongatus, and 67.3% vs. 68.5% for R. sphaeroides). Furthermore, 11% of all reads corresponding to the S. elongatus genome had a GC content of ≥69% (which is the average for R. sphaeroides), demonstrating that SeAgo can select guide DNAs independently of their GC-composition. Yet, the results demonstrate that the majority of small DNAs that are co-purified with SeAgo associate with it in the cells of S. elongatus prior to cell lysis.
Targeting of plasmid and chromosomal DNA by SeAgo
Previous studies suggested that pAgo proteins can preferentially target plasmid DNA (see Introduction). S. elongatus cells contain the main 2.695 Mb circular chromosome and the endogenous 46,366 bp plasmid pANL, present in ~3 copies per chromosome [32]. Analysis of the SeAgo-bound small DNAs demonstrated that 3.0 ± 1.5% of all reads mapped to the pANL plasmid, and 97% to the chromosome (mean and standard deviation from four replicates). These numbers roughly correspond to the relative lengths of the plasmid and chromosomal DNA after correction for the copy number. Therefore, SeAgo has no preference for plasmid DNA and randomly samples the endogenous plasmid and the main chromosome to generate guide DNAs. Mapping of SeAgo-bound small DNAs to the pANL sequence revealed their uneven distribution along the plasmid, with about 1.5-fold variations in the rpkm values (number of reads per kilobase per million reads in the library) (Fig. 4D). These variations do not correlate with the positioning of the plasmid replication origin, and their causes remain to be established.
To reveal whether SeAgo could target foreign plasmids in the heterologous E. coli system, we analysed plasmid maintenance in E. coli co-transformed with an SeAgo-expressing plasmid (or a corresponding control plasmid without SeAgo) and another plasmid, containing different antibiotic resistance markers (Fig. S6A). Cultivation of the E. coli cells without the antibiotic corresponding to the second plasmid resulted in its gradual loss during several passages. At the same time, the efficiency of plasmid loss was the same independently of SeAgo expression, suggesting that it cannot efficiently fight plasmids in E. coli (Fig. S6).
Mapping of SeAgo-bound small DNAs to the main chromosome showed that they are generated from all genomic regions, with the majority of regions having the density of small DNAs in the range of 325–375 rpkm (Fig. 4E). At the same time, three broad peaks with increased read density (up to 700 rpkm) were detected, likely corresponding to preferable sites of small DNA processing (Fig. 4E). The first peak coincides with the origin of replication in the S. elongatus chromosome [33]. The second peak is located at the opposite side of the chromosome and may correspond to the proposed region of replication termination [34]. While the origin of the third smaller peak remains unknown, we speculate that it may indicate another site of DNA processing (see Discussion). Overall, it can be concluded that SeAgo randomly samples DNA replicons present in the cell to generate guides, with some preference for the regions of replication origin and termination in the chromosome.
Analysis of SeAgo-associated proteins
Cellular partners, if any, of pAgo proteins remain unknown. To identify binding partners of SeAgo, we performed single-step purification of His-tagged SeAgo from the ↑AgoWT strain of S. elongatus by Co2+-chromatography followed by a mass-spectrometry analysis of associated proteins (see Materials and Methods for details). The empty strain without His-tagged SeAgo was used as a control. Among top 15 proteins identified by mass-spectrometry (Table S2, sorted in decreasing number of total independent spectra) the first two were single-stranded binding proteins of S. elongatus, the canonical SeSSB (Synpcc7942_0301) and its distant homolog (Synpcc7942_0079). Since no DNase treatment was performed during SeAgo purification, it cannot be excluded that the observed interactions with SSB proteins are mediated by single-stranded DNA fragments bound by SeAgo. No strong hits were observed in control samples isolated from S. elongatus cells without expression of His6-tagged SeAgo. Thus, SSB proteins may be involved in genomic DNA sampling by SeAgo (see Discussion). Other identified proteins in the top list do not include any known factors potentially involved in DNA processing (Table S2).
Discussion
In vitro activities of SeAgo
In contrast to eukaryotic Agos that target RNA, prokaryotic members of this protein family can act on DNA targets, resulting in their processing in either guide-dependent or guide-independent ways. This raises intriguing questions about their functions in prokaryotic cells, which contain naked DNA potentially sensitive to their action [3,4,35,36]. The ability to program pAgos with small DNA or RNA guides also makes them a potential tool for genome editing applications [15,16,37]. However, the majority of previously studied pAgos were isolated from thermophilic species and were poorly active at physiological temperatures in biochemical tests in vitro or during their expression in the heterologous E. coli system [9,10,12,14,17]. Recently, CbAgo and LrAgo from mesophilic bacteria were shown to be active at much lower temperatures; however, their in vivo activities have not been studied [15,16]. At the same time, not a single catalytically active mesophilic pAgo has been characterized in its native host bacterium. Here, we describe the in vitro properties and in vivo activities of the SeAgo protein from the photosynthetic cyanobacterium S. elongatus. We show that it acts as a programmable DNA-guided DNA nuclease in vitro and can also target genomic and plasmid DNA in vivo but without obviously affecting cell survival.
Analysis of the in vitro activities of SeAgo demonstrated that it is active as endonuclease only with DNA guides and DNA targets, and prefers manganese ions for cleavage. Manganese is an essential component of photosystem II and its concentration is high in cyanobacteria [38,39], suggesting that manganese ions may be naturally used as cofactors by SeAgo in vivo. The nucleic acid specificity of SeAgo is similar to other previously characterized pAgos from thermophilic and mesophilic prokaryotes, including TtAgo, PfAgo, MjAgo, CbAgo and LrAgo [9,12,15–17]. Two pAgos – mesophilic RsAgo and thermophilic MpAgo – were shown to use RNA guides for target recognition; however, only the latter protein contains an intact catalytic site and can perform DNA or RNA cleavage [7,22]. Thus, mesophilic pAgo proteins that could naturally use RNA guides and/or recognize RNA targets remain to be discovered.
Many Ago proteins show a preference for a specific nucleotide residue at the first position of the guide molecule and require the presence of a phosphate group at its 5ʹ-end [12,17,22,40–42]. In contrast, SeAgo can perform ssDNA cleavage in vitro using guide DNAs with all four nucleotides at the first position, similarly to TtAgo, PfAgo, CbAgo, and LrAgo [9,15–17]. In agreement with this, no significant nucleotide bias is observed for small guide DNAs associated with SeAgo in vivo. Similarly, to CbAgo and LrAgo [16], SeAgo can also utilize guide DNAs lacking the 5ʹ-phosphate, resulting in a one-nucleotide shift in the cut site position.
In the case of eukaryotic Agos, mismatches in the seed region of guide RNAs (positions 2–8) can dramatically change the efficiency of target recognition and cleavage [25,27,28,43]. In contrast, DNA-targeting pAgos – TtAgo, CbAgo, and LrAgo – are more strongly affected by mismatches in the 3ʹ-supplementary region of guide molecules, downstream of the cleavage site [16,30]. Similarly, the activity of SeAgo towards ssDNA targets is inhibited by mismatches between guide positions 12 and 15, but is stimulated by mismatches in the seed region. Therefore, these proteins form a distinct group of Ago nucleases, whose activity depends on correct base-paring within the 3ʹ-part of the guide nucleic acid.
We found that guide-free SeAgo can also target double-stranded circular plasmid DNA in vitro, resulting in its linearization and subsequent processing into a smear of shorter fragments. Such ‘chopping’ activity, previously observed for several other pAgos [13,16,18], may possibly lead to the generation of small guide molecules further loaded into the pAgo. This activity was not observed with the catalytically dead SeAgo mutant suggesting that it is initiated by SeAgo-dependent plasmid cleavage. Furthermore, the catalytically dead SeAgo lacks bound DNA guides when expressed in bacterial cells (Fig. 4) indicating that the endonuclease activity is required to generate guides in vivo. It cannot be excluded that the gradual degradation of plasmid DNA after its initial linearization by SeAgo can be assisted by other cellular nucleases in vivo (as well as by traces of exonucleases in the SeAgo preparations in vitro).
Several previously studied pAgo proteins, including TtAgo, CbAgo and LrAgo, were shown to cleave double-stranded DNA substrates at specific sites defined by guide DNAs, depending on the presence of AT-rich or partially melted regions in the target DNA [15–17]. In contrast, the chopping activity of SeAgo towards dsDNA substrates was not significantly affected by the presence of specific guide molecules, suggesting that guide-independent DNA processing may be the primary activity of SeAgo in the cell. Indeed, in S. elongatus SeAgo is loaded with small DNAs that map uniformly across the whole genome suggesting that it does not have strong site preferences.
Possible in vivo functions of SeAgo
pAgo proteins were proposed to function in cellular defence against invading DNA elements, such as plasmids, transposons, and phages [3–5,17,22,35]. In addition, it was speculated that pAgos might play roles in the regulation of gene expression, DNA processing and repair [4,22].
The in vivo activities of only two pAgo proteins, RsAgo and TtAgo, were previously analysed in detail [17,22]. RsAgo is a catalytically inactive mesophilic pAgo that uses small RNA guides derived from various mRNA sequences to target plasmid DNA and foreign DNA elements in the chromosome (including transposons and prophages). RsAgo can also inhibit the expression of plasmid-encoded genes even without plasmid degradation. TtAgo is a catalytically active thermophilic protein that has strong preferences for plasmid sequences when it is expressed in E. coli. Both RsAgo and TtAgo can reduce plasmid content and decrease plasmid transformation efficiency in their host bacteria and in heterologous E. coli, suggesting that plasmids are their natural targets in vivo [17,22].
In contrast, analysis of the genomic distribution of small DNAs associated with SeAgo revealed no preferences for endogenous plasmid DNA or specific chromosomal elements such as transposons, arguing against specific targeting of these genetic elements. Furthermore, SeAgo expression did not have measurable effects on plasmid maintenance in the heterologous E. coli system. The low level of endogenous expression of SeAgo in S. elongatus may prevent extensive degradation of genomic and plasmid DNA by its nuclease activity. At the same time, even a hundred-fold higher level of SeAgo expression in the engineered strain was not toxic for cell growth, suggesting that possible genomic DNA damage caused by SeAgo can be efficiently repaired. In the case of M. jannaschii, chromatin histone proteins were shown to protect genomic DNA from the action of MjAgo [13]; however, it is unknown whether any factors might have a similar protective role in S. elongatus. It also remains to be investigated whether SeAgo might specifically target invading DNA during cell transformation with exogenous plasmids or upon phage infections, and whether these events might activate SeAgo expression in S. elongatus.
The uniform distribution of SeAgo-bound small DNAs suggests that it is unlikely to be involved in specific regulation of gene expression. At the same time, SeAgo has a certain preference for the chromosomal regions involved in the initiation and termination of replication. The first peak of small DNAs associated with SeAgo corresponds to the ori site of the chromosome. The second peak of small DNAs is located exactly opposite the ori region in the middle of the chromosome (1.315 Mb, Fig. 4E). While the position of the ter site in S. elongatus remains unknown, analysis of the genomic GC-skew and marker segregation between daughter cells indicated that it is located between 0.75 and 1.75 Mb of chromosomal coordinates [34]. Furthermore, S. elongatus encodes the XerC recombinase (located on the endogenous plasmid) that is responsible for the resolution of chromosomal and plasmid dimers in many bacteria, and dif-sites recognized by this recombinase often define the position of replication termination [44]. Our search of dif-like sites in the S. elongatus chromosome did not reveal strong dif consensus but the closest match to the dif sequence was found at position 1.275 Mb (CTGCCAAATAATTGTGATTATTGGTGCG in comparison with the DBBBCSBATAATRTAYATTATGTHAANT consensus). Thus, the observed peak of small DNAs associated with SeAgo may identify the exact position of the termination site in the S. elongatus genome.
In addition, we detected a third lower peak of small DNAs bound to SeAgo between the ori and ter regions (0.715 Mb, Fig. 4D). The genome of S. elongatus has an unusual cumulative GC-skew plot with no single peak, suggesting its recombinogenic origin [34]. Interestingly, the third peak of small DNAs coincides with the beginning of a long region (0.75–1.75 Mb) with the highest GC skew values [34]. We therefore speculate that this peak may correspond to an additional site of replication termination or DNA recombination.
The preference of SeAgo to the sites of replication initiation and termination parallels recently discovered targeting of the chromosomal ter regions during spacer acquisition by type 1 and type 2 CRISPR-Cas systems in E. coli and Staphylococcus aureus [45,46]. It was suggested that this might reflect interactions of the spacer acquisition machinery with the products of double-strand DNA end degradation by cellular DNA recombination machineries at the sites of termination. Similarly, SeAgo might preferentially interact with DNA intermediates during replication initiation and termination and participate in their processing.
Analysis of SeAgo protein partners revealed its association with single-stranded DNA binding proteins. Recent analysis indicated that the activity of TtAgo in vitro can be stimulated by proteins involved in DNA processing, including heterologous SSB and the UvrD helicase [30]. The role of SSBs in the SeAgo function may be in the stabilization of transiently formed single-stranded DNA regions in the context of the double-stranded genome, even though SSB does not promote DNA processing by SeAgo in vitro. This may also indicate that small DNAs are preferably produced by SeAgo from single-stranded DNA regions during DNA replication. Thus, further experiments are required to understand possible functions of SeAgo and other pAgos in DNA metabolism and reveal their interplay with other DNA processing enzymes in their host cells.
Funding Statement
This work was supported by the Russian Science Foundation [16-14-10377];Russian Foundation for Basic Research [18-29-07086].
Acknowledgments
We thank Denis Yudin for helpful discussions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Ghildiyal M, Zamore PD.. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10:94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hutvagner G, Simard MJ.. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32. [DOI] [PubMed] [Google Scholar]
- [3].Swarts DC, Makarova K, Wang Y, et al. The evolutionary journey of Argonaute proteins. Nat Struct Mol Biol. 2014;21:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lisitskaya L, Aravin AA, Kulbachinskiy A. DNA interference and beyond: structure and functions of prokaryotic Argonaute proteins. Nat Commun. 2018;9:5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Makarova KS, Wolf YI, van der Oost J, et al. Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol Direct. 2009;4:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ryazansky S, Kulbachinskiy A, Aravin AA. The expanded universe of prokaryotic Argonaute proteins. mBio. 2018;9:e01935–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kaya E, Doxzen KW, Knoll KR, et al. A bacterial Argonaute with noncanonical guide RNA specificity. Proc Natl Acad Sci U S A. 2016;113:4057–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sheng G, Zhao H, Wang J, et al. Structure-based cleavage mechanism of Thermus thermophilus Argonaute DNA guide strand-mediated DNA target cleavage. Proc Natl Acad Sci U S A. 2014;111:652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Swarts DC, Hegge JW, Hinojo I, et al. Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 2015;43:5120–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang Y, Juranek S, Li H, et al. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature. 2008;456:921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang Y, Juranek S, Li H, et al. Nucleation, propagation and cleavage of target RNAs in Ago silencing complexes. Nature. 2009;461:754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Willkomm S, Oellig CA, Zander A, et al. Structural and mechanistic insights into an archaeal DNA-guided Argonaute protein. Nat Microbiol. 2017;2:17035. [DOI] [PubMed] [Google Scholar]
- [13].Zander A, Willkomm S, Ofer S, et al. Guide-independent DNA cleavage by archaeal Argonaute from Methanocaldococcus jannaschii. Nat Microbiol. 2017;2:17034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yuan YR, Pei Y, Ma JB, et al. Crystal structure of A. aeolicus argonaute, a site-specific DNA-guided endoribonuclease, provides insights into RISC-mediated mRNA cleavage. Mol Cell. 2005;19:405–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hegge JW, Swarts DC, Chandradoss SD, et al. DNA-guided DNA cleavage at moderate temperatures by Clostridium butyricum Argonaute. Nucleic Acids Res. 2019;47:5809–5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kuzmenko A, Yudin D, Ryazansky S, et al. Programmable DNA cleavage by Ago nucleases from mesophilic bacteria Clostridium butyricum and Limnothrix rosea. Nucleic Acids Res. 2019;47:5822–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Swarts DC, Jore MM, Westra ER, et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature. 2014;507:258–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Swarts DC, Szczepaniak M, Sheng G, et al. Autonomous generation and loading of DNA guides by bacterial Argonaute. Mol Cell. 2017;65:985–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rippka R, Deruelles J, Waterbury JB, et al. Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J Gen Microbiol. 1979;111:1–61. [Google Scholar]
- [21].de Wet JR, Wood KV, DeLuca M, et al. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol. 1987;7:725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Olovnikov I, Chan K, Sachidanandam R, et al. Bacterial argonaute samples the transcriptome to identify foreign DNA. Mol Cell. 2013;51:594–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016. [Google Scholar]
- [24].MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bartel DP. Metazoan MicroRNAs. Cell. 2018;173:20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu Y, Esyunina D, Olovnikov I, et al. Accommodation of helical imperfections in Rhodobacter sphaeroides Argonaute ternary complexes with guide RNA and target DNA. Cell Rep. 2018;24:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Salomon WE, Jolly SM, Moore MJ, et al. Single-molecule imaging reveals that argonaute reshapes the binding properties of its nucleic acid guides. Cell. 2015;162:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wee LM, Flores-Jasso CF, Salomon WE, et al. Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell. 2012;151:1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lapinaite A, Doudna JA, Cate JHD. Programmable RNA recognition using a CRISPR-associated Argonaute. Proc Natl Acad Sci U S A. 2018;115:3368–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hunt EA, Evans TC Jr., Tanner NA. Single-stranded binding proteins and helicase enhance the activity of prokaryotic argonautes in vitro. PloS One. 2018;13:e0203073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen Y, Holtman CK, Magnuson RD, et al. The complete sequence and functional analysis of pANL, the large plasmid of the unicellular freshwater cyanobacterium Synechococcus elongatus PCC 7942. Plasmid. 2008;59:176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen Y, Taton A, Go M, et al. Self-replicating shuttle vectors based on pANS, a small endogenous plasmid of the unicellular cyanobacterium Synechococcus elongatus PCC 7942. Microbiology. 2016;162:2029–2041. [DOI] [PubMed] [Google Scholar]
- [33].Watanabe S, Ohbayashi R, Shiwa Y, et al. Light-dependent and asynchronous replication of cyanobacterial multi-copy chromosomes. Mol Microbiol. 2012;83:856–865. [DOI] [PubMed] [Google Scholar]
- [34].Chen AH, Afonso B, Silver PA, et al. Spatial and temporal organization of chromosome duplication and segregation in the cyanobacterium Synechococcus elongatus PCC 7942. PloS One. 2012;7:e47837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Koonin EV. Evolution of RNA- and DNA-guided antivirus defense systems in prokaryotes and eukaryotes: common ancestry vs convergence. Biol Direct. 2017;12:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Willkomm S, Makarova K, Grohmann D. DNA-silencing by prokaryotic Argonaute proteins adds a new layer of defence against invading nucleic acids. FEMS Microbiol Rev. 2018;42:376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hegge JW, Swarts DC, van der Oost J. Prokaryotic Argonaute proteins: novel genome-editing tools? Nature Rev Microbiol. 2017;16:5–11. [DOI] [PubMed] [Google Scholar]
- [38].Eisenhut M. Manganese homeostasis in cyanobacteria. Plants. 2019;9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Keren N, Kidd MJ, Penner-Hahn JE, et al. A light-dependent mechanism for massive accumulation of manganese in the photosynthetic bacterium Synechocystis sp. PCC 6803. Biochemistry. 2002;41:15085–15092. [DOI] [PubMed] [Google Scholar]
- [40].Elkayam E, Kuhn CD, Tocilj A, et al. The structure of human argonaute-2 in complex with miR-20a. Cell. 2012;150:100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nakanishi K, Weinberg DE, Bartel DP, et al. Structure of yeast Argonaute with guide RNA. Nature. 2012;486:368–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Matsumoto N, Nishimasu H, Sakakibara K, et al. Crystal structure of silkworm PIWI-clade argonaute siwi bound to piRNA. Cell. 2016;167:484–497. [DOI] [PubMed] [Google Scholar]
- [43].Becker WR, Ober-Reynolds B, Jouravleva K, et al. High-throughput analysis reveals rules for target RNA binding and cleavage by AGO2. Mol Cell. 2019;75:741–755.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cassier-Chauvat C, Veaudor T, Chauvat F. Comparative genomics of DNA recombination and repair in cyanobacteria: biotechnological implications. Front Microbiol. 2016;7:1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Levy A, Goren MG, Yosef I, et al. CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature. 2015;520:505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Modell JW, Jiang W, Marraffini LA. CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature. 2017;544:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.