

Abstract
Hepatocellular carcinoma (HCC) is the most common type of liver cancer and has limited treatment options. Snail family transcriptional repressor 1 (SNAI1) is a master regulator of epithelial–mesenchymal transition (EMT) and has been implicated in HCC initiation and progression. However, the precise role of SNAI1 and the way it contributes to hepatocarcinogenesis have not been investigated in depth, especially in vivo. Here, we analyzed the functional relevance of SNAI1 in promoting hepatocarcinogenesis in the context of the AKT/c-Met–driven mouse liver tumor model (AKT/c-Met/SNAI1). Overexpression of SNAI1 did not accelerate AKT/c-Met–induced HCC development or induce metastasis in mice. Elevated SNAI1 expression rather led to the formation of cholangiocellular (CCA) lesions in the mouse liver, a phenotype that was paralleled by increased activation of Yap and Notch. Ablation of Yap strongly inhibited AKT/c-Met/SNAI-induced HCC and CCA development, whereas inhibition of the Notch pathway specifically blocked the CCA-like phenotype in mice. Intriguingly, overexpression of SNAI1 failed to induce EMT, indicated by strong E-cadherin expression and lack of vimentin expression by AKT/c-Met/SNAI tumor cells. SNAI1 mRNA levels strongly correlated with the expression of CCA markers, including SOX9, CK19, and EPCAM, but not with EMT markers such as E-CADHERIN and ZO-1, in human HCC samples. Overall, our findings suggest SNAI1 regulates the CCA-like phenotype in hepatocarcinogenesis via regulation of Yap and Notch.
Significance:
These findings report a new function of SNAI1 to promote cholangiocellular transdifferentiation instead of epithelial–mesenchymal transition in hepatocellular carcinoma.
Introduction
Liver cancer is one of the most common tumors and ranks second as a cause of cancer mortality in the world (1, 2). Hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA) are the two major histotypes of liver cancer. The diagnosis for most HCC and CCA patients is achieved in the advanced stage of these tumors, when only very limited treatment options are available (3, 4). Better understanding of the mechanisms underlying HCC and CCA molecular pathogenesis is obviously of high importance for the development of novel drugs able to treat efficiently these deadly malignancies.
Recently, several studies have demonstrated the plasticity of liver cells. Specifically, mounting evidence indicates that mature hepatocytes can transform into cholangiocytes and vice versa (5-7). Besides the stem cell and cholangiocyte compartments, intrahepatic CCA can derive from adult hepatocytes in mice (8, 9). Studies in human HCC also identified a subset of HCC known as cholangiocellular (CC) like HCC, characterized by the expression of biliary markers such as CK19 and EPCAM (10,11). In addition, mixed/combined HCC and CCA represent a liver cancer entity in humans. Specifically, these mixed tumors have a prognosis similar to CCA and worse than HCC. A recent genomic profile analysis indicated that mixed HCC/CCA possess molecular features similar to HCC, even in the CCA component (12). The precise mechanisms underlying this conversion remain poorly understood. Recent studies suggest that the Hippo effector Yap and its downstream target Notch are major regulators of cell fate in the liver (13). In mice, overexpression of the Hippo kinase Lats2 prevents CC-like lesions formation in mouse liver tumor models induced by activated AKT and N-Ras oncogenes (14). Furthermore, activated forms of Yap cooperate with AKT to promote CCA development in mice (15-17). Notch signaling has been identified as a major cascade downstream of Yap. Specifically, Yap directly induces the expression of the Notch2 and Jag1 genes (13, 18), and ablation of Notch2 completely prevents Yap-dependent CC formation in vivo (19). Overall, these studies suggest that, during hepatocarcinogenesis, the Yap/Notch signaling cascade may promote a CC-like phenotype in the liver.
Epithelial-to-mesenchymal transition (EMT) is a cellular process where epithelial cells lose their cell polarity and transform into mesenchymal-like cells. In tumors, EMT is frequently observed and associated with increased tumor cell proliferation, invasion, and metastasis (20). EMT is recognized by the gain of expression of mesenchymal marker, such as vimentin and N-cadherin, as well as loss of epithelial markers, including E-cadherin, ZO-1 (TJP1), and occludin, in tumor cells. It is well established that Snail family transcriptional repressor 1 (SNAI1) is a master regulator of EMT during tumor progression (21). It induces EMT via binding to the three E-boxes of the CDH1 (E-cadherin) promoter region, leading to the suppression of CDH1 expression (22, 23). Multiple studies have shown that SNAI1 activation is able to induce EMT in vitro (22, 24-26). However, whether overexpression of SNAI1 promotes EMT and metastasis in HCC in vivo remains undefined.
Recently, we developed a clinically relevant murine HCC model via stable overexpression of an activated form of AKT (myr-AKT) together with the c-Met protooncogene in mouse hepatocytes via sleeping beauty mediated somatic integration and hydrodynamic injection (27, 28). In the current study, we investigated whether overexpression of SNAI1 suffices to promote EMT and metastasis in the context of AKT/c-Met–driven hepatocarcinogenesis. We found that concomitant expression of SNAI1 and AKT/c-Met (AKT/c-Met/SNAI1) neither resulted in EMT in mouse HCC, nor led to distant metastases. Overexpression of SNAI1 was able instead to promote CC-like lesion formation in AKT/c-Met mice.
Materials and Methods
Constructs and reagents
Plasmids, including pT3-EF1α, pT3-EF1α-HA-myr-AKT (mouse), pT3-EF1α-c-Met (human), pT3-EF1α-c-Myc (human), pT3-EF1α-dnRBPJ (human), and pCMV/sleeping beauty (SB) transposase, were used as previously described (15, 19, 28, 29). pT2/C-Luc//PGK-SB13 (cat. #20207) and Flag-SNAI1 (human; cat. #16218) were obtained from Addgene. They were used as the template to generate pT3-EF1α-Luc (Luc), pT3-EF1α-Flag-SNAI1 (SNAI1), andpT3-EF1α-SNAI1-V5 (SNAI1-V5) constructs via the Gateway cloning strategy. All plasmids were purified utilizing the Endotoxin-Free Maxiprep kit for in vivo studies (Sigma- Aldrich). SNAI1 with C-terminal HA-tag was also cloned into pLenti-puro vector for in vitro studies. EGFP/pLenti-puro was obtained from Addgene (cat. #26431) and used as control. d-Luciferin, potassium salt (cat. #L2916) was obtained from Life Technologies Corporation.
Hydrodynamic tail-vein injection
FVB/N mice were purchased from The Jackson Laboratory. Yapflox/flox mice were kindly provided by Dr. Eric Olson of the University of Texas Southwestern Medical Center (Dallas, TX). Six-week-old FVB/N mice were injected with SNAI1, AKT/pT3, AKT/SNAI1, AKT/c-Met/pT3, AKT/c-Met/SNAI1, AKT/c-Met/pT3/Luc, AKT/c-Met/SNAI1/Luc, AKT/c-Met/SNAI1/pT3, AKT/c-Met/SNAI1/dnRBPJ, c-Myc/pT3, or c-Myc/SNAI1-V5 construct. Six-week-old Yapflox/flox mice were injected with AKT/c-Met/SNAI1/pCMV or AKT/c-Met/SNAI1/Cre. The hydrodynamic injection procedure was performed as previously described (27). Detailed plasmid mixture information used in the mouse studies is listed in Supplementary Table S1. Mice were monitored three times a week for liver tumor development and euthanized when they developed large abdominal masses. All animal studies were performed according to protocols approved by the Committee for Animal Research at the University of California San Francisco (San Francisco, CA).
Human tissue samples
A collection of 73 frozen HCC and corresponding nontumorous surrounding livers was used. Tumors were divided in HCC with shorter/poorer (HCCP; n = 32) and longer/better (HCCB; n = 41) survival, characterized by <3 and ≥3 years' survival following partial liver resection, respectively. The clinicopathologic features of liver cancer patients are summarized in Supplementary Table S2. HCC specimens were collected at the Medical Universities of Greifswald (Greifswald, Germany) and Sassari (Sassari, Italy). Institutional Review Board approval was obtained at the local Ethical Committee of the Medical Universities of Greifswald and Sassari. Informed written consent was obtained from all individuals.
Additional information for material and methods can be found in Supplementary File. Primary and secondary antibodies are listed in Supplementary Tables S3 and S4. Primer sequences are listed in Supplementary Table S5.
Results
Overexpression of SNAI1 neither accelerates AKT/c-Met–induced liver cancer development nor induces distant metastases in mice
To study the oncogenic potential of SNAI1, we overexpressed SNAI1 in the mouse liver via hydrodynamic transfection. We found that SNAI1 alone is unable to induce liver tumor formation in mice (Supplementary Fig. S1). The resulting liver tissues appeared to be completely normal, undistinguishable from uninjected liver from the wild-type mice (Supplementary Fig. S1). Thus, SNAI1 presumably contributes to tumor progression rather than to tumor onset in the liver.
Previously, we established a clinically relevant murine HCC model by hydrodynamic overexpression of AKT and c-Met protooncogenes (AKT/c-Met). AKT/c-Met induced well-to-moderately differentiated HCC in mice within 6 to 8 weeks after hydrodynamic injection without metastatic dissemination (28). As SNAI1 is a master regulator of EMT, we hypothesized that overexpression of SNAI1 might induce EMT and promote distant metastatization in the AKT/c-Met–driven HCC model. To test this hypothesis, we hydrodynamically coinjected SNAI1 (with Flag tag) together with myr-AKT (with HA-tag) and c-Met as well as the SB transposase into the mouse liver (AKT/c-Met/SNAI1). Additional mice were coinjected with AKT/c-Met together with pT3-EF1α (empty vector) as control (AKT/c-Met/pT3; Fig. 1A). Similar to what we have reported previously on the AKT/c-Met HCC model, AKT/c-Met/pT3 injected mice developed lethal burden of liver tumors around 8 weeks after injection. Coexpression of SNAI1 did not significantly accelerate or delay this process, and all AKT/c-Met/SNAI1 mice developed high tumor burden by 8 weeks after injection (Fig. 1B and C). The two cohorts of mice demonstrated similar liver tumor burden as measured by total liver weight or liver/body ratio (Fig. 1D). To search for possible metastatic lesions, we examined additional organs in the mice, including lung, kidney, intestine, and hilar lymph nodes. However, no metastases could be detected in AKT/c-Met/SNAI1 mice, by macroscopic examination or by histologic analysis of the various organs (Supplementary Fig. S2).
Figure 1.

SNAI1 neither accelerates nor delays tumor formation in AKT/c-Met-induced HCC in mice. A, Study design. B, Representative gross images of livers from mice injected with AKT/c-Met/pT3 or AKT/c-Met/SNAI1 plasmids. C, Survival curve. D, Liver weight and liver/body ratio of the mice. W.p.i., weeks post injection; N.S, not significant.
To facilitate the monitoring of metastatic lesions in vivo, we generated a luciferase containing pT3-EF1α vector (pT3-EF1α-Luc). We coinjected AKT/c-Met/pT3 and AKT/c-Met/SNAI1 with pT3-EF1α-Luc and monitored tumor development in mice using bioluminescence imaging (Supplementary Fig. S3). As expected, high luciferase signal was detected in the mouse liver, but no signal was revealed in any other body region/organ (Supplementary Fig. S3).
In summary, our results indicate that coexpression of SNAI1 with AKT/c-Met is accompanied neither by accelerated HCC development nor by the formation of distant metastases in mice.
SNAI1 overexpression promotes the formation of CCA-like lesions in the mouse liver
Next, we performed histologic analysis of liver tumor lesions from AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mice. Consistent with our previous report (28), AKT/c-Met/pT3 liver parenchyma was extensively occupied by HCC lesions, consisting of lipid-rich and lipid-poor nodules, the latter consisting of basophilic cells with high cytologic atypia (Fig. 2A). No cholangiocellular (CC) lesions were detected in AKT/c-Met/pT3 mice, in accordance with our earlier investigation (28). As expected, strong immunostaining for HA-tag for ectopically expressed AKT as well as Ki-67 for rapid tumor cell proliferation was detected in AKT/c-Met/pT3 tumor cells (Fig. 2A). The expression of biliary marker cytokeratin 19 (Ck19) was restricted to bile duct epithelia cells of the nontumor liver tissues (Fig. 2B). In contrast, in addition to some HCC lesions equivalent to those seen in AKT/c-Met/pT3 mice, AKT/c-Met/SNAI1 mouse livers displayed the occurrence of CC-like tumor lesions (Fig. 2A and B). All tumor cells in AKT/c-Met/SNAI1 mice, including HCC and CCA cells, were positive for HA-tag (for AKT), and demonstrated nuclear Flag tag staining (for SNAI1), supporting their origin by the ectopically injected oncogenes (Fig. 2A). When compared with AKT/c-Met/pT3 liver tumors, the expression levels of biliary epithelial cell (BEC) markers, including Sry-box containing gene 9 (Sox9) and Ck19, were significantly increased, whereas the expression of the hepatocyte-specific marker Hnf-4α was absent in CC-like lesions from AKT/c-Met/SNAI1 mice (Fig. 2B).
Figure 2.

Histology analysis of AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse livers. A, Representative images of hematoxylin and eosin (H&E), HA-tag, Flag tag, and Ki-67 staining in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. B, Representative images of CK19, Sox9, Hnf4α, and Yap staining in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. C, Representative images of EMT markers, including E-cadherin, vimentin, Sirius Red, S100a4, and α-Sma staining in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. D, Percentage of Ki-67–positive cells in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues and percentage of Ck19-positive area fraction in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mice liver tumor. Scale bars, 100 μm. N.S., not significant; ****, P < 0.0001.
Next, we performed detailed analysis to determine whether overexpression of SNAI1 promoted EMT. For this purpose, we performed IHC for the EMT markers E-cadherin and vimentin. All HCC lesions in AKT/c-Met/pT3 were positive for E-cadherin, whereas vimentin staining was limited to fibroblasts scattered within the HCC nodules (Fig. 2C). In AKT/c-Met/SNAI1 liver tissues, strong membrane staining of E-cadherin could also be found in both HCC and CCA lesions. Vimentin staining was, again, limited to the fibroblasts within the tumor nodules. Consistent with the observation that CCA is highly desmoplastic (30), there was a significant increased number of cancer-associated fibroblasts within CC-like lesions in AKT/c-Met/SNAI1 mice. These cells stained positive for vimentin, S100a4, and α-Sma (Fig. 2C). This observation was validated using Sirius Red Staining (Fig. 2C). Quantification of Ck19-positive areas showed that CC-like lesions occupied ~20% of liver tissues in AKT/c-Met/SNAI1 mice. All the tumor lesions from AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mice displayed similar, high levels of Ki-67 staining (Fig. 2A and D).
To further substantiate this finding, we performed double immunofluorescence on the liver tissues. Specifically, HA-tag and E-cadherin were costained in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 liver tissues. We found that all tumor cells demonstrated positiveness for HA (+) and E-cadherin (+) immunoreactivity in both cohorts of mice (Fig. 3A). Subsequently, we costained E-cadherin and vimentin in the same liver specimens. In both AKT/c-Met/pT3 and AKT/c-Met/SNAI1 liver tissues, E-cadherin and vimentin staining did not overlap (Fig. 3B). Next, we focused on the CC-like lesions in AKT/c-Met/SNAI1 tissues and the costaining of Ck19 with E-cadherin and Ck19 with vimentin was used. Again, we found that Ck19 (+) cells were E-cadherin (+), but vimentin (−) in AKT/c-Met/SNAI1 CC-like tumors (Fig. 3C and D).
Figure 3.

Expression of EMT markers, Ck19, and HA-tag levels in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. A, Representative immunofluorescence staining images of E-cadherin and HA-tag in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. B, Representative immunofluorescence staining images of E-cadherin and vimentin in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. C, Representative immunofluorescence staining images of E-cadherin and Ck19 in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. D, Representative immunofluorescence staining images of vimentin and Ck19 in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. Scale bars, 50 μm.
Finally, to investigate how SNAI1 promotes CCA formation in the mouse liver, we injected additional mice with AKT/c-Met/pT3 or AKT/c-Met/SNAI1 constructs and harvested the mice 2 weeks later. Histologic evaluation revealed the presence of liver steatosis, which is consistent with overexpression of AKT. Intriguingly, although Ck19(+) cells were only found in the bile ducts at the portal tract in AKT/c-Met/pT3 mouse livers, sporadic Ck19(+) cells could already be observed in AKT/c-Met/SNAI1 liver parenchyma away from the portal tract. All cells exhibited membranous E-cadherin staining and no fibrosis could be appreciated (Supplementary Fig. S4). These results indicate that SNAI1 most likely promotes the CC-like phenotype via cell-autonomous mechanisms.
In summary, our analysis demonstrates that overexpression of SNAI1 triggers the CC phenotype in AKT/c-Met mouse livers. However, overexpression of SNAI1 is unable to induce EMT in vivo.
Overexpression of SNAI1 induces Yap and Notch activation in mouse liver tumors
To unravel the molecular mechanisms whereby SNAI1 overexpression contributes to hepatocarcinogenesis, we analyzed the expression of major players in hepatocarcinogenesis in normal livers from wild-type mice and in liver tumors from AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mice using Western blot analysis. As expected, AKT and c-Met proteins were highly expressed in the two tumor cohorts when compared with normal liver, whereas Flag tag (part of the SNAI1 construct) could only be detected in AKT/c-Met/SNAI1 liver tumors (Fig. 4A). E-Cadherin expression was expressed in all normal liver and liver tumor tissues, with more pronounced levels occurring in AKT/c-Met/SNAI1 CC-like tumors (Fig. 4A). Expression of vimentin was higher in AKT/c-Met/pT3 liver tumors when compared with normal livers, and the highest levels were observed in AKT/c-Met/SNAI1 liver tumors (Fig. 4A). Increased expression of the BEC marker Sox9 was also detected in AKT/c-Met/SNAI1 liver lesions (Fig. 4A). Thus, the molecular results were all consistent with the histologic findings.
Figure 4.
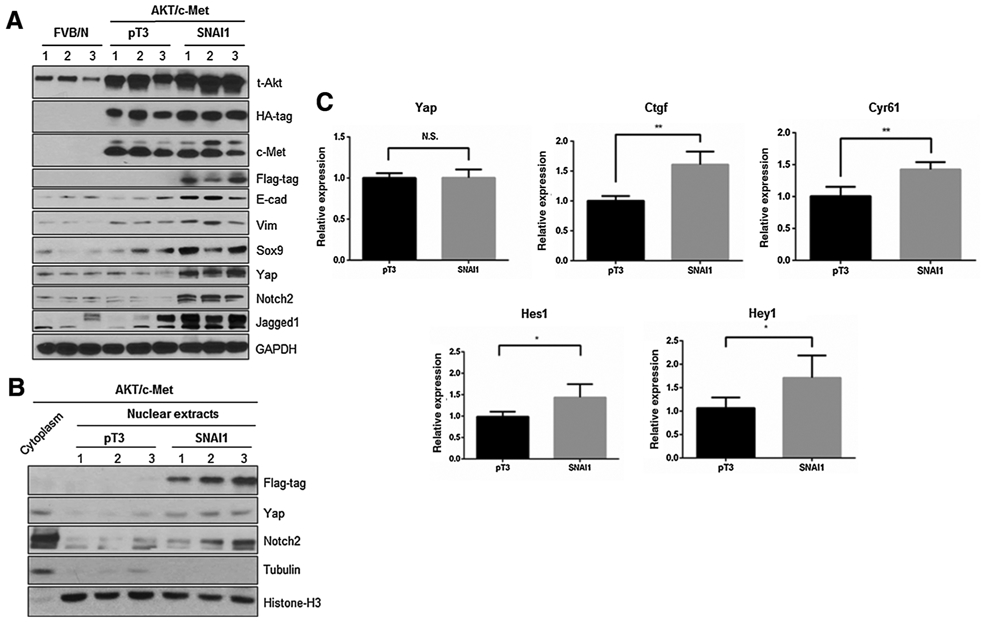
Yap signaling activity is increased in AKT/c-Met/SNAI1 mouse liver tissues. A, Western blot analysis of relative protein expression in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. B, Western blot analysis was performed in nuclear and cytoplasmic protein extracts from FVB/N normal mouse livers and AKT/c-Met/pT3, AKT/c-Met/SNAI1 mouse liver tumor samples. C, Detection of Yap and Notch target genes by qRT-PCR in AKT/c-Met/pT3 and AKT/c-Met/SNAI1 mouse liver tissues. *, P < 0.05; **, P < 0.01. N.S., not significant.
Previous studies from our group indicate that Yap is a major determinant factor for CCA development in the liver, with Notch2 and Jagged1 being the prominent signaling molecules downstream of Yap in CCA development and progression (15, 19). Thus, we examined the protein levels of Yap, Notch2, and Jagged1 in mouse liver tissues. Consistent with the increased CCA phenotype, Yap, Notch2, and Jagged1 protein levels were exclusively upregulated in AKT/c-Met/SNAI1 liver tumor tissues (Fig. 4A). Importantly, an augmented nuclear expression of Yap and Notch2 was detected in AKT/c-Met/SNAI1 liver tissues, implying an increased Yap and Notch activity (Figs. 2B and 4B). In addition, the mRNA levels of Yap target genes Ctgf and Cγr61, and Notch target genes Hes1 and Heγ1 were increased, whereas YAP mRNA level was not affected, upon SNAI1 overexpression (Fig. 4C).
Next, the findings were confirmed in HLE and SNU449 HCC cell lines, which were stably transfected with HA-tagged SNAI1 cDNA (Supplementary Fig. S5). In these cells, forced overexpression of SNAI1 increased HCC cell migration and invasion without affecting long-term cell growth (Supplementary Fig. S5). At the molecular level, transfection of SNAI1 triggered activation of the YAP pathway, as indicated by increased nuclear translocation of YAP protein and upregulation of YAP/NOTCH target genes (CYR61, CTGF, NOTCH2, JAG1, and SOX9), whereas levels of YAP mRNA remained unchanged (Supplementary Fig. S6).
Altogether, our data indicate that overexpression of SNAI1 promotes Yap and Notch activation in AKT/c-Met induced liver tumors.
Ablation of Yap impairs AKT/c-Met/SNAI1 HCC and CCA development in mice
Subsequently, we investigated the functional importance of Yap in AKT/c-Met/SNAI1-induced tumor using conditional Yapflox/flox mice. Specifically, we hydrodynamically injected AKT/c-Met/SNAI1 and Cre into Yapflox/flox mice (AKT/c-Met/SNAI1/Cre). This allows the deletion of Yap in the mouse hepatocytes that overexpress AKT/c-Met/SNAI1. Additional Yapflox/flox mice injected with AKT/c-Met/SNAI1 and pCMV empty vector (AKT/c-Met/SNAI1/pCMV) were used as controls (Fig. 5A). We found that Cre-dependent ablation of Yap strongly inhibited AKT/c-Met/SNAI1-induced liver tumor development in mice. Indeed, although all AKT/c-Met/SNAI1/pCMV Yapflox/flox mice exhibited high tumor burden, AKT/c-Met/SNAI1/Cre Yapflox/flox mice showed the absence of macroscopic appreciable tumor nodules, and displayed normal liver weight as well as normal liver/body weight ratio at 8 weeks after injection (Fig. 5B). Histologically, both CCA and HCC lesions could be detected in the liver parenchyma of AKT/c-Met/SNAI1/pCMV Yapflox/flox mice. In striking contrast, no tumor lesions were identified in AKT/c-Met/SNAI1/Cre mouse livers at this time point (Fig. 5C and D). Over long term, one AKT/c-Met/SNAI1/Cre Yapflox/flox mouse developed liver tumors (Supplementary Fig. S7). Histologically, HCC lesions were detected, and tumor cells were negative for Yap and Ck19, but positive for the proliferation marker Ki67 (Supplementary Fig. S7).
Figure 5.

Yap ablation inhibits AKT/c-Met/SNAI1-induced liver tumor formation. A, Study design. B, Survival curve. C, Liver weight and liver/body ratio of Yapflox/flox mice injected with AKT/c-Met/pT3 or AKT/c-Met/SNAI1. D, Representative gross and hematoxylin and eosin (H&E), CK19, and Ki-67 staining images of livers from Yapflox/flox mice injected with AKT/c-Met/SNAI1/pCMV or AKT/c-Met/SNAI1/Cre plasmids. Scale bar, 100 μm. ***, P < 0.01; ****, P < 0.0001. W.p.i., weeks post injection.
The importance of YAP as a downstream effector of SNAI1 was further confirmed in HLE and SNU449 cell lines stably transfected with SNAI1 (Supplementary Fig. S8). Treatment with super-TDU, a peptide blocking YAP-TEAD binding (31), led to decreased proliferation and increased apoptosis in SNAI1-over-expressing cell lines, whereas absent/lower antigrowth and anti-survival effects were detected in EGFP- (control) transfected cell lines (Supplementary Fig. S8). As expected, treatment with super-TDU reduced/abolished the levels of YAP target genes (CTGF and CYR61) in SNAI1-transfected cells, while not having an appreciable effect on control counterparts (Supplementary Fig. S9).
Overall, the present data indicate that ablation of Yap strongly impairs AKT/c-Met/SNAI1-induced HCC and CCA formation in vivo as well as tumor growth in vitro.
Blocking of the Notch cascade suppresses AKT/c-Met/SNAI1-induced CC-like lesion development
Next, we investigated the role of the canonical Notch cascade in AKT/c-Met/SNAI1-driven liver tumor development. For this purpose, we coexpressed the dominant-negative form of RBP-J (dnRBPJ) (19) with AKT/c-Met/SNAI1 plasmids into the mouse liver (AKT/c-Met/SNAI1/dnRBPJ). As control, AKT/c-Met/SNAI1 plasmids were coinjected with pT3 empty vector (AKT/c-Met/SNAI1/pT3; Fig. 6A). Mice from both cohorts needed to be harvested ~10 weeks after injection (Fig. 6B). Liver weight and liver/body ratio were similar in AKT/c-Met/SNAI1/pT3 and AKT/c-Met/SNAI1/dnRBPJ mice (Fig. 6C). Histologic evaluation revealed both HCC and CC-like lesions in AKT/c-Met/SNAI1/pT3 liver tissues. In striking contrast, only HCCs were found in AKT/c-Met/SNAI1/dnRBPJ mice (Fig. 6D). This observation was validated via Ck19 immunostaining (Fig. 6D and E). Tumor cells from both cohorts of mice were highly proliferative, with similar proliferation rates (Fig. 6D and E).
Figure 6.

Inhibition of the canonical Notch signaling suppresses AKT/c-Met/SNAI1-induced CC-like phonotype in mice. A, Study design. B, Survival curve. C, Liver weight and liver/body ratio of the mice. D, Representative gross images, hematoxylin and eosin (H&E), CK19, and Ki-67 staining images of livers from mice injected with AKT/c-Met/SNAI1/pT3 or AKT/c-Met/SNAI1/dnRBPJ plasmids. Scale bar, 100 μm. E, Percentage of Ki-67–positive cells in liver tissues and percentage of Ck19-positive area fraction in mice liver tumor injected with AKT/c-Met/SNAI1/pT3 or AKT/c-Met/SNAI1/dnRBPJ plasmids. ***, P < 0.001. N.S., not significant; W.p.i., weeks post injection.
The relevance of the NOTCH pathway in SNAI1-mediated oncogenesis was also evaluated in HLE and SNU449 cells (Supplementary Fig. S10). For this purpose, SAHM1, a peptide that prevents NOTCH transcriptional activity (32), was administered to EGFP (control)- and SNAI1-transfected cells. As expected, the highest decrease of proliferation and the most pronounced induction of apoptosis as well as downregulation of the NOTCH target gene HES1 was induced only in SNAI1-overexpressing cells (Supplementary Fig. S11).
In summary, our study implies that blocking of the canonical Notch signaling specifically abolishes SNAI1-induced CC development in vivo and reduces SNAI1-dependent growth in vitro.
Development of CC-like lesions by SNAI1 is oncogene dependent
To determine whether the induction of a CC-like phenotype is universally driven by SNAI1 overexpression in liver cancer, we tested this possibility in additional mouse models driven by other oncogenes. First, we used a well-established c-Myc mouse model, prone to develop HCC following overexpression of the c-Myc oncogene by hydrodynamic transfection (29). In this model, we found that concomitant overexpression of c-Myc and SNAI1 neither accelerated HCC growth, nor induced CC formation (Supplementary Fig. S12 and S13). Subsequently, as AKT/mTOR signaling has been implicated in CCA development (15), we investigated whether activation of AKT may prime the hepatocytes for SNAI1-induced CC-like phenotype development. Thus, we coexpressed myr-AKT with SNAI1 in the mouse liver. Although AKT overexpression alone only induced hepatic steatosis by 16 weeks after injection, AKT/SNAI1 coexpression triggered the formation of CC-like lesions (Supplementary Fig. S14), recapitulating the effects induced by SNAI1 in AKT/c-Met mice.
Overall, the present findings suggest that the potential of SNAI1 to induce the CC phenotype in HCC depends on the oncogenes driving the carcinogenic process.
SNAI1 positively correlates with markers of BECs and Yap targets in human HCC samples
Our investigation implies that overexpression of SNAI1 promotes CC-like phenotype in the mouse liver via activation of the YAP and Notch cascades. To determine whether the same phenotype applies to human HCC, we analyzed SNAI1 gene expression in relationship with EMT and CCA markers in human samples using The Cancer Genome Atlas (TCGA) data set (33). We found that SNAI1 expression positively correlates with CC markers, including SOX9, CK19, and EPCAM in the data set (Fig. 7A). No correlation between SNAI1 with YAP mRNA levels was observed. Nonetheless, SNAI1 expression positively correlated with the levels of YAP target genes (CTGF and CYR61) as well as Notch pathway members (NOTCH2 and JAG1) in the same human HCC samples (Fig. 7B). As concerns the EMT markers, SNAI1 mRNA positively correlated with the expression of mesenchymal markers, including VIMENTIN, α-SMA, and S100A4. However, surprisingly, a statistically significant positive correlation between SNAI1 and E-CADHERIN (CDH1) expression was found (Fig. 7C and D). There was also a weak positive correlation between SNAI1 and the epithelial marker ZO-1 (TJP1), whereas no correlation was found between SNAI1 and KRT18, OCCLUDIN or CLAUDIN.
Figure 7.

Analysis on the relationship of SNAI1 mRNA expression with EMT and CC markers in human HCC samples using TCGA data set. A, SNAI1 expression positively correlates with CK19, SOX9, and EPCAM mRNA levels. B, SNAI1 expression positively correlates with YAP target genes, but not with YAP mRNA levels. C, Correlation between SNAI1 and epithelium markers levels. D, Correlation between SNAI1 and mesenchymal markers levels. E, Genomic background of human HCC samples using the TCGA data set.
Next, we analyzed SNAI1 expression and its correlation with mutations commonly observed in human HCC samples from the TCGA data set (Fig. 7E; Supplementary Fig. S15). Intriguingly, we discovered that CTNNB1 and AXIN1 mutations were enriched in HCC samples with low SNAI1 expression. It has been reported that human HCCs with CTNNB1 mutations are well differentiated (34), with low genomic instability (35) and better prognosis (36). In contrast, the CC-like gene-expression signature has been linked to an aggressive HCC phenotype and poor prognosis (11). Therefore, the present data indicate that CTNNB1/AXIN1-mutant tumors and those with SNAI1-elevated expression and a CC-like phenotype represent two distinct subclasses of human HCC.
Finally, we validated some of the data extracted from the TCGA data set in our HCC collection (n = 73). We found that mRNA levels of SNAI1 were significantly higher in HCC when compared with nontumorous surrounding livers, with the most pronounced upregulation of SNAI1 occurring in human HCC with poorer prognosis (HCCP; Supplementary Fig. S16). When determining the relation between SNAI1 and patients' clinicopathologic data, we found that higher expression of the SNAI1 gene correlates with lower HCC survival rate (Supplementary Fig. S16). This association remained strongly significant after multivariate Cox regression analysis (P < 0.0001; Supplementary Information). Thus, SNAI1 mRNA expression is an independent prognostic factor for HCC. No relationship between the mRNA levels of SNAI1 and other clinicopathologic features of the patients, including age, gender, etiology, presence of cirrhosis, tumor size, and tumor grade, was detected (Supplementary Information). Similar to the TCGA data set, a significant, direct correlation was detected between expression levels of SNAI1 and those of CC markers SOX9, CK19, and EPCAM. In addition, SNAI1 expression correlated with that of YAP (CYR61, CTGF) and Notch (JAG1) targets, whereas no correlation was observed with YAP levels. We also found a positive correlation between SNAI1 and VIMENTIN (VIM), E-CADHERIN (CDH1), and ZO-1 (TJP1) levels, thus closely recapitulating the TCGA findings (Supplementary Fig. S17; Supplementary Information).
It is important to underline that, because mesenchymal markers could be detected in both EMT cells and cancer-associated fibroblasts, one cannot determine whether the correlation between SNAI1 expression and these mesenchymal markers is due to EMT or increased numbers of cancer-associated fibroblasts. In contrast, epithelial makers such as E-CADHERIN and ZO-1 are expressed only in the tumor cells, and loss of these markers is a more reliable marker indicating EMT.
Altogether, the present findings indicate that SNAI1 levels correlate with a CC-like gene expression, but not an EMT signature, in human HCC.
Discussion
Previous evidence supports the role of SNAI1 as a master regulator of EMT during tumor progression, an event that leads to increased tumor metastatization (21, 37). However, most of the studies implying SNAI1 in the promotion of EMT were conducted in vitro using cell lines. More recent investigations using genetically engineered mouse models, including transgenic mice and conditional knockout mice, have challenged this prevailing hypothesis (38). Indeed, in vivo studies have shown that SNAI1 and other EMT inducing transcription factors have other major roles independent of EMT, including the regulation of cell-fate specification and transition, stem cell plasticity, and resistance to therapies (38).
SNAI1 has been implicated in EMT and metastasis in liver cancer, including both HCC and CCA (26, 39). However, the precise mechanisms played by SNAI1 in these processes have not been well characterized in vivo. In the current article, we tested the hypothesis that overexpression of SNAI1 promotes EMT and distant metastases of HCC in mice. Unexpectedly, we found that concomitant expression of SNAI1 with AKT/c-Met oncogenes neither accelerated liver tumor development nor promoted tumor metastases. In accordance with our data, some studies suggested that SNAI1 and related EMT may not be required for metastasis (40, 41). However, it is important to underline that liver tumors developed rapidly in AKT/c-Met/SNAI1 mice, and all mice were required to be euthanized by 8 weeks after injection due to high liver tumor burden based on the IACUC protocol. Thus, we cannot exclude that tumor cells are eventually able to metastasize if additional survival time would be available for the mice. Additional studies using slow-growing liver tumor models are required to further clarify this important issue.
A major discovery of our study was the finding that overexpression of SNAI1 promotes the development of a CC-like phenotype in AKT/c-Met mouse lesions. The association between SNAI1 and CC markers in human HCC samples was further validated using the TCGA data set and our HCC sample collection. In accordance with our findings, it has recently been reported that HCCs with elevated CK19 levels also exhibit high SNAI1 expression (42). In addition, it has been shown that overexpression of SNAI1 cooperates with activated TAZ to induce liver tumors with mixed HCC and CC features in mice (43). Thus, these previous results provide independent validation of our observations. However, it is important to underline that the induction of a CC-like phenotype in the liver might be context/oncogene dependent. Indeed, we found in this study that SNAI1 overexpression drives CC conversion in AKT mouse lesions but not in c-Myc mice.
Mechanistically, we discovered that the increased CC development in AKT/c-Met/SNAI1 mice was accompanied by an increased activation of Yap and Notch in the AKT/c-Met/SNAI1 mouse liver tumor tissues. Using genetic approaches, we demonstrated that Yap is required for AKT/c-Met/SNAI1-induced HCC and CC development, whereas Notch cascade is specifically required for CC formation in vivo. These observations are consistent with the reports that Notch is a major target of Yap, and Yap is likely to function via Notch and additional signaling pathways (13, 14, 18). Therefore, ablation of Yap is likely to have more profound effects in liver tumor development than only blocking the Notch cascade.
As SNAI1 is a transcription factor, it could be envisaged that SNAI1 may directly induce YAP gene expression. However, our data indicate that overexpression of SNAI1 does not lead to increased Yap mRNA expression in AKT/c-Met/SNAI1 liver tumor tissues, but rather to nuclear accumulation of the YAP protein and increased expression of its downstream effectors. Using human HCC samples, we confirmed a strong positive correlation between SNAI1 mRNA levels with YAP downstream targets but not YAP mRNA. Consistently, overexpression of SNAI1 in human HCC cells promoted YAP activation and higher mRNA levels of YAP target genes, without changes in YAP mRNA expression. Although the precise mechanisms whereby SNAI1 modulates YAP activity require further investigation, the present data indicate that SNAI1 regulates YAP at posttranscriptional level. Furthermore, our present in vitro findings suggest the inhibition of YAP and/or Notch pathways as a potentially effective strategy in human HCC exhibiting elevated SNAI1 levels.
A second major discovery of our study is that overexpression of SNAI1 failed to promote EMT in the context of AKT/c-Met-driven hepatocarcinogenesis. Consistently, in the c-Myc HCC mouse model, overexpression of SNAI1 did not lead to EMT either. This was demonstrated by the fact that tumor cells showed an intact E-cadherin membranous staining, and vimentin expression was found to be restricted to fibroblasts surrounding the tumor cells. Importantly, in the human HCC specimens, we discovered that SNAI1 mRNA levels paradoxically display a positive correlation with epithelial markers, such as E-CADHERIN and ZO-1, mRNA expression. It is worth to mention that in cell culture systems, overexpression of SNAI1 suppresses E-CADHERIN expression and induces VIMENTIN upregulation. Noticeably, we were able to accurately reproduce these molecular changes in two human HCC cell lines (Supplementary Fig. S6). Therefore, our data suggest that SNAI1 is capable of inducing EMT in vitro, but it might have limited potential in driving EMT in vivo, especially alone. The present results further underlie the notion that critical differences exist between in vivo and in vitro growing cells. Thus, based on this body of evidence, our study suggests either that SNAI1 requires other partners to drive EMT or that other transcription factors, such as SLUG, ZEB1, and TWIST, are the pivotal regulators of EMT along hepatocarcinogenesis in humans and mice.
Supplementary Material
Acknowledgments
This study was supported by the NIH (R01CA136606, R01CA190606, R01CA228483, and P30DK026743) and scholarship from the China Scholarship Council (contract numbers: 201606280273, 201706240075, and 201703170154). We thank Dr. Eric Olson of the University of Texas Southwestern Medical Center for Yapflox/flox mice.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–86. [DOI] [PubMed] [Google Scholar]
- 2.Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017; 152:745–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet 2018;391: 1301–14. [DOI] [PubMed] [Google Scholar]
- 4.Malek NP, Schmidt S, Huber P, Manns MP, Greten TF. The diagnosis and treatment of hepatocellular carcinoma. Dtsch Arztebl Int 2014;111:101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O'Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017;547:350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaub JR, Huppert KA, Kurial SNT, Hsu BY, Cast AE, Donnelly B, et al. De novo formation of the biliary system by TGFbeta-mediated hepatocyte transdifferentiation. Nature 2018;557:247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ober EA, Lemaigre FP. Development of the liver: insights into organ and tissue morphogenesis. J Hepatol 2018;68:1049–62. [DOI] [PubMed] [Google Scholar]
- 8.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sekiya S, Suzuki A.Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Invest 2012;122:3914–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo RC, Leung CO, Chok KS, Ng IO. Variation of stemness markers expression in tumor nodules from synchronous multi-focal hepatocellular carcinoma – an immunohistochemical study. Diagn Pathol 2017;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woo HG, Lee JH, Yoon JH, Kim CY, Lee HS, Jang JJ, et al. Identification of a cholangiocarcinoma-like gene expression trait in hepatocellular carcinoma. Cancer Res 2010;70:3034–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joseph NM, Tsokos CG, Umetsu SE, Shain AH, Kelley RK, Onodera C, et al. Genomic profiling of combined hepatocellular-cholangiocarcinoma reveals similar genetics to hepatocellular carcinoma. J Pathol 2019;248: 164–78. [DOI] [PubMed] [Google Scholar]
- 13.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell 2014;157:1324–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang S, Wang J, Wang H, Fan L, Fan B, Zeng B, et al. Hippo cascade controls lineage commitment of liver tumors in mice and humans. Am J Pathol 2018;188:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang S, Song X, Cao D, Xu Z, Fan B, Che L, et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J Hepatol 2017;67:1194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song X, Liu X, Wang H, Wang J, Qiao Y, Cigliano A, et al. Combined CDK4/6 and pan-mTOR inhibition is synergistic against intrahepatic cholangiocarcinoma. Clin Cancer Res 2018. DOI: 10.1158/1078-0432.CCR-18-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada D,Rizvi S,Razumilava N,Bronk SF,Davila JI,Champion MD,et al. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 2015;61:1627–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tschaharganeh DF, Chen X, Latzko P, Malz M, Gaida MM, Felix K, et al. Yes-associated proteinup-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology 2013;144:1530–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Dong M, Xu Z, Song X, Zhang S, Qiao Y, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37:3229–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer 2018;18:128–34. [DOI] [PubMed] [Google Scholar]
- 21.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007;7:415–28. [DOI] [PubMed] [Google Scholar]
- 22.Lee DG, Kim HS, Lee YS, Kim S, Cha SY, Ota I, et al. Helicobacter pylori CagA promotes Snail-mediated epithelial-mesenchymal transition by reducing GSK-3 activity. Nat Commun 2014;5:4423. [DOI] [PubMed] [Google Scholar]
- 23.Dong C, Wu Y, Yao J, Wang Y, Yu Y, Rychahou PG, et al. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J Clin Invest 2012;122:1469–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye X, Tam WL, Shibue T, Kaygusuz Y, Reinhardt F, NgEaton E, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015;525:256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qian Y, Yao W, Yang T, Yang Y, Liu Y, Shen Q, et al. aPKC-iota/P-Sp1/Snail signaling induces epithelial-mesenchymal transition and immunosuppression in cholangiocarcinoma. Hepatology 2017;66:1165–82. [DOI] [PubMed] [Google Scholar]
- 26.Yang MH, Chen CL, Chau GY, Chiou SH, Su CW, Chou TY, et al. Comprehensive analysis of the independent effect of twist and snail in promoting metastasis of hepatocellular carcinoma. Hepatology 2009; 50:1464–74. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014;184:912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu J, Che L, Li L, Pilo MG, Cigliano A, Ribback S, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep 2016;6:20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu P, Ge M, Hu J, Li X, Che L, Sun K, et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017;66:167–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sirica AE, Gores GJ. Desmoplastic stroma and cholangiocarcinoma: clinical implications and therapeutic targeting. Hepatology 2014;59: 2397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014;25:166–80. [DOI] [PubMed] [Google Scholar]
- 32.Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, et al. Direct inhibition of the NOTCH transcription factor complex. Nature 2009;462:182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169: 1327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calderaro J, Couchy G, Imbeaud S, Amaddeo G, Letouze E, Blanc JF, et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J Hepatol 2017; 67:727–38. [DOI] [PubMed] [Google Scholar]
- 35.Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001;120: 1763–73. [DOI] [PubMed] [Google Scholar]
- 36.Hsu HC, Jeng YM, Mao TL, Chu JS, Lai PL, Peng SY. Beta-catenin mutations are associated with a subset of low-stage hepatocellular carcinoma negative for hepatitis B virus and with favorable prognosis. Am J Pathol 2000;157: 763–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding W, You H, Dang H, LeBlanc F, Galicia V, Lu SC, et al. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology 2010;52:945–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goossens S, Vandamme N, Van Vlierberghe P, Berx G. EMT transcription factors in cancer development re-evaluated: beyond EMT and MET. Biochim Biophys Acta Rev Cancer 2017;1868:584–91. [DOI] [PubMed] [Google Scholar]
- 39.Vaquero J, Guedj N, Claperon A, Nguyen Ho-Bouldoires TH, Paradis V, Fouassier L. Epithelial-mesenchymal transition in cholangiocarcinoma: from clinical evidence to regulatory networks. J Hepatol 2017; 66:424–41. [DOI] [PubMed] [Google Scholar]
- 40.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015;527:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong ST, et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015;527:472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim H, Choi GH, Na DC, Ahn EY, Kim GI, Lee JE, et al. Human hepatocellular carcinomas with "stemness"-related marker expression: keratin 19 expression and a poor prognosis. Hepatology 2011;54:1707–17. [DOI] [PubMed] [Google Scholar]
- 43.Moon H, Ju HL, Chung SI, Cho KJ, Eun JW, Nam SW, et al. Transforming growth factor-beta promotes liver tumorigenesis in mice via up-regulation of snail. Gastroenterology 2017;153:1378–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.