

Abstract
The mechanism of action of quaternary ammonium compound (QAC) antiseptics has long been assumed to be straightforward membrane disruption, although the process of approaching and entering the membrane has little modeling precedent. Furthermore, questions have more recently arisen regarding bacterial resistance mechanisms, and why select classes of QACs (specifically, multicationic QACs) are less prone to resistance. In order to better understand such subtleties, a series of molecular dynamics simulations were utilized to help identify these molecular determinants, directly comparing mono-, bis-, and triscationic QACs in simulated membrane intercalation models. Three distinct membranes were simulated, mimicking the surfaces of Escherichia coli and Staphylococcus aureus, as well as a neutral phospholipid control. By analyzing the resulting trajectories in the form of a timeseries analysis, insight was gleaned regarding the significant steps and interactions involved in the destabilization of phospholipid bilayers within the bacterial membranes. Finally, to more specifically probe the effect of the hydrophobic section of the amphiphile that presumably penetrates the membrane, a series of alkyl- and ester-based biscationic quaternary ammonium compounds were prepared, tested for antimicrobial activity against both Gram-positive and Gram-negative bacteria, and modeled.
Keywords: antibacterial compounds, lipids, molecular dynamics, quaternary ammonium compounds, simulations
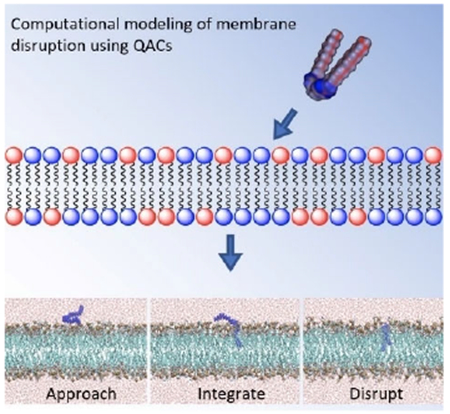
The steps of QAC activity:
Quaternary ammonium compounds (QACs) have long been assumed to disrupt bacterial membranes through electrostatic attraction followed by intercalation and subsequent disruption, but little modeling evidence has been presented to support this; the mechanism of action of multiQACs was even less clear. In a holistic study featuring computational modeling and SAR, we examine how QACs bearing multiple cations disrupt bacterial membranes.
Graphical Abstract
Introduction
Quaternary ammonium compounds (QACs) are a staple of modern antiseptics and are ubiquitously employed in private and industrial settings.[1] Since the mid 1930s, many QACs such as benzalkonium chloride (BAC) and cetylpyridinium chloride (CPC) have been heavily utilized as broad spectrum antibacterial agents,[2,3] killing a variety of pathogenic bacteria such as Escherichia coli and Staphylococcus aureus, while having only modest toxicity to humans.[4] Chemically stable and easily prepared, QACs have been some of the dominant products on the antibacterial market, leaning on their amphiphilic nature to effect bacterial membrane lysis. However, over the course of the past few decades, bacterial resistance to many of these QACs has risen, causing a great concern for human health.[5]
Much of the bacterial resistance in QACs has been attributed to their overuse and persistent environmental exposure. It is estimated that approximately 700 000 tons of QACs are used and subsequently released into the environment on an annual basis.[6,7] BAC in particular has been reported to have an approximately 9 month half-life in the environment, and is thus likely to promote greater resistance against traditional QAC antibacterials[8]
This loss of potency has long been attributed to the action of efflux pumps ejecting QACs from the bacterial inner membrane; however, this is unlikely to be the complete explanation for bacterial resistance to antiseptics, as differential composition of membrane lipids is just one of many possible contributing factors.[1,9,10] QACs with monocationic charges have been demonstrated to be significantly susceptible to resistant bacterial strains, jeopardizing their utility as antibacterial agents. However, over the past decade, bis- and triscationic QACs have been shown to be highly active against resistant bacteria,[3,11–13] establishing a strong correlation between charge and potency. A representative set of compounds featuring analogous mono-, bis-, and triscationic pyridinium compounds is represented in Figure 1. The well-known CPC, a common mouthwash ingredient, is a simple pyridinium-based amphiphile featuring a single 16-carbon substituent;[14] the 2Pyr series (and related bispyridinium compounds) have been reported;[15] the 3Pyr series was reported in our labs in 2016.[3] In that report, it was noted that while CPC showed up to a 32-fold decline in activity versus S. aureus strains bearing resistance genes (e.g., CA-methicillin-resistant S. aureus (MRSA) strain USA300-0114), compounds such as M3Pyr-12,12,12 showed no differential antimicrobial activity against methicillin-resistant versus methicillin-susceptible S. aureus strains (MIC≈1 μm). Molecular-level explanations for this differential, however, have been elusive; hypotheses range from an inability of multiQACs to enter the intracellular space of bacteria, to more rapid membrane disruption, to cooperative binding of the multicationic species to the net anionic bacterial cell surface.
Figure 1.
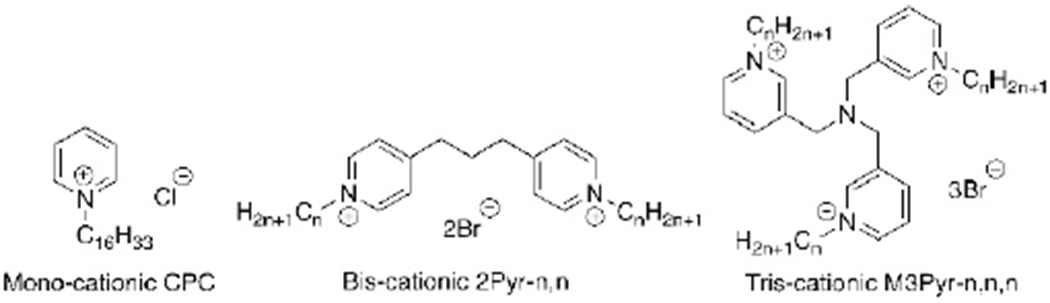
Representative mono-, bis- and trisQAC compounds: CPC, 2Pyr and 3Pyr.
Although much of the research in QAC bacterial resistance has focused on the negative transcriptional regulator QacR found in bacterial cells,[16,17] little exploration of the computational interactions of multicationic QACs with the phospholipid bilayers in bacterial membranes has thus far been conducted.[18,19] It is possible that elucidating the molecular interactions that QACs have with the bacterial bilayer can improve our understanding of the differing antimicrobial efficacies amongst QAC structures and amongst bacterial strains. Such insights may assist in the development of new generations of potent QACs, while mitigating the bacterial resistance involved with antiseptics. Importantly, several attempts had already been made in order to reduce the proliferation of bacterial resistance, particularly the development of “soft QACs” amphiphiles bearing cleavable side chains that hydrolyze into non-potent residues.[20–23] Both ester- and amide-based side chains have been previously installed within a variety of QACs, as they allowed for faster decomposition in environmental settings (Figure 2). Many soft multiQACs still offer strong antibacterial properties relative to BAC; however, their potency has been reported to be much lower than that of more traditional QACs with linear alkyl hydrophobic residues, despite identical cation number and placement.[24,25] Given that the effectiveness of efflux pumps and QacR have already been linked to charge of ligands, we hypothesized that perhaps the phospholipid bilayers may play a significant role in dictating potency based on other parameters.
Figure 2.
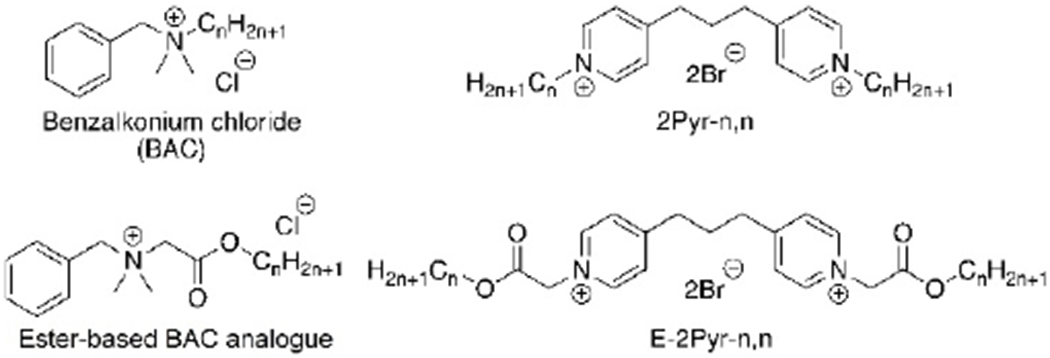
A comparison of alkyl- and ester-based BAC and bispyridinium compounds.
To advance our understanding of the differential antibacterial activity of QACs, we set out to elucidate three unanswered questions regarding the behavior of QACs in bacterial membranes: 1)The mechanistic pathway QACs utilize in the penetration of a phospholipid bilayer, with an eye towards any differentiation of pathway due to charge state and number of hydrophobic residues, 2) the initial molecular events leading to bilayer destabilization by QACs of varying charge, and 3) the molecular determinants of potency in the ester- and alkyl-based multiQACs. In order to address these concerns, we decided to pursue both an experimental and in silico approach. Experimentally, we set out to prepare analogous sets of biscationic alkyl- and ester-based QACs to determine their potency. We then embarked on a series of molecular dynamics (MD) simulations for a variety of QACs and a variety of membrane mimics in order to model these fundamental interactions.
Results and Discussion
Bioactivity analysis showed results consistent with previous publications; a full MIC table is presented in the Supporting Information, and representative data is presented below. As expected, the three main factors driving QAC potency are the length of the hydrophobic tails (with optimal activity at ≈12 carbon chains), the type of side chain (with alkyl chains more active than their ester counterparts), and the number of cationic residues (singly cationic species are less potent). As suggested by previous reports, the ester-based QACs showed significantly higher MIC values against both Gram-positive and Gram-negative bacteria than the straight alkyl compounds. Interestingly, though, biscationic esterQACs showed little drop-off in activity when comparing S. aureus strains; MRSA strains were equally inhibited compared to their MSSA (methicillin-susceptible S. aureus) counterparts (MIC = 4–8 μm). This was not true for the monocationic CPC, which showed 32x loss in potency when comparing S. aureus to CA-MRSA. Likewise, the other multi-cationic amphiphiles (2Pyr-12,12 and M-3Pyr-12,12,12) maintained activity against resistant strains. Finally, there was no advantage in the triscationic species as compared to the bisQAC; in fact, 2Pyr-12,12 held a ≈2× edge in activity over M-3Pyr-12,12,12 across the board.
In order to investigate the mechanism of QAC integration within the various membranes, a series of MD simulations were performed, placing a different QAC at approximately 5 Å above a pre-equilibrated phospholipid bilayer (Figure 3A; E. coli membrane mimic). Despite differing cationic charges and hydrophobic tail lengths, a similar mechanism was observed for all QACs tested whereby the QAC approaches the bilayer and integrates a non-polar side chains into the membrane (Figure 3B). Upon further equilibration, the second side chain of multiQACs subsequently integrates into the bilayer (Figure 3C); importantly, a noticeable undulation develops as a result of this second integration event. Supporting Information contains links to a video of the simulation, and sample parameter files.
Figure 3.

Mechanism of QAC integration into the bacterial bilayer of modeled E. coli. Molecular configurations sampled along the molecular dynamics trajectory resulting in the insertion of the QAC. Note how, after approaching the membrane, the QAC immerses one tail at a time in the lipid bilayer.
This mechanism of integration illustrated in Figure 3 was also observed with the mono-cationic CPC as well as the triscationic derivatives. It was determined that integration of QACs into the bilayer proceeds stepwise, one tail at a time. Upon insertion into the bilayer, noticeable undulations within the z-axis were observed as lipids laterally translated across the y-axis in order to accommodate the new compound (Figure 3C). We attempted to integrate several QACs such as 2Pyr-12,12 into a neutral POPC bilayer; however, after 200ns the compound had still failed to penetrate the membrane (we define penetration as the insertion of at least two tail carbons within the bilayer). We hypothesize that the charged character of the E. coli membrane is a prerequisite for QAC binding; by contrast, the insertion is most likely driven by lipid–QAC hydrophobic interaction. Since QACs are not expected to penetrate once the minimum free energy configuration is achieved, a fine-tuning between electrostatics and hydrophobic interactions is perhaps crucial to optimize potency. This also held true when attempting to integrate multiple QACs into the same bilayer.
In order to glean insights in the process of membrane destabilization, we analyzed the structure, specifically via charge density, of the bilayer throughout the simulation and up to 80ns after insertion of the QAC. As QACs possess a positive charge localized on the quaternary nitrogen atoms, we postulated that QACs are able to affect the lateral organization of the components of bacterial membranes. The bacterial membranes contain negatively charged lipids such as phosphatidyl-glycerol (DLPG) and cardiolipin (TMCL), therefore it is possible that QACs attract these lipids, thereby promoting their clustering. This would imply a crucial role for negatively charged lipids and might rationalize a selective action of QACs on bacterial membranes, which are rich in negatively charged lipids.
Accordingly, a series of 12 simulations were conducted in order to analyze the interactions between QACs and bacterial membranes. As a reference, we monitored the clustering tendency of DLPG and DLPE (1,2-dilauroyl-sn-glycero-3-phosphoethanolamine) in the bacterial membrane of E. coli without inclusion of any ligand. The relative densities of DLPG and DLPE were calculated using the GROMACS plugin g_mydensity (Figure 4). Densities calculated on the first and last 20 ns of the simulation show no difference, indicating that negatively charged lipids are normally dispersed and do not form aggregates.
Figure 4.
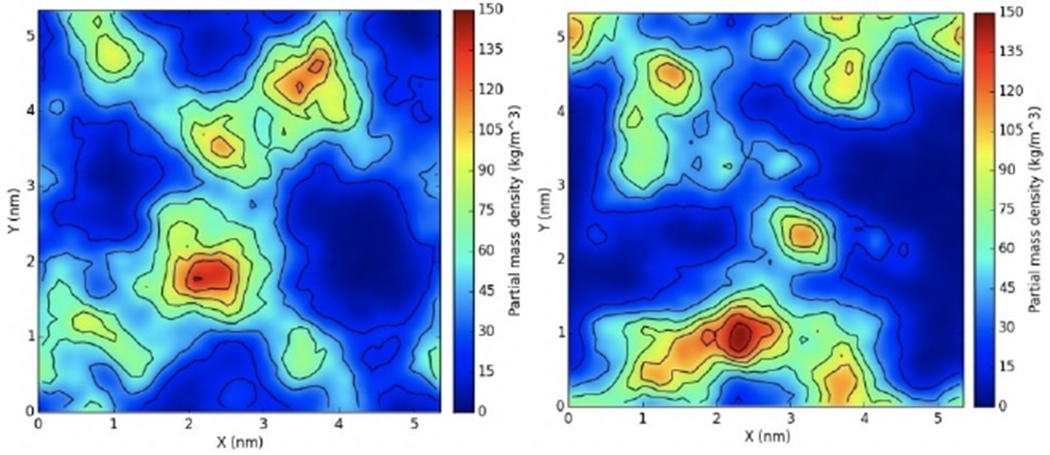
Surface density of DLPG in the membrane. The first (left) and last (right) 20 ns of the trajectory of an E. coli membrane mimic were used to calculate averages. Note that the lateral distribution of DLPG is similar in the two cases.
We then repeated the calculations after the addition of a single QAC moiety. A series of mono-, bis-, and triscationic QACs were equilibrated in the E. coli membrane and their DLPG, DLPE, and ligand densities were computed for the first and last 10% of the simulation (equating to approximately 20 ns each).
To begin, CPC was allowed to equilibrate for 200 ns and integrate in the membrane. The density of DLPG lipids was then calculated for the first 20 ns, and the last 20 ns of the simulation and compared for each ligand. The highest concentration of DLPG in the first 20 ns was reported to be lower than 90 kg m−3, whereas the concentration in the last 20 ns was reported to be as high as 150 kg m−3 and adjacent to the ligand (Figure 5). CPC bears only one positive charge localized on its quaternary nitrogen atom, therefore the lack of a great density of DLPG near the CPC ligand is to be expected.
Figure 5.
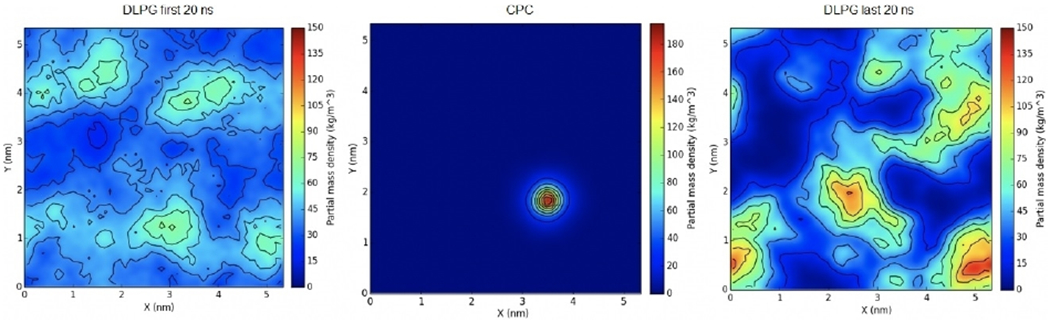
Surface density of mono-cationic CPC in an E. coli membrane mimic. The first (left) and last (right) 20 ns of the trajectory were used to calculate averages. Note that DLPG lipids accumulate around the position of the QAC (middle) as a result of the insertion.
This simulation was followed by the biscationic 2Pyr-12,12 ligand, which was equilibrated in a similar fashion for 350 ns. The density of DLPG in the first 20 ns of the simulation indicate that the highest concentration does not exceed 120 kg m−3 and is not localized in any particular area adjacent to the ligand (Figure 6). On the other hand, the density of DLPG in the last 20 ns of the simulation does seem to localize near the ligand with a maximum concentration of 180 kg m−3, thus indicating that 2Pyr-12,12 attracted the negatively charged lipids, somewhat more strongly than as shown by CPC.
Figure 6.
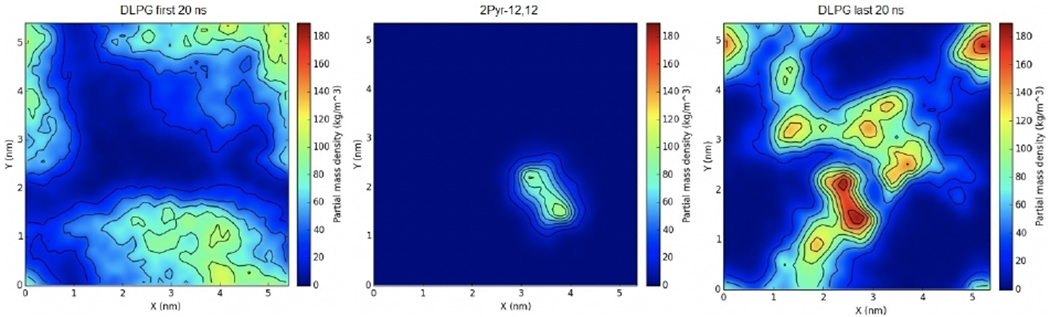
Surface density of biscationic 2Pyr-12,12 in an E. coli membrane mimic. The first (left) and last (right) 20 ns of the trajectory were used to calculate averages. The distribution of the QAC is shown in the middle panel.
Similarly, another simulation with the tris-cationic M3Pyr-12,12,12 was also equilibrated in the same fashion for 120 ns, in which it similarly integrated itself within the bilayer. The first and last 20 ns of the simulation were analyzed for DLPG density showing similar results (Figure 7). The M3Pyr-12,12,12 ligand was surrounded by the negatively charged DLPG lipids towards the end of the simulation relative to the beginning.
Figure 7.
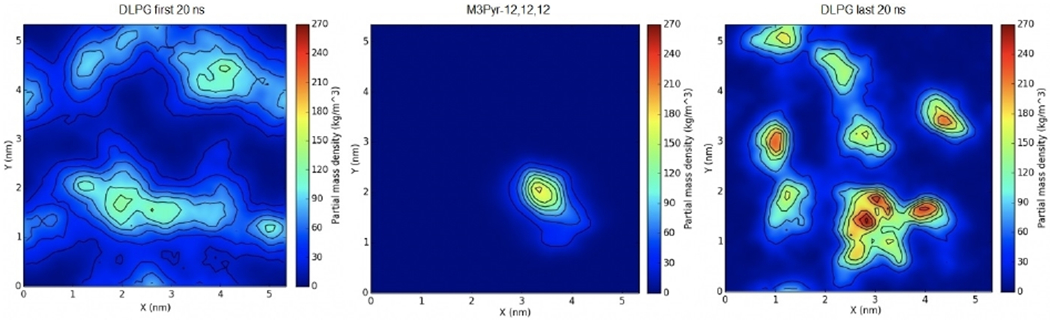
Surface density of tris-cationic M3Pyr-12,12,12 in an E. coli membrane mimic. The first (left) and last (right) 20 ns of the trajectory were used to calculate averages. The distribution of the QAC is shown in the middle panel.
Given the promising results shown with 2Pyr-12,12 and the E. coli membrane mimic, a similar analysis was then performed for 2Pyr-12,12 within a S. aureus membrane (Figure 8). The main difference that should be noted relative to E. coli is the fact that S. aureus contains a much higher concentration of negatively charged lipids, therefore a larger clustering of lipids is expected at the end of the simulation. Density analysis of 2Pyr-12,12 in S. aureus showed, indeed, higher concentrations of DLPG and TMCL lipids surrounding the QAC in the later part of the simulation relative to the beginning. A comparison of the same ligand in E. coli further showed that the positively charged lipids did not cluster around the ligand.
Figure 8.
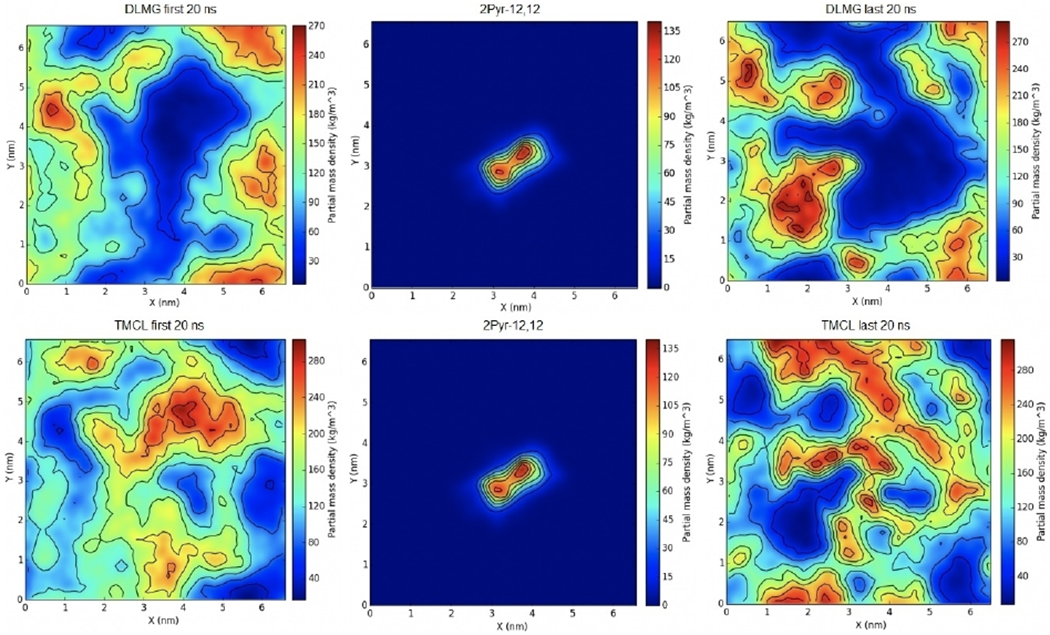
Surface density of 2Pyr-12,12 in an S. aureus membrane mimic. The first (left) and last (right) 20 ns of the trajectory were used to calculate averages. The distribution of the QAC is shown in the middle panel. The density of DLMG and TMCL is reported in the top and bottom row, respectively.
The clustering of the DLMG and TMCL lipids near the QAC in the S. aureus system confirmed that negatively charged components of the system do in fact cluster near the ammonium salt; much more so that their positively charged counterparts. It should be noted that the concentration of negatively charged entities was determined to be approximately 150 kg m−3 for the mono-cationic CPC, 180 kg m−3 for the biscationic 2Pyr-12,12, and almost 270 kg m−3 for the tris-cationic 3Pyr-12,12,12, clear indication that charge does in fact affect the clustering of these entities.
Finally, to understand the differences between ester- and alkyl-based QACs, E-2Pyr12,12 was minimized and equilibrated on an E. coli membrane. Subsequently, an analogous series of the alkyl- and ester-based forms of the 2Pyr series bearing 10, 11, 12, or 13 linear atoms in the side chain were simulated in the same conditions for nearly 80 ns. The results of the simulation showed that all compounds eventually integrate within the lipid bilayer; however, a notable difference was observed in the kinetics of integration: ligands belonging to the alkyl series penetrated the membrane much faster than their ester-based counter parts. Interestingly, these same compounds have a lower MIC compared to the ester derivatives (Table 1 and Figure 9), suggesting a link between kinetics of adsorption and MIC. While the durations of the ester-based QACs remained consistent, those of the Alkyl-based QACs did not. However, differences are not statistically significant except for the case of C-13, which shows faster kinetics compared to all the other chain lengths.
Table 1.
Minimum inhibitory concentrations.
| Compound | MIC [μm] | |||||
|---|---|---|---|---|---|---|
| S. aureus | E. faecalis | E. coli | P. aeruginosa | CA-MRSA | HA-MRSA | |
| CPC | 0.5 | 1 | 8 | 63 | 16 | 1 |
| 2Pyr-12,12 | 0.5 | 0.5 | 1 | 4 | 0.25 | 0.5 |
| E-2Pyr-12,12 | 8 | 8 | 16 | 63 | 4 | 4 |
| M-3Pyr-12,12,12 | 1 | 1 | 1 | 8 | 0.5 | 1 |
Figure 9.
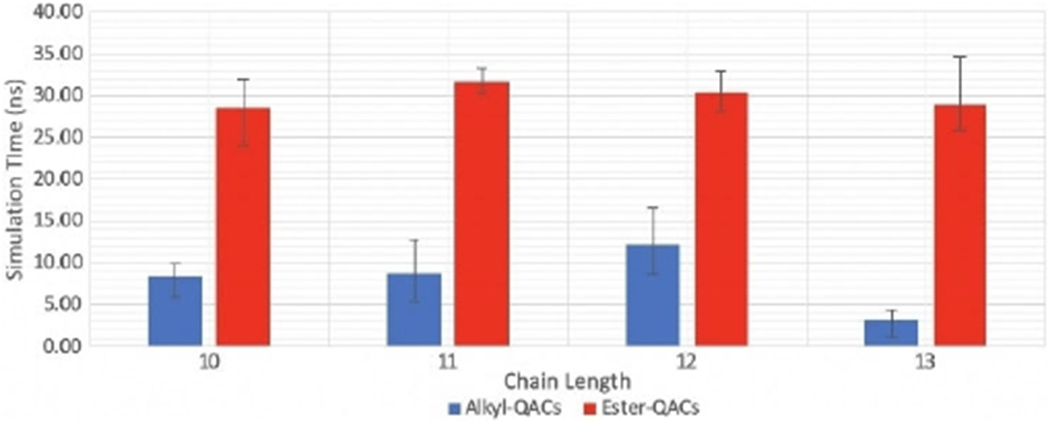
Kinetics of insertion of alkyl- and ester-QACs with different chain length.
Conclusion
Quaternary ammonium compounds have long been assumed to disrupt bacterial membranes through electrostatic attraction followed by intercalation and subsequent disruption, but little modeling evidence has been presented to support this; the mechanism of action of multiQACs was even less clear. Herein we have used molecular dynamics to investigate this issue for a varied set of QACs: mono-, bis-, and triscationic species, as well as ester-substituted analogs. Our results suggest a uniform method of integration into bacterial bilayers: all QACs modeled used the electrostatic behavior of the negatively charged lipids in order to approach the membrane, followed by membrane integration of the hydrophobic tails, leading to undulations due to the lateral diffusion of lipids. The congregation of negatively charged lipids within the membrane towards the QAC is an important and quantitative component in the overall mechanism of likely membrane destabilization; this observation was noted for all QACs that integrated into the membrane. Interestingly, faster integration and higher congregation of lipids was observed going from the monocationic CPC to its biscationic 2Pyr-12,12 counterpart. And while multiple attempts were made to investigate the mechanistic differences between ester- and alkyl-based compounds, the only significant property that recognized a difference was the time it took for an alkyl-based QAC to enter the membrane, which is consistent with the superior antimicrobial activity of the alkyl-based compounds. Although multiple attempts were made to distinguish between the ester- and alkyl-based QACs, further analysis will be required to identify the specific molecular interactions that dictate potency. It is possible that chain length plays a significant role; however, our current data does not allow us to draw any definite conclusions concerning this matter. Therefore, further investigation will be required.
Experimental Section
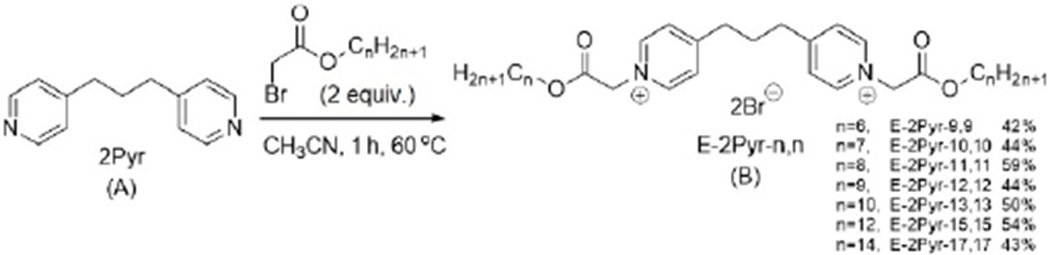
The preparation of pyridinium compounds 2Pyr-n,n and M3Pyr-n,n compounds were accomplished according to known methods.[26] The synthesis of ester-based pyridinium bisQACs (dubbed the E-2Pyr-n,n series) consisted of alkylation of 1,3-bispyridyl propane with a series of bromo-esters ranging from 6 to 14 carbons in chain length, as shown in Scheme 1. Resulting compounds were named according to the number of linear atoms in the side chain; yields ranged from 42 to 59%.[27] Specific details concerning the preparation of these compounds as well as their characterization can be found in the supporting information.
Scheme 1.
Preparation of esters-bromides and ester QACs.
Antimicrobial activity was assessed following standard protocols.[28] MIC values against six bacteria [S. aureus, Enterococcus faecalis, E. coli, Pseudomonas aeruginosa, community-acquired MRSA (USA300-0114), hospital-acquired MRSA (ATCC 3359a)], were determined, along with red blood cell (RBC) lysis (presented as lysis20, the highest concentration at which <20% of RBCs are lysed) as a toxicity estimation.
Membrane preparation was performed using the CHARMM-GUI lipid generator. The lipid composition of the bilayer was chosen to mimic membranes from E. coli and S. aureus.[29] As a reference system, we also generated a lipid bilayer containing only the neutral species 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC). Thus the three membranes were composed of: 1) 100% POPC; 2)80% 1,2-dilauroyl-sn-glycero-3-phosphoethanolamine (DLPE) and 20% 1,2-Dilauroyl-sn-glycero-3-phospho-sn-glycerol (phosphatidyl glycerol, DLPG) to mimic E. coli; or 3) 58% DLPG and 42 % 1′,3′-bis[1,2-dimyristoyl-sn-glycero-3-phospho]-sn-glycerol (cardiolipin, TMLC) for S. aureus.[30] The membranes were inserted into individual boxes containing water molecules and Na+ and Cl− ions. This system underwent energy minimization for 10000 steps prior to the molecular dynamics simulations.
Ligand preparation was performed using first Avogadro 1.90 to generate atomic coordinates[31] and then CGenFF[32] to generate topology files. The ligands were inserted in a box of water and underwent 20 ns of MD simulation to explore the conformational states accessible at room temperature. Clustering analysis was then performed to extract the most representative structure using the VMD clustering plugin with a cutoff of 1.0; an example of minimized E-2Pyr-12,12 is an example illustrated in Figure 5. The most populated clusters were used, for each compound, as starting configuration in the subsequent MD simulations. In particular, the ligands were inserted into the equilibrated membrane systems (after initial membrane equilibration) by placing a QAC parallel to the membrane at a distance of 5 Å from the head groups of the lipid bilayer (Figure 3).
Molecular dynamics simulations were performed with NAMD 2.12,[33] using the energy function CHARMM36[33] and the TIP3P potential for water molecules.[35] Periodic boundaries conditions were applied, and long-range electrostatic interactions were treated by the particle mesh Ewald algorithm with a grid spacing of 1.20 Å and a cutoff in real space of 12 Å (the same cutoff was used to truncate Lennard Jones interactions).[36]. All trajectories are generated with a time step of 2 fs at constant normal pressure (1 atm) controlled by a Langevin piston and constant temperature (323 K) using a Nosé-Hoover thermostat.[37] A total trajectory of approximately 380 ns was collected for each system (200 ns for equilibration). Data were compiled and analyzed by using Python 3.6 (Figure 10).
Figure 10.
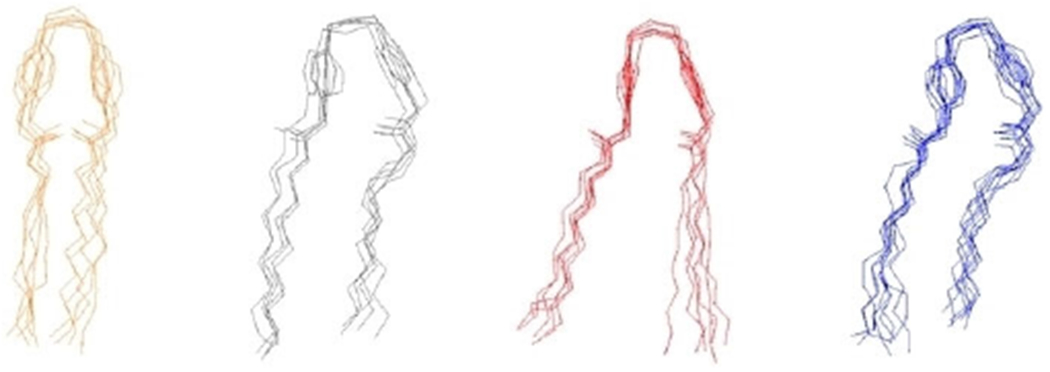
Clustering Results of biscationic E-2Pyr-12,12. Selected molecular configurations from the four most populated clusters are shown.
Supplementary Material
Acknowledgements
This work was funded by the US National Institute of General Medical Sciences (R35 GM119426 to W.M.W.), as well as Temple and Villanova Universities. M.C.J. acknowledges a US National Science Foundation fellowship (DGE1144462). This research includes calculations carried out on Temple University’s HPC resources and thus was supported by the National Science Foundation through major research instrumentation grant number 1625061, by the US Army Research Laboratory under contract number W911NF-16-2-0189, and by the National Institute of General Medical Sciences of the National Institutes of Health through grant S10OD020095.
Footnotes
Supporting information and the ORCID identification numbers for the authors of this article can be found under https://dx.doi.org/10.1002/cbic.201900398.
Conflicts of Interest
W.M.W. and K.P.C.M have intellectual property claims on the multiQAC compounds disclosed and modeled.
References
- [1].Walker EB, Paulson D, Quaternary Ammonium Compounds, New York, Marcel Dekker, 2002. [Google Scholar]
- [2].Domagk G, Dtsch. Med. Wochenschr 1935, 61, 829–832. [Google Scholar]
- [3].Al-Khalifa SE, Jennings MC, Wuest WM, Minbiole KPC, ChemMedChem 2017, 12, 280–283. [DOI] [PubMed] [Google Scholar]
- [4].“Registry of Toxic Effects of Chemical Substances (RTECS)”, US Center for Disease Control, 2019: https://www.cdc.gov/niosh-rtecs/bo3010b0.html. [Google Scholar]
- [5].Jennings MC, Buttaro BA, Minbiole KPC, Wuest WM, ACS Infect. Dis 2015, 1, 304–309. [DOI] [PubMed] [Google Scholar]
- [6].Martinez-Carballo E, Sitka A, Gonzalez-Barreiro C, Kreuzinger N, Furhacker M, Scharf S, Gans O, Environ. Pollut 2007, 145, 489–496. [DOI] [PubMed] [Google Scholar]
- [7].Ivankovic T, Hrenovic J, Arh. Hig. Rada Toksikol 2010, 61, 95–110. [DOI] [PubMed] [Google Scholar]
- [8].Kummerer K, Eitel A, Braun U, Hubner P, Daschner F, Mascart G, Milandri M, Reinthaler F, Verhoef J, Chromatogr J. 1997, 744, 281 −286. [DOI] [PubMed] [Google Scholar]
- [9].Jennings MC, Forman M, Duggan S, Minbiole KPC, Wuest WM, ChemBioChem 2017, 18, 1573–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tischer M, Pradel G, Ohlsen K, Holzgrabe U, ChemMedChem 2012, 7, 22–31. [DOI] [PubMed] [Google Scholar]
- [11].Jimenez A, Garcia P, Puente S, Modrona A, Camarasa M, Perez M, Quintela J, Portillo F, San-Felix A, Molecules 2018, 23, 1513–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang N, Ma S, Eur. J. Med. Chem 2019, 184, 111743. [DOI] [PubMed] [Google Scholar]
- [13].Shrestha JP, Baker C, Kawasaki Y, Subedi YP, Vincent de Paul NN, Takemoto JY, Chang C-WT, Eur. J. Med. Chem 2017, 126, 696–704. [DOI] [PubMed] [Google Scholar]
- [14].Latimer J, Munday JL, Buzza KM, Forbes S, Sreenivasan PK, McBain AJ, BMC Microbiol. 2015, 15, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen S, Biswas C, Bartley R, Widmer F, Pantarat N, Obando D, Djordjevic J, Ellis D, Jolliffe K, Sorrell T, Antimicrob. Agents Chemother 2010, 3233–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Costa S, Viveiros M, Amaral L, Couta I, Open Microbiol. J 2013, 7, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Grkovic S, Brown M, Skurray RA, Microbiol. Mol. Bio. Rev 2002, 66, 671–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhao W, Gurtovenko A, Vattulainen I, Karttunen M, J. Phys. Chem. B 2012, 116, 269–276. [DOI] [PubMed] [Google Scholar]
- [19].Palermo E, Lee D-K, Ramamoorthy A, Kuroda K, J. Phys. Chem. B 2012, 115, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hoque J, Konai MM, Sequeira SS, Samaddar S, Haldar J, J. Med. Chem 2016, 59, 10750–10762. [DOI] [PubMed] [Google Scholar]
- [21].Bodor N, J. Med. Chem 1980, 23, 469–474. [DOI] [PubMed] [Google Scholar]
- [22].Lindstedt M, Allenmark S, Thompson RA, Edebo L, Antimicrob. Agents Chemother 1990, 34, 1949–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tezel U, Pavlostathis SG, Curr. Opin. Biotechnol 2015, 26, 296–304. [DOI] [PubMed] [Google Scholar]
- [24].Allen RA, Jennings MC, Mitchell MA, Al-Khalifa SE, Wuest WM, Minbiole KPC, Bioorg. Med. Chem. Lett 2017, 27, 2107–2112. [DOI] [PubMed] [Google Scholar]
- [25].Lindstedt N, Allenmark S, Thompson RA, Edebo L, Antimicrob. Agents Chemother 1990, 34, 1949–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fromkerz K, Spiegelberg H, Helv. Physiol. Pharmacol. Acta 1948, 42–54. [PubMed] [Google Scholar]
- [27].Patial P, Shaheen A, Ahmad I, J. Surfactants Deterg 2013, 16, 49–56. [Google Scholar]
- [28].“Methods for Dilution Antimicrobial Tests for Bacteria that Grow Aerobically”, CLSI Document M07-A9, Approved Standard, 9th ed., 23, 2012. [Google Scholar]
- [29].Wu E, Jo S, Huan R, Song K, Davila-Contreras E, Qi Y, Lee J, Monje-Galvan V, Veneble R, Klauda J, Im W, J. Comput. Chem 2014, 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Epand RF, Savage PB, Epand RM, Biochim. Biophys. Acta Biomembr 2007, 1768, 2500–2509. [DOI] [PubMed] [Google Scholar]
- [31].Hanwell M, Curtis D, Lonie D, Vandermeersch T, Zurek E, Hutchison G, J. Cheminf 2012, 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, MacKerell A, J. Comput. Chem 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Phillips J, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel R, Kale L, Schulten K, J. Comput. Chem 2005, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].MacKerell AD, Feig M, Brooks LC, J. Comput. Chem 2004, 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- [35].Jorgensen WL, Chandrasekhar J, Madura J, Impey R, Klein M, J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- [36].Essmann U, Perera L, Berkowitz ML, J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- [37].Feller SE, Zhang Y, Pastor RW, Brooks BR, J. Chem. Phys 1995, 103, 4613–4621. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.