

Abstract
The field of organic chemistry has recently witnessed a rapid rise in the use of chemoenzymatic strategies for the synthesis of complex molecules. Under this paradigm, biocatalytic methods and contemporary synthetic methods are used synergistically in a multi-step approach towards a target molecule. In light of the unparalleled regio- and stereoselectivity of enzymatic transformations and the reaction diversity of contemporary organic chemistry, chemoenzymatic strategies hold enormous potential for streamlining access to important bioactive molecules. This review covers recent demonstrations of chemoenzymatic approaches in chemical synthesis, with special emphasis on the preparation of medicinally relevant natural products.
Introduction
Biocatalysis is increasingly becoming an indispensable tool in chemical synthesis. Due to their unique selectivity profiles, enzymes are especially useful for the preparation of novel building blocks or late-stage modification of complex molecules. However, enzymes only catalyze a small subset of organic transformations and designing novel non-natural transformations using biological systems remains non-trivial [1]. Furthermore, producing synthetic small molecules using purely biological means is still highly challenging as biological systems are highly complex and their refactoring often results in a dramatic decrease in product titer or complete abolition of product formation [2].
In recent years, organic chemists have begun exploring the combination of biocatalytic and synthetic organic methods to provide efficient access to important bioactive small molecules [3,4,5]. Such a chemoenzymatic approach allows practitioners of the field to harness the unparalleled site- and stereoselectivity of enzymatic transformations, while also taking advantage of various organic transformations to forge chemical bonds that are otherwise unattainable using purely enzymatic means. Herein, we highlight recent developments in the chemoenzymatic synthesis of bioactive small molecules, paying special attention to cases in natural product synthesis, whereby the use of this hybrid approach has provided significant improvement in synthesis economy. The select case studies are organized according to types of chemical bonds (C–C or C–X) that are formed enzymatically. For each case study, we also provide a brief discussion on the main advantages that the chemoenzymatic approach has to offer over contemporary chemical approaches.
Chemoenzymatic Synthesis Featuring Enzymatic C–C Bond Formation
Jorunnamycin A and saframycin A
Tetrahydroisoquinoline (THIQ) alkaloids, represented by the saframycins, jorunnamycins and ecteinascidins, are a family of tyrosine-derived molecules with potent antitumor activity [6]. Recently, the Oikawa and Oguri groups collaborated to accomplish a concise chemoenzymatic synthesis of these alkaloids [7]. The authors utilized a phosphopantetheinylated SfmC, a dual Pictet-Spengler enzyme discovered by the Oikawa group [8], to construct the common pentacyclic core of these molecules from a tyrosine analogue (1) and corresponding aldehydes 2 and 3 (Figure 1A) . Strikingly, two C–C bonds, three C–N bonds, and reduction of the thioester were accomplished in one enzymatic step. The secondary amine was methylated by treatment of the crude mixture with excess formaldehyde and 2-picoline borane (4) to give 18% and 13% yield of 5 and 6 from tyrosine. Further hydrolysis to remove the fatty acid and oxidation state adjustment gave jorunnamycin A (7) and saframycin A (8), respectively. Through a combination of chemical and enzymatic steps, this work achieved the shortest syntheses of jorunnamycin A and saframycin A to date.
Figure 1.
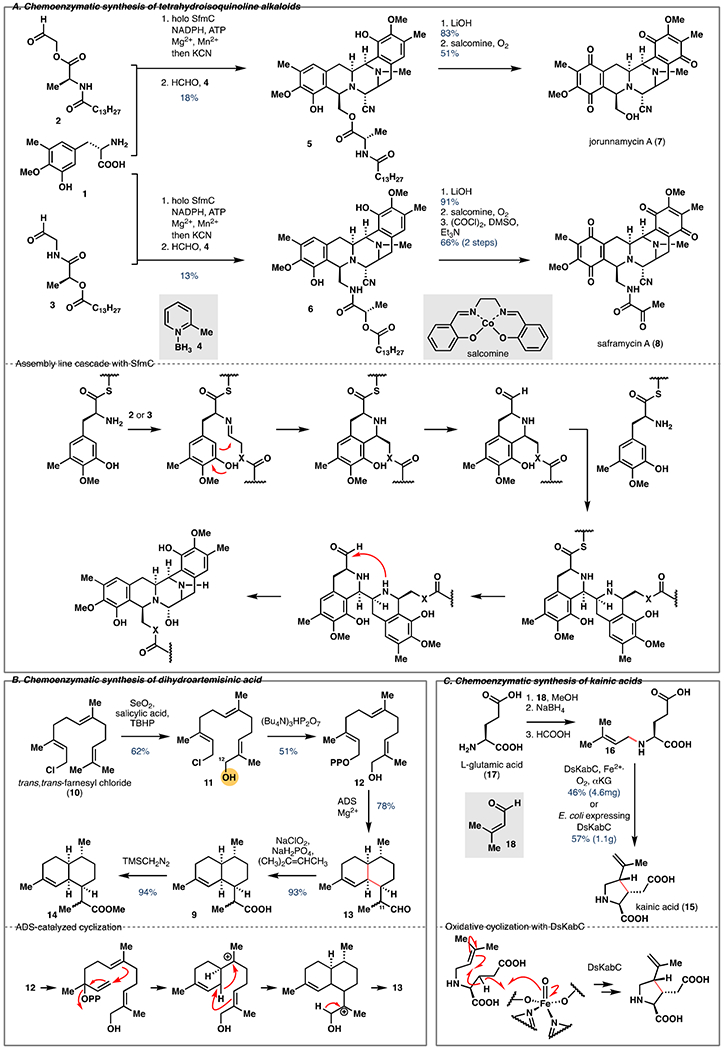
A. Chemoenzymatic synthesis of jorunnamycin A (7) and saframycin A (8) featuring construction of their pentacyclic core with SfmC. B. Chemoenzymatic synthesis of dihydroartemisinic acid (9) via enzymatic cyclization of 12 with amorphadiene synthase (ADS). C. Chemoenzymatic synthesis of kainic acid (15) via oxidative cyclization of prekainic acid (16) with DsKabC.
Dihydroartemisinic acid
Artemisinin is a sesquiterpenoid endoperoxide widely used to treat malaria [9]. Recently, Allemann and co-workers reported a chemoenzymatic route to dihydroartemisinic acid (9) [10], a precursor for the production of artemisinin [11]. The synthesis commenced with a Riley oxidation of commercially available trans,trans-farnesyl chloride (10) to provide 11 in 62% yield (Figure 1B) . Diphosphorylation of chloride 11 was accomplished in 51% yield by SN2 substitution with (Bu4N)3HP2O7. In the biosynthesis of artemisinin, the sesquiterpene cyclase amorphadiene synthase (ADS) acts upon (E,E)-farnesyl diphosphate to regio- and stereoselectively form two 6-membered rings, four stereocenters and two double bonds. Oxidation of the C11 methyl group to the corresponding acid occurs later in the biosynthesis. Allemann and co-workers believed a more concise synthesis of dihydroartemisinic acid (9) could be achieved by reversing the reaction order. To this end, ADS was optimized for the cyclization of non-native substrate 12 which yielded 13 in 78% yield as a 3:2 mixture of C11 epimers. Pinnick oxidation and methylation gave dihydroartemisinic acid and dihydroartemisinic acid methyl ester (14) in high yields. This work is complementary to a previous synthetic biology approach [12] and offers an alternative “reversed” chemoenzymatic approach for the production of artemisinin.
Kainic acid
Isolated from the Japanese marine alga Digenea simplex in 1953 [13], kainic acid (15) is an important neuropharmacological agent due to its agonism of kainate receptors. Although kainic acid is a monocyclic compound, it poses a significant synthetic challenge due to the presence of three contiguous stereocenters [14]. Moore and co-workers recently reported a concise chemoenzymatic synthesis of kainic acid [15] through an α-ketoglutarate(αKG)-dependent dioxygenase-induced oxidative cyclization. In previous studies [16], the authors had identified an αKG-dependent dioxygenase DabC that catalyzes the conversion of N-geranyl-L-glutamic acid to domoic acid. Here, they identified a DabC homolog, DsKabC, that catalyzes similar conversion of “prekainic acid” (16) to kainic acid (15). To rapidly access 16, a simple solution to the synthesis of prekainic acid was developed (Figure 1C), featuring a reductive amination between L-glutamic acid (17) and 3-methylcrotonaldehyde (18). Purified DsKabC could convert 16 to kainic acid in 46% yield on 10 mg scale. To improve the scalability of this key reaction, the authors used E. coli expressing DsKabC to directly convert the crude “prekainic acid” to kainic acid in 57% yield on gram-scale. This two-step sequence represents a significant improvement over previous synthetic approaches, which required at least six steps to complete.
Podophyllotoxin
Podophyllotoxin (19) is an aryltetralin lignan with potent tubulin depolymerizing activity [17]. This activity has inspired the development of semi-synthetic derivatives etoposide and teniposide as immunotherapy agents. Earlier this year, Chien and co-workers confirmed the role of an αKG-dependent dioxygenase 2-ODD-PH in the biosynthesis of deoxypodophyllotoxin (20) from yatein (21) [18]. Shortly after, two different groups reported two independent strategies to synthesize podophyllotoxin [19,20]. Fuchs’ strategy relied on a biocatalytic kinetic resolution of racemic hydroxyyatein with 2-ODD (Figure 2A). Reductive allylation of aldehyde 22 with bromide 23 afforded homoallylic alcohol 24, which was converted to rac-hydroxyyatein (rac-25) via Rh-catalyzed 1,4-additon. The oxidative kinetic resolution of rac-25 with 2-ODD-PH gave 39% yield of 26 in 95% ee on gram-scale and 45% recovery of the unreactive enantiomer in 66% ee. An oxidation/reduction sequence was used to invert the stereocenter of the C7 alcohol to give podophyllotoxin. Contemporaneously, our laboratory employed Baran’s oxidative enolate coupling [21] to synthesize enantiopure yatein (20, Figure 2B). Using lysate of E. coli expressing 2-ODD-PH, the biotransformation of enantiopure yatein provided 21 in 95% yield on gram-scale. Benzylic oxidation with CrO3 and subsequent reduction completed the synthesis in 58% yield. Both groups tested the substrate scope of 2-ODD-PH and synthesized analogs and related natural products. Both chemoenzymatic approaches provide improved overall yields and superior stereocontrol relative to previously developed synthetic strategies to the aryltetralin lignans.
Figure 2.
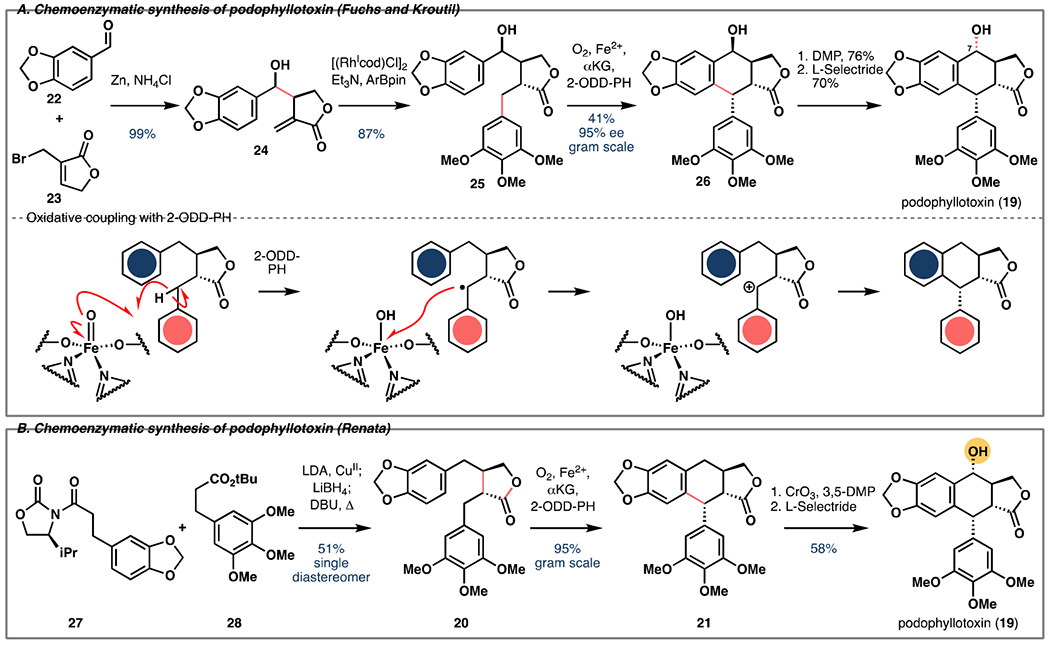
A. Chemoenzymatic synthesis of podophyllotoxin (19) via oxidative kinetic resolution of 26 with 2-ODD-PH. B. Chemoenzymatic synthesis of podophyllotoxin (19) via oxidative cyclization of enantiopure yatein (20) with 2-ODD-PH and benzylic oxidation.
Chemoenzymatic Synthesis Featuring Enzymatic C–X Bond Formation
Sorbicillinoids
The sorbicillinoids are a family of fungal natural products with promising antiviral activities [22]. They also possess intriguing molecular architectures that arise from an asymmetric oxidative dearomatization of highly substituted phenols and subsequent coupling reactions. Significant advances have been made in achieving the former transformation chemically, but they require stoichiometric chiral hypervalent iodine reagents or chiral CuI salts [23,24]. Independent reports from the Gulder and Narayan groups [25,26] showed that an FAD-dependent monooxygenase from the sorbicillinol biosynthesis can catalyze a highly regioselective oxidative dearomatization of a suite of highly-substituted phenols (e.g., 29 to 30) with exquisite enantioselectivity (Figure 3A). Both groups further showed that the resulting products constitute useful intermediates in the chemoenzymatic synthesis of various sorbicillinoids, including rezishanone C (31), sorbicatechol A (32), epoxysorbicillinol (33) and urea sorbicillinoid (34). For example, the quinone moiety of 30 readily underwent a Diels-Alder cycloaddition to give 31, 32 or 34, and the tertiary alcohol of 30 could direct a Weitz-Scheffer epoxidation to give rise to 33. Overall, the use of this biocatalytic oxidation strategy results in a dramatic improvement in synthesis economy over previous approaches to the sorbicillinoids, both in terms of step counts and reaction yields. Furthermore, the biocatalytic dearomatization step obviates the need for stoichiometric chiral reagents in the synthesis of key intermediate 30.
Figure 3.
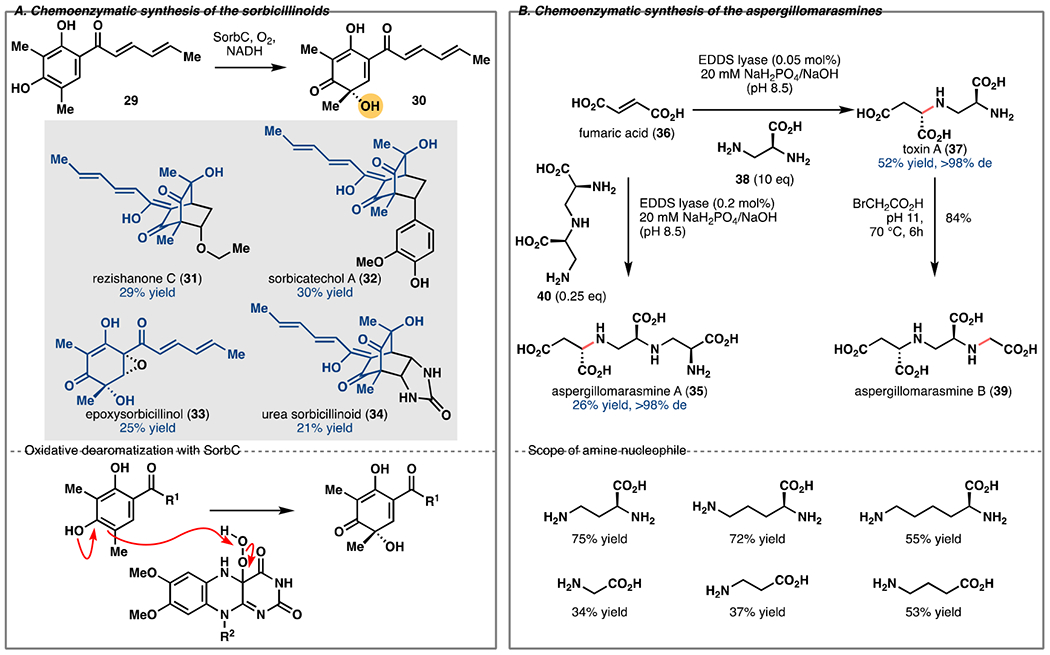
A. Chemoenzymatic synthesis of the sorbicillinoids via enantioselective oxidative dearomatization of 29 with SorbC. B. Chemoenzymatic synthesis of the aspergillomarasmines featuring biocatalytic amination of fumaric acid (36) using EDDS lyase.
Aspergillomarasmine A and Related Aminocarboxylic Acids
The fungal natural product aspergillomarasmine A (AMA, 35) was identified to potently and selectively inhibit New Delhi metallo-β-lactamase-1 via zinc chelation. AMA and related compounds are therefore promising candidates for the recovery or potentiation of β-lactam antibiotic activity [27]. Fu et al. recently developed the first chemoenzymatic route to a suite of these challenging aminocarboxylic acids using a promiscuous ethylenediamine-N,N’-disuccinic acid (EDDS) lyase [28].
EDDS lyase catalyzes the stepwise, stereoselective C–N bond formation between ethylenediamine and two molecules of fumaric acid (36). Toxin A (37), an intermediate in the biosynthesis of AMA, bears close resemblance to the native intermediate of EDDS lyase, prompting the evaluation of this enzyme’s substrate scope. Indeed, 0.05 mol % of EDDS lyase catalyzed the regio- and stereoselective amination of fumaric acid by 38 to provide toxin A in 52% yield and >98% de (Figure 2B). Compound 38 was then elaborated to aspergillomarasmine B (AMB, 39) by regioselective N-alkylation in 84% yield. Finally, AMA could also be directly prepared via amination of fumaric acid with dipeptide 40 (Figure 3B). Thus, the exceptional promiscuity of EDDS lyase allows for the expedient asymmetric synthesis of diverse aminocarboxylic acids for further biological studies.
Tetrahydroquinoline Alkaloids
The tetrahydroquinoline (THQ) motif is present in many biologically active natural products and pharmaceuticals, often with a defined stereoconfiguration at the 2 position [29]. Cosgrove et al. directly synthesized a variety of 2-substituted dihydroquinolines (DHQs) by a rhodium-catalyzed conjugate addition/condensation of ortho-amino phenylboronic acids with enone acceptors [30]. The intermediate DHQs were reduced in one pot with NaBH(OAc)3 to provide the racemic natural product precursors (rac-41–44) for biocatalytic kinetic resolution (Figure 4A).
Figure 4.
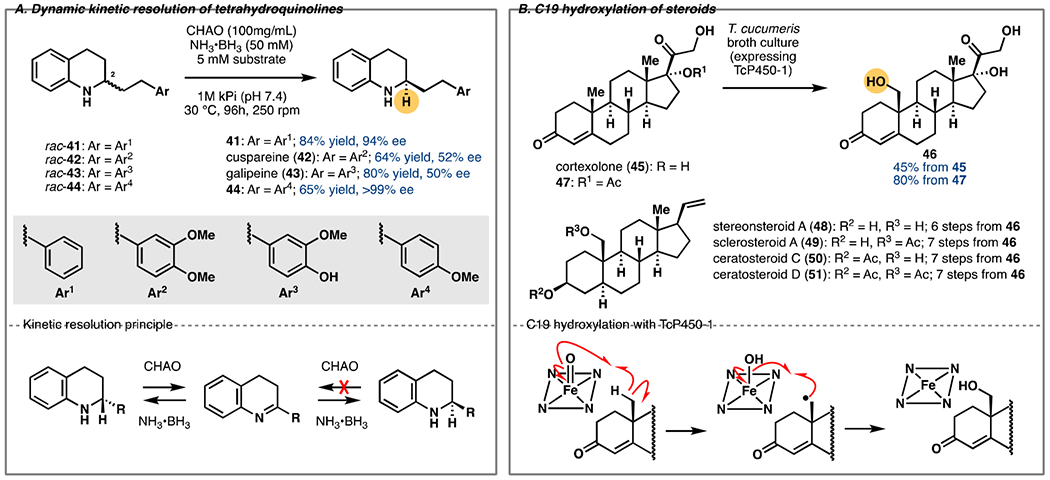
A. Asymmetric synthesis of tetrahydroquinoline alkaloids via dynamic kinetic resolution with CHAO. B. Chemoenzymatic synthesis of 19-hydroxy steroids (e.g., 48–51) via enzymatic hydroxylation with Tc450-1.
In recent years, flavin-dependent amine oxidases have been used for the deracemization of acyclic and cyclic amines [31,32]. Namely, a cyclohexylamine oxidase (CHAO) was engineered by Lau et al. to provide an array of 2-substituted THQs via in situ ammonia-borane reduction to accumulate the desired amine enantiomer [33]. Here, whole-cell E. coli expressing CHAO was used to perform the deracemization of 41–44 in 64–84% yield and up to >99% ee. Rac-cuspareine and rac-galipeine (42 and 43) stalled at ~50% e.e. while 44 gave full deracemization, suggesting that the 3-methoxy group is a limiting factor. The merging of chemocatalytic C–C bond formation and biocatalytic deracemization provides rapid access to these high-value alkaloids at preparative scale.
C19-Hydroxylated Steroids
Direct C19 functionalization of steroids is an unsolved challenge in the chemical literature that has traditionally required multiple steps to realize [34]. Recently, Wang et al. reported the multi-gram scale C19 hydroxylation of cortexolone (45, Figure 4B) [35]. Biotransformation of 45 to 19-OH-45 (46) using Thanatephorus cucumeris NBRC 6298 has been reported [36,37], but the conversion rate was low (20%), prompting cloning of the responsible P450 genes. While providing improved yield of the C19-hydroxylated product (48%), biotransformation of 45 with heterologously expressed TcP450-1 also resulted in the formation of multiple byproducts. As a workaround, the authors optimized the biotransformation conditions with T. cucumeris to increase conversion to 45%. Subsequent screening identified 17-acetyl-cortexolone (47) as an optimal substrate, providing significantly higher conversion (80%) in the biotransformation and a drastically reduced amount of side products. Furthermore, the product underwent in situ hydrolysis to the desired product 46. Thus, over 5 g of 46 could be prepared and its elaboration to six C19-hydroxylated pregnanes (e.g., 48–51) could be achieved in 4–9 steps. This work set the stage for rapid access to numerous C19-functionalized steroids and future engineering studies to further expand the substrate scope of the enzymatic reaction.
Conclusions
The case studies outlined above demonstrate the transformative power of chemoenzymatic approaches in simplifying synthetic access to bioactive small molecules. Recent explosion in genomic data [38] and advances in bioinformatics [39], analytical techniques [40] and enzyme engineering [41] should provide even more opportunities to develop novel enzymatic transformations and integrate them strategically in multi-step syntheses. Many times, however, practitioners of organic chemistry are not fully aware of the types of enzymatic transformations that are at their disposal. Further formulation of key guidelines for applying biocatalytic retrosynthetic analysis [42,43] in synthesis planning and general education in biocatalysis will prove invaluable in addressing this gap. These efforts will foster the next generation of chemists who are well-versed in both chemical and biological synthesis and ultimately allow us to realize the full potential of chemoenzymatic techniques.
Acknowledgments
The authors acknowledge the support of the National Institute of Health (1R35GM128895) and The Scripps Research Institute. The content is solely the responsibility of the authors and does not represent the official views of any of the funding agencies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
Nothing declared.
References
- 1.Renata H, Wang JZ, Arnold FH: Expanding the enzyme universe: Accessing non-natural reactions by mechanism-guided directed evolution. Angew Chem Int Ed 2015, 54:3351–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pfleger BF, Prather KLJ: Biological synthesis unbounded? Nature Biotechnol 2015, 33:1148–1149. [DOI] [PubMed] [Google Scholar]
- 3.Hönig M, Sondermann P, Turner NJ, Carreira EM: Enantioselective chemo- and biocatalysis: Partners in retrosynthesis Angew Chem Int Ed 2017, 56: 8942–8973. [DOI] [PubMed] [Google Scholar]
- 4.Rudroff F, Mihovilovic MD, Gröger H, Snajdrova R, Iding H, Bornscheuer UT: Opportunities and challenges for combining chemo- and biocatalysis Nature Catal 2018, 1: 12–22. [Google Scholar]
- 5.Doyon T, Narayan ARH: Synthetic utility of one-pot chemoenzymatic reaction sequences Synlett 2019, 30: A–G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott JD, Williams RM: Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem Rev 2002,102:1669–1730. [DOI] [PubMed] [Google Scholar]
- 7.Tanifuji R, Koketsu K, Takakura M, Asano R, Minami A, Oikawa H, Oguri H: Chemoenzymatic total syntheses of jorunnamycin A, saframycin A, and N-Fmoc saframycin Y3. J Am Chem Soc 2018, 140:10705–10709. [DOI] [PubMed] [Google Scholar]; •• The shortest synthesis of two tetrahydroisoquinoline alkaloids was achieved by using a chemoenzymatic approach that centers on the application of a ‘Pictet-Spenglerase’ to construct the pentacyclic core of the natural products.
- 8.Koketsu K, Watanabe K, Suda H, Oguri H, Oikawa H: Reconstruction of the saframycin core scaffold defines dual Pictet-Spengler mechanisms. Nat Chem Biol 2010, 6:408–410., [DOI] [PubMed] [Google Scholar]
- 9.Krishna S, Bustamante L, Haynes RK, Staines HM: Artemisinins: their growing importance in medicine. Trends Pharmacol Sci 2008, 29:520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demiray M, Tang X, Wirth T, Faraldos JA, Allemann RK: An efficient chemoenzymatic synthesis of dihydroartemisinic aldehyde. Angew Chem Int Ed 2017, 56:4347–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]; • The use of amorphadiene synthase on a modified farnesyl pyrophosphate enabled rapid construction of the core skeleton of artemisinin and efficient synthesis of dihydroartemisinic acid.
- 11.Lévesque F, Seeberger PH: Continuous-flow synthesis of the anti-Malaria drug artemisinin. Angew Chem Int Ed 2012, 51:1706–1709. [DOI] [PubMed] [Google Scholar]
- 12.Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MCY, Withers ST, Shiba Y, Sarpong R, Keasling JD: Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440:940–943. [DOI] [PubMed] [Google Scholar]
- 13.Nitta I, Watase H, Tomiie Y: Structure of kainic acid and its isomer, allokainic Acid. Nature 1958,181:761–762. [DOI] [PubMed] [Google Scholar]
- 14.Cl Stathakis, Yioti EG, Gallos JK: Total syntheses of (−)- α -kainic acid. Ear J Org Chem 2012, 4661–4673. [Google Scholar]
- 15.Chekan JR, McKinnie SMK, Moore ML, Poplawski SG, Michael TP, Moore BS: Scalable biosynthesis of the seaweed neurochemical, kainic acid. Angew Chem Int Ed 2019, 58:8454–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The discovery of an αKG-dependent dioxygenase that catalyzes an oxidative C–C bond construction in kainic acid biosynthesis facilitated the development of an efficient synthesis of the natural product via chemoenzymatic means.
- 16.Brunson JK, McKinnie SMK, Chekan JR, McCrow JP, Miles ZD, Bertrand EM, Bielinski VA, Luhavaya H, Obomik M, Smith GJ, Hutchins DA, Allen AE, Moore BS: Biosynthesis of the neurotoxin domoic acid in a bloom-forming diatom. Science 2018, 361:1356–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Botta B, Monache GD, Misiti D, Vitali A, Zappia G: Aryltetralin lignans: chemistry, pharmacology and biotransformations. Carr Med Chem 2001, 8:1363–1381. [DOI] [PubMed] [Google Scholar]
- 18.Chang WC, Yang ZJ, Tu YH, Chien TC: Reaction mechanism of a nonheme iron enzyme catalyzed oxidative cyclization via C–C bond formation. Org Lett 2019, 21:228–232. [DOI] [PubMed] [Google Scholar]
- 19.Lazzarotto M, Hammerer L, Hetmann M, Borg A, Schmennund L. Steiner L, Hartmann P, Belaj F, Kroutil W, Gruber W, Fuchs M: Chemoenzymatic total synthesis of deoxy-, epi-, and podophyllotoxin and a biocatalytic kinetic resolution of dibenzylbutyrolactones. Angew Chem Int Ed 2019, 58:8226–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]; • An asymmetric synthesis of (−)-podophyllotoxin was achieved by biocatalytic kinetic resolution of a racemic precursor with 2-ODD-PH, an αKG-dependent dioxygenase from podophyllotoxin biosynthesis.
- 20.Li J, Zhang X, Renata H: Asymmetric chemoenzymatic synthesis of (−)-podophyllotoxin and related aryltetralin lignans. Angew Chem Int Ed 2019, 58:11657–11660. [DOI] [PMC free article] [PubMed] [Google Scholar]; • A short synthesis of (−)-podophyllotoxin was achieved by combining oxidative enolate coupling and biocatalytic benzylic coupling with 2-ODD-PH.
- 21.DeMartino MP, Chen K, Baran PS: Intermolecular enolate heterocoupling: scope, mechanism, and application. J Am Chem Soc 2008,130:11546–11560. [DOI] [PubMed] [Google Scholar]
- 22.Meng J, Wang X, Xu D, Fu X, Zhang X, Lai D, Zhou L, Zhang G: Sorbicillinoids from fungi and their bioactivities.Molecules 2016, 21:715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bosset C, Coffinier R, Peixoto PA, El Assal M, Miqueu K, Sotiropoulos JM, Pouysegu L, Quideau S: Asymmetric hydroxylative phenol dearomatization promoted by chiral binaphthylic and biphenylic iodanes. Angew Chem Int Ed 2014, 53:9860–9864. [DOI] [PubMed] [Google Scholar]
- 24.Zhu JL, Grigoriadis NP, Lee JP, Porco JA: Synthesis of the azaphilones using copper-mediated enantioselective oxidative dearomatization. J Am Chem Soc 2005, 127:9342–9343. [DOI] [PubMed] [Google Scholar]
- 25.Sib A, Gulder TAM: Chemo-enzymatic total synthesis of oxosorbicillinol, sorrentanone, rezishanones B and C, sorbicatechol A, bisvertinolone, and (+)-epoxysorbicillinol. Angew Chem Int Ed 2018, 57:14650–14653. [DOI] [PubMed] [Google Scholar]; • Enantioselective oxidative dearomatization of sorbicillin-type synthetic precursors with the monooxygenase SorbC facilitated a divergent access to seven sorbicillinoid natural products.
- 26.Dockrey SAB, Lukowski AL, Becker MR, Narayan ARH: Biocatalytic site- and enantioselective oxidative dearomatization of phenols. Nature Chem 2018, 10:119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Substrate profiling of three FAD-dependent monooxygenases enabled enantioselective oxidative dearomatization of a suite of phenolic substrates, culminating in the synthesis of several complex molecules and natural products.
- 27.King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, Wright GD: Aspergillomarasmine A overcomes metallo-β 2-lactamase antibiotic resistance. Nature 2014, 510:503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu H, Zhang J, Saifuddin M, Cruiming G, Tepper PG, Poelarends GJ: Chemoenzymatic asymmetric synthesis of the metallβ-lactamase inhibitor aspergillomarasmine A and related aminocarboxylic acids. Nat Catal 2018, 1:186–191. [Google Scholar]; • A short synthesis of the aspergillomarasmines was achieved by the use of biocatalytic hydroamination of fumaric acid with the appropriate amino acid or dipeptide coupling partner.
- 29.Sridharan V, Suryavanshi PA, Menendez JC: Advances in the chemistry of tetrahydroquinolines. Chem Rev 2011, 111:7157–7259. [DOI] [PubMed] [Google Scholar]
- 30.Cosgrove SC, Hussain S, Turner NJ, Marsden SP: Synergistic Chemo/Biocatalytic Synthesis of Alkaloidal Tetrahydroquinolines. ACS Catal 2018, 8:5570–5573. [Google Scholar]; • An efficient synthesis of several tetrahydroquinoline alkaloids was achieved by the use of biocatalytic dynamic kinetic resolution on their racemic precursors.
- 31.Ghislieri D, Green AP, Pontini M, Willies SC, Rowles I, Frank A, Grogan G, Turner NJ: Engineering an enantioselective amine oxidase for the synthesis of pharmaceutical building blocks and alkaloid natural products. J Am Chem Soc 2013, 135:10863–10869. [DOI] [PubMed] [Google Scholar]
- 32.Deng G, Wan N, Qin L, Cui B, An M, Han W, Chen Y: Deracemization of Phenyl-Substituted 2-Methyl-1,2,3,4-Tetrahydroquinolines by a Recombinant Monoamine Oxidase from Pseudomonas monteilii ZMU-T01. ChemCatChem 2018, 10:2374–2377. [Google Scholar]
- 33.Yao P, Cong P, Gong R, Li J, Li G, Ren J, Feng J, Lin J, Lau PCK, Wu Q, et al. : Biocatalytic Route to Chiral 2-Substituted-1,2,3,4-Tetrahydroquinolines Using Cyclohexylamine Oxidase Muteins. ACS Catal 2018, 8:1648–1652. [Google Scholar]
- 34.Renata H, Zhou Q, Dünstl G, Felding J, Merchant RR, Yeh CH, Baran PS: Development of a concise synthesis of ouabagenin and hydroxylated corticosteroid analogues. J Am Chem Soc 2015, 137:1330–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Zhang Y, Liu H, Shang Y, Zhou L, Wei P, Yin W-B, Deng Z, Qu X, Zhou Q: A biocatalytic hydroxylation-enabled unified approach to C19-hydroxylated steroids. Nat Commun 2019, 10:3378. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• The discovery of a steroid C19-hydroxylase provided an elegant solution for the functionalization of the C19 methyl group of steroids and enabled efficient synthesis of six C19-hydroxylated pregnanes.
- 36.Clark TA, Chong R, Maddox IS: An investigation into the 19-hydroxylation of androstenedione, cortexolone and progesterone by selected fungi. Appl Microbiol Biotechnol 1985, 21:132–134. [Google Scholar]
- 37.Wang Y, Zhang F, Chen X, Feng J, Wu Q, Zhu D, Ma Y: Optimization of the transformation conditions for the 19-hydroxylation of cortexolone by Thanatephorus cucumeris. Chin J Appl Env Biol 2016, 22:860–864. [Google Scholar]
- 38.Mardis ER: The impact of next-generation sequencing technology on genetics. Trends Genet 2008, 24:133–141. [DOI] [PubMed] [Google Scholar]
- 39.Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, Whalen KL: Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim Biophys Acta 2015, 1854:1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gielen F, Hours R, Emond S, Fischlechner M, Schell U, Hollfelder F: Ultrahigh-throughput-directed enzyme evolution by absorbance-activated droplet sorting (AADS). Proc Natl Acad Sci USA 2016, 113:E7383–E7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y, Cirino PC: Recent advances in engineering proteins for biocatalysis. Biotechnol Bioeng 2014, 111:1273–1287. [DOI] [PubMed] [Google Scholar]
- 42.De Souza ROMA, Miranda LSM, Bornscheuer UT: A retrosynthesis approach for biocatalysis in organic synthesis. Chem Eur J 2017, 12040–12063. [DOI] [PubMed] [Google Scholar]
- 43.Turner NJ, O’Reilly E: Biocatalytic retrosynthesis. Nature Chem Biol 2013, 9:285–288. [DOI] [PubMed] [Google Scholar]