

Abstract
Genetic code expansion (GCE) has revolutionized the field of protein chemistry. Over the past several decades more than 150 different non-canonical amino acids (ncAAs) have been co-translationally installed into proteins within various host organisms. The vast majority of these ncAAs have been incorporated between the start and stop codons within an open reading frame. This requires that the ncAA be able to form a peptide bond at the α-amine, limiting the types of molecules that can be genetically encoded. In contrast, the α-amine of the initiating amino acid is not required for peptide bond formation. Therefore, including the initiator position in GCE allows for co-translational insertion of more diverse molecules that are modified, or completely lacking an α-amine. This review explores various methods which have been used to initiate protein synthesis with diverse molecules both in vitro and in vivo.
Keywords: synthetic biology, chemical biology, translation initiation, genetic code expansion, non-canonical amino acids
Graphical Abstract
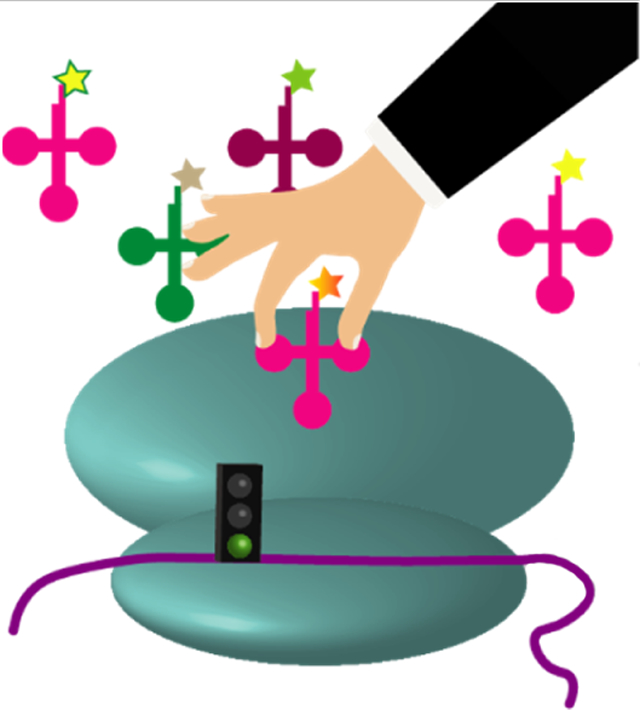
Taking control! Translation initiation can be engineered to occur at any non-AUG codon using several different strategies both in vitro and in vivo. The α-amine of the initiating amino acid is not required for elongation which allows for a diverse range of molecules to be incorporated.
1. Introduction
Over the past several decades great progress has been made towards expanding the genetic code to co-translationally install non-canonical amino acids (ncAAs) bearing diverse sidechains. These ncAAs have been used for a myriad of applications, from being used as tools to study the structure and function of biomolecules, to generating designer proteins with new-to-nature chemistry.[1] Most efforts towards genetic code expansion have focused on incorporating ncAAs at internal positions, i.e., between the initiating and terminating codons within an open reading frame. However, by mis-acylating the initiator tRNA, translation initiation can also be reprogrammed to install ncAAs at the N-terminus of proteins both in vitro and in vivo. A key feature of the initiating amino acid is that, unlike elongating amino acids, the α-amine is not involved in peptide bond formation. Therefore, in principle, amino acids that are modified at the α-amine and carboxylic acids that completely lack an α-amine can be encoded at this position. This has been exploited in vitro to generate polypeptides containing diverse molecules co-translationally installed at the N-terminus. Such molecules would be difficult or impossible to encode elsewhere within a protein. Non-canonical initiation (NCI), however, comes with a set of unique challenges that have prevented initiation with more diverse molecules in vivo. In this review, we discuss strategies and survey recent progress towards expanding the genetic code through reprogramming translation initiation in vitro and in vivo.
2. Translation Initiation in Prokaryotes
Initiation of mRNA translation is the rate limiting step of protein biosynthesis in bacteria.[2] In E. coli initiation involves several components including the initiator tRNA (itRNA), mRNA, three initiation factors IF1, IF2, and IF3, and the 30S and 50S ribosomal subunits. These components work in a coordinated fashion to assemble a translationally competent 70S initiation complex (70SIC) the details of which have been described in recent reviews.[3] Here we will summarize the mechanism of translation initiation in E. coli to provide appropriate background for discussing NCI in later sections.
Translation is universally initiated with the amino acid methionine (Met). In E. coli and other eubacteria, translation initiates with N-modified Met in the form of Nα-formylmethionine (fMet). Two types of methionine tRNAs exist in nature: elongator and initiator methionine tRNA (tRNAMet and tRNAfMet, respectively, Figure 1). Both of these tRNAs are aminoacylated with Met by methionyl-tRNA synthetase (MetRS); however, following aminoacylation, Met-tRNAfMet is further modified to fMet-tRNAfMet. Of the two methionine tRNAs, tRNAMet exclusively incorporates Met in response to internal (elongating) AUG codons, while tRNAfMet is used to initiate translation.[4] Since the overall three-dimensional structures of tRNAfMet and tRNAMet are similar, the functional specialization of these tRNAs arises from defined sequence motifs located in tRNAfMet. Most important among these motifs is the presence of three consecutive G:C base pairs in the anticodon-stem and the lack of a Watson-Crick base pair at the first position (C1xA72) in the acceptor-stem of tRNAfMet (Figure 1).[2, 4] The role of these two features in governing itRNA function is highlighted by the fact that transplanting these nucleotides into elongator tRNAs is sufficient to convert an elongator tRNA to an itRNA.[5] Both of these sequence motifs have well-defined functions that allow tRNAfMet to participate in initiation. The three GC base pairs in the anticodon-stem, a feature that is conserved in itRNAs from all domains of life, allows direct binding of tRNAfMet to the ribosomal P-site.[6] The lack of a Watson-Crick base pair at the first position (1×72), along with the identity of nucleotides at the second (2:71) and third (3:70) base pair positions, allows recognition of tRNAfMet by the methionyl-tRNA transformylase (FMT). FMT is responsible for transferring a formyl group to the α-amine of the amino acid, converting Met-tRNAfMet to fMet-tRNAfMet.[7] As the latter is chemically equivalent to a peptidyl-tRNA, the mismatch (C1xA72) is also essential for protecting the aminoacyl-tRNA from hydrolysis by peptidyl-tRNA hydrolase.[8] Formylation of Met-tRNAfMet has two functions: (i) precluding fMet-tRNAfMet from elongation, and (ii) increasing the affinity of the aminoacyl-itRNA for initiation factor 2 (IF2).[4, 9]
Figure 1.
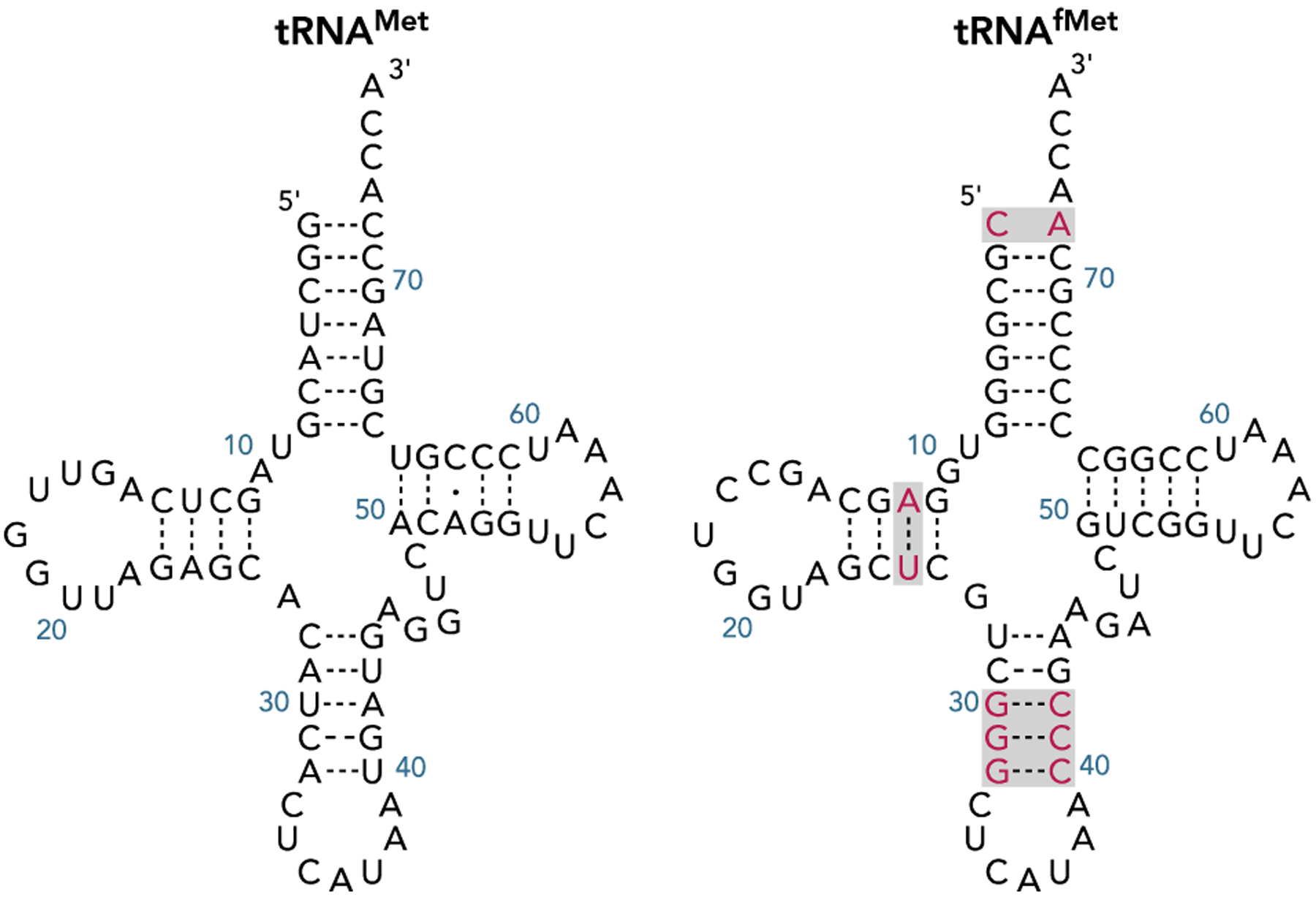
The cloverleaf structures of E. coli elongator (tRNAMet) and initiator (tRNAfMet) methionine tRNA. Nucleotides in tRNAfMet that contribute to its ability to initiate are highlighted.
Three initiation factors (IF1–3) are required for translation initiation in E. coli. IF1 binds to the A-site of the 30S subunit, blocking binding of aminoacyl-tRNAs until after formation of the 70SIC. Binding of IF1 to the 30S subunit also has a stimulatory effect on the activities of both IF2 and IF3.[2–3] IF2 is a GTPase that binds the 30S subunit in the form of IF2-GTP. IF2 was once thought to carry fMet-tRNAfMet to the ribosome in a manner analogous to EF-Tu; however, more recent data indicate that IF2 binds the 30S subunit first and promotes subsequent binding of fMet-tRNAfMet.[10] On the ribosome, the binding interaction of IF2 and fMet-tRNAfMet is localized to the six 3’-terminal nucleotides (3’ACCAAC) of tRNAfMet and to the fMet moiety which fits into a well-defined binding pocket. This binding may also be facilitated by the 1×72 base pair mismatch in tRNAfMet.[11] Therefore, IF2 helps to ensure correct itRNA selection, discriminating based on both the identity of the aminoacyl-tRNA and formylation state.[12] Multiple functions have been attributed to IF3 including preventing premature ribosome subunit assembly, ensuring the fidelity and rapid formation of itRNA and mRNA codon-anticodon interaction in the P-site, and facilitating subunit recycling following termination.[2, 3b]
Prior to assembly of the 70SIC, the 30S ribosomal subunit acts as a scaffold for assembly of the 30S preinitiation complex (30SPIC). The first step in assembly of the 30SPIC is binding of IF2-GTP and IF3 to the 30S subunit. This is followed by binding of IF1 and then either fMet-tRNAfMet or the mRNA.[3a, 13] Once both mRNA and fMet-tRNAfMet are recruited, IF-mediated mRNA codon-anticodon pairing in the P-site gives way to the more stable 30S Initiation Complex (30SIC, Figure 2) which is rapidly docked by the 50S ribosomal subunit to yield the 70SIC. 70SIC assembly and the stimulation of GTP hydrolysis within IF2, resulting in a conformational change, together lead to dissociation of the initiation factors. Initiation factor dissociation allows for EF-Tu binding, dipeptide formation, and subsequent elongation.[2–3]
Figure 2.
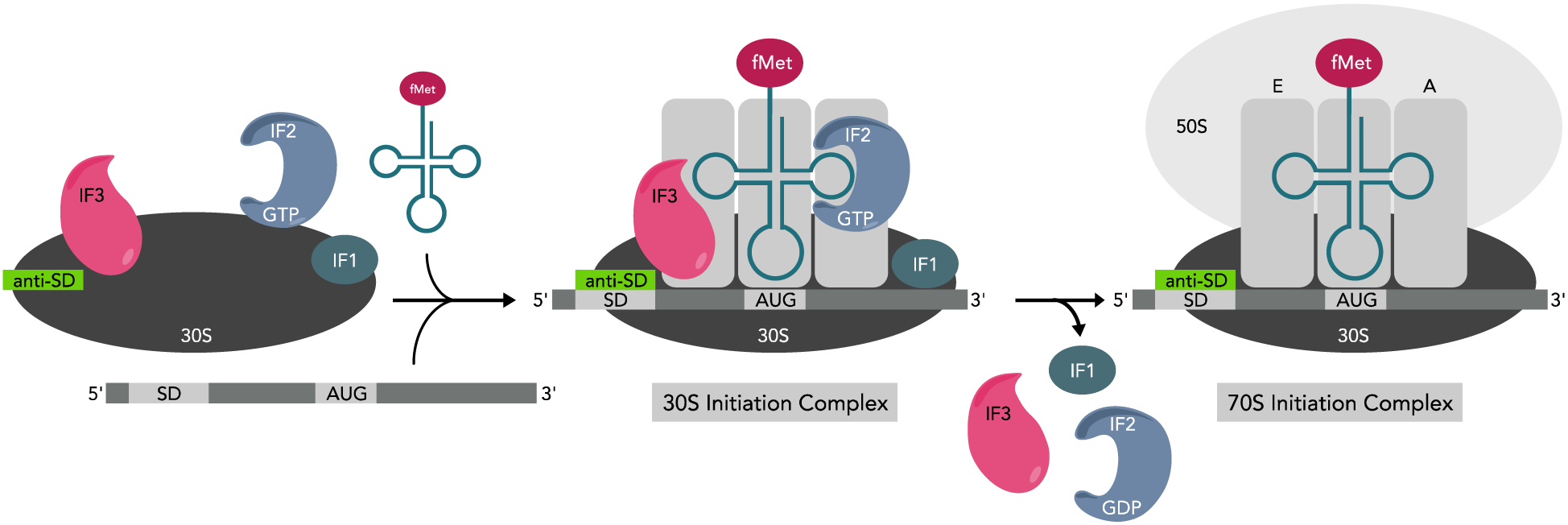
Model of translation initiation. The first step in initiation is binding of IF3 and IF2, followed by IF1, mRNA, and fMet-tRNAfMet to the 30S ribosomal subunit giving way to the 30S pre-initiation complex. Initiation factor-mediated anticodon-codon pairing between mRNA and fMet-tRNAfMet gives rise to a more stable 30S initiation complex. 50S ribosomal subunit binding stimulates GTP hydrolysis by IF2 resulting in a conformational change and initiation factor dissociation, giving rise the translationally competent 70S initiation complex.
3. Non-AUG Initiation
While translation is traditionally thought to start at AUG, genomic searches are revealing an increasing number of genes that initiate at non-AUG codons. For example in bacteria, 81.8% of genes are reported to initiate at AUG while GUG (13.8%), UUG (4.35%), CUG (0.02%), AUU (0.018%), AUC (0.006%), and AUA (0.004%) account for the rest.[14] These non-canonical initiating codons are recognized primarily by tRNAfMet, initiating with fMet and not the amino acid predicted by the codon. However, because of poor codon recognition, initiation from non-AUG codons with wildtype tRNAfMet is relatively inefficient. Of the naturally occurring non-canonical initiator codons identified, GUG and UUG initiate at greater than 10% of the efficiency of AUG, while CUG, AUU, AUC, and AUA initiate with only 0.1–1% AUG efficiency. Interestingly, a systematic study which measured initiation at all 64 codons found low, but detectable levels of initiation from the majority of codons in E. coli. This indicates that the translational machinery has a wide tolerance for initiation at codons other than AUG.[14d]
Regardless of the codon used for initiation, in nature translation always initiates with Met/fMet. Phylogenetic analyses have revealed a distant ancestral itRNA which split off into the three domains of life. Because of its early origin, the amino acid used for initiation must have been able to be synthesized via one-carbon metabolism. Met and N10-formyltetrahydrofolate, the precursors of fMet, are perfect candidates providing a possible explanation for Met as the initiating amino acid.[9, 15]
While initiation is constrained to Met in nature, a number of studies (discussed in the following sections) have shown that mis-acylated tRNAfMet can be used to initiate protein synthesis with non-Met amino acids, including ncAAs. However, to initiate translation with ncAAs the mis-acylated tRNAfMet must compete with endogenous fMet-tRNAfMet for initiation at AUG. Inefficient competition for AUG can significantly decrease the efficiency of NCI both in vitro and in vivo. One strategy for improving NCI is to use an orthogonal initiator codon that is poorly recognized by tRNAfMet, or other endogenous tRNAs. Because of its low usage in E. coli and lack of recognition by endogenous tRNAs, the amber (UAG) nonsense codon has been widely used for genetic code expansion (GCE) to incorporate ncAAs at internal positions in proteins. More than thirty years ago it was shown that a mutant of tRNAfMet bearing a CUA anticodon (carrying fGln) could efficiently initiate translation of genes in which the initiator AUG was mutated to UAG.[16] The efficiency and orthogonality of amber codon initiation has since been demonstrated in several common laboratory strains of E. coli and in Mycobacterium smegmatis.[17] Recently the effects of amber-reader tRNAfMet (tRNAfMetCUA) expression were thoroughly characterized in a genomically recoded organism in which all occurrences of UAG were removed.[18] While tRNAfMetCUA expression was found to upregulate expression of proteins associated with tRNA degradation and amino acid biosynthesis, as well as ribosome-associated proteins, proteomic analyses provided no evidence for off-target translation initiation. These data demonstrate high orthogonality of tRNAfMetCUA. Therefore, amber codon initiation is a good strategy for maintaining orthogonality and efficiency of NCI.
4. Initiation with Non-canonical Amino Acids In Vitro
In vitro translation (IVT) using mis-acylated E. coli tRNAfMet has been used to install a wide variety of diverse molecules at the N-terminus of polypeptides revealing incredible plasticity of the translational machinery for initiation with amino acids other than Met. The major hurdle for NCI in vitro is generating tRNAfMet that is mis-acylated with the desired molecule for initiation. Several methods for generating mis-acylated itRNAs for IVT reactions have been developed and are described below.
The most straightforward method for generating mis-acylated tRNAfMet takes advantage of the inherent substrate promiscuity of MetRS to acylate tRNAfMet with structural analogs of Met.[19] A major benefit of this strategy is that the aminoacyl-tRNAfMet can be formed in situ without the need for purification prior to IVT. However, this method is limited to a relatively small pool of Met analogs that are substrates for MetRS. A related strategy uses a combination of enzymatic and chemical modification in which MetRS is first used to acylate tRNAfMet with Met, followed by chemical modification of the aminoacyl-tRNAfMet at the α-amine using an appropriate electrophile (Figure 3A).[20] Following purification, the Nα-modified aminoacyl-tRNAfMet is added to IVT reactions to generate polypeptides labelled at the N-terminus. This strategy has been used to prepare Met-tRNAfMet labelled at the α-amine with biotin (1) for co-translational installation of an N-terminal affinity tag,[21] or fluorescent dyes (2-3) which afford a fluorescently labeled translation product and are useful tools for studying nascent polypeptide dynamics on the ribosome (Figure 4A).[22]
Figure 3.
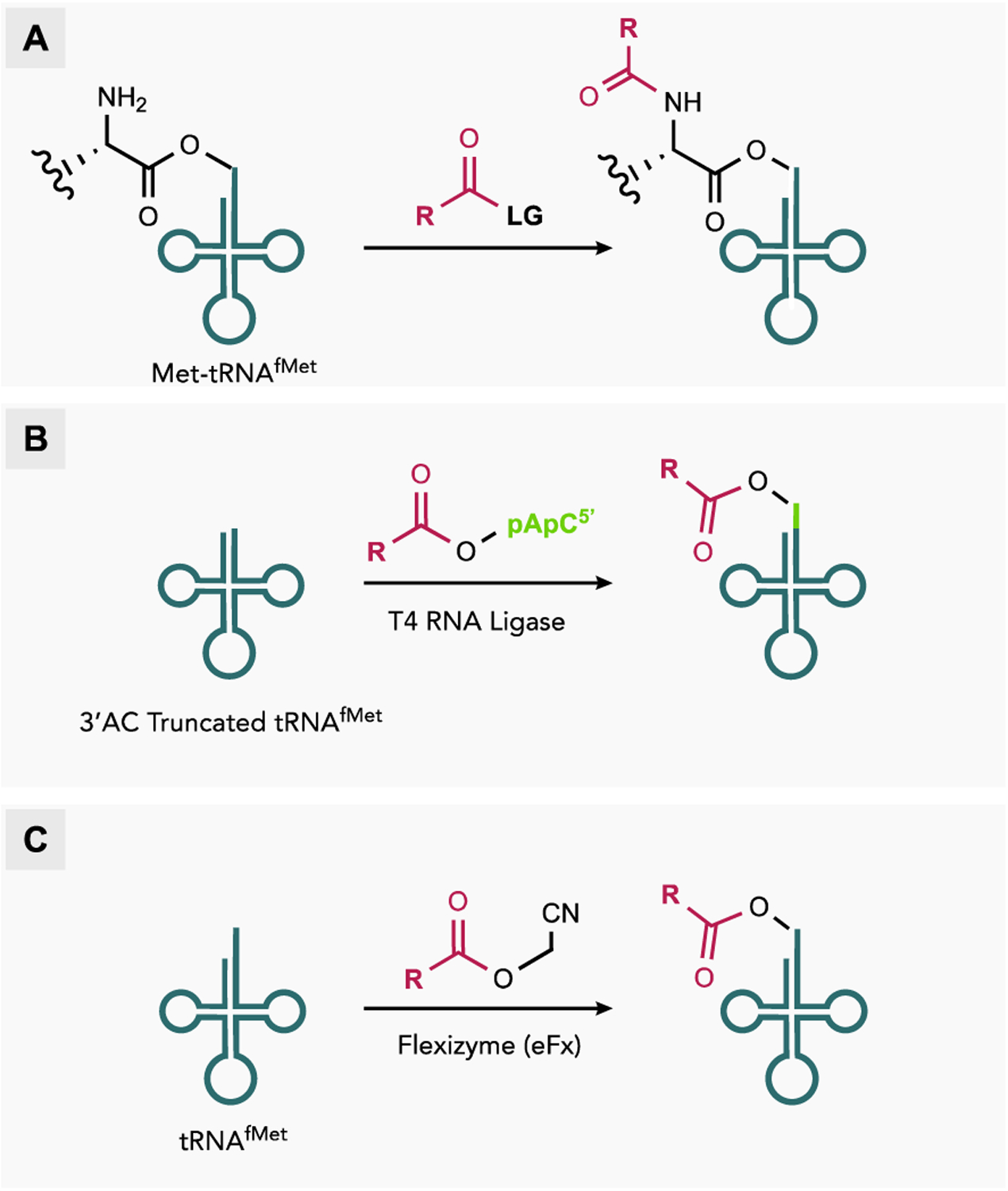
Strategies for generating mis-acylated tRNAfMet for non-canonical initiation in vitro. (A) Aminoacylation by MetRS followed by chemical modification of the α-amine. (B) Enzymatic ligation of a synthetic 2’,(3’)-O-acyl-pCpA to a truncated tRNAfMet transcript. (C) Flexizyme-catalyzed acylation.
Figure 4.
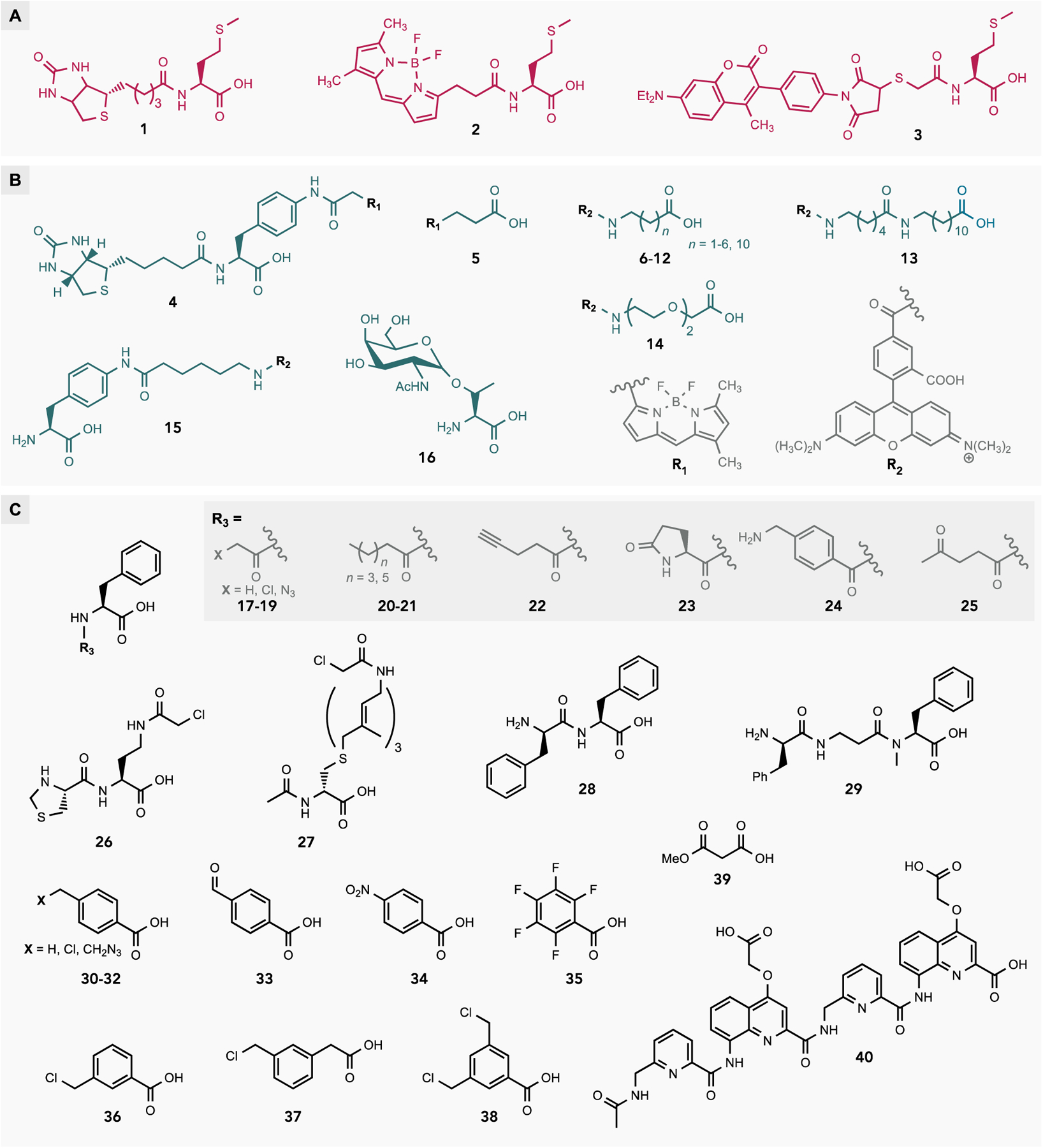
Representative carboxylic acids used to initiate translation in vitro. Mis-acylated tRNAfMets for in vitro translation were prepared by acylation with MetRS followed by chemical modification of the α-amine (A), enzymatic ligation of synthetic 2’,(3’)-O-acyl-pCpA to a 3’-truncated tRNAfMet (B), or flexizyme catalyzed acylation (C).
Chemical synthesis is another strategy to generate mis-acylated tRNAfMet for NCI with IVT. Although this technique is more laborious, it can be used to generate mis-acylated tRNAfMet bearing diverse molecules which no aaRS can recognize. Chemical synthesis of mis-acylated tRNAfMet is performed using a tRNAfMet transcript lacking the 3’-terminal pCpA dinucleotide. Alternatively, the pCpA dinucleotide of tRNAfMet purified from E. coli can be selectively removed by treatment with snake venom phosphodiesterase I to generate the truncated tRNAfMet. In parallel, the pCpA dinucleotide acylated with the desired compound is chemically synthesized and purified. In lieu of pCpA the deoxy pdCpA dinucleotide can be used. Using pdCpA greatly simplifies the synthesis of the acylated dinucleotide and yields similar translation results.[23] The truncated tRNAfMet transcript is then ligated to the synthetic 2’,(3’)-O-acyl-pCpA using T4 RNA ligase to afford the full-length mis-acylated tRNAfMet (Figure 3B).[24] Chemical synthesis methods have been used to initiate translation with various carboxylic and non-α amino acids (5-14) demonstrating unequivocally that the α-amine—modified or otherwise—is not essential for translation initiation (Figure 4B).[25] This method has also been used to initiate translation with structurally complex amino acids such as 4, a doubly-labeled amino acid used for simultaneous installation of biotin and a fluorophore at the N-terminus,[26] or 16, a glycosylated amino acid that can be used to prepare homogenously glycosylated proteins for research and pharmaceutical purposes.[27]
Flexizymes, ribozymes that catalyze tRNA acylation, are also widely used to generate mis-acylated tRNAfMet for IVT. In one fascinating study, flexizymes were used to generate tRNAfMet mis-acylated with each of the canonical amino acids which were then tested for initiation efficiency. Interestingly, all of the canonical amino acids were shown to be able to initiate translation with the majority initiating at greater than 50% the efficiency of fMet.[28] In general, the efficiency of initiation with canonical amino acids was found to correlate with their ability to be formylated by FMT. It was further shown that the efficiency of initiation could be improved by pre-acylating the α-amine. Flexizymes have also been used to acylate tRNAfMet and initiate translation with a myriad of non-canonical molecules for various applications (Figure 4C). For example, 17-26 are Nα-modified amino acids, several of which contain reactive moieties that have been used to generate side-chain-to-backbone and backbone-to-backbone cyclic peptides.[28–29] D-amino acids, which are frequently components of natural cyclic peptides, have also been used to initiate translation via flexizyme-catalyzed acylation of tRNAfMet.[29d, 30] One such example is 27, whose terpene side chain containing a reactive chloroacetamide was used to generate a macrocyclic peptide inspired by the structure of the antifungal natural product amphotericin B.[29e] Flexizymes can also be used to acylate itRNAs with short peptides, such as 28-29, a useful strategy for installing multiple non-canonical, D-, and β-amino acids.[31] In addition to amino acids and peptides, flexizymes have been used to acylate itRNAs with small aromatic carboxylic acids such as 30-38, 1,3-dicarbonyls such as 39, and large foldamers composed of quinoline and pyridine, such as 40, to generate foldamer-peptide hybrids with unique structural properties.[29b, 32] Thus, at least in vitro, the translational apparatus shows remarkable versatility with regard to the types of molecules that can be used to initiate translation.
The type of IVT system and choice of initiating codon are two factors that have significant influence on the efficiency of NCI. Early studies on NCI utilized E. coli S30 cell-free extracts supplemented with the mis-acylated tRNAfMet. Initiation with non-Met amino acids in S30 extracts is very inefficient because of competition with endogenous fMet-tRNAfMet.[22b, 22d, 24d] As discussed in the previous section, competition from endogenous fMet-tRNAfMet can be prevented if the anticodon of the mis-acylated tRNAfMet is mutated from CAU to CUA, resulting in initiation at UAG codons. In this way, the efficiency of NCI can be improved from ~1–2% initiating at AUG to 27–67% initiation at UAG.[24d] Although there is no competition from endogenous fMet-tRNAfMet with UAG initiation, initiation with undesired amino acids can still occur by recycling of the mis-acylated tRNAfMetCUA. After one cycle, the itRNA can be aminoacylated by endogenous aaRSs. To circumvent this misincorporation, reconstituted translation systems have grown in popularity. Reconstituted translation systems contain individually prepared translation components (ribosomes, aaRSs, tRNAs, etc.) which are recombined for IVT, allowing precise control of the reaction components.[33] Therefore, by excluding Met, tRNAfMet, or MetRS quantitative NCI with mis-acylated tRNAfMet can be achieved.[19a, 34] While vastly more efficient than cell extracts for NCI, reconstituted systems require numerous components that must be individually purified and thus, are significantly more costly.[35]
5. Initiation with Noncanonical Amino Acids In Vivo
In vitro studies have demonstrated remarkable versatility of the translational machinery for initiation with diverse amino and non-α amino acids. However, in vivo NCI remains a significant challenge. The fundamental hurdles for in vitro and in vivo initiation are the same: generating the mis-acylated tRNAfMet and overcoming competition with fMet-tRNAfMet. However, unlike IVT, there is far less control of the individual reaction components in vivo. Two widely used strategies for GCE, residue-specific incorporation and site-specific incorporation have both been used for NCI in vivo. Each of these techniques has unique benefits and challenges which are discussed below.
Residue-specific incorporation (RSI) is a method for GCE that is used to incorporate ncAAs which are structural analogs of canonical amino acids. This method takes advantage of the inability of aaRSs to distinguish between their native substrate and an isostructural non-canonical analog.[36] When provided in the growth media, the aaRS will aminoacylate its cognate tRNA with the analog and the aminoacyl-tRNA will then insert the analog co-translationally in response to the corresponding sense codon (Figure 5A). Two features of E. coli MetRS, high conformational flexibility of its active site and the lack of an editing domain, make it particularly well-suited for the RSI technique.[37] More than sixty years ago it was shown that selenomethionine (41) could efficiently replace Met for incorporation into proteins when supplemented into the growth media of Met auxotrophic E. coli.[38] Since then, this strategy has been used to incorporate numerous ncAAs, that are structural analogs of Met, using both wildtype and mutant variants of MetRS (Figure 6).[37, 39] Of these amino acids, 41 and 42 contain heavy atoms for use in X-ray crystallography, 45 and 46 are fluorinated amino acids useful for their environmentally sensitive 19F NMR signal, 54-58 contain azide or alkyne moieties for azide-alkyne click labelling, while 59 with its diazirine moiety is potentially a useful photo-crosslinker. For Met analogs that are poorly activated by MetRS, the efficiency of their incorporation can be improved by overexpressing MetRS and carefully controlling expression conditions, such as induction time and concentrations of Met and ncAA.[40] Biochemical analyses of proteins expressed in the presence of Met analogs have unequivocally demonstrated incorporation of Met analogs at the N-terminal position, indicating that these analogs are charged to tRNAfMet and initiate translation in vivo.[37, 39b, 39c] However, with RSI, unlike site-specific incorporation (vide infra), all instances of the canonical amino acid are replaced with the non-canonical analog. Therefore, Met analogs will also be incorporated at internal positions by the elongator tRNAMet in response to internal AUG codons.
Figure 5.
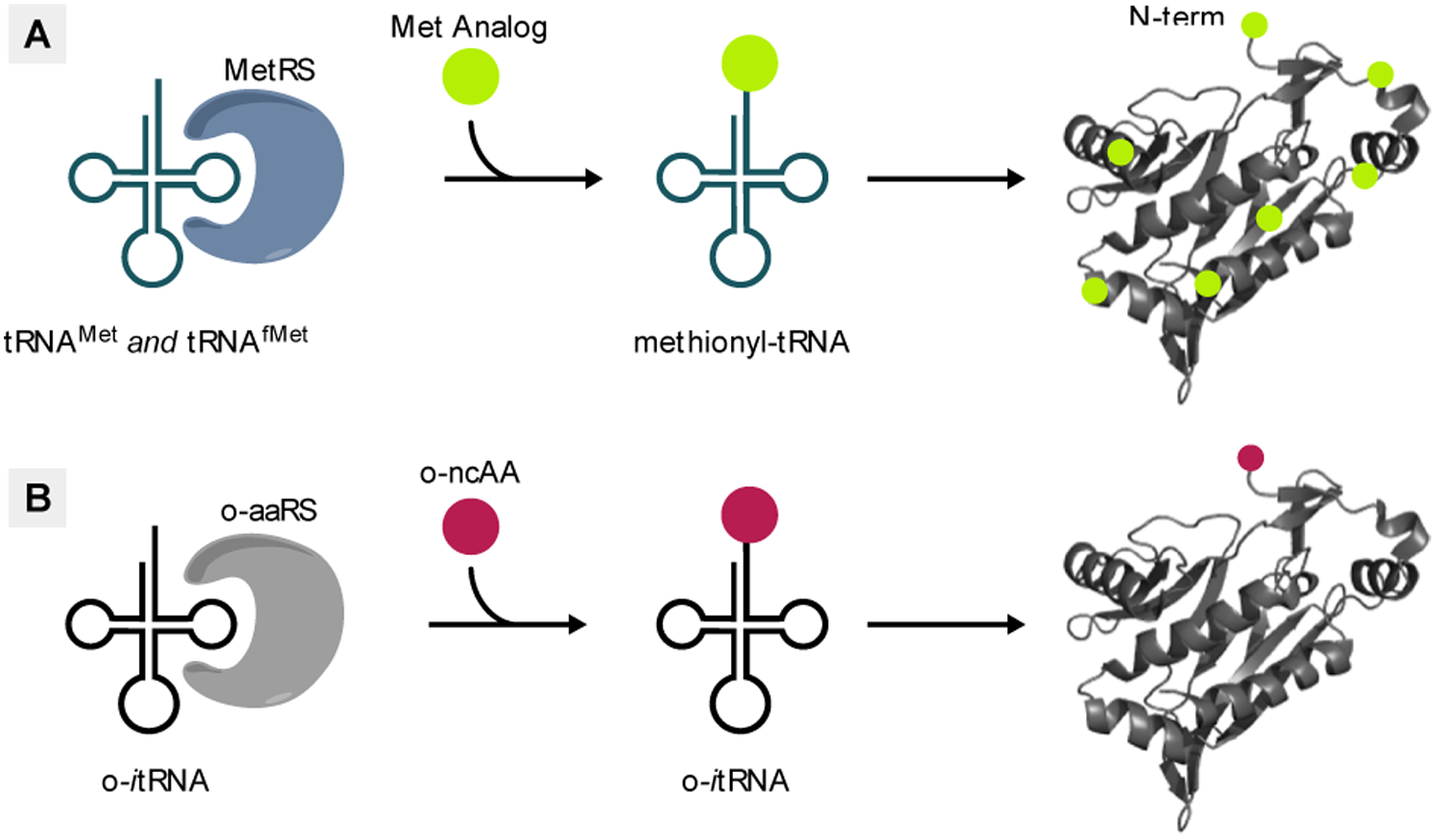
Methods for genetic code expansion. (A) Residue-specific incorporation uses endogenous aaRS/tRNA pairs to incorporate ncAAs that are structural analogs of canonical amino acids. This method replaces all instances of a canonical amino acid with the ncAA. (B) Site-specific incorporation uses orthogonal aaRS/tRNA pairs to incorporate ncAAs that are not recognized by endogenous aaRSs. This method allows for incorporation of ncAAs at defined sites within the protein.
Figure 6.
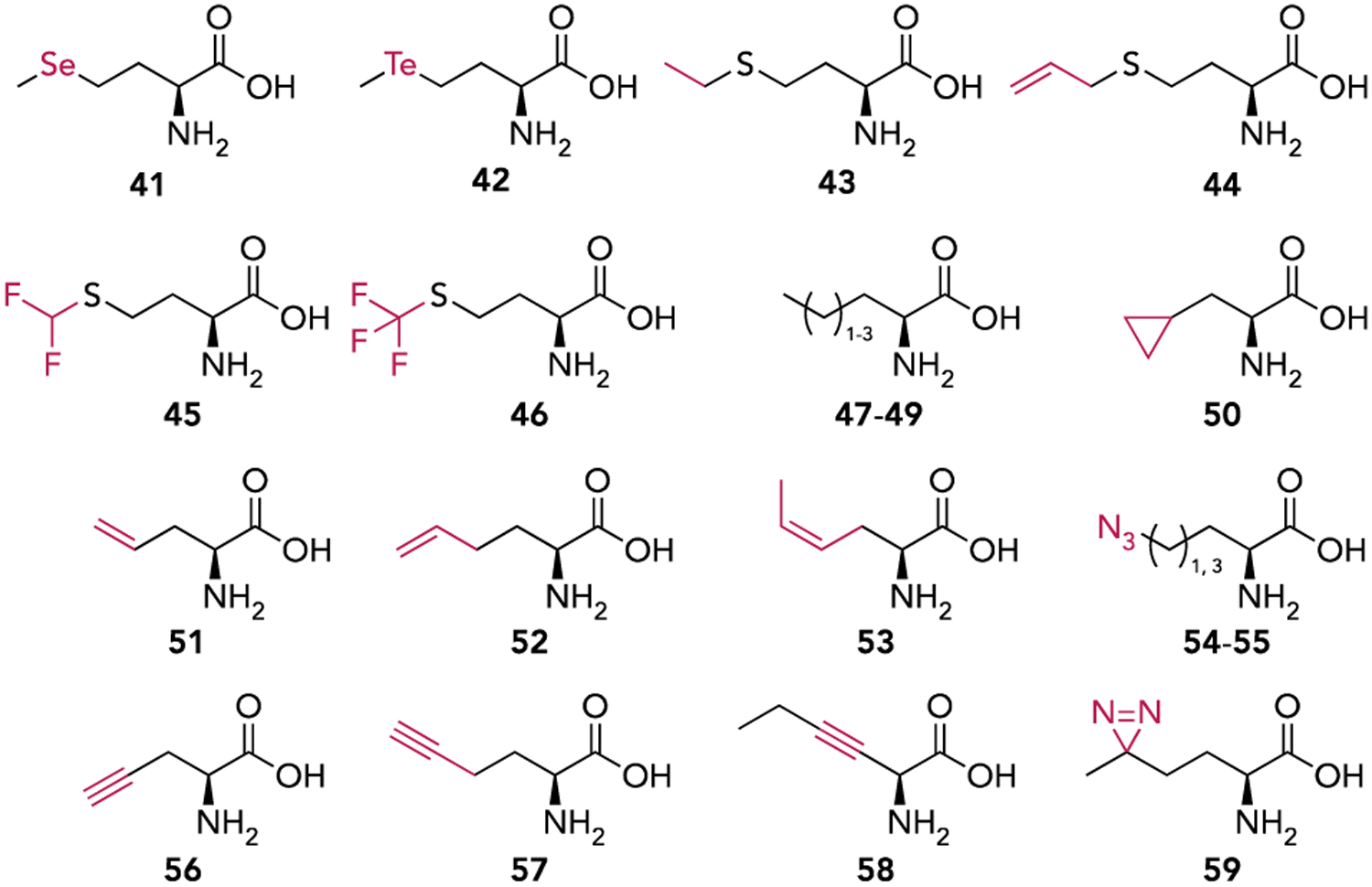
The structures of methionine analogs used to initiate translation in vivo via the residue-specific incorporation technique.
While RSI generally affords high protein yields, there are two significant limitations of this methodology. The first is the heterogeneity of products that are usually obtained. As discussed above, fMet-tRNAfMet competes with the non-canonical aminoacyl-tRNAfMet for initiation at AUG codons resulting in a mixture of products containing both Met and the ncAA at the initiating position. The use of Met auxotrophic E. coli strains and chemically defined growth media, which allows precise control of the concentration of ncAA and canonical amino acids, can limit competition from fMet-tRNAfMet and has allowed for near quantitative initiation with certain ncAAs.[39b] However, the incorporation efficiency differs with each ncAA, and most analogs afford proteins with significant Met incorporation. The second major limitation of this method results from lack of site-selectivity arising from the fact that Met analogs are incorporated in response to both initiating and elongating AUG codons. Several strategies have been employed to limit incorporation of Met analogs to the initiating position. One straight forward approach is to replace Met residues at internal positions with structurally similar canonical amino acids.[41] However, this results in a mutant target protein and therefore is not applicable if internal Met residues are necessary for protein structure or function. An alternative approach for site-selectivity uses two MetRSs with non-overlapping substrate recognition. For example, a mutant E. coli MetRS which recognizes the analog azidonorleucine (55, Anl), but not Met, was shown to be able to aminoacylate mammalian initiator tRNAMet but not elongator tRNAMet. Expressing this mutant E. coli MetRS in HEK293T afforded specific incorporation of Anl at the initiating position and Met at internal AUG positions.[42] A similar strategy was used in E. coli employing what was thought to be an orthogonal MetRS from Sulfolobus acidocaldarius (Sa-MetRS) with unique ncAA recognition.[43] It was hypothesized that deleting elongator tRNAMet from the E. coli genome with rescue by Sa-MetRS/Sa-tRNAMet would allow for specific incorporation of ethionine (43, Eth) at elongator AUG codons facilitated by Sa-MetRS/Sa-tRNAMet while incorporation of azidohomoalanine (54, Aha) could be restricted to the initiator position, incorporated by endogenous MetRS/tRNAfMet. However, this strategy was met with limited success as it was found that the E. coli and S. acidocaldarius MetRS enzymes were not completely orthogonal. To overcome this problem, it was proposed that a demonstrated orthogonal aaRS/tRNA pair, such as the pyrrolysyl-tRNA synthetase (PylRS)/tRNAPyl could be evolved for Met recognition and used to replace elongator tRNAMet.[43] Regardless of the method used to improve site-selectivity, RSI will still cause global integration of the amino acid analog into the host proteome. This global reassignment of, e.g., Met, may have undesired effects on gene expression and growth of the host organism.
The above method is limited to ncAAs that can be recognized by MetRS. In order to expand the ncAAs which can be used to initiate translation in vivo, the endogenous translation machinery can also potentially be used. More than thirty years ago it was shown that anticodon mutants of tRNAfMet are aminoacylated by aaRSs other than MetRS.[46] Furthermore, it was shown that these mis-acylated tRNAs can initiate translation of mRNA carrying the corresponding mutation at the initiating position. In this way, in vivo initiation with glutamine, valine, isoleucine, phenylalanine, and lysine has been demonstrated.[16, 44] Glutamine and valine initiation has also been demonstrated in eukaryotic cells.[45] As with in vitro NCI, the efficiency of initiation in vivo correlated with how well the amino acid was formylated by FMT.[44a] With this in mind, RSI could be used to incorporate other amino acid analogs through aaRSs other than MetRS.[36a] While this strategy would suffer the same limitation discussed for Met analog incorporation, it would significantly broaden the scope of ncAAs that can be used to initiate in vivo. However, to our knowledge this has not yet been reported.
An alternative GCE strategy is site-specific incorporation (SSI). This strategy utilizes aaRS/tRNA pairs that are orthogonal to the host translation system (o-aaRS/o-tRNA) which incorporate ncAAs that are themselves orthogonal (i.e. not substrates for endogenous aaRSs). The o-aaRS is used to aminoacylate its cognate o-tRNA which then incorporates the ncAA site-specifically, usually in response to a redefined nonsense codon (Figure 5B). The tyrosyl-tRNA synthetase/tRNATyr from Methanocaldococcus jannaschii (Mj-TyrRS/Mj-tRNATyr) and the PylRS/tRNAPyl from Methanosarcina barkeri or mazei (and more recently Methanomethylophilus alvus) are two such o-aaRS/o-tRNA pairs that are widely used for GCE.[46] As both pairs originate from archaea, they are orthogonal in E. coli and the PylRS pair is additionally orthogonal in eukaryotic cells. Wildtype and mutant Mj-TyrRS and PylRS have been used to encode more than 150 different ncAAs in E. coli and a variety of other host organisms.[46a, 46d] However, as both Mj-tRNATyr and tRNAPyl are elongator tRNAs, they lack sequence motifs that allow translation initiation. Recently, we engineered an amber-reader tRNA (itRNATy2) that is aminoacylated by Mj-TyrRS and functions both as an elongator and initiator tRNA in E. coli.[47] Using itRNATy2, in conjunction with mutant Mj-TyrRSs we demonstrated NCI at a UAG codon with sixteen different aromatic ncAAs (60-75), bearing diverse sidechains, in vivo (Figure 7). We further found that deleting three of the four copies of tRNAfMet from the genome of E. coli significantly improved the efficiency of NCI. This strategy is analogous to using reconstituted translation systems in vitro in which the concentration of tRNAfMet is reduced. To our knowledge, this work was the first to utilize an o-aaRS/o-tRNA pair to initiate translation site-specifically using ncAAs unrelated to Met. In theory, an itRNA substrate for other aaRSs could be developed using a similar strategy.
Figure 7.
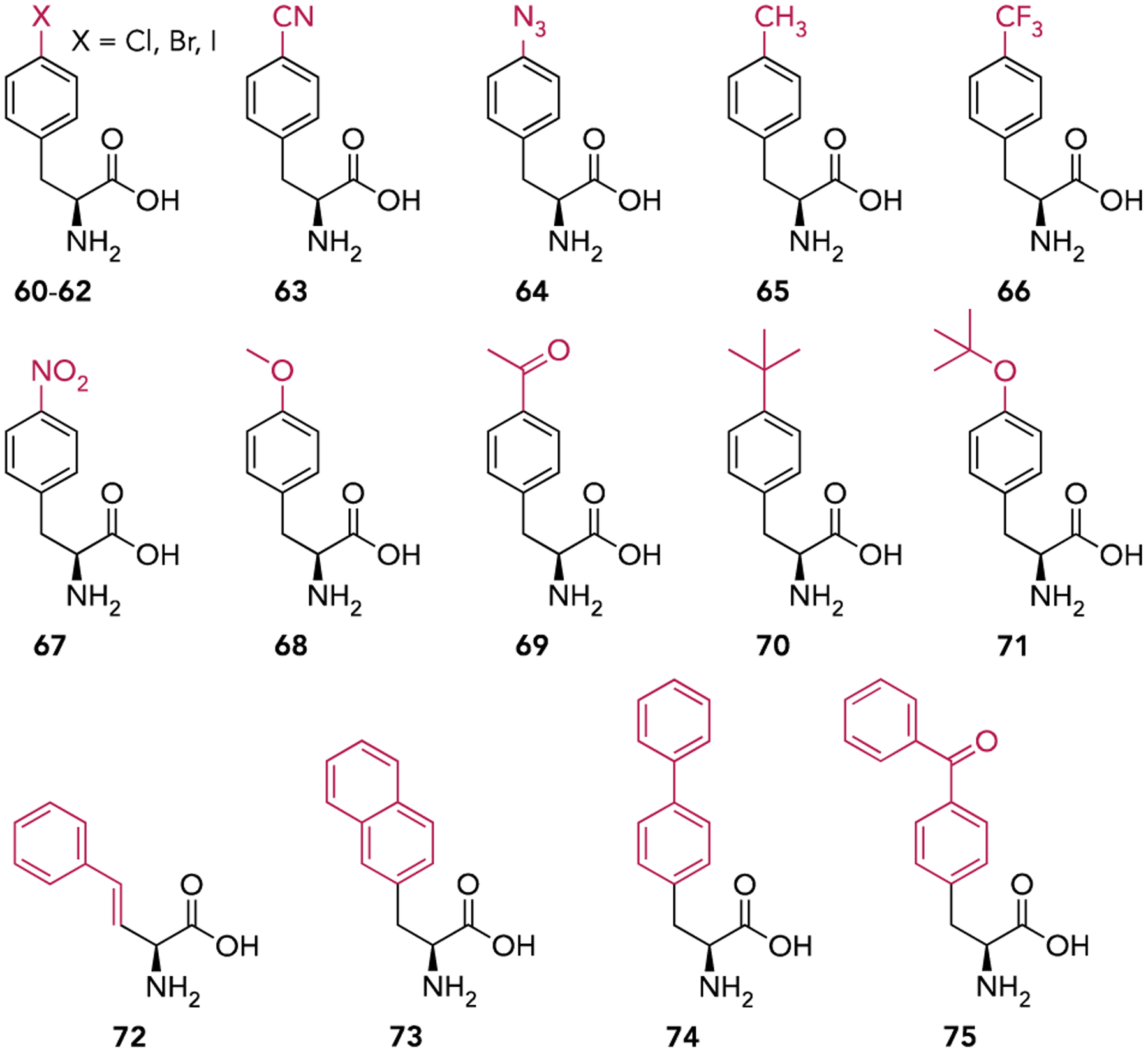
The structures of non-canonical amino acids used to initiate translation in vivo via site-specific incorporation.
In vivo NCI has also reportedly been achieved using chemically synthesized aminoacyl-itRNAs in human cells.[48] To generate the mis-acylated itRNA, a human itRNA transcript was enzymatically aminoacylated with the Nα-modified Met derivative Cy5-Met. After purification, the aminoacyl-itRNA was transfected into HeLa cells which allowed for SSI with Cy5-Met. NCI was confirmed with two reporter proteins, EGFP and the HIV-Tat protein[48]
While NCI is a convenient tool for producing N-terminally modified recombinant proteins in vivo, there are several important considerations that are not applicable when ncAAs are incorporated at an internal position of a protein. The first is how, and to what extent, the N-terminal residue will be modified. For example, in E. coli, canonical initiation occurs with Nα-formylmethionine but the formyl group is removed from most proteins by peptide deformylase.[49] However, in our studies we found that a significant fraction of superfolder green fluorescent protein which initiated with the aromatic ncAA 4-methyl-l-phenylalanine (65, MeF) retains the formyl group—likely a consequence of inefficient recognition of MeF by the peptide deformylase. Furthermore, it was shown that, like Met, Anl is co-translationally Nα-acetylated in eukaryotic cells.[42] However, whether more structurally diverse ncAAs will carry this modification is unknown. A second consideration is the extent of N-terminal residue excision. In E. coli, the initiating Met residue is excised from ~60% of proteins.[50] It has been shown that Met analogs are also removed in a similar fashion; however, the behavior of more diverse ncAAs is not known.[41b, 51] It should be noted that N-terminal Met removal largely depends on the identity of the second amino acid.[52] Inefficient ncAA incorporation resulting from poor formylation may be overcome by overproduction of the o-itRNA.[53] Likewise, changes in Shine-Dalgarno sequence in the mRNA or use of an orthogonal ribosome evolved to reduce initiation with endogenous tRNAfMet may allow for enhanced initiation with ncAAs.[54] Finally, the identity of the N-terminal residue strongly influences the in vivo half-life of proteins.[55] Current studies have shown that ncAAs incorporated at the N-terminus of proteins have varying effects on the stability of proteins in vivo.[56] However, protein stability may be difficult to predict when more diverse molecules are used for initiation in vivo.
6. Summary and Outlook
In this review we discuss the different methods that have been used to introduce diverse molecules into the initiator position of a protein both in vitro and in vivo. Initiating with diverse molecules expands the platform for protein engineers, allowing modification at any position in the protein and potential for an increased substrate range at the initiator position. Reliable methods for non-canonical initiation are slowly emerging with our results providing insight into new strategies for in vivo SSI. However, information on the modification and processing of these diverse molecules at the initiator position is not well understood and will likely be the subject of future studies.
Acknowledgements
We thank Andreas Ehnbom for his help preparing this manuscript. Work in the authors’ laboratories was supported by the Center for Genetically Encoded Materials, an NSF Center for Chemical Innovation (NSF CHE-1740549), the National Institute of General Medical Sciences (R35GM122560), from the Division of Chemical Sciences, Geosciences and Biosciences, Office of Basic Energy Sciences of the Department of Energy (DE-FG02-98ER20311), and the Department of Science and Technology, Ministry of Science and Technology, Government of India.
Biographies

Jeffery Tharp obtained his BS in chemistry from Indiana State University in 2012. He completed a PhD in chemistry at Texas A&M University with Professor Wenshe Liu in 2018. His PhD work utilized genetic code expansion to screen phage-displayed peptide libraries containing diverse non-canonical amino acids. In 2018 he joined the laboratory of Dieter Söll and the Center for Genetically Encoded Materials at Yale University where he is engineering translational components to encode non-α amino acids.

Natalie Krahn obtained her BSc and PhD from the University of Manitoba under the supervision of Professor Jörg Stetefeld. Her PhD work investigated the structural and biophysical characterization of protein-protein and protein-DNA interactions. In 2018 she joined the Department of Molecular Biophysics and Biochemistry at Yale University under the supervision of Professor Dieter Söll where she is currently working on selenocysteine incorporation and genetic code expansion.

Umesh Varshney is Professor of Microbiology and Cell Biology at the Indian Institute of Science, Bangalore. The primary interest of his laboratory is to study mechanistic aspects of protein synthesis and DNA repair using Escherichia coli and mycobacteria as model organisms. He is a fellow of The World Academy of Sciences, Trieste; Indian National Science Academy, New Delhi; Indian Academy of Sciences, Bangalore; and The National Academy of Sciences (India), Allahabad.

Dieter Söll is Sterling Profesor of Molecular Biophyics and Biochemistry at Yale University. The recurring theme of his research is the understanding of the role of transfer RNA and aminoacyl-tRNA synthetases in interpreting the genetic code. He is a member of the US National Academy of Sciences, an Associate Member of the European Molecular Biology Organization (EMBO), a fellow of the American Academy of Microbiology, and a fellow of the American Asocition for the Advancement of Science.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted after peer review and appears as an Accepted Article online prior to editing, proofing, and formal publication of the final Version of Record (VoR). This work is currently citable by using the Digital Object Identifier (DOI) given below. The VoR will be published online in Early View as soon as possible and may be different to this Accepted Article as a result of editing. Readers should obtain the VoR from the journal website shown below when it is published to ensure accuracy of information. The authors are responsible for the content of this Accepted Article.
References
- [1].a) Agostini F, Völler JS, Koksch B, Acevedo-Rocha CG, Kubyshkin V, Budisa N, Angew. Chem. Int. Ed 2017, 56, 9680–9703; [DOI] [PubMed] [Google Scholar]; b) Drienovská I, Mayer C, Dulson C, Roelfes G, Nat. Chem 2018, 10, 946–952; [DOI] [PubMed] [Google Scholar]; c) Chin JW, Nature 2017, 550, 53–60. [DOI] [PubMed] [Google Scholar]
- [2].Laursen BS, Sørensen HP, Mortensen KK, Sperling-Petersen HU, Microbiol. Mol. Biol. Rev 2005, 69, 101–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Gualerzi CO, Pon CL, Cell Mol. Life Sci 2015, 72, 4341–4367; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rodnina MV, Cold Spring Harb. Perspect. Biol 2018, 10, a032664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].RajBhandary UL, J. Bacteriol 1994, 176, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Varshney U, Lee CP, RajBhandary UL, Proc. Natl. Acad. Sci. USA 1993, 90, 2305–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Seong BL, RajBhandary UL, Proc. Natl. Acad. Sci. USA 1987, 84, 334–338; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Berk V, Zhang W, Pai RD, Cate JH, Proc. Natl. Acad. Sci. USA 2006, 103, 15830–15834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Lee CP, Seong BL, RajBhandary UL, J. Biol. Chem 1991, 266, 18012–18017; [PubMed] [Google Scholar]; b) Guillon JM, Meinnel T, Mechulam Y, Lazennec C, Blanquet S, Fayat G, J. Mol. Biol 1992, 224, 359–367. [DOI] [PubMed] [Google Scholar]
- [8].a) Schulman LH, Pelka H, J. Biol. Chem 1975, 250, 542–547; [PubMed] [Google Scholar]; b) Kössel H, RajBhandary UL, J. Mol. Biol 1968, 35, 539–560. [DOI] [PubMed] [Google Scholar]
- [9].Shah RA, Varada R, Sah S, Shetty S, Lahry K, Singh S, Varshney U, Nucleic Acids Res 2019, 47, 1908–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Milon P, Carotti M, Konevega AL, Wintermeyer W, Rodnina MV, Gualerzi CO, EMBO Rep 2010, 11, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Guenneugues M, Caserta E, Brandi L, Spurio R, Meunier S, Pon CL, Boelens R, Gualerzi CO, EMBO J 2000, 19, 5233–5240; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sprink T, Ramrath DJ, Yamamoto H, Yamamoto K, Loerke J, Ismer J, Hildebrand PW, Scheerer P, Bürger J, Mielke T, Spahn CM, Sci. Adv 2016, 2, e1501502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Wu XQ, RajBhandary UL, J. Biol. Chem 1997, 272, 1891–1895; [DOI] [PubMed] [Google Scholar]; b) Mayer C, Köhrer C, Kenny E, Prusko C, RajBhandary UL, Biochemistry 2003, 42, 4787–4799. [DOI] [PubMed] [Google Scholar]
- [13].López-Alonso JP, Fabbretti A, Kaminishi T, Iturrioz I, Brandi L, Gil-Carton D, Gualerzi CO, Fucini P, Connell SR, Nucleic Acids Res 2017, 45, 2179–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Blattner FR, Plunkett G 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y, Science 1997, 277, 1453–1462; [DOI] [PubMed] [Google Scholar]; b) Binns N, Masters M, Mol. Microbiol 2002, 44, 1287–1298; [DOI] [PubMed] [Google Scholar]; c) Sacerdot C, Fayat G, Dessen P, Springer M, Plumbridge JA, Grunberg-Manago M, Blanquet S, EMBO J 1982, 1, 311–315; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hecht A, Glasgow J, Jaschke PR, Bawazer LA, Munson MS, Cochran JR, Endy D, Salit M, Nucleic Acids Res 2017, 45, 3615–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bhattacharyya S, Varshney U, RNA Biol. 2016, 13, 810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Varshney U, RajBhandary UL, Proc. Natl. Acad. Sci. USA 1990, 87, 1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Vincent RM, Yiasemides P, Jaschke PR, Matters 2019; [Google Scholar]; b) Govindan A, Miryala S, Mondal S, Varshney U, J. Bacteriol 2018, 200, e00372–00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Vincent RM, Wright BW, Jaschke PR, ACS Synth. Biol 2019, 8, 675–685. [DOI] [PubMed] [Google Scholar]
- [19].a) Josephson K, Hartman MC, Szostak JW, J. Am. Chem. Soc 2005, 127, 11727–11735; [DOI] [PubMed] [Google Scholar]; b) Hartman MC, Josephson K, Lin CW, Szostak JW, PLoS One 2007, 2, e972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Odom OW, Kudlicki W, Hardesty B, Methods Mol. Biol 1998, 77, 93–103. [DOI] [PubMed] [Google Scholar]
- [21].Taki M, Sawata SY, Taira K, J. Biosci. Bioeng 2001, 92, 149–153. [DOI] [PubMed] [Google Scholar]
- [22].a) Kudlicki W, Odom OW, Kramer G, Hardesty B, J. Mol. Biol 1994, 244, 319–331; [DOI] [PubMed] [Google Scholar]; b) Gite S, Mamaev S, Olejnik J, Rothschild K, Anal. Biochem 2000, 279, 218–225; [DOI] [PubMed] [Google Scholar]; c) Ramachandiran V, Willms C, Kramer G, Hardesty B, J. Biol. Chem 2000, 275, 1781–1786; [DOI] [PubMed] [Google Scholar]; d) McIntosh B, Ramachandiran V, Kramer G, Hardesty B, Biochimie 2000, 82, 167–174; [DOI] [PubMed] [Google Scholar]; e) Ellis JP, Bakke CK, Kirchdoerfer RN, Jungbaue LM, Cavagnero S, ACS Chem. Biol 2008, 3, 555–566; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Tsalkova T, Odom OW, Kramer G, Hardesty B, J. Mol. Biol 1998, 278, 713–723. [DOI] [PubMed] [Google Scholar]
- [23].Robertson SA, Noren CJ, Anthony-Cahill SJ, Griffith MC, Schultz PG, Nucleic Acids Res 1989, 17, 9649–9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Heckler TG, Chang LH, Zama Y, Naka T, Chorghade MS, Hecht SM, Biochemistry 1984, 23, 1468–1473; [DOI] [PubMed] [Google Scholar]; b) Hohsaka T, Methods Mol. Biol 2012, 794, 229–239; [DOI] [PubMed] [Google Scholar]; c) Robertson SA, Ellman JA, Schultz PG, J. Am. Chem. Soc 1991, 113, 2722–2729; [Google Scholar]; d) Mamaev S, Olejnik J, Olejnik EK, Rothschild KJ, Anal. Biochem 2004, 326, 25–32. [DOI] [PubMed] [Google Scholar]
- [25].a) Muranaka N, Miura M, Taira H, Hohsaka T, ChemBioChem 2007, 8, 1650–1653; [DOI] [PubMed] [Google Scholar]; b) Masanori M, Norihito M, Ryoji A, Takahiro H, Bull. Chem. Soc. Jpn 2010, 83, 546–553. [Google Scholar]
- [26].Tadayoshi E, Kaori S, Takuya C, Yasunori T, Issei I, Takahiro H, Bull. Chem. Soc. Jpn 2010, 83, 176–181. [Google Scholar]
- [27].Matsubara T, Iijima K, Watanabe T, Hohsaka T, Sato T, Bioorg. Med. Chem. Lett 2013, 23, 5634–5636. [DOI] [PubMed] [Google Scholar]
- [28].Goto Y, Ohta A, Sako Y, Yamagishi Y, Murakami H, Suga H, ACS Chem. Biol 2008, 3, 120–129. [DOI] [PubMed] [Google Scholar]
- [29].a) Takatsuji R, Shinbara K, Katoh T, Goto Y, Passioura T, Yajima R, Komatsu Y, Suga H, J. Am. Chem. Soc 2019, 141, 2279–2287; [DOI] [PubMed] [Google Scholar]; b) Kawakami T, Ogawa K, Hatta T, Goshima N, Natsume T, ACS Chem. Biol 2016, 11, 1569–1577; [DOI] [PubMed] [Google Scholar]; c) Yamagishi Y, Ashigai H, Goto Y, Murakami H, Suga H, ChemBioChem 2009, 10, 1469–1472; [DOI] [PubMed] [Google Scholar]; d) Goto Y, Iseki M, Hitomi A, Murakami H, Suga H, ACS Chem. Biol 2013, 8, 2630–2634; [DOI] [PubMed] [Google Scholar]; e) Torikai K, Suga H, J. Am. Chem. Soc 2014, 136, 17359–17361. [DOI] [PubMed] [Google Scholar]
- [30].Goto Y, Murakami H, Suga H, Rna 2008, 14, 1390–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Goto Y, Suga H, J. Am. Chem. Soc 2009, 131, 5040–5041. [DOI] [PubMed] [Google Scholar]
- [32].a) Rogers JM, Kwon S, Dawson SJ, Mandal PK, Suga H, Huc I, Nat. Chem 2018, 10, 405–412; [DOI] [PubMed] [Google Scholar]; b) Tsiamantas C, Kwon S, Douat C, Huc I, Suga H, Chem. Commun 2019, 55, 7366–7369; [DOI] [PubMed] [Google Scholar]; c) Ad O, Hoffman KS, Cairns AG, Featherston AL, Miller SJ, Söll D, Schepartz A, ACS Cent. Sci 2019, 5, 1289–1294; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lee J, Schwieter KE, Watkins AM, Kim DS, Yu H, Schwarz KJ, Lim J, Coronado J, Byrom M, Anslyn EV, Ellington AD, Moore JS, Jewett MC, Nat. Commun 2019, 10, 5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T, Nat. Biotechnol 2001, 19, 751–755; [DOI] [PubMed] [Google Scholar]; b) Murakami H, Ohta A, Ashigai H, Suga H, Nat. Methods 2006, 3, 357–359. [DOI] [PubMed] [Google Scholar]
- [34].Goto Y, Katoh T, Suga H, Nat. Protoc 2011, 6, 779–790. [DOI] [PubMed] [Google Scholar]
- [35].Passioura T, Suga H, Chem. Commun 2017, 53, 1931–1940. [DOI] [PubMed] [Google Scholar]
- [36].a) Johnson JA, Lu YY, Van Deventer JA, Tirrell DA, Curr. Opin. Chem. Biol 2010, 14, 774–780; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hoesl MG, Budisa N, Curr. Opin. Biotech 2012, 23, 751–757. [DOI] [PubMed] [Google Scholar]
- [37].Kiick KL, Weberskirch R, Tirrell DA, FEBS Lett 2001, 502, 25–30. [DOI] [PubMed] [Google Scholar]
- [38].Cowie DB, Cohen GN, Biochim. Biophys. Acta 1957, 26, 252–261. [DOI] [PubMed] [Google Scholar]
- [39].a) Boles JO, Lewinski K, Kunkle M, Odom JD, Dunlap RB, Lebioda L, Hatada M, Nat. Struct. Biol 1994, 1, 283–284; [DOI] [PubMed] [Google Scholar]; b) Budisa N, Steipe B, Demange P, Eckerskorn C, Kellermann J, Huber R, Eur. J. Biochem 1995, 230, 788–796; [DOI] [PubMed] [Google Scholar]; c) van Hest JCM, Kiick KL, Tirrell DA, J. Am. Chem. Soc 2000, 122, 1282–1288; [Google Scholar]; d) Kiick KL, Saxon E, Tirrell DA, Bertozzi CR, Proc. Natl. Acad. Sci. USA 2002, 99, 19–24; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tanrikulu IC, Schmitt E, Mechulam Y, Goddard WA, Tirrell DA, Proc. Natl. Acad. Sci. USA 2009, 106, 15285–15290; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wilson MJ, Hatfield DL, Poirier LA, FEBS Lett 1981, 128, 157–160; [DOI] [PubMed] [Google Scholar]; g) Vaughan MD, Cleve P, Robinson V, Duewel HS, Honek JF, J. Am. Chem. Soc 1999, 121, 8475–8478; [Google Scholar]; h) Duewel H, Daub E, Robinson V, Honek JF, Biochemistry 1997, 36, 3404–3416. [DOI] [PubMed] [Google Scholar]
- [40].Kiick KL, Tirrell DA, Tetrahedron 2000, 56, 9487–9493. [Google Scholar]
- [41].a) Merkel L, Beckmann HSG, Wittmann V, Budisa N, ChemBioChem 2008, 9, 1220–1224; [DOI] [PubMed] [Google Scholar]; b) Wang A, Nairn NW, Johnson RS, Tirrell DA, Grabstein K, ChemBioChem 2008, 9, 324–330. [DOI] [PubMed] [Google Scholar]
- [42].Ngo JT, Schuman EM, Tirrell DA, Proc. Natl. Acad. Sci. USA 2013, 110, 4992–4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].De Simone A, Acevedo-Rocha CG, Hoesl MG, Budisa N, Croat. Chem. Acta 2016, 89, 243–253. [Google Scholar]
- [44].a) Li SH, Kumar NV, Varshney U, RajBhandary UL, J. Biol. Chem 1996, 271, 1022–1028; [DOI] [PubMed] [Google Scholar]; b) Chattapadhyay R, Pelka H, Schulman LH, Biochemistry 1990, 29, 4263–4268; [DOI] [PubMed] [Google Scholar]; c) Pallanck L, Schulman LH, Proc. Natl. Acad. Sci. USA 1991, 88, 3872–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Drabkin HJ, RajBhandary UL, Mol. Cell Biol 1998, 18, 5140–5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].a) Dumas A, Lercher L, Spicer CD, Davis BG, Chem. Sci 2015, 6, 50–69; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Willis JCW, Chin JW, Nat. Chem 2018, 10, 831–837; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Young DD, Schultz PG, ACS Chem. Biol 2018, 13, 854–870; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wan W, Tharp JM, Liu WR, Biochim. Biophys. Acta 2014, 1844, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tharp JM, Ad O, Amikura K, Ward FR, Garcia EM, Cate JHD, Schepartz A, Söll D, Angew. Chem. Int. Ed 2019, In Press. [DOI] [PubMed] [Google Scholar]
- [48].Kim JM, Seong BL, Sci. Rep 2017, 7, 11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rajagopalan PTR, Datta A, Pei DH, Biochemistry 1997, 36, 13910–13918. [DOI] [PubMed] [Google Scholar]
- [50].Waller JP, J. Mol. Biol 1963, 7, 483–496. [DOI] [PubMed] [Google Scholar]
- [51].Wiltschi B, Merkel L, Budisa N, ChemBioChem 2009, 10, 217–220. [DOI] [PubMed] [Google Scholar]
- [52].Hirel PH, Schmitter JM, Dessen P, Fayat G, Blanquet S, Proc. Natl. Acad. Sci. USA 1989, 86, 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shetty S, Shah RA, Chembazhi UV, Sah S, Varshney U, Nucleic Acids Res 2017, 45, 2040–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].a) Shetty S, Nadimpalli H, Shah RA, Arora S, Das G, Varshney U, Proc. Natl. Acad. Sci. USA 2014, 111, 4224–4233; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schmied WH, Tnimov Z, Uttamapinant C, Rae CD, Fried SD, Chin JW, Nature 2018, 564, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].a) Bachmair A, Finley D, Varshavsky A, Science 1986, 234, 179–186; [DOI] [PubMed] [Google Scholar]; b) Tobias JW, Shrader TE, Rocap G, Varshavsky A, Science 1991, 254, 1374–1377. [DOI] [PubMed] [Google Scholar]
- [56].Kunjapur AM, Stork DA, Kuru E, Vargas-Rodriguez O, Landon M, Söll D, Church GM, Proc. Natl. Acad. Sci. USA 2018, 115, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]