

Abstract
Recent advances in the chemistry of peptides containing fluorinated phenylalanines (Phe) represents a hot topic in drug research over the last few decades. ᴅ- or ʟ-fluorinated phenylalanines have had considerable industrial and pharmaceutical applications and they have been expanded also to play an important role as potential enzyme inhibitors as well as therapeutic agents and topography imaging of tumor ecosystems using PET. Incorporation of fluorinated aromatic amino acids into proteins increases their catabolic stability especially in therapeutic proteins and peptide-based vaccines. This review seeks to summarize the different synthetic approaches in the literature to prepare ᴅ- or ʟ-fluorinated phenylalanines and their pharmaceutical applications with a focus on published synthetic methods that introduce fluorine into the phenyl, the β-carbon or the α-carbon of ᴅ-or ʟ-phenylalanines.
Keywords: α-fluorophenylalanine; β- and β,β-difluorophenylalanine; fluorinated phenylalanines; PET; pharmaceuticals application
Introduction
Major efforts have been focused on the synthesis of fluorinated organic molecules particularly for drug development. The replacement of hydrogen by fluorine has been used in the development to improve the biophysical and chemical properties of bioactives. Such tuning in properties arises from the small size of fluorine, the next in size to hydrogen. However, the high electronegativity of the fluorine leads to low polarizability and a strong covalent bond to carbon [1–4]. Therefore, the introduction of fluorine into phenylalanine (Phe) can modulate the acidity, basicity, hydrophobicity, geometry, conformation, reactivity, and moreover the bioavailability of the analogue [1]. Fluorinated amino acids (FAAs) have considerable industrial and pharmaceutical potential [2]. Also, they have played an important role as enzyme inhibitors as well as therapeutic agents [3–4]. Moreover, they modulate the properties of peptides and proteins [5–7], influencing aspects such as protein folding, protein–protein interactions, ribosomal translation, lipophilicity, acidity/basicity, optimal pH, stability, thermal stability, and therapeutic properties [8–10]. This extends to metabolic properties of membrane permeability and reactivity [11–14]. The effect of peptide structure and stability has been found to depend on the position and number of fluorine atoms within the amino acid chains [15–17]. Incorporation of fluorinated aromatic amino acids into proteins can increase their shelf life, especially in therapeutic proteins and peptide-based vaccines [18]. Enhanced catabolic stability [6] can arise from the role of particular aromatic amino acids in membrane–protein interactions [19]. Furthermore, fluorinated aromatic amino acids can alter enzymatic activity as a result of enhanced protein stability [5]. Also fluorinated aromatic amino acids can destabilize ΙΙ-cation interactions whereas their increased hydrophobicity enhances binding affinity [19]. Moreover, the incorporation of fluorinated amino acids into proteins provides the opportunity for probing structure (by NMR techniques) including protein–protein and protein–ligand interactions and consequently metabolic processes [20–21].
Fluorinated phenylalanines (FPhe) have been incorporated into various proteins and enzymes [22–25] with advantageous biophysical, chemical, and biological properties, and their effect on the stability and activity of peptides in therapeutic vaccines and enzymes has been studied [19,26–33].
In this review we provide an overview for the various syntheses of FPhes and analogues. Five different categories of FPhe are represented and are classified I–V according to the position of the fluorine(s) (Figure 1).
Figure 1.

Categories I–V of fluorinated phenylalanines.
Review
1. Synthesis of fluorinated phenylalanine of type I and II
Direct attachment of the fluorine atom to the aryl ring of Phe or fluorinated groups directly attached to a spacer extending from the aryl ring constitute types I and II (Figure 1), accordingly we reported herein different methods for their synthesis.
1.1. Negishi cross coupling of aryl halide and organozinc compounds
Jackson and co-workers reported the synthesis of a range of phenylalanine derivatives via Negishi cross-coupling reactions of aryl halides and Zn homoenolates of the protected (R)-iodoalanine 2. The reaction was activated using Pd(0) as a catalyst.
A palladium-catalyzed cross-coupling reaction between an organozinc iodide and aryl halides offers a convenient method for the direct preparation of protected fluorinated Phe analogues 3. Thus, cross coupling of the protected iodoalanine 2 with 4-fluorobromobenzene (1a) or 2- and 4-fluoroiodobenzene (1b and 1c), respectively, was accomplished using the reported coupling conditions in Scheme 1 to give the N-Boc-protected 2- or 4-fluoro-ʟ-phenylalanine esters 3a,b [34–35].
Scheme 1.

Synthesis of fluorinated phenylalanines via Jackson’s method.
On the other hand, attempted cross-coupling of fluoroiodobenzenes 1b and 1c with iodoalanine 2 at room temperature using Pd2(dba)3 (2.5 mol %) and SPhos (5.0 mol %) provided excellent yields of (S)-phenylalanines 3a (70% yield) and 3b (80% yield), respectively. Such an efficiency improvement testifies to the suitability of SPhos as a ligand for these coupling reactions, rather than the less-reactive organozinc reagents [36] (Scheme 1). Decreasing the molar ratio of Pd2(dba)3/SPhos to 0.25:0.5 mol % provided a lower yield of 3a (21%), whereas 3b showed only a slight decrease in yield (77%).
The two-inseparable para/meta isomers of all-cis-2,3,5,6-tetrafluorocyclohexylphenyliodide 4 and 5 were subjected to Jackson’s methodology for the synthesis of the appropriate amino acid products. Thus, the coupling of the zinc homoenolate of (R)-iodoalanine 2 with a mixture of 4 and 5 in the presence of Pd(dba)3 and SPhos resulted in an excellent conversion to the fully protected amino acid isomers 6 and 7, which were readily separated from each other by chromatography. Deprotection of isomers 6 and 7 gave the individual free amino acids p-(S)-8 and m-(S)-9 [37–38] (Scheme 2).
Scheme 2.

Synthesis of all-cis-tetrafluorocyclohexylphenylalanines.
Coupling of one molar equivalent of neopentyl sulfonate ester 10a or trichloroethyl ester 10b with two molar equivalents of the zincate of Fmoc-3-iodo-ʟ-alanine methyl ester 11, to form the protected ʟ-4-[sulfono(difluoromethyl)]phenylalanines 12a and 12b, was carried out using 5 mol % (PPh3)2PdCl2 and 10 mol % DIBAL in THF/DMAC 1:1 at 65–70 °C for 6 h. Partial deprotection by alkaline hydrolysis of 12a and 12b afforded ʟ-4-[sulfono(difluoromethyl)]phenylalanine derivatives 13a and 13b, respectively [39] (Scheme 3).
Scheme 3.

Synthesis of ʟ-4-[sulfono(difluoromethyl)]phenylalanine (nPt: neopentyl, TCE: trichloroethyl).
1.2. Alkylations of fluorinated aryl halides with a chiral auxiliary
Alternatively, the coupling of the bis(dimethoxybenzyl)-protected sulfonamide 14, instead of the esters 10a and 10b with zincate 11 using a variety of catalysts and different reaction conditions, was unsuccessful. However, coupling of 4-[bis(dimethoxybenzyl)difluoromethyl]benzyl bromide (14) with the lithium enolate of William`s lactone 15 gave the protected amino acid 16 in 80% yield. The desired protected amino acid 17 was readily obtained after reduction of 16 using PdCl2 as a catalyst, followed by treating the product with Fmoc-OSu in dioxane/aq Na2CO3 (99%, two steps) [40] as illustrated in Scheme 4.
Scheme 4.

Synthesis of ʟ-4-[sulfono(difluoromethyl)]phenylalanine derivatives 17.
The reaction of aminomalonate 18 with fluorinated benzyl bromides 19a–c afforded the corresponding aralkyl diesters 20a–c. Partial hydrolysis followed by decarboxylation gave the N-benzyloxycarbonyl ᴅʟ-amino acid esters 22a–c, which upon enzymatic hydrolysis of the ester group using the subtilisin-type Carlsberg enzyme led to ᴅ-amino acid esters 23a–c and the corresponding Cbz-protected p, m-fluoro-, or pentafluoro-ʟ-phenylalanine derivatives 24a–c, respectively [41] (Scheme 5).
Scheme 5.

Synthesis of fluorinated Phe analogues from Cbz-protected aminomalonates.
The stereoselective benzylation of (S)-imidazolidinone ((S)-Boc-BMI) 25 with tetrafluorobenzyl bromides 26a,b afforded the benzylated imidazolidinones 27a,b. The acidic hydrolysis of 27a,b with simultaneous deprotection to release the free amines gave amides 28a,b. Treatment of the latter with aqueous potassium hydroxide finally afforded (S)-2-amino-3-(2,3,4,5-tetrafluorophenyl)propionic acid (29a) and (S)-2-amino-3-(2,3,5,6-tetrafluorophenyl)propionic acid (29b) [42] (Scheme 6).
Scheme 6.

Synthesis of tetrafluorophenylalanine analogues via the 3-methyl-4-imidazolidinone auxiliary 25.
Alternatively, phenylalanines 29a,b were also synthesized by alkylation of 26a,b with the chiral auxiliary 31, which was obtained by reaction of the cyclic dipeptide 30 with triethyloxonium tetrafluoroborate. The alkylation reaction of 26a,b was carried out with n-BuLi in THF at −78 °C to give 32a,b. Acid hydrolysis of the alkylated product 32a,b afforded the ethyl esters of tetrafluorophenylalanine 33a,b, which by alkaline hydrolysis afforded the tetrafluoro derivatives 29a and 29b, respectively [43] (Scheme 7).
Scheme 7.

Synthesis of tetrafluoro-Phe derivatives via chiral auxiliary 31.
The syntheses of (R)-38a and (R)-38b were carried out by alkylation of the Schöllkopf reagent ((2S)-(+)-2,5-dihydro-3,6-dimethoxy-2-isopropylpyrazine, 34) with the corresponding fluorinated benzyl bromides 35a,b via intermediates 36a,b. The alkylation products were then hydrolyzed to generate the amino acid esters which were directly Boc protected to give the N-Boc-protected amino acid methyl esters 37a,b. Finally, ester hydrolysis afforded the useful Boc-(R)-amino acids 38a,b [44] (Scheme 8).
Scheme 8.

Synthesis of 2,5-difluoro-Phe and 2,4,5-trifluoro-Phe via Schöllkopf reagent 34.
A one-pot double alkylation of the chiral auxiliary 39 with benzyl iodides 40a,b gave cis-dialkyl derivatives 41a,b in 70–72% yield. The subsequent removal of the auxiliary followed by treatment with Fmoc-OSu gave the N-protected 2-fluoro- and 2,6-difluorophenylalanine derivatives 42a,b in quantitative yields [45] (Scheme 9).
Scheme 9.

Synthesis of 2-fluoro- and 2,6-difluoro Fmoc-Phe derivatives starting from chiral auxiliary 39.
The radiolabeled 2-[18F]-fluoro-ʟ-phenylalanine 46 was synthesized as a promising radiopharmaceutical agent for molecular imaging by positron emission tomography (PET). The three-step synthesis of 46 started from [18F]-fluoride exchange in 43 to generate 44. The isotope exchange was explored by using [18F]-TBA in DMF at 130 °C for 10 min to give [18F]-44. Decarbonylation of 44 was achieved by treatment with Rh(PPh3)3Cl to afford 45 and the subsequent removal of protecting groups gave 46. Conventional reactions yielded the desired product 2-[18F]FPhe 46 in 43% yield, whereas under microwave irradiation a 34% yield was obtained. Under the optimized conditions, the enantiomeric purity was reported to be ≥94% ee [46] (Scheme 10).
Scheme 10.

Synthesis of 2-[18F]FPhe via chiral auxiliary 43.
1.3. Photooxidative cyanation of fluorinated benzylamine
A convenient, protecting group-free, and semicontinuous process was reported for the synthesis of racemic fluorinated phenylalanine·HCl starting from benzylamines 47a–c. Thus, a singlet oxygen-driven photooxidative cyanation of amines 47a–c using tetraphenylporphyrin (Tpp), followed by an acid-mediated hydrolysis of the intermediate fluorinated α-amino nitrile 48a with 30% HCl aq/acetic acid, gave the 4-fluorophenylalanine·HCl 49a in a good overall yield (67%) [47] (Scheme 11).
Scheme 11.

Synthesis of FPhe 49a via photooxidative cyanation.
1.4. Hydrolysis of Erlenmeyer’s azalactone
A multistep Erlenmeyer azalactone synthesis was reported as an important method for the synthesis of fluorinated α-amino acids 53a–h. Thus, a three-component condensation of a series of fluorinated benzaldehydes 50a–h, N-acetyl- or N-benzoylglycine 51a or 51b, respectively, and an excess of acetic anhydride in the presence of sodium acetate afforded the oxazolones 52a–h. The subsequent reductive ring cleavage of 52a–h without isolation, was carried out with red phosphorus in hydroiodic acid to give the fluorinated phenylalanine analogues 53a–h. Alternatively, a two-step sequence to generate amino acids 53a–h was attempted by first hydrolysis of 52a–h to form acids 54a–h which then were reduced with P/HI to the desired products 53a–h [48]. The free amino acid 53i was prepared by the same protocol [49] (Scheme 12).
Scheme 12.

Synthesis of FPhe derivatives via Erlenmeyer azalactone synthesis.
2,5-Difluorophenylalanines with either R or S configuration were synthesized also via the Erlenmeyer azalactone method. The synthesis started with the multicomponent reaction of aldehyde 55, acetylglycine 51a and acetic anhydride to give the azalactone 56. The subsequent basic hydrolysis of 56 gave 57 that, on catalytic hydrogenation, afforded racemic difluorinated Phe 58. The isomers were separated by selective hydrolysis using a protease from Bacillius sp to generate the (S)-N-acetyl acid 59 with >99.5% ee and the corresponding (R)-N-acetyl ester 60 with >99.5% ee [50] (Scheme 13).
Scheme 13.

Synthesis of (R)- and (S)-2,5-difluoro Phe via the azalactone method.
The synthesis of 3-bromo-4-fluoro-(S)-Phe (65) was carried out by reacting 3-bromo-4-fluorobenzaldehyde (61) and N-acetylglycine (51a) to provide intermediate 63 in high yield and without purification. Then, the transition-metal-catalyzed asymmetric hydrogenation of the α-amidocinnamic acid 63 using the less frequently used ferrocene-based ligand Me-BoPhoz led to the N-acetyl-ʟ-phenylalanine derivative 64 with complete conversion and with 94% ee. The desired enantiomer (S)-65 was obtained as a single isomer (>99% ee) after selective enzymatic hydrolysis of 64 using an acylase under mild conditions (pH 8.0, 40 °C, 4 d, with CoCl2 as co-factor) [51] (Scheme 14).
Scheme 14.

Synthesis of 3-bromo-4-fluoro-(S)-Phe (65).
1.5. Direct radiofluorination of ʟ-phenylalanine
The direct radiofluorination of ʟ-phenylalanine (66) with either [18F]F2 or [18F]AcOF in trifluoroacetic acid (TFA) afforded the three isomeric o, m, and p-fluoro-ʟ-phenylalanines 46, 67, and 68, in ratio 72.5:13.9:13.6, respectively. In this reaction, [18F]AcOF showed a higher regioselectivity and less side product formation compared with [18F]F2 [52] (Scheme 15).
Scheme 15.

Synthesis of [18F]FPhe via radiofluorination of phenylalanine with [18F]F2 or [18F]AcOF.
The radiolabeled compound, 4-borono-2-[18F]fluoro-ʟ-phenylalanine (70) was prepared by direct fluorination of 4-borono-ʟ-phenylalanine (BPA, 69) with [18F]AcOF or [18F]F2. The reaction was followed by a HPLC separation using a Delta-Pak Cl8 cartridge 0.1% acetic acid as the mobile phase at a flow rate of 10 mL/min. The product was isolated in radiochemical yields of 25–35% and with a radiochemical purity of more than 99%. The 18F-labeling of 4-borono-ᴅ,ʟ-phenylalanine (BPA) provided a potential tool for cancer treatment by boron neutron capture therapy [53] (Scheme 16).
Scheme 16.

Synthesis of 4-borono-2-[18F]FPhe.
The syntheses of a variety of clinically relevant radiotracers including protected 4-[18F]fluorophenylalanines 72a,b [54] were achieved by a copper-mediated nucleophilic radiofluorination of arylstannanes 71a,b with [18F]KF (Scheme 17).
Scheme 17.

Synthesis of protected 4-[18F]FPhe via arylstannane derivatives.
1.6. Alkylation of benzophenone imine of glycine ester
Pentafluoro-ʟ-phenylalanine (77a) and 2,4-ditrifluoromethyl-ʟ-phenylalanine (77b) were synthesized through alkylation of the benzophenone imine of glycine ester 73, with perfluorinated benzylbromide 19c or 2,4-bis(trifluoromethyl)benzyl bromide (74) in the presence of 2,7-bis[O(9)-allylhydrocinchonidinium-N-methyl]naphthalene dibromide, to afford the fluorinated phenylalanine imines 75a,b with ees < 98%. The products were hydrolyzed and deprotected in a two-step protocol to afford the desired products 77a,b [55] (Scheme 18).
Scheme 18.

Synthesis of FPhe derivatives via intermediate imine formation.
Interestingly substitution of Phe by either 77b or 77a in the proteasome inhibitors bortezomib or epoxymicin, led to an increase in the efficiency as anticancer proteasome inhibitors. The fluorinated amino acids 77a and 77b were used mainly for two reasons, i.e., the ready availability and hydrophobicity [55].
Further, (S)-pentafluorophenylalanine (Pff, 77a) was used to stabilize proteins for potential applications in various protein-based biotechnologies. To improve protein stability, natural hydrocarbon amino acids were replaced with Pff 77a. The effect of enhanced protein stability upon this replacement is referred as to ‘fluoro-stabilization effect’ [56].
1.7. Knoevenagel condensation of methyl isocyanoacetate
Three isomers of fluorinated phenylalanines 53a,b and 81 were synthesized by Knoevenagel condensation of methyl isocyanoacetate (79) and the corresponding fluorinated benzaldehyde derivatives 50a,b, and m-fluorobenzaldehyde (78) as electrophiles in the presence of catalytic amounts of Cu(I) and base. The cinnamate derivatives 80a–c obtained were hydrogenated either under homogeneous or heterogeneous conditions followed by deprotection of both the amide and the ester moieties to give the racemic fluorinated phenylalanines 53a,b, or m-fluorophenylalanine (81) with good yields [57] (Scheme 19).
Scheme 19.

Synthesis of FPhe derivatives via Knoevenagel condensation.
1.8. Coupling of N-hydroxytetrachlorophthalimide esters with boronic acids
4-(2-Fluoroethyl)-ʟ-phenylalanine and 3-(2-fluoroethyl)-ʟ-phenylalanine (88a,b), respectively, were synthesized starting from partially protected ʟ- or ᴅ-aspartic acid derivatives 82 which were activated as the N-hydroxytetrachlorophthalimide esters 83. The treatment of the esters 83 with boronic acids 84a,b afforded the substituted phenylalanine derivatives 85a,b, respectively [58]. Deprotection of the hydroxy group was achieved by treatment with TBAF in THF to give 86a,b. Finally, fluorination of the alcohols 86a,b with DAST followed by deprotection gave the targeted compounds 88a,b (Scheme 20).
Scheme 20.

Synthesis of FPhe derivatives 88a,b from aspartic acid derivatives.
Cross-coupling reactions with boronic acids were found to be successful only for the synthesis of para and meta-derivatives. Several attempts were made to prepare the ortho-substituted derivatives 93 and 95. The synthesis of ᴅ,ʟ-93 or ʟ-95 was achieved by vinylation of the protected ᴅ,ʟ-N-Boc-2-bromophenylalanine (89) using a Stille coupling reaction to give the o-vinyl derivative 90 as key intermediate. A hydroboration reaction of compound 90 afforded the primary alcohol 91, which was directly fluorinated and deprotected to give the free amino acids 93 (ᴅ and ʟ). Alternatively, alcohol 91 was activated by tosylation to give 94 as a precursor for radiofluorination that was achieved to give 2-[18F]FELP ʟ-95 using [18F]-fluoride complexed with Kryptofix®/K+ followed by deprotection with HCl and purification. This product emerged as promising new PET tracer for brain tumor imaging [58] (Scheme 21).
Scheme 21.

Synthesis of 2-(2-fluoroethyl)phenylalanine derivatives 93 and 95.
1.9. Alkylation and hydrolysis of Ni(II) or Zn(II) complexes
The synthesis of a series of fluorinated phenylalanines was achieved by transamination reactions between (R) or (S)-(15-aminomethyl-14-hydroxy-5,5-dimethyl-2,8-dithia[9](2,5)pyridinophanes) 96 and the sodium salt of o, m, or p-fluoro or (trifluoromethyl)phenylpyruvic acids 97a–f. The reaction was carried out in presence of 0.5 equiv of zinc(II) acetate in the presence of NaOMe. The initially formed complexes 98a–f underwent isomerization to 99a–f. Acid hydrolysis then gave the FPhe derivatives 53a,b, 53i, 81, and 101c,d with modest enantiomeric excesses (33–66% ee) and in moderate yields [59] (Scheme 22).
Scheme 22.

Synthesis of FPhe derivatives via Zn2+ complexes.
A convenient preparative method for the synthesis of enantiomerically pure o-, m-, and p- or pentafluorinated phenylalanines 53a, 81, 101c, and 107 was carried out by the alkylation of glycine. The Ni(II) complex 104 was obtained through the reaction of 102 with glycine (103) and Ni(NO3)2. The subsequent alkylation of complex 104 with fluorine-containing benzyl chlorides 105a–d followed by hydrolysis with HCl afforded enantiomerically enriched (<90% ee) (S)-fluorinated phenylalanine derivatives 53a, 81, 101c, and 107 [60–61] (Scheme 23).
Scheme 23.

Synthesis of FPhe derivatives via Ni2+ complexes.
Following the previous method, the chiral auxiliary 108 was readily cleaved under mild acidic conditions to afford the hydrochloride salt of 3,4,5-trifluoro-Phe 109 in 86% yield and 95% ee, indicating very low racemization [62] (Scheme 24).
Scheme 24.

Synthesis of 3,4,5-trifluorophenylalanine hydrochloride (109).
1.10. PAM enzymatic catalytic amination of (E)-cinnamic acid
The enzyme phenylalanine aminomutase (PAM) from Taxus chinensis catalyzes the stereoselective isomerization of α-phenylalanine to β-phenyalanine 111a–c. Mechanistic studies showed that (E)-cinnamic acid is an intermediate in this transformation [63]. Accordingly, addition of ammonia to o, m, or p-fluoro-(E)-cinnamic acids 110a–c catalyzed by PAM afforded (R)-fluoro-β-phenylalanines 111a–c and o, m, and p-(S)-fluorophenylalanines 53a, 81, 101c, respectively, with excellent enantioselectivities (<99% ee) (Scheme 25).
Scheme 25.

Synthesis of FPhe derivatives via phenylalanine aminomutase (PAM).
1.11. From enamine intermediates
The synthesis (R)-2,5-difluorophenylalanine derivative 115 was carried out by coupling the commercially available aldehyde 55 and N-Boc phosphonate glycinate 112 to generate the enamino ester intermediate 113. The asymmetric hydrogenation of this enamine afforded the N-Boc-protected (R)-2,5-difluorophenylalanine ester 114 with >99% ee. A following alkaline hydrolysis of the ester 114 gave N-Boc-(R)-2,5-difluorophenylalanine 115 (Scheme 26) [50].
Scheme 26.

Synthesis of (R)-2,5-difluorophenylalanine 115.
After compiling the above synthetic methods, a number of conclusions can be drawn regarding the synthesis of FPhe analogues of type I and II. The most convenient method involved a Negishi cross coupling of an aryl halide and the Zn homoenolate of the protected (R)-iodoalanine 2 using a Pd(0) catalyst. This method provided a versatile range of fluorinated phenylalanine products with high enantioselectivities and in acceptable yields.
2. Synthesis of β-fluorophenylalanines of type III
2.1. Fluorination of protected (1R,2R)-2-amino-ʟ-phenylpropane-1,3-diol
Recently, Okuda et al. reported the synthesis of (3R)-3-fluoro-ʟ-phenylalanine (121) from (1R,2R)-2-amino-ʟ-phenylpropane-1,3-diol (116). Thus, Boc-protection of the amine group in 116 followed by the protection of the primary hydroxy group (Alloc) gave alcohol 117 in good yield. The fluorination of 117 was achieved by treatment with DAST to form 118. Then, selective removal of the Alloc protecting group using Pd(PPh3)4, was followed by oxidation of the resulting Boc-protected amino alcohol 119 to give the N-Boc-protected acid 120 in good yield. Finally, removal of the Boc group then generated the free amino acid (3R)-3-fluoro-ʟ-phenylalanine (121) [64] (Scheme 27). Okuda’s group used 121 in the synthesis of a nucleoside that could be used to assess the transition state of a ribosome-catalyzed peptide-bond formation [64].
Scheme 27.

Synthesis of β-fluorophenylalanine via 2-amino-1,3-diol derivatives.
2.2. Stereoselective benzylic fluorination of N-(2-phenylacetyl)oxazolidin-2-one using NFSI
Treatment of oxazolidinone 122 with N-fluorobenzenesulfonimide (NFSI) in the presence of NaHMDS afforded the fluorinated oxazolidinone derivative 123. The reductive removal of the chiral auxiliary with LiBH4 resulted in alcohol 124 which was oxidized by Dess–Martin periodinane to give (S)-(−)-2-fluoro-2-phenylacetaldehyde (125). This aldehyde is prone to racemization and decomposition and therefore was directly converted to the arylidene derivative 127, by treatment with p-toluenesulfinamide (126). Then, reaction of 127 (1.0 mmol) with 1.5/1.0 equiv of diethylaluminum cyanide (Et2AlCN)/iPrOH at −78 °C in THF gave nitrile 128. Deprotection of the latter, followed by hydrolysis of the nitrile group afforded syn-(2S,3S)-(+)-3-fluorophenylalanine (129) [65] (Scheme 28) .
Scheme 28.

Synthesis of β-fluorophenylalanine derivatives via the oxazolidinone chiral auxiliary 122.
2.3. Multistep synthesis from ethyl trifluoropyruvate hemiketal
The reaction of ethyl trifluoropyruvate hemiketal 130 with thionyl chloride in pyridine afforded the chlorinated derivative 131, which upon treatment with zinc powder in DMF, afforded the dihalogenated olefin 132. The substitution of one fluorine atom in 132 with a tributylstannyl group to give 133 was accomplished by the reaction with (Bu3Sn)2CuLi in THF at −78 °C. The reaction took place following an addition–elimination mechanism. Then, coupling of 133 with iodobenzene in the presence of Pd(PPh3)4 and CuI as the co-catalyst afforded ethyl (E)-3-phenyl-3-fluoro-2-methoxypropenoate (134) which was converted into the corresponding α-ketoacid 135 by treatment with trimethylsilyl iodide. Finally, the reaction of 135 with aqueous ammonia followed by reduction with sodium borohydride gave the racemic erythro-β-fluorophenylalanine 136 in 40% yield [66] (Scheme 29).
Scheme 29.

Synthesis of β-fluorophenylalanine from pyruvate hemiketal 130.
2.4. Fluorodehydroxylation of β-hydroxyphenylalanine
Alternatively, Kollonitach et al. prepared racemic 3-fluorophenylalanine (136) through the fluorodehydroxylation of 3-hydroxyphenylalanine (137) using sulfur tetrafluoride (SF4) in HF [67] (Scheme 30).
Scheme 30.

Synthesis of β-fluorophenylalanine (136) via fluorination of β-hydroxyphenylalanine (137).
2.5. Ring opening of aziridine derivatives by HF/Py
The ring opening reaction of aziridines 138a,b by treatment with hydrogen fluoride in pyridine afforded 3-fluorophenylalanine esters 139a,b. The subsequent enzymatic hydrolysis of esters 139a,b gave the threo-isomer 136 in an enantiomerically pure form [68–69] (Scheme 31).
Scheme 31.

Synthesis of β-fluorophenylalanine from aziridine derivatives.
On the other hand, Wade et al. reported that ester 138b (R = iPr) afforded the isopropyl 3-fluorophenylalaninate (139b) as racemate in 45–50% yield [70] under similar reaction conditions (Scheme 31).
2.6. Fluorination and reductive amination of phenylpyruvate
A direct fluorination of the ester derivatives of phenylpyruvic acids 140a,b with F2 followed by hydrolysis of the resulting fluoropyruvates in 50% isopropanol in the presence of NaHCO3 gave 3-fluoro-3-phenylpyruvate 141 in 40–50% yields [68]. The direct reductive amination gave a partially racemized mixture of threo and erythro-136 with the erythro stereoisomer 136 as the major product (Scheme 32).
Scheme 32.

Synthesis of β-fluorophenylalanine 136 via direct fluorination of pyruvate esters.
The reductive amination of 3-fluoro-3-phenylpyruvic acid (144) obtained by the fluorodehydroxylation of the enol form of ethyl 3-phenylpyruvate 142, using DAST instead of SF4 followed by hydrolysis, produced both threo and erythro-diastereomers of 136 [68] (Scheme 33).
Scheme 33.

Synthesis of β-fluorophenylalanine via fluorination of ethyl 3-phenylpyruvate enol using DAST.
2.7. Photocatalyzed benzylic fluorination of N-phthalimido phenylalanine
The photocatalyzed benzylic fluorination of phthalimide-protected phenylalanine methyl ester 145, using the photosensitizer 1,2,4,5-tetracyanobenzene (TCB), and Selectfluor in acetonitrile was carried out using a pen lamp (λmax = 302 nm). By this route, the β-fluoro derivative 146 was obtained in 62% yield as racemic mixture [71] (Scheme 34). Recently, Egami and coworker also synthesized compound 146 in 43% yield (dr = 1:1) via the fluorination of 145, however without TCB as photosensitizer, but instead using an LED light source (365 nm) and Selectfluor in MeCN [72].
Scheme 34.

Synthesis of β-fluorophenylalanine derivatives using photosensitizer TCB.
Alternatively, a visible light (14 Watt CFL) mediated benzylic fluorination of a series of N- and C-terminally protected phenylalanines 147 using Selectfluor and dibenzosuberenone in acetonitrile, afforded the β-fluorophenylalanine derivatives 148 in variable yields with partial racemization. Phthalimido and trifluoroacetyl N-terminal protecting groups (R1 = Phth or TFA) and unprotected C-terminal derivatives (R2 = H) provided the most efficient outcomes (80 and 67% yield, respectively). An N-acetyl group was also suitable as protecting group for the reaction providing the desired product with 57% yield. Also, methyl and ethyl esters as C-terminal protecting groups in combination with phthalimino as the N-terminal protecting group, were both successfully explored. However, when the trifluoroacetyl amide was used as a substrate the methyl ester performed better than the ethyl ester (74% versus 60% yield). However, N-protecting groups such as Boc, Fmoc, and Cbz were not compatible with the fluorination (0–10% yield). Moreover, when tert-butyl, trityl, and adamantyl protecting groups were installed for C-terminal protection additional fluorination, decomposition, and consequently low yields of the β-fluorinated derivatives 148 were observed [73] (Scheme 35).
Scheme 35.

Synthesis of β-fluorophenylalanine derivatives using Selectflour and dibenzosuberenone.
2.8. Fluorination of aziridinium derivatives
The N,N-dibenzylated 3-fluorophenylalanine derivative 151 was prepared with excellent diastereoisomeric ratio (dr > 99:1) from α-hydroxy-β-amino ester 142. In this case, XtalFluor-E was used to activate the OH group in the substrate and displaced by neighboring amino-group participation creating an aziridinium intermediate 150. The latter then was opened stereo- and regioselectively by fluoride to give 151 in good yield and high diastereoisomeric purity (Scheme 36). The subsequent deprotection of 151 had to be achieved with BrO3−, because hydrogenolysis resulted in defluorination [74].
Scheme 36.

Synthesis of protected β-fluorophenylalanine via aziridinium intermediate 150.
Alternatively, a series of substituted anti-β-fluorophenylalanine derivatives 154a–d was obtained from the corresponding enantiopure α‑hydroxy-β-aminophenylalanine esters [75–76] 152a–d using XtalFluor-E. The reaction also included an aziridinium ion rearrangement as the key step. Deprotection of the resultant β-fluoro-α-amino acid esters 153a–d afforded the corresponding enantiopure anti-β-fluorophenylalanines 154a–d in good yield and high diastereoisomeric purities [74] (Scheme 37).
Scheme 37.

Synthesis of β-fluorophenylalanine derivatives via fluorination of α-hydroxy-β-aminophenylalanine derivatives 152.
The deoxyfluorination of the enantiopure α-hydroxy-β-amino ester 152a or α-amino-β-hydroxyphenylalanine ester 155 [74–76] under the same conditions proceeded via aziridinium ion 156 and generated β-fluoro-α-amino ester 153a in good yield and high diastereoisomeric purity (dr > 99:1) (Scheme 38).
Scheme 38.

Synthesis of β-fluorophenylalanine derivatives from α- or β-hydroxy esters 152a and 155.
2.9. Direct fluorination of β-methylene C(sp3)−H
The direct fluorination of β-methylene C(sp3)−H bonds of Phe derivatives 157a–v having installed the bidentate auxiliary, 2-(pyridine-2-yl)isopropylamine (PIP-amine) 158, was attempted using Selectfluor in the presence of Pd(OAc)2 as a catalyst. The corresponding β-fluoro derivatives 159a–v were obtained with high stereoselectivity using DCM and iPrCN as co-solvent (30:1 v/v). Interestingly reducing the Pd(OAc)2 concentration from 10 mol % to 6 mol % led to an improved yield of 73% for 159a, 159g, and 159t. Selectfluor was found superior to other fluorination reagents such as fluoropyridinium tetrafluoroborate, 2,4,6-trimethylfluoropyridinium tetrafluoroborate, or NFSI. The fluorination process was explored with a broad range of substituted Phe derivatives. The removal of the PIP auxiliary group without affecting the newly introduced fluorine atom was attempted by a two-step, one-pot protocol involving an in situ esterification of a highly electrophilic pyridinium triflate intermediate [77] and afforded the anti-β-fluoro-α-amino acid methyl ester 160a in 52% yield and with 98.8% ee (Scheme 39).
Scheme 39.

Synthesis of a series of β-fluoro-Phe derivatives via Pd-catalyzed direct fluorination of β-methylene C(sp3)–H bonds in Phe substrates functionalized with the PIP auxiliary group.
On the other hand, when the quinoline-based ligand 162 was used, it was shown to promote the palladium-catalyzed direct electrophilic fluorination of β-methylene C(sp3)–H bonds. Thus, fluorinations of ʟ-phenylalanine 4-trifluoromethylphenylamides 161a–l carrying a range of functional groups such as fluoro, chloro, bromo, methoxy, acetyl, cyano, nitro, and trifluoromethyl, were well-tolerated and afforded the corresponding anti-β-fluoro-α-amino acids 163a–l in moderate to excellent yields [78] (Scheme 40).
Scheme 40.

Synthesis of series of β-fluorinated Phe derivatives using quinoline-based ligand 162 in the Pd-catalyzed direct fluorination of β-methylene C(sp3)–H bonds.
3. Synthesis of β,β-difluorophenylalanine derivatives of type IV via 2-phenyl-2,2-difluoroacetaldehyde derivatives
2-Phenyl- and 2-(4-fluorophenyl)-2,2-difluoroacetaldehyde 164a and 164b proved to be key starting points for the synthesis of β,β-difluorophenylalanine analogs 168a,b. The conversion of 164a,b into their respective cyanohydrins 165a,b followed by acid hydrolysis with gaseous HCl in ethanol afforded the α-hydroxy esters 166a,b. Dess–Martin oxidation [79–80] of the latter, followed by hydrolysis of the ester gave keto acids 167a,b. Finally, the reductive amination of 167a,b with 25% aqueous ammonia and NaBH4 afforded the racemic β,β-difluorophenylalanine derivatives 168a,b in 67% yield [81] (Scheme 41).
Scheme 41.

Synthesis of β,β-difluorophenylalanine derivatives from 2,2-difluoroacetaldehyde derivatives 164a,b.
An alternative approach to the difluorinated compound 168a was achieved by the condensation of 164a with (S)-1-phenylethylamine (169), to give the imine 170. Heating of imine 170 with TMSCN in the presence of zinc iodide [82] generated the nitrile 171 as a 1:1 mixture of diastereoisomers which was immediately hydrolyzed to provide the racemic carboxamide 172. The subsequent removal of the chiral auxiliary by catalytic hydrogenation then afforded the carboxamide 173. Finally, an acid-mediated hydrolysis of the carboxamide 173 to generate the free amino acids ʟ- or ᴅ-168a, was carried out with aqueous H2SO4. However, the acid hydrolysis step was accompanied with extensive racemization [81,83] (Scheme 42).
Scheme 42.

Synthesis of β,β-difluorophenylalanine derivatives via an imine chiral auxiliary.
4. Synthesis of α-fluorophenylalanine of type V via α-fluorination of Phe derivatives
The successful α-fluorination of phenylalanine derivative 174 carrying a picolinamide auxiliary to give 176 was carried out using Selectfluor as the fluorination reagent. The direct α-C(sp3)–H fluorination of the starting compound 174 was catalyzed by Cu(OAc)2 with (R)-3-hydroxyquinuclidine ligand 175. Comparative studies demonstrated that using ligand 175 rather than other ligands gave higher yields and no β-elimination products. The effective removal of the auxiliary using triflic anhydride with LiOH as nucleophile, gave product 177a in good yield. Alternative nucleophiles such as EtOH or methyl esters of amino acids, in the presence of catalytic amounts of CoCl2, afforded product 177b or fluorinated Phe dipeptides [84] 177c–g as racemic mixtures (Scheme 43).
Scheme 43.

Synthesis of α-fluorophenylalanine derivatives via direct fluorination of protected Phe 174.
5. Pharmaceutical applications of fluorinated phenylalanine derivatives
Peptides and proteins containing FPhe are important tools to identify enzyme–substrate complexes, mechanisms of protein aggregation, and modifying the chemical and thermal stabilities of proteins. The properties of protein were preserved, when low levels of fluorine are incorporated into the constituent amino acids, and were comparable with that of the original proteins. Helpfully, fluorine incorporation may favorably adjust protein function including improved stability and substrate selectivity.
5.1. Applications of FPhe derivatives in positron emission tomography (PET)
The molecular imaging technique positron emission tomography (PET) provides information on tumor metabolism, which allows for a more accurate diagnostic and therapy response in neuro-oncology, compared to, for example, magnetic resonance imaging (MRI). PET is particularly well-suited to differentiate neoplastic tissue from non-specific changes induced by chemotherapy treatments [85]. PET is particularly used for the early detection of tumors and metastases, and is an established tool for the diagnosis, staging, and the treatment planning of various malignancies. The selective imaging of tumors using PET exploits radiotracers that target aberrant cellular metabolism or increased protein expression [86–87]. Here, the 18F isotope is particularly useful for the preparation of radiotracers to be used in PET due to its relatively long half-life (109 min). In this section we highlight two selected 18FPhe derivatives which are used for PET tumor detection. 4-Borono-2-[18F]fluoro-ᴅ,ʟ-phenylalanine ([18F]FBPA, 70), is a fluorinated derivative of the parent compound designed for boron neutron capture therapy (BNCT) [53,88–92]. This compound was used for PET imaging of melanoma in animal models.
The low affinity of 178 for the ʟ-type amino acid transporter1 (LAT1), however, limited the use of this compound as PET radiotracer for brain tumor imaging [93–96]. Therefore, for further analysis and comparison with 178 (performed in vitro), as the most promising candidate 2-[18F]-2-fluoroethyl-ʟ-phenylalanine (2-[18F]FELP, 95) was selected. In a F98 glioblastoma rat model, 2-[18F]FELP exhibited improved in vitro characteristics over [18F]FET 178, especially in view of the affinity and specificity for system L [58]. Accordingly, 2-[18F]FELP 95 emerged as a promising PET radiotracer for brain tumor imaging [97–101] (Figure 2).
Figure 2.

Structures of PET radiotracers of 18FPhe derivatives.
5.2. Incorporation of FPhe for the synthesis of fluorinated drugs
5.2.1. Melflufen, an anticancer drug: 4-Fluoro-ʟ-phenylalanine ester is required for the synthesis of melflufen (179), an anticancer drug currently being in clinical trials for the treatment of relapsed and refractory multiple myeloma (RRMM) [102–103]. Melflufen is a next generation form of the more historical drug, melphalan 180 (Figure 3).
Figure 3.

Structures of melfufen (179) and melphalan (180) anticancer drugs.
5.2.2. Gastrazole (JB95008), a CCK2 receptor antagonist: 2-Fluoro-ʟ-phenylalanine derivatives are required for the synthesis of gastrazole (JB95008, 181), a potent and highly selective cholecystokinin-2 (CCK2) receptor antagonist, originally developed at the James Black Foundation [104–109]. Roberts et al. demonstrated its inhibitory activity of gastrin-stimulated growth of pancreatic cancer both in vitro and in vivo studies [110] (Figure 4).
Figure 4.

Structure of gastrazole (JB95008, 181), a CCK2 receptor antagonist.
5.2.3. Dual CCK1/CCK2 receptor antagonists: Johnson & Johnson identified compound 182 as a dual CCK1/CCK2 receptor antagonist with desirable pharmacologic properties [51,111–113] (Figure 5). As can be seen from the structure of 182, 3-bromo-4-fluoro-ʟ-phenylalanine (65) is required for the synthesis.
Figure 5.
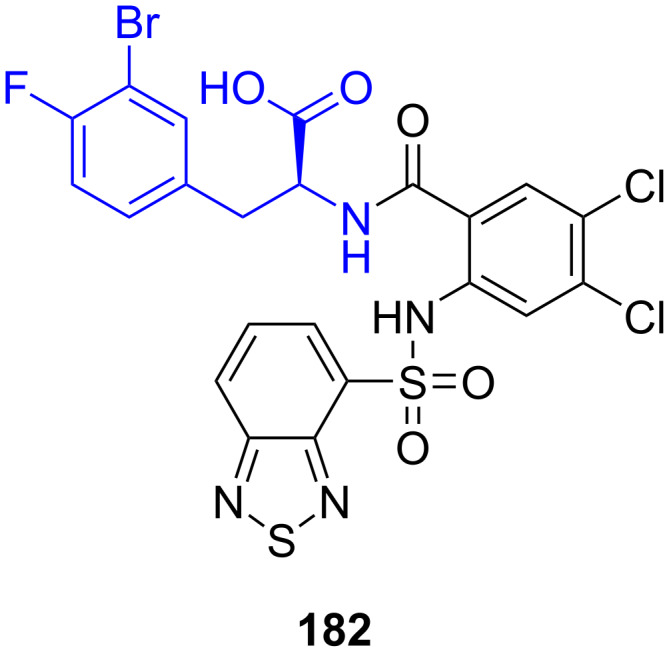
Dual CCK1/CCK2 antagonist 182.
5.2.4. Antidiabetes drugs, sitagliptin: (R)-2,4,5-Trifluorophenylalanine 38b is a constituent of sitagliptin (183, Figure 6). Sitagliptin is used to decrease the level of blood sugar in patients with type 2 diabetes and belongs to the dipeptidyl peptidase-4 (DPP-4) class of inhibitors [114–115]. This enzyme breaks down the incretins GLP-1 and GIP, gastrointestinal hormones released in response to a meal. By preventing the breakdown of GLP-1 and GIP, they are able to increase the secretion of insulin by the pancreas that modulates blood sugar level when it is high. Sitagliptin was granted FDA approval in October, 2006 [116].
Figure 6.
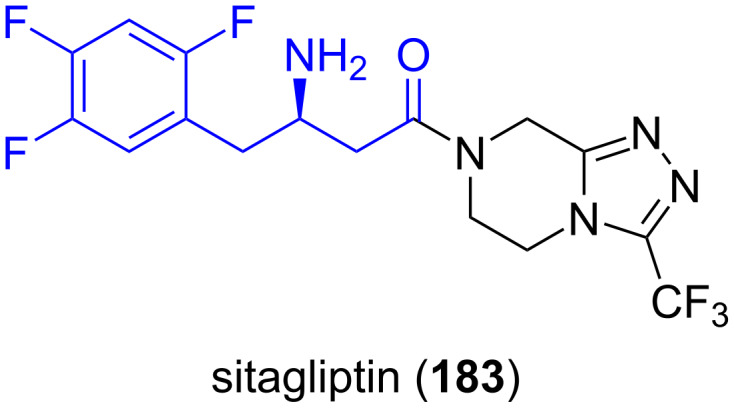
Structure of sitagliptin (183), an antidiabetic drug.
Retagliptin phosphate: Retagliptin phosphate (184) is under investigation as a DPP-4 inhibitor for treating type-2 diabetes. It is an analogue of sitagliptin which was developed for the same application [109,117], but compound 184 appears to have an improved activity [118]. Retagliptin showed efficacy in clinical trials and is now entering phase III studies. (R)-2,4,5-Trifluorophenylalanine 38b is used as a building block in the synthesis of compound 184 [119–120] (Figure 7).
Figure 7.

Structure of retaglpitin (184) and antidiabetic drug.
Evogliptin: (R)-2,4,5-Trifluorophenylalanine 38b is required for the synthesis of evogliptin (185, Figure 8), an antidiabetic drug in the dipeptidyl peptidase-4 (DPP-4) inhibitor or "gliptin" class of drugs. The South Korean pharmaceutical company Dong-A ST developed evogliptin (185) and it is currently approved for use in South Korea [121–123].
Figure 8.
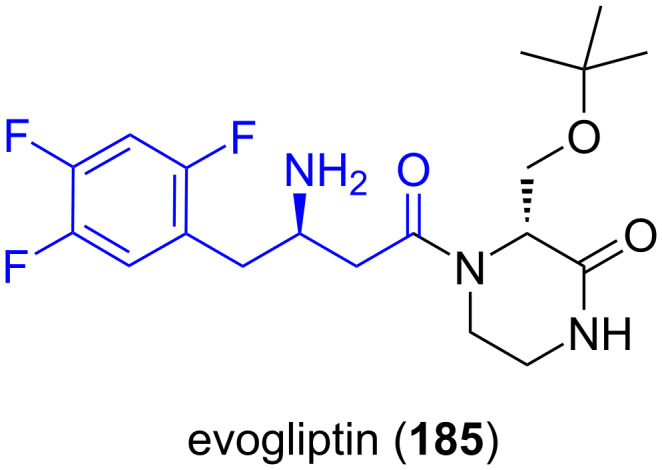
Structure of evogliptin (185), an antidiabetic drug.
LY2497282: Eli Lilly identified LY2497282 (186) as a potent and selective DPP-4 inhibitor, also for the treatment of type II diabetes. The inhibition of GLP-1 degradation by dipeptidyl peptidase IV (DPP-4) has emerged as a promising approach for treatment. (R)-2,5-Difluorophenylalanine is a required building block for the synthesis of LY2497282 [50,124] (Figure 9).
Figure 9.
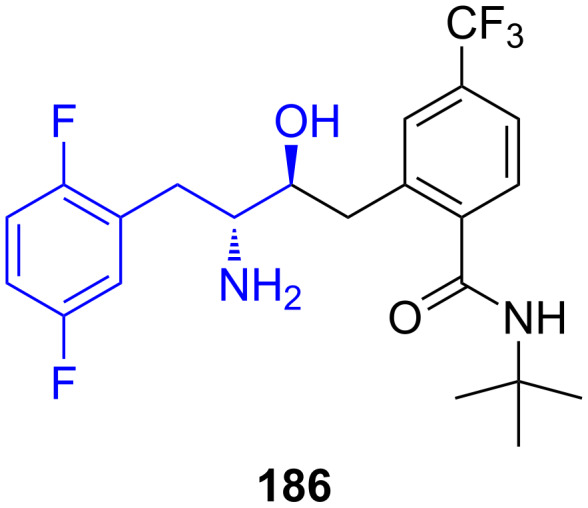
Structure of LY2497282 (186) a DPP-4 inhibitor for the treatment of type II diabetes.
5.2.5. Ulimorelin: Ulimorelin (187) is a small cyclic peptide containing ᴅ-4-FPhe. Ulimorelin acts as a selective agonist of the ghrelin/growth hormone secretagogue receptor (GHSR-1A), which is currently being developed by Tranzyme Pharma (code name TZP-101) as a first-in-class treatment for both, postoperative ileus (POI) and diabetic gastroparesis. POI describes a deceleration or arrest in intestinal motility following surgery [109,125–127] (Figure 10).
Figure 10.
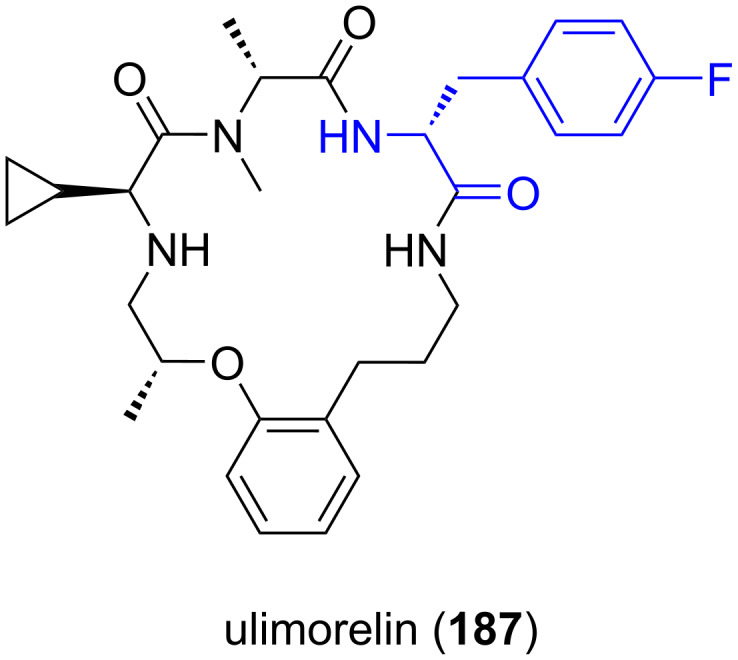
Structure of ulimorelin (187).
5.2.6. The glucagon-like peptide-1 receptor (GLP1R): 3’-Fluorophenylalanine is a key motif in the structure of the glucagon-like peptide-1 receptor (GLP1R, 188, Figure 11). GLP1R is a receptor protein found on beta cells of the pancreas and on neurons of the brain. It is participating in the modulation of blood sugar levels by increasing insulin secretion. Consequently, GLP1R plays a key role in the development of drugs to treat diabetes mellitus [128–130].
Figure 11.

Structure of GLP1R (188).
5.2.7. Sodium channel blockers (benzazepinone Nav1.7 blocker): Sodium channel blockers are used in the treatment of neuropathic pain. This is a chronic, debilitating condition that results from injury of the peripheral or central nervous system and can be triggered by a variety of events or conditions, including diabetes, shingles and chemotherapy [131]. Merck reported [132] the discovery of a structurally novel class of benzazepinone hNav1.7 voltage-gated sodium channel blockers containing 2-trifluoromethoxy-ʟ-phenylalanine derivative 189 and 3-ʟ-FPhe 190 (Figure 12) [133]. Compounds 189 and 190 were investigated as potential drugs for the treatment of neuropathic pain because they inhibited action potential firing. It was suggested, based on genetic studies, that a selective Nav1.7 inhibition, will produce robust inhibition of pain without significant side effects [134–135].
Figure 12.
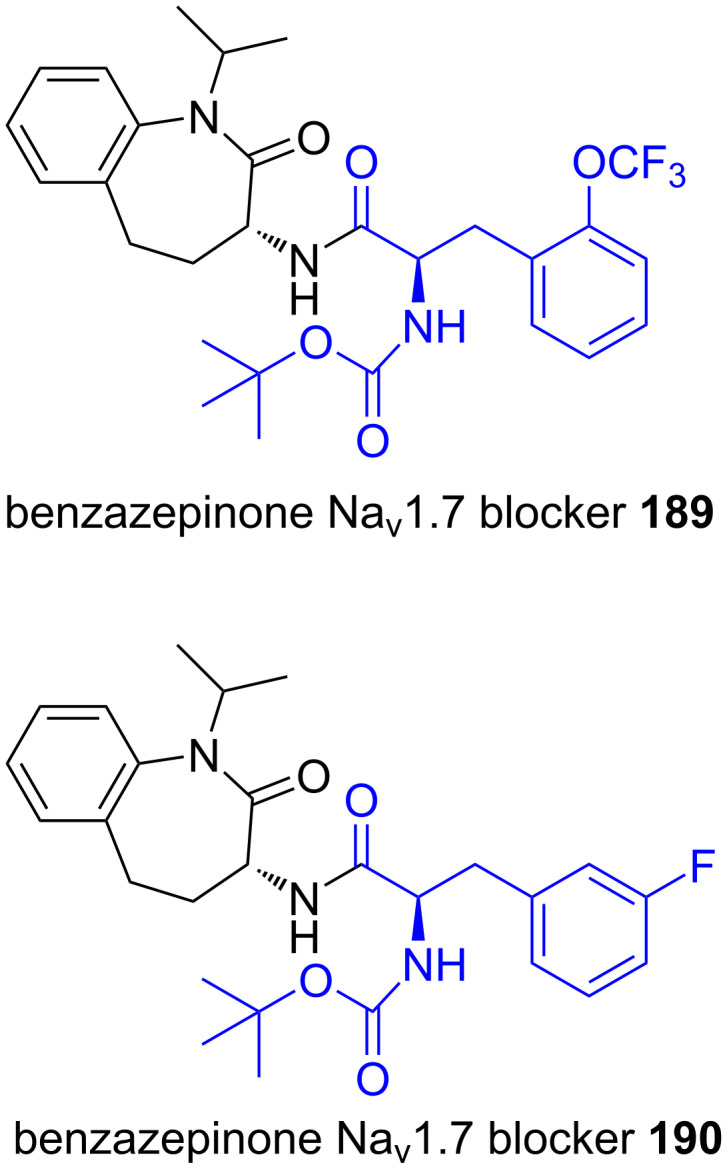
Structures of Nav1.7 blockers 189 and 190.
Conclusion
In view of the increased significance of FAAs in the development of bioactive compounds, considerable efforts were dedicated to the development of efficient synthetic protocols to FAAs. Among them, a range of fluorinated phenylalanines emerged, that have enhanced the biophysical, chemical and biological properties of bioactives. Accordingly, synthetic approaches to five distinct classes of fluorinated analogues were reviewed here. Synthetic protocols and strategies varied according to the position of the fluorine substituent. Also included were 18FPhe derivatives, some of which emerged as promising radiotracers in positron emission tomography (PET). Finally, it is notable that there are a significant number of FPhe derivatives which are nowadays incorporated into drug scaffolds of compounds either licensed or currently being studied in clinical trials.
Table 1.
List of abbreviations.
| Ada | adamantyl |
| Alloc | alloxycarbonyl |
| BNCT | boron neutron capture therapy |
| Boc | tert-butoxycarbonyl |
| Cbz | benzyloxycarbonyl |
| CCK | cholecystokinin |
| DAST | diethylaminosulfur trifluoride |
| DIBAL | diisobutylaluminium hydride |
| DMB | dimethoxybenzyl |
| DMAc | dimethylacetamide |
| DPP-4 | dipeptidyl peptidase-4 |
| DMPU | N,N′-dimethylpropyleneurea |
| FAAs | fluorinated amino acids |
| FPhe | fluorinated phenylalanine |
| Fmoc-Osu | N-(9-fluorenylmethoxycarbonyloxy)succinimide |
| GLP1R | glucagon-like peptide-1 receptor |
| kryptofix®222 | 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane |
| LAT1 | L-type amino acid transporter1 |
| LHMDS | lithium bis(trimethylsilyl)amide |
| LDA | lithium diisopropylamide |
| NaHDMS | sodium bis(trimethylsilyl)amide |
| NFSI | N-fluorobenzenesulfonimide |
| PAM | phenylalanine aminomutase |
| PET | positron emission tomography |
| PIP | 2-(pyridin-2-yl)isopropylamine |
| Phth | phthalimido |
| RRMM | relapsed and refractory multiple myeloma |
| Selectfluor | 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) |
| SPhos | 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl |
| TBAF | tetrabutylammonium fluoride |
| TMSI | trimethylsilyl iodide |
| TMSCN | trimethylsilyl cyanide |
| TCE | trichloroethyl |
| TCB | 1,2,4,5-tetracyanobenzene |
| Tr | triphenylmethyl |
| TEMPO | 2,2,6,6-tetramethylpiperidin-1-yl)oxyl or (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl |
| TBTA | tert-butyl-2,2,2-trichloroacetimidate |
| TCNHPI | N-hydroxytetrachlorophthalimide |
| XtalFluor-E | N,N-diethylamino-S,S-difluorosulfinium tetrafluoroborate |
Acknowledgments
L.F.A and M.S.A express sincere gratitude to Prof David O'Hagan for his support, also we are grateful to Faculty of Science, Alexandria University, Egypt.
This article is part of the thematic issue "Organo-fluorine chemistry V".
Contributor Information
Laila Fathy Awad, Email: laila.fathy@yahoo.com.
Mohammed Salah Ayoup, Email: mohammedsalahayoup@gmail.com.
References
- 1.Remete A M, Nonn M, Fustero S, Fülöp F, Kiss L. Tetrahedron. 2018;74:6367–6418. doi: 10.1016/j.tet.2018.09.021. [DOI] [Google Scholar]
- 2.Salwiczek M, Nyakatura E K, Gerling U I M, Ye S, Koksch B. Chem Soc Rev. 2012;41:2135–2171. doi: 10.1039/c1cs15241f. [DOI] [PubMed] [Google Scholar]
- 3.Merkel L, Budisa N. Org Biomol Chem. 2012;10:7241–7261. doi: 10.1039/c2ob06922a. [DOI] [PubMed] [Google Scholar]
- 4.Purser S, Moore P R, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 5.Odar C, Winkler M, Wiltschi B. Biotechnol J. 2015;10:427–446. doi: 10.1002/biot.201400587. [DOI] [PubMed] [Google Scholar]
- 6.Gómez-Nuñez M, Haro K J, Dao T, Chau D, Won A, Escobar-Alvarez S, Zakhaleva V, Korontsvit T, Gin D Y, Scheinberg D A. PLoS One. 2008;3:e3938. doi: 10.1371/journal.pone.0003938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noren C, Anthony-Cahill S, Griffith M, Schultz P. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 8.Ye S, Berger A A, Petzold D, Reimann O, Matt B, Koksch B. Beilstein J Org Chem. 2010;6:No. 40. doi: 10.3762/bjoc.6.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang Y, Ghirlanda G, Petka W A, Nakajima T, DeGrado W F, Tirrell D A. Angew Chem, Int Ed. 2001;40:1494–1496. doi: 10.1002/1521-3773(20010417)40:8<1494::aid-anie1494>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 10.Bilgiçer B, Fichera A, Kumar K. J Am Chem Soc. 2001;123:4393–4399. doi: 10.1021/ja002961j. [DOI] [PubMed] [Google Scholar]
- 11.Kohlmann J, Braun T, Laubenstein R, Herrmann R. Chem – Eur J. 2017;23:12218–12232. doi: 10.1002/chem.201700549. [DOI] [PubMed] [Google Scholar]
- 12.Hoffmann W, Langenhan J, Huhmann S, Moschner J, Chang R, Accorsi M, Seo J, Rademann J, Meijer G, Koksch B, et al. Angew Chem, Int Ed. 2019;58(24):8216–8220. doi: 10.1002/anie.201813954. [DOI] [PubMed] [Google Scholar]
- 13.Firooznia F, Gude C, Chan K, Fink C A, Qiao Y, Satoh Y, Marcopoulos N, Savage P, Beil M E, Bruseo C W, et al. Bioorg Med Chem Lett. 2001;11(3):375–378. doi: 10.1016/s0960-894x(00)00657-0. [DOI] [PubMed] [Google Scholar]
- 14.Lee K-H, Lee H-Y, Slutsky M M, Anderson J T, Marsh E N G. Biochemistry. 2004;43:16277–16284. doi: 10.1021/bi049086p. [DOI] [PubMed] [Google Scholar]
- 15.Jäckel C, Koksch B. Eur J Org Chem. 2005;(21):4483–4503. doi: 10.1002/ejoc.200500205. [DOI] [Google Scholar]
- 16.Jäckel C, Salwiczek M, Koksch B. Angew Chem, Int Ed. 2006;45:4198–4203. doi: 10.1002/anie.200504387. [DOI] [PubMed] [Google Scholar]
- 17.Salwiczek M, Samsonov S, Vagt T, Nyakatura E, Fleige E, Numata J, Cölfen H, Pisabarro M T, Koksch B. Chem – Eur J. 2009;15:7628–7636. doi: 10.1002/chem.200802136. [DOI] [PubMed] [Google Scholar]
- 18.Budisa N, Wenger W, Wiltschi B. Mol BioSyst. 2010;6:1630–1639. doi: 10.1039/c002256j. [DOI] [PubMed] [Google Scholar]
- 19.He T, Gershenson A, Eyles S J, Lee Y-J, Liu W R, Wang J, Gao J, Roberts M F. J Biol Chem. 2015;290:19334–19342. doi: 10.1074/jbc.m115.668343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammill J T, Miyake-Stoner S, Hazen J L, Jackson J C, Mehl R A. Nat Protoc. 2007;2:2601–2607. doi: 10.1038/nprot.2007.379. [DOI] [PubMed] [Google Scholar]
- 21.Jackson J C, Hammill J T, Mehl R A. J Am Chem Soc. 2007;129:1160–1166. doi: 10.1021/ja064661t. [DOI] [PubMed] [Google Scholar]
- 22.Wiltschi B. Expressed Protein Modifications: Making Synthetic Proteins. In: Weber W, Fussenegger M, editors. Synthetic Gene Networks: Methods and Protocols. Totowa, NJ, USA: Humana Press; 2012. pp. 211–225. [DOI] [PubMed] [Google Scholar]
- 23.Furter R. Protein Sci. 1998;7:419–426. doi: 10.1002/pro.5560070223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Munier R, Cohen G N. Biochim Biophys Acta. 1959;31:378–391. doi: 10.1016/0006-3002(59)90011-3. [DOI] [PubMed] [Google Scholar]
- 25.Moschner J, Stulberg V, Fernandes R, Huhmann S, Leppkes J, Koksch B. Chem Rev. 2019;119:10718–10801. doi: 10.1021/acs.chemrev.9b00024. [DOI] [PubMed] [Google Scholar]
- 26.Andra K K. Biochem Anal Biochem. 2015;4:No. 1000235. doi: 10.4172/2161-1009.1000235. [DOI] [Google Scholar]
- 27.Mallareddy J R, Tóth G, Fazakas C, Molnár J, Nagyőszi P, Lipkowski A W, Krizbai I A, Wilhelm I. Chem Biol Drug Des. 2012;79:507–513. doi: 10.1111/j.1747-0285.2011.01306.x. [DOI] [PubMed] [Google Scholar]
- 28.Mallareddy J R, Borics A, Keresztes A, Kövér K E, Tourwé D, Tóth G. J Med Chem. 2011;54:1462–1472. doi: 10.1021/jm101515v. [DOI] [PubMed] [Google Scholar]
- 29.Piepenbrink K H, Borbulevych O Y, Sommese R F, Clemens J, Armstrong K M, Desmond C, Do P, Baker B M. Biochem J. 2009;423:353–361. doi: 10.1042/bj20090732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doubrovina E S, Doubrovin M M, Lee S, Shieh J-H, Heller G, Pamer E, O’Reilly R J. Clin Cancer Res. 2004;10:7207–7219. doi: 10.1158/1078-0432.ccr-04-1040. [DOI] [PubMed] [Google Scholar]
- 31.Dominguez M A, Jr, Thornton K C, Melendez M G, Dupureur C M. Proteins: Struct, Funct, Genet. 2001;45:55–61. doi: 10.1002/prot.1123. [DOI] [PubMed] [Google Scholar]
- 32.Pace C J, Gao J. Acc Chem Res. 2013;46(4):907–915. doi: 10.1021/ar300086n. [DOI] [PubMed] [Google Scholar]
- 33.Dougherty D A. Acc Chem Res. 2013;46(4):885–893. doi: 10.1021/ar300265y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oswald C L, Carrillo-Márquez T, Caggiano L, Jackson R F W. Tetrahedron. 2008;64:681–687. doi: 10.1016/j.tet.2007.11.031. [DOI] [Google Scholar]
- 35.Jackson R F W, Moore R J, Dexter C S, Elliott J, Mowbray C E. J Org Chem. 1998;63:7875–7884. doi: 10.1021/jo981133u. [DOI] [Google Scholar]
- 36.Ross A J, Lang H L, Jackson R F W. J Org Chem. 2010;75:245–248. doi: 10.1021/jo902238n. [DOI] [PubMed] [Google Scholar]
- 37.Ayoup M S, Cordes D B, Slawin A M Z, O'Hagan D. Org Biomol Chem. 2015;13:5621–5624. doi: 10.1039/c5ob00650c. [DOI] [PubMed] [Google Scholar]
- 38.Ayoup M S, Cordes D B, Slawin A M Z, O'Hagan D. Beilstein J Org Chem. 2015;11:2671–2676. doi: 10.3762/bjoc.11.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu S, Dockendorff C, Taylor S D. Org Lett. 2001;3:1571–1574. doi: 10.1021/ol0158664. [DOI] [PubMed] [Google Scholar]
- 40.Hill B, Ahmed V, Bates D, Taylor S D. J Org Chem. 2006;71:8190–8197. doi: 10.1021/jo061496r. [DOI] [PubMed] [Google Scholar]
- 41.Bosshard H R, Berger A. Helv Chim Acta. 1973;56:1838–1845. doi: 10.1002/hlca.19730560603. [DOI] [PubMed] [Google Scholar]
- 42.Zheng H, Comeforo K, Gao J. J Am Chem Soc. 2009;131:18–19. doi: 10.1021/ja8062309. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Xiao W, He G, Zheng W, Yu N, Tan M. Colloids Surf, A. 2012;408:79–86. doi: 10.1016/j.colsurfa.2012.05.034. [DOI] [Google Scholar]
- 44.Kim D, Wang L, Beconi M, Eiermann G J, Fisher M H, He H, Hickey G J, Kowalchick J E, Leiting B, Lyons K, et al. J Med Chem. 2005;48:141–151. doi: 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- 45.Setty S C, Horam S, Pasupuleti M, Haq W. Int J Pept Res Ther. 2017;23:213–225. doi: 10.1007/s10989-016-9553-5. [DOI] [Google Scholar]
- 46.Castillo Meleán J, Ermert J, Coenen H H. Org Biomol Chem. 2011;9:765–769. doi: 10.1039/c0ob00440e. [DOI] [PubMed] [Google Scholar]
- 47.Vukelić S, Ushakov D B, Gilmore K, Koksch B, Seeberger P H. Eur J Org Chem. 2015;(14):3036–3039. doi: 10.1002/ejoc.201500300. [DOI] [Google Scholar]
- 48.Samet A V, Coughlin D J, Buchanan A C, III, Gakh A A. Synth Commun. 2002;32(6):941–946. doi: 10.1081/scc-120002709. [DOI] [Google Scholar]
- 49.FILLER R, NOVAR H. J Org Chem. 1960;25:733–736. doi: 10.1021/jo01075a015. [DOI] [Google Scholar]
- 50.Yu H, Richey R N, Stout J R, LaPack M A, Gu R, Khau V V, Frank S A, Ott J P, Miller R D, Carr M A, et al. Org Process Res Dev. 2008;12(2):218–225. doi: 10.1021/op700235c. [DOI] [Google Scholar]
- 51.Liu J, Deng X, Fitzgerald A E, Sales Z S, Venkatesan H, Mani N S. Org Biomol Chem. 2011;9:2654–2660. doi: 10.1039/c0ob01004a. [DOI] [PubMed] [Google Scholar]
- 52.Coenen H H, Franken K, Kling P, Stöcklin G. Int J Radiat Appl Instrum, Part A. 1988;39(12):1243–1250. doi: 10.1016/0883-2889(88)90107-4. [DOI] [Google Scholar]
- 53.Ishiwata K, Ido T, Mejia A A, Ichihashi M, Mishima Y. Int J Radiat Appl Instrum, Part A. 1991;42:325–328. doi: 10.1016/0883-2889(91)90133-l. [DOI] [PubMed] [Google Scholar]
- 54.Makaravage K J, Brooks A F, Mossine A V, Sanford M S, Scott P J H. Org Lett. 2016;18(20):5440–5443. doi: 10.1021/acs.orglett.6b02911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Geurink P P, Liu N, Spaans M P, Downey S L, van den Nieuwendijk A M C H, van der Marel G A, Kisselev A F, Florea B I, Overkleeft H S. J Med Chem. 2010;53:2319–2323. doi: 10.1021/jm9015685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiu H-P, Suzuki Y, Gullickson D, Ahmad R, Kokona B, Fairman R, Cheng R P. J Am Chem Soc. 2006;128:15556–15557. doi: 10.1021/ja0640445. [DOI] [PubMed] [Google Scholar]
- 57.Panella L, Aleixandre A M, Kruidhof G J, Robertus J, Feringa B L, de Vries J G, Minnaard A J. J Org Chem. 2006;71:2026–2036. doi: 10.1021/jo052451d. [DOI] [PubMed] [Google Scholar]
- 58.Verhoeven J, Hulpia F, Kersemans K, Bolcaen J, De Lombaerde S, Goeman J, Descamps B, Hallaert G, Van den Broecke C, Deblaere K, et al. Sci Rep. 2019;9(1):2878. doi: 10.1038/s41598-019-40013-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ando M, Kuzuhara H. Bull Chem Soc Jpn. 1990;63:1925–1928. doi: 10.1246/bcsj.63.1925. [DOI] [Google Scholar]
- 60.Kukhar V P, Belokon Y N, Soloshonok V, Svistunova N Yu, Rozhenko A B, Kuz'mina N A. Synthesis. 1993:117–120. doi: 10.1055/s-1993-25812. [DOI] [Google Scholar]
- 61.Soloshonok V A, Belokon' Yu N, Kukhar' V P, Chernoglazova N I, Saporovskaya M B, Bakhmutov V I, Kolycheva M T, Belikov V M. Russ Chem Bull. 1990;39:1479–1485. doi: 10.1007/bf00957865. [DOI] [Google Scholar]
- 62.Drouet F, Noisier A F M, Harris C S, Furkert D P, Brimble M A. Eur J Org Chem. 2014;(6):1195–1201. doi: 10.1002/ejoc.201301718. [DOI] [Google Scholar]
- 63.Szymanski W, Wu B, Weiner B, de Wildeman S, Feringa B L, Janssen D B. J Org Chem. 2009;74:9152–9157. doi: 10.1021/jo901833y. [DOI] [PubMed] [Google Scholar]
- 64.Okuda K, Hirota T, Kingery D A, Nagasawa H. J Org Chem. 2009;74:2609–2612. doi: 10.1021/jo802611t. [DOI] [PubMed] [Google Scholar]
- 65.Davis F A, Srirajan V, Titus D D. J Org Chem. 1999;64:6931–6934. doi: 10.1021/jo990947n. [DOI] [PubMed] [Google Scholar]
- 66.Shi G-q, Zhao Z-y, Zhang X-b. J Org Chem. 1995;60:6608–6611. doi: 10.1021/jo00125a060. [DOI] [Google Scholar]
- 67.Kollonitsch J, Marburg S, Perkins L. J Org Chem. 1975;40:3808–3809. doi: 10.1021/jo00913a900. [DOI] [Google Scholar]
- 68.Tsushima T, Kawada K, Nishikawa J, Sato T, Tori K, Tsuji T, Misaki S. J Org Chem. 1984;49:1163–1169. doi: 10.1021/jo00181a003. [DOI] [Google Scholar]
- 69.Tsushima T, Sato T, Tsuji T. Tetrahedron Lett. 1980;21:3591–3592. doi: 10.1016/0040-4039(80)80243-7. [DOI] [Google Scholar]
- 70.Wade T N, Gaymard F, Guedj R. Tetrahedron Lett. 1979;20:2681–2682. doi: 10.1016/s0040-4039(01)86385-1. [DOI] [Google Scholar]
- 71.Bloom S, McCann M, Lectka T. Org Lett. 2014;16:6338–6341. doi: 10.1021/ol503094m. [DOI] [PubMed] [Google Scholar]
- 72.Egami H, Masuda S, Kawato Y, Hamashima Y. Org Lett. 2018;20:1367–1370. doi: 10.1021/acs.orglett.8b00133. [DOI] [PubMed] [Google Scholar]
- 73.Bume D D, Pitts C R, Jokhai R T, Lectka T. Tetrahedron. 2016;72:6031–6036. doi: 10.1016/j.tet.2016.08.018. [DOI] [Google Scholar]
- 74.Davies S G, Fletcher A M, Frost A B, Roberts P M, Thomson J E. Org Lett. 2015;17:2254–2257. doi: 10.1021/acs.orglett.5b00880. [DOI] [PubMed] [Google Scholar]
- 75.Davies S G, Fletcher A M, Frost A B, Roberts P M, Thomson J E. Tetrahedron. 2014;70:5849–5862. doi: 10.1016/j.tet.2014.06.057. [DOI] [Google Scholar]
- 76.Davies S G, Fletcher A M, Frost A B, Lee J A, Roberts P M, Thomson J E. Tetrahedron. 2013;69:8885–8898. doi: 10.1016/j.tet.2013.08.007. [DOI] [Google Scholar]
- 77.Zhang Q, Yin X-S, Chen K, Zhang S-Q, Shi B-F. J Am Chem Soc. 2015;137:8219–8226. doi: 10.1021/jacs.5b03989. [DOI] [PubMed] [Google Scholar]
- 78.Zhu R-Y, Tanaka K, Li G-C, He J, Fu H-Y, Li S-H, Yu J-Q. J Am Chem Soc. 2015;137:7067–7070. doi: 10.1021/jacs.5b04088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meyer S D, Schreiber S L. J Org Chem. 1994;59:7549–7552. doi: 10.1021/jo00103a067. [DOI] [Google Scholar]
- 80.Dess D B, Martin J C. J Org Chem. 1983;48(22):4155–4156. doi: 10.1021/jo00170a070. [DOI] [Google Scholar]
- 81.Schlosser M, Brügger N, Schmidt W, Amrhein N. Tetrahedron. 2004;60:7731–7742. doi: 10.1016/j.tet.2004.06.086. [DOI] [Google Scholar]
- 82.Harada K, Fox S W. Naturwissenschaften. 1964;51:106–107. doi: 10.1007/bf00629604. [DOI] [Google Scholar]
- 83.Schlosser M. The Chemical and Physiological size of Fluorine. In: Soloshonok V A, editor. Enantiocontrolled Synthesis of Fluoro-organic Compounds: Stereochemical Challenges and Biomedicinal Targets. Chichester, UK: John Wiley & Sons; 1999. pp. 613–659. [Google Scholar]
- 84.Wei Q, Ma Y, Li L, Liu Q, Liu Z, Liu G. Org Lett. 2018;20:7100–7103. doi: 10.1021/acs.orglett.8b03044. [DOI] [PubMed] [Google Scholar]
- 85.Vandenberghe S, Marsden P K. Phys Med Biol. 2015;60:R115–R154. doi: 10.1088/0031-9155/60/4/r115. [DOI] [PubMed] [Google Scholar]
- 86.Martarello L, Schaffrath C, Deng H, Gee A D, Lockhart A, O'Hagan D. J Labelled Compd Radiopharm. 2003;46:1181–1189. doi: 10.1002/jlcr.779. [DOI] [Google Scholar]
- 87.Deng H, Cobb S L, Gee A D, Lockhart A, Martarello L, McGlinchey R P, O'Hagan D, Onega M. Chem Commun. 2006;(6):652–654. doi: 10.1039/b516861a. [DOI] [PubMed] [Google Scholar]
- 88.Snyder H R, Reedy A J, Lennarz W J. J Am Chem Soc. 1958;80(4):835–838. doi: 10.1021/ja01537a021. [DOI] [Google Scholar]
- 89.Ishiwata K, Ido T, Kawamura M, Kubota K, Ichihashi M, Mishima Y. Int J Radiat Appl Instrum, Part B. 1991;18:745–751. doi: 10.1016/0883-2897(91)90013-b. [DOI] [PubMed] [Google Scholar]
- 90.Ishiwata K. Ann Nucl Med. 2019;33:223–236. doi: 10.1007/s12149-019-01347-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ishiwata K, Ido T, Honda C, Kawamura M, Ichihashi M, Mishima Y. Int J Radiat Appl Instrum, Part B. 1992;19:311–318. doi: 10.1016/0883-2897(92)90116-g. [DOI] [PubMed] [Google Scholar]
- 92.Chandra S, Kabalka G W, Lorey D R, Smith D R, Coderre J A. Clin Cancer Res. 2002;8:2675–2683. [PubMed] [Google Scholar]
- 93.Tscherpel C, Dunkl V, Ceccon G, Stoffels G, Judov N, Rapp M, Meyer P T, Kops E R, Ermert J, Fink G R, et al. Neuro-Oncology (Cary, NC, U S) 2017;19:710–718. doi: 10.1093/neuonc/now243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Verhoeven J, Bolcaen J, Kersemans K, De Meulenaere V, Deblaere K, Kalala J, Van den Broecke C, Vanhove C, Goethals I, De Vos F. Neuro-Oncology (Cary, NC, U S) 2017;19(Suppl 3):iii46–iii46. doi: 10.1093/neuonc/nox036.166. [DOI] [Google Scholar]
- 95.Chiaravalloti A, Filippi L, Ricci M, Cimini A, Schillaci O. Cancers. 2019;11:1853. doi: 10.3390/cancers11121853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Moreau A, Febvey O, Mognetti T, Frappaz D, Kryza D. Front Oncol. 2019;9:No. 1134. doi: 10.3389/fonc.2019.01134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lahoutte T, Caveliers V, Camargo S M, Franca R, Ramadan T, Veljkovic E, Mertens J, Bossuyt A, Verrey F. J Nucl Med. 2004;45:1591–1596. [PubMed] [Google Scholar]
- 98.Bolcaen J, Descamps B, Deblaere K, Boterberg T, De Vos Pharm F, Kalala J-P, Van den Broecke C, Decrock E, Leybaert L, Vanhove C, et al. Nucl Med Biol. 2015;42:38–45. doi: 10.1016/j.nucmedbio.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 99.Feral C C, Tissot F S, Tosello L, Fakhry N, Sebag F, Pacak K, Taïeb D. Eur J Nucl Med Mol Imaging. 2017;44(5):812–821. doi: 10.1007/s00259-016-3586-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun A, Liu X, Tang G. Front Chem (Lausanne, Switz) 2018;5:No. 124. doi: 10.3389/fchem.2017.00124. [DOI] [Google Scholar]
- 101.Chiotellis A, Müller Herde A, Rössler S L, Brekalo A, Gedeonova E, Mu L, Keller C, Schibli R, Krämer S D, Ametamey S M. J Med Chem. 2016;59(11):5324–5340. doi: 10.1021/acs.jmedchem.6b00057. [DOI] [PubMed] [Google Scholar]
- 102.Strese S. Anticancer Activity of Melflufen: Preclinical Studies of a Novel Peptidase-Potentiated Alkylator. Acta Universitatis Upsaliensis; 2015. [Google Scholar]
- 103.Carlier C, Strese S, Viktorsson K, Velander E, Nygren P, Uustalu M, Juntti T, Lewensohn R, Larsson R, Spira J, et al. Oncotarget. 2016;7(37):59322–59335. doi: 10.18632/oncotarget.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ormerod D, Willemsens B, Mermans R, Langens J, Winderickx G, Kalindjian S B, Buck I M, McDonald I M. Org Process Res Dev. 2005;9:499–507. doi: 10.1021/op0500638. [DOI] [Google Scholar]
- 105.Chau I, Cunningham D, Russell C, Norman A R, Kurzawinski T, Harper P, Harrison P, Middleton G, Daniels F, Hickish T, et al. Br J Cancer. 2006;94(8):1107–1115. doi: 10.1038/sj.bjc.6603058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Harper E A, Roberts S P, Kalindjian S B. Br J Pharmacol. 2007;151(8):1352–1367. doi: 10.1038/sj.bjp.0707355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McDonald I M, Black J W, Buck I M, Dunstone D J, Griffin E P, Harper E A, Hull R A D, Kalindjian S B, Lilley E J, Linney I D, et al. J Med Chem. 2007;50(13):3101–3112. doi: 10.1021/jm070139l. [DOI] [PubMed] [Google Scholar]
- 108.Harper E A, Mitchell E A, Griffin E P, Kalindjian S B. Eur J Pharmacol. 2008;581:1–12. doi: 10.1016/j.ejphar.2007.11.055. [DOI] [PubMed] [Google Scholar]
- 109.Mei H, Han J, Klika K D, Izawa K, Sato T, Meanwell N A, Soloshonok V A. Eur J Med Chem. 2020;186:111826. doi: 10.1016/j.ejmech.2019.111826. [DOI] [PubMed] [Google Scholar]
- 110.Roberts S, Griffin E, Harper E, Hull R, Kalindjian S, Lilley E, Kotecha A, Shankley N, Sykes D, Watt G. Pharmacologist. 2002;44(Suppl 1):30. [Google Scholar]
- 111.Herranz R. Med Res Rev. 2003;23:559–605. doi: 10.1002/med.10042. [DOI] [PubMed] [Google Scholar]
- 112.Berna M J, Tapia J A, Sancho V, Jensen R T. Curr Opin Pharmacol. 2007;7:583–592. doi: 10.1016/j.coph.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pippel M, Boyce K, Venkatesan H, Phuong V K, Yan W, Barrett T D, Lagaud G, Li L, Morton M F, Prendergast C, et al. Bioorg Med Chem Lett. 2009;19:6376–6378. doi: 10.1016/j.bmcl.2009.09.065. [DOI] [PubMed] [Google Scholar]
- 114.Herman G A, Stevens C, Van Dyck K, Bergman A, Yi B, De Smet M, Snyder K, Hilliard D, Tanen M, Tanaka W, et al. Clin Pharmacol Ther (Hoboken, NJ, U S) 2005;78:675–688. doi: 10.1016/j.clpt.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 115.Kushwaha R N, Haq W, Katti S. Chem Biol Interface. 2014;4:137–162. [Google Scholar]
- 116.Baksh S N, McAdams-DeMarco M, Segal J B, Alexander G C. Pharmacoepidemiol Drug Saf. 2018;27:660–667. doi: 10.1002/pds.4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaihong Y, Piaoyang S, inventors. Pharmaceutical composition for treatment of type 2 diabetes. US 8,476,272 B2. U.S. Patent. 2013 Jul 2;
- 118. [Feb 25;2020 ];Drugbank, Retagliptin. Available from: https://www.drugbank.ca/drugs/DB14898.
- 119.Notte G T. Annu Rep Med Chem. 2014;49:417–436. doi: 10.1016/b978-0-12-800167-7.00026-2. [DOI] [Google Scholar]
- 120.Yong X, Hu T, Feng S, Du X, Shi H, Feng W. Trop J Pharm Res. 2015;14:1481–1486. doi: 10.4314/tjpr.v14i8.22. [DOI] [Google Scholar]
- 121.McCormack P L. Drugs. 2015;75:2045–2049. doi: 10.1007/s40265-015-0496-5. [DOI] [PubMed] [Google Scholar]
- 122.Flick A C, Ding H X, Leverett C A, Kyne R E, Jr, Liu K K-C, Fink S J, O’Donnell C J. J Med Chem. 2017;60(15):6480–6515. doi: 10.1021/acs.jmedchem.7b00010. [DOI] [PubMed] [Google Scholar]
- 123.Oh J, Kim A H, Lee S, Cho H, Kim Y S, Bahng M Y, Yoon S H, Cho J-Y, Jang I-J, Yu K-S. Diabetes Obes Metab. 2017;19:294–298. doi: 10.1111/dom.12813. [DOI] [PubMed] [Google Scholar]
- 124.Zhao W. Handbook for Chemical Process Research and Development. Boca Raton, FL, USA: CRC Press; 2016. [DOI] [Google Scholar]
- 125.Fraser G L, Hoveyda H R, Tannenbaum G S. Endocrinology. 2008;149:6280–6288. doi: 10.1210/en.2008-0804. [DOI] [PubMed] [Google Scholar]
- 126.Lasseter K C, Shaughnessy L, Cummings D, Pezzullo J C, Wargin W, Gagnon R, Oliva J, Kosutic G. J Clin Pharmacol. 2008;48:193–202. doi: 10.1177/0091270007310380. [DOI] [PubMed] [Google Scholar]
- 127.Hoveyda H R, Marsault E, Gagnon R, Mathieu A P, Vézina M, Landry A, Wang Z, Benakli K, Beaubien S, Saint-Louis C, et al. J Med Chem. 2011;54:8305–8320. doi: 10.1021/jm2007062. [DOI] [PubMed] [Google Scholar]
- 128.Gallwitz B. Drugs. 2011;71:1675–1688. doi: 10.2165/11592810-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 129.Trujillo J M, Nuffer W. Pharmacotherapy. 2014;34:1174–1186. doi: 10.1002/phar.1507. [DOI] [PubMed] [Google Scholar]
- 130.Gallwitz B. Glucagon-like peptide-1 receptor agonists. In: Gough S, editor. Handbook of Incretin-based Therapies in Type 2 Diabetes. Cham, Switzerland: Springer International Publishing; 2016. pp. 31–43. [DOI] [Google Scholar]
- 131.Theile J W, Cummins T R. Front Pharmacol. 2011;2:No. 54. doi: 10.3389/fphar.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hoyt S B, London C, Abbadie C, Felix J P, Garcia M L, Jochnowitz N, Karanam B V, Li X, Lyons K A, McGowan E, et al. Bioorg Med Chem Lett. 2013;23:3640–3645. doi: 10.1016/j.bmcl.2013.03.121. [DOI] [PubMed] [Google Scholar]
- 133.Hoyt Scott B, London C, Ok D, Parsons W H, inventors. Benzaepinones as sodium channel blockers. US 7,888,345 B2. U.S. Patent. 2011 Feb 15;
- 134.Hoyt S B, London C, Gorin D, Wyvratt M J, Fisher M H, Abbadie C, Felix J P, Garcia M L, Li X, Lyons K A, et al. Bioorg Med Chem Lett. 2007;17:4630–4634. doi: 10.1016/j.bmcl.2007.05.076. [DOI] [PubMed] [Google Scholar]
- 135.Hoyt S B, London C, Ok H, Gonzalez E, Duffy J L, Abbadie C, Dean B, Felix J P, Garcia M L, Jochnowitz N, et al. Bioorg Med Chem Lett. 2007;17:6172–6177. doi: 10.1016/j.bmcl.2007.09.032. [DOI] [PubMed] [Google Scholar]