

Abstract
The 78-kDa glucose-regulated protein (GRP78), an endoplasmic reticulum (ER) chaperone, is a master regulator of the ER stress. A number of studies revealed that high levels of GRP78 protein in cancer cells confer multidrug resistance (MDR) to therapeutic treatment. Therefore, drug candidate that reduces GRP78 may represent a novel approach to eliminate MDR cancer cells. Our earlier studies showed that a set of 4H-chromene derivatives induced selective cytotoxicity in MDR cancer cells. In the present study, we elucidated its selective mechanism in four MDR cancer cell lines with one lead candidate (CXL146). Cytotoxicity results confirmed the selective cytotoxicity of CXL146 toward the MDR cancer cell lines. We noted significant overexpression of GRP78 in all four MDR cell lines compared with the parental cell lines. Unexpectedly, CXL146 treatment rapidly and dose-dependently reduced GRP78 protein in MDR cancer cell lines. Using human leukemia (HL) 60/mitoxantrone (MX) 2 cell line as the model, we demonstrated that CXL146 treatment activated the unfolded protein response (UPR); as evidenced by the activation of inositol-requiring enzyme 1α, protein kinase R–like ER kinase, and activating transcription factor 6. CXL146-induced UPR activation led to a series of downstream events, including extracellular signal-regulated kinase 1/2 and c-Jun N-terminal kinase activation, which contributed to CXL146-induced apoptosis. Targeted reduction in GRP78 resulted in reduced sensitivity of HL60/MX2 toward CXL146. Long-term sublethal CXL146 exposure also led to reduction in GRP78 in HL60/MX2. These data collectively support GRP78 as the target of CXL146 in MDR treatment. Interestingly, HL60/MX2 upon long-term sublethal CXL146 exposure regained sensitivity to mitoxantrone treatment. Therefore, further exploration of CXL146 as a novel therapy in treating MDR cancer cells is warranted.
SIGNIFICANCE STATEMENT
Multidrug resistance is one major challenge to cancer treatment. This study provides evidence that cancer cells overexpress 78-kDa glucose-regulated protein (GRP78) as a mechanism to acquire resistance to standard cancer therapies. A chromene-based small molecule, CXL146, selectively eliminates cancer cells with GRP78 overexpression via activating unfolded protein response–mediated apoptosis. Further characterization indicates that CXL146 and standard therapies complementarily target different populations of cancer cells, supporting the potential of CXL146 to overcome multidrug resistance in cancer treatment.
Introduction
Despite the wide range of available cancer therapies and many candidates in the development pipeline, cancer treatment typically suffers from one significant and common challenge—cancer cells can acquire resistance, particularly multidrug resistance (MDR), through various mechanisms that substantially limit the efficacy of cancer treatment. Therapeutic candidates selectively targeting MDR cancer cells are urgently needed to complement current cancer therapies for better treatment outcome.
Through the combination of rational design and phenotypic screening, we identified and developed a series of 4H-chromene–based compounds, namely CXL series, which induce selective cytotoxicity toward a range of MDR cancer cells in comparison with the corresponding parental cancer cells and untransformed cells (Tian et al., 2008; Das et al., 2009, 2011; Aridoss et al., 2012; Bian et al., 2018). Mechanistically, these compounds induce cell death via apoptosis through an endoplasmic reticulum (ER) calcium-signaling pathway instead of the traditional mitochondria pathway (Hermanson et al., 2009; Bleeker et al., 2013; Bian et al., 2018). Most excitingly, MDR cancer cells, upon chronic exposure to CXL compounds, not only failed to acquire drug resistance toward CXL compound but also regained sensitivity toward standard therapies (Das et al., 2013). These unique characteristics strongly support the potential clinical impact of CXL series. The mechanism of action of CXL compounds, however, remains to be fully characterized, particularly an understanding of the molecular basis for the selective cytotoxicity toward MDR cancer cells. Such knowledge is critical to guide its translational evaluation.
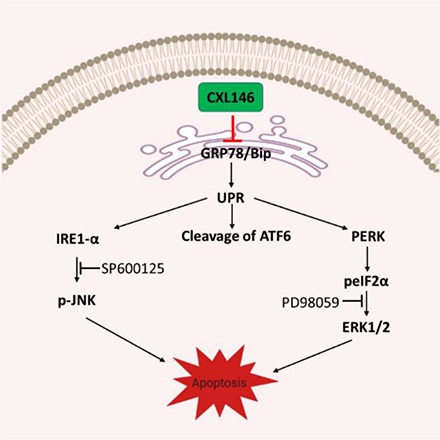
The 78-kDa glucose-regulated protein (GRP78), also referred to as heat shock protein A5 or binding immunoglobin protein, is an ER chaperone protein that regulates ER function and is involved in the unfolded protein response (UPR) (Malhotra and Kaufman, 2007). In brief, GRP78 resides in ER lumen to facilitate the proper folding and assembly of newly synthesized polypeptides into functional proteins, retain unassembled precursors to the ER, and direct misfolded proteins for degradation. Under normal ER stress, UPR engages adaptive pathways to alleviate ER stress by arresting protein translation, upregulating chaperones and folding enzymes, and enhancing the degradation of misfolded proteins. Intense or persistent activation of UPR in response to continued ER stress, however, triggers apoptosis, which involves three signaling pathways mediated by the key proteins inositol-requiring enzyme 1α (IRE1α), activating transcription factor (ATF) 6, and protein kinase R–like ER kinase (PERK) (Kaufman, 1999; Sano and Reed, 2013). IRE1α activation stimulates the c-Jun N-terminal kinase (JNK). ATF6, a transcriptional factor, translocates to the Golgi compartment upon ER stress and is proteolytically cleaved into an active form, which subsequently is translocated to the nucleus, where it activates its target gene transcription. PERK attenuates protein translation by phosphorylating eIF2α. Phosphorylation of eIF2α induces preferential translation of UPR-dependent genes, such as ATF4, which then activate the apoptotic pathway through extracellular signal-regulated kinase 1/2 (ERK1/2) (Ron and Hubbard, 2008). GRP78 has been reported to be overexpressed in several types of cancer (Pootrakul et al., 2006; Mozos et al., 2011; Roller and Maddalo, 2013; Gifford et al., 2016), some of which are resistant to chemotherapies (Reddy et al., 2003; Pootrakul et al., 2006; Pyrko et al., 2007; Mozos et al., 2011; Roller and Maddalo, 2013; Abdel Malek et al., 2015; Jagannathan et al., 2015; Gifford et al., 2016).
In this study, we provide evidence that four independently developed MDR cancer cell lines acquired by exposure to different chemotherapeutics have higher expression of GRP78 compared with their parental cell lines. CXL146 treatment led to the reduction of GRP78 in these cancer cells. CXL146 treatment also activated all three arms of UPR signaling in the MDR cells, thus contributing to the induction of apoptosis. Targeted depletion of GRP78 via CRISPR knockout supports our hypothesis that GRP78 overexpression in MDR cancer cells was responsible for CXL146’s selective cytotoxicity. Long-term CXL146 exposure resulted in reduced GRP78 in MDR cancer cells and increased the sensitivity of MDR cancer cells to mitoxantrone. These results support a role for GRP78 as a key target for CXL146’s selective cytotoxicity toward MDR cancer cells.
Materials and Methods
Cell Culture, Chemicals, and Reagents.
HL60, HL60/MX 2, HL60/DOX, K562, K562/DOX, and K562/HHT300 cell lines were purchased from American Type Culture Collection or provided through our collaborators (Aridoss et al., 2012). HL60/MX2 and HL60/DOX were developed from HL60 upon chronic exposure to sublethal mitoxantrone and doxorubicin, respectively. K562/DOX and K562/HHT300 were obtained from K562 upon chronic exposure to sublethal doxorubicin and homoharringtonine, respectively. They were authenticated via the Cell Line Authentication Service provided by Genetica DNA Laboratories (Burlington, NC). They were cultured in RPMI1640 medium supplemented with 10% FBS (Gibco). Antibodies for GRP78, PERK, p-IRE1α, IRE1α, ATF6, ATF4, JNK, p-JNK, ERK1/2, p-ERK1/2, cyclin D1, cleaved PARP, and anti-rabbit IgG secondary antibody were purchased from Cell Signaling Technology (Beverly, MA). Anti–β-actin was purchased from Sigma-Aldrich (St. Louis, MO). CellTiter-Blue reagent was purchased from Promega (Madison, WI). CXL146 was synthesized and characterized after our reported procedures (Bian et al., 2018).
Cytotoxicity Assay.
The effect of CXL146 and mitoxantrone on the growth of cancer cell lines was determined using CellTiter-Blue cell viability assay (G9241; Promega). Briefly, HL60, HL60/MX2, HL60/DOX, K562, K562/DOX, and K562/HHT300 cells were plated in a 96-well plate (1 × 104 cells/well) and treated with CXL146 or mitoxantrone in a series of 1% DMSO dilutions in triplicate. Cells treated with 1% DMSO medium served as the control. After 48-hour treatment, CellTiter-Blue reagent was added according to the manufacturer protocol with the fluorescence intensity measured typically 1 to 2 hours after. Concentration for 50% of maximal growth inhibition (GI50) was calculated by fitting the relative viability of the cells to the compound concentration using a dose-response curve in the GraphPad Prism Software, Inc. (San Diego, CA). A minimum of three biologic replicates were performed on different days, and data were reported as mean GI50 ± S.D.
Western Blot Analysis.
Cells with or without treatment were harvested and lysed using radioimmunoprecipitation assay buffer with protease inhibitor cocktail and cleared by centrifugation at 10,000 rpm for 10 minutes at 4°C. Protein concentrations were determined by Bio-Rad Protein Assay. Equal amount of protein (50 µg) was electrophoresed on SDS-polyacrylamide gels and transferred onto polyvinylidene fluoride membrane. Membranes were blocked with 5% milk in Tris-buffered saline containing Tween-20 before probing with primary antibody according to the instructions of the manufacturer. Subsequently, the membranes were incubated with the corresponding horseradish peroxidase–conjugated secondary antibody for 1 hour. Protein bands were detected by enhanced ECL reagent (Thermo Scientific) and visualized by Bio-Rad Imaging system. For reprobing, blots were stripped with Restore Western Blot stripping buffer (Thermo Scientific). A minimum of three biologic replicates were performed on different days.
CXL146 or Mitoxantrone Long-Term Exposure.
HL60/MX2 cell lines were exposed to varying concentrations of CXL146 or mitoxantrone or 1.0% DMSO medium for 3 months. The medium was replaced every 3 days with new compound replenished, and the cell density was adjusted so that the medium was sufficient for 3-day culturing. The cell viability was measured using CellTiter-Blue assay. The concentrations of CXL146 or mitoxantrone were adjusted so that there would be no more than 25% growth inhibition during the experimental period (the cells were continuously exposed to subtoxic levels).
GRP78 Downregulation by CRISPR/Cas9.
Three guide RNAs targeting GRP78 (NM_005347.4) were chosen from the Brunello genome-wide CRISPR pooled gRNAs library (Doench et al., 2016).
GRP78 KO-1 (CAGACGGGTCATTCCACGTG), GRP78 KO-2 (AATGGCAAGGAACCATCCCG), and GRP78 KO-3 (GGTGAGAAGAGACACATCGA) gRNAs were cloned into the lentiviral vector LentiCRISPRv2 (Plasmid 52961; Addgene) using the Golden Gate assembly strategy as described (Joung et al., 2017). To generate lentiviruses for each GRP78 gRNA, LentiCRISPRv2 vector harboring each of the GRP78 gRNA was LipofectAMINE-cotransfected in human embryonic kidney 293T cells with the envelop PMD2.G and packaging psPAX2 plasmid after our standard procedures. HL60/MX2 cells were transduced separately with each of the three GRP78 gRNAs using the spinoculation method. To select the cells that integrated the lentiviral genome (lentiCRISPRv2 harboring the GRP78 gRNA), the transduced cells were cultured in medium supplemented with 2 µg/ml puromycin for 6 days.
Statistical Analysis.
One-way ANOVA followed by Dunnett’s test was used for comparisons of the data between different groups. P value ≤ 0.05 was considered statistically significant. All analyses were conducted in GraphPad Prism4 (GraphPad Software, Inc.).
Results
CXL146 Showed Preferential Cytotoxicity toward MDR Cancer Cells.
The sensitivity of HL60, HL60/MX2, HL60/DOX, K562, K562/DOX, and K562/HHT300 cells toward CXL146 or mitoxantrone was evaluated via the CellTiter-Blue assay. As shown in Table 1, MDR cancer cells (HL60/MX2, HL60/DOX, K562/DOX, and K562/HHT300) (Aridoss et al., 2012) are less sensitive to mitoxantrone in comparison with the corresponding parental cancer cell lines (HL60 and K562, respectively). In contrast, with CXL146 exposure, all MDR cancer cells were more sensitive to CXL146 relative to the corresponding parental cancer cell lines. These results were consistent with our previous observations that the CXL derivatives selectively kill MDR cancer cells (Tian et al., 2008; Das et al., 2009, 2011; Aridoss et al., 2012; Bian et al., 2018.
TABLE 1.
GI50s of CXL146 and mitoxantrone toward parental and MDR cancer cell lines
| Selectivity is the ratio of GI50 of the parental cell line to that of the MDR cell line. |
 CXL146 CXL146 |
Mitoxantrone | ||
|---|---|---|---|---|
| Cell line | GI50 (µM) | Selectivity | GI50 (µM) | Selectivity |
| HL60 | 0.48 ± 0.07 | 1 | 0.0040 ± 0.0003 | 1 |
| HL60/MX2 | 0.17 ± 0.03 | 2.8 | 0.18 ± 0.01 | 0.02 |
| HL60/DOX | 0.30 ± 0.07 | 1.6 | 0.007 ± 0.001 | 0.57 |
| K562 | 0.75 ± 0.09 | 1 | 0.31 ± 0.05 | 1 |
| K562/DOX | 0.49 ± 0.11 | 1.5 | 0.89 ± 0.04 | 0.35 |
| K562/HHT300 | 0.51 ± 0.03 | 1.5 | 3.8 ± 0.2 | 0.08 |
Characterization of GRP78 Proteins in Parental and MDR Cancer Cell Lines and the Effect of CXL146 Treatment.
Given the role of GRP78 in modulating ER stress; its overexpression in cancer cells, particularly in the context of MDR; the increased sensitivity of MDR cells to CXL146; and the mechanism of CXL in inducing ER stress (Hermanson et al., 2009; Bleeker et al., 2013), we characterized the levels of GRP78 in the parental and MDR cancer cell lines. GRP78 was overexpressed in all four MDR cancer cell lines relative to the parental control cell lines (Fig. 1A). Since CXL146 showed enhanced cytotoxicity to the MDR cancer cells, we characterized the effect of CXL146 on GRP78 in these cell lines. Upon 24-hour exposure, CXL146 treatment dose-dependently and significantly reduced GRP78 levels in all of these cell lines (Fig. 1B). Overall, lower concentrations of CXL146 were required to reduce GRP78 in MDR cells as compared with the parental cell lines. It should be noted that the CXL146 concentrations that resulted in the loss of GRP78 were comparable to its growth inhibition concentrations in these cell lines (Table 1), corroborating the potential mechanistic relevance.
Fig. 1.

(A) Expression of GRP78 in different MDR and parental cancer cell lines. (B) CXL146 treatment resulted in a dose-dependent reduction of GRP78 in all cell lines tested with lower concentrations of CXL146 in MDR cell lines relative to the corresponding parental cell lines. These images are representative of at least three independent replicates.
CXL146 Reduced the Expression of GRP78 and Activated the Three Arms of UPR in HL60/MX2 Cell Line.
We next used HL60/MX2 as the model to further assess the effect of CXL146. As shown in Fig. 2A, the expression of the GRP78-binding partners, IRE1α, PERK, and ATF6, was substantially higher in HL60/MX2 cells relative to the parental HL60 cells. A 24-hour CXL146 treatment dose-dependently increased IRE1α, p-IRE1α, PERK, and the cleavage of ATF6 (Fig. 2B), which was consistent with UPR activation. Given that CXL146 rapidly (within minutes) induces cytosolic calcium increase by blocking ER calcium uptake (Bleeker et al., 2013; Bian et al., 2018), we characterized the time-course effect of CXL146 treatment on GRP78. As expected, CXL146 treatment induced the reduction in GRP78 even with a 4-hour exposure, and the effect persisted for up to 36 hours (Fig. 2C).
Fig. 2.

(A) Characterization of GRP78 and other UPR markers in HL60 and HL60/MX2 cell lines. (B) CXL146 treatment activated the three arms of UPR in HL60/MX2 cells. (C) CXL146 rapidly induced GRP78 reduction in HL60/MX2 cells. These images are representative of at least three independent replicates.
CXL146 Induced Apoptosis through UPR Downstream Signaling in HL60/MX2 Cells.
Among the three UPR arms, PERK mediates eIF2α phosphorylation, leading to the preferential translation of specific mRNAs, one of which encodes the ATF4 protein. ATF4 subsequently activates the proapoptotic factor and initiates ER stress–associated apoptosis (Blais et al., 2004; Sano and Reed, 2013). Hence, we examined the expression of p-eIF2α and ATF4 upon 24-hour CXL146 treatment. We observed increased phosphorylation of eIF2α with the reduction of total eIF2α upon CXL146 treatment at various concentrations (Fig. 3A). The level of ATF4 increased upon CXL146 treatment in a dose-dependent manner as well. CXL146 also dose-dependently induced phosphorylation of ERK1/2 (Fig. 3A), which was reported to facilitate ER stress–induced apoptosis. Previous studies revealed that the reduction of GRP78 could lead to the phosphorylation of JNK, a downstream target of IRE1α, which also regulates cell death (Urano et al., 2000; Lei and Davis, 2003). In concordance with this, we noticed a dose-dependent phosphorylation of JNK (Fig. 3B). Additionally, GRP78 can interact with procaspase-2 and procaspase-7, suppressing their activation (Reddy et al., 2003; Davidson et al., 2005; Ermakova et al., 2006). In agreement with this, as shown in Fig. 3A and B, CXL146 treatment reduced the levels of full-length caspase 2 and 7 and increased PARP cleavage and caspase 9–cleaved products. In addition to the induction of the caspase-dependent apoptotic pathway, expression of cyclin D1 also plays an important role in the activation of UPR and induces ER stress–mediated apoptosis (Bustany et al., 2015). Furthermore, cyclin D1 is positively regulated by the activation of ERK1/2 (Lavoie et al., 1996). Since we observed the activation of UPR and ERK1/2 by CXL146, we characterized the effect of CXL146 on the expression of cyclin D1. Cyclin D1 expression was induced by CXL146 in a dose-dependent manner (Fig. 3B). Lastly, inhibitors specific for ERK1/2 (PD98059) and JNK (SP600125) were able to partially block CXL146-induced PARP cleavage (Fig. 3C), supporting the role of ERK1/2 and JNK activation in CXL146-induced apoptosis.
Fig. 3.

(A) CXL146 activated UPRs. (B) CXL146 induced apoptosis. (C) CXL146 induced-apoptosis could be partially blocked by ERK1/2 and JNK inhibition. These images are representative of at least three independent replicates.
CXL146 Selectively Inhibited HL60/MX2 Cell Proliferation Because of GRP78 Overexpression.
To confirm the role of GRP78 in the selective mechanism of CXL146, we targeted the HPSA5 gene encoding GRP78 in HL60/MX2 with three independent single gRNA/Cas9 vectors. We evaluated the pools of heat shock protein A5 knockouts corresponding to each single gRNA instead of individual isolated clones because previous work suggested that the pooled approach provides a robust evaluation of the functional importance of the gene and avoids clone-specific effects (Shalem et al., 2014; Parnas et al., 2015; Potting et al., 2018). We first characterized GRP78 expression in the pooled knockouts by Western blotting. Among the three constructs, two (KO-1 and KO-2) were effective, whereas the third construct (KO-3) did not significantly reduce GRP78 in HL60/MX2 (Fig. 4A). We then evaluated their sensitivity to CXL146. As expected, HL60/MX2 cells with reduced levels of GRP78 (KO-1 and KO-2) showed decreased sensitivity to CXL146, whereas the third construct showed no changes in sensitivity relative to the control cells (Fig. 4B). These results strongly support that CXL146 selectively targets cancer cells with increased levels of GRP78, which are typically drug-resistant.
Fig. 4.

(A) Confirmation of GRP78 downregulation via CRISPR/Cas9 in HL60/MX2 cell line. (B) Relative sensitivity of HL60/MX2 cell lines of varied GRP78 levels toward CXL146. The results are representative of at least three independent replicates. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
HL60/MX2 Reduced GRP78 upon CXL146 Chronic Exposure and Regained Sensitivity toward Mitoxantrone.
To further explore the relevance of GRP78 as the cellular target of CXL146, HL60/MX2 cells were exposed to a sublethal concentration of CXL146 for 3 months (mitoxantrone was used as a standard cancer therapy control). We hypothesized that if the high level of GRP78 accounts for the enhanced sensitivity of HL60/MX2 cells to CXL146, CXL146 chronic exposure would select HL60/MX2 cell populations with reduced levels of GRP78 for survival. On the other hand, if GRP78 overexpression confers HL60/MX2 resistance to mitoxantrone, mitoxantrone exposure would select cells with higher levels of GRP78. Indeed, chronic exposure of HL60/MX2 cells to CXL146 resulted in cells with reduced GRP78, whereas chronic exposure of mitoxantrone resulted in further overexpression of GRP78 in HL60/MX2 cells (Fig. 5A). HL60/MX2 cells upon chronic CXL146 treatment regained sensitivity toward mitoxantrone (Figs. 5B and 6), potentially because of the reduction in GRP78.
Fig. 5.

(A) The effect of long-term exposure of CXL146 and mitoxantrone on GRP78 protein level in HL60/MX2. (B) The relative sensitivity of these cell lines toward mitoxantrone. The results are representative of at least three independent replicates. **P ≤ 0.01.
Fig. 6.

Proposed mechanism of action for CXL146.
Discussion
MDR is one major barrier to the success of cancer treatment. Anticancer agents selectively targeting MDR cancers are critical for more effective cancer management. We developed a series of 4H-chromene derivatives, which demonstrated preferential cytotoxicity toward MDR cells (Tian et al., 2008; Das et al., 2009, 2011; Aridoss et al., 2012; Bian et al., 2018). Understanding the mechanism of action, particularly selective anticancer activity toward MDR cancer cells, is important for guiding its future translational development and potentially clinical evaluation. This study focused on CXL146 as the lead compound to investigate its mechanism in MDR cells.
Among mechanisms that potentially underlie MDR in cancer cells, we focused on GRP78 for two reasons. First, GRP78 has been well-documented to be overexpressed in various malignancies. One recent study reported that GRP78 regulates ATP-binding cassette transporter activity in pancreatic cancers to confer drug resistance (Dauer et al., 2018). Because ATP-binding cassette transporter upregulation is one major MDR mechanism, GRP78 overexpression may be a general mechanism of cancer cells to acquire MDR. Our CXL compounds demonstrated selective cytotoxicity toward a wide range of MDR cancer cells (Aridoss et al., 2012; Bian et al., 2018), suggesting that they may be targeting a common MDR mechanism. Second, our earlier studies suggested that ER stress and cytosolic calcium influx contribute to the mechanism of action for CXL compounds (Hermanson et al., 2009; Bleeker et al., 2013; Bian et al., 2018). GRP78 is a key protein on ER in regulating ER stress response and buffering ER calcium (Leustek et al., 1991; Hammadi et al., 2013). These together suggest that GRP78 is a logical target candidate of CXL compounds.
In this study, we first showed that GRP78 is highly expressed in four MDR cell lines compared with their parental cell lines. These MDR cell lines were selected for study because they have increased sensitivity toward CXL146, although they are resistant to standard therapies, such as mitoxantrone. Interestingly, CXL treatment resulted in a rapid and dose-dependent reduction in GRP78, providing the first piece of evidence that GRP78 is mechanistically relevant to CXL146. Although CXL146 treatment led to a dose-dependent reduction of GRP78 in both parental and MDR cancer cells, the concentrations of CXL146 needed to induce the reduction in GRP78 in MDR cells were generally lower than those for the parental cell lines. These findings are consistent with the preferential cytotoxicity of CXL toward MDR cancer cell lines and support a role for GRP78 in the preferential cytotoxicity of CXL compounds to MDR cells. Decreased expression of GRP78 via CRISPR Cas9 targeting indeed reduced the sensitivity of HL60/MX2 to CXL146 treatment, providing evidence that GRP78 is essential for the selective mechanism of CXL146. Furthermore, natural selection data demonstrated that GRP78 is integral to the cytotoxicity of CXL146, as chronic sublethal exposure of HL60/MX2 to CXL146 selected cells with reduced GRP78 level. These acute, chronic, and genetic findings collectively support that GRP78 is necessary for the MDR phenotype in the studied cells and that CXL146 selectively kills cancer cells with elevated GRP78 levels, demonstrating its unique anticancer potential (Fig. 6). Notably, the HL60/MX2 cells selected by sublethal CXL146 exposure showed increased sensitivity toward mitoxantrone consistent with our earlier results (Das et al., 2013), providing strong evidence that CXL146 may be complementary to standard therapies to mitigate the risk of MDR development in cancer treatment.
Using HL60/MX2 as the MDR model, we showed that in addition to GRP78 expression, its partners (IRE1α, ATF6, and PERK) were also highly expressed. Importantly, CXL146 treatment impaired the function of GRP78 to sequester IRE1α, ATF6, and PERK and activated all of the three UPR transducers pathways. Several downstream markers provided further evidence that CXL146 treatment activated UPR (Urano et al., 2000; Lei and Davis, 2003). Based on the fact that small molecules could bind to the ATPase domain of GRP78 and facilitate GRP78 dissociation from its UPR partners, CXL146 may target GRP78 via the same mechanism. Further studies are required to verify this hypothesis.
In summary, our results showed that CXL146 effectively reduces GRP78 expression, which is commonly upregulated among various malignancies and necessary for MDR in some cancer cells. Importantly, CXL146 and standard therapies appear to target different populations of cancer cells on the basis of the GRP78 levels. CXL146 therefore may be complementary to standard cancer therapies for effective cancer management. CXL146 has also demonstrated acceptable pharmacokinetics in vivo. Its preferential anticancer potential toward MDR cancer cells will be evaluated in vivo to validate its anticancer potential.
Acknowledgments
None.
Abbreviations
- ATF
activating transcription factor
- DOX
doxorubicin
- eIF2α
eukaryotic initiation factor 2α
- ER
endoplasmic reticulum
- ERK1/2
extracellular signal-regulated kinase 1/2
- GI50
concentration for 50% of maximal growth inhibition
- gRNA
guide RNA
- GRP78
78-kDa glucose-regulated protein
- HL
human leukemia
- IRE1α
inositol-requiring enzyme 1α
- JNK
c-Jun N-terminal kinase
- KO
knockout
- MDR
multidrug resistance
- MX
mitoxantrone
- p-
phosphorylated
- PARP
poly(ADP-ribose) polymerase
- PERK
protein kinase R–like ER kinase
- UPR
unfolded protein response
Authorship Contributions
Participated in research design: Bian, Tagmount, Vulpe, Vijendra, Xing.
Conducted experiments: Bian, Tagmount, Vijendra.
Performed data analysis: Bian, Tagmount, Vulpe, Vijendra, Xing.
Wrote or contributed to the writing of the manuscript: Bian, Tagmount, Vulpe, Vijendra, Xing.
Footnotes
This work was supported by grants from the Harry T. Mangurian Jr. Foundation [C.X.], National Institutes of Health (NIH) National Cancer Institute [R01CA163864, C.X.], the Frank Duckworth Endowment College of Pharmacy University of Florida [C.X.], the Startup Fund University of Florida Health Cancer Center [C.X.], and the Raman Postdoctoral Fellowship from UGC, India [F. 5/92 IC]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or any funding agencies.
References
- Abdel Malek MA, Jagannathan S, Malek E, Sayed DM, Elgammal SA, Abd El-Azeem HG, Thabet NM, Driscoll JJ. (2015) Molecular chaperone GRP78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 6:3098–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridoss G, Zhou B, Hermanson DL, Bleeker NP, Xing C. (2012) Structure-activity relationship (SAR) study of ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) and the potential of the lead against multidrug resistance in cancer treatment. J Med Chem 55:5566–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian T, Chandagirikoppal Vijendra K, Wang Y, Meacham A, Hati S, Cogle CR, Sun H, Xing C. (2018) Exploring the structure-activity relationship and mechanism of a chromene scaffold (CXL series) for its selective antiproliferative activity toward multidrug-resistant cancer cells. J Med Chem 61:6892–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, Wouters BG, Bell JC. (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24:7469–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleeker NP, Cornea RL, Thomas DD, Xing C. (2013) A novel SERCA inhibitor demonstrates synergy with classic SERCA inhibitors and targets multidrug-resistant AML. Mol Pharm 10:4358–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustany S, Cahu J, Guardiola P, Sola B. (2015) Cyclin D1 sensitizes myeloma cells to endoplasmic reticulum stress-mediated apoptosis by activating the unfolded protein response pathway. BMC Cancer 15:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SG, Doshi JM, Tian D, Addo SN, Srinivasan B, Hermanson DL, Xing C. (2009) Structure-activity relationship and molecular mechanisms of ethyl 2-amino-4-(2-ethoxy-2-oxoethyl)-6-phenyl-4h-chromene-3-carboxylate (sha 14-1) and its analogues. J Med Chem 52:5937–5949. [DOI] [PubMed] [Google Scholar]
- Das SG, Hermanson DL, Bleeker N, Lowman X, Li Y, Kelekar A, Xing C. (2013) Ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017): a novel scaffold that resensitizes multidrug resistant leukemia cells to chemotherapy. ACS Chem Biol 8:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das SG, Srinivasan B, Hermanson DL, Bleeker NP, Doshi JM, Tang R, Beck WT, Xing C. (2011) Structure-activity relationship and molecular mechanisms of ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) and its analogues. J Med Chem 54:5937–5948. [DOI] [PubMed] [Google Scholar]
- Dauer P, Sharma NS, Gupta VK, Nomura A, Dudeja V, Saluja A, Banerjee S. (2018) GRP78-mediated antioxidant response and ABC transporter activity confers chemoresistance to pancreatic cancer cells. Mol Oncol 12:1498–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson DJ, Haskell C, Majest S, Kherzai A, Egan DA, Walter KA, Schneider A, Gubbins EF, Solomon L, Chen Z, et al. (2005) Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res 65:4663–4672. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermakova SP, Kang BS, Choi BY, Choi HS, Schuster TF, Ma WY, Bode AM, Dong Z. (2006) (-)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res 66:9260–9269. [DOI] [PubMed] [Google Scholar]
- Gifford JB, Huang W, Zeleniak AE, Hindoyan A, Wu H, Donahue TR, Hill R. (2016) Expression of GRP78, master regulator of the unfolded protein response, increases chemoresistance in pancreatic ductal adenocarcinoma. Mol Cancer Ther 15:1043–1052. [DOI] [PubMed] [Google Scholar]
- Hammadi M, Oulidi A, Gackière F, Katsogiannou M, Slomianny C, Roudbaraki M, Dewailly E, Delcourt P, Lepage G, Lotteau S, et al. (2013) Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: involvement of GRP78. FASEB J 27:1600–1609. [DOI] [PubMed] [Google Scholar]
- Hermanson D, Addo SN, Bajer AA, Marchant JS, Das SG, Srinivasan B, Al-Mousa F, Michelangeli F, Thomas DD, Lebien TW, et al. (2009) Dual mechanisms of sHA 14-1 in inducing cell death through endoplasmic reticulum and mitochondria. Mol Pharmacol 76:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan S, Abdel-Malek MA, Malek E, Vad N, Latif T, Anderson KC, Driscoll JJ. (2015) Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia 29:2184–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, Zhang F. (2017) Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12:828–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. (1999) Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev 13:1211–1233. [DOI] [PubMed] [Google Scholar]
- Lavoie JN, L’Allemain G, Brunet A, Müller R, Pouysségur J. (1996) Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 271:20608–20616. [DOI] [PubMed] [Google Scholar]
- Lei K, Davis RJ. (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100:2432–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leustek T, Toledo H, Brot N, Weissbach H. (1991) Calcium-dependent autophosphorylation of the glucose-regulated protein, Grp78. Arch Biochem Biophys 289:256–261. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. (2007) The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18:716–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozos A, Roué G, López-Guillermo A, Jares P, Campo E, Colomer D, Martinez A. (2011) The expression of the endoplasmic reticulum stress sensor BiP/GRP78 predicts response to chemotherapy and determines the efficacy of proteasome inhibitors in diffuse large b-cell lymphoma. Am J Pathol 179:2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, Przybylski D, Platt RJ, Tirosh I, Sanjana NE, et al. (2015) A genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162:675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pootrakul L, Datar RH, Shi SR, Cai J, Hawes D, Groshen SG, Lee AS, Cote RJ. (2006) Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin Cancer Res 12:5987–5993. [DOI] [PubMed] [Google Scholar]
- Potting C, Crochemore C, Moretti F, Nigsch F, Schmidt I, Manneville C, Carbone W, Knehr J, DeJesus R, Lindeman A, et al. (2018) Genome-wide CRISPR screen for PARKIN regulators reveals transcriptional repression as a determinant of mitophagy. Proc Natl Acad Sci USA 115:E180–E189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrko P, Schönthal AH, Hofman FM, Chen TC, Lee AS. (2007) The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res 67:9809–9816. [DOI] [PubMed] [Google Scholar]
- Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. (2003) Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem 278:20915–20924. [DOI] [PubMed] [Google Scholar]
- Roller C, Maddalo D. (2013) The molecular chaperone GRP78/BiP in the development of chemoresistance: mechanism and possible treatment. Front Pharmacol 4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Hubbard SR. (2008) How IRE1 reacts to ER stress. Cell 132:24–26. [DOI] [PubMed] [Google Scholar]
- Sano R, Reed JC. (2013) ER stress-induced cell death mechanisms. Biochim Biophys Acta 1833:3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Das SG, Doshi JM, Peng J, Lin J, Xing C. (2008) sHA 14-1, a stable and ROS-free antagonist against anti-apoptotic Bcl-2 proteins, bypasses drug resistances and synergizes cancer therapies in human leukemia cell. Cancer Lett 259:198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664–666. [DOI] [PubMed] [Google Scholar]