

Summary
Cells sense elevated temperatures and mount an adaptive heat shock response that involves changes in gene expression, but the underlying mechanisms, particularly on the level of translation, remain unknown. Here we report that, in budding yeast, the essential translation initiation factor Ded1p undergoes heat-induced phase separation into gel-like condensates. Using ribosome profiling and an in vitro translation assay, we reveal that condensate formation inactivates Ded1p and represses translation of housekeeping mRNAs while promoting translation of stress mRNAs. Testing a variant of Ded1p with altered phase behavior as well as Ded1p homologs from diverse species, we demonstrate that Ded1p condensation is adaptive and fine-tuned to the maximum growth temperature of the respective organism. We conclude that Ded1p condensation is an integral part of an extended heat shock response that selectively represses translation of housekeeping mRNAs to promote survival under conditions of severe heat stress.
Keywords: phase separation, heat stress, evolutionary adaptation, heat shock response, 5′, UTR, ribosomal scanning, Ded1p, condensate, chaperone, cytosolic pH
Graphical Abstract
Highlights
-
•
Ded1p phase-separates in response to heat and pH to form gel condensates
-
•
Condensation inactivates Ded1p and represses housekeeping mRNAs
-
•
Ded1p condensation promotes stress protein production and limits cell growth
-
•
Ded1p condensation is adapted to the maximum growth temperature of a species
Heat-induced phase separation of a helicase promotes a switch in translation from housekeeping transcripts to stress-response transcripts.
Introduction
To survive a heat shock, all organisms activate a process known as the heat shock response (Lindquist, 1986, Lindquist and Craig, 1988). This stress response program represses expression of housekeeping proteins and promotes production of stress-protective proteins (Causton et al., 2001, Gasch et al., 2000). In eukaryotes, the transcription factor Hsf1 promotes transcription of a wide range of stress-protective genes (Craig and Gross, 1991, Morano et al., 2012, Trotter et al., 2002, Zheng et al., 2016) that are then translated by ribosomes in the cytoplasm to produce stress-protective proteins.
The heat shock response involves changes on the level of transcription but also on the level of translation. For example, exposure of yeast to heat shock induces a rapid and long-lasting translational shutdown (Lindquist et al., 1982). Moreover, extracts made from heat-shocked Drosophila cells preferentially translate heat shock transcripts, whereas extracts made from unstressed cells indiscriminately translate housekeeping and heat shock transcripts (Krüger and Benecke, 1981, Lindquist, 1986, Storti et al., 1980). This led to the proposal that there is a specific factor that discriminates between these two types of transcripts (Lindquist et al., 1982). However, the nature of the responsible factor and the features that distinguish housekeeping and stress transcripts have remained unknown. Recent studies revealed that heat-induced translation regulation coincides with assembly of large ribonucleoprotein granules called stress granules (SGs), which efficiently inhibit protein synthesis by sequestering mRNAs and translation factors (Cherkasov et al., 2013, Grousl et al., 2009).
In recent years, the principle of phase separation has emerged as a way to describe the assembly of SGs. Phase separation is a process by which a homogeneous solution of components, such as proteins, separates to form a dense phase (or condensate) that coexists with a dilute phase (Banani et al., 2017; Shin and Brangwynne, 2017). Condensate assembly appears to be an ideal mechanism for stress adaptation for two reasons: (1) it is very sensitive to changes in physical-chemical conditions as they occur during stress, and (2) it can specifically regulate protein activities (Franzmann and Alberti, 2019). In agreement with this idea, many proteins assemble into higher-order structures upon heat stress (Cherkasov et al., 2015, Leuenberger et al., 2017, Wallace et al., 2015). The predominant view is that accumulation of insoluble proteins during heat stress is a result of uncontrolled protein misfolding. However, recent studies have suggested that some of the assemblies may be adaptive condensates (Kroschwald et al., 2018, Riback et al., 2017). Similar findings were made in yeast subjected to starvation or pH stress (Franzmann et al., 2018, Kroschwald et al., 2018, Munder et al., 2016, Narayanaswamy et al., 2009, Riback et al., 2017). Importantly, preventing condensate assembly is associated with fitness defects (Franzmann et al., 2018; Kroschwald et al., 2018, Munder et al., 2016, Petrovska et al., 2014, Riback et al., 2017). Why and how the condensates protect cells from stress, however, is still unknown.
One component of yeast SGs is the essential translation initiation factor Ded1p (Hilliker et al., 2011). Ded1p is an ATP-dependent Asp-Glu-Ala-Asp (DEAD)-box RNA helicase. It resolves secondary structure in the 5′ untranslated regions (UTRs) of mRNAs to facilitate ribosomal scanning and identification of the start codon (Berthelot et al., 2004, Guenther et al., 2018, Sen et al., 2015). Accordingly, changes in cellular Ded1p levels have dramatic effects on gene expression (Firczuk et al., 2013). Interestingly, Ded1p rapidly becomes insoluble upon heat shock (Wallace et al., 2015), but the nature and function of stress-induced Ded1p assemblies have remained unclear.
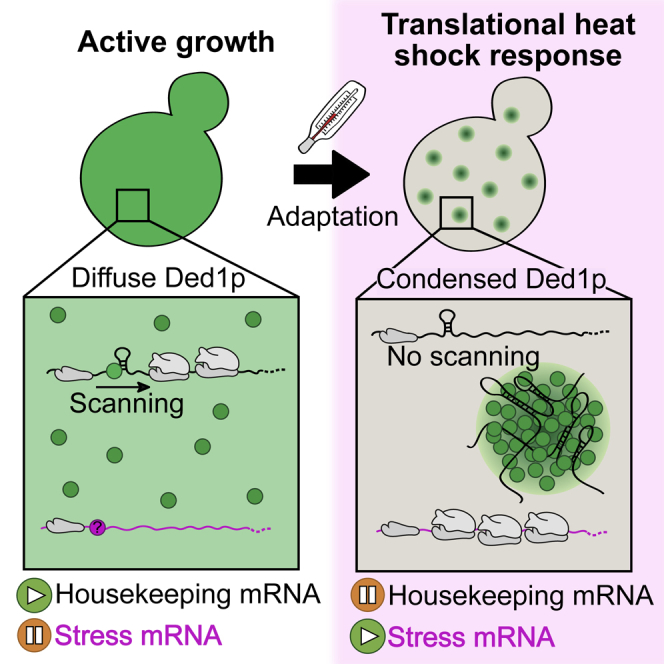
Here we show that Ded1p acts as a stress sensor that directly responds to sudden changes in environmental conditions. We find that Ded1p phase separation is strongly correlated with the magnitude and duration of a heat stress stimulus and that Ded1p condensation occurs rapidly at temperatures above 39°C. Using time-lapse fluorescence microscopy and in vitro reconstitution biochemistry, we show that the heterotypic interaction of Ded1p and mRNA results in assembly of soft gel-like condensates that are reversible upon cessation of stress. We further demonstrate that condensate assembly represses translation of structurally complex housekeeping mRNAs, whereas structurally simple stress mRNAs, including those encoding heat shock proteins, escape translational repression. We propose that heat-induced phase separation of Ded1p drives an evolutionarily conserved extended heat shock response program that selectively downregulates translation of housekeeping transcripts and arrests cell growth.
Results
Heat Shock Promotes a Switch in Protein Synthesis Dependent on 5′ UTR Complexity
Many proteins become insoluble when budding yeast is exposed to heat shock (Cherkasov et al., 2015, Leuenberger et al., 2017, Wallace et al., 2015). Among these proteins are components of SGs and proteins involved in mRNA translation (Cherkasov et al., 2015, Wallace et al., 2015). To test whether heat-induced assembly of these proteins promotes a switch in gene expression at the level of translation that may complement the Hsf1-mediated transcriptional heat shock response, we performed ribosome profiling on yeast exposed for 10 min to 30°C (normal growth temperature), 40°C, or 42°C (maximum growth temperature) (see STAR Methods and Figures S1A and S1B for quality control of ribosome profiling). Because protein assembly is more prominent at 42°C (Wallace et al., 2015), and gene expression changes are predominantly translational between 40°C and 42°C (Figure 1A), we performed a detailed analysis between 40°C and 42°C by analyzing translation efficiencies (TEs). This revealed 113 significantly induced and 299 repressed genes at 42°C compared with 40°C (Figure S1C).
Figure S1.

Extended Data and Quality Control for Ribosome Profiling and RNAseq Experiments, Related to Figure 1
(A) Principal component analysis (PCA) of the normalized count data from duplicate ribosome profiling reads (left) and RNaseq samples (right) taken from yeast treated for 10 min at 30°C, 40°C and 42°C. Plot shows the first two principal components accounting for 59% and 26% of the total data variance, respectively.
(B) Clustering distance heatmap of the projection of the Euclidean distance between samples, which approximately corresponds to the biological variation coefficient.
(C) Volcano plots displaying differentially translated genes using translation efficiencies.
(D) Fraction of soluble Ded1-mCherry in ribosome profiling yeast cell lysate samples after treatment at 30°C, 40°C and 42°C as assessed by immunoblotting.
(E) Distribution of the minimum free energy of the 5′UTR of all yeast mRNAs compared to significantly repressed mRNAs at 40°C (in comparison to 30°C) or 42°C (in comparison to 40°C) in wild-type (WT) yeast cells. Significance was confirmed using a two-sided Wilcoxon test.
Figure 1.

Heat Shock Promotes a Switch in Gene Expression that Mimics Ded1p Inactivation
(A) Genome-wide distribution of log2-fold-changes of ribosome profiling and RNA abundance between 30°C and 40°C (top left) and 40°C and 42°C (top right). Differentially expressed genes (adjusted p value [p adjust] < 0.05) at the level of transcription (orange) and translation (blue), concordant (transcription and translation in the same direction, green), or in opposite directions (red) are highlighted and identified at the bottom right. Stacked bar charts (bottom left) indicate the absolute numbers of transcriptionally and translationally differentially expressed genes.
(B) Distribution of the MFE of the 5′ UTR of all yeast mRNAs compared with the translational efficiencies of mRNAs significantly induced or repressed at 42°C versus 40°C in wild-type (WT) yeast cells.
(C) Analysis of the data from Sen et al. (2015), comparing the TE of yeast expressing WT Ded1 or Ded1ts at 37°C. The distribution of the MFE of all yeast mRNAs was compared with that induced or repressed with a log2-fold-change of at least 0.5 at the restrictive temperature.
Statistical significance was calculated with a two-sided Wilcoxon test. See also Figure S1 and Table S1.
The 5′ UTRs of mRNAs are known to regulate translation (Vega Laso et al., 1993). We thus analyzed the 5′ UTRs of the differentially expressed mRNAs by comparing the length with the predicted secondary structure as calculated by the minimum free energy (MFE) using the software ViennaRNA (Lorenz et al., 2011). 5′ UTRs of genes induced at 42°C were shorter compared with all yeast 5′ UTRs (median length, 53 nt [all mRNAs] and 34 nt [induced mRNAs]). Likewise, the 5′ UTRs of induced genes were less structured (median MFE, −5 kcal/mol [all mRNAs] and −1.61 kcal/mol [induced mRNAs]) (Figure 1B; Table S1). In contrast, the 5′ UTRs of genes repressed at 42°C were longer (median length, 78 nt) and more structured (median MFE, −7.1 kcal/mol) (Figure 1B; Table S1). These data revealed that, between 40°C and 42°C, yeast cells switch translation from mRNAs with long and structured 5′ UTRs to mRNAs with short and unstructured 5′ UTRs.
Next we sought to determine which cellular factors could account for translational repression of mRNAs with longer and more structured 5′ UTRs during heat stress. The translation initiation factors Ded1p and eIF4B are required for ribosomal scanning of long transcripts with structured 5′ UTRs (Sen et al., 2016, Sen et al., 2015). Both proteins become insoluble and relocalize to SGs upon heat stress (Cherkasov et al., 2015, Wallace et al., 2015). Ded1p was classified as a “super-aggregator” because it aggregated with faster kinetics than eIF4B upon heat stress (Wallace et al., 2015). Because the 5′ UTRs of Ded1p-dependent mRNAs tend to be longer and more structured (Guenther et al., 2018, Sen et al., 2015), we hypothesized that repression of mRNAs during heat stress may be driven by sequestration of Ded1p into SGs. With this in mind, we re-analyzed the 5′ UTR complexity of mRNAs that were differentially translated in a previous ribosome profiling study that used a temperature-sensitive Ded1p variant (Ded1ts) (Sen et al., 2015) with our set of MFEs. As reported by Sen et al. (2015), when Ded1p was inactivated, mRNAs with shorter and less structured 5′ UTRs were induced (median length, 54 nt [all mRNAs] and 47 nt [induced mRNAs]; median MFE, −4.9 kcal/mol [all mRNAs] and −3.6 kcal/mol [induced mRNAs]), and mRNAs with longer and more structured 5′ UTRs were repressed (median length, 102 nt; median MFE, −14.4 kcal/mol) (Figure 1C). Thus, inactivation of Ded1ts recapitulates the switch in translation between 40°C and 42°C (Figure 1B), a temperature range in which Ded1p becomes insoluble (Figure S1D), and structured mRNAs are repressed (Figure S1E). Taken together, the data suggest that heat-induced assembly inactivates Ded1p, which coincides with a switch in translation of mRNAs with different 5′ UTR complexities.
Ded1p Condensates Assemble in Response to Heat Shock
To investigate Ded1p assembly upon heat shock, we first visualized the localization of Ded1p using live-cell fluorescence microscopy. Upon heat shock, diffusely distributed Ded1p assembled into punctate structures that colocalized with the SG marker Pab1p (Figure 2A). Ded1p also assembled during starvation and acidification of the cytosol (Figures S2A and S2B). This demonstrates that Ded1p assembly is sensitive to various physical-chemical parameters, including temperature and pH, and that the assemblies colocalize with SGs.
Figure 2.

The RNA Helicase Ded1p Assembles in Response to Elevated Temperature
(A) Fluorescence images of S. cerevisiae expressing Ded1-GFP and the SG marker protein Pab1-mCherry. Left: yeast before exposure to heat stress. Right: yeast after 10 min at 46°C (right). Cells were imaged at 46°C to observe colocalization with Pab1-mCherry, which only assembles above 44°C (yellow arrows). Scale bar, 5 μm.
(B) Quantification of Ded1-GFP foci per cell following a temperature ramp of 10°C/min from a starting temperature of 35°C using live-cell microscopy. Representative images of cells after 5 min are shown on the right. Scale bar, 5 μm.
(C). Fraction of soluble Ded1p in cell lysates after 10-min incubation at the indicated temperatures (mean, SD, n = 3).
(D) Distribution of the growth rate (h−1) of W303 yeast cells at different temperatures. The trend line (gray) is shown as a guide.
See also Figure S2.
Figure S2.

The RNA Helicase Ded1p Assembles in Response to Elevated Temperature, Starvation Conditions, and Lowered pH, Related to Figure 2
(A) S. cerevisiae expressing endogenous levels of Ded1-GFP and Pab1-mCherry during exponential growth (left) and after exposure to 60 min of glucose starvation (right). Yellow arrows highlight Ded1p and Pab1p assemblies. Scale bar 5 μm.
(B) S. cerevisiae expressing Ded1-GFP incubated in phosphate buffer pH 7.0 (top right) or pH 5.8 (bottom right) for 60 min in the presence of DNP. Yeast prior to DNP treatment shown left. Yellow arrows highlight Ded1p assemblies. Scale bar 5 μm.
(C) Representative immunoblot for the sedimentation analysis of Ded1-mCherry after 10 min of heat stress in yeast cells (see Figure 2C for quantification). PGK as a loading control.
(D) Representative immunoblot (top) used for the quantification (bottom) of the soluble fraction of Ded1-mCherry after 30 min of heat stress at indicated temperatures (Mean, SD, n = 3).
To investigate the temperature dependence of Ded1p assembly in living yeast cells, we used live-cell microscopy combined with a temperature stage (Mittasch et al., 2018). Although no Ded1p assemblies were observed at 35°C, the number of Ded1p foci as well as the apparent assembly rates increased as a function of temperature (Figure 2B). Accordingly, the fraction of soluble Ded1p decreased with increasing temperature and time (Figures 2C, S2C, and S2D). The fraction of soluble Ded1p was correlated with the growth rate of yeast (Figure 2D). Between 30°C and 37°C, the growth rate increased, but at temperatures above 37°C, the growth rate decreased sharply, a temperature range in which almost all Ded1p became insoluble after 30-min exposure to heat (Figure S2D). We conclude that the fraction of assembled Ded1p correlates with the duration and magnitude of the stress stimulus as well as the ability of yeast cells to grow.
Ded1p Condensates Assemble in a pH- and Temperature-Dependent Manner In Vitro
To provide a mechanistic understanding of temperature-dependent assembly, we purified Ded1p as well as Ded1p fused to monomeric GFP from insect cells and characterized the phase behavior in vitro. Purified Ded1p was diffuse at physiological concentrations of 2 μM. Increasing the temperature to 42°C induced Ded1p condensates (Figures 3A and S3A). Condensates also formed when the pH was lowered from 7.0 to 6.0 (Figure S3A). Although the condensates formed at low pH were spherical, condensates formed at elevated temperature resembled clusters of small spherical structures, suggesting that they form by phase separation but then gel rapidly. Heat-shocked cells experience a decrease in cytosolic pH to approximately 6.5 (Kroschwald et al., 2018, Weitzel et al., 1985). We thus tested for the effect of pH on temperature-induced Ded1p condensation. Indeed, Ded1p condensation occurred at a lower temperature when the pH was adjusted to that of the heat-stressed cytosol (Figure 3B). We thus conclude that Ded1p condensation is sensitive to temperature and pH, two parameters that frequently change in the life cycle of yeast cells.
Figure 3.

Ded1p Has an Intrinsic Property to Form Condensates upon Elevated Temperature
(A) Fluorescence image of 2 μM purified Ded1-GFP in piperazine-N,N′-bis(2-ethanesulfonic acid (PIPES)/KOH buffer (pH 7) and 100 mM KCl buffer at 25°C (left image) and after 10 min at 42°C (right image). Scale bar, 3 μm.
(B) Phase diagram of Ded1p in PIPES/KOH and 100 mM KCl buffer with the control parameters pH and temperature. Analysis was performed after 30 min of incubation. Green dots indicate condensates, and “x” indicates no condensates.
(C) Images from a time-lapse video monitoring Ded1p assembly at pH 6.8. Scale bar, 15 μm.
(D) Hydrodynamic radius of 0.5, 1, and 2 μM Ded1-GFP in PIPES/KOH (pH 6.8) and 100 mM KCl buffer as a function of temperature.
(E) Kinetic analysis of the apparent Ded1-GFP and Pab1-GFP assembly reaction, measured by DLS in PIPES/KOH (pH 6.8) and 100 mM KCl buffer. Plotted is the natural logarithm of the apparent rate constant (ln(k)) as a function of the reciprocal temperature.
(F) Analysis of the transition temperature midpoints of Ded1p condensation as determined by light scattering (TM scattering) and changes in tertiary structure as determined by nano-differential scanning fluorimetry (DSF) (TM tertiary structure) at varying Ded1p concentrations.
See also Figure S3.
Figure S3.

Ded1p Has an Intrinsic Property to Form Condensates upon Elevated Temperature and Lowered pH, Related to Figure 3
(A) Brightfield images of 2 μM purified Ded1p protein without fluorescent tag imaged at 25°C in PIPES/KOH pH 7.0, 100mM KCl buffer (left image), after exposure to 42°C for 10 min (top right) or in PIPES/KOH pH 6.0, 100 mM KCl buffer (bottom right). Scale bar 3 μm.
(B) Representative nano-DSF measurement monitoring changes in the intrinsic tryptophan/tyrosine fluorescence ratio (F350/330 nm). Dashed line depicts TM.
(C) Light scattering plot of the same sample shown in B. Dashed line depicts TM.
(D) Left: CD spectra of Ded1p at different temperatures starting from 21°C (green) to 45°C (pink). Right: Normalized CD spectra.
(E) Left: CD spectra of Ded1p in PIPES/KOH pH 6 and pH 7, 100 mM KCl. Right: Normalized CD spectra.
To assess the temperature dependence of Ded1p assembly, we determined the temperature of condensation onset (Tonset), which we define as the temperature at which Ded1p condenses upon a rapid change in temperature. Ded1p was initially diffuse at 39°C but formed condensates at 40°C (Figure 3C). A similar Tonset of ∼40°C was determined by dynamic light scattering (DLS) (Figure 3D). Next we determined the apparent assembly rates of Ded1p as a function of temperature. For comparison, we also determined the apparent assembly rates of Pab1p (Riback et al., 2017). The natural logarithm of the apparent reaction rate (ln(k)) for both proteins depended linearly on the reciprocal temperature (Figure 3E). Ded1p assembly was 2-fold more temperature dependent and occurred faster compared with Pab1p (Ded1p, activation energy (EA) 680 kJ∙mol−1; Pab1p, EA 365 kJ∙mol−1). This suggests that Ded1p undergoes a highly cooperative assembly transition when exposed to heat stress.
The highly cooperative assembly suggests that Ded1p may undergo temperature-induced structural changes. To gain information about tertiary structure changes, we monitored the intrinsic protein fluorescence of the 7 tryptophan and 16 tyrosine residues in Ded1p. Plotting the 350/330-nm ratio over a temperature gradient of 1°C/min, we were able to observe structural rearrangements in Ded1p. The fluorescence ratio decreased initially but increased sharply at temperatures above Tonset (∼40°C) (Figure S3B), indicating changes in protein tertiary structure. This structural change coincided with Ded1p condensation, as monitored by light scattering (Figure S3C). Moreover, the transition midpoint (TM) of the tertiary structure change and the TM of Ded1p condensation were very similar at various tested concentrations (Figure 3F). Next we used circular dichroism (CD) to test for secondary structure changes (Greenfield, 2006). Upon a temperature increase (Figure S3D) or a decrease in pH (Figure S3E), the α-helical (negative bands at 222 nm and 208 nm and a positive band at 193 nm) and β sheet content (negative bands at 218 nm and positive bands at 195 nm) remained largely unchanged, except for an overall decrease in CD signal. The signal loss was mostly due to depolarization of the polarized light by formation of light-scattering condensates, and normalization led to overlap of the CD curves (Figures S3D and S3E).
Taken together, these results suggest that the secondary structure of Ded1p remains largely unchanged within the tested temperature and pH ranges, whereas Ded1p condensation appears to coincide with tertiary structure rearrangements.
mRNA Partitions into Ded1p Condensates and Modifies their Properties
Ded1p is an RNA helicase. Hence, we compared Ded1p condensates formed in the presence or absence of different RNAs. tRNA and rRNA did not affect Ded1p heat-induced condensates. In contrast, condensates formed in the presence of mRNA were spherical (Figure 4A; Video S1), and a substantial mRNA fraction became insoluble (Figures 4C, 4D, and S4A). Additionally, mRNA decreased the Tonset of Ded1p condensation from ∼40°C to ∼36°C (Figure 4B). Ded1p-mRNA condensates initially exhibited liquid-like properties but hardened after a few minutes of heating (Figure 4E; Video S1). Importantly, hardened mRNA-containing condensates dissolved upon increasing the salt concentration (Figure 4F). Condensation of Ded1p was promoted by mRNAs that were reported previously to be hyper-dependent on Ded1p with respect to assembly of the 48S preinitiation complex (Gupta et al., 2018). Accordingly, a greater fraction of hyper-dependent mRNAs was enriched in condensates compared with hypo-dependent Ded1p mRNAs (Figures S4B–S4E). In summary, these data suggest that mRNA is specifically enriched in Ded1p condensates and that it renders Ded1p condensates softer and reversible.
Figure 4.

mRNA Is Sequestered into Ded1p Heat-Induced Condensates and Affects Condensate Physical Properties
(A) Images of 2.5 μM purified Ded1-GFP mixed with water, tRNA, rRNA, or mRNA (capped and poly(A)-tailed coding for nano-luciferase with the 5′ UTR of PAB1) at a final RNA concentration of 45 ng/μL in PIPES/KOH (pH 6.8) and 200 mM KCl buffer and incubated at 42°C for 15 min. Scale bar, 5 μm.
(B) Mean hydrodynamic radius of Ded1p in the presence or absence of mRNA as a function of temperature in PIPES/KOH (pH 6.8) and 200 mM KCl buffer.
(C) Analysis of the soluble RNA fraction after co-incubation with Ded1-GFP for 15 min at 42°C in PIPES/KOH (pH 6.8) and 200 mM KCl buffer. 260-nm values were normalized to the fraction of soluble RNA at 25°C (mean, SD, n = 3).
(D) Mean and standard deviation of fluorescence intensities of n = 40 condensates of Ded1 with and without mRNA formed for 10 min at 42°C in PIPES/KOH (pH 6.8) and 200 mM KCl buffer and labeled with Sybr Green II RNA dye.
(E) Condensates formed by 5-min incubation at 42°C in PIPES/KOH (pH 6.8) and 200 mM KCl buffer were made to fuse with optical tweezers at room temperature. Top: schematics of controlled condensate fusion probed with optical tweezers. Center: bright-field images of a fusion time course. Scale bars, 2 μm. Bottom: force curve (laser signal [ arbitrary units, AU]) as a function of time. The relaxation time was derived from a single exponential fit (pink line) of the fusion trace.
(F) Ded1p condensates with and without mRNA formed after 5 min at 42°C in PIPES/KOH (pH 6.8) and 200 mM KCl buffer (left) and upon increasing the NaCl concentration to 1 M (right). Scale bar, 3 μm.
Figure S4.

Ded1p Hyper-Dependent mRNAs Are Preferentially Sequestered into Ded1p Heat-Induced Condensates, Related to Figure 4
(A) Controls for Figure 4C. The fraction of soluble mRNA after heat treatment in the presence of GFP or PIPES/KOH pH 6.8, 200mM KCl buffer was measured by nanodrop using an absorbance of 260 nm and normalized to the fraction of soluble mRNA prior to treatment at 25°C (Mean, SD, n = 3).
(B) Schematic representation of mRNAs used in microscopy experiment shown in (C). To circumvent effects of mRNA length on Ded1p condensation, mRNAs of similar length were synthesized: full length for the RPL41A mRNA and the first 350 nucleotides of the 5′UTR and coding sequence of PMA1 and SFT2 mRNAs.
(C) Representative images of 1 μM Ded1-GFP and 18 ng/μL Cy3 labeled mRNAs after 10 min at 42°C in PIPES/KOH pH 6.8, 150mM KCl buffer in the presence of absence of 18 ng/μL tRNA (n = 3).
(D and E) Quantification of the condensed Ded1p fraction and partitioning (Iin/Iout) of mRNA into Ded1p condensates from multiple fields of view are shown in D and E, respectively.
4 μM Ded1-GFP and synthesized mRNA containing the 5′UTR from PAB1, were mixed at room temperature in PIPES/KOH pH 6.8 buffer, 200 mM KCl and imaged on the temperature stage with Pico-Surf to prevent aberrant surface effects, as described in the STAR Methods. The temperature was increased to 40°C while imaging.
Condensation of Ded1p Is Modified by Its Intrinsically Disordered Regions
The central helicase domain of Ded1p is conserved (Figure 5A) and flanked by two intrinsically disordered regions (IDRs), with the N-terminal IDR having a prion-like amino acid composition (Lancaster et al., 2014). To determine the role of the N- and C-terminal IDRs in condensation, we investigated the phase behavior of two truncation variants (Ded1ΔC and Ded1ΔN) in vitro. Compared with full-length Ded1p, Ded1ΔN had a lowered Tonset (Figures 5B, 5C, and S5B) and formed irregularly shaped clusters at low pH (Figure S5A), suggesting that the N-terminal IDR increases solubility and inhibits Ded1p phase separation. Ded1ΔC was less sensitive to changes in pH (Figure S5A) and temperature (Figure 5B). In fact, Ded1ΔC did not assemble into condensates upon heating when using the same protein concentrations and buffer conditions as for full-length Ded1p (Figure 5B). Heat-induced assembly of Ded1ΔC was observed at higher protein concentrations (Figure S5B) and at low pH (Figure 5C), indicating that the C-terminal IDR drives Ded1p condensation.
Figure 5.

The Phase Behavior of Ded1p Is Modulated by Its IDRs
(A) Top: domain structure of Ded1p. Bottom: disorder plot of Ded1p (VSL2 function of the online tool Predictor of Natural Disordered Regions (PONDR); Peng et al., 2005).
(B) Mean hydrodynamic radii of 3 μM GFP-tagged Ded1p (Ded1p), Ded1ΔN, and Ded1ΔC as a function of temperature in phosphate (pH 7.4) and 200 mM KCl buffer.
(C) Phase diagram of Ded1p and Ded1ΔN in PIPES/KOH (pH 7.5) and 200 mM KCl buffer (left) and Ded1p and Ded1ΔC in PIPES/KOH (pH 6.8) and 200 mM KCl buffer (right) over a temperature and concentration range as determined by the scattering function of nano-DSF. The data of three technical replicates were plotted. Trendlines are shown as a guide.
(D) Mean hydrodynamic radii of 4 μM GFP-tagged Ded1p (wild-type) and Ded1-IDRm (IDRm) as a function of temperature in phosphate (pH 7.4) and 100 mM KCl buffer.
(E) Images of S. cerevisiae expressing Ded1-mCherry and Ded1-IDRm-mCherry after 30 min at the indicated temperatures. Yellow arrows point to the temperature at which assemblies were first observed. Scale bar, 5 μm.
(F) Spot titer assay with 5-fold serial dilutions of strains expressing Ded1-mCherry or Ded1-IDRm-mCherry grown at 22°C, 30°C, 39°C, and 41°C.
Shown are representative images of 3 independent repeats. See also Figure S5.
Figure S5.

The Condensation of Ded1p in Response to Elevated Temperature and Lowered pH is Regulated by Its IDRs, Related to Figure 5
(A) Representative images of 2 μM Ded1p, Ded1ΔN and Ded1ΔC in PIPES/KOH buffers ranging from pH 6.0 to 7.5 in 100 mM KCl. Scale bar, 5 μm.
(B) Light scattering of GFP-tagged 3 μM Ded1p (blue), 3 μM Ded1ΔN (green), 3 μM (magenta) and 10 μM Ded1ΔC (light magenta) measured using nano-DSF as a function of temperature in PIPES/KOH pH 7.5, 200 mM KCl buffer. Data were normalized to the minimum light scattering value of the corresponding protein.
(C) Alignment of the N-terminal region of Ded1p and Ded1-IDRm with the ClustalW algorithm (Chenna et al., 2003). (∗): identical amino acids, (:): highly similar amino acid properties, (.): similar amino acids.
(D) Quantification of the condensed fraction of Ded1p (wild-type) and Ded1-IDRm (IDRm) as a function of temperature as determined by microscopy data shown in (E). (Mean, SD).
(E) Representative images of 4 μM GFP-labeled Ded1p (wild-type) and Ded1-IDRm (IDRm) in PIPES/KOH pH 6.8, 200 mM KCl buffer at indicated temperatures. Scale bar, 10 μm.
Altering the Temperature Sensitivity of Ded1p Limits Cell Growth
To investigate the functional significance of temperature-induced Ded1p assembly in cells, we aimed to generate a variant of Ded1p exhibiting altered Tonset while maintaining the essential function(s) of Ded1p under normal growth conditions. We sought to decrease the Tonset of Ded1p rather than increasing it because other factors are likely to become limiting at elevated temperatures. We targeted the N-terminal IDR for mutagenesis because its deletion decreased the Tonset of Ded1p (Figure 5B). To identify sites for mutagenesis, we searched for molecular features that are evolutionarily preserved in otherwise highly diverged disordered sequences (Zarin et al., 2019; see STAR Methods for details). Residues 35–54 showed conservation of glycine residues and polar residues such as asparagine. We replaced the corresponding region in the DED1 gene with a sequence lacking the conserved features, yielding the variant DED1-IDRm (Figure S5C). Purified Ded1-IDRm had a lower Tonset compared with wild-type Ded1p (Figures 5D, S5D, and S5E). Moreover, a yeast strain expressing mCherry-tagged Ded1-IDRm from the endogenous locus formed condensates at a lower temperature compared with Ded1p (Figure 5E). Accordingly, 80% of Ded1-IDRm was insoluble after heat treatment at 42°C compared with 50% of wild-type Ded1p (Figure S6D).
Figure S6.

Extended Data and Quality Control for Ribosome Footprinting and RNAseq Experiments of IDRm and Wildtype Ded1p, Related to Figure 6
(A) Clustering distance heatmap of the projection of the Euclidean distance between ribosome profiling samples (left) and RNaseq samples (right).
(B) Principal component analysis (PCA) of the normalized count data from the ribosome profiling reads (left) and RNaseq reads (right) of the duplicate samples.
(C) Volcano plots showing differentially translated genes.
(D) Fraction of soluble Ded1-mCherry and Ded1-IDRm-mCherry from yeast incubated at 30°C, 40°C and 42°C for 10 min as assessed by immunoblotting (Mean, SD, n = 3).
(E) Analysis of the soluble (supernatant) and insoluble (pellet) fractions of yeast expressing Ded1p and Ded1-IDRm incubated at 30°C, 40°C and 42°C from RNaseq read analysis. Boxplot displays the distribution of the 5′UTR minimum free energies of transcripts differentially enriched in the pellet over the supernatant (left) and supernatant over pellet (right) compared to the 5′UTR of all genes. Significance was confirmed using a two-sided Wilcoxon test.
(F) Microscopy images of 0.8 μM Ded1-GFP condensates after 30 min at 40°C or 42°C in pH 6.8 PIPES/KOH, 100mM KCl buffer before (left) and after (right) addition to the translation assays at 25°C. Images were taken right after the addition of the condensates (0 min) and at the end of the assay (90 min). Scale bar, 5 μm.
We next compared the growth of yeast expressing Ded1-IDRm with yeast expressing wild-type Ded1p using serial dilution spot titer assays. At 22°C, both yeast strains grew indistinguishably well (Figure 5F). However, at higher temperatures, cells expressing Ded1-IDRm exhibited a temperature-dependent growth defect (Figure 5F). This growth defect was already visible at 30°C, suggesting that a small fraction of Ded1-IDRm already becomes insoluble at normal temperatures, as also seen in our in vitro experiments (Figures 5D, S5D, and S5E). At 41°C, the Ded1-IDRm strain exhibited a strong growth defect (Figure 5F). Combined with our in vitro results, this suggests that condensate formation of Ded1p limits cell growth.
Ded1p Condensation Represses Translation of Housekeeping Transcripts
What could be the physiological relevance of temperature-induced Ded1p condensation? Unlike other yeast proteins, which only become insoluble at temperatures above 50°C (Leuenberger et al., 2017), Ded1p already forms condensates at ∼40°C (Figures 3C and 3D). We reasoned that condensation of Ded1p may inhibit its function in promoting translation of mRNAs with longer and more structured 5′ UTRs (Berthelot et al., 2004, Guenther et al., 2018, Sen et al., 2015; Figure S1D) while minimally affecting and even promoting translation of mRNAs with unstructured 5′ UTRs (Figure 1B).
One group of proteins strongly induced upon heat shock is the group of heat shock proteins (Hsps), suggesting that the mRNAs encoding Hsps have short and unstructured 5′ UTRs. To test this idea, we assessed the complexity of their 5′ UTRs by determining their length and MFE. We found that many Hsp-encoding transcripts (HSP104, SSA4, SSA2, HSP10, HSP12, HSP26, HSP78, HSP82, HSC82, and SSE2) have short 5′ UTRs with a MFE close to 0 kcal/mol (Figure 6A). An exception is SSA3, which has a long and highly structured 5′ UTR (−24.5 kcal/mol) that contains several upstream start codons, suggesting another form of translational regulation. This is consistent with the idea that heat shock transcripts may evade translational repression by Ded1p condensation. Conversely, mRNAs with more structurally complex 5′ UTRs become repressed upon Ded1p condensation. Such mRNAs could, for example, code for housekeeping proteins, which we define as proteins required for housekeeping functions during normal growth under non-stress conditions.
Figure 6.

Heat-Induced Condensation of Ded1p Facilitates a Switch in Translation to Selectively Repress Housekeeping mRNAs
(A) Length versus MFE for all yeast 5′ UTR sequences as determined by Kertesz et al. (2010). Heat shock factors and housekeeping proteins are color labeled.
(B) Boxplots showing the distribution of the MFE of all yeast mRNAs and those induced or repressed in yeast expressing Ded1-IDRm compared with WT cells at 42°C. Significance was confirmed with a two-sided Wilcoxon test. Distance clustering heatmaps and systematic comparison by principal-component analysis (PCA) are shown in Figures S6A and S6B.
(C) Log2-fold-change of the TEs of each gene plotted against the MFE. Heat shock factors and housekeeping proteins are color labeled.
(D) Schematics of reporter transcripts. 5′ UTRs from mRNAs with structurally complex 5′ UTRs (housekeeping transcripts) or 5′ UTRs with little structure (stress transcripts) were fused to nano-luciferase, a short 3′ UTR and a poly(A) tail.
(E) In vitro translation of stress reporter transcripts and housekeeping transcripts as measured by luminescence in the presence of different Ded1p concentrations. Luminescence measurements were normalized to the reaction containing no added Ded1p (mean, SD, n = 3).
(F) In vitro translation of different reporter transcripts in the presence of diffuse 0.8 μM Ded1p (25°C) or Ded1p condensates formed at 40°C or 42°C for 30 min in PIPES/KOH (pH 6.8) and 100 mM KCl buffer. Luminescence measurements were normalized to the reaction with the unstructured GIS2 5′ UTR reporter (mean, SD, n = 3).
To provide experimental evidence, we compared ribosome footprints of wild-type cells and cells expressing Ded1-IDRm after 10 min of heat stress (see Figures S6A and S6B for quality control of ribosome profiling). We reasoned that increased Ded1p condensation in the Ded1-IDRm strain would lead to stronger repression of Ded1p-dependent transcripts (Guenther et al., 2018, Sen et al., 2015). This effect should be most pronounced at 42°C, where we observed the largest difference in Ded1p condensation between wild-type and Ded1-IDRm cells (Figure S6D). Indeed, 270 genes were induced and 286 genes were repressed at 42°C in the Ded1-IDRm variant compared with wild-type cells (Figure S6C). The 5′ UTRs of repressed genes were longer, with a higher propensity for secondary structure (median length, 74 nt; median MFE, −9.3 kcal/mol) than all mRNAs (median length, 53 nt; median MFE, −5 kcal/mol) (Figure 6B; Table S1). Thus, increased condensation of Ded1-IDRm leads to reduced expression of mRNAs with longer and more structured 5′ UTRs. Importantly, heat shock factors were generally upregulated in the Ded1-IDRm strain compared with wild-type cells at 42°C (Figure 6C).
Given that mRNA partitions into Ded1p condensates (Figures 4C, S4C, and S4E), we determined whether the lower Tonset of Ded1-IDRm affects the solubility of mRNAs in living cells. We sequenced the soluble and insoluble RNA fractions of wild-type and Ded1-IDRm cells. The insoluble fractions of wild-type and Ded1-IDRm yeast were enriched for mRNAs with structurally complex 5′ UTRs (Figure S6E). Although many of the insoluble mRNAs were shared between wild-type and Ded1-IDRm (1,135 at 30°C, 1,131 at 40°C, and 1,086 at 42°C), the mRNAs that were specific to the Ded1-IDRm strain were enriched for structurally complex 5′ UTRs and those specific to the WT strain were not (Figure S6E). Notably, mRNAs with more structurally complex 5′ UTRs were most prominently enriched in the insoluble fraction of the Ded1-IDRm strain at 30°C (Figure S6E). This may be a consequence of the downshifted Tonset of Ded1-IDRm (Figures 5D, 5E, S5D, and S5E) and may contribute to the growth phenotype of the Ded1-IDRm strain at 30°C (Figure 5F). Overall, our results suggest that lowering the Tonset of Ded1p exacerbates translational repression of Ded1p-dependent mRNAs.
To further investigate the role and specificity of Ded1p condensation in regulating protein synthesis, we set up a yeast in vitro translation assay with different reporter transcripts in which the coding sequence of nano-luciferase was fused to short 5′ UTRs with little structure (MFE, 0 to −0.8 kcal/mol) of a control and stress transcripts or longer and more structured 5′ UTRs (MFE, −2.7 to −16.83 kcal/mol) of housekeeping transcripts (Figure 6D). We did not select more structured 5′ UTRs to remain close to the median complexity of all yeast 5′ UTRs, which corresponds to −5 kcal/mol (Figures 6A–6C; Table S2). Addition of Ded1p increased the translation of housekeeping transcripts 15- to 25-fold (Figure 6E). In contrast, stress transcripts and a control transcript with no secondary structure in the 5′ UTR (GIS2) were marginally affected by additional Ded1p (Figure 6E). Notably, SSA1, a constitutive member of the yeast Hsp70 family that harbors a longer and more structured 5′ UTR (MFE, −2.7 kcal/mol) than other heat shock factors, behaved similar to housekeeping transcripts and showed high Ded1p dependence. This agrees with previous findings that SSA1 functions as a housekeeping chaperone in growing cells (Werner-Washburne et al., 1987). In contrast, SSA4, a stress-inducible member of the Hsp70 family with a short 5′ UTR with no predicted secondary structure, was largely insensitive to increasing Ded1p concentrations (Figure 6E).
We next tested the effect of adding Ded1p condensates formed at different temperatures to our in vitro translation system. To this end, we incubated Ded1-GFP at 25°C, 40°C, or 42°C and added the diffuse or condensed protein to the translation assay. Ded1-GFP condensates persisted for the duration of the assay (Figure S6F). Translation of all mRNAs decreased with increasing incubation temperature of Ded1p, but translation of housekeeping transcripts was more strongly repressed than that of stress transcripts (Figure 6F). Thus, we conclude that condensate assembly inactivates Ded1p and that this represses translation of mRNAs with longer and more structured 5′ UTRs while minimally affecting stress mRNAs with short and unstructured 5′ UTRs.
Ded1p Homologs from Species Adapted to Different Thermal Niches Show Distinct Temperature Sensitivity
Our data suggest that Ded1p condensation promotes an extended heat shock response program for preferential production of stress proteins at temperatures above 39°C. One could envision that such a mechanism should be adapted to the environment in which an organism lives. We thus replaced the S. cerevisiae DED1 allele with the homologous DED1 sequences from two fungi that have adapted their growth to lower or higher temperatures compared with that of S. cerevisiae: (1) cold-adapted Saccharomyces kudriavzevii (Salvadó et al., 2011, Scannell et al., 2011), which belongs to the Saccharomyces sensu stricto genus that evolved ∼20 million years ago (Dujon, 2006) and has a maximum growth temperature of approximately 36°C (Salvadó et al., 2011), and (2) the evolutionarily more distinct thermophilic fungus Thielavia terrestris (Berka et al., 2011, Samson et al., 1977), which diverged from S. cerevisiae around 590 million years ago (Kumar et al., 2017) and has a maximum growth temperature of ∼50°C (Figure 7A; Table S3).
Figure 7.

Ded1p Homologs from Different Fungi Exhibit Distinct Temperature Sensitivity
(A) Schematic representation of the growth temperature ranges for cold-adapted S. kudriavzevii, mesophilic S. cerevisiae, and thermophilic T. terrestris. The temperatures at which the organisms experience heat stress are marked in magenta.
(B) Representative images of S. cerevisiae in which endogenous Ded1 was allele replaced with GFP-tagged DED1 from S. kudriavzevii (S.k.), T. terrestris (T.t.), or mock replaced (S.c.) at the indicated temperatures. Yellow arrows highlight the temperature at which Ded1p assemblies were first observed. Scale bar, 5 μm.
(C) Mean hydrodynamic radius of Ded1-GFP homologs as a function of temperature in PIPES/KOH (pH 6.8) and 100 mM KCl buffer at 2 μM protein concentration.
(D) Microscopy images of 2 μM Ded1-GFP homologs incubated in PIPES/KOH and 100 mM KCl buffer for 10 min at the indicated temperatures. Scale bar, 5 μm.
(E) Model. Under growing conditions, Ded1p facilitates scanning of housekeeping mRNAs that harbor a complex secondary structure in their 5′ UTR, whereas it is not needed for stress mRNAs that harbor simple 5′ UTRs. Other factors may work together with Ded1p to regulate translation of structurally complex housekeeping mRNAs. In addition, stress mRNAs could be specifically regulated by factors that have yet to be identified (factor X). Upon heat stress, Ded1p and potentially other factors condense, and, consequently, Ded1p-dependent housekeeping mRNAs are silenced, whereas stress mRNAs are preferentially translated.
Cold-adapted S. kudriavzevii Ded1p expressed in S. cerevisiae had a lowered Tonset, whereas the thermophilic Ded1p variant had an increased Tonset (Figure 7B). Using recombinant purified Ded1p from the three species (Figure S7A), we found that temperature-dependent assembly of Ded1p followed the heat sensitivity of the respective organism (Figures 7C, 7D, and S7B). Tonset occurred at 34°C for S. kudriavzevii, at 40°C for S. cerevisiae, and at 46°C for T. terrestris Ded1p. Remarkably, these temperatures are just below the threshold temperature at which these three species enter a growth-arrested state (Table S3). We conclude that the condensation of the Ded1p homologs is adapted to the thermal niche of the respective organism and that occurs in a protein-autonomous manner.
Figure S7.

Condensation of Ded1p in Response to Elevated Temperature and Lowered pH Is Conserved in Three Fungi, Related to Figure 7
(A) Coomassie stained SDS-PAGE of purified Ded1-GFP from S. kudriavzevii (S.k.), S. cerevisiae (S.c.) and T. terrestris (T.t.).
(B) Light scattering of purified Ded1-GFP homologs in PIPES/KOH pH 6.8, 100 mM KCl buffer. Temperature ramp 1°C/min.
(C) Representative microscopy images of Ded1-GFP homologs in PIPES/KOH pH 6.8 or PIPES/KOH pH 5.7, 100 mM KCl buffer. Scale bar, 3 μm.
Discussion
Our understanding of how stress signals are detected and converted into a response that promotes selective expression of stress-protective proteins is limited. In this paper, we show that the essential translation initiation factor Ded1p has an intrinsic ability to respond to high temperature by phase separation and subsequent gelation (summarized in Figure 7E). Given the essential role of Ded1p in translation initiation, condensate assembly of Ded1p together with mRNA promotes a switch in protein synthesis that silences production of housekeeping proteins and frees up necessary resources for production of stress proteins. Thus, condensate assembly by Ded1p adds an important molecular step to the heat shock response that combines the ability to detect changes in the environment with the ability of cells to mount an effective stress response.
To survive, cells need mechanisms that sense the intensity and duration of heat stress to mount an appropriate stress response (Morano et al., 2012). Because phase separation is sensitive to small changes, it is an ideal mechanism to detect and respond to temperature changes in the environment (Alberti and Hyman, 2016, Franzmann and Alberti, 2019). The phase behavior of Ded1p is such an example. In the temperature range between 37°C and 42°C, the amount of condensed Ded1p correlates with the intensity and duration of heat stress. At temperatures at and above 42°C, the kinetics of Ded1p phase separation are fast, and Ded1p becomes almost completely insoluble within 30 min (Figure S2D). At this temperature, cells cease growth, suggesting that the amount of soluble Ded1p is a major determinant of cell growth.
Our data suggest that Ded1p condensation is an important mechanism underlying the response of yeast cells to severe heat stress. This response, which we refer to as the extended heat shock response, is increasingly activated at temperatures above 39°C (Figures 2C and 3D), is predominantly translational (Figure 1A), and is characterized by downregulation of housekeeping proteins (Figures 1B, 6C, and S1E). We speculate that the extended heat shock response is a specific adaptation to severe temperature stress that may help reduce the load of misfolding-prone proteins generated during translation. This is in contrast to the transcriptional heat shock response driven by Hsf1p, which is already turned on at 33°C and maximally activated at temperatures above 39°C (Hahn et al., 2004, Morano et al., 2012). In this temperature range, yeast cells show robust growth (Figure 2D), suggesting that one of the main roles of the transcriptional response could be to ensure growth at elevated temperatures. In contrast to the transcriptional heat shock response, which is driven primarily by Hsf1p, the translational response appears to involve several different factors working together in a coordinated manner (Franzmann and Alberti, 2019, Kroschwald et al., 2018, Riback et al., 2017, Wallace et al., 2015). Ded1p appears to adopt a key role, as suggested by the observation that increased condensation (mediated by Ded1-IDRm) is sufficient to increase stress protein translation (Figure 6C) and cause a slow-growth phenotype at lower temperatures (Figure 5F).
One striking observation is that temperature-induced Ded1p condensation is only slightly dependent on protein concentration. This suggests that Ded1p has evolved a specific mechanism that couples detection of temperature changes to condensate assembly. This is reminiscent of the polymer Poly(N-isopropylacrylamide) (PNIPAM), which phase-separates in a largely concentration-independent manner at high temperatures. In the case of PNIPAM, phase separation is thought to be promoted by rapid collapse of random coils into globules (Okada and Tanaka, 2005). We propose that Ded1p may use a similar mechanism. The C-terminal IDR of Ded1 is rich in hydrophobic residues and has been shown to promote Ded1 oligomerization (Gao et al., 2016). In agreement, we find that the C-terminal IDR greatly affects the Tonset of Ded1p condensation (Figures 5B, 5C, and S5B). Moreover, condensation coincides with a change in Ded1p protein tertiary structure (Figures 3F, S3B, and S3C), suggesting that a cooperative conformational rearrangement promotes condensation. We speculate that this rearrangement regulates the availability of necessary valences for condensation, presumably located within the hydrophobic and tryptophan-rich C-terminal IDR. Such a mechanism could ensure that Ded1p condensation is robust and occurs within a narrow temperature range. Understanding the contribution of individual amino acids to driving Ded1p condensation requires further study.
Previous studies have proposed that Pab1p and Pub1p form gel-like condensates upon increased temperature (Kroschwald et al., 2018, Riback et al., 2017). These studies observed large clusters of spherical structures, supportive of liquid phases that gel rapidly. Using temperature-controlled fluorescence microscopy and optical tweezers, we were able to provide direct evidence of the liquid-like state of heat-induced Ded1p-mRNA condensates. These liquid-like condensates harden quickly into gels. Importantly, Ded1p-mRNA condensates were reversible whereas Ded1p-only condensates were not (Figure 4F). This points to an important role of mRNA in determining the material properties of the condensates. Taken together, our data suggest that a gel transition of Ded1p-mRNA condensates prevents Ded1p from fulfilling its function in translation initiation. The reversibility of the Ded1p-mRNA gel condensates could facilitate their dissolution for reuse of Ded1p and mRNA in the stress recovery phase.
We propose that Ded1p condensation drives a switch in gene expression on the level of translation by silencing the expression of mRNAs with longer and more highly structured 5′ UTRs, such as those found in housekeeping transcripts. The direct and indirect consequences of this switch in translation remain to be determined, but we entertain the possibility of two major functional roles: (1) reducing the burden of misfolding-prone proteins produced during translation, and (2) freeing up resources for production of stress factors encoded by transcripts with less structured 5′ UTRs. Another potential function could be translational regulation of upstream open reading frames (uORFs), whereby decreased Ded1p levels enhance uORF translation (Guenther et al., 2018). More detailed studies investigating ribosomal initiation in the 5′ UTRs of various different mRNAs are needed to determine precisely how stress-induced condensation of Ded1p affects translational regulation.
Our results also suggest that Ded1p-dependent housekeeping mRNAs are sequestered in Ded1p condensates (Figures 4C, 4D, and S4A–S4E). Sequestration of housekeeping mRNAs away from the pool of actively translated mRNAs reduces the number of mRNAs that compete for translation factors, supporting translational reprogramming during stress. In addition, mRNA sequestration may help protect these mRNAs from degradation and allow storage for later use. Release of stored mRNAs from condensates would then make housekeeping mRNAs available for reentry into the cell cycle in the stress recovery phase. In agreement with this, mRNAs recruited to mammalian SGs have more secondary structure in their 5′ UTRs and poor translational efficiency (Khong et al., 2017). Moreover, we find that the 5′ UTRs of mRNAs enriched in the insoluble fraction of the Ded1-IDRm strain are structurally more complex (Figure S6E). Whether Ded1p alone provides the specificity for mRNA selection or operates together with other translation initiation factors remains to be determined.
In summary, we propose that temperature-driven phase separation of Ded1p is an evolutionarily tuned physiological process and an integral part of an extended heat stress response program that represses translation of housekeeping mRNAs and facilitates preferential translation of stress mRNAs. This mechanism of stress-induced phase separation is likely harnessed by numerous other translation factors in yeast and other organisms. We suspect that cell adaptation by phase separation is a widespread process in nature that helps organisms detect sudden fluctuations in the environment and mount stress-specific responses.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti mCherry | Biovision | Cat#5993-100; RRID:AB_1975001 |
| Mouse monoclonal anti PGK | Invitrogen | Cat#459250; RRID:AB_2532235 |
| Goat IgG anti-Rabbit IgG (H+L)-Alexa Fluor 790 | Jackson Immuno Research | Cat#111-655-144; RRID:AB_2338086 |
| Goat anti-Mouse IgG (H+L)-Alexa Fluor 680 | Dianova | Cat#115-625-146; RRID:AB_2338935 |
| Bacterial and Virus Strains | ||
| Sf9 cells | Expression Systems | Cat#94-001F |
| Subcloning Efficiency DH5α Competent Cells | Invitrogen | Cat#18265017 |
| NEB 5-alpha Competent E. coli | NEB | Cat#C2987I |
| E. coli XL1-Blue | Agilent | Cat#200249 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RNaseOUT | Invitrogen | Cat#10777019 |
| cOmplete Protease Inhibitor Cocktail | Roche | Cat#11836145001 |
| cOmplete Protease Inhibitor Cocktail, EDTA-free | Roche | Cat#5056489001 |
| Bond-Breaker TCEP Solution | ThermoFisher Scientific | Cat#77720 |
| Magnesium acetate | Sigma | Cat#M-2545 |
| Potassium acetate | Merck | Cat#104820 |
| Potassium chloride | Merck | Cat#104935 |
| HEPES | Carl Roth | Cat# 9105.2 |
| DTT | Fermentas Life Sciences | Cat#R0862 |
| ATP | Applichem | Cat#A1348,0005 5g |
| GTP | Thermo Fisher | Cat#18332015 |
| Creatine phosphate | Merck | Cat#2380 |
| Creatine phosphokinase | Sigma | Cat#C3755 |
| Micrococcal nuclease | NEB | Cat#M0247S |
| 2,4-dinitrophenol (DNP) | Sigma-Aldrich | Cat#D199303 |
| Pipes | Applichem | Cat# A1079 |
| Mannitol | Sigma | Cat#M4125 |
| Zeba Desalt Spin columns | Thermo Fisher | Cat# 89894 |
| Tris | Carl Roth | Cat# 5429 |
| Triton X-100 | Serva | Cat# 39795 |
| Benzonase | In-house produced | N/A |
| Amylose resin | NEB | Cat# E8021S |
| EDTA | Roche | Cat# 105063 |
| GST-3C Protease | (PreScission Protease; inhouse produced) | N/A |
| Acid-washed glass beads (425-600 μm) | Sigma-Aldrich | Cat#G8772 |
| DNase I | Roche | Cat#4716728001 |
| RNase I | Ambion | Cat#AM2295 |
| 25 μm polystyrene beads (Polybead) | Polysciences | Cat#30 μm polystyrene beads (Polybead) |
| Pico-SurfTM 1 (2% in NovecTM 7500) | Sphere Fluidics | Cat#F001 |
| Sybr Green II | Invitrogen | Cat#S7564 |
| T4 RNA ligase | NEB | Cat#M0204S |
| pCp-Cy3 | Jena Bioscience | Cat#NU-1706-Cy3 |
| MicroSpin G-25 columns | GE Healthcare Life Sciences | Cat#27532501 |
| Ded1-monoGFP | This paper | N/A |
| Ded1ΔC-monoGFP | This paper | N/A |
| Ded1ΔN-monoGFP | This paper | N/A |
| Ded1_S.k.-monoGFP | This paper | N/A |
| Ded1_T.t.-monoGFP | This paper | N/A |
| Ded1-IDRm-monoGFP | This paper | N/A |
| Ded1 | This paper | N/A |
| Pab1-monoGFP | This paper | N/A |
| monoGFP | In house produced | N/A |
| Critical Commercial Assays | ||
| mMESSAGE mMACHINE T7 Transcription Kit | ThermoFisher | Cat#AM1344 |
| Poly(A) Tailing Kit | ThermoFisher | Cat#AM1350 |
| Nano-Glo Luciferase Assay | Promega | Cat#N1110 |
| QuikChange II Site-Directed Mutagenesis Kit | Agilent | Cat#200523 |
| RNA 6000 Nano Kit | Agilent | Cat# 5067-1511 |
| Q5 Site-Directed Mutagenesis Kit | NEB | Cat#E0554S |
| Deposited Data | ||
| 5′UTR sequences | Kertesz et al., 2010 | https://genie.weizmann.ac.il/pubs/PARS10/ |
| Genome of S288C (SGD: R64-2-1) | Saccharomyces Genome Database | https://www.yeastgenome.org/ |
| Genome of W303 GenBank: JRIU00000000) | Saccharomyces Genome Database | https://www.yeastgenome.org/ |
| Ribosome profiling data (GEO: GSE131176) | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131176 |
| RNA-Seq data (GEO: GSE141029) | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141029 |
| Experimental Models: Organisms/Strains | ||
| W303 ADE+ | Gift, Zachariae lab | #2952 |
| W303 ded1::HIS::s.k.Ded1-sfGFP-KanMX | This paper | #4322 |
| W303 ded1::HIS::ded1-sfGFP-KanMX | This paper | N/A |
| W303 ded1::HIS::T.t.Ded1-sfGFP-KanMX | This paper | #4255 |
| W303 ADE+ Ded1-sfGFP-KanMX, Pab1-mCherry-Hyg | This paper | #4096 |
| W303 ADE+ Ded1-mCherry-His3MX6 | This paper | #4194 |
| W303 ADE+ Ded1-sfGFP-KanMX | This paper | #4253 |
| W303 ADE+ Ded1-IDR-mutant-mCherry-His3MX6 | This paper | #4314 |
| Oligonucleotides | ||
| Ribosomal RNA | Bioworld | Cat#11020001-2 |
| tRNA | Invitrogen | Cat#AM7119 |
| pUC57-kan-fw-seq: CCTCTTCGCTATTACGC | This paper | N/A |
| pUC57-kan-rev-seq: AGTTAGCTCACTCATTAGGC | This paper | N/A |
| Ded1-237-256-fw: ATCTAACGGCCGTTCTGGTG | This paper | N/A |
| Ded1-1663-1644-rev: CTCTGTTGTTGTTGCGACCG | This paper | N/A |
| Seq-Ded1-fw: CTAACGGTAGAGACTTGATGG | This paper | N/A |
| Ded1ΔN-NotI-fw-1: AGTACGCGGCCGCAACCGAA TTTACGTCTCCCCCTTTG |
This paper | N/A |
| Ded1ΔN-rev: CTACGCATGCCTTAACCCATGATCTG | This paper | N/A |
| Ded1ΔC-fw: ACCCGACAGCTGTGATTATGGC | This paper | N/A |
| Ded1dC-AscI-rev: AGTCAGGCGCGCCCACTT CTTGGTTAGCCTCTGTCAGG |
This paper | N/A |
| Ded1-C1-Guide-fw: CGGGTGGCGAATGGGACTTTTTTA CCTCACCTCCATTGGAGTTTTAGAGCTAGAAATAGC |
This paper | N/A |
| Ded1-C1-Guide-rev: GCTATTTCTAGCTCTAAAACTCCA ATGGAGGTGAGGTAAAAAAGTCCCATTCGCCACCCG |
This paper | N/A |
| Ded1-C1-mutant: GGACGTTTCTGGTAAGGATGTTCCT GAACCAATCACAGAATTTACCTCTCCTCCATTGGA |
This paper | N/A |
| Ded1-C1-A-fw: AATTTCCAATCTTCTGGTATTAACTTCG ATAACTACGATGATATTCCAGTGGACGTTTCT |
This paper | N/A |
| Ded1-C1-A-rev: GTGTTGGCTTGGTGAAACGGGCCAATTT GATGTTTTCCAATAACAATCCGTCCAATGGAG |
This paper | N/A |
| Ded1-yeast-rv: ATCAGAGTTTGTCTTTCACC | This paper | N/A |
| Ded1-yeast-seq-rv: CAACAGGAGTCATATCACAG | This paper | N/A |
| Ded1-5UTR-amp-fw: CAGAGGCTAGCAGAATTACCCTCC | This paper | N/A |
| Ded1-5UTR-amp-rev: AATATGAAATGCTTT TCTTGTTGTTCTTACGG |
This paper | N/A |
| Ded1-3UTR-amp-fw: GTGCTGGTGCTGGTTTAATTAACATG | This paper | N/A |
| Ded1-3UTR-amp-rev: TTTACACTTTATGCTTCCGGCTCGTATG | This paper | N/A |
| Ded1-T_t-amp-fw: TAGGTGGCAAGTGGTATTCCGTAAGAACA ACAAGAAAAGCATTTCATATTATGGCTGAGCAATTGTCAGGATC |
This paper | N/A |
| Ded1-T_tamp-rev: ACAACACCAGTGAATAATTCTTCACCTTT AGACATGTTAATTAAACCAGCACCAGCACCC |
This paper | N/A |
| Ded1-S_k-amp-fw: TAGGTGGCAAGTGGTATT CCGTAAGAACAACAAGAAAAGCATTTCATATT ATGGCTGAGCTGTCTGAACAGG |
This paper | N/A |
| Ded1-sk-seq1-fw: CTGTGATAATGGCTCCTAC | This paper | N/A |
| Ded1-sk-seq2-fw: GCAGTCAGAGAGAGAAAG | This paper | N/A |
| Ded1-Sc-seq-c-term-fw: TGCATGAGATCTTGACTG | This paper | N/A |
| Ded1-seqc-fw: TGCATGAAATTTTGACTG | This paper | N/A |
| Ded1-5UTR-IVT-fw: ATTTCATATTATGGTTTT TACTTTAGAAGATTTTG |
This paper | N/A |
| Ded1-5UTR-IVT-rev: GCTTTTCTTGTCCCTAT AGTGAGTCGTATTAG |
This paper | N/A |
| Ded1-T_t-seq-fw: TGAGCAACATAGAACTAGC | This paper | N/A |
| Ded1-T_t-seq-fw2: TACGTCAGAGAATATCACC | This paper | N/A |
| Tt-Ded1-AR-seq3-fw: ATGGCTGAGCAATTGTCAG | This paper | N/A |
| Tt-Ded1-AR-seq4-rev: TTTCCACTACCAGTTTGAGC | This paper | N/A |
| Gis2-5UTR-IVT-fw: GAAAAAGATGGTTTTTA CTTTAGAAGATTTTG |
This paper | N/A |
| Gis2-5UTR-IVT-rev: GTATTCTCCCTAT AGTGAGTCGTATTAG |
This paper | N/A |
| Pab1-sense: GTCTTTCAAAAAGGAGCAAGAACAACAAA CTGAGCAAGCTGGTGACGGTGCTGGTTTA |
This paper | N/A |
| Pab1-asense: TAAGTTTGTTGAGTAGGGAAGTAGGTGAT TACATAGAGCATCGATGAATTCGAGCTCG |
This paper | N/A |
| tz_yor204w_35-53_guide_F: CGGGTGGCGAATGGGACTTT AACTACAATAACAACAACGGGTTTTAGAGCTAGAAATAGC |
This paper | N/A |
| tz_yor204w_35-53_guide_R: GCTATTTCTAG CTCTAAAACCCGTTGTTGTTATTGTAGTTAAA GTCCCATTCGCCACCCG |
This paper | N/A |
| tz_yor204w_35-53_sim_351_repair_mol: AGTGAGAACCTAAACAGGTTATTGATCT TCCTAAACCCTGAGTATTCAAGCAATCTTGGG |
This paper | N/A |
| tz_yor204w_35-53_sim_351_repair_F: AGAAT GGTTATGTTCCTCCTCACTTAAGAGGAAAAC CAAGAAGTGCCAGAAGTGAGAACC |
This paper | N/A |
| tz_yor204w_35-53_sim_351_repair_R: AAGAAAC CACCGTTGCCGTAACCACCACGACGGTTGTT GCTAAAGAAGCTCCCAAGATTG |
This paper | N/A |
| Recombinant DNA | ||
| pKT127-sfGFP | This paper | L-269 |
| pKT128-sfGFP | This paper | L-270 |
| pBS35 | Yeast Resource Center Washington | N/A |
| pKT128-mCherry | This paper | L-295 |
| pUC57-kan-Pab1 | Genscript | L-609 |
| pUC57-kan-Ded1 | Genscript | L-605 |
| pUC57-kan-Ded1ΔC | This paper | L-606 |
| pUC57-kan-T.t.-Ded1-sc_opt | Genscript | L-535 |
| pUC57-kan-S.k.-Ded1-sc_opt | Genscript | L-536 |
| pUC57-kan-Ded1-plus-for allele replacement | Genscript | L-534 |
| pUC57-kan-T.t.-Ded1 | Genscript | L-527 |
| pUC57-kan-S.k.-Ded1 | Genscript | L-528 |
| pOCC120 Ded1ΔC | This paper | L-617 |
| pOCC120-Pab1 | This paper | L-612 |
| pOCC97-Ded1 | This paper | L-613 |
| pOCC120-Ded1 | This paper | L-604 |
| pOCC177-kan-Ded1-S.k | This paper | L-558 |
| pOCC120-kan-T.t.-Ded1 | This paper | L-538 |
| pOCC120-kan-S.k.-Ded1 | This paper | L-539 |
| pOCC120-Ded1ΔN | This paper | L-726 |
| pOCC120-Ded1-IDRm | This paper | L-729 |
| pUC57-kan-Ssa1-5UTR-Nanoluc | Genscript | L-638 |
| pUC57-kan-Ssa4-5UTR-Nanoluc | Genscript | L-635 |
| pUC57-kan-Hsp12-5UTR-Nanoluc | Genscript | L-652 |
| pUC57-kan-Hsp104-5UTR-Nanoluc | Genscript | L-636 |
| pUC57-kan-Ssk2-5UTR-Nanoluc | Genscript | L-639 |
| pUC57-kan-Sbe22-5UTR-Nanoluc | Genscript | L-646 |
| pUC57-kan-Pab1-5UTR-Nanoluc | Genscript | L-645 |
| pUC57-kan-Gis2-5UTR-Nanoluc | This paper | L-647 |
| pUC57-kan-Ded1-5UTR-Nanoluc | This paper | L-649 |
| pCas | Ryan et al., 2016 | Addgene plasmid # 60847 |
| Software and Algorithms | ||
| Fiji | NIH | https://fiji.sc/ |
| KNIME | KNIME.com AG | https://www.knime.com |
| FastQC (v0.11.2) | Babraham Institute | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| R / RStudio | R Core Team |
https://www.r-project.org/ https://www.rstudio.com/ |
| cutadapt (v1.17) | PyPI | https://cutadapt.readthedocs.io/en/stable/installation.html |
| ClustalW | EMBL-EBI | https://www.ebi.ac.uk/Tools/msa/clustalw2/ |
| STAR 2.5.2b | GitHub | https://github.com/alexdobin/STAR/releases |
| PR. ThermControl software | Nanotemper | https://nanotempertech.com/prometheus-pr-thermcontrol-software/ |
| DESeq2 (v1.22.2) | Bioconductor | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| clusterProfiler (v3.10.1) | Bioconductor | http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
| RNAfold (ViennaRNA Package v2.4.11) | TBI (University of Vienna) | https://www.tbi.univie.ac.at/RNA/ |
| Bowtie 2 (v2.2.6) | bioconda | https://anaconda.org/bioconda/bowtie2/files |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Prof. Dr. Simon Alberti (simon.alberti@tudresden.de).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Ribosome profiling data
The ribosome profiling data used for analysis are deposited in the GEO repository and can be accessed with the following link:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131176
RNAseq data
The RNaseq data used for analysis are deposited in the GEO repository and can be accessed with the following link:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141029
Experimental Model and Subject Details
Yeast genetic techniques, strains, and media
S. cerevisiae cells were cultured in standard synthetic complete (SC), synthetic dropout (SD), or yeast extract peptone dextrose (YPD) media containing 2% D-glucose. The yeast strain background used was W303 Ade+ (MAT alphacan1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1). Yeast cells were grown in cultures of 50-100 mL at 25°C or 30°C to an optical density 600 (OD600) that was not greater than 0.5. For a list of yeast strains used, see the Key Resources table.
Generating a variant with altered phase behavior using CRISPR/Cas9
To generate a variant with altered phase behavior, we simulated the evolution of the Ded1p N-terminal disordered region according to a substitution matrix specific for disordered regions and retained existing conserved motifs (such as binding sites) but not any higher order molecular features (such as sequence composition or repeats) (Nguyen Ba et al., 2014, Nguyen Ba et al., 2012, Zarin et al., 2019, Zarin et al., 2017). We identified a sequence spanning residues 35-54 that showed a conservation of glycine residues and polar residues such as asparagine. We replaced the corresponding region (NNSSNYNNNNGGYNGGRGG) in wild-type DED1 with one of our simulated sequences (SENLNRLLIFLNPEYSSNLG) yielding the mutant DED1-IDRm. The replacement was performed using a modified CRISPR/Cas9 method for site-directed mutagenesis in budding yeast as described (Ryan et al., 2016), with the modification that guide-RNA plasmids (Ryan et al., 2014) were created using the QuikChange II Site-Directed Mutagenesis Kit.
Allele replacement
Alleles of S. cerevisiae DED1 were replaced by homologous recombination. For this purpose, three DNA pieces were PCR amplified from different plasmids:
-
1)
500 bp upstream of the S. cerevisiae DED1 coding region,
-
2)
coding DNA for replacement,
-
3)
sfGFP, a G418 selection cassette and 500 bp downstream of the S. cerevisiae DED1 coding region.
The three PCR pieces were each equipped with overlapping ends and subsequently co-transformed in S. cerevisiae using a standard lithium acetate/single-stranded carrier DNA method of transformation. Correctly replaced genes were confirmed by microscopy, immunoblotting and sequencing of the complete coding and sfGFP sequence.
Yeast growth assay in liquid medium
W303 yeast cells grown to exponential phase at 30°C were diluted to an OD600 of 0.1 in YPD liquid medium. 200 μL of the yeast culture was transferred to 16 wells in a 96-well transparent flat bottom plate (Greiner AG, Kremsmünster, Austria) and placed in a SparkⓇ multimode microplate reader (Tecan, Männedorf, Switzerland) at different temperatures (30°C, 33°C, 37°C, 39°C, 40°C, 41°C and 42°C) with shaking. Absorbance measurements at 600 nm were made every 10 min for 15 hours.
Yeast spot titer assay
Yeast cultures were grown to exponential phase in YPD medium at 30°C. Cell density of yeast cultures as assessed by OD600 measurements were adjusted to equal concentrations. Cultures were diluted in 5-fold serial dilutions with distilled water in 96 well-plates (Multiple Plates Vee Bottom with Lid, Sarstedt, Nümbrecht). Diluted cultures were spotted on YPD agar plates using a sterilized multi-blot-replicator with 48 pins. The plates were incubated at the indicated temperatures and photographed once per day with a standard digital camera.
pH treatment of yeast cells
The intracellular pH of S. cerevisiae cells was adjusted by incubation in 0.1 M potassium phosphate buffer of pH 7.0 or 5.8 in the presence of 2 mM 2,4-dinitrophenol (DNP) as described previously (Petrovska et al., 2014).
Starvation treatment of yeast cells
Yeast cultures were starved by removal of SC-medium, followed by 3 washes in water and incubation in SC-medium lacking glucose for 60 min.
Heat treatment of yeast cells
Yeast cultures were grown to exponential phase in SC-medium with 2% D-glucose, subsequently concentrated and transferred to a thermomixer or water bath set to the respective temperature.
Method Details
Yeast live cell imaging
Yeast samples were prepared in 4-chamber glass-bottom dishes (Greiner Bio-One, Frickenhausen, Germany). Dishes were coated with concanavalin A coating solution (2.2 mL PBS, 125 μL phosphate buffer pH 6, 2.5 μL 1 M CaCl2, 5 μL azide, 250 μL 5 mg/mL concanavalin A (Sigma-Aldrich, Steinheim, Germany) solution in PBS) for 30 min. The coating solution was then removed and the dish was washed twice with water. 500 μL of cell suspension (OD ∼0.5) was added to the chambers and the cells were allowed to settle for ∼10 min. The supernatant was removed and the cells were washed twice with SC/SD medium containing 2% D-glucose. Time-lapse microscopy using the DeltaVison Core (Olympus, Hamburg, Germany) was done using a 100x/1.4 NA U Plan Super-Apochromat oil objective (Olympus, Tokyo, Japan). Fiji was used to create maximum Z-projections, crop images and to create scale bars.
Protein sedimentation assay after heat treatment of yeast cells
Yeast cells were grown to exponential phase in YPD liquid medium. 16-20 OD units of cells in 400 uL of YPD and placed at the indicated temperature for a specified amount of time (10 or 30 min). The cell pellets were resuspended in 800 μL of lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 2.5 mM EDTA, 0.5% (v/v) Triton X-100, and protease inhibitors (1 mM PMSF, 1.25 mM benzamidine, 10 μg/mL pepstatin, 10 μg/mL chymostatin, 10 μg/mL aprotinin, 10 μg/mL leupeptin and 10 μg/mL E-64)). Cells were subsequently lysed using glass beads (acid-washed, 425-600 μm) and the TissueLyser II (setting: 20 min, 25/sec) with a 24-well adaptor (QIAGEN, Hilden, Germany) at 4°C. Cell debris was removed by centrifugation (1 min at 380 x g) and the supernatant was taken as the total protein fraction. The total fraction was subsequently centrifuged for 5 min at 8,000 x g at 4°C and the supernatant was taken as the supernatant fraction containing soluble Ded1p. The supernatant and total fraction were analyzed by SDS-PAGE and immunoblotting.
Sample preparation for ribosome profiling and RNaseq
Duplicate 1 L yeast cultures were grown at 30°C to an OD600 of 0.5 in YPD medium and split into three 200 mL cultures that were incubated for 10 min at 30°C, 40°C or 42°C. Subsequently, the cultures were rapidly filtered (All-Glass Filter 90mm, Millipore), flash frozen in liquid nitrogen, mixed with 600 μL of frozen lysis buffer (20 mM Tris-HCl pH 8.0, 140 mM KCl, 6 mM MgCl2, 0.1% NP-40, 0.1 mg/mL cycloheximide, 1 mM PMSF, 2x cOmplete EDTA-free protease inhibitors (Roche, Penzberg, Germany), 0.02 U/mL DNase I, 20 mg/mL leupeptin, 20 mg/mL aprotinin, 10 mg/mL E-64, 40 mg/mL bestatin) and pulverized by mixer milling (2 min, 30 Hz, MM400, Retsch). Thawed cell lysates were cleared by centrifugation (1 min at 2,000 rpm) at 4°C. From these samples, 200 μg total RNA in 100 μL lysis buffer were used for ribosome profiling and 800 μg of total RNA in 400 μL lysis buffer were further processed for RNaseq.
Further processing for ribosome profiling
Lysates for ribosome profiling were subsequently digested by RNase I (50 U/200 μg nucleic acid) for 30 min at 4°C to obtain ribosome footprints. The digested lysates were diluted to 400 μL and loaded onto 800 μL sucrose cushions (25% sucrose, 20 mM Tris-HCl pH 8.0, 140 mM KCl, 10 mM MgCl2, 0.1 mg/mL cycloheximide, 1 mM PMSF, 1x Complete EDTA-free protease inhibitors). Gradients were centrifuged for 1.5 h at 75,000 rpm at 4°C. After removal of the supernatant, the pellets were resuspended in 200 μL lysis buffer and used for RNA purification. RNA was purified by hot acid-phenol chloroform extraction (Becker et al., 2013). Deep sequencing libraries were prepared following the protocol described in Shiber et al. (2018), with adaptor sequences given in McGlincy and Ingolia (2017) and sequenced on a NextSeq 500 (Illumina).
Further processing for RNA sequencing
200 μL of the lysate samples for RNaseq were directly used for RNA purification and represent the total fraction. The remaining 200 μL were centrifuged for 5 min at 18,000 x g and 4°C. Supernatants were transferred to fresh tubes, RNA of these samples was purified and represents the supernatant fraction. Pellets were resuspended in 200 μL lysis buffer, centrifuged for 5 min at 18,000 x g and 4°C and resuspended again in 200 μL lysis buffer. These samples were transferred to RNA purification and represent the pellet fraction. RNA was purified for all above described samples by hot acid-phenol chloroform extraction (Becker et al., 2013). mRNA was isolated from 2 μg DNase treated total RNA using the RiboMinus Kit Kit (yeast) from Thermo Fisher Scientific according to the manufacturer’s instructions. Final elution was done in 5 μl nuclease free water. Samples were then directly subjected to the workflow for strand specific RNA-Seq library preparation (Ultra Directional RNA Library Prep II, NEB). For ligation custom adaptors were used 1: (Adaptor-Oligo 5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT-3′, Adaptor-Oligo 2: 5′-P-GAT CGG AAG AGC ACA CGT CTG AAC TCC AGT CAC-3′). After ligaton adapters were depleted by an XP bead purification (Beckman Coulter) adding bead in a ratio of 1:1. Indexing was done during the following PCR enrichment (15 cycles) using custom amplification primers carrying the index sequence indicated with ‘NNNNNN’. (Primer1: Oligo_Seq AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC T, primer2: GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC T, primer3: CAA GCA GAA GAC GGC ATA CGA GAT NNNNNN GTG ACT GGA GTT. After two more XP beads purifications (1:1) libraries were quantified using Qubit dsDNA HS Assay Kit (Invitrogen). For Illumina flowcell production, samples were equimolarly pooled and distributed on Illumina NextSeq 500.
Constructs, protein expression and protein purification
The protein expression and purification facility of MPI-CBG kindly provided plasmid backbones for virus production, viruses and SF9 ESF cells for the purification of Ded1p, Ded1p variants and Pab1p. The facility also provided purified GFP protein. Wild-type and variant forms of Ded1p as well as Pab1p coding sequences (optimized for Spodoptera frugiperda cell expression) were cloned using classical cloning techniques. For fluorescently labeled protein, the fluorescent tag was positioned on the opposite side of the prion-like-domain (C terminus for Ded1p, N terminus for Pab1p) to avoid potential adverse effects of the fluorescent tag on phase separation. SF9 ESF insect cells (1 × 106 cells/mL) were transfected with recombinant baculovirus stocks (1:100) for the expression of proteins and incubated for four days. Cells were harvested by centrifugation (15 min 500 x g) and resuspended in 30 mL of lysis buffer (50 mM Tris/HCl, pH 7.6, 1 M KCl, 1 mM DTT, 1 protease inhibitor tablet with EDTA/50 mL buffer, 2 mM EDTA, 10 μL benzonase (250 U/mL)) and lysed using the EmulsiFlex-C5 (Avestin, Ottawa, Canada). Lysates were clarified by centrifugation at 16,000 rpm for 1 h at 4°C (rotor JA 25.50, Beckman Coulter, Brea, California, USA). The supernatant was incubated with amylose resin for 1 h at 4°C and loaded onto a 20 mL chromatography column (Bio-Rad, Hercules, CA, USA). After washing 3 column volumes with wash buffer (50 mM Tris/HCl, pH 7.6, 1 M KCl, 1 mM DTT, 2 mM EDTA), the protein was eluted with elution buffer (50 mM Tris, pH 7.6, 1 M KCl, 1 mM DTT, 2 mM EDTA, 20 mM maltose). MBP was cleaved using 0.01 mg/mL GST-3C Protease. Before size exclusion chromatography (SEC), protein aggregates were removed by centrifugation in Falcon Tubes for 4 min at 3452 x g. If necessary, the protein was concentrated using Amicon Ultra-15-30k centrifugal filters at 3452 x g. Proteins were loaded onto an Äkta Pure chromatography setup (GE Healthcare, Uppsala, Sweden) equipped with a Superdex 200 26/60 column or Superdex 200 16/60 column (GE Healthcare, Piscataway, NJ, United States). After SEC, the protein was concentrated by centrifugation using Amicon Ultra 15-30k centrifugal filters (Merck, Kenilworth, NJ, USA) at 3452 x g. The purity of the protein was assessed using a Coomassie gel to exclude protein-based impurities and degradation bands. Protein concentration was determined at 280 nm and 488 nm for GFP-tagged variants using the Nanodrop (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The protein was flash-frozen in a protein storage buffer (50 mM Tris, pH 7.6, 1 M KCl, 1 mM DTT, 2 mM EDTA) using liquid-nitrogen and stored at −80°C.
Calculation of Ded1p concentration
Ded1p is expressed at around 31800 molecules (N) per cell (Kulak et al., 2014) in the nucleus and cytoplasm. Taking an average haploid cell volume of 42 μm3 (Uchida et al., 2011) from which 70% is composed of the cytoplasm and 6%–11% of the nucleus, we estimated the total molar concentration of Ded1p to be 1.3 μM with the formula C = N/(NA∗Vcell) where NA is Avogadro’s number (Uchida et al., 2011). Since the used cytosolic volume includes the cytoskeleton, large macromolecular complexes and the ribosome, the actual cytosolic volume is most likely smaller, which means that we are underestimating the concentration of Ded1p. Thus, we assume a cytosolic and nuclear Ded1p concentration to be approximately 2 μM.
Phase separation assays
Proteins were diluted to various concentrations in buffers with a final concentration of 100 mM or 200 mM KCl and 1mM TCEP. Exact buffer conditions are highlighted in the figure legends. In most experiments, 20 mM PIPES/KOH buffer at various pH was used. Alternatively, 0.1 M MES/KOH, 0.1 M HEPES/KOH and 0.1 M phosphate buffers were used and Ded1p followed a highly similar behavior. All of the assays were done with untagged Ded1p or Ded1p C-terminally tagged with GFP. When applicable, RNA was added in the phase separation assays to a final concentration of ∼80 ng/μL in the presence of 4 μM Ded1p.
Dynamic light scattering (DLS)
For DLS measurements using the Zetasizer NANO (Malvern Panalytical, Malvern, UK), proteins were diluted to ∼2-4 μM in 20 mM PIPES/KOH buffer pH 6.8 or 0.1 M potassium phosphate buffer pH 7.4 (for Ded1p truncation mutants and Ded1-IDRm) containing 1 mM DTT to obtain a final concentration of 100 or 200 mM KCl. The samples were heated in 1-2°C increments per min (including a 30 s settling time) in a Zen 2112 quartz cuvette (Hellma) from 25°C to 50°C or 60°C.
Circular dichroism (CD)
For CD measurements, using Chirascan Plus (Applied Photophysics, Leatherhead, UK), proteins were diluted to ∼2 μM in 20 mM PIPES/KOH, 100 mM KCl. If applicable, the samples were heated in 1°C increments per min from 20°C to 60°C. At each temperature step, the sample was equilibrated for 30 s and 3 readings were obtained. Spectra were recorded from 200 to 250 nm with 0.5 s integration time, a step size of 1 nm and a bandwidth of 1 nm.
Nano-differential scanning fluorimetry (nanoDSF)
For nano-DSF measurements using the Prometheus NT.48 (Nanotemper, Munich), proteins were diluted to varying concentrations in 20 mM PIPES/KOH buffer pH 6.8 or pH 7.5 to a final concentration of 200 mM KCl and in the presence of 1 mM TCEP. Samples were heated in high sensitivity capillaries (Nanotemper, Munich) with 1°C increments per min from 20°C to 95°C. NanoDSF was also used to measure light scattering.
Protein sample preparation for imaging of Ded1p condensates
10-20 μL of protein samples were incubated at the indicated temperatures and times and subsequently added into 384 well non-binding microplates (Greiner bio-one, Kremsmünster, Austria). Microscopy images were taken after droplets had settled to the bottom of the plate. Images were taken with either a confocal microscope (STORM spinning disc, Andor, Belfast, UK) with an Apo 100x/1.49 NA oil objective (Nikon Instruments, Amsterdam, Netherlands) or with a Plan Apochromat VC 60x/1.2 NA water objective (Nikon Instruments, Amsterdam, Netherlands) or widefield microscopes (DeltaVision Elite; Olympus, Tokyo, Japan and Zeiss Axiovert 200M, Jena, Germany) with an UPlan Super-Apochromat 100x 1.4 NA oil objective (Olympus, Tokyo, Japan) or with a Plan-Apochromat 40x 0.95 NA air objective (Zeiss, Jena, Germany).
Automated microscopy
To create the pH x temperature phase diagram in Figure 3B, Ded1p in PIPES/KOH buffer solutions with 100 mM KCl, 1 mM TCEP were added to a PCR plate. The PCR plate was heated for 30 min using a temperature gradient in a standard PCR machine. Next, a pipetting robot (freedom evo, TECAN, Männedorf, Switzerland) was used to mix the protein/buffer samples, split them in two, and transfer them to a low volume 384-well imaging plate (Greiner bio-one, Kremsmünster, Austria). An automated fluorescence microscopy imaging system (CV7000, Yokogawa, Tokyo, Japan) was used to image the samples.
Temperature stage
The stage was set up as described in Mittasch et al. (2018). For imaging of yeast cells, the height of the buffer-filled chamber was controlled using thin strips of parafilm to form chambers that were coated with concanavalin A coating solution and subsequently, yeast cells. Yeast cells were subsequently exposed to a fast temperature ramp of 10°C/min from 35°C to 37°C, 39°C, 41°C, or 44°C and cells were imaged using a confocal microscope (Spinning disc; Andor, Belfast, UK) with an Apochromat 100x/1.49 NA Oil objective (Nikon Instruments, Amsterdam, Netherland) using 1.5x Optovar magnification. An objective heater (Objective heater, large collar; Bioptechs, Butler, PA, USA) was set to 35°C to avoid the objective from becoming a heat sink. Finally, the assemblies per cell were counted over all time points and temperatures using automated analysis with a Fiji script and plotted using R.
For imaging of protein condensates, the height of the buffer-filled chamber was set with 20-30 μm polystyrene beads. Ded1p or Ded1p/mRNA was mixed with Pico-Surf™ 1 (2% in NovecTM 7500) so that condensates were protected from aberrant surface effects. Protein samples were subsequently exposed to a fast temperature ramp of 10°C/min from 35°C to 40°C and the Ded1-GFP assembly formation was imaged using the same confocal microscope set-up described above.
Optical tweezer measurements
Controlled condensate fusion experiments were done using a custom-built dual-trap optical tweezer microscope with two movable traps (Jahnel et al., 2011). Ded1p/mRNA in PIPES/KOH buffer pH 7.5, 200 mM KCl, 1mM TCEP was heated to 42°C for 5 min in a PCR machine. All subsequent steps were carried out at room temperature. 10 μL of the reaction volume were applied and sealed in a static flow chamber. Sample preparation and mounting was carried out within 3 min. Protein condensates were trapped due to a mismatch in the index of refraction between condensates and buffer. The laser power of the 1064 nm trapping laser was kept at minimum (< 70 mW) to prevent heating artifacts. Keeping one optical trap stationary, the other optical trap was moved until the condensates touched, after which condensate fusion was recorded with a temporal resolution of 1 ms (1 kHz).
In vitro transcription and poly(A)-tailing
Template DNA for the in vitro transcription reaction was prepared by PCR from the template plasmids pUC57-kan-varying-5UTR-Nanoluc (Genscript, Piscataway, NJ). The 5′UTR lengths were derived from (Kertesz et al., 2010). Primers used for amplification were pUC57-kan-fw-seq and pUC57-kan-rev-seq. See the Key Resources Table for the list of plasmids and primers used.
Capped mRNAs were synthesized using the mMESSAGE mMACHINE T7 Transcription Kit by following the manufacturer’s proceeding with a transcription time of 2 hours at 37°C. Poly(A)-tailing of synthesized mRNAs was done using the Poly(A) Tailing Kit by following the manufacturer’s proceedings with an incubation time of 1 hour. Lithium chloride was used to precipitate the mRNAs as described in the mMESSAGE mMACHINE T7 Transcription Kit and resuspended in nuclease-free water.
Quality assessment of mRNA
The quality and concentration of mRNAs was assessed using the bioanalyzer 2100 (Agilent, Santa Clara, CA, USA) with the RNA 6000 Nano Kit.
Sedimentation assay with RNA
Ded1-GFP or GFP protein was diluted to 2.5 μM with 45 ng/μL RNA in pH 6.8 PIPES/KOH, 200 mM KCl, 1 mM TCEP buffer. RNAs used include capped and poly(A)-tailed mRNA, yeast tRNA and rRNA. Ded1p/RNA samples were incubated for 15 min at 42°C followed by centrifugation at 8,000 x g for 1 min. The concentration of RNA in the supernatant before and after heat treatment was measured by Nanodrop at an absorbance of 260 nm. Sedimentation of Ded1p condensates was confirmed by microscopy.
Fluorescent labeling of mRNA for microscopy
Synthesized mRNAs were 3′ end labeled with T4 RNA ligase and pCp-Cy3 (Jena Bioscience) using the manufacturer’s proceedings. Unbound pCp-Cy3 was removed with the use of MicroSpin G-25 columns (GE Healthcare Life Sciences). For microscopy, 18 ng/μL of labeled mRNA was incubated with 1 μM Ded1-GFP in the presence or absence of 18 ng/μL tRNA.
Production of yeast cell-free translation extract
Yeast cell-free translation extracts were prepared from W303 yeast cells using the protocol from Wu and Sachs (2014). In short: 800 mL of yeast cells grown to an OD600 of 1.5 were harvested in 30 mL of BufferA/Mannitol (60 mM HEPES/KOH pH 7.6, 200 mM potassium acetate, 6 mM magnesium acetate, 2 mM DTT, 8.5% w/v Mannitol). Cells were subsequently washed 4 times with BufferA/Mannitol. Cells were resuspended in 1.5 mL of BufferA/Mannitol with EDTA-free cOmplete protease inhibitors per gram of cell weight (Roche, Penzberg, Germany). Cells were broken using the TissueLyser II (20 min, 25/sec). For this purpose, 800 μL of cell/buffer suspension were added to Eppendorf tubes containing glass beads (acid-washed, 425-600 μm) and placed into a 24-well adaptor (QIAGEN, Hilden, Germany) at 4°C. After a quick spin at 650 x g for 2 min, the lysate was cleared by ultracentrifugation at 38,000 x g for 6 min at 4°C. The cleared lysate was desalted with Zebra desalt spin columns with a 7 kDa molecular weight cut-off (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Yeast cell-free translation extracts were then flash-frozen and placed at −80°C.
In vitro translation
The protocol was adapted from Wu et al., 2007. In short: Thawed yeast cell-free translation extract was treated with 1 U of micrococcal nuclease (NEB, Ipswich, Massachusetts, United States) in the presence of 1 mM CaCl2 to remove single-stranded nucleic acids. After 10 min incubation at 25°C, nuclease activity was stopped by the addition of 2 mM EGTA. Treated yeast extracts were then placed on ice. An mRNA of interest (coding for Nano-Luciferase and fused to different 5′UTRs from various genes) was added to a reaction mix (60 mM HEPES/KOH pH 7.6, 1 mM ATP, 0.05 mM GTP, 80 mM creatine phosphate, 10 mM magnesium acetate, 15 mM potassium acetate, 2 mM complete amino acid mix (Promega, Madison, Wisconsin, United States), 0.5 U RNaseOUT (Thermo Fisher Scientific, Waltham, Massachusetts, USA), 2 mM DTT) in the presence of tRNA. For one reaction, 8.8 μL of mRNA/tRNA/reaction mix was used with a concentration of 50 ng/μL mRNA and 80 ng/μL tRNA. Different concentrations of 8 μL of protein in 20 mM PIPES/KOH pH 6.8, 100 mM KCl, 1 mM TCEP were added to the mRNA/tRNA/reaction mix on ice prior to the addition of 8 μL of the nuclease-free treated yeast translation extract. In vitro translation reactions were placed at 25°C for 90 min. The translation activity was determined with the use of a Nano-Glo® Luciferase assay (Promega, Madison, Wisconsin, United States). The in vitro translation reactions were thawed on ice and the Nano-Glo® Luciferase Assay components were added according to the manufacturers proceedings. The PerkinElmer Envision plate reader (PerkinElmer, Waltham, Massachusetts, United States) was used to measure luminescence at 460 nm with an ultra-sensitive luminescence protocol and a 1 s measurement time. Alternatively, the Glomax 96 Microplate luminometer was also used to measure luminescence with a 1 s measurement time.
Quantification and Statistical Analysis
General data analysis
Quantification and statistical analysis were performed using R/Rstudio. Significance levels: ∗ < 0.05, ∗∗ < 0.01, ∗∗∗ < 0.001. Boxplot: horizontal line depicts the median, the box is the 25 and 75 percentile and the whiskers denote the min. and max. values considered for analysis. Further statistical details can be found in the figure legends and STAR Methods.
Calculation of the growth rate at different temperatures
Growth curves of W303 yeast cells at different temperatures were analyzed by fitting the absorbance at 600 nm as a function of time to a general logistic growth model Abs = Amax / (1 + ((Amax - Amin) / Amin) ∗ exp(-r ∗ t)), where Amax is the maximum growth density, Amin is the initial growth density and r is the growth rate. In Figure 2D, the growth rate is depicted as the inverse of the doubling time in hours (h-1). All calculations were carried out using R.
Western blot quantification
The relative amounts of protein in each well were quantified using the gel analysis package in Fiji (Schindelin et al., 2012). The Ded1-mCherry signal in each lane was first normalized to the PGK loading control prior to the calculation of the ratio of protein in the supernatant versus in the total fractions (fraction soluble). The average ± standard deviation of three independent samples were analyzed and plotted.
Determination of the mean hydrodynamic radius (DLS)
The mean hydrodynamic radii were determined using the Zetasizer software (Zetasizer NANO (Malvern Panalytical, Malvern, UK).
Determination of Tonset and TM using nanoDSF
The Tonset of scattering (for the phase diagrams in Figure 5C) were determined by PR.ThermControl software (Nanotemper, Munich) and plotted over protein concentration. The TM (Figure 3F) of scattering and tertiary structure change was derived manually by determination of the local maximum of the first derivative of the scattering and (350, 330 nm) ratio change respectively.
Quantification of Ded1p and mRNA condensates from microscopy images
Images were analyzed with a custom semi-automated workflow in Fiji. For the segmentation of condensates, the image was illuminationation corrected by normalizing the image to its own blurred image using a Gaussian blur of 50 pixels. Objects with intensities larger than 1 or 5% of the mean intensity of the normalized image were segmented and selected. The selection was placed to the original image for measurement. The condensed fraction was calculated using (ICondensate ∗ ACondensate / (ITotal ∗ ATotal) where ICondensateis the mean condensate fluorescence intensity, ACondensate is the condensate area and ITotal is the mean fluorescence intensity of the image and ATotal is the size of the image. For the measurement of mRNA partitioning in Ded1p condensates, the ImRNA inside of Ded1p condensates was divided by the ImRNA outside of Ded1p condensates. Data was plotted with R/RStudio.
Calculation of soluble RNA in vitro
The fraction of soluble RNA was determined by calculating the ratio of absorbance at 260 nm in the supernatant before and after heating with Ded1p. Statistical significance between samples was determined with a Student’s t test.
Arrhenius Plot
An Arrhenius plot for the apparent assembly rates of Ded1p and Pab1p was created by plotting ln(k) over (1/T) using DLS data. From this plot, the activation energy of the reaction was determined, which is defined to be - [universal gas constant] times the slope of the plot of ln(k) versus (1/T).
Data analysis ribosome profiling and RNaseq
Quality of reads was assessed using FastQC (v0.11.2). Ribosome profiling (Ribo-Seq) reads were trimmed with cutadapt (v1.17) (Martin, 2011) to remove adapters and mapped against the RNA coding genes of the S288C reference genome (release R64-2-1) using bowtie2 (v2.2.6) (Langmead and Salzberg, 2012). Non-mapping reads were mapped to the genomes of W303 (Genbank: JRIU00000000) and S288C (SGD: R64-2-1) obtained from the Saccharomyces Genome Database using STAR 2.5.2b (Dobin et al., 2013). Although similar read mapping efficiencies were observed for both genomes, differences in the genome annotation quality resulted in a considerably higher number of gene-mapping reads for the reference genome S288C, which was therefore used for further downstream analyses. RNA-seq reads were mapped to the genome of S288C using STAR 2.5.2b retaining only reads that map to single genomic position (Dobin et al., 2013). Systematic comparison of the log-normalized gene counts by principal component analysis (PCA) and Euclidean distance clustering revealed that the ribosome occupancy and the RNaseq read distribution was distinct between the samples exposed to different temperatures (Figures S1A, S1B, S6A, and S6B). Translational efficiency was calculated with DESeq2 (v1.22.2) (Love et al., 2014) using the design “∼assay+condition+assay:condition” and the total RNA fraction as RNaseq input. Differentially translated genes (DTGs) were chosen using an adjusted p value threshold of 0.05.
Differences in RNA abundance between pellet and supernatant were calculated with DESeq2 pairwise comparisons, pellet versus supernatant for each genotype and temperature. To compare mRNA expression changes (from total fraction) with changes in ribosome profiling, differences between conditions were calculated with DESeq2 pairwise comparisons. Differentially regulated genes were chosen using an adjusted p value threshold of 0.05.
Publicly available information regarding the positions of 5′UTRs in yeast (Kertesz et al., 2010) were used to extract 5′-UTR sequences from the S288C reference genome. Secondary structure prediction was then performed using RNAfold (ViennaRNA Package v2.4.11, Lorenz et al., 2011). Differences in the minimum free energy of 5′ UTRs of DTGs and non-DTGs were assessed using the Wilcoxon rank sum test.
Quantification of fusion dynamics from optical tweezer experiments
To characterize fusion dynamics, time traces of the tweezer signal, S(t), were fitted with an exponential decay model:
Where τ denotes the relaxation time, tstart the onset of fusion, Soffset the signal offset on the detector, and Splateau the final signal value after coalescence finished.
Acknowledgments
We thank the following MPI-CBG facilities: Protein Expression Purification and Characterization, Light Microscopy, Technology Development Studio, FACS, and Scientific Computing. We thank the MPI-CBG staff for comments on the manuscript, C. Roden for helpful discussions, A. Pozniakovsky for guidance with cloning, E. Nüske for image acquisition, the DRESDEN-concept Genome Center for sample analyses, and the Light Microscopy Facility at BIOTEC (TU Dresden) for support. We acknowledge funding from the Max Planck Society, the TU Dresden, the MaxSynBio Consortium, and the European Research Council (725836). This work was further supported by the Human Frontier Science Program (RGP0034/2017) and by a Life? grant of the Volkswagen Foundation. C.J. was supported by the Boehringer Ingelheim Fonds. A.W.F. was supported by the ELBE postdoctoral fellows program.
Author Contributions
This paper is the result of the doctorate project of C.I., who performed a large part of the experiments under supervision of S.A. Analysis and interpretation of the data was largely performed by C.I., C.D.A., and S.A. U.F. and G.K. helped with the ribosome profiling experiments. L.H., A.D., U.-P.G., and M.W.H. helped with analysis of ribosome profiling and RNA-seq data. T.Z. and A.M.M. designed the Ded1-IDRm variant. M.J. helped with the tweezer experiments. A.W.F., M.M., and M.K. helped with the temperature stage experiments. T.M.F. performed the experiments shown in Figures 3D, 3E, and 4B and helped with interpretation of the CD experiments and yeast growth rates shown in Figure 2D. C.D.A. helped with the experiments shown in Figures 2C, 2D, 4A, 4C, 5F, 6D, 6E, S2C, S4A–S4E, and S6E and with protein purification. C.J. helped with the experiments shown in Figures 5B–5D and S5B–S5E. D.R. helped with cloning, CRISPR/Cas9 strain engineering, and immunoblotting. C.I. and S.A. drafted the manuscript. C.I., C.D.A., C.J., U.-P.G., T.M.F., A.A.H., and S.A. edited the manuscript. Funding was acquired by S.A.
Declaration of Interests
S.A. is an advisor on the scientific advisory board of Dewpoint Therapeutics. A.A.H. is a co-founder of Dewpoint Therapeutics. U.-P.G. is an employee of DKMS Life Science Lab GmbH.
Published: April 30, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.04.009.
Supplemental Information
References
- Alberti S., Hyman A.A. Are aberrant phase transitions a driver of cellular aging? BioEssays. 2016;38:959–968. doi: 10.1002/bies.201600042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banani S.F., Lee H.O., Hyman A.A., Rosen M.K. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–289. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker A.H., Oh E., Weissman J.S., Kramer G., Bukau B. Selective ribosome profiling as a tool for studying the interaction of chaperones and targeting factors with nascent polypeptide chains and ribosomes. Nat. Protoc. 2013;8:2212–2239. doi: 10.1038/nprot.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berka R.M., Grigoriev I.V., Otillar R., Salamov A., Grimwood J., Reid I., Ishmael N., John T., Darmond C., Moisan M.-C. Comparative genomic analysis of the thermophilic biomass-degrading fungi Myceliophthora thermophila and Thielavia terrestris. Nat. Biotechnol. 2011;29:922–927. doi: 10.1038/nbt.1976. [DOI] [PubMed] [Google Scholar]
- Berthelot K., Muldoon M., Rajkowitsch L., Hughes J., McCarthy J.E.G. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol. Microbiol. 2004;51:987–1001. doi: 10.1046/j.1365-2958.2003.03898.x. [DOI] [PubMed] [Google Scholar]
- Causton H.C., Ren B., Koh S.S., Harbison C.T., Kanin E., Jennings E.G., Lee T.I., True H.L., Lander E.S., Young R.A. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell. 2001;12:323–337. doi: 10.1091/mbc.12.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenna R., Sugawara H., Koike T., Lopez R., Gibson T.J., Higgins D.G., Thompson J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkasov V., Hofmann S., Druffel-Augustin S., Mogk A., Tyedmers J., Stoecklin G., Bukau B. Coordination of translational control and protein homeostasis during severe heat stress. Curr. Biol. 2013;23:2452–2462. doi: 10.1016/j.cub.2013.09.058. [DOI] [PubMed] [Google Scholar]
- Cherkasov V., Grousl T., Theer P., Vainshtein Y., Glässer C., Mongis C., Kramer G., Stoecklin G., Knop M., Mogk A., Bukau B. Systemic control of protein synthesis through sequestration of translation and ribosome biogenesis factors during severe heat stress. FEBS Lett. 2015;589:3654–3664. doi: 10.1016/j.febslet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- Craig E.A., Gross C.A. Is hsp70 the cellular thermometer? Trends Biochem. Sci. 1991;16:135–140. doi: 10.1016/0968-0004(91)90055-z. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B. Yeasts illustrate the molecular mechanisms of eukaryotic genome evolution. Trends Genet. 2006;22:375–387. doi: 10.1016/j.tig.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Firczuk H., Kannambath S., Pahle J., Claydon A., Beynon R., Duncan J., Westerhoff H., Mendes P., McCarthy J.E. An in vivo control map for the eukaryotic mRNA translation machinery. Mol. Syst. Biol. 2013;9:635. doi: 10.1038/msb.2012.73. 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmann T.M., Alberti S. Protein Phase Separation as a Stress Survival Strategy. Cold Spring Harb. Perspect. Biol. 2019;11 doi: 10.1101/cshperspect.a034058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzmann T.M., Jahnel M., Pozniakovsky A., Mahamid J., Holehouse A.S., Nüske E., Richter D., Baumeister W., Grill S.W., Pappu R.V. Phase separation of a yeast prion protein promotes cellular fitness. Science. 2018;359:eaao5654. doi: 10.1126/science.aao5654. [DOI] [PubMed] [Google Scholar]
- Gao Z., Putnam A.A., Bowers H.A., Guenther U.-P., Ye X., Kindsfather A., Hilliker A.K., Jankowsky E. Coupling between the DEAD-box RNA helicases Ded1p and eIF4A. eLife. 2016;5 doi: 10.7554/eLife.16408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch A.P., Spellman P.T., Kao C.M., Carmel-Harel O., Eisen M.B., Storz G., Botstein D., Brown P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006;1:2876–2890. doi: 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grousl T., Ivanov P., Frýdlová I., Vasicová P., Janda F., Vojtová J., Malínská K., Malcová I., Nováková L., Janosková D. Robust heat shock induces eIF2alpha-phosphorylation-independent assembly of stress granules containing eIF3 and 40S ribosomal subunits in budding yeast, Saccharomyces cerevisiae. J. Cell Sci. 2009;122:2078–2088. doi: 10.1242/jcs.045104. [DOI] [PubMed] [Google Scholar]
- Guenther U.-P., Weinberg D.E., Zubradt M.M., Tedeschi F.A., Stawicki B.N., Zagore L.L., Brar G.A., Licatalosi D.D., Bartel D.P., Weissman J.S., Jankowsky E. The helicase Ded1p controls use of near-cognate translation initiation codons in 5′ UTRs. Nature. 2018;559:130–134. doi: 10.1038/s41586-018-0258-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N., Lorsch J.R., Hinnebusch A.G. Yeast Ded1 promotes 48S translation pre-initiation complex assembly in an mRNA-specific and eIF4F-dependent manner. eLife. 2018;7 doi: 10.7554/eLife.38892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J.-S., Hu Z., Thiele D.J., Iyer V.R. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell. Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliker A., Gao Z., Jankowsky E., Parker R. The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol. Cell. 2011;43:962–972. doi: 10.1016/j.molcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnel M., Behrndt M., Jannasch A., Schäffer E., Grill S.W. Measuring the complete force field of an optical trap. Opt. Lett. 2011;36:1260–1262. doi: 10.1364/OL.36.001260. [DOI] [PubMed] [Google Scholar]
- Kertesz M., Wan Y., Mazor E., Rinn J.L., Nutter R.C., Chang H.Y., Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong A., Matheny T., Jain S., Mitchell S.F., Wheeler J.R., Parker R. The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Mol. Cell. 2017;68:808–820.e5. doi: 10.1016/j.molcel.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroschwald S., Munder M.C., Maharana S., Franzmann T.M., Richter D., Ruer M., Hyman A.A., Alberti S. Different Material States of Pub1 Condensates Define Distinct Modes of Stress Adaptation and Recovery. Cell Rep. 2018;23:3327–3339. doi: 10.1016/j.celrep.2018.05.041. [DOI] [PubMed] [Google Scholar]
- Krüger C., Benecke B.J. In vitro translation of Drosophila heat-shock and non--heat-shock mRNAs in heterologous and homologous cell-free systems. Cell. 1981;23:595–603. doi: 10.1016/0092-8674(81)90155-0. [DOI] [PubMed] [Google Scholar]
- Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Suleski M., Hedges S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017;34:1812–1819. doi: 10.1093/molbev/msx116. [DOI] [PubMed] [Google Scholar]
- Lancaster A.K., Nutter-Upham A., Lindquist S., King O.D. PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics. 2014;30:2501–2502. doi: 10.1093/bioinformatics/btu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuenberger P., Ganscha S., Kahraman A., Cappelletti V., Boersema P.J., von Mering C., Claassen M., Picotti P. Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science. 2017;355 doi: 10.1126/science.aai7825. [DOI] [PubMed] [Google Scholar]
- Lindquist S. The heat-shock response. Annu. Rev. Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- Lindquist S., Craig E.A. The heat-shock proteins. Annu. Rev. Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Lindquist S., DiDomenico B., Bugaisky G., Kurtz S. Regulation of the heat-shock response. In: Schlesinger M.J., Ashburner M., Tissieres A., editors. Heat Shock: From Bacteria to Man. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1982. [Google Scholar]
- Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17 [Google Scholar]
- McGlincy N.J., Ingolia N.T. Transcriptome-wide measurement of translation by ribosome profiling. Methods. 2017;126:112–129. doi: 10.1016/j.ymeth.2017.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittasch M., Gross P., Nestler M., Fritsch A.W., Iserman C., Kar M., Munder M., Voigt A., Alberti S., Grill S.W., Kreysing M. Non-invasive perturbations of intracellular flow reveal physical principles of cell organization. Nat. Cell Biol. 2018;20:344–351. doi: 10.1038/s41556-017-0032-9. [DOI] [PubMed] [Google Scholar]
- Morano K.A., Grant C.M., Moye-Rowley W.S. The response to heat shock and oxidative stress in Saccharomyces cerevisiae. Genetics. 2012;190:1157–1195. doi: 10.1534/genetics.111.128033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Ba A.N., Yeh B.J., van Dyk D., Davidson A.R., Andrews B.J., Weiss E.L., Moses A.M. Proteome-wide discovery of evolutionary conserved sequences in disordered regions. Sci. Signal. 2012;5:rs1. doi: 10.1126/scisignal.2002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munder M.C., Midtvedt D., Franzmann T., Nüske E., Otto O., Herbig M., Ulbricht E., Müller P., Taubenberger A., Maharana S. A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. Elife. 2016;5:e09347. doi: 10.7554/eLife.09347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanaswamy R., Levy M., Tsechansky M., Stovall G.M., O’Connell J.D., Mirrielees J., Ellington A.D., Marcotte E.M. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. 2009;106:10147–10152. doi: 10.1073/pnas.0812771106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Ba A.N., Strome B., Hua J.J., Desmond J., Gagnon-Arsenault I., Weiss E.L., Landry C.R., Moses A.M. Detecting functional divergence after gene duplication through evolutionary changes in posttranslational regulatory sequences. PLoS Comput. Biol. 2014;10:e1003977. doi: 10.1371/journal.pcbi.1003977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y., Tanaka F. Cooperative Hydration, Chain Collapse, and Flat LCST Behavior in Aqueous Poly(N-isopropylacrylamide) Solutions. Macromolecules. 2005;38:4465–4471. [Google Scholar]
- Peng K., Vucetic S., Radivojac P., Brown C.J., Dunker A.K., Obradovic Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005;3:35–60. doi: 10.1142/s0219720005000886. [DOI] [PubMed] [Google Scholar]
- Petrovska I., Nüske E., Munder M.C., Kulasegaran G., Malinovska L., Kroschwald S., Richter D., Fahmy K., Gibson K., Verbavatz J.-M., Alberti S. Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. eLife. 2014;3 doi: 10.7554/eLife.02409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riback J.A., Katanski C.D., Kear-Scott J.L., Pilipenko E.V., Rojek A.E., Sosnick T.R., Drummond D.A. Stress-Triggered Phase Separation Is an Adaptive, Evolutionarily Tuned Response. Cell. 2017;168:1028–1040. doi: 10.1016/j.cell.2017.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan O.W., Skerker J.M., Maurer M.J., Li X., Tsai J.C., Poddar S., Lee M.E., DeLoache W., Dueber J.E., Arkin A.P., Cate J.H.D. Selection of chromosomal DNA libraries using a multiplex CRISPR system. eLife. 2014;3:3. doi: 10.7554/eLife.03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan O.W., Poddar S., Cate J.H.D. CRISPR-Cas9 Genome Engineering in Saccharomyces cerevisiae Cells. Cold Spring Harb. Protoc. 2016;2016 doi: 10.1101/pdb.prot086827. [DOI] [PubMed] [Google Scholar]
- Salvadó Z., Arroyo-López F.N., Guillamón J.M., Salazar G., Querol A., Barrio E. Temperature adaptation markedly determines evolution within the genus Saccharomyces. Appl. Environ. Microbiol. 2011;77:2292–2302. doi: 10.1128/AEM.01861-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson R.A., Jack Crisman M., Tansey M.R. Observations on the thermophilous ascomycete Thielavia terrestris. Trans. Br. Mycol. Soc. 1977;69:417–423. [Google Scholar]
- Scannell D.R., Zill O.A., Rokas A., Payen C., Dunham M.J., Eisen M.B., Rine J., Johnston M., Hittinger C.T. The Awesome Power of Yeast Evolutionary Genetics: New Genome Sequences and Strain Resources for the Saccharomyces sensu stricto Genus. G3 (Bethesda) 2011;1:11–25, 11–25. doi: 10.1534/g3.111.000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N.D., Zhou F., Ingolia N.T., Hinnebusch A.G. Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases Ded1 and eIF4A. Genome Res. 2015;25:1196–1205. doi: 10.1101/gr.191601.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N.D., Zhou F., Harris M.S., Ingolia N.T., Hinnebusch A.G. eIF4B stimulates translation of long mRNAs with structured 5′ UTRs and low closed-loop potential but weak dependence on eIF4G. Proc. Natl. Acad. Sci. USA. 2016;113:10464–10472. doi: 10.1073/pnas.1612398113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiber A., Döring K., Friedrich U., Klann K., Merker D., Zedan M., Tippmann F., Kramer G., Bukau B. Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature. 2018;561:268–272. doi: 10.1038/s41586-018-0462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y., Brangwynne C.P. Liquid phase condensation in cell physiology and disease. Science. 2017;357:eaaf4382. doi: 10.1126/science.aaf4382. [DOI] [PubMed] [Google Scholar]
- Storti R.V., Scott M.P., Rich A., Pardue M.L. Translational control of protein synthesis in response to heat shock in D. melanogaster cells. Cell. 1980;22:825–834. doi: 10.1016/0092-8674(80)90559-0. [DOI] [PubMed] [Google Scholar]
- Trotter E.W., Kao C.M.-F., Berenfeld L., Botstein D., Petsko G.A., Gray J.V. Misfolded proteins are competent to mediate a subset of the responses to heat shock in Saccharomyces cerevisiae. J. Biol. Chem. 2002;277:44817–44825. doi: 10.1074/jbc.M204686200. [DOI] [PubMed] [Google Scholar]
- Uchida M., Sun Y., McDermott G., Knoechel C., Le Gros M.A., Parkinson D., Drubin D.G., Larabell C.A. Quantitative analysis of yeast internal architecture using soft X-ray tomography. Yeast. 2011;28:227–236. doi: 10.1002/yea.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega Laso M.R., Zhu D., Sagliocco F., Brown A.J., Tuite M.F., McCarthy J.E. Inhibition of translational initiation in the yeast Saccharomyces cerevisiae as a function of the stability and position of hairpin structures in the mRNA leader. J. Biol. Chem. 1993;268:6453–6462. [PubMed] [Google Scholar]
- Wallace E.W.J., Kear-Scott J.L., Pilipenko E.V., Schwartz M.H., Laskowski P.R., Rojek A.E., Katanski C.D., Riback J.A., Dion M.F., Franks A.M. Reversible, Specific, Active Aggregates of Endogenous Proteins Assemble upon Heat Stress. Cell. 2015;162:1286–1298. doi: 10.1016/j.cell.2015.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzel G., Pilatus U., Rensing L. Similar dose response of heat shock protein synthesis and intracellular pH change in yeast. Exp. Cell Res. 1985;159:252–256. doi: 10.1016/s0014-4827(85)80054-9. [DOI] [PubMed] [Google Scholar]
- Werner-Washburne M., Stone D.E., Craig E.A. Complex interactions among members of an essential subfamily of hsp70 genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 1987;7:2568–2577. doi: 10.1128/mcb.7.7.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Amrani N., Jacobson A., Sachs M.S. The use of fungal in vitro systems for studying translational regulation. Methods Enzymol. 2007;429:203–225. doi: 10.1016/S0076-6879(07)29010-X. [DOI] [PubMed] [Google Scholar]
- Wu C., Sachs M.S. Preparation of a Saccharomyces cerevisiae cell-free extract for in vitro translation. Methods Enzymol. 2014;539:17–28. doi: 10.1016/B978-0-12-420120-0.00002-5. [DOI] [PubMed] [Google Scholar]
- Zarin T., Tsai C.N., Nguyen Ba A.N., Moses A.M. Selection maintains signaling function of a highly diverged intrinsically disordered region. Proc. Natl. Acad. Sci. USA. 2017;114:E1450–E1459. doi: 10.1073/pnas.1614787114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarin T., Strome B., Nguyen Ba A.N., Alberti S., Forman-Kay J.D., Moses A.M. Proteome-wide signatures of function in highly diverged intrinsically disordered regions. eLife. 2019;8 doi: 10.7554/eLife.46883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Krakowiak J., Patel N., Beyzavi A., Ezike J., Khalil A.S., Pincus D. Dynamic control of Hsf1 during heat shock by a chaperone switch and phosphorylation. eLife. 2016;5 doi: 10.7554/eLife.18638. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
4 μM Ded1-GFP and synthesized mRNA containing the 5′UTR from PAB1, were mixed at room temperature in PIPES/KOH pH 6.8 buffer, 200 mM KCl and imaged on the temperature stage with Pico-Surf to prevent aberrant surface effects, as described in the STAR Methods. The temperature was increased to 40°C while imaging.
Data Availability Statement
Ribosome profiling data
The ribosome profiling data used for analysis are deposited in the GEO repository and can be accessed with the following link:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131176
RNAseq data
The RNaseq data used for analysis are deposited in the GEO repository and can be accessed with the following link:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE141029