

Abstract
CHARGE syndrome is a rare and complex disorder, causing multiple birth defects and sensory deficits. The CHD7 gene was proved to be the major pathogenic gene in CHARGE syndrome. To date, the phenotype of neonatal CHARGE syndrome is still poorly recognized. In this paper, we report a Chinese neonate with typical CHARGE syndrome. During his stay in the neonatal intensive care unit of our hospital, the patient presented with various appearance abnormalities, severe dyspnea, dysphagia and recurrent infection. Integrated analysis of the clinical manifestations and examinations suggested a diagnosis of CHARGE syndrome. Later, the genetic analysis revealed a de novo null heterozygous pathogenic mutation in the patient’s CHD7 gene [c.6292C>T (p.Arg2098*)]. Taken together, the patient was diagnostic confirmed as typical CHARGE syndrome. The physicians provided symptomatic treatments for the patient which significantly alleviated his condition, including infection control, laryngoplasty, nasogastric tube feeding and respiratory support. To our knowledge, this case broadens the clinical phenotypic spectrum of typical CHARGE syndrome in neonatal period due to the null mutation of CHD7 gene [c.6292C>T (p.Arg2098*)]. It also demonstrates that genetic analysis is essential in the diagnosis of CHARGE syndrome early in life. Clinicians should focus on providing supportive and corrective therapies in early treatment, particularly in controlling infection, and improving breathing and feeding.
Keywords: CHARGE syndrome, phenotypic spectrum, neonate, CHD7, case report
Introduction
CHARGE syndrome is a rare disorder characterized by a combination of multiple congenital anomalies and inherited in an autosomal dominant manner (1). It was first described and reported by Hall in 1979 and officially named CHARGE syndrome by Pagon et al. in 1981 (2,3). CHARGE is an acronym for coloboma, heart disease, atresia choanae, retarded growth, genital hypoplasia and, ear anomalies and deafness (3). The chromodomain helicase DNA binding protein 7 (CHD7) gene was discovered in 2004 and has been proved to be the main pathogenic gene involved in CHARGE syndrome (4). It has been reported that the prevalence of CHARGE syndrome was about 1:8,500 to 1:15,000 and most individuals with CHARGE syndrome are sporadic (1,5,6). Blake et al. proposed the diagnostic criteria based on clinical manifestations in 1998, and Verloes updated the criteria in 2005 (7,8). Considering currently reported prevalence worldwide, it is reasonable to believe that there is a high rate of underdiagnosis due to limited clinical recognition during neonatal period. In this report, we described a unique case of typical CHARGE syndrome in a Chinese neonate who presented with various clinical manifestations, some of which hadn’t been reported in the case of the same site mutation. We present the following case in accordance with the CARE Guideline (9).
Case presentation
A male neonate developed dyspnea and laryngeal stridor shortly after birth. After receiving oxygen and anti-infective treatment at the local hospital, his conditions did not improve. Thus, he was admitted to the neonatal intensive care unit (NICU) of our hospital 1 day after birth. The newborn was delivered naturally at 38+2 weeks. His Apgar scores were normal. He was the second child born to a gravida 3, para 2 mother. His mother denied the history of consuming any toxic substances during her pregnancy and the prenatal examinations were normal. He had a 7-year-old elder sister. His parents and sister were healthy, and his parents denied consanguineous marriage and any family history of genetic disease. On admission to the NICU, the patient’s weight was 3,520 g, height was 47 cm and occipitofrontal circumference was 37 cm. Many abnormalities were observed during the physical examination, including ocular hypertelorism, flat nasal bridge, high-arched palate, retrognathia, low-set ears, micropenis and cryptorchidism.
A number of targeted laboratory and imaging examinations were performed. The routine blood examination showed a white blood cell count of 9.24×109/L, and the C reactive protein concentration was up to 15 mg/L. Sputum culture indicated positive Klebsiella and Escherichia coli. Blood culture was negative. The results of immune system test suggested immune deficiency (Table 1). Cervical CT images revealed irregular morphology of laryngoventricular. Fiberoptic bronchoscopy demonstrated an epiglottic cartilage malformation or absence, and endobronchitis. Cardiac ultrasound revealed an atrial septal defect (1.5 mm). Hypoplastic semi-circular canals were observed in ear MRI (Figure 1). The results of cranial MRI suggested the possibility of brain dysplasia (Figure 2). The Chest CT indicated pneumonia and a small thymus (Figure 3). The evoked potential report showed severe hearing impairment on the left side. The eye fundus examination revealed the absence of left optic disc (Figure 4). All the clinical examinations suggested a diagnosis of CHARGE syndrome. Later, genetic analysis revealed a heterozygous null mutation in the patient’s CHD7 gene [c.6292C>T(p.Arg2098*)], which was located in 29th exon. Sanger sequencing results showed that the heterozygous nonsense mutation was not detected in the patient’s parents, indicating that it was a de novo mutation (Figure 5). Combined with the above results, the patient was diagnosed as typical CHARGE syndrome.
Table 1. Results of immune function testing.
| Items | Results | Reference |
|---|---|---|
| IgG | 2.43 g/L | 5.70–13.70 g/L |
| IgA | <0.0647 g/L | <0.06 g/L |
| IgM | 0.20 g/L | 0.06–0.20 g/L |
| IgE | <4.34 IU/mL | <15 IU/mL |
| CD3+ | 1.35% | 61.04–71.82% |
| CD8+ | 0.39% | 20.36–32.78% |
| CD4+ | 0.91% | 29.78–39.94% |
| CD16+56+/CD3− | 8.14% | 8.35–18.23% |
| CD19+ | 89.08% | 14.35–22.65% |
| CD4+/CD8+ | 2.33% | 0.94–1.86% |
| CD3+ absolute | 0.01×109/L | 1.41–3.38×109/L |
| CD8+ absolute | 0.003×109/L | 0.54–1.36×109/L |
| CD4+ absolute | 0.006×109/L | 0.71–1.84×109/L |
| Lymphocytes absolute | 0.731×109/L | 2.34–4.81×109/L |
| CD16+56+/CD3− absolute | 0.061×109/L | 0.28–0.63×109/L |
| CD19+ absolute | 0.672×109/L | 0.45–0.82×109/L |
Figure 1.
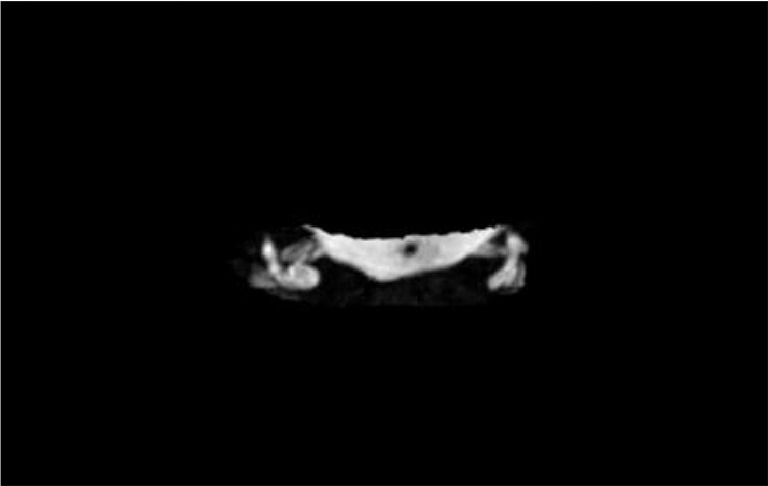
Ear MRI imaging of the patient. Hypoplastic semi-circular canals on both sides were showed in the ear MRI.
Figure 2.
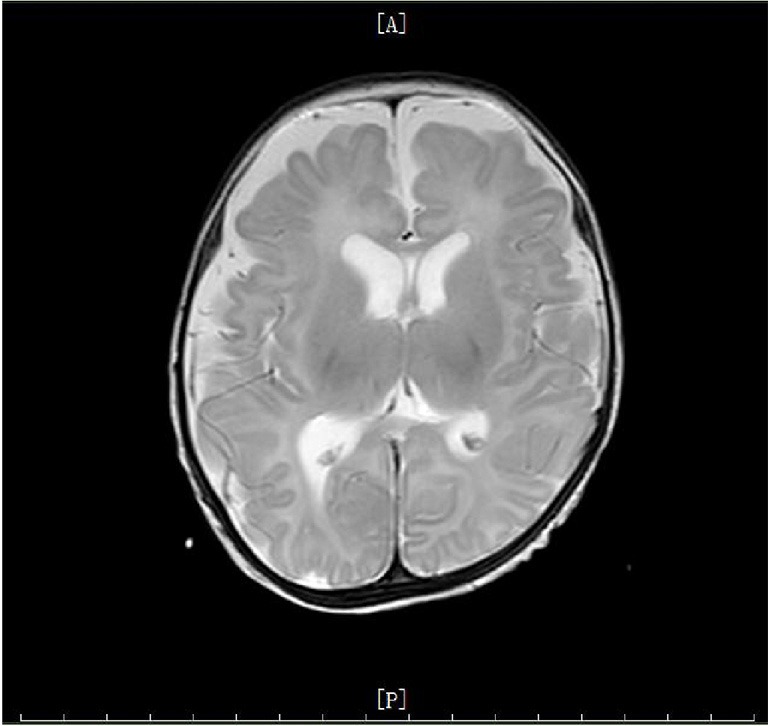
Cranial MRI imaging of the patient. The lateral frontotemporal parietal space on both sides was widened, and sulcus fissure was widened and increased. All of these suggested a possibility of brain dysplasia.
Figure 3.
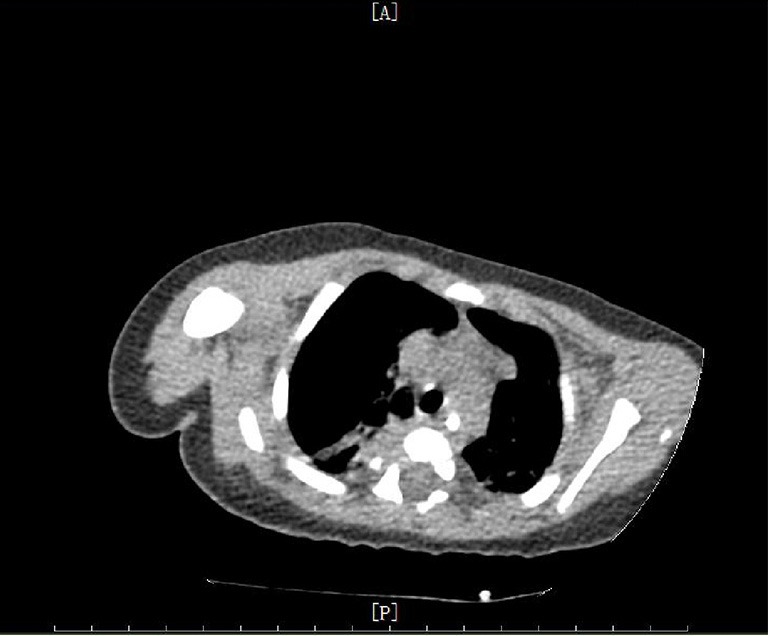
Chest CT imaging of the patient. The chest CT showed that the thymus was smaller than normal.
Figure 4.

Eye fundus examination imaging of the patient. The imaging revealed the absence of left optic disc.
Figure 5.
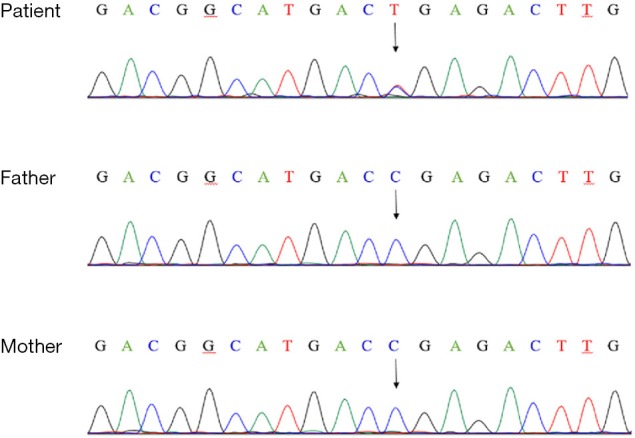
Genomic sequence of the patient. Genetic analysis revealed a heterozygous null mutation in the patient’s CHD7 gene [c.6292C>T(p.Arg2098*)]. No genetic mutations were found in his parents. The arrows indicated the mutation site.
During his stay in the NICU, the patient presented with recurrent infection, severe dyspnea and dysphagia. Various treatments were provided for the patient. Respiratory support was provided to improve dyspnea. Ampicillin sodium and sulbactam sodium, ceftazidime, cefoperazone sodium and sulbactam sodium were used to control the infection respectively. At 18 days of age, the patient received a laryngoplasty. During the 2 months of hospital stay, the patient was fed by a nasogastric tube due to the dysphagia.
Considering the complicated conditions and possibility of poor prognosis, his parents gave up all the treatment and discharged automatically (Figure 6). The patient was followed up by telephone. In the days that followed, the patient inevitably suffered from severe malnutrition, growth delay and recurrent infections. The patient eventually died of infection and malnutrition at 5 months of age.
Figure 6.

Timeline of this case.
Discussion
CHARGE syndrome is an autosomal dominant congenital disorder with a variety of clinical manifestations. The diagnostic criteria was proposed by Blake et al. in 1998, and updated by Verloes in 2005 (7,8). Eight key features were proposed in the 2005 criteria, including three major signs and five minor signs (Table 2). Patients diagnosed as typical CHARGE syndrome are those who presented three major signs, or two major signs and two minor signs. Patients who have two major signs but only one minor sign can be diagnosed as partial CHARGE syndrome, whereas those who have two major and no minor, or one major sign and at least three minors can be diagnosed as atypical CHARGE syndrome (8). According to the 2005 criteria, the various clinical manifestations of our patient suggested a clinical diagnosis of typical CHARGE syndrome (Table 2). Later, genetic analysis was performed to confirm the diagnosis. The genetic analysis identified a heterozygous null mutation in CHD7 gene [c.6292C>T (p.Arg2098*)]. It has been proven that CHARGE syndrome is mainly caused by the heterozygous mutation in the CHD7 on chromosome 8q12, which is first discovered in 2004 by Vissers et al. (4). Over 90% of CHARGE patients who meet the clinical diagnostic criteria of Blake et al. and/or Verloes occur due to the CHD7 mutation (6,10). A recent study showed that CHARGE syndrome was characterized by co-occurrence of multiple malformations due to abnormal development of neural crest cells (11). CHD7 is a key regulator of maintenance of the lineage-specific epigenome and cell type-specific gene expression that is integral to neuroepithelial (NE) cell fate and central nervous system (CNS) lineage commitment. It has been found that collapse of a cell type-specific gene expression program in the NE progenitor population is sufficient to affect a wide range of neural and NC derivatives throughout human fetal development, which may be the pathogenesis of multiple anomalies in CHARGE syndrome caused by CHD7 (12). Combined with all the test results, our patient was diagnosed as typical CHARGE syndrome. Hale et al. has proposed to add pathogenic CHD7 variant status as a major criterion which highlights the importance of genetic analysis (13). Our case also suggested that the identification of CHD7 mutation is necessary for the early diagnosis of CHARGE syndrome.
Table 2. Diagnostic criteria and the patient’s anomalies.
| Diagnostic criteria | Patient’s anomalies |
|---|---|
| Major signs (‘‘the 3 C’’) | |
| Coloboma | Absence of left optic disc |
| Atresia of choanae | N/A |
| Hypoplastic semi-circular canals | Hypoplastic semi-circular canals |
| Minor signs | |
| Rhombencephalic dysfunction | Dysphagia and deafness |
| Hypothalamo-hypophyseal dysfunction | Micropenis and cryptorchidism |
| Abnormal middle or external ear | Low-set ears |
| Malformation of mediastinal organs | Atrial septal defect and small thymus |
| Mental retardation | Brain dysplasia |
N/A, not available.
Through literature review, we found that the mutation in the CHD7 gene locus mentioned in our case has been reported in a patient with CHARGE syndrome (14). In that case, the patient was diagnosed with truncus arteriosus shortly after birth and had dysmorphic features which were consistent with the phenotype of CHARGE syndrome (14). She shared some of the similar clinical features with our patient. Both patients had severe immune deficiency. In that case, the patient did not have T lymphocytes initially, but developed lymphocytosis in conjunction with other clinical features and eosinophilia (14). The lymphocyte absolute of our patient, especially T lymphocyte absolute, are significantly lower than normal. It’s worth noting that there are still notable differences between these two patients (Table 3). Unlike the patient reported, our patient had bilateral semi-circular canals hypoplasia and hearing impairment on the left side, which were important clinical features of individuals with CHARGE syndrome. Another significant difference is that our patient had severe laryngomalacia and cranial nerve dysfunction, which resulted in dyspnea, dysphagia and feeding difficulties. Our patient received a laryngoplasty during the early months of life and was fed by nasogastric tube until discharge. Laryngomalacia and cranial nerve dysfunction are prevalent in patients with CHARGE syndrome and most of them need to be fed by tube at some time (15,16). Chen et al. retrospectively analyzed 12 Chinese newborns with CHD7 pathogenic variants and found that feeding difficulty was a dominant clinical feature, which is consistent with the clinical manifestations of our patient (17). In summary, the identification of CHD7 resulted in broadening of the clinical phenotype spectrum of typical CHARGE syndrome in neonatal period.
Table 3. Clinical features of two patients with same mutant of CHD7.
| Clinical features | Patient 1 | Patient 2 |
|---|---|---|
| Gender | Male | Female |
| Gestational age | 38+2 weeks | 36 weeks |
| Appearance characteristics | ||
| Eyes | Ocular hypertelorism | N/A |
| Nose | Flat nasal bridge | N/A |
| Mouth | High-arched palate | Small mouth |
| Ears | Low-set ears | Low-set ears with primitive helical and pattern pre-auricular tags |
| Mandible | Retrognathia | Retrognathia |
| Genitals | Micropenis and cryptorchidism | N/A |
| Other characteristics | N/A | Sacral dimple |
| Laboratory examinations | Immune deficiency | Immune deficiency |
| Hypocalcemia | ||
| Imaging examinations | Atrial septal defect | Atrial septal defect |
| Small thymus | Ventricular septal defect | |
| Absence of left optic disc | Truncus arteriosus | |
| Bilateral semi-circular canals hypoplasia | Absent thymus | |
| Brain dysplasia | Bilateral optic disc hypoplasia | |
| Hearing impairment on the left side | ||
| Laryngomalacia and abnormal epiglottis | ||
| Gene analysis | c.6292C>T (p.Arg2098*) | c.6292C>T (p.Arg2098*) |
| Outcome | Died of infection and malnutrition | Died from cardiorespiratory insufficiency |
Patient 1, the patient in our case; Patient 2, the patient reported by Gennery et al. (14). N/A, not available.
CHARGE syndrome has been studied for almost 40 years, but there is still lack of effective and curative treatment. The prognosis of patients with CHARGE syndrome was different. Infant mortality is high in individuals with CHARGE syndrome (1). To date, patients with CHARGE syndrome often receive various supportive and corrective therapies, such as surgical repair of cardiac, repair of retinal detachments, tracheostomies, hernia repairs, etc. Therapies targeted toward augmentation of existing skills are also highly beneficial, including hearing aids and cochlear implants (5). In addition, dynamic detection of hormone levels, endocrine therapy timely, and psychological assessment are necessary. Esposito and his colleagues have found that hormone replacement therapy may be effective in treating growth delay (18). These therapies are important for survival and favorable outcomes in patients with CHARGE syndrome, but none of them is considered curative. In hospitalization, our patient received a wide variety of therapies, including infection control, laryngoplasty, nasogastric tube feeding and respiratory support. These treatments significantly alleviated the patient's condition. However, his parents decided to end all treatment and released from hospital after 2 months of in-hospitalization. The patient in our case died of infection and malnutrition at 5 months of age. In view of this, it requires a long-term multidisciplinary comprehensive management to the patients.
In this case, we took full advantages of the genetic testing to diagnose a complex congenital disease in a newborn, which help us make targeted treatment decision. However, it’s difficult for physicians to cure genetic disease due to technique limitation. The patient was discharged automatically after two months of in-hospitalization and we couldn’t provide further effective therapies for him.
Conclusions
In conclusion, we report a Chinese neonate diagnosed as typical CHARGE syndrome with a wide variety of clinical manifestations, which broadens the clinical phenotype of typical CHARGE syndrome caused by the heterozygous null mutation in CHD7 gene [c.6292C>T (p.Arg2098*)]. It’s worth noting that early genetic analysis plays an important role in the diagnosis of CHARGE syndrome. Supportive and corrective therapies in early life, particularly in controlling infection, and improving breathing and feeding, are essential for the prognosis of patients.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: We acknowledge the support by Shanghai Jiao Tong University “Jiao Tong Star” Medical-Industrial Cross-Research Fund (YG2019ZDA01) and Key Discipline of Top Priority in Shanghai (2017ZZ02019).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Written informed consent was obtained from patient’s guardian.
Footnotes
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/tp.2020.03.09). The authors have no conflicts of interest to declare.
References
- 1.Issekutz KA, Graham JM, Jr, Prasad C, et al. An epidemiological analysis of CHARGE syndrome: Preliminary results from a Canadian study. Am J Med Genet A 2005;133A:309-17. 10.1002/ajmg.a.30560 [DOI] [PubMed] [Google Scholar]
- 2.Hall BD. Choanal atresia and associated multiple anomalies. J Pediatr 1979;95:395-8. 10.1016/S0022-3476(79)80513-2 [DOI] [PubMed] [Google Scholar]
- 3.Pagon RA, Graham JM, Jr, Zonana J, et al. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr 1981;99:223-7. 10.1016/S0022-3476(81)80454-4 [DOI] [PubMed] [Google Scholar]
- 4.Vissers LE, van Ravenswaaij CM, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet 2004;36:955-7. 10.1038/ng1407 [DOI] [PubMed] [Google Scholar]
- 5.van Ravenswaaij-Arts C, Martin DM. New insights and advances in CHARGE syndrome: Diagnosis, etiologies, treatments, and research discoveries. Am J Med Genet C Semin Med Genet 2017;175:397-406. 10.1002/ajmg.c.31592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sánchez N, Hernández M, Cruz JP, et al. Phenotypic spectrum of neonatal CHARGE syndrome. Rev Chil Pediatr 2019;90:533-8. 10.32641/rchped.v90i5.1080 [DOI] [PubMed] [Google Scholar]
- 7.Blake KD, Davenport SL, Hall BD, et al. CHARGE association: an update and review for the primary pediatrician. Clin Pediatr (Phila) 1998;37:159-73. 10.1177/000992289803700302 [DOI] [PubMed] [Google Scholar]
- 8.Verloes A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am J Med Genet A 2005;133A:306-08. 10.1002/ajmg.a.30559 [DOI] [PubMed] [Google Scholar]
- 9.Riley DS, Barber MS, Kienle GS, et al. CARE guidelines for case reports: explanation and elaboration document. J Clin Epidemiol 2017;89:218-35. 10.1016/j.jclinepi.2017.04.026 [DOI] [PubMed] [Google Scholar]
- 10.Janssen N, Bergman JE, Swertz MA, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat 2012;33:1149-60. 10.1002/humu.22086 [DOI] [PubMed] [Google Scholar]
- 11.Bérubé-Simard FA, Pilon N. Molecular dissection of CHARGE syndrome highlights the vulnerability of neural crest cells to problems with alternative splicing and other transcription-related processes. Transcription 2019;10:21-8. 10.1080/21541264.2018.1521213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chai M, Sanosaka T, Okuno H, et al. Chromatin remodeler CHD7 regulates the stem cell identity of human neural progenitors. Gene Dev 2018;32:165-80. 10.1101/gad.301887.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hale CL, Niederriter AN, Green GE, et al. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A 2016;170A:344-54. 10.1002/ajmg.a.37435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gennery AR, Slatter MA, Rice J, et al. Mutations in CHD7 in patients with CHARGE syndrome cause T-B + natural killer cell + severe combined immune deficiency and may cause Omenn-like syndrome. Clin Exp Immunol 2008;153:75-80. 10.1111/j.1365-2249.2008.03681.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu P, Ma A, Wilson M, et al. CHARGE syndrome: a review. J Paediatr Child Health 2014;50:504-11. 10.1111/jpc.12497 [DOI] [PubMed] [Google Scholar]
- 16.Aramaki M, Udaka T, Kosaki R, et al. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr 2006;148:410-4. 10.1016/j.jpeds.2005.10.044 [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Yan K, Gao Y, et al. Feeding difficulty is the dominant feature in 12 Chinese newborns with CHD7 pathogenic variants. BMC Med Genet 2019;20:93. 10.1186/s12881-019-0813-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esposito A, Tufano M, Di Donato I, et al. Effect of long-term GH treatment in a patient with CHARGE association. Ital J Pediatr 2014;40:51. 10.1186/1824-7288-40-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as