

Abstract
CO2-reactivity and neurovascular coupling are sequentially lost within the first 24 h after subarachnoid hemorrhage (SAH). Whether and when these impairments recover is not known. Therefore, we investigated the reactivity of pial and intraparenchymal vessels by in vivo two-photon microscopy one month after experimental SAH.
C57BL/6 mice were subjected to either sham surgery or SAH by filament perforation. One month later, cerebral blood flow following CO2-challenge and forepaw stimulation was assessed by laser Doppler fluxmetry. Diameters of pial and intraparenchymal arterioles were quantified by in vivo two-photon microscopy.
One month after SAH, pial and parenchymal vessels dilated in response to CO2. Neurovascular coupling was almost completely absent after SAH: vessel diameter did not change upon forepaw stimulation compared to a 20% increase in sham-operated mice.
The current results demonstrate that neurovascular function differentially recovers after SAH: while CO2-reactivity normalizes within one month after SAH, neurovascular coupling is still absent. These findings show an acute and persistent loss of neurovascular coupling after SAH that may serve as a link between early brain injury and delayed cerebral ischemia, two distinct pathophysiological phenomena after SAH that were so far believed not to be directly related.
Keywords: Subarachnoid hemorrhage, neurovascular coupling, mice, in vivo, two-photon microscopy
Introduction
Subarachnoid hemorrhage (SAH) is a rare but severe subtype of stroke1 with a mortality rate of almost 50%.2 Moreover, 30% of SAH survivors are unable to resume their premorbid lifestyle.3–5
In 85% of cases, SAH is caused by a spontaneous rupture of a cerebral aneurysm located at the skull base, with subsequent bleeding into the subarachnoid space. Bleeding causes a rapid increase in intracranial pressure (ICP) and global cerebral ischemia, which may be fatal within minutes.6 While advanced interventional and microsurgical techniques allow for safe and efficient occlusion of aneurysms, morbidity and mortality are still high for patients who survive the initial phase after SAH. A major feature associated with adverse outcome is the occurrence of delayed cerebral ischemia (DCI). While DCI has been attributed to vasospasm of larger brain arteries, increasing evidence suggests that a pronounced dysfunction of the cerebral microcirculation in the early phase after hemorrhage may also be involved. Furthermore, autoregulatory mechanisms are often impaired after SAH.7,8 This results in the inability of the brain to compensate reductions in cerebral perfusion and to adequately match blood supply to its metabolic demand.
Cortical spreading depolarizations (CSDs) are waves of electrical depression that occur frequently after SAH9 and are thought to be associated with post-hemorrhagic brain damage and DCI.9–12 CSDs may lead to local ischemia, as the necessary energy reserves to increase cerebral blood flow are not available after SAH.11,13–15 Therefore, impaired regulatory mechanisms may be key factors in the development of DCI after subarachnoid hemorrage.8,16,17 Impaired cerebrovascular CO2-reactivity has been previously described in clinical18 and experimental studies19 and seems to be associated with adverse outcome and development of DCI.18
Neurovascular coupling (NVC) is another important regulatory mechanism responsible for increasing blood flow to active regions of the brain in order to provide sufficient amounts of oxygen and glucose20 through the coordinated action of neurons, astrocytes, pericytes, and endothelial cells.21 We recently demonstrated for the first time in vivo that within 3 h after SAH, pial and parenchymal cerebral vessels lose their ability to respond to the selective cerebral vasodilator CO2, while NVC was fully functional.22 When CO2-reactivity and NVC were investigated 24 h after SAH, both processes were massively impaired, indicating that post-hemorrhagic neurovascular dysfunction is a progressive disorder.22,23 This miscommunication between neurons and vessels following SAH may lead to an imbalance between metabolic demand and blood flow therefore inducing or aggravating regional ischemia and post-hemorrhagic brain damage.
So far, however, the temporal profile of neurovascular dysfunction after SAH has not been investigated beyond one day after SAH. Hence, it remains unknown whether and when CO2 reactivity and NVC recover after SAH. Therefore, we investigated neurovascular reactivity one month after experimental SAH using in vivo two-photon microscopy.
Materials and methods
Animal breeding, housing and all experimental procedures were reviewed and approved by the Veterinary Office and Ethical Review Board of the Government of Upper Bavaria (protocol number 136-11) based on paragraph seven to nine of the German Animal Protection Act (TierSchG). Male C57BL/6 mice six to eight weeks old (20 to 23 g body weight Charles River Laboratory, Sulzfeld, Germany) were used for this study. Experiments were reported according to the Animal Research: Reporting of in Vivo Experiments (ARRIVE) guidelines.24
Randomization and blinding
All animals were randomly assigned to surgery, group allocation was done by drawing lots shortly before induction of hemorrhage, i.e. the surgical preparation was performed in a blinded manner. Two-photon microscopy one month after SAH and subsequent data analysis were performed by a researcher blinded towards group allocation of the animals.
Animal preparation and monitoring
Experimental animals had free access to food and water before and after surgery. For induction of SAH, anesthesia was induced by intraperitoneal injection of midazolam (5 mg/kg; Braun, Melsungen, Germany), fentanyl (0.05 mg/kg; Jansen-Cilag, Neuss, Germany), and medetomidine (0.5 mg/kg; Pfizer, Karlsruhe, Germany) as previously described.25,26 Mice underwent orotracheal intubation and were then mechanically ventilated (Minivent, Hugo Sachs, Hugstetten, Germany). End-tidal pCO2 was measured with a microcapnometer (Capnograph, Hugo Sachs, Hugstetten, Germany) and kept constant between 30 and 40 mmHg by respective adjustments to the ventilation. To maintain body temperature at 37℃ a thermostatically regulated, feedback-controlled heating pad (FHC, Bowdoin, ME, USA) was used. ICP was measured in each animal for 15 min after SAH using a microsensor-based ICP probe (Codman & Shurteff Inc., Raynham, MA) to prove successful induction and severity of SAH as described before.25 For continuous monitoring of regional cerebral blood flow (rCBF), a flexible laser-Doppler probe (Periflux 4001 Master, Perimed, Stockholm, Sweden) was glued onto the skull above the territory of the left middle cerebral artery (MCA). Blood gases and electrolytes were measured at the end of each experiment (Supplementary Table 1).
For experiments on neurovascular reactivity, mice were initially anesthetized with 2% isoflurane in 70% N2O and 30% O2. Later on, isoflurane was gradually reduced over the course of 10 min to a range of 0.5 to 0.9% in 70% room air and 30% O2. At the same time, a continuous intra-arterial infusion of ketamine (30 mg/kg/h, Inresa, Freiburg, Germany) was administered.22,23,27
Induction of SAH
SAH was induced using the filament perforation model as previously described.28–30 Briefly, a 5–0 monofilament was introduced via the left external carotid artery into the internal carotid artery and advanced towards the Circle of Willis. SAH induction was marked by a sudden increase of ICP. Immediately thereafter, the filament was withdrawn, the external carotid artery was ligated, the clip removed from the common carotid artery. In sham-operated mice, the filament was not advanced far enough to induce hemorrhage. Anesthesia was terminated by intraperitoneal injection of atipamezole (2.5 mg/kg), naloxone (1.2 mg/kg), and flumazenil (0.5 mg/kg). Thereafter, mice were kept in an incubator heated at 33℃ for 2.5 h.
Nutritional support protocol
In order to reduce post-operative mortality, we used a nutritional support protocol previously shown to reduce mortality after focal cerebral ischemia.31 Jelly food was prepared daily by mixing food powder containing xanthan gum (ssnif-Spezialdiaten GmbH, Soest, Germany) with water at a ratio of 1:3. Mice were gently restrained and given 1 ml of jelly food three times a day by oral feeding for seven days starting 48 h after SAH. Body weight and body temperature were measured daily.
Forepaw-evoked NVC
Mice were anesthetized 30 days after SAH and the CBF response after NVC was evaluated as previously described.22,23,27 Briefly, the left forepaw was stimulated with two subdermal needle electrodes with a diameter of 0.2 mm (Hwato, Suzhou, China) at an intensity of 2 mA for 0.3 ms (Digitimer Ltd, Hertfordshire, England). One stimulation cycle contained 96 stimulations and lasted for 16 s (6 Hz). The interval between two stimulation cycles was 40 s. CBF was assessed at different locations covering the whole somatosensory cortex. The region with the strongest CBF response was continuously stimulated (2 mA) for 1 min in order to evaluate the response to a tonic stimulus. This region was later analyzed by two-photon microscopy. A graphical representation of the experimental protocol is shown in Figure 1(a).
Figure 1.

Experimental design and SAH induction. (a) Schematic representation of the experimental design for both SAH and sham-operated animals. Neurovascular coupling and endothelium-dependent response were assessed one month after the surgical procedure. (b) Cerebral blood flow and (c) intracranial pressure recordings starting 5 min before SAH or sham-operated induction and continuing for subsequent 15 min. A sudden decrease in CBF and sharp increase in ICP confirms SAH inductions. Mean ± SD; n = 10 in sham and n = 9 in SAH group.
Two-photon microscopy
A cranial window (2 × 1 mm) was drilled under constant cooling with isotonic saline above the area of the somatosensory cortex associated to the forepaw leaving the dura mater intact. Mice were placed under a two-photon microscope (Zeiss LSM-7 MP, Oberkochen, Germany) equipped with a Li:Ti laser (Chameleon, Coherent, USA) as described previously,32 and the exposed dura mater was covered with isotonic saline. The fluorescent plasma dye fluorescein isothiocyanate (FITC-dextran; molecular weight 150 kDa) was injected via a femoral artery catheter (0.05 ml of a 0.5% solution; Sigma, Deisenhofen, Germany) and all parenchymal (diameter: 5–20 µm; depth: circa 150 µm) and pial arterioles (diameter: 20 to 40 µm) in the region previously selected (see above) were visualized using 10 × Zeiss EC Plan-NeoFluar objective. Pial arterioles were followed into the parenchyma along an axis normal to the brain surface. Arterioles were distinguished from venules on the basis of velocity and direction of blood flow. Vessel diameter was quantified using a user defined, full-width measurement approach.
Neurovascular reactivity to CO2
Diameters of both parenchymal and pial arterioles were examined under tight control of end-tidal pCO2 and maintenance of physiological conditions in order to obtain baseline values. Thereafter, arteriolar diameter was observed during inhalation of 10% CO2 for 10 min. The amount of inhaled CO2 was measured by microcapnometry (Hugo-Sachs Elektronik, March-Hugstetten, Germany). Arteriolar diameters were quantified with a calibrated image analysis software (Zen, Zeiss, Oberkochen, Germany) and expressed in percentage of baseline as previously described.27
Vessel diameter during forepaw-evoked NVC
For evaluation of changes in vessel diameter after electrical forepaw stimulation, the same protocol was used as for assessment of CBF. Briefly, the region yielding the most pronounced CBF response was stimulated with one stimulation cycle that contained 96 stimulations and lasted for 16 s (6 Hz). The interval between two stimulation cycles was 40 s. Thereafter, the same region was continuously stimulated (2 mA) for 1 min in order to evaluate the response to a tonic stimulus.
Statistical analysis
Sample size calculation was performed with a standard statistical software package (Sigma Plot 12.5; Systat Software, Erkrath, Germany). For sample size calculation, we used the following parameters: alpha error = 0.05, beta error = 0.2. Statistical analysis was performed using R Statistical Software (Foundation for Statistical Computing, Vienna, Austria). Mixed-design ANOVA was used to test the fixed effects of time (within-group) or surgery and time (between-group) with random effects of mouse ID (CBF analysis) or baseline vessel diameter within mouse ID and interaction between time and baseline within mouse ID (vessel diameter analysis). P was considered significant at < 0.05.
Results
In vivo physiological parameters and mortality
Following SAH, the resulting increase in ICP peaked at values of around 60 mmHg and led to a reduction in cerebral perfusion pressure, which then caused a reduction of cerebral blood flow of around 80%. Within 5 min, ICP decreased to values of around 20 mmHg, and CBF normalized. Both values remained stable for the remaining observation period of 15 min (Figure 1(b) and (c)). Following surgery, all mice showed reduced motor activity. Two mice from each group (4/20) died before imaging could be performed. Physiological parameters known to have strong effects on CBF, i.e. body temperature, systemic blood pressure, and pCO2, were carefully monitored and kept within physiological limits before and after SAH induction. Parameters did not differ between SAH and sham-operated mice (Supplementary Table S1).
In vivo CO2 reactivity is restored one month after SAH in parenchymal arterioles
Direct visualization of pial and parenchymal vessels with two-photon microscopy one month after sham or SAH surgery showed differential alterations in CO2-response. In pial vessels, there was no significant effect of surgery (F(1, 17.197) = 0.1283; p = 0.724596) or interaction between surgery and time (F(1, 168.882) = 0.4336; p = 0.511102) on vessel dilation in percentage of baseline in response to the increase in pCO2 (Figure 2(a) upper panel and Figure 2(b), white and gray symbols). However, while there was a significant increase in vessel diameter in sham-operated mice relative to baseline (F(1, 63.134) = 9.9671; p = 0.002444), the increase in vessel diameter in SAH mice relative to baseline was not significant (F(1, 59) = 2.3145; p = 0.1335). Thus, the status of CO2 reactivity in pial arterioles cannot be conclusively ascertained from these observations.
Figure 2.

Parenchymal arterioles respond normally to hypercapnia one month after SAH. (a) Representative two-photon microscopy images of pial (top panels) and parenchymal (bottom panels) arterioles of sham and SAH mice before and during 10% CO2 inhalation. (b) Pial and (c) parenchymal artery diameter quantification during hypercapnia in mice subjected to sham surgery (white symbols) or SAH (grey symbols). Arteriolar dilation in response to hypercapnia relative to baseline was verified in pial and parenchymal arterioles in sham-operated mice. Parenchymal arterioles in SAH mice also dilated in response to hypercapnia. Mean ± SD. 12 to 24 arterioles in (b) and 12 to 15 arterioles in (c) in n = 7 to 8 mice per group.
In parenchymal vessels, no significant effect of surgery (F(1, 12.005) = 1.5493; p = 0.23699) or interaction between surgery and time (F(1, 135.989) = 1.9751; p = 0.16219) on vessel dilation in percentage of baseline was observed in response to the increase in pCO2 (Figure 2(a) bottom panel and (c) white and gray symbols). Furthermore, while the increase in vessel diameter in sham-operated mice shows a trend effect relative to baseline (F(1, 71.664) = 3.6162; p = 0.0868), there is a significant increase in vessel diameter relative to baseline (F(1, 58.057) = 8.5949; p = 0.004816) in the SAH group. These results demonstrate that the vascular response elicited by CO2-inhalation, which we previously showed to be absent in both pial and parenchymal vessels at 3 and 24 h following SAH,19,22,23 is restored one month after the initial bleeding in parenchymal vessels.
In vivo NVC is compromised one month after SAH
NVC one month after SAH induction was evaluated using two different approaches: sensory stimulation of the forepaw was induced by either 10 discrete stimuli or one continuous stimulus for 60 s.
Visualization of parenchymal arterioles of up to 20 µm in diameter with two-photon microscopy revealed a clear impairment of the vessel response to discrete stimulation in SAH mice relative to sham (F(1, 34.15) = 9.0103; p = 0.004991). While artery diameter in sham-operated mice increased in response to sensory stimulation relative to baseline (Figure 3(a) upper panels and (b), white symbols. F(10, 300.79) = 3.4726; p = 0.000243), arterioles after SAH failed to dilate (Figure 3(a) bottom panels and (b), gray symbols. F(10, 161.86) = 0.9234; p = 0.5132). However, continuous stimulation of the forepaw for 1 min showed no significant effect of surgery (F(1, 22.425) = 0.7504; p = 0.395537) or interaction between surgery and time (F(1, 127.424) = 1.1105; p = 0.293967) on vessel dilation. Continuous stimulation led to a significant increase in artery diameter of about 25% relative to baseline in the sham group (Figure 4(a) upper panels and (b), white symbols. F(1, 66.431) = 5.1792; p = 0.02609). In SAH mice, arterial response to continuous stimulation showed a trend to dilation of only about 7% relative to baseline (Figure 4(a) bottom panels and (b), gray symbols. F(1, 59.883) = 3.798; p = 0.056). These results suggest that NVC remains partially compromised one month after SAH. The range of vessel dilation across stimulation modalities is shown in Figure 5.
Figure 3.

Two-photon imaging of arteriolar diameter in response to discrete stimulation one month after SAH. (a) Representative two-photon microscopy images of parenchymal arterioles in sham (top panels) and SAH mice (bottom panels) at baseline and after stimulation. (b) Arteriolar dilation in response to discrete stimulation was verified in sham-operated mice, but found to be impaired after SAH; 17 to 30 arterioles in n = 7 to 8 mice per group.
Figure 4.

Two-photon imaging of arteriolar diameter in response to a continuous stimulation one month after SAH. (a) Representative two-photon microscopy images of parenchymal arterioles in sham operated (top panels) and SAH mice (bottom panels) at baseline and after sensory stimulation. (b) Arteriolar dilation in response to continuous stimulation was verified in sham-operated mice, but found to be attenuated in SAH mice. SAH mice showed a trend to dilation relative to baseline. Mean ± SD; 10 to 13 arterioles in 7 to 8 mice per group.
Figure 5.
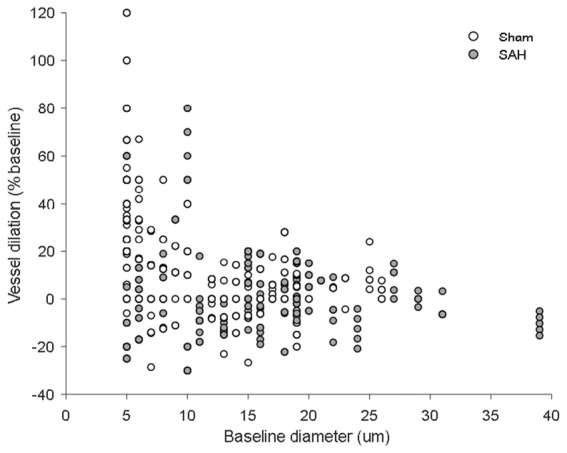
Scatterplot of vessel dilation vs. baseline diameter. Vessel dilation at every time point across stimulation modalities plotted against baseline vessel diameter for both sham- and SAH-operated mice.
NVC was also evaluated on the basis of the CBF response to forepaw stimulation. One month after hemorrhage, the increase in CBF response to discrete sensory stimulation of the forepaw was comparable between sham and SAH mice, reaching an increase in response of around 10% (data not shown). In the SAH group, CBF response slightly decreased after the 6th stimulus without showing a significant change (data not shown). Continuous sensory stimulation of the forepaw for 1 min increased CBF by approximately 20% in sham mice (Figure 6, white symbols), while in SAH mice there was no increase in CBF-response, (Figure 6, gray symbols). While lower at all-time points, due to the variability of the response this was not statistically significant (F(1,72) = 0.6188, p = 0.4467).
Figure 6.
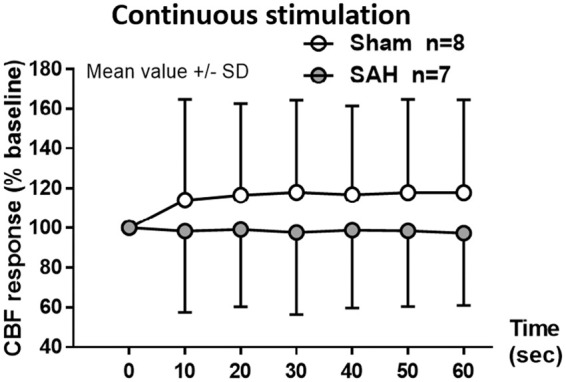
Changes in CBF response to neurovascular coupling one month after SAH. Continuous electrical stimulation at the forepaw leads to an increase in CBF in sham-operated mice (white symbols) of approximately 20% compared to baseline while in the SAH group (grey symbols) the response was lower but not significantly different. Mean ± SD; n = 7 to 8 per group.
Discussion
SAH leads to cerebral microvasospasms and a loss of CO2 reactivity and NVC within 24 h following the initial insult.19,22,23,27 The goal of the present study was to investigate whether and when these vascular dysfunctions resolve. Our investigation provides evidence that one month after SAH, parenchymal arterioles restore their ability to dilate in response to CO2, a brain-specific vasodilator. However, our data show also that NVC is still almost absent one month after SAH induction.
Despite early evidence of post-hemorrhagic endothelium-dependent dysfunction in vivo,30,33,34 only recently has two-photon microscopy allowed the first in vivo measurements of parenchymal arteriolar functionality, both at 3 and 24 h after SAH. The earliest study investigated the very acute phase after hemorrhage (3 h),22 a time of particular interest in the pathophysiology of SAH due to the occurrence of pial microvasospasms and microthrombosis, as well as a decrease of CBF in both experimental SAH and SAH patients.6,30 An endothelium-dependent dysfunction was shown in pial19,33,34 and parenchymal arterioles.22 This early loss of CO2-reactivity in pial and parenchymal vessels was shown to last at least 24 h after hemorrhage in a follow-up study.23 Additionally, in vivo and ex vivo investigation of the NVC revealed a severe dysfunction 24 h following SAH: neuronal stimulation resulted in vasoconstriction rather than vasodilatation.13,23 In the current study, we show that one month following SAH induction, parenchymal vessels restore their reactivity to CO2. The ability of blood vessels to dilate in response to changes in CO2 partial pressure is mediated at least partially by nitric oxide (NO), whose concentration acutely decreases after SAH due to decreased production by endothelial NO-synthase (eNOS).35–38 Recovery of CO2-reactivity in parenchymal arterioles may thus indicate a partial recovery of endothelial NO signaling one month after experimental SAH.
We previously showed a deterioration of NVC early after SAH both in vivo and ex vivo.13,22,23 The present data indicate that microvascular dysfunction persists long-term and may lead to a long-lasting mismatch of neuronal activity and cerebral blood flow. The impaired CBF-response to neuronal activation most likely increases the susceptibility of the brain to ischemic damage even when global cerebral perfusion is sufficiently restored after hemorrhage (e. g. by pharmacological or surgical therapy) and may be the pathophysiological basis for long-lasting neurocognitive and executive deficits in SAH-patients.39
Early brain injury (EBI) describes a variety of pathological mechanisms occurring at the level of cerebral microcirculation within the first 72 h after SAH40–43 that have been associated with decreased CBF, disturbances of cerebral metabolism, and subsequent brain damage. While it becomes increasingly evident that these early pathophysiological changes decisively impact neurological outcome, the mechanisms that link EBI to later phenomena such as DCI are unclear. DCI has been repeatedly shown to correlate with prognosis and neurological outcome after SAH.44–48 The previously held notion that DCI is caused by delayed onset vasospasm of Circle of Willis arteries has been abandoned in the face of recent results of clinical studies where vasospasm was completely reversed, but the frequency and severity of DCI were not affected.49–51 Our results point to a possible link between EBI and DCI: microcirculatory disturbances that originate early after SAH may partially persist for a time period extending well over the range postulated for EBI.
A limitation of our study is the impossibility to examine NVC in the same animals over time. Systemic variables such as arterial pCO2 and blood pressure need to be closely monitored during imaging and such monitoring requires an arterial catheter and orotracheal intubation: procedures that are not compatible with post-operative survival.
Further efforts must be made to characterize the causes of long-term SAH-induced microvascular dysfunction and the underlying cellular and molecular mechanisms. These investigations may provide the basis for novel prognostic and therapeutic innovations that may improve the outcome of patients after SAH.
Supplemental Material
Supplemental Material for Long-term impairment of neurovascular coupling following experimental subarachnoid hemorrhage by Matilde Balbi, Max Jativa Vega, Athanasios Lourbopoulos, Nicole A Terpolilli and Nikolaus Plesnila in Journal of Cerebral Blood Flow & Metabolism
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Solorz-Żak foundation. We thank Mrs. Nicole Heumos for technical assistance. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy – ID 390857198).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
MB designed the study, performed all in vivo experiments, analyzed the data, and wrote the manuscript, MJV analyzed the data and edited the manuscript, AL designed the feeding protocol, helped feeding the mice, NAT edited the manuscript, NP designed the study, analyzed the data, wrote and edited the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
- 1.Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction? Stroke 2009; 40(3 Suppl): S86–S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weaver JP, Fisher M. Subarachnoid hemorrhage: an update of pathogenesis, diagnosis and management. J Neurol Sci 1994; 125: 119–131. [DOI] [PubMed] [Google Scholar]
- 3.van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain 2001; 124(Pt 2): 249–278. [DOI] [PubMed] [Google Scholar]
- 4.Mayer SA, Kreiter KT, Copeland D, et al. Global and domain-specific cognitive impairment and outcome after subarachnoid hemorrhage. Neurology 2002; 59: 1750–1758. [DOI] [PubMed] [Google Scholar]
- 5.Passier PE, Visser-Meily JM, Rinkel GJ, et al. Life satisfaction and return to work after aneurysmal subarachnoid hemorrhage. J Stroke Cerebrovasc Dis 2011; 20: 324–329. [DOI] [PubMed] [Google Scholar]
- 6.Schubert GA, Seiz M, Hegewald AA, et al. Acute hypoperfusion Immediately after subarachnoid hemorrhage: a xenon contrast-enhanced CT study. J Neurotrauma 2009; 26: 2225–2231. [DOI] [PubMed] [Google Scholar]
- 7.Budohoski KP, Czosnyka M, Smielewski P, et al. Impairment of cerebral autoregulation predicts delayed cerebral ischemia after subarachnoid hemorrhage: a prospective observational study. Stroke 2012; 43: 3230–3237. [DOI] [PubMed] [Google Scholar]
- 8.Santos GA, Petersen N, Zamani AA, et al. Pathophysiologic differences in cerebral autoregulation after subarachnoid hemorrhage. Neurology 2016; 86: 1950–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dreier JP, Woitzik J, Fabricius M, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain 2006; 129(Pt 12): 3224–3237. [DOI] [PubMed] [Google Scholar]
- 10.Dreier JP, Major S, Manning A, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009; 132(Pt 7): 1866–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartings JA, York J, Carroll CP, et al. Subarachnoid blood acutely induces spreading depolarizations and early cortical infarction. Brain 2017; 140: 2673–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woitzik J, Dreier JP, Hecht N, et al. Delayed cerebral ischemia and spreading depolarization in absence of angiographic vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab 2012; 32: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koide M, Sukhotinsky I, Ayata C, et al. Subarachnoid hemorrhage, spreading depolarizations and impaired neurovascular coupling. Stroke Res Treat 2013; 2013: 819340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosche B, Graf R, Ernestus RI, et al. Recurrent spreading depolarizations after subarachnoid hemorrhage decreases oxygen availability in human cerebral cortex. Ann Neurol 2010; 67: 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarrafzadeh A, Santos E, Wiesenthal D, et al. Cerebral glucose and spreading depolarization in patients with aneurysmal subarachnoid hemorrhage. Acta Neurochir Suppl 2013; 115: 143–147. [DOI] [PubMed] [Google Scholar]
- 16.Gaasch M, Schiefecker AJ, Kofler M, et al. Cerebral autoregulation in the prediction of delayed cerebral ischemia and clinical outcome in poor-grade aneurysmal subarachnoid hemorrhage patients. Crit Care Med 2018; 46: 774–780. [DOI] [PubMed] [Google Scholar]
- 17.Otite F, Mink S, Tan CO, et al. Impaired cerebral autoregulation is associated with vasospasm and delayed cerebral ischemia in subarachnoid hemorrhage. Stroke 2014; 45: 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrera E, Kurtz P, Badjatia N, et al. Cerebrovascular carbon dioxide reactivity and delayed cerebral ischemia after subarachnoid hemorrhage. Arch. Neurol 2010; 67: 434–439. [DOI] [PubMed] [Google Scholar]
- 19.Friedrich B, Michalik R, Oniszczuk A, et al. CO2 has no therapeutic effect on early microvasospasm after experimental subarachnoid hemorrhage. J. Cereb. Blood Flow Metab 2014; 34: e1–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore CI, Cao R. The hemo-neural hypothesis: on the role of blood flow in information processing. J Neurophysiol 2008; 99: 2035–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Attwell D, Buchan AM, Charpak S, et al. Glial and neuronal control of brain blood flow. Nature 2010; 468: 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balbi M, Koide M, Schwarzmaier SM, et al. Acute changes in neurovascular reactivity after subarachnoid hemorrhage in vivo. J Cereb Blood Flow Metab 2017; 37: 178–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balbi M, Koide M, Wellman GC, et al. Inversion of neurovascular coupling after subarachnoid hemorrhage in vivo. J Cereb Blood Flow Metab 2017; 37: 3625–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drummond GB, Paterson DJ, McGrath JC. ARRIVE: new guidelines for reporting animal research. Exp Physiol 2010; 95: 841. [DOI] [PubMed] [Google Scholar]
- 25.Feiler S, Friedrich B, Scholler K, et al. Standardized induction of subarachnoid hemorrhage in mice by intracranial pressure monitoring. J Neurosci Methods 2010; 190: 164–170. [DOI] [PubMed] [Google Scholar]
- 26.Thal SC, Plesnila N. Non-invasive intraoperative monitoring of blood pressure and arterial pCO2 during surgical anesthesia in mice. J Neurosci Methods 2007; 159: 261–267. [DOI] [PubMed] [Google Scholar]
- 27.Balbi M, Ghosh M, Longden TA, et al. Dysfunction of mouse cerebral arteries during early aging. J Cereb Blood Flow Metab 2015; 35: 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feiler S, Friedrich B, Scholler K, et al. Standardized induction of subarachnoid hemorrhage in mice by intracranial pressure monitoring. J Neurosci Methods 2010; 190: 164–170. [DOI] [PubMed] [Google Scholar]
- 29.Buhler D, Schuller K, Plesnila N. Protocol for the induction of subarachnoid hemorrhage in mice by perforation of the Circle of Willis with an endovascular filament. Transl Stroke Res 2014; 5: 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Friedrich B, Muller F, Feiler S, et al. Experimental subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: an in-vivo microscopy study. J Cereb Blood Flow Metab 2012; 32: 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lourbopoulos A, Mamrak U, Roth S, et al. Inadequate food and water intake determine mortality following stroke in mice. J Cereb Blood Flow Metab 2017; 37: 2084–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwarzmaier SM, Zimmermann R, McGarry NB, et al. In vivo temporal and spatial profile of leukocyte adhesion and migration after experimental traumatic brain injury in mice. J Neuroinflammation 2013; 10: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun BL, Zheng CB, Yang MF, et al. Dynamic alterations of cerebral pial microcirculation during experimental subarachnoid hemorrhage. Cell Mol Neurobiol 2009; 29: 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishikawa M, Kusaka G, Yamaguchi N, et al. Platelet and leukocyte adhesion in the microvasculature at the cerebral surface immediately after subarachnoid hemorrhage. Neurosurgery 2009; 64: 546–553. [DOI] [PubMed] [Google Scholar]
- 35.Sehba FA, Bederson JB. Nitric oxide in early brain injury after subarachnoid hemorrhage. Acta Neurochir Suppl 2011; 110(Pt 1): 99–103. [DOI] [PubMed] [Google Scholar]
- 36.Sehba FA, Chereshnev I, Maayani S, et al. Nitric oxide synthase in acute alteration of nitric oxide levels after subarachnoid hemorrhage. Neurosurgery 2004; 55: 671–677. [DOI] [PubMed] [Google Scholar]
- 37.Sehba FA, Schwartz AY, Chereshnev I, et al. Acute decrease in cerebral nitric oxide levels after subarachnoid hemorrhage. J Cereb Blood Flow Metab 2000; 20: 604–611. [DOI] [PubMed] [Google Scholar]
- 38.Sun BL, Zhang SM, Xia ZL, et al. L-arginine improves cerebral blood perfusion and vasomotion of microvessels following subarachnoid hemorrhage in rats. Clin Hemorheol Microcirc 2003; 29: 391–400. [PubMed] [Google Scholar]
- 39.Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke 2010; 41: e519–e536. [DOI] [PubMed] [Google Scholar]
- 40.Cahill J, Calvert JW, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab 2006; 26: 1341–1353. [DOI] [PubMed] [Google Scholar]
- 41.Caner B, Hou J, Altay O, et al. Transition of research focus from vasospasm to early brain injury after subarachnoid hemorrhage. J Neurochem 2012; 123(Suppl 2): 12–21. [DOI] [PubMed] [Google Scholar]
- 42.Sehba FA, Hou J, Pluta RM, et al. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol 2012; 97: 14–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sehba FA, Pluta RM, Zhang JH. Metamorphosis of subarachnoid hemorrhage research: from delayed vasospasm to early brain injury. Mol Neurobiol 2011; 43: 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foreman B. The pathophysiology of Delayed Cerebral Ischemia. J Clin Neurophysiol 2016; 33: 174–182. [DOI] [PubMed] [Google Scholar]
- 45.Hop JW, Rinkel GJ, Algra A, et al. Initial loss of consciousness and risk of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Stroke 1999; 30: 2268–2271. [DOI] [PubMed] [Google Scholar]
- 46.Juvela S, Siironen J, Varis J, et al. Risk factors for ischemic lesions following aneurysmal subarachnoid hemorrhage. J Neurosurg 2005; 102: 194–201. [DOI] [PubMed] [Google Scholar]
- 47.Siesjo BK. Pathophysiology and treatment of focal cerebral ischemia. Part II: mechanisms of damage and treatment. J Neurosurg 1992; 77: 337–354. [DOI] [PubMed] [Google Scholar]
- 48.Springer MV, Schmidt JM, Wartenberg KE, et al. Predictors of global cognitive impairment 1 year after subarachnoid hemorrhage. Neurosurgery 2009; 65: 1043–1050. [DOI] [PubMed] [Google Scholar]
- 49.Macdonald RL, Higashida RT, Keller E, et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol 2011; 10: 618–625. [DOI] [PubMed] [Google Scholar]
- 50.Macdonald RL, Kassell NF, Mayer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke 2008; 39: 3015–3021. [DOI] [PubMed] [Google Scholar]
- 51.Macdonald RL, Pluta RM, Zhang JH. Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat Clin Pract Neurol 2007; 3: 256–263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material for Long-term impairment of neurovascular coupling following experimental subarachnoid hemorrhage by Matilde Balbi, Max Jativa Vega, Athanasios Lourbopoulos, Nicole A Terpolilli and Nikolaus Plesnila in Journal of Cerebral Blood Flow & Metabolism