

Abstract
The transmembrane (TM) anchors of cell surface proteins had been one of the “blind spots” in structural biology because they are generally very hydrophobic and sometimes dynamics, and are thus difficult targets for structural characterization. A plethora of examples showed that these membrane anchors are not merely anchors but can multimerize specifically to activate signaling receptors on the cell surface or to stabilize the envelope proteins in viruses. Through a series of studies of the TM domains of immune receptors and viral membrane proteins, we have established a robust protocol for determining atomic resolution structures of TM oligomers by nuclear magnetic resonance (NMR) in bicelles that closely mimic a lipid bilayer. Here, we provide the details of the protocol consisting of five major sections: 1) general expression, purification, and bicelle reconstitution of hydrophobic TM and membrane-proximal domains; 2) determination of the oligomeric state of TM domains in bicelles; 3) detection of inter-molecular contacts; 4) structure determination; and 5) characterization of the protein TM partition. This protocol is broadly applicable to filling the structural gaps in membrane for many Type I/II membrane proteins.
Introduction
There have been increasing experimental data indicating that what had been commonly thought of as the transmembrane (TM) anchors of many signaling receptors actually play critical roles in receptor signaling, and the diversity of mechanism with which the TM regions can promote signaling is beyond the traditional views in receptor biology. For example, matured assembly of the TCR/CD3 complex is primarily mediated by interactions among the TM domains1,2. Different modes of TM helix dimerization appear to contribute to the ‘on’ and ‘off’ states of the epidermal growth factor (EGF) receptor3,4. TM domain trimerization is required for the Fas death receptor, a member of the tumor necrosis factor (TNF) receptor family, to signal and this appears to apply to other members of the TNF receptor superfamily5. These examples are only the tip of the iceberg, as there remain a vast number of Type I and II TM receptors whose membrane regions are unknown. Revealing the structures of these membrane regions is required to gain thorough understanding of receptor activation for many of the immune co-stimulators currently being targeted for cancer immunotherapy. The membrane regions of cell surface proteins, however, have been difficult targets for crystallography, because they are generally very hydrophobic and often dynamic; they are also too small for cryo-electron microscopy (cryo-EM) at the moment.
As a versatile spectroscopic tool capable of determining atomic resolution structures, solution NMR has often been the go-to method for tackling small TM domains of Type I and II membrane proteins. The application of solution NMR to TM helix oligomer was demonstrated more than two decades ago on the TM domain of glycophorin A6. In that study, the NMR structure, solved in dodecyl-phosphocholine micelles, revealed the structural role of the GXXXG signature sequence in mediating TM helix dimer formation, and recently, this NMR structure has been independently validated by a crystal structure of the same TM domain determined in the lipidic cubic phase (LCP)7. NMR has since been widely applied to investigating the structures of small TM domains in detergent micelles and small bicelles1,3,5,8–14. Despite the powerful utility, a general and robust protocol for determining TM structures by NMR is still lacking. We have integrated the most effective and practical methods from a series of our recent applications to generate a protocol for general structural characterization of the small membrane-embedded and membrane-proximal (MP) regions of TM proteins in bicelles that are sufficiently large to mimic a lipid bilayer.
Overview of the Protocol
This protocol addresses the major technical challenges associated with structural analysis of the small TM/MP complexes, including the production of highly hydrophobic protein fragments with suitable isotope labeling for NMR, protein reconstitution in appropriate membrane-mimetic media, determination of the protein oligomerization state, detection of inter-chain contacts for structure determination, as well as characterization of the protein partition in lipid bilayer. A conceptual overview of the protocol is illustrated in Fig. 1.
Figure 1. Protocol overview.
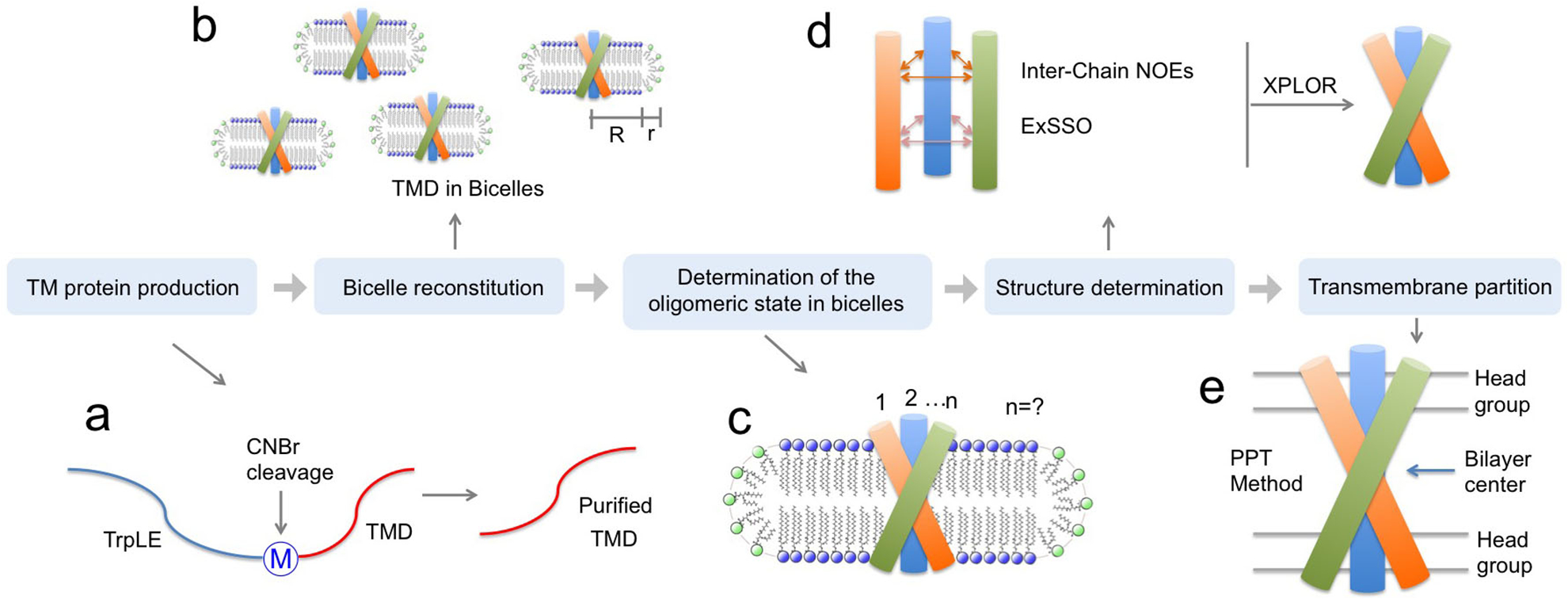
(a) TM protein production, (b) bicelle reconstitution, (c) oligomeric state determination, (d) structure determination, and (e) TM partition determination.
Specifically, we show that by using targeted expression into inclusion bodies in bacteria along with highly optimized affinity and high-pressure reverse-phase lipid chromatography, hydrophobic peptides with a range of sizes (10 – 200 residues) and hydrophobicity (GRAVY score 0.5 – 2)15,16 can be produced. The peptides can be uniformly isotopically labeled for NMR measurements. In the protocol, we reconstitute the peptides into bicelles that are sufficiently large to mimic the membrane, because the small TM/MP domains often require a lipid bilayer environment to form stable oligomeric complexes. We found that when using bicelles with molar ratio of lipid/detergent (q) greater than or equal to 0.5 (e.g., 0.5 ≤ q ≤ 0.6; the bilayer region size is between 44 and 50 Å), with which the bicelles are very close to lipid discs17–20, the TM proteins still yield high quality NMR spectra feasible for structure determination. A challenge associated with the use of bicelles is the accurate determination of the oligomeric state of the small TM domains, as the dynamic self-assembly of bicelles is incompatible with standard methods for measuring molecular mass of protein complexes such as size-exclusion chromatography and equilibrium sedimentation. We note that the use of NMR relaxation parameters to infer molecular mass is potentially misleading, because membrane proteins usually exhibit very heterogeneous dynamics. Hence, our approach is to use the intermolecular paramagnetic resonance enhancement (PRE) analysis to confirm multimeric assembly and the oligomer label (OG-label) method to determine the oligomeric state of the TM complex. NMR-based structure determination of small membrane proteins has been rather controversial, as different sources of structural information such as nuclear Overhauser enhancement (NOE), PRE, or residual dipolar coupling (RDC) have been used to solve structures. This protocol focuses almost entirely on the use of the NOE for structure determination, because the NOE remains the most direct NMR probe of internuclear distances in a molecule; it makes full use of isotope labeling strategy and strand-selective experiment for detecting inter-chain NOEs that define the oligomeric structure. Finally, probably the most unique feature of the protocol is the incorporation of the paramagnetic probe titration (PPT) method that allows for accurate characterization of the membrane partition of TM proteins. This method is based on the notion that when bicelles are sufficiently wide (q ≥ 0.5), so that the protein resides essentially in the bilayer region of the bicelles, simple titration of either water-soluble or lipophilic paramagnetic probes such as Gd-DOTA or nitroxide-labeled fatty acids (e.g. 16-DSA), respectively, can be used to accurately determine residue-specific depth immersion along the bicelle normal18,21–23. When the structure of a TM oligomer is known, the PPT data can thus be used to determine the membrane partition of the protein. The above techniques constitute a comprehensive analysis of the structure, oligomerization state, and membrane partition of TM and MP regions of cell surface proteins.
Advantages
In addition to solution NMR, other techniques have been used to determine structures of TM oligomers. For example, there have been several cases in which LCP crystallization generated crystals of small TM fragments that were good enough for high resolution structure determination7,24–26. A more recent crystallographic study also managed to capture the entire structure of the viral envelope protein (including the membrane region) from herpes simplex virus27. Solid-state NMR is another attractive alternative, as it allows structural study in a completely lipid bilayer environment. Achieving atomic resolution structures by solid-state NMR relies on proteoliposome samples that yield NMR spectra with high resolution, and this usually require that the TM fragments form microcrystals28,29. Small TM domains are obviously too small for current cryo-EM application; they need to be analyzed by cryo-EM as a part of the much larger full-length proteins, which would be the best approach, if feasible, to study the TM structures. The success of this type of cryo-EM application, however, depends on whether the large extra- or intra- cellular domains are rigidly connected to the TM domain and such property is case dependent.
The main strength of the reported protocol is that it provides a practical, robust, and general solution for obtaining TM structures in near lipid bilayer environment. First, this protocol provides a rather complete solution to determining TM oligomer structures in bicelles that are sufficiently large to mimic a lipid bilayer, including the robust OG-label method for determining the protein oligomeric state in bicelles. Second, the use of ideal bicelles enables the implementation of the PPT method to determine membrane partition of the TM domains. This is a distinct advantage over previous NMR methods, as the new protocol not only provides the TM structure, but also information of how the TM protein resides in membrane. Third, the protocol is very rigorous from sample preparation to structure determination; it provides practical strategies for directly detecting inter-molecular contacts that are critical for structure determination of TM oligomers.
Limitations
The key limitation of the protocol is due to the fundamental molecular mass limitation of the solution NMR technique, that the TM domains cannot be studied in the context of the full-length transmembrane proteins. Empirically, we found that TM/MP oligomers with monomeric chain ~10 kD, reconstituted in bicelles with q = 0.5, is close to the size limitation within which NMR-based structure determination is feasible, but large deviations are possible depending on the protein oligomeric-state and dynamics. Further, bicelle solutions still contain free detergent, e.g., the concentration of free DHPC in DMPC/DHPC bicelles is ~ 5 mM30 and this amount of free detergent, while not affecting the TM domain, could potentially generate structural artifacts in the MP regions.
Future Applications
Our protocol should be useful for studying TM domain oligomerization of many immunoreceptors and receptor tyrosine kinases, because for many of these Type I/II membrane proteins the TM domain plays essential roles in receptor assembly and possibly in receptor clustering as well. Detailed structural information of TM domain oligomerization in membrane-like environment would be valuable clues for elucidating how the connected intracellular signaling domains are clustered to activate the downstream signaling. The protocol is equally applicable to the unknown membrane regions of the many viral membrane fusion proteins. Although our earlier NMR applications have focused primarily on the TM domains, the current protocol has been proven effective also in revealing the structures of MP domains, as demonstrated recently for the membrane-proximal external region (MPER) of the HIV-1 gp41 fusion protein23. In addition to the Type I/II membrane proteins, the protocol is in principle applicable to TM domain that spans the membrane multiple times, as long as the size of the overall protein complex in bicelles is within the limitation of solution NMR. For example, bacteria have developed two-component systems (receptor histidine kinases) for sensing all kinds of environmental factors such as cellular cytokines, osmotic pressure, pH, membrane curvature, etc31–34. These receptors have a small TM domain usually consisting of two TM helices, that is believed to physically transmit signals from extracellular environmental interaction to activate intracellular kinase. We believe our protocol can be used effectively on these interesting systems as well.
Experimental Design
TM protein production.
Transmembrane domains (TMDs) of cell surface proteins are very hydrophobic, which makes protein expression and purification difficult. We express the small hydrophobic TMDs in E. coli as C-terminal fusion to the TrpLE sequence, which is a fragmented protein that drives inclusion body formation upon synthesis. As such, the hydrophobic TMDs, usually toxic to the cells per se, are sequestered in the inclusion bodies and thus can be expressed to high quantities. This is done using a plasmid named pMM-LR6, originally from Stephen Blacklow35,36. Based on the pMM-LR6, a His9-tag is added to the N-terminus of TrpLE to facilitate purification. A methionine is inserted between the TrpLE and the TMD to enable cleavage at this position with Cyanogen Bromide (CNBr) so that the two fragments can be separated (Box 1). Hence, the TM sequence cannot contain any methionine. The purification of the TMD can be achieved with 3 major steps, all under denaturing conditions: 1) purification of the TrpLE-TMD fusion from inclusion bodies by Ni-NTA affinity, 2) CNBr cleavage to separate TrpLE and TMD, 3) purification of the TMD by reverse phase high-pressure liquid phase chromatography (HPLC).
Box 1. The pMM-LR6 vector for TM domain expression.
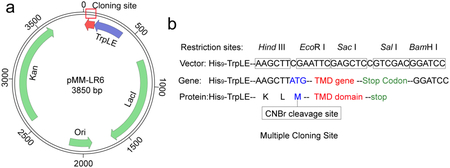
(a) Schematic diagram of the pMM-LR6 vector. Kan: Kanamycin resistance gene; Ori: The origin of replication; LacI: Lactose operon repressor; TrpLE: Fragment gene from Anthranilate Synthase, which cannot be folded well in cell and will drive inclusion bodies formation.
(b) Multiple cloning sites for TM domain gene insertion, including the methionine code (ATG) between TrpLE and TM sequence (for CNBr cleavage) and the stop codons (TAA, TGA, TAG) at the end of the TM sequence. The TrpLE sequence is:
KAIFVLKGSLDRDLDSRIELELRTDHKELSEHLLLVDLARNDLARIATPGSRYVADLTKVDRYSYVLHLVSRVVGELRHDLDALHAYRAALNLGTLSGAPKVRAKL
For certain NMR experiments, very high level of deuteration (e.g., > 98%) is needed. We note that using the above expression vector in BL21(DE3) E. coli cell line, this level of deuteration is achievable by using 99% D2O and deuterated (98% 2H) glucose (details in Part 1 procedures).
For every target TMD, we suggest performing a sequence analysis to check for methionines and cysteines, and to evaluate the overall hydrophobicity of the TMD. Since methionine is the CNBr cleavage site, additional methionines, if any, must be mutated to an amino acid of similar hydropathicity. Cysteines might form non-native disulfides during purification. If they are not conserved, we suggest mutating them to serine or alanine, which would simplify the purification and reconstitution procedures. The overall hydrophobicity of the TMD sequence can be evaluated using the program ProtParam (https://web.expasy.org/protparam/), which calculates the grand average of hydropathicity (GRAVY) score. The GRAVY score for a typical TMD is 0.5 – 1.5; less than 0.5 means hydrophilic, and greater than 1.5 means extremely hydrophobic. If a TMD has very high GRAVY score (> 1.5) and its purification failed, one can consider the option of mutating non-conserved hydrophobic residues to decrease the GRAVY score.
Reconstitution in bicelles.
The target TMDs are reconstituted in bicelles that are sufficiently large to mimic the bilayer environment of the membrane. When the lipid/detergent ratio (q) is greater than 0.5, DMPC/DHPC bicelles are known to become disc-like in which the lipids and detergents are largely segregated17,18,37. When a protein is reconstituted in such bicelles, typically with 0.5 ≤ q ≤ 0.6, it is essentially in a lipid bilayer environment while still amenable for high resolution solution NMR spectroscopy. This approach has been successful in for several important systems, including the TMDs of HIV-1 Env12,23, Fas5, and the intact p7 channel of HCV22. Although DMPC has been the most common lipid for bicelles, other lipids such as POPC, POPG, and POPE are equally compatible with DHPC in forming bicelles. A general protocol for incorporating TMDs into bicelles is to first denature and completely solubilize the protein in the presences of lipid and detergent and then slowly remove the denaturant to allow self-assembly of bicelles around the protein. Specifically, the purified and lyophilized TMD is dissolved in hexafluoro-isopropanal (HFIP) with suitable amount of lipid, followed by drying of the solution under nitrogen stream to achieve thin films. The thin films are then dissolved in 8 M urea solution containing calculated amount of detergent. Reconstitution begins as the denaturant is removed by dialysis. Some detergents are lost during dialysis and therefore need to be added back to the sample to maintain the desired bicelle q. The q value of the sample can be accurately measured using 1D 1H NMR.
Determination of oligomeric state in bicelles.
Characterizing the oligomeric state for small TMDs remains difficult, especially in dynamically assembled systems such as lipid/detergent bicelles. The two-component bicelles makes it extremely difficult to perform experiments such as SEC-MALS and equilibrium sedimentation. In addition, direct chemical crosslinking of membrane proteins using Lomant’s reagents is inefficient and can generate non-specific ladder patterns at higher crosslinker concentrations, probably because the proteins are mostly buried in bicelles and/or their primary amine groups are usually not well positioned for crosslinking. We have thus developed a solution to this problem by first labeling, non-covalently, each protomer in an oligomer with a small soluble protein and then crosslinking the soluble protein to determine the oligomeric state22. In this method, named oligomer labeling (OG-label), the soluble crosslinkable protein (SCP) used is a small protein named GB1 (M.W. = 8.4 kDa). Its N-terminus is linked to a TriNTA molecule via a crosslinker to form the TriNTA-GB1 conjugate. The target TM protein to be examined has a His6-tag. The TriNTA molecule has extremely high binding affinity to His6-tag sequence (20 ± 10 nM)38, which can strongly attach GB1 to the individual protomers of the TMD oligomer in bicelles. Then, the concentration of stoichiometric amount of GB1 to the membrane protein oligomer allows for more efficient crosslinking than the free GB1 in solution. The crosslinked GB1 can be released from the oligomer by addition of EDTA or imidazole and analyzed by SDS-PAGE to determine the oligomerization number.
Inter-protomer structural restraints and structural calculation.
Structure determination of homo-oligomers is challenging because NOEs between structurally equivalent subunits having the same chemical shifts are needed as inter-protomer distance restraints. To solve this problem, we use mixed samples in which half of the monomers are (15N, 2H)-labeled and the other half 13C-labeled, which allows us to detect exclusively NOEs between the 15N-attached protons of one subunit and the 13C-attached protons of the neighboring subunits using the JCH-modulated NOE experiment5,23. These restraints are then used to build a model of the oligomeric TMD, and for this, we need a fast and efficient way to assemble the symmetric TM oligomer and calculate all the conformations that fit the confirmed NOEs. We developed a new program named ExSSO to achieve this goal39. This program performs an exhaustive search to find all oligomeric assemblies that satisfy the inter-protomer NOE restraints. If a unique oligomeric packing solution has converged, a representative structure is used as the starting model for further structural refinement in standard program such as XPLOR-NIH40. This refinement process involves 1) identifying self-consistent backbone-sidechain and sidechain-sidechain NOE restraints in conventional 15N- and 13C-edited NOESYs, and 2) updating the structure with the new NOE restraints. The above two steps are performed iteratively until the desired RMSD of the structural ensemble is reached.
Transmembrane partition.
An important aspect of a TM or MP structure is its interaction with the membrane, which can provide clues to its structural and functional roles. It is well known that TMDs have the ability to modulate the thickness of a lipid bilayer22,41,42, while MP regions can cause membrane curvature or deformation to facilitate their function43,44. Therefore, accurate determination of the protein membrane partition is very important. We have developed a method named paramagnetic probe titration (PPT)18,21, which provides PRE data that globally reflect the protein partition in the bicelles. To be applicable, the method assumes that the protein structure is known and requires the protein to be reconstituted in wide or “ideal” bicelles (q ≥ 0.5). This requirement, in addition to providing a near-membrane environment for the protein, greatly simplifies the data analysis by ensuring that the measurable PRE is proportional only to the residue position along the bicelle normal. As such, the sample is titrated with either a water-soluble or lipophilic paramagnetic agent (e.g. Gd-DOTA or 16-DSA, respectively) while recording a 2D 1H-15N TROSY-HSQC spectrum at each titration point. The analysis of the peak intensity decay vs. the paramagnetic probe concentration derives residue-specific PRE amplitudes (PREamp), which are reporters of residue-specific membrane immersion depths. By exploiting the knowledge of the protein structure, which provides the relative position of each residue along the protein symmetry axis, parallel to the bilayer normal, it is then possible to determine the position of the protein relative to the bilayer center that yield the best fit to the experimental PREamp, and such placement represents the membrane partition of the protein. This partition analysis can address which protein regions are outside the membrane, measure variations in the membrane thickness around the protein, and, in cases of complex TM proteins, can provide information on the relative orientation of different protein segments.
Part 1. Expression, purification and reconstitution of TM domains
Materials
BIOLOGICAL MATERIALS
BL21 (DE3) competent cells (New England biolabs, cat. no. C2527). Store them at −80°C.
Expression plasmid (pMM-LR6 vector) coding for the target protein fused to the C-terminus of TrpLE tag. The plasmid carries antibiotic resistance to Kanamycin. Store it at −20°C. (Box 1)
REAGENTS
1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP, 99.5%, Oakwood Products, Inc. cat. no. 003409)
1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC, Avanti Polar Lipids, Inc., cat. no. 850305)
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC, Avanti Polar Lipids, Inc., cat. no. 850345)
1,2-dimyristoyl-d54-sn-glycero-3-phosphocholine (deuterated DMPC, d54, 99%, Avanti Polar Lipids, Inc., cat. no. 860345)
1,2-dihexanoyl-d22-sn-glycero-3-phosphocholine (deuterated DHPC, d22, 99%, Avanti Polar Lipids, Inc., cat. no. 790427)
2-(N-Morpholino)ethanesulfonic acid hydrate (MES, Oakwood Chemical, cat. no. M05729)
2,2,2-Trifluoroethanol (TFE, 99%, STREM Chemicals, Inc., cat. no. 09–7310)
4-(1,1,3,3-Tetramethylbutyl)phenyl-polyethylene glycol (TritonX-100, Sigma-Aldrich, cat. no. X100)
Acetonitrile (EMD Millipore, cat. no. EM-AX0151–1)
Ammonium chloride (NH4Cl, Sigma-Aldrich, cat. no. A9434)
Ammonium chloride (15N, 99%, Cambridge Isotope Laboratories, Inc. cat. no. NLM-467)
Calcium chloride (CaCl2, Sigma-Aldrich, cat. no. C1016)
Cyanogen bromide (CNBr, Sigma-Aldrich, cat. no. 57654055) CAUTION. Fatal if swallowed, in contact with skin or if inhaled. Causes severe skin burns and eye damage. Contact with acids liberates very toxic gas. A cyanide antidote kit MUST be rapidly available and ingredients replaced every 1 to 2 years to ensure freshness.
D-Glucose (Sigma-Aldrich, cat. no. G8270)
D-Glucose (U-13C6, 99%, Cambridge Isotope Laboratories, Inc. cat. no. CLM-1396)
D-Glucose (1,2,3,4,5,6,6-D7, 98%, Cambridge Isotope Laboratories, Inc. cat. no. DLM-2062)
Deuterium oxide (D2O, 99.96%, Cambridge Isotope Laboratories, Inc., cat. no. DLM-6-PK)
Dichloromethane (99.9%, EMD Millipore Corporation, cat. no. 75-09-2)
Disodium hydrogen phosphate (Na2HPO4, Sigma-Aldrich, cat. no. S3264)
Formic acid (FA, 90%, VWR International, cat. no. JT0129–1) CAUTION. Combustible liquid. Cause severe skin burns and eye damage. Toxic if inhaled.
Guanidine hydrocholoride (VWR International, cat. no. 71003)
HisPur Ni-NTA resin (Thermo Fisher, cat. no. 88223).
Isopropyl alcohol (lPA, EMD Millipore, cat. no. EM-PX1838P-1)
Isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma-Aldrich, cat. no. 329815691)
Kanamycin sulfate (K, Sigma-Aldrich, cat. no. 60615)
Luria-Bertani (LB) agar, granulated (RPI Research Products International, cat. no. L24033–500)
Luria-Bertani broth (LB, VWR life science, cat. no. J106)
Magnesium sulfate (MgSO4, Sigma-Aldrich, cat. no. M7506)
Sodium chloride (NaCl, Sigma-Aldrich, cat. no. S7653)
Sodium phosphate monobasic (NaH2PO4, Sigma-Aldrich, cat. no. S3139)
SOC medium (New England Biolabs, cat. no. B9020S)
Trifluoroacitic acid (TFA, EMD Millipore, cat. no. EM-TX1275–3) CAUTION. Cause severe skin burns and eye damage. Harmful if inhaled.
Tris base (Tris, Calbiochem, cat. no. 648310)
Urea (Thermo Fisher Scientific, cat. no. 15505050)
EQUIPMENT
All the standard equipment necessary for handling of recombinant proteins
HPLC instrument (e.g. Bio-Rad Duo Flow system) consisting of a degasser, sampler, pumps, and detectors (to measure conductivity and UV absorbance at 214 and 280)
Zorbax SB-C3 column (Agilent Technologies, cat. no. 880995–209)
NMR spectrometer and related NMR data-processing software (e.g., TOPSPIN, VNMR, NMRPIPE, etc.)
REAGENT SETUP
LB medium (K 50 μg/mL).
Dissolve 25 g of LB broth in 1 L of dH2O and autoclave the solution. Once the solution has cooled down to RT, add 50 mg of Kanamycin (50 μg/mL). Operate close to the flame to avoid contamination of the medium. Store the solution at RT.
LB agar plates (K 50 μg/mL).
Approximately 10 plates are obtained per 200 mL of prepared solution. Dissolve LB agar to the desired volume of dH2O so that its concentration is 37 g/L. Autoclave the solution and let it cool down under gentle shaking to prevent solidification of the agar. When the solution temperature reaches approximately 50°C, add K to the final concentration of 50 μg/mL and mix well. Operate close to the flame to avoid buffer contamination. Aliquot the medium and pour it into sterile plates. After gel coagulation, store the plates at 4°C.
CaCl2, 1 M.
Dissolve 14.7 g of CaCl2 in 100 mL of dH2O. Store the solution at RT. CRITICAL. For perdeuterated samples, dissolve in D2O, 99.96% instead of dH2O. Store at RT.
MgSO4, 1 M.
Dissolve 24 g of MgSO4 in 200 mL of dH2O. Store the solution at RT. CRITICAL. For perdeuterated samples, dissolve in D2O, 99.96% instead of dH2O. Store at RT.
M9 medium (1 L).
Dissolve 6 g of Na2HPO4, 3 g of KH2PO4, 0.5 g of NaCl and 1 g of NH4Cl (15N, 99%, for 15N-labeled samples) in 1 L of dH2O (D2O, 99.96%, for perdeuterated samples). Autoclave the solution, let it cool down to RT, then add 4 of g Glucose (U-13C6, 99%, for 13C-labeled samples; 1,2,3,4,5,6,6-D7, 98%, for perdeuterated samples), 2 mL of 1 M MgSO4 (in 99.96% D2O for perdeuterated samples), 100 μL of 1 M CaCl2 (in 99.96% D2O for perdeuterated samples) and 50 mg of K (50 μg/mL). Mix well until complete dissolution of the chemicals. Store the medium at RT and use it within a couple of days. CRITICAL. When using D2O, 99.96% (perdeuterated samples), do not autoclave the medium to prevent moisture contamination, but only autoclave the empty flask before preparing the solution.
IPTG, 1 M.
Dissolve 2.4 g of IPTG in 10 mL of dH2O. Split the solution in 1 mL aliquots and store them at −20°C. CRITICAL. For perdeuterated samples, dissolve in D2O, 99.96% instead of dH2O.
Lysis buffer (50 mM Tris, pH 8.0, 200 mM NaCl).
Dissolve 6.1 g of Tris base and 11.8 g of NaCl in 1 L of dH2O. Adjust the pH to 8.0. Store the solution at RT.
Guanidine buffer (6M Guanidine-HCl, 50mM Tris, pH 8.0, 200 mM NaCl, 1% vol/vol TritonX-100).
Dissolve 573 g of Guanidine HCl, 6 g of Tris base and 11.8 g of NaCl in 1 L of dH2O. Adjust the pH to 8.0. Add 10 mL of Triton X-100. Store the solution at RT.
Urea, 8 M.
Dissolve 480.5 g of Urea in dH2O to reach the final volume of 1 L. The solution can be heated to aid the Urea dissolution. Store the solution at RT.
HPLC buffer A (5% (vol/vol) IPA, 95% (vol/vol) H2O, 0.1% (vol/vol) TFA).
For 1 L solution, mix 50 mL of IPA, 950 mL of dH2O and 1 mL of TFA. Before use, degas and filter the solution with a 0.2 μM membrane. Store the solution at RT.
HPLC buffer B (75% (vol/vol) IPA, 25% (vol/vol) Acetonitrile, 0.1% (vol/vol) TFA).
For 1 L solution, mix 750 mL of IPA, 250 mL of Acetonitrile and 1 mL of TFA. Before use, degas and filter the solution with a 0.2 μM membrane. Store the solution at RT.
HPLC buffer C (95% (vol/vol) IPA, 5% (vol/vol) H2O, 0.1% (vol/vol) TFA).
For 1 L solution, mix 950 mL of IPA, 50 mL of dH2O and 1 mL of TFA. Before use, degas and filter the solution with a 0.2 μM membrane. Store the solution at RT.
NMR buffer.
The NMR buffer can be optimized for the specific sample under study. Typically, it consists of ~20–50 mM Phosphate or MES buffer at pH ~6.7–6.8. The DMPC/DHPC bicelle system is stable within 6.0 ≤ pH ≤ 7.5.
DHPC (100 mg/mL).
Dissolve 100 mg of DHPC (protonated or deuterated) in 1 mL of NMR buffer. Split the solution in 50 μL aliquots and store them at −20°C.
EQUIPMENT SETUP
“Cleaning” HPLC method.
Isocratic flow: HPLC Buffer A (100%). Flow rate: 2 mL/min. Total volume: 30 mL
Linear gradient: from HPLC Buffer A (100%)/HPLC Buffer B (0%) to HPLC Buffer A (0%)/HPLC Buffer B (100%). Flow rate: 2 mL/min. Total volume: 50 mL
Isocratic flow: HPLC Buffer B (100%). Flow rate: 2 mL/min. Total volume: 30 mL
“Sample purification” HPLC method.
Sample injection
Isocratic flow: HPLC Buffer A (100%). Flow rate: 2 mL/min. Total volume: 15 mL
Linear gradient: from HPLC Buffer A (100%)/HPLC Buffer B (0%) to HPLC Buffer A (70%)/HPLC Buffer B (30%). Flow rate: 2 mL/min. Total volume: 5 mL
Linear gradient: from HPLC Buffer A (70%)/HPLC Buffer B (30%) to HPLC Buffer A (0%)/HPLC Buffer B (100%). Flow rate: 2 mL/min. Total volume: 150 mL
Isocratic flow: HPLC Buffer B (100%). Flow rate: 2 mL/min. Total volume: 20 mL
Procedures
Protein expression
The following protocol (Steps 1–21) describes the procedure for expression of perdeuterated proteins. For protonated samples, Steps 10–13 can be omitted and dH2O should be used instead of D2O in Steps 14–15.
-
1
Take one vial (50 μL) of BL21(DE3) competent E. coli cells and let them thaw on ice.
-
2
Add 1 μL of plasmid coding for the target protein to the competent cells and mix well. Incubate the mixture on ice for 30 minutes.
-
3
Move the mixture into a warm bath or incubator at 42°C. Incubate for 50 seconds.
-
4
Transfer the mixture into ice and incubate for 2 minutes.
-
5
Add 200 μL of SOC medium to the cells and move them in an incubator shaker at 37°C for 30 min. Operate close to the flame to avoid contamination.
-
6
Spread the cells on a LB agar plate (K 50 μg/mL) using a sterile glass pipette or plating beads. Operate close to the flame to avoid contamination.
-
7
Incubate the plate at 37°C for 16 hours. PAUSE POINT. This step is typically carried overnight. The plate can then be stored at 4°C, but the E.coli colonies should be grown within 1–2 days after transformation.
-
8
Pick a single colony from the plate and inoculate it into 5 mL of LB medium (K 50 μg/mL). Move the tube into an incubator shaker at 37°C and shake at 220 rpm for 8 hours.
-
9
Spin down the cells centrifuging at 3,000 rpm for 5 minutes.
-
10
Dump the supernatant, inoculate the cells into 100 mL of M9 media (H2O) and incubate the culture at 37°C shaking at 220 rpm until the OD600 is close to 0.4 (about 2 hours).
-
11
Spin down the cells centrifuging at 3,000 rpm for 5 minutes.
-
12
Dump the supernatant, inoculate the cells into 10 mL of M9 media (50% H2O, 50% D2O (vol/vol)) and incubate the culture at 37°C shaking at 220 rpm until the OD600 is close to 1.0 (about 2 hours).
-
13
Centrifuge 2 mL of culture at 3,000 rpm for 5 minutes to spin down cells.
-
14
Dump the supernatant, inoculate the cells into 100 mL of M9 media (D2O) and incubate the culture at 37°C shaking at 220 rpm until the OD600 is close to 1.0 (about 8 hours).
-
15
Inoculate the entire culture into 1 L of M9 media (D2O). Incubate the culture at 37°C shaking at 220 rpm until the OD600 reaches to 0.6~0.8 (about 6 hours). Collect 100 μL of culture, measure its OD600 and store it at 4°C, to be later used as control for evaluating the protein expression level.
-
16
Change the incubator shaker temperature to the desired temperature for protein expression. Wait about 30 minutes until the culture reaches the desired temperature.
-
17
Induce protein expression by adding the required amount of IPTG from the 1 M stock solution.
-
18
Grow the culture shaking at 220 rpm for about 16 hours. Collect 100 μL of culture, measure its OD600 and store it at 4°C, to be used later to evaluate the protein expression level.
-
19
Collect the cells centrifuging at 4,000 rpm for 30 minutes at 4°C.
-
20
Dump the supernatant and suspend the cell pellet in 50 mL of Lysis buffer at RT. PAUSE POINT. The cell suspension can be stored at −80°C.
-
21
Evaluate the protein expression level by SDS-PAGE analysis of the samples collected at Steps 15 (before induction) and 18 (after expression). For a better comparison, use the measured OD600 to determine the amount of samples to be used for the analysis so that all samples contain a comparable number of cells in the gel. ? TROUBLESHOOTING
Ni-NTA purification
-
22
Disrupt the cell suspension by sonicating for 10 minutes at intervals of 1 second; apply pulses of 1 second at 40% of the maximum power. Keep the sample in ice during the procedure.
-
23
Centrifuge the suspension at 18,000 rpm for 20 minutes at 4°C to spin down the inclusion bodies.
-
24
Take 10 μL of supernatant and precipitate for SDS-PAGE analysis. Make sure that the target protein is localized in the latter.
-
25
Dump the supernatant and dissolve the precipitate in 50 mL of Guanidine buffer using a 50 mL glass tissue grinder.
-
26
Centrifuge the solution at 18,000 rpm for 30 minutes at 4°C.
-
27
Retain the supernatant and add 4 mL of HisPur Ni-NTA resin. Mix them well by continuously gently stirring the mixture on a rotator for ~2–8 hours at RT. CRITICAL. Before use, wash several times the HisPur Ni-NTA resin with dH2O to completely remove the ethanol used for its storage. PAUSE POINT. The mixture can be left stirring overnight.
-
28
Transfer the mixture into a glass chromatography column for gravity flow purification. The target protein, bound to the Ni-NTA resin, remains trapped in the column. Discard the flow-through.
-
29
Wash the column twice with 50 mL of 8 M Urea. Discard the flow-through.
-
30
Wash the column twice with 20 mL of dH2O. Discard the flow-through. Do not let the resin dry.
-
31
To release the target protein from the Ni-NTA resin, add 4 mL of FA, 90%. Let the acid react at RT for about 2–5 minutes. Collect the eluate in a 50 mL Falcon tube.
-
32
Repeat Step 31 two more times, eluting the protein in a total volume of 12 mL of FA, 90%.
Cyanogen bromide cleavage
-
33
Before use, let the CNBr warm up to RT (about 30 minutes). CRITICAL. CNBr is very toxic, so it should be always manipulated under the hood wearing the appropriate PPE. All the equipment and waste contaminated by CNBr should be collected in a dedicated hazardous waste container for proper disposal.
-
34
Add ~1.5 g of CNBr to the 12 mL protein solution in FA, 90%. Use a vortex mixer to completely dissolve the CNBr.
-
35
Cover the tube with aluminum foil to shield it from light and let the reaction occur under gentle nitrogen gas stream for 1 hour. CRITICAL. Do not increase significantly the reaction time, as this may increase the occurrence of side reactions, e.g. formylation. ? TROUBLESHOOTING
-
36
After the reaction is completed, transfer the solution into a 3.0–12.0 mL dialysis cassette (3,500 MWCO) and dialyze it versus 4 L of dH2O for 40 minutes.
-
37
Dialyze the sample a second time versus 4 L of dH2O for 40 minutes.
-
38
Transfer the solution into a 50 mL Falcon tube and prepare the sample for lyophilization by making a few holes in the tube cap.
-
39
Freeze the solution in liquid nitrogen (−196°C) for 10–15 minutes, making sure the solution is well-frozen. Transfer the tube in the lyophilizer (−80°C, ~10–20 mBar) and lyophilize the protein (about 1 day). PAUSE POINT. After lyophilization, the protein powder can be stored at −20°C.
Reverse phase HPLC
-
40
Switch on the HPLC instrument and annexed detectors. Connect a Zorbax SB-C3 column to the instrument.
-
41
Every time a new buffer is used, wash the system and related pump for 1–2 minutes (flow rate: 10 mL/min). CRITICAL. Make sure that no bubbles are present in the system.
-
42Wash and equilibrate the column with the following buffers, adjusting the flow rate depending on each buffer viscosity to maintain the column pressure of ~1,500–2,000 psi.
- dH2O
- HPLC buffer A
- HPLC buffer C
- Dichloromethane
- HPLC buffer C
- HPLC buffer B
- HPLC buffer A
-
43
Wash the column by injecting 5 mL of FA, 90% and running the “Cleaning” HPLC method. CRITICAL. Remove all bubbles before loading the FA, 90%; remove the air from the loop before running the HPLC method. ? TROUBLESHOOTING
-
44
After the column is clean, equilibrate it with Buffer A until a steady conductivity value (~5 mS/cm) is reached.
-
45
Dissolve the dried protein powder (from Step 39) in 4 mL of FA, 90%. Load the sample in the column and run the “Sample purification” HPLC method. Collect fractions of 2 mL volume. ? TROUBLESHOOTING
-
46
Save the fractions exhibiting UV280 and UV214 absorbances. For each elution peak, lyophilize a small aliquot (100–200 μL) for subsequent SDS-PAGE analysis to identify which contains the target protein. ? TROUBLESHOOTING
-
47
Lyophilize the fractions containing the purified target protein as described in Steps 38–39. Acetonitrile or dH2O can be added 1:2 to the HPLC fractions to avoid melting of the solution in the lyophilizer. PAUSE POINT. After lyophilization, the protein powder can be stored at −20°C.
Protein reconstitution in DMPC/DHPC bicelles
-
48
Dissolve the lyophilized protein powder (1–2 mg) in 1 mL of HFIP. Mix it with approximately 9 mg of DMPC (protonated or deuterated) and 27 mg of DHPC (protonated or deuterated).
-
49
Dry the solution under a gentle nitrogen stream until a thin film is obtained, then lyophilize overnight. PAUSE POINT. The dried mixture can be stored at −20°C.
-
50
Dissolve the dried solution in 3 mL of 8 M Urea. If the solution is not clear, add additional DHPC (~5 mg). Mix well until the solution becomes clear. If needed, add more DHPC in small amount (~2–5 mg).
-
51
Transfer the solution to a 0.5–3.0 mL dialysis cassette (3,500 MWCO) and dialyze it for 3 hours versus 1 L of NMR buffer. Stir gently. If the sample starts becoming cloudy, add 50 μL of DHPC (100 mg/mL) directly into the dialysis cassette. CRITICAL STEP. During the dialysis, the DHPC gradually diffuses outside the cassette, while the DMPC remains trapped inside. When approaching the liposome state, it is important to replenish the lost DHPC to prevent possible protein aggregation, which may occur especially for highly concentrated samples.
-
52
Perform a second dialysis versus 1 L of NMR buffer for 3 hours. If the sample starts becoming cloudy, add 50 μL of DHPC (100 mg/mL) directly into the dialysis cassette. Adjust the DHPC amount so that the bicelle q is close to 0.5.
-
53
Transfer the sample into a 4 mL concentrator (3,000 MWCO) and concentrate it to a volume of ~300–350 μL by centrifuging at 3,000 rpm at RT. Add 10% D2O (vol/vol) and transfer the sample into a Shigemi NMR tube.
-
54
Determine the exact bicelle q of the sample by recording a 1H NMR spectrum (2H NMR spectrum if deuterated DMPC and DHPC are used). The relative amount of DHPC and DMPC (~0.90 p.p.m. and ~0.85 p.p.m., respectively) is quantified by comparing the integral of their NMR signals. If needed, adjust the q to the desired value. ? TROUBLESHOOTING
Timing
Steps 1–21, Protein expression, 4 days
Steps 22–32, Ni-NTA purification, 1 day
Steps 33–39, Cyanogen bromide cleavage, 1 day
Steps 40–47, Reverse phase HPLC, 1 day
Steps 48–54, Protein reconstitution in DMPC/DHPC bicelles, 2 days
Troubleshooting
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
| 21 | Weak or absent protein expression | The amount of IPTG used for induction and/or the expression temperature are not optimal | Induce cells at OD600 within the range 0.6–0.8; screen for the best protein expression conditions varying the temperature in the range 16–35°C and the [IPTG] from 0.1 to 0.5 mM. |
| 35 | CNBr cleavage is not efficient | Too much oxygen is present in the reaction system | Before the reaction starts, pretreat the solution bowing a gentle nitrogen gas stream for 5 minutes; maintain the gas stream during the entire reaction |
| 43 | The column is not clean even after several washings | FA, 90% may be not strong enough to completely clean the column | Run the “cleaning” HPLC method loading 1 mL of TFE, instead of 5 mL of FA, 90%. Repeat the washing 2–3 times if needed |
| 45 | Elution peaks are not resolved | The HPLC method used to purify the protein is not optimized | Change the composition of HPLC buffers A and B varying the amount of IPA and Acetonitrile; change the HPLC method by using slower gradients |
| 46 | Excessive protein formylation occurred | The protein has been dissolved too much time in FA, 90% | Reduce the CNBr cleavage reaction time (Step 35) or the FA concentration (Steps 31–32) |
| 54 | The bicelle q is higher than 0.5 | Insufficient DHPC is present | Calculate the amount of DHPC present in the sample based on the actual q and the amount of lipids used in Step 48. Calculate the amount of DHPC that would be needed to obtain q = 0.5 for the lipids used. Add DHPC (100 mg/mL) to cover the difference. Measure a 1D 1H NMR spectrum (Step 54) to ensure the q is now 0.5. |
| 54 | The bicelle q is lower than 0.5 | Too much DHPC is present | Calculate the amount of DHPC present in the sample based on the actual q and the amount of lipids used in Step 48. Calculate the amount of DHPC that would be needed to obtain q = 0.5 for the lipids used. Dilute the sample with the amount of NMR buffer required to retain the excess of detergent, knowing that in solution the free [DHPC] is ~5 mM. Repeat Steps 53–54. |
Anticipated Results
The protocol describes the steps needed to prepare bicelle-reconstituted TMD samples suitable for structural studies by NMR; it is demonstrated below for the TMD of Fas (Fas-TMD), a member of the TNF receptor superfamily. Although only shown here for the Fas-TMD as an example, the applicability of the protocol is very broad as it has been demonstrated in numerous studies, including ones that involve MP regions1,8,12,23,45–49.
The Fas-TMD was expressed in E. coli as a fusion to the C terminus of the TrpLE sequence with a methionine residue added in between (Box 1, Fig. 1b)47. The expression level of the fusion protein was approximately 2 mg/L, which is comparable to that of most other hydrophobic protein fragments expressed using the same system (Fig. 2b)8,48. The methionine residue was used as a cleavage site for cyanogen bromide (CNBr), separating the TMD from the TrpLE (Fig. 1b, 2b). Importantly, the fusion protein is cleaved right after the methionine such that the TMD consists only of its native amino acid sequence. After cleavage, the Fas-TMD was separated by HPLC using the Zorbax SB-C3 column (HPLC, Fig. 2c). The HPLC profile and SDS-PAGE analysis both indicated high protein purity (Fig. 2c). The expression protocol above can be used to achieve extremely high level of protein deuteration. Measurements from analytical liquid chromatography-mass spectrometry of our 2H-labeled Fas-TMD sample show that protein deuteration percentage was about 98.3% (Box 2). This high level of deuteration is ideal for the inter-chain NOE experiment (described in Part 3) because the intra-chain NOEs from aliphatic groups would be invisible, allowing for exclusive detection of inter-chain contacts.
Figure 2. Expression, purification and bicelle reconstitution of TM domains.
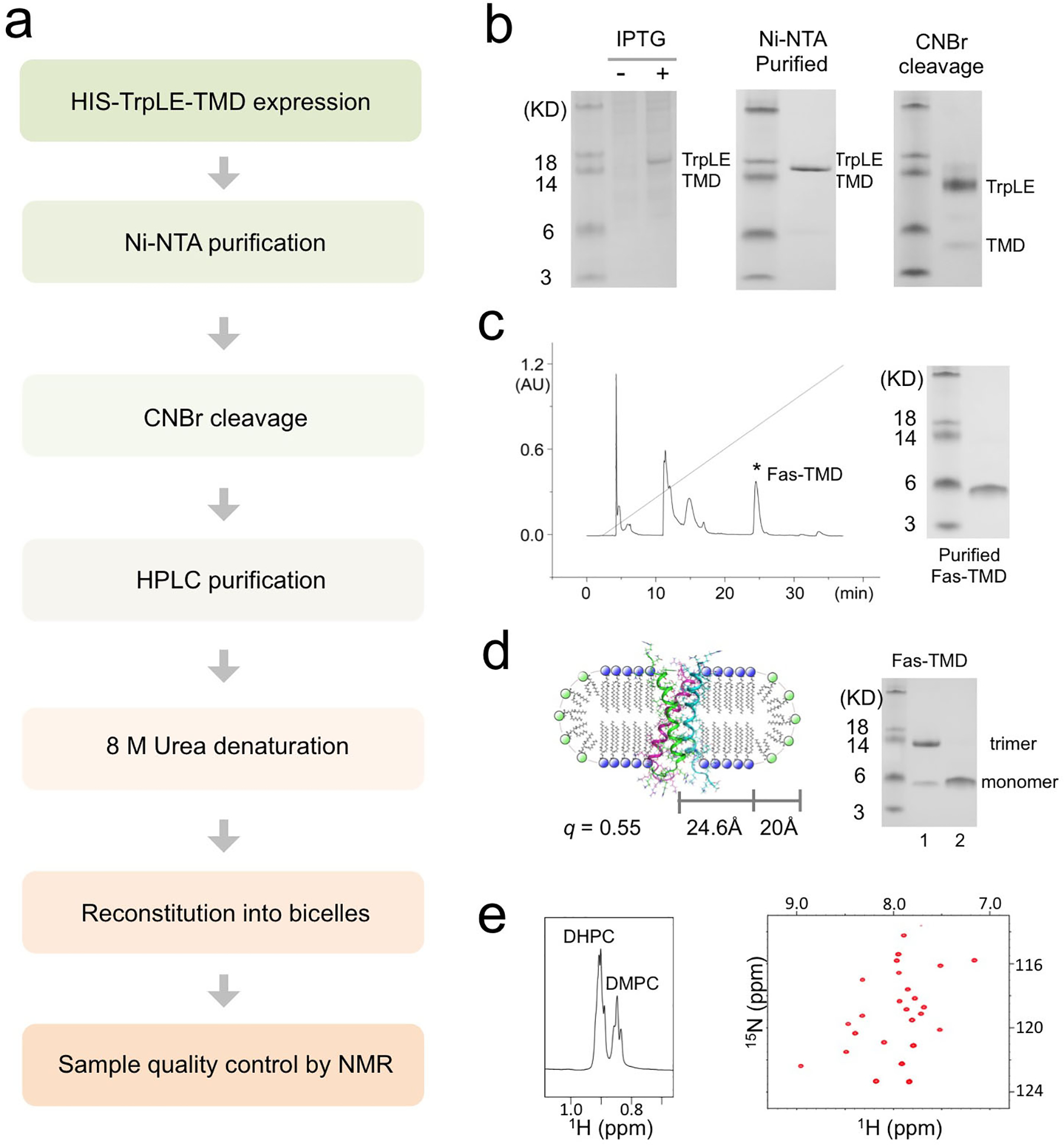
The protocol was demonstrated for the Fas-TMD. (a) Flow chart of the steps required for TM/MP domain expression, purification and reconstitution into bicelles with 0.5 ≤ q ≤ 0.6. (b) SDS-PAGE analysis of expression and purification. Left: Cell lysate before (lane 1) and after (lane 2) induction with IPTG; Middle: The TrpLE-TMD fusion protein after Ni-NTA affinity purification; Right: Purified TrpLE-TMD after CNBr cleavage. (c) HPLC chromatogram of cleaved TrpLE-TMD (left) and SDS-PAGE analysis of the Fas-TMD elution peak. (d) Schematic illustration (Left) of the Fas-TMD reconstituted in bicelles with q = 0.55 (q is the molar ratio of DMPC to DHPC). The radii of the planar region of the bicelle (R) and the DHPC rim (r) are 24.6 Å and 20 Å, respectively. Right: SDS-PAGE analysis of (1) the Fas-TMD after bicelle-reconstitution and (2) the Fas-TMD before reconstitution (dried powder in loading buffer). (e) The 1H NMR spectrum of a typical bicelle-reconstituted Fas-TMD sample, showing the DHPC and DMPC methyl peaks used for calculating the bicelle q (left), and the corresponding 2D 1H-15N TROSY-HSQC spectrum (right). The spectra were acquired at 600 MHz at 303K.
Box 2. Quantification of sample deuteration level.
The expression strategy described in Part 1 of our protocol allows high level of protein deuteration. For example, the deuteration level for the Fas-TMD was shown to be ~98.3% as analyzed by LC Mass Spectroscopy (Fig. 4b). The high level of deuteration permits the use of regular NOESY to detect inter-chain NOEs because the intra-chain NOEs from non-labile aliphatic groups are 50 times weaker, which would be in the noise.
The LC/MS procedure used to quantify Fas-TMD deuteration is provided below:
100 μg Fas-TMD powder was dissolved in 100 μL of 0.2% Formic acid.
A BioBasic™ 18 LC column (2.1 × 100 mm, 5 μm particle size, 300 Å pore size; Thermo Fisher Scientific) was directly connected to the standard electrospray ionization source of LTQ-Qrbitrap XL Fourier transform mass spectrometer (Thermo Fisher Scientific).
5 μL of sample was injected through an Agilent 1200 autosampler. The gradient was set from 5% elution buffer (98% Acetonitrile, 0.2% Formic acid) to 80% elution buffer over 20 minutes at a flow rate of 250 μL/min.
The mass spectrometer parameters were set as follows: spray voltage = 4.5 kV, capillary voltage = 26 V, tube lens voltage = 120 V, capillary temperature = 275°C, sheath flow rate = 40, auxiliary gas flow = 25.
All Fourier transform mass spectra were acquired at resolution 60,000 with 300–2,000 Da mass ranges. Mass accuracy was higher than 3 ppm after external calibration.
The mass spectra were de-convoluted using Xcaliber (Thermo Fisher Scientific).
Before reconstitution in bicelles, the SDS-PAGE analysis of the Fas-TMD powder resolved the protein as a monomer (~4 kDa). However, after reconstitution in DMPC/DHPC bicelles with q ~ 0.55, which yields lipid discs with diameter of about 50 Å (Fig. 2d), the Fas-TMD migrated on SDS-PAGE as a trimer (~13 kDa), indicating that the Fas-TMD spontaneously trimerize in lipid bilayer and the trimers apparently resisted the denaturing power of SDS-PAGE (Fig.2d). After reconstitution into bicelles, the sample quality was further evaluated using 2D 1H-15N TROSY-HSQC NMR spectrum. The NMR spectrum has good chemical shift dispersion and one peak per residue (Fig. 2e), indicating that the protein is well folded and adopts a single trimeric conformation. We emphasize that some TMD oligomers do not survive the denaturing environment of SDS-PAGE. It is therefore crucial that TMD oligomerization is examined using methods in Part 2 below.
Part 2. Determination of the oligomeric state of TM domains in bicelles
a). Inter-molecular PRE analysis
Materials
BIOLOGICAL MATERIALS
~1:1 mixture (wt/wt) of lyophilized 15N-labeled protein and Cys-mutant of the same protein. Store them at −20°C. CRITICAL. Purify the protein mixture as described in Part 1 but with 5 mM DTT (or TCEP) added to the Lysis and Guanidine buffers.
REAGENTS
1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP, 99.5%, Oakwood Products, Inc. cat. no. 003409)
(1-Oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)methanethiosulfonate (MTSL; Santa Cruz Biotechnology, cat. no. sc-208677) CAUTION. Light sensitive
1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC, Avanti Polar Lipids, Inc., cat. no. 850305)
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC, Avanti Polar Lipids, Inc., cat. no. 850345)
1,4-Dithiothreitol (DTT, VWR, cat. no. 97061) CAUTION. May cause skin and eye irritation.
Deuterium oxide, 99.96% (D2O, Cambridge Isotope Laboratories, Inc., cat. no. DLM-6-PK)
Dimethyl sulfoxide (DMSO, Sigma-Aldrich, cat. no. 276855)
Disodium hydrogen phosphate (Na2HPO4, Sigma-Aldrich, cat. no. S3264)
MES hydrate (Oakwood Chemical, cat. no. M05729)
L-Ascorbic acid (Sigma-Aldrich, cat. no. A92902)
Sodium phosphate monobasic (NaH2PO4, Sigma-Aldrich, cat. no. S3139)
Urea (Thermo Fisher Scientific, cat. no. 15505050)
EQUIPMENT
All the equipment necessary for handling of recombinant proteins
NMR spectrometer fully-equipped for triple-resonance experiments of biological macromolecules equipped with a cryogenically cooled probe head with z-field gradients
NMR data-processing software (e.g., TOPSPIN, VNMR, NMRPIPE, etc.)
Visualization and analysis software for NMR spectra (e.g., TOPSPIN, CARA, Sparky, NMRView, NmrDraw, CCPN, etc.)
Data analysis software (e.g., ORIGIN, MATLAB, etc.)
REAGENT SETUP
Urea, 8 M.
Dissolve 480.5 g of Urea in dH2O to reach the final volume of 1 L. The solution can be heated to aid the Urea dissolution. Store the solution at RT.
DTT, 1 M.
Dissolve 1.6 g of DTT in dH2O to reach the final volume of 10 mL. Split the solution in 1 mL aliquots and store them at −20°C. Use the solution within 1 year.
MTSL, 100 mM.
Dissolve 5.3 mg of MTSL in 200 μL of DMSO. Split the solution in 20 μL aliquots and store them at −20°C. Use the solution within 1 year. CAUTION. Store the aliquots in dark vials or cover them in aluminum foil.
NMR buffer (pH 6.7).
The NMR buffer can be optimized for the specific sample under study. Typically, it consists of ~20–50 mM Phosphate or MES buffer close to physiological pH, e.g. 6.7. Store at RT.
NMR buffer (pH 6.2).
Prepare an identical buffer to the NMR buffer (pH 6.7), but at pH 6.2. Store at RT. Degas right before use.
Pi, 400 mM (pH 7.5).
Dissolve 4.6 g of Na2HPO4 and 1 g of NaH2PO4 in 100 mL of dH2O. Adjust the pH to 7.5. Store the solution at RT.
DHPC, 100 mg/mL.
Dissolve 150 mg of DHPC in 1.5 mL of NMR buffer. Split the solution in 50 μL aliquots and store them at −20°C.
Ascorbic acid, 0.5 M.
Dissolve 0.9 g of ascorbic acid in 10 mL of previously degassed NMR buffer. Adjust the pH to match that of the NMR buffer (e.g. 6.7). Prepare fresh, discard after use. CRITICAL STEP. Adjust the pH of the solution to be as close as possible to that of the final NMR sample.
Procedures
Sample preparation
-
1
Dissolve the lyophilized protein/Cys-mutant mixture in 1 mL of HFIP. Mix it with approximately 9 mg of DMPC and 27 mg of DHPC.
-
2
Dry the solution under a gentle nitrogen gas stream until a thin film is obtained, then lyophilize overnight. PAUSE POINT. The dried mixture can be stored at −20°C.
-
3
Dissolve the dried solution in 3 mL of 8 M Urea. If the solution is not clear, add additional DHPC (~5 mg). Mix well until the solution becomes clear. If needed, add more DHPC in small amount (~2–5 mg).
-
4
Add 30 μL of 1 M DTT to the solution. Mix well. Incubate for approximately 10 minutes, then gently spin down the solution by centrifuging at 1,000 rpm for 2 minutes at RT.
-
5
Transfer the solution in a 0.5–3.0 mL dialysis cassette (3,500 MWCO) and dialyze it for 3 hours versus 1 L of NMR buffer (pH 6.7), to which 1 mL of 1 M DTT has been added. Stir gently. If the sample starts becoming cloudy, add 50 μL of DHPC (100 mg/mL) directly into the dialysis cassette. CRITICAL STEP. During the dialysis, the DHPC gradually diffuses outside the cassette, while the DMPC remains trapped inside. When approaching the liposome state, it is important to replenish the lost DHPC to prevent possible protein aggregation, which may occur especially for highly concentrated samples.
-
6
After the dialysis, add 15 μL of 1 M DTT directly into the dialysis cassette. Perform a second dialysis for 3 hours versus 1 L of NMR buffer (pH 6.7), to which 1 mL of 1 M DTT has been added. When the sample starts becoming cloudy, add 50 μL of DHPC (100 mg/mL) directly into the dialysis cassette.
-
7
Perform a third dialysis versus 1 L of degassed NMR buffer (pH 6.2) for 3 hours. When the sample starts becoming cloudy, add 50 μL of DHPC (100 mg/mL) directly into the dialysis cassette.
-
8
While dialyzing the sample, use 3.5 mL of degassed NMR buffer (pH 6.2) (this volume corresponds approximately to the actual sample volume in the dialysis cassette) to determine the amount of 400 mM Pi buffer (pH 7.5) required to raise its pH to ~7.2–7.3. Note the volume added.
-
9
5–10 minutes before the end of the dialysis, thaw one aliquot of 100 mM MTSL solution. CRITICAL. Shield the vial from light.
-
10
After the dialysis is finished, transfer the sample into a 15 mL Falcon tube. Quickly raise the sample pH to ~7.2–7.3 by adding the same amount of 400 mM Pi buffer (pH 7.5) as determined in Step 8, then immediately add 10 μL of 100 mM MTSL (the MTSL concentration should be ~10 times in excess with respect to that of the Cys-mutant). Mix well, seal the tube with parafilm and cover it with aluminum foil. Let the reaction proceed overnight at RT. CRITICAL STEP. Make sure that the tube is shielded from light during the reaction. PAUSE POINT. The sample can be left overnight at RT. ? TROUBLESHOOTING
-
11
Transfer the sample in a 0.5–3.0 mL dialysis cassette (3,500 MWCO) and dialyze it for 3 hours versus 1 L of NMR buffer (pH 6.7). When the sample starts becoming cloudy, add 50 μL of DHPC (100 mg/mL) directly into the dialysis cassette. CRITICAL STEP. Maintain the dialysis apparatus shielded from light.
-
12
Perform three additional dialysis as in Step 11, always keeping the dialysis apparatus shielded from light. CRITICAL STEP. The excess of free MTSL left from the reaction in Step 10 gradually diffuses outside the dialysis cassette together with the detergent. To ensure that the free MTSL has been completely removed from the sample, perform at least four dialysis (Steps 11–12) so that the amount of lost DHPC is ≥ 60 mg. ? TROUBLESHOOTING
-
13
Transfer the sample into a 4 mL concentrator (3,000 MWCO) and concentrate it to a volume of ~300–350 μL by centrifuging at 3,000 rpm at RT. Add 10% D2O (vol/vol) and transfer the sample into a Shigemi NMR tube. CRITICAL STEP. Cover the NMR tube with aluminum foil to shield it from light.
NMR experiments
-
14
Set the probe temperature to the desired value (e.g. 308 K). Insert the sample in the magnet and wait ~15 minutes for the sample to achieve temperature stability. Lock, tune and shim the magnet. Activate the auto-shim.
-
15
Determine the pulse lengths of hard 90° pulses for 1H and 15N (in the direct and indirect acquisition mode setup, respectively).
-
16
Measure a 1D 1H NMR spectrum to determine the actual bicelle q of the sample. If needed, adjust it to 0.5 as explained in Part 1, Step 54.
-
17
Acquire a high-resolution 2D 1H-15N TROSY-HSQC spectrum of the paramagnetic state. CRITICAL STEP. The recovery delay should be set at least to 3.5 s to prevent the occurrence of significant longitudinal PRE.
-
18
Process the spectrum with standard parameters. Typically, raw data are multiplied by an apodization function, zero-filled and Fourier-transformed in both dimensions. Proper zero-and first-order phase correction and baseline correction in both dimensions are applied after the Fourier transformation.
-
19
Remove the sample from the magnet. Add 15 μL of 0.5 M ascorbic acid solution to the sample. Mix well. CRITICAL STEP. Check that the sample pH has remained identical after the addition of the ascorbic acid solution. If required, restore the pH to the previous value.
-
20
Repeat Steps 14–18 to acquire the 2D 1H-15N TROSY-HSQC spectrum of the diamagnetic state. Use identical acquisition and processing parameters as for the paramagnetic state.
Data analysis
-
21
Load the assignment peak list on top of both 2D 1H-15N TROSY-HSQC spectra. Fine-adjust the peak positions to exactly match the top of the NMR cross-peaks. Export the NMR peak intensities of the paramagnetic and diamagnetic states. Discard overlapping peaks from the analysis.
-
22
Divide the peak intensities of the paramagnetic state (I) by those of the diamagnetic state (I0).
-
23
Plot (I/I0) vs. (residue number). For structure validation purpose, map the I/I0 values on the protein structure.
Timing
Steps 1–13, Sample preparation, 3 days
Steps 14–20, NMR experiments, 3–6 days depending on the sample concentration and spectrometer sensitivity
Steps 21–23, Data analysis, 1 day
Troubleshooting
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
| 10 | MTSL labeling is not efficient | The sample pH is not sufficiently high | Increase the sample pH to ~7.5 before adding MTSL |
| The cysteine residue of the Cys-mutant is partially or completely oxidized | Increase the amount of DTT in Steps 4 and 6 or use a stronger reducing agent (e.g. TCEP); lower the sample pH to ~6.0 while removing the DTT by dialysis (Step 7) | ||
| The amount of MTSL used is not sufficient | Increase the excess of MTSL to ~20–30 times | ||
| 12 | Free MTSL is not removed completely | The DHPC removal rate is too slow | Increase the stirring speed to aid DHPC removal; increase the number of dialysis |
Anticipated Results
The protocol describes an efficient strategy for site-specific paramagnetic labeling of TMD to detect inter-chain PREs if the TMD forms oligomer. For this experiment, two protein preparations are needed: one for 15N-labeled protein that serves as the NMR readout, and the other for spin-labeled protein that would influence the NMR signals of the 15N-labeled protein if the two mix and oligomerize. For the spin-labeled protein, a cysteine mutation is introduced (usually in regions predicted to be accessible, e.g. one of the protein termini) for labeling with MTSL. The two preparations are then mixed at a ratio of 1:1 for optimal measurement of inter-chain PREs. The main purpose of the experiment is to address whether the TMD is monomeric or multimeric in bicelles, although it can also be used later to validate NOE-derived oligomeric structures.
The results expected from this type of analysis are exemplified here by the application to the Fas-TMD. The 15N-labeled Fas-TMD and the unlabeled mutant (with a cysteine introduced at the C-terminus) were mixed at 1:1 ratio right after HPLC purification. As elaborated in the protocol above, the mixed protein was reconstituted into DMPC/DHPC bicelles (q = 0.5), followed by linking the cysteine and MTSL via disulfide bonding. After recording the two NMR spectra, one before (paramagnetic) and one after ascorbic acid addition (diamagnetic), the peak intensity ratios of paramagnetic (I) to diamagnetic (I0) state (defined here as the PRE) were measured for all residues. The PRE vs. residue plot (Fig.3a) shows strong PREs for the C-terminal region of the 15N-labeled Fas-TMD but not for the N-terminal region, indicating that the PREs are due to specific, parallel oligomerization of the TMD. Moreover, average signal loss of the affected region is ~50%, which is expected from a 1:1 mixed sample. The inter-chain PRE results indicate that the Fas-TMD is oligomeric.
Figure 3. Characterization of oligomeric state of TM domains in bicelles.
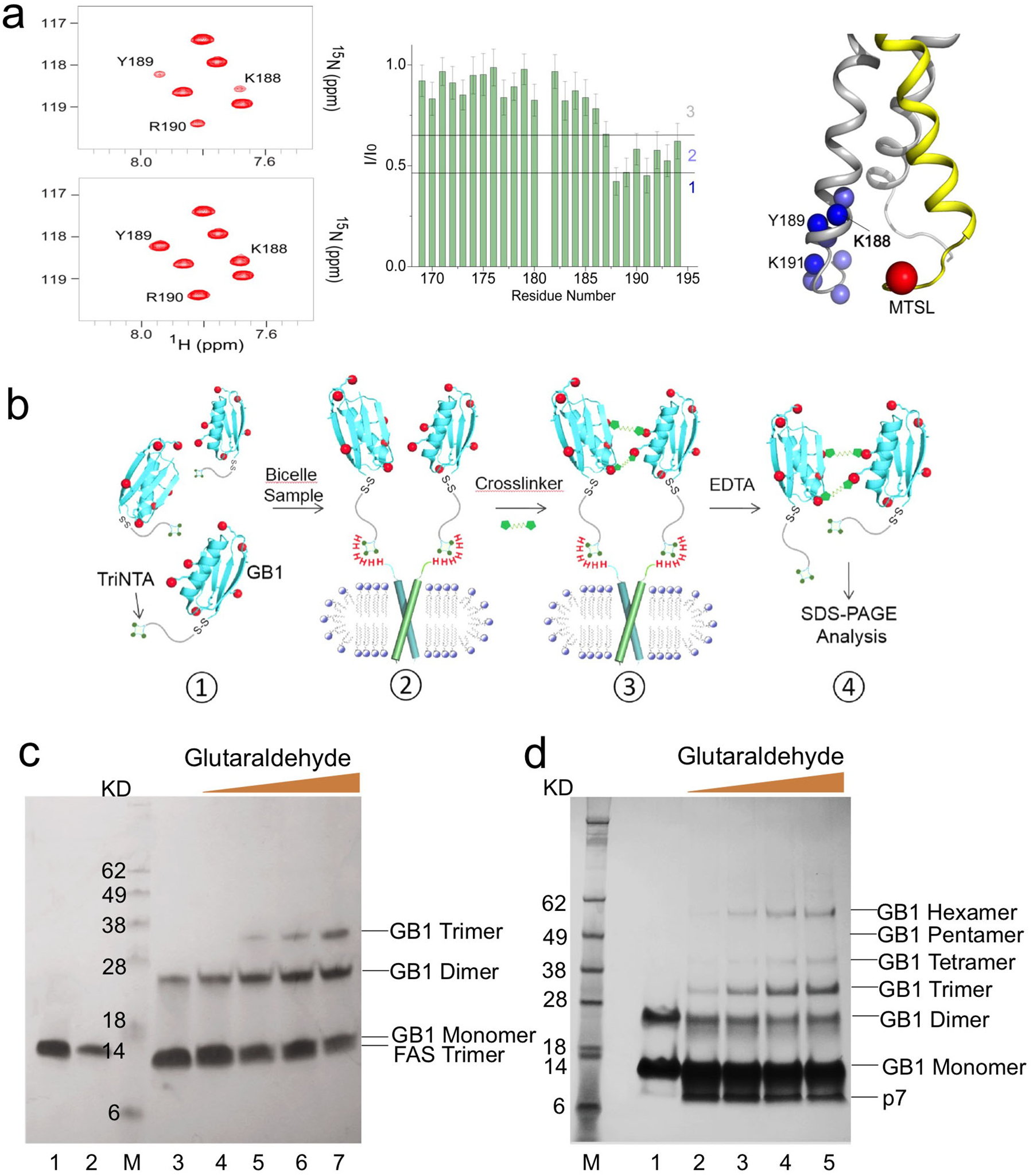
(a) Inter-chain PRE analysis demonstrated here for the Fas-TMD reconstituted in bicelles with q = 0.55. The sample consisted of a ~1:1 mixture of 15N-labeled Fas-TMD and 14N Fas-TMD spin-labeled with MTSL at the C-terminus (Cys194). Left: 2D 1H-15N TROSY-HSQC spectra before (top) and after (bottom) reducing the nitroxide with ascorbic acid, acquired at 600 MHz and 303 K. Middle: Residue-specific PRE, defined as the ratio of the peak intensity before (I) to that after (I0) ascorbic acid addition. Right: Mapping PRE onto the Fas-TMD trimer structure, showing the backbone amide protons (blue spheres) with strong PREs and the spin-label Cβ position (red sphere). Amide protons are colored based on the PRE regions in middle panel: 1 – dark blue; 2 – light blue. 15N-labeled and MTSL-labeled chains are shown as gray and yellow ribbons, respectively. (b) Schematic illustration of the OG-label procedure for characterizing oligomerization state. (1) TriNTA-GB1 is sufficiently dilute (less than 100 μM) for minimizing non-specific crosslinking. (2) In the presence of bicelle-reconstituted TM oligomers (containing one His6-tag per protomer), the TriNTA-GB1 is recruited to the TM oligomer in stoichiometric amount via strong affinity between the His6-tag and the TriNTA. Prior to incubation with the TriNTA-GB1, the primary amines of the TM protein are blocked. (3) Addition of crosslinkers (first BS3 and then Glutaraldehyde) to crosslink the GB1s. (4) TriNTA-GB1 is released from the TM protein by stripping Ni2+ with EDTA, followed by SDS-PAGE analysis. (c) OG-label application to the Fas-TMD reconstituted in DMPC/DHPC bicelles with q = 0.5. As a control, 45 μM TriNTA-GB1 was treated with 1 mM BS3 and then with 1 mM glutaraldehyde for 5 min (lane 3). In the presence of 30 μM bicelle-reconstituted Fas-TMD, 45 μM TriNTA-GB1 was treated first with 1 mM BS3 and then with increasing amount of glutaraldehyde for 5 min: 0.1 mM (lane 4), 0.3 mM (lane 5), 1 mM (lane 6), and 3 mM (lane 7). The bands of TriNTA-GB1 and bicelle-reconstituted Fas-TMD (trimer) alone are shown in lane 1 and 2, respectively. The SDS-PAGE gel used was a 12% Bis-Tris protein gel. (d) OG-label application to the HCV p7 reconstituted in DMPC/DHPC bicelles with q = 0.6. As a control, 30 μM TriNTA-GB1 was treated first with 0.6 mM DTSSP and then with 0.5 mM glutaraldehyde for 5 min (lane 1). In the presence of 20 μM bicelle-reconstituted p7, 30 μM TriNTA-GB1 was treated first with 0.6 mM DTSSP and then with increasing amount of glutaraldehyde for 5 min: 0.1 mM (lane 2), 0.5 mM (lane 3), 1 mM (lane 4), and 2.5 mM (lane 5). The SDS-PAGE gel used was a 4–12% Bis-Tris protein gel.
Several potential pitfalls deserve special attention. The mixed PRE analysis is only meaningful when two differently labeled chains are adequately mixed within oligomers, but this can be problematic for certain TMDs that oligomerize very strongly. For example, the TMD of HIV-1 gp41 fusion protein trimerizes so strongly that once the trimer is formed the monomers can no longer be mixed between the trimers12,23. In that case, the two differently labeled species were mixed before CNBr cleavage, as the TrpLE-TMD fusion protein is monomeric in formic acid. Another potential pitfall is the incomplete removal of MTSL labels. MTSL is hydrophobic and partitions weakly in micelles or bicelles. We found that the sample must be dialyzed extensively (e.g., 4 times) to completely remove the free MTSL. Otherwise, even tiny residual amount of MTSL could generate significant but non-specific PREs. A simple way to test the robustness of the MTSL removal protocol is to test the protocol on a control sample containing only the 15N-labeled protein (without cysteine) and the same amount of MTSL used for labeling. If free MTSLs are completely removed, no PRE should be observed. Finally, caution must be taken when reducing the spin-label with ascorbic acid. Since ascorbic acid is acidic, it is important to ensure that the ascorbic acid solution is buffered to the same pH as the protein sample, as reducing the sample pH could change NMR peak intensity by changing solvent exchange. A final pH check after ascorbic acid addition should be performed before recording the TROSY-HSQC spectrum.
b). OG-label for characterizing the oligomerization state
Materials
BIOLOGICAL MATERIALS
BL21 (DE3) competent cells (New England biolabs, cat. no. C2527). Store them at −80°C.
TM protein reconstituted in DMPC/DHPC bicelles (q = 0.5) in the OG-label buffer (see Reagents setup). The protein needs to feature a His6-tag.
Expression plasmid (pET15b vector) coding for GB1. A cysteine, additional lysines, and a Strep-tag sequence (CKDKDKWSHPQFEK) are added to the N-terminus of the GB1 sequence. The plasmid carries antibiotic resistance to Ampicillin. Store it at −20°C.
Expression plasmid (pET15b vector) coding for Foldon. A His6-tag need is added to the protein. The plasmid carries antibiotic resistance to Ampicillin. Store it at −20°C.
REAGENTS
1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC, Avanti Polar Lipids, Inc., cat. no. 850305)
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC, Avanti Polar Lipids, Inc., cat. no. 850345)
Acetonitrile (ACN, EMD Millipore, cat. no. EM-AX0151–1)
Ampicillin (A, Sigma-Aldrich, cat. no. A9393)
BS-3 crosslinker (Thermo Fisher, cat. no. 21580)
d-Desthiobiotin (Sigma-Aldrich, cat. no. D1411)
Disodium hydrogen phosphate (Na2HPO4, Sigma-Aldrich, cat. no. S3264)
DL-Dithiothreitol (DTT, Sigma-Aldrich, cat. no. 43819) CAUTION. May cause skin and eye irritation.
Ethylenediaminetetraacetic acid disodium salt dehydrate (EDTA, Sigma-Aldrich, cat. no. E6635)
Glutaraldehyde (Sigma-Aldrich, cat. no. G7776)
HEPES (Sigma-Aldrich, cat. no. H3375)
Hispur Ni-NTA Resin (Thermo Fisher, cat. no. 88223)
Isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma-Aldrich, cat. no. 329815691)
Luria-Bertani (LB) agar, granulated (RPI Research Products International, cat. no. L24033–500)
NHS-activated agrose resin (Thermo Fisher, cat. no. 26196)
SM(PEG)2 (PEGylated SMCC crosslinker) (Thermo Fisher, cat. no. 22103)
Sodium bicarbonate (NaHCO3, Sigma-Aldrich, cat. no. S5761)
Sodium chloride (NaCl, Sigma-Aldrich, cat. no. S7653)
Sodium phosphate monobasic (NaH2PO4, Sigma-Aldrich, cat. no. S3139)
SOC medium (New England Biolabs, cat. no. B9020S)
Sulfo-NHS-Acetate (ThermoFisher, cat. no. 26777)
TriNTA (synthesized by Medicilon In.(Shanghai, China) upon request). The detailed description of synthesis was previously published38.
Triethylammonium Acetate (TEAA, Calbiochem, cat. no. 625718)
Tris base (Sigma-Aldrich, cat. no. T1503)
EQUIPMENT
All the standard equipment necessary for handling of recombinant proteins
Amicon Ultra-15 Centrifugal Filter Unit (EMD Millipore, cat. no. UFC900324)
FPLC instrucment (e.g. AKTA pure protein purification system) consisting of multiple pumps, sampler, detectors, and fraction collector
HPLC instrument (e.g. Bio-Rad Duo Flow system) consisting of a degasser, sampler, pumps, and detectors (to measure conductivity and UV absorbance at 214 and 280)
PD-10 column (GE Healthcare, cat. no. G17085101)
StrepTrap HP column (GE Healthcare, cat. no. 28907547)
Superdex S75 26/60 (GE Healthcare, cat. no. 28989334)
Zorbax SB-C18 column (Agilent Technologies, cat. no. 880995–202)
REAGENT SETUP
LB medium (A 100 μg/mL).
Dissolve 25 g of LB broth in 1 L of dH2O and autoclave the solution. Once the solution has cooled down to RT, add 100 mg of ampicillin (100 μg/mL). Operate close to the flame to avoid contamination of the medium. Store the solution at RT.
LB agar plates (A 100 μg/mL).
Approximately 10 plates are obtained per 200 mL of prepared solution. Dissolve 37 g of LB agar to the 1 L of dH2O. Autoclave the solution and let it cool down under gentle shaking to prevent solidification of the agar. When the solution temperature reaches approximately 50°C, add ampicillin to the final concentration of 100 μg/mL and mix. Operate close to the flame to avoid buffer contamination. Aliquot the medium and pour it into sterile plates. After gel coagulated, store the plates at 4°C.
IPTG, 1M.
Dissolve 2.4 g of IPTG in 10 mL of dH2O. Split the solution in 1 mL aliquots and store them at −20°C.
GB1 storage buffer (25 mM HEPES, pH 7.2, 10 mM DTT).
Dissolve 5.95 g of HEPES and 1.54 g of DTT in 1 L of dH2O. Adjust the pH to 7.2. Make the buffer fresh, and store the solution at 4˚C.
HEPES buffer (25 mM HEPES, pH 7.5).
Dissolve 5.95 g of HEPES in 1 L of dH2O. Adjust the pH to 7.5. Store the solution at RT.
HPLC buffer D (5% (vol/vol) ACN, 0.1 M TEAA).
For 1 L solution, mix 50 mL of ACN, 100 mL of 1M TEAA and 850 mL of dH2O. Before use, degas and filter the solution with a 0.2 μM membrane. Store the solution at RT.
HPLC buffer E (60% (vol/vol) ACN, 0.1 M TEAA).
For 1 L solution, mix 600 mL of IPA, 100 mL of 1M TEAA and 300 mL of dH2O. Before use, degas and filter the solution with a 0.2 μM membrane. Store the solution at RT.
Ni-NTA binding buffer (25 mM Na2HPO4, pH 7.4, 100 mM NaCl).
Dissolve 3.55 g of Na2HPO4 and 5.85 g of NaCl in 1 L of dH2O. Adjust the pH to 7.4. Store the solution at RT.
Ni-NTA elution buffer (25 mM Na2HPO4, pH 7.4, 100 mM NaCl, 500 mM Imidazole).
Dissolve 3.55 g of Na2HPO4, 5.85 g of NaCl, and 34 g of imidazole in 1 L of dH2O. Adjust the pH to 7.4. Store the solution at RT.
OG-label buffer (25 mM Na2HPO4, pH 7.5, 200 mM NaCl)
Dissolve 3.55 g of Na2HPO4 and 11.7 g of NaCl in 1 L of dH2O. Adjust the pH to 7.5. Store the solution at RT.
Phosphate NMR buffer (25 mM Na2HPO4, pH 7.2).
Dissolve 3.55 g of Na2HPO4 in 1 L of dH2O. Adjust the pH to 7.2. Store the solution at RT.
StrepTrap binding buffer (100 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 10 mM DTT).
Dissolve 12.1 g of Tris base, 8.8 g of NaCl, 0.372 g of EDTA, and 1.54 g of DTT in 1 L of dH2O. Adjust the pH to 8.0. Make the buffer fresh, and store the solution at 4˚C.
StrepTrap elution buffer (100 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 10 mM DTT, 2.5 mM desthiobiotin).
Dissolve 12.1 g of Tris base, 8.8 g of NaCl, 0.372 g of EDTA, 1.54 g of DTT, and 0.535 g of desthiobiotin in 1 L of dH2O. Adjust the pH to 8.0. Make the buffer fresh, and store the solution at 4˚C.
TE buffer (10 mM Tris, pH 8.0, 1mM EDTA).
Dissolve 1.21 g of Tris base, and 0.372 g of EDTA in 1 L of dH2O. Adjust the pH to 8.0. Store the solution at RT.
TriNTA reaction buffer (100 mM Sodium bicarbonate, pH 8.0).
Dissolve 8.4 g of sodium bicarbonate in 1 L of dH2O.Adjust the pH to 8.0. Make the buffer fresh.
Tris buffer (1 M Tris, pH 7.5).
Dissolve 121 g of Tris base in 1L of dH2O. Adjust the pH to 7.5. Store the solution at RT.
EQUIPMENT SETUP
“Protein refinement” program
Connect the Superdex S75 26/60 to the FPLC system.
Connect the pump to the GB1 storage buffer/Ni-NTA binding buffer.
Connect the 5 mL loop.
Empty the loop. Flow rate: 1 mL/min. Total volume: 10 mL.
Wash Superdex S75 26/60 column. Flow rate: 1 mL/min. Total volume: 150 mL.
Collect fractions from 10th mL, 1 mL per fraction.
“SMCC-TriNTA purification” program
Connect the C18 column to the HPLC system.
Connect the HPLC Buffer D to pump A and the HPLC Buffer E to the pump B.
Isocratic flow to empty the sample loop: HPLC Buffer D (100%). Flow rate: 3 mL/min. Total volume: 15 mL.
Linear gradient: from HPLC Buffer D (100%)/HPLC Buffer E (0%) to HPLC Buffer D (50%)/HPLC Buffer E (50%). Flow rate: 3 mL/min. Total volume: 120 mL.
Linear gradient: from HPLC Buffer D (50%)/HPLC Buffer E (50%) to HPLC Buffer D (0%)/HPLC Buffer E (100%). Flow rate: 3 mL/min. Total volume: 30 mL.
Isocratic flow: HPLC Buffer B (100%). Flow rate: 3 mL/min. Total volume: 30 mL.
Procedures
GB1 expression and purification
-
1
Dissolve 4 μg of GB1-pET15b plasmid in 20 μl of TE buffer.
-
2
Thaw one tube (50 μl) of BL21(DE3) competent cells on ice.
-
3
Add 1 μl of GB1-pET15b plasmid to 50 μl of competent cells, mix, incubate the tube on ice for 30 minutes.
-
4
Heat shock the cells at 42°C for 50 seconds.
-
5
Let the mixture cool in ice for 2 minutes.
-
6
Add 350 μl of SOC media to the tube and put it in an incubator at 37°C with 220 rpm shaking speed for 1 hour.
-
7
Add 60 μl of the culture from step 6 to LB agar plate (A 100 μg/mL), spread the media.
-
8
Incubate the plate at 37°C for about 16 hours. PAUSE POINT. This step is typically carried overnight. The plate can then be stored at 4°C, but the E.coli colonies should be grown within 1–2 days after transformation.
-
9
Pick a single colony from the plate, inoculate it into 200 ml of LB media (A 100 μm/mL), and move the media to a 37 °C incubator with 220 rpm shaking speed for about 16 hours. PAUSE point. This step is typically carried overnight.
-
10
Measure the OD600 of the overnight culture, add the appropriate amount of overnight culture to 1 L of LB media (A 100 μm/mL) so that the starting OD600 of 1 L culture is about 0.1 (e.g., add 25 mL of culture of OD600 = 4 to 1 L LB media).
-
11
Incubate the 1 L culture in 37 °C incubator with 220 rpm shaking speed. Induce expression by adding 1 mL of 1 M IPTG to the culture when the OD600 reaches 0.6.
-
12
After 4–6 hours of growing at 37°C, harvest cells by spinning down the culture at 4,600 rpm for 30 minutes. PAUSE POINT. The cell pellet can be stored at −80°C for further use.
-
13
Resuspend the cell pellet in 50 mL of StrepTrap binding buffer, disrupt the cell suspension by sonicating the solution on ice for 5 minutes at intervals of 1.5 seconds at 60% of the maximum power. Repeat the sonication step a second time.
-
14
Spin down the cell suspension from step 13 by centrifuging at 22,000 rpm for 30 minutes at 4˚C, collect the supernatant and filter it via a 0.45 μm filtration device.
-
15
Connect the StrepTrap HP column to the AKTA FPLC system, equilibrate the StrepTrap HP column by washing it with 5 column volumes of StrepTrap binding buffe at 5 mL/min. Multiple StrepTrap columns can be used simultaneously to increase the yield of GB1.
-
16
Manually slowly inject the GB1 supernatant from step 14 to the StrepTrap HP column. CRITICAL. Make sure that no bubbles are injected into the column.
-
17
Wash the StrepTrap HP column with the StrepTrap binding buffer for 5 column volumes. ? TROUBLESHOOTING
-
18
Elute the GB1 protein through StrepTrap elution buffer for 5 column volumes, collect the elution. PAUSE POINT. The GB1 elution can be stored at 4˚C, but it should be processed as described in Steps 19–20 within 1–2 days.
-
19
Connect the Superdex S75 26/60 column to the AKTA FPLC system, equilibrate the column with 150 mL of GB1 storage buffer.
-
20
Concentrate the GB1 elution from Step 19 to about 2.5 mL. Inject the concentrated GB1 elution into the AKTA injection loop, run the “Protein refinement” program. Collect the GB1 and store it at −80°C for future use.
TriNTA-GB1 conjugation
-
21
Dissolve the TriNTA in the TriNTA reaction buffer to achieve a 1 mM TriNTA solution (e.g., dissolve 1.05 mg of TriNTA in 1 mL of buffer). CRITICAL. Use fresh TriNTA reaction buffer.
-
22
Add SM(PEG)2 4 times in excess with respect to the TriNTA solution (e.g., add 16 μl of 250 mM SM(PEG)2 to 1 mL of 1 mM TriNTA). Leave the reaction mixture at RT for 1 hour or at 4°C overnight. ? TROUBLESHOOTING
-
23
Connect the Zorbax 300SB-C18 column to the HPLC system. Equilibrate the column with HPLC buffer D (wash for 5 column volumes).
-
24
Inject the reaction mixture from Step 22 into the HPLC sample loop, run the “SMCC-TriNTA purification” program and collect all fractions.
-
25
Save the fractions exhibiting strong UV280 and UV214 absorbances, lyophilize all the different fractions.
-
26
Dissolve 20% of dried powder of each fraction in phosphate NMR buffer, acquire a 1D 1H NMR spectrum of each fraction to identify fractions that contain the SMCC-TriNTA product.
-
27
Leave part of the SMCC-TriNTA product at RT for immediate use. PAUSE POINT. The rest of SMCC-TriNTA dry powder can be stored at −80°C.
-
28
Load the GB1 protein (Step 20) in a PD-10 column. Remove the DTT using HEPES buffer.
-
29
Immediately after the GB1 elution, add two times excess of SMCC-TriNTA powder from Step 26. Degas the mixture for 30 minutes and incubate the reaction for additional 30 minutes at RT. CRITICAL. The reaction mixture must be degassed to prevent GB1 from dimerizing via formation of disulfide bonds. ? TROUBLESHOOTING
-
30
Transfer the reaction solution in a dialysis cassette (7,000 MWCO) and dialyze it for 3 hours versus 1 L of Ni-NTA biding buffer, stirring the solution. Perform two additional dialysis to achieve complete removal of the non-reacted SMCC-TriNTA in excess.
-
31
Measure the concentration of the TriNTA-GB1 and GB1 mixture via nanodrop, then add 3 times excess of Ni2+ to the TriNTA-GB1 conjugate. PAUSE POINT. The mixture should be stored at −80˚C.
Conjugated TriNTA-GB1 purification
CRITICAL: free GB1 (not conjugated with TriNTA) must be removed from the mixture before performing the OG-label experiment. A His-tag column is used to separate the conjugated from the free GB1. The column is obtained by covalent linking of a His-tag containing Foldon protein to a NHS-activated agarose resin.
-
32
Express the Foldon protein using the same procedures and experimental conditions as for the GB1 protein (Steps 3–12).
-
33
Resuspend the Foldon pellet in the Ni-NTA binding buffer and use the same sonication protocol as in Step 13 to lyse cells.
-
34
Centrifuge the suspension at 22,000 rpm for 30 minutes at 4˚C. Collect and filter the supernatant.
-
35
Add HisPur Ni-NTA resin to a chromatography column (1 mL of HisPur Ni-NTA per liter of expressed Foldon culture). Wash the Ni-NTA resin with 5 resin volumes of Ni-NTA binding buffer.
-
36
Add the supernatant from Step 34 to the His-pur Ni-NTA resin and incubate the mixture on a rotator for 30 minutes.
-
37
Wash the Ni-NTA resin with Ni-NTA binding buffer with 10 times the resin volume, then elute the Foldon protein with 5 times the resin volume of Ni-NTA elution buffer.
-
38
Use 3 kDa cut-off Amicon centrifugal filter to concentrate the Foldon solution to 2.5 mL and purify the concentrated foldon with a Superdex S75 26/60 column using the “Protein refinement” program with Ni-NTA binding buffer.
-
39
Collect the foldon fractions and measure the protein centration via nanodrop. Store the foldon fractions at 4°C. PAUSE POINT.
-
40
Take an aliquot of the Foldon solution containing 20 mg of protein. Add 600 mg of NHS-activated agarose to the solution and adjust the volume with Ni-NTA binding buffer to ~8 mL. Mix the reaction end-over-end for 1 hour.
-
41
Add 2 mL of 1 M Tris (pH 7.5) to quench the reaction. Incubate for 20 minutes at RT.
-
42
Wash the Foldon-resin with 10 resin volumes of Ni-NTA binding buffer. Store the resin in Ni-NTA binding buffer at 4°C.
-
43
Add the TriNTA-GB1 and GB1 mixture from Step 31 to the Foldon-resin. Incubate the mixture on a rotator at RT for 30 minutes. Wash the resin with 10 resin volumes of Ni-NTA binding buffer, then elute the TriNTA-GB1 using 5 resin volumes of Ni-NTA elution buffer. Collect all the wash and elution fractions. ? TROUBLESHOOTING
-
44
Confirm the purity of the TriNTA-GB1 by SDS-PAGE analysis.
-
45
Transfer the fraction containing the TriNTA-GB1 to a dialysis cassette (7,000 MWCO) and dialyze it for 3 hours versus 1 L of OG-label buffer, always stirring the solution. Repeat the dialysis step three more times.
-
46
Use 3 kDa cut-off Amicon centrifugal filter to concentrate the TriNTA-GB1 to about 125 μM. PAUSE POINT. The purified TriNTA-GB1 should be stored at −80°C.
OG-label
-
47
Reconstitute in OG-label buffer the His6-tag containing TM protein as indicated in Part 1 (Steps 48–52).
-
48
Add 100-fold molar excess of Sulfo-NHS Acetate to the TM protein sample. Incubate the mixture for 1 hour at RT. CRITICAL. Primary amines of the reconstituted TM protein must be blocked with Sulfo-NHS Acetate prior to performing the OG-label experiment, to prevent unspecific cross-linking between the TM protein and GB1.
-
49
Add 5% (vol/vol) of Tris buffer to quench the reaction. Incubate for 20 minutes at RT.
-
50
Transfer the reaction solution to a dialysis cassette (3,000 MWCO) and dialyze it for 3 hours against 1 L of OG-label buffer, stirring the solution. Perform two additional dialysis to achieve complete removal of the Sulfo-NHS Acetate in excess. During the dialysis, add 3 mg of DHPC per hour directly into the dialysis cassette. CRITICAL STEP. During the dialysis, the DHPC gradually diffuses outside the cassette, while the DMPC remains trapped inside. Replenish the lost DHPC to maintain the bicelle q ~ 0.5–0.7.
-
51
Transfer the sample into a 4 mL concentrator (3,000 MWCO) and concentrate it to about 50 μM by centrifuging at 3,000 rpm at RT.
-
52
Mix the TM sample with TriNTA-GB1 in the molar ratio of 1:1.5. Incubate for 10 minutes at RT. After mixing, the concentration of the TM protein (monomeric) is about 30 μM. Split the mixture into 4 tubes. Add BS3 cross-linker to each tube so that its concentration is 1 mM, then incubate for 30 minutes at RT. Add Glutaraldehyde to the tubes so that its final concentration is 0.1, 0.3, 1 and 3 mM (one condition per tube). Incubate for 5 minutes at RT. ? TROUBLESHOOTING
-
53
Prepare a negative control by adding OG-label buffer (use the same volume as for the TM protein in Step 52) to the TriNTA-GB1. Add BS3 cross-linker so that its concentration is 1 mM, then incubate for 30 minutes at RT. Add Glutaraldehyde so that its final concentration is 1 mM and incubate for 5 minutes at RT.
-
54
Add 5% (vol/vol) of Tris buffer to each tube to quench the reaction. Incubate for 20 minutes at RT.
-
55
Add EDTA to each tube to a final concentration of 25 mM to release the TriNTA-GB1 from the TM protein.
-
56
Quantify the oligomeric state of TriNTA-GB1 via SDS-PAGE analysis.
Timing
Steps 1–20, GB1 expression and purification, 4 days
Steps 21–31, TriNTA-GB1 conjugation, 3 days
Steps 32–46, Conjugated TriNTA purification, 2 weeks
Steps 47–56, OG-label, 3 days
Troubleshooting
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
| 17 | Only asmall fraction of GB1 binds to the StrepTRap HP column | The sample injection is too fast | Inject the sample to the column slowly (less than 1 mL/min); connect a few columns together; clean the columns after each use |
| 22 | The efficiency of the reaction between TriNTA and SMCC is low | Incorrect buffer conditions | Make fresh sodium carbonate buffer (pH 8.0). The pH of sodium carbonate can increase to 10 after about one week |
| 29 | The efficiency of the reaction between SMCC-TriNTA and GB1 is low | GB1 is not completely reduced; GB1 is oxidized by air during the reaction; the amount of SMCC-TriNTA is not enough | Make sure that the GB1 is completely reduced before the reaction; degas the buffer; use a higher excess of SMCC-TriNTA |
| 43 | The binding between the TriNTA-GB1 and Foldon resin is weak | The resin is not saturated by Foldon; the incubation time is too short; residual imidazole is present in the buffer | Use at least 20 mg of Foldon per 150 mg of dry resin; incubate the TriNTA-GB1 and the Foldon resin for at least 30 minutes; make sure that no imidazole is present on the resin nor in the TriNTA-GB1 buffer; repeat the purification procedure a few times to achieve high TriNTA-GB1 purity |
| 52 | The efficiency of the cross-linking reaction is low | The His6-tags of the TM protein are too close in space; the protein concentration is low; the cross-linker is too short | When multiple His6-tags are too close in space, the binding stoichiometry between His6-tag and TriNTA may not be 1:1 anymore. Introduce the His6-tag based on the protein structure so that the His6-tags are fairly distant; add charged residues in the proximity of the His6-tag to introduce electrostatic repulsion near the latter; use concentrated protein; use longer cross-linkers |
Anticipated Results
The mixed PRE experiment above (Part 2a) can only address whether the TMD is oligomer, but cannot determine the oligomerization number, which is an essential parameter for NMR structure determination. The OG-label method (Fig. 3b) was developed as a standard tool for addressing TMD oligomeric state in bicelles. It can be considered as a standard because the use of a soluble crosslinkable protein (SCP) as the crosslinking readout of the TMD oligomer makes the method much less sensitive to variations in the ability of the target TMDs to be crosslinked. This method is demonstrated here for the trimeric Fas-TMD.
The GB1 protein was the SCP used, and was linked to a TriNTA molecule via a PEG-2-SMCC (succinimidyl 4-(N-maleinidomethyl)cysclohexane-1-carboxylate); the efficiency of conjugating GB1 and TriNTA was ~90%. The TriNTA-GB1 was purified using the His6-tag column and the purified TriNTA-GB1 migrated as a 10 kDa band on SDS-PAGE. For the OG-label experiment, the Fas-TMD with a C-terminal His6-tag was expressed, purified, and reconstituted as described above (see Part 1). To prevent undesirable crosslinking between Fas-TMD and GB1, we first blocked all the active amine groups with the addition of 100-fold molar excess of Sulfo-NHS Acetate to the bicelle sample. Excessive Sulfo-NHS Acetate was removed by dialysis while tightly controlling the bicelle q. After dialysis, 30 μM Fas-TMD (monomer concentration) was mixed with 45 μM TriNTA-GB1 to ensure the His6-tagged Fas-TMDs were saturated with TriNTA-GB1. The mixture was then treated with 1 mM of BS3 for 30 min, followed by incubation with various concentration (0.1, 0.3, 1.0, and 3 mM) of glutaraldehyde for 5 min. The crosslinking reaction was quenched with a 20 mM Tris buffer (pH 7.5). As a negative control, 1 mM of BS3 and 1 mM of glutaraldehyde were sequentially added to 45 μM TriNTA-GB1 in the absence of the His6-tagged Fas-TMD. SDS-PAGE of the crosslinked GB1 showed oligomeric species up to trimer and that the trimer band intensified with increased glutaraldehyde. The results indicate that the Fas-TMD in bicelles forms trimers (Fig. 3c). In addition to the Fas-TMD, the OG-labeled has been demonstrated for higher-order oligomers such as the p7 protein from the Hepatitis C virus (HCV)22. In that case, OG-label managed to detect hexamers (Fig. 3d).
The major potential pitfall of the OG-label method is the situation when multiple His6-tags in the TMD oligomer are very close, as this could result in the binding of one TriNTA-GB1 to multiple His6-tags and thereby underestimate the oligomeric state. An effective solution to this problem is to insert a linker between the His6-tag and the protein to spread the His6-tags from different protomers. If the 1:1 binding between TriNTA-GB1 and His6-tag still cannot be achieved for extremely tight TMD oligomers, charged residues such as argines can be added before and/or after the His6-tag sequence to separate His6-tags by electrostatic repulsion.
Part 3. JCH-modulated NOE experiment for detecting unambiguous inter-protomer NOEs
Materials
BIOLOGICAL MATERIALS
~1:1 mixture (wt/wt) of 2H,15N-labeled protein (perdeuterated or partially deuterated) and 13C-labeled protein reconstituted in deuterated DMPC/DHPC bicelles (q = 0.5) in the appropriate NMR buffer.
EQUIPMENT
All the equipment necessary for handling of recombinant proteins
NMR spectrometer fully-equipped for triple-resonance experiments of biological macromolecules equipped with a cryogenically cooled probe head with z-field gradients. For measuring NOESY experiments, the use of high magnetic field is recommended (800–900 MHz)
NMR data-processing software (e.g., TOPSPIN, VNMR, NMRPIPE, etc.)
Visualization and analysis software for NMR spectra (e.g., TOPSPIN, CARA, Sparky, NMRView, NmrDraw, CCPN, etc.)
Procedures
Sample preparation
-
1
Reconstitute the protein sample as indicated in Part 1 (Steps 48–53).
NMR experiments
-
2
Set the probe temperature to the desired value (e.g. 308 K). Insert the sample in the magnet and wait ~15 minutes for the sample to achieve temperature stability. Lock, tune and shim the magnet. Activate the auto-shim.
-
3
Determine the pulse lengths of hard 90° pulses for 1H, 13C and 15N (in the direct, indirect and indirect acquisition mode setup, respectively).
-
4
Measure a 1D 1H NMR spectrum to determine the actual bicelle q of the sample. If needed, adjust it to 0.5 as explained in Part 1 (Step 54).
-
5
Acquire a high resolution 2D 1H-15N TROSY-HSQC spectrum as 2D reference for the subsequent NOESY analysis and process it with standard parameters. Typically, raw data are multiplied by an apodization function, zero-filled and Fourier-transformed in both dimensions. Proper zero-and first-order phase correction and baseline correction in both dimensions are applied after the Fourier transformation.
-
6
Acquire a high resolution 2D 1H-13C HSQC spectrum as 2D reference for the NOESY analysis and process it with standard parameters. Typically, raw data are multiplied by an apodization function, zero-filled and Fourier-transformed in both dimensions. Proper zero-and first-order phase correction and baseline correction in both dimensions are applied after the Fourier transformation.
-
7
Create a new experiment for acquiring the 3D JCH-modulated NOE spectrum. Set the experimental parameters as described in Box 3. Based on the previously acquired 2D 1H-15N TROSY-HSQC spectrum (Step 5), set the proper carrier positions and spectral windows. Set the recovery delay to at least 1.2 s and the NOE mixing time (τmix) to 200 ms.
-
8
Acquire the 15H-1H and 1H-1H planes to verify that all the parameters have been set correctly and that the experiment is working as expected. CAUTION. The two interleaved spectra are stored in the 15N dimension; therefore, to correctly process the 15H-1H plane it is necessary to first split the FID in the two sub-spectra.
-
9
Set the number of 1H and 15N increments to a resolution of ~8–10 and ~30–35 ms, respectively. CAUTION. The actual number of points along the 15N indirect dimension is half than what set, because the two interleaved spectra are stored in this dimension.
-
10
Acquire the 3D JCH-modulated NOE spectrum.
Box 3. Pulse sequence of the 3D JCH-modulated NOE experiment.
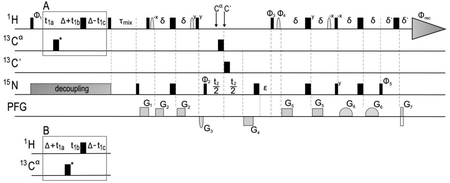
The experiment is based on the 3D 15N-edited NOESY-TROSY-HSQC. Narrow and wide bars represent 90° and 180° pulses, respectively. The pulses are applied along the x-axis unless indicated otherwise. The arrows indicate switching of the 13C carrier frequency to 56 (Cα) or 176 (C’) ppm. The 13C pulse with asterisk (23.5 μs @ 800 MHz) is applied at 60 ppm and inverts aliphatic carbons; the other 13C pulses (35.6 μs @ 800 MHz) selectively invert either Cα or Cʹ on carrier. 15N decoupling is achieved using the WALTZ-16 (1.5 kHz) sequence. The Gaussian shaped 1H pulses for rotating water are 1 ms long applied on-resonance. The pulse field gradients (PFGs) are applied along the z-axis. The gradient durations and strengths are: G1 (2000 μs, 48%), G2 (1200 μs, 35%), G3 (1000 μs, −45%), G4 (2500 μs, −50%), G5 (1200 μs, 33%), G6 (2200 μs, 35%), and G7 (251 μs, 50%), with 100% = 0.56 T/m. G1, G2, G4, G5 and G7 are rectangular, and G3 and G6 have a sine shape. The delays are: Δ = 4.0 ms; δ = 2.4 ms; ε = the duration of G4; δʹ = 0.5 ms; τmix = 200 ms. The phase cycles are: Φ1 = x, -x; Φ2 = 2(y), 2(-y); Φ3 = -y; Φ4 = y; Φ5 = -x; Φrec = x, 2(-x), x. The boxes A and B show two interleaved variants during 1H frequency labeling that result in 0 and 8 ms J(1H-13C) evolution, respectively. The semi-constant time evolution delays during 1H frequency labeling are: t1a = t1/2; t1b = t1a - t1c; t1c = Δ/N1 (where N1 is the number of total increments in the t1 dimension). Frequency discrimination in the 1H indirect dimension is achieved with States-TPPI incrementation of Φ1 and Φrec. For frequency discrimination in the 15N dimension, the first FID is acquired with G4 and G7 having the opposite polarity, then a second FID is acquired for each t2 delay, with Φ3, Φ4, Φ5 incremented by 180° and G4 and G7 having the same polarity. The data are processed in the echo/anti-echo fashion.
Spectra processing
-
11
Split the free induction decay (FID) along the 15N dimension to obtain the two sub-data sets, which follow the pathway A and B in Box 3. Process the two spectra with standard parameters. Typically, raw data are multiplied by an apodization function, zero-filled and Fourier-transformed in all dimensions. Proper zero-and first-order phase correction and baseline correction in both dimensions are applied after the Fourier transformation. ? TROUBLESHOOTING
-
12
Combine the two sub-data sets by summing and subtracting their FIDs to obtain the spectra selective for the intra- (A+B) and inter-NOEs (A-B), respectively. Process the two spectra with identical parameters as used for those in Step 11.
Timing
Steps 1, Sample preparation, 2 days
Steps 2–10, NMR experiments, 10–12 days depending on the sample concentration and spectrometer sensitivity
Steps 11–12, Spectra processing, 1 hour
Troubleshooting
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
| 11 | No inter-NOEs are observed | The 2H,15N- and 13C-label proteins have not been mixed adequately | Mix the two preparations before performing CNBr cleavage |
| Positive inter-NOEs are present in the sub-spectrum A, but negative inter-NOEs are weak or not present in the sub-spectrum B | NMR relaxation is more pronounced in the pathway B of the experiment | Perform the analysis as described. Inter-NOEs in the inter-NOEs selective spectrum (A-B) may appear weaker, but intra-NOEs will still be completely canceled |
Anticipated Results
Since the regular 15N- or 13C- edited NOESY spectrum is usually very complex, it is crucial to first detect exclusively inter-chain NOEs using mixed sample consisting of differently isotopically labeled chains as described in the protocol. We again use the Fas-TMD as an example to illustrate the expected results of this experiment.
We used a mixed sample in which half of the Fas-TMD were (15N/2H)-labeled and the other half 15% 13C-labeled to measure exclusively NOEs between the 15N-attached protons of one subunit and aliphatic protons of the neighboring subunits (Fig. 4a). The non-deuterated protein was 15% 13C-labeled for recording the 1H-13C HSQC spectrum as an internal aliphatic proton chemical shift reference while providing stereospecific assignment of leucine and valine methyl groups50. As elaborated above in Part 2a, adequate mixing of two differently labeled proteins is critical for this type of experiment. We found that HPLC profile can provide useful information of the miscibility of the TMDs. For example, the HPLC elution peaks of deuterated and protonated Fas-TMD are separated (Box 4a), indicating that the Fas-TMD is monomeric in the HPLC buffer. Therefore, mixing separately purified Fas-TMD immediately after HPLC elution should achieve thorough mixing. In contrast, the deuterated and protonated TMDs of HIV-1 gp41 eluted as one peak (Box 4b), suggesting that the protein oligomerized even in the HPLC organic solvent. For gp41 TMD, it is thus important to mix the differently labeled protein before CNBr cleavage. For the mixed Fas-TMD sample, the simple 15N-edited NOESY-TROSY-HSQC spectrum was recorded and showed obvious inter-chain NOE peaks between backbone amide and methyl groups (Fig.4c). These visible NOEs must be inter-chain because the 15N labeled Fas-TMD is > 98% deuterated as indicated by the mass spec analysis (Fig.4b).
Figure 4. Detection of inter-chain NOEs.
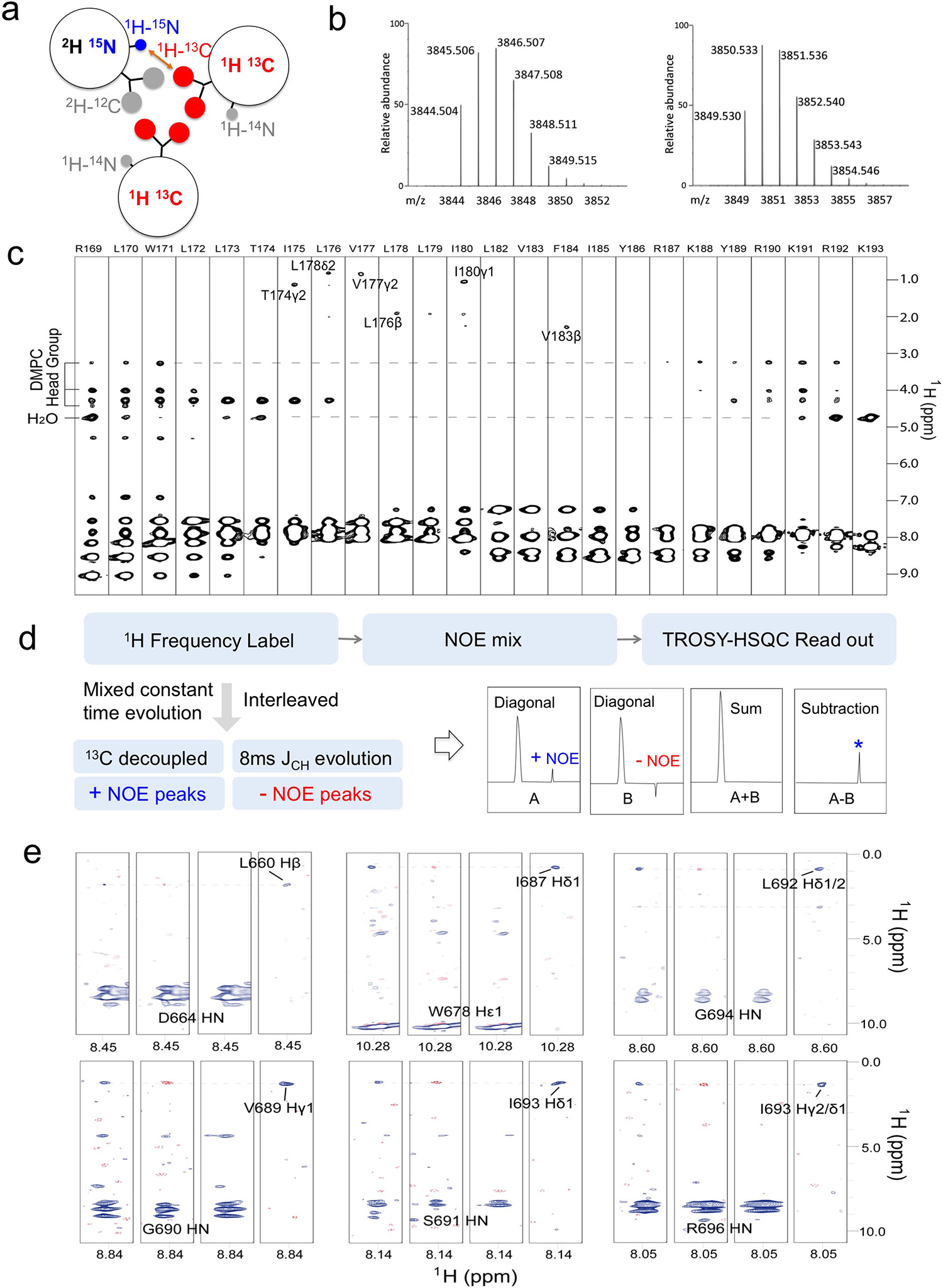
(a) Isotopic labeling scheme for detecting inter-chain NOEs. For regular NOESY experiments, the (15N, 2H)-labeled chain is perdeuterated (>98%) while the other half is either (1H, 15% 13C)- or (1H,13C)-labeled. When using the JCH-modulated NOE experiment, the (15N, 2H)-labeled chain may have a lower deuteration level, while the other half is uniformly 13C-labeled. (b) LC/MS analysis of a perdeuterated Fas-TMD sample (see Box 2), showing the calculated mass distribution assuming 100% deuteration at non-labile positions (right) and experimental mass distribution (left). The two dominant peaks in the theoretical spectrum are at 3850 Da and 3851 Da, while the corresponding peaks in the experimental spectrum are at 3845 Da and 3846 Da. The 5 Da difference means that on average only 5 out of 288 deuteration positions of the Fas-TMD are protonated, corresponding to a deuteration level of 98.3%. (c) Residue-specific strips from the 3D 15N-edited NOESY-TROSY-HSQC spectrum (NOE mixing time = 200 ms) recorded at 800 MHz and 303 K using a Fas-TMD sample containing 50% (15N, 2H)-labeled and 50% (1H, 15% 13C)-labeled chains. The labeled cross-peaks are inter-chain NOEs between the backbone amide and side-chain aliphatic protons. (d) Flow diagram of the interleaved, JCH-modulated NOE experiment (Box 3). Inter-chain NOEs are positive in A (JCH evolution = 0) and negative in B (JCH evolution = 8 ms). A+B selects for the intra-chain NOEs and A-B selects for the inter-chain NOEs. (e) Residue-specific strips from the JCH-modulated NOESY (NOE mixing time = 200 ms) recorded at 800 MHz and 308 K using a HIV-1 gp41 MPER-TMD sample containing 50% (15N, 2H)-labeled and 50% (1H, 13C)-labeled chains. For each selected residue, four strips are shown from left to right: A (positive inter-NOEs, blue), B (negative inter-NOEs, red), A+B (inter-NOEs canceled), and A-B (inter-NOEs selected). In this example, the inter-chain NOEs selected are: D664HN-L660Hβ, W678Hε1-I687Hδ1, G694HN-L692Hδ1/2, G690HN-V689Hγ1, S691HN-I693Hδ1, and R696HN-I693Hγ2/δ1.
Box 4. Evaluation of the tendency of TM chains to mix within oligomers by HPLC.
In this experiment, 1:1 ratio of (15N, 2H)-labeled and (1H, 15% 13C)-labeled TrpLE-TMDs are mixed before CNBr cleavage. After CNBr cleavage, the tendency of the differently labeled TMDs to stay oligomeric in strong organic solvent can be evaluated by HPLC elution profile.
(a) HPLC chromatogram (left) of a mixed sample of the Fas-TMD, showing that the deuterated and the protonated proteins are completely separated. MALDI-TOF mass spectroscopy analysis of the two peaks (right) confirmed that peaks 1 and 2 are the deuterated and protonated Fas-TMD, respectively. (b) HPLC chromatogram (left) of a mixed sample of HIV-1 gp41 TMD, showing that the deuterated and the protonated proteins are still associated even in the highly denaturing HPLC solvent. MALDI-TOF mass spectroscopy (right) confirmed that both labeled species are in the same elution peak.
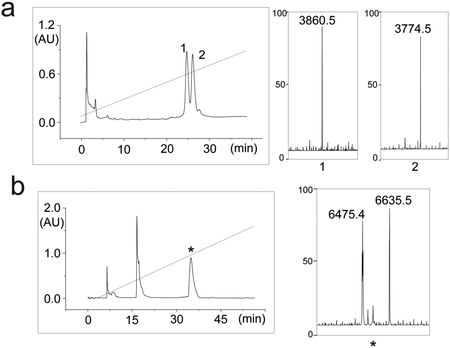
For certain applications where the protein cannot be perdeuterated due to high cost, the JCH-modulated NOE experiment23 can be used to selectively detect NOEs between 15N-attached 1H and 13C-attached 1H. For this experiment, the mixed sample should contain 50% (15N, ~85% 2H)-labeled and 50% (13C, 1H)-labeled chains (Fig.4a). The JCH-modulated NOE experiment is based on the regular 3D 15N-edited NOESY-TROSY-HSQC, in which the 1H evolution period before the NOE mixing is changed to a “mixed constant-time” evolution to introduce 1H-13C J evolutions (Fig.4d). Two interleaved 3D spectra are recorded (Box 3). In one case, the total J evolution is 0 ms, and the inter- and intra- molecular NOE peaks are both positive (Fig.4d). In the other case, the total J evolution is 8 ms, making inter-molecular NOE peaks negative and intra-molecular NOE peaks positive (Fig.4d). Collecting the two data sets interleaved with each other allow us to add the two spectra to see only intra-molecular NOEs or subtract the two spectra to see only inter-molecular NOEs. This method was applied to a fragment of the HIV-1 gp41 protein containing the MPER (membrane-proximal external region) and TMD, designated MPER-TMD. As shown in Fig.4e, the inter- and intra- molecular NOEs could be distinguished by simple addition and subtraction of the interleaved spectra23.
Part 4. Oligomerization solution and structure determination
Materials
EQUIPMENT
TALOS+ program (https://spin.niddk.nih.gov/bax/software/TALOS/)
XPLOR-NIH program (https://nmr.cit.nih.gov/xplor-nih/)
ExSSO program (http://www.csbio.sjtu.edu.cn/bioinf/ExSSO/)
Molecular graphics software (e.g. PyMOL, UCSF Chimera, etc.)
Procedures
Generation of the monomer structure
-
1
Run TALOS+ to predict the protein phi and psi backbone torsion angles providing the protein residue sequence and the available assigned chemical shifts (HN, Hα, Cα, Cβ, Cʹ, N). ? TROUBLESHOOTING
-
2
Use XPLOR-NIH to build the protein monomer structure using the dihedral restraints from Step 1. CRITICAL. Include only the dihedral restraints classified as “Good” by TALOS+.
Exhaustive search to determine the mode of oligomeric assembly
-
3
Use the ExSSO program to perform an exhaustive search of the symmetric conformational space guided by inter-protomer NOEs to identify the mode of assembly of the symmetric oligomer (Steps 4–7).
-
4
Set up the parameters of the configure.py script by providing i) the protein name ($proName), ii) the file path of the protomer structure (from Step 2), iii) the file path of the inter-NOE restraints table (determined using the 3D JCH-modulated NOE experiment, see Part 3), iv) the number of protomers in the oligomer (as determined by OG-label, see Part 2b). Instructions on the format to be used for ii) and iii) can be seen by typing $ python check_format.py -p $homeDir/$proName/param -t pdb and $ python check_format.py -p $homeDir/$proName/param -t noe, respectively.
-
5
Run the script by typing: $ python configure.py. The file param, containing all the user defined parameters, is created in $homeDir/$proName/.
-
6
Perform the exhaustive search of the symmetric conformational space by running the following script: $ python exhaustive_search.py -p $homeDir/$proName/param. Representative conformations of the solution(s) found are stored in $homeDir/$proName/$proName.cs. ? TROUBLESHOOTING
-
7
Generate a representative structure for each solution found in Step 6 by running the following script: $ python get_structure.py -p $homeDir/$proName/param. The calculated structures ($proName_*.pdb) are stored in $homeDir/$proName/ and sorted from best to worst according to the ExSSO scoring system.
Final structural refinement
-
8
Use the best scorer from Step 7 as starting model for structural refinement with XPLOR-NIH.
-
9
Identify more self-consistent inter-chain NOEs from the regular 15N-edited and 13C-edited NOESY spectra. Run XPLOR-NIH with the new NOE restraints to improve the structural ensemble.
-
10
Update the starting model with the average structure of the new structural ensemble calculated in Step 9.
-
11
Repeat Steps 9–10 iteratively until no new NOEs are found or until the desired RMSD of the structural ensemble (15 lowest energy structures out of 150 calculated) is reached.
Timing
Steps 1–2, Generation of the monomer structure, 1 day
Steps 3–7, Exhaustive search to determine the mode of oligomeric assembly, 1 day
Steps 8–12, Final structural refinement, days to weeks depending on the quality of the NMR spectra and the user experience
Troubleshooting
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
| 1 | TALOS+ predictions are not classified as “Good” | Chemical shift may be not calibrated; chemical shift assignments may be too incomplete | Calibrate the chemical shifts against DSS; acquire additional NMR spectra to improve the chemical shift assignments |
| 6 | More than one solution is found | The number of inter-NOEs may be insufficient | Improve the NOESY assignment. |
Anticipated Results
The main purpose of using the ExSSO program is to examine whether the inter-chain NOE restraints from Part 3 above are sufficient to derive a unique solution of oligomeric assembly, and if so, to provide a starting model of the oligomer for further structural refinement. As explained in the original ExSSO paper39, for TMD oligomers that are higher than dimer, the inter-chain NOE restraints all have two-fold directional ambiguity. The ExSSO program performs exhaustive search of the symmetric conformational space to resolve such ambiguity; it is demonstrated below for the Fas-TMD. In the ExSSO algorithm, each Fas-TMD monomer is treated as a rigid body whose orientation and position relative to the symmetry axis were evaluated in the context of the symmetric oligomer and against the inter-chain NOE restraints. The monomer structure (Fig. 5a) was initially constructed with the backbone dihedral angles derived from chemical shifts (using the TALOS+ program51). The oligomerization number was determined to be 3 by SDS-PAGE (Fig. 2d) and OG-label (Fig. 3b). The algorithm assigns the symmetry axis (Z-axis) and samples the orientation of each protomer by performing an Euler rotation around its center-of-mass. The reoriented protomers are placed at distance r between the Z-axis and their center-of mass. Then, the oligomer structure is evaluated against the inter-chain restraints (from Fig. 4c and 4e) using the ExSSO scoring system.
Figure 5. Structure determination of TM domains.
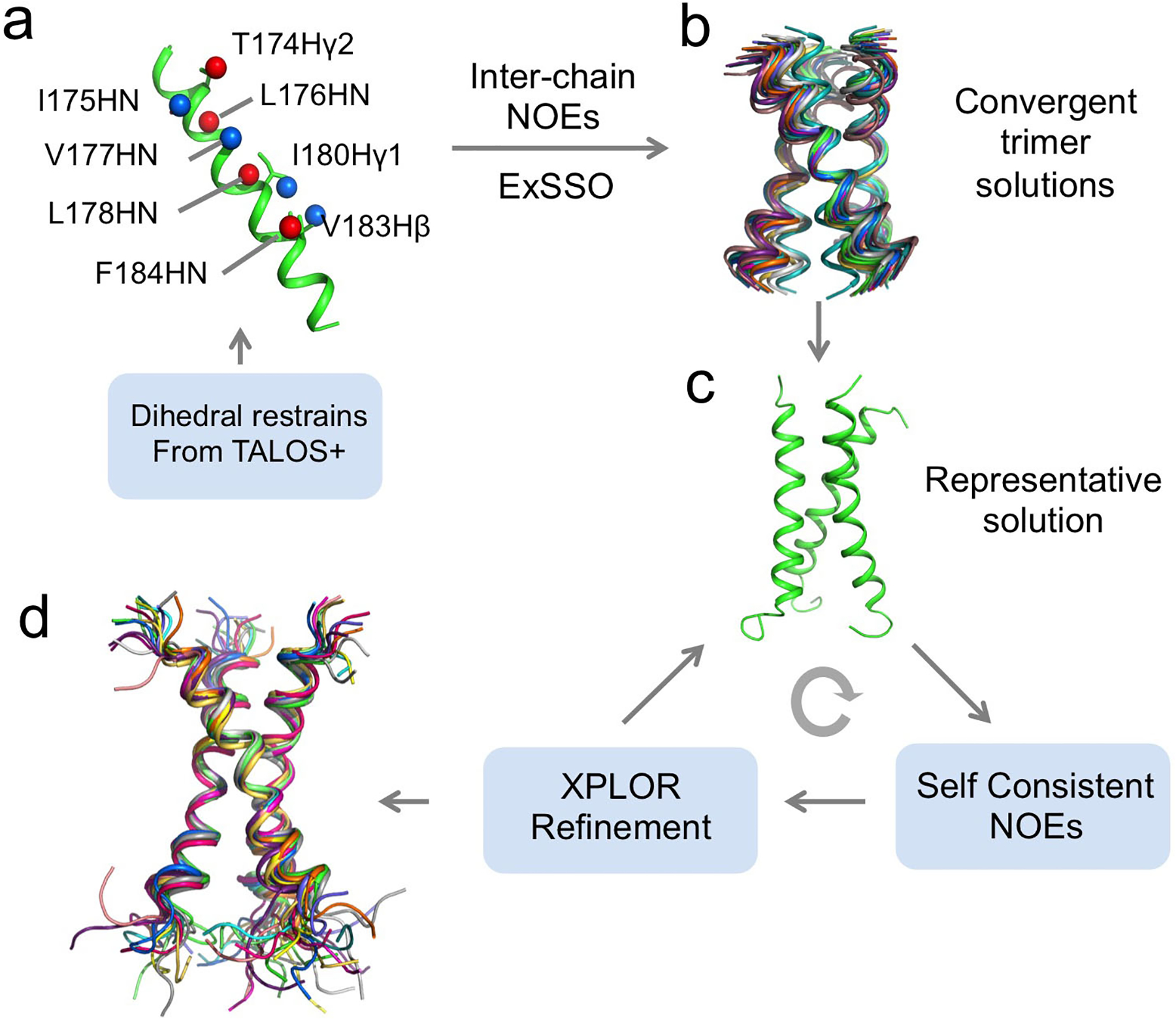
The protocol used to calculate the structure of oligomeric TM domains is demonstrated for the Fas-TMD. (a) The monomer structure was generated using backbone dihedral angles restraints derived from chemical shifts using TALOS+. Backbone amide and side chain protons exhibiting inter-chain NOEs are highlighted as spheres. Red and blue spheres represent inter-protomer pairs. (b) Exhaustive search performed (ExSSO program) to resolve the directional ambiguity of inter-chain NOE restraints and to derive convergent assembly solutions. Trimer was used for the Fas-TMD based on the OG-label result in Fig. 3C. (c) A representative trimer structure was selected and used as the starting model for further structural refinement using the XPLOR-NIH program. Additional self-consistent NOEs were added and the structure was updated. The process was repeated iteratively. (d) The final structural ensemble of 15 low-energy structures has backbone and heavy atom RMSDs of 0.829 Å and 1.392 Å, respectively.
The procedure was repeated for the Fas-TMD trimer to cover the entire symmetry space, and showed that the top conformation clusters converged to the same mode of trimeric assembly (Fig. 5b). The best scorer (Fig. 5c) was used as the starting model for further structural refinement, which involved 1) identifying more self-consistent inter-chain NOEs from the regular 15N-edited and 13C-edited NOESY spectra, and 2) updating the structure in XPLOR-NIH40 with the new NOE restraints. The above two steps were performed iteratively until the structural ensemble had a root-mean-square deviation (RMSD) of ~0.829Å and ~1.392Å for backbone and heavy atoms, respectively (Fig. 5d).
Part 5. Characterization of the protein transmembrane partition
Materials
BIOLOGICAL MATERIALS
15N-labeled protein reconstituted in DMPC/DHPC bicelles (q = 0.5) in the appropriate NMR buffer
REAGENTS
1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC, Avanti Polar Lipids, Inc., cat. no. 850305)
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC, Avanti Polar Lipids, Inc., cat. no. 850345)
2-(14-Carboxytetradecyl)-2-ethyl-4,4-dimethyl-3-oxazolidinyloxy, free radical (16-DSA, Sigma-Aldrich, cat. no. 253596)
Gadolinium (III) 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetate (Gd-DOTA; Macrocyclics, cat. no. M-147). CAUTION. May cause respiratory and skin irritation.
EQUIPMENT
All the equipment necessary for handling of recombinant proteins
NMR spectrometer fully-equipped for triple-resonance experiments of biological macromolecules equipped with a cryogenically cooled probe head with z-field gradients
NMR data-processing software (e.g., TOPSPIN, VNMR, NMRPIPE, etc.)
Visualization and analysis software for NMR spectra (e.g., TOPSPIN, CARA, Sparky, NMRView, NmrDraw, CCPN, etc.)
Data analysis software (e.g., ORIGIN, MATLAB, etc.)
REAGENT SETUP
Gd-DOTA, 600 mM.
Dissolve 97.9 mg of Gd-DOTA in 250 μL of NMR buffer. Split the solution in 25 μL aliquots and store them at −20°C. Use the solution within 1 year.
Gd-DOTA, 200 mM.
Dilute 1:3 the 600 mM Gd-DOTA mM solution with the NMR buffer (e.g. 10 μL of 600 mM Gd-DOTA in 20 μL of NMR buffer). Store at RT and use for the duration of the NMR titration.
16-DSA, 20 mM.
Dissolve 1.0 mg of 16-DSA in a bicelle solution of q = 0.5 previously prepared dissolving 9 mg of DMPC and 12.04 mg of DHPC in 130 μL of NMR buffer. The solubilization of 16-DSA in the bicelles can be aided by flash-freezing and then thawing the solution a couple of times, as well as by gentle sonication. Store the solution at RT and use for the duration of the NMR titration.
Procedures
Sample preparation
-
1
Reconstitute the protein sample as indicated in Part 1 (Steps 48–53).
NMR experiments
-
2
Set the probe temperature to the desired value (e.g. 308 K). Insert the sample in the magnet and wait ~15 minutes for the sample to achieve temperature stability. Lock, tune and shim the magnet. Activate the auto-shim.
-
3
Determine the pulse lengths of hard 90° pulses for 1H and 15N (in the direct and indirect acquisition mode setup, respectively).
-
4
Measure a 1D 1H NMR spectrum to determine the actual bicelle q of the sample. If needed, adjust it to 0.5 as explained in Part 1 (Step 54).
-
5
Acquire a high-resolution 2D 1H-15N TROSY-HSQC spectrum that would serve as reference for the titration. CRITICAL STEP. The recovery delay should be set at least to 3.5 s to prevent the occurrence of significant longitudinal PRE.
-
6
Process the spectrum with standard parameters. Typically, raw data are multiplied by an apodization function, zero-filled and Fourier-transformed in both dimensions. Proper zero-and first-order phase correction and baseline correction in both dimensions are applied after the Fourier transformation.
-
7
Remove the sample from the magnet. Add the desired amount of paramagnetic probe (Gd-DOTA or 16-DSA) to the sample (see Step 9 for quantities) and mix well.
-
8
Repeat Steps 2–3 and 5–6 to acquire a 2D 1H-15N TROSY-HSQC spectrum for the actual paramagnetic probe concentration. Use identical acquisition and processing parameters as for the reference spectrum. CRITICAL STEP. When performing the titration with Gd-DOTA, high concentration of the latter (~10–20 mM) broadens the water signal and hinders the shimming of the magnet. Make sure a good shimming is achieved before recording the spectra. ? TROUBLESHOOTING
-
9Repeat Steps 7–8 to acquire all the points of the titration. After the reference spectrum, the following points are typically measured:
- [Gd-DOTA]: 2, 4, 6, 8, 10, 15, 20 mM (e.g. for a 360 μL sample, add 3.6 μL of 200 mM Gd-DOTA solution for the first five points, then 3.0 μL of 600 mM Gd-DOTA solution for the last two. Dilution can be neglected in the calculation).
- [16-DSA]: 0.6, 1.2, 1.8, 2.4, 3.0, 3.6, 4.2 mM (e.g. for a 360 μL sample, add for each point 10.8 μL of 20 mM 16-DSA solution. Dilution can be neglected in the calculation). ? TROUBLESHOOTING
Data analysis
-
10
Load the assignment peak list on top of the 2D 1H-15N TROSY-HSQC spectra. Fine-adjust the peak positions to exactly match the top of the NMR cross-peaks. Export the NMR peak intensities of all the spectra. Discard overlapping peaks from the analysis.
-
11
Normalize the peak intensities of all the spectra with respect to those of the reference spectrum (I/I0).
-
12For each residue, generate a (I/I0) vs. [paramagnetic probe] plot and fit it to the following equation with two unknown parameters (PREamp and τ):
where I and I0 are the peak intensities in the presence and absence of the paramagnetic agent, respectively, [paramagnetic probe] is the concentration of the paramagnetic agent (Gd-DOTA or 16-DSA), τ is the decay constant, and PREamp is the PRE amplitude.(Eq. 1) -
13
Plot PREamp vs. (residue number) to obtain information on the topology of the protein (e.g. how many times the protein crosses the bilayer center) or for obtaining structural information for structure validation/refinement (the slopes of the plot reflect the relative orientation of the different protein segments with respect to the bicelle normal axis).
-
14
Determine the protein symmetry axis. Rotate the protein structure so that its symmetry axis is parallel to the bicelle bilayer normal (rZ axis). Place arbitrarily the protein along the axis, assigning rZ = 0 to the position of a chosen protein amide proton. Calculate the position of all the other amide protons along the axis (rZ, Å) with respect to the assigned rZ = 0.
-
15
Apply an offset to the previously assigned rZ = 0 (e.g. ±1, ±2, ±3 Å, etc.) and recalculate the position of all the amide protons along the rZ axis based on the new rZ = 0. Repeat the procedure for several offsets, generating enough rZ data sets to probe the entire protein.
-
16
For each rZ data set, calculate the absolute values of the measured rZ (|rZ|) and plot PREamp vs. |rZ|.
-
17Fit each PREamp vs. |rZ| curve to the following sigmoidal function with four unknown parameters (PREampmin, PREampmax, rzI and SLOPE):
where PREampmin and PREampmax are the limits within which PREamp can vary for a given protein system, rzI is the inflection point (the distance from the bilayer center at which PREamp is halfway between PREampmin and PREampmax), and SLOPE is a parameter which reports the steepness of the curve at the inflection point.(Eq. 2) -
18
Compare the adjusted coefficient of determination (R2adj) of each fitting and identify which rZ data set (or which offset applied to the initially assigned rZ = 0 (Step 14)) yields the best fitting (highest R2adj). The corresponding protein position would be very close to the actual protein partition in the bicelle.
-
19
Starting from the best rZ data set identified in Step 18, repeat Step 15 but applying smaller offsets to achieve better accuracy (e.g. ±0.1, ±0.2, ±0.3 Å, etc.).
-
20
Repeat Steps 16–18. The best fitting of PREamp vs. |rZ| to Eq. 2 (highest R2adj) provides the accurate membrane partition of the protein (rZ data set), with the bilayer center corresponding to the assigned rZ = 0.
Timing
Steps 1, Sample preparation, 2 days
Steps 2–9, NMR experiments, 3–6 days depending on the sample concentration and spectrometer sensitivity
Steps 10–20, Data analysis, 1 day
Troubleshooting
| Step | Problem | Possible reason | Possible solution |
|---|---|---|---|
| 8 | Gradient shim fails at high concentration of Gd-DOTA | Gd-DOTA generates a strong PRE effect on the D2O signal | Adjust the field to center the deuterium signal, lock and load the shim map used for the previous point of the titration. Perform the gradient shim. If it still fails, shim manually on Z, X, Y, XZ, YZ, and Z. |
| 9 | The 20 mM 16-DSA solution turns unclear after a few hours | The [16-DSA] is close to its maximum solubility in bicelles | Sonicate the solution for 5 minutes right before use; prepare a new 16-DSA solution at lower concentration (~15 mM) |
Anticipated Results
The PPT protocol can be used to generate rather quantitative information about protein partition in the bilayer region of the bicelles, and it is demonstrated here for the Fas-TMD trimer in bicelles (Box 5). For this application, (15N, 2H)-labeled Fas-TMD was reconstituted in DMPC/DHPC bicelles with q = 0.5 (Box 5a), and Gd-DOTA was used to generate solvent PRE. We emphasize that for the PPT method to be applicable, bicelles with q ≥ 0.5 should be used, because when bicelles are sufficiently wide, the lateral contribution to the measurable PRE becomes negligible, so that the observed PREs can be used directly to probe residue-specific depth immersion of the protein along the bicelle normal axis. Another critical aspect of PPT is to add the paramagnetic agent via serial titration rather than a single addition, as a wide range of paramagnetic field strengths (from very weak to strong) are required to probe both solvent-exposed and buried protein regions.
Box 5. The PPT method for determining membrane partition.
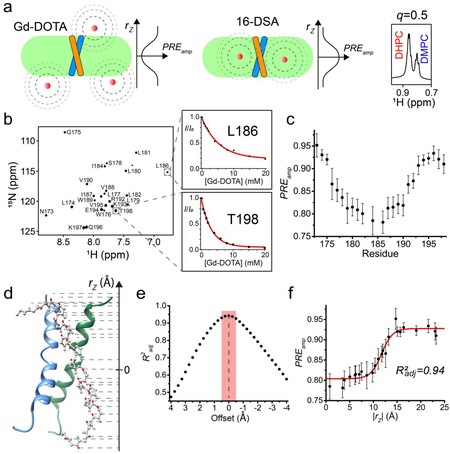
The stepwise results of the PPT analysis are shown here for the Fas-TMD trimer in bicelles. (a) Schematic illustration of bicelle-reconstituted TM domain titrated with either a water-soluble paramagnetic probe such as Gd-DOTA (left) or a lipophilic paramagnetic probe such as 16-DSA (middle). Next to each bicelle, the expected PRE profile along the bicelle normal is shown. 1H NMR spectrum of the bicelle-reconstituted Fas-TMD showed that the bicelle q was 0.5. (b) The 2D 1H-15N TROSY-HSQC spectrum of the Fas-TMD in bicelles (left). As examples, the I/I0 vs. [Gd-DOTA] plots of L186 (buried) and T198 (exposed) are shown. Data fitting (red line) to the exponential decay function (Eq. 1) yielded PREamp for the two residues. (c) The residue-specific PREamp plot. (d) The NMR structure of the Fas-TMD trimer with the symmetry axis aligned with the bicelle normal, showing the amide protons (red spheres) for which the PREamp has been determined. The dashed lines point to the projected positions (rZ) of the amide protons onto the bicelle normal. (e) The adjusted coefficient of determination (R2adj), from fitting the PREamp vs. rZ data to the sigmoidal function (Eq. 2), is used to evaluate the agreement between the assigned protein position and the PRE data. The position yielding the best fit (highest R2adj) represents the protein position relative to the bilayer center, with uncertainty estimated to be within ± 0.5 Å (shaded in red). (f) The best fit of the PREamp vs. rZ data to Eq. 2 from (e). All NMR spectra were acquired at 600 MHz at 303 K.
At each titration point, a 2D 1H-15N TROSY-HSQC spectrum was recorded to measure residue-specific PRE, defined here as I/I0, in which I and I0 are the intensities of a peak in the presence and absence of the paramagnetic agent, respectively (Box 5b). For each of the residues, the PRE titration curve was then fitted to the exponential decay function (Eq. 1) to derive the residue-specific PREamp (Box 5c). To determine the Fas-TMD position relative to the bilayer center, the PPT method exploits the knowledge of the protein structure and of its symmetry axis, which is parallel to the bicelle normal and defines the protein orientation. The only degree of freedom, the protein position along the bicelle normal, is determined by translating the protein as a rigid body until its position agrees with the measured PREamp. Specifically, the PREamp vs. residue number plot was converted to PREamp vs. bilayer immersion depth by calculating, for each residue i, the distance along the bilayer normal (rz) from the amide proton to an arbitrary reference point based on the protein structure (Box 5d). The PREamp vs. rz plot showed three main PRE regimes: i) a PRE-saturated regime near the bicelle surface; ii) a PRE-insensitive regime in the lipid core; iii) a PRE-sensitive regime connecting the previous two. Owing to the symmetric nature of the fast tumbling bicelles, the PREamp vs. |rz| can be described by the symmetric sigmoidal function (Eq. 2). Likewise, if the protein position in the bicelle is correctly assigned, the PREamp should fit well to the symmetric function. Based on the above rationale, the Fas-TMD trimer was systematically slid along the bicelle normal by moving the reference point (rz = 0) until the best fit of PREamp to Eq. 2 was reached (Box 5e). The Fas-TMD position yielding the highest adjusted coefficient of determination (R2adj) represents the membrane partition of the protein (Box 5f).
The PPT is not limited to solvent PRE. Lipophilic probe such as 16-DSA is an equally powerful probe that provides reciprocal PRE profile to that of the Gd-DOTA. In practice, Gd-DOTA is better suited for investigating residues well-buried in the bicelle, whereas 16-DSA is better at probing residues in the bicelle head-group region (Fig. 6a).
Figure 6. Analysis of transmembrane partition.
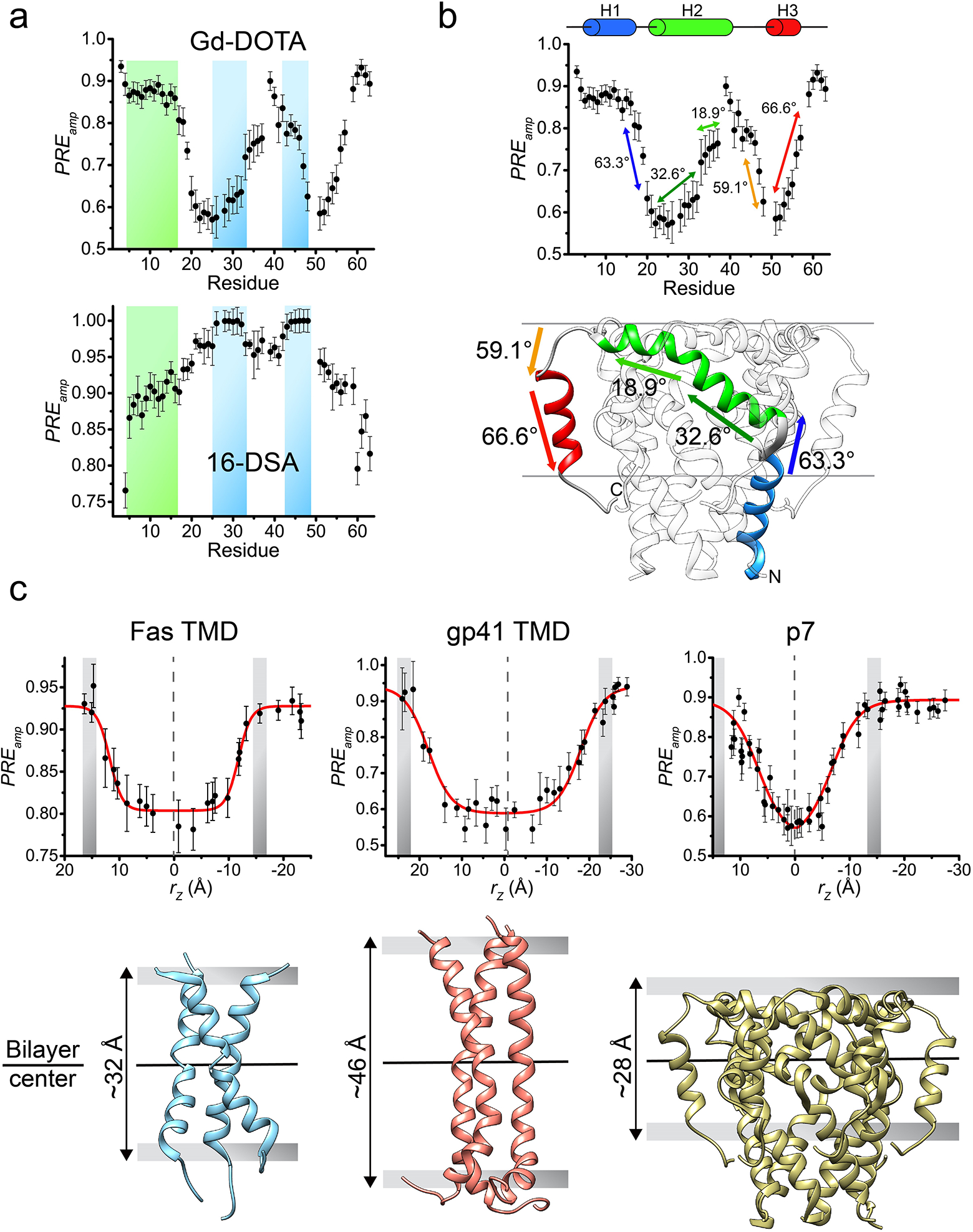
(a) Comparison between residue-specific PREamp derived from titrating Gd-DOTA (top) and 16-DSA (bottom), shown for the HCV p7 in bicelles with q = 0.6. The two plots are reciprocal and their analysis (as in Box 5) yielded identical membrane partition. As can be seen, Gd-DOTA is better suited for investigating residues well-buried in the bicelle (shaded in blue), showing steeper slope in the Gd-DOTA plot, whereas 16-DSA is more senstive at probing residues in the bicelle head-group region (shaded in green). (b) The PREamp plot from Gd-DOTA titration in (a) for showing correlation with the p7 structure. The arrows (in different colors) indicate the fragments of the p7 structure (bottom) for which the slope of PREamp correlates well with the steepness of the fragment in the hexamer structure. The steepness is reported as the angle of the fragment relative to the bilayer plane. (c) Membrane partition calibration curves obtained for the Fas-TMD (left), HIV-1 gp41 TMD (middle) and HCV p7 (right), describing the position of the proteins in the bicelle. The gray bars represent the bicelle boundaries, identified as the position at which the PREamp begin reaching saturation. At the bottom, the membrane partitions of the three proteins show different lipid bilayer thickness around the proteins (32, 46 and 28 Å for Fas-TMD, HIV-1 gp41 TMD and HCV p7, respectively).
The PPT method can also be useful for studying the topology of a TM protein with unknown structure, or for obtaining structural information for structure validation/refinement. In fact, the slopes of the PREamp versus residue number plot reflect the relative orientation of the different protein segments with respect to the bicelle normal axis. For example, the PREamp vs. residue number plot for the HCV p7 shows a “W” or “M” shape (for Gd-DOTA or 16-DSA, respectively), indicating that the protein crosses the bilayer center two times. Importantly, the slopes of the plot correlate very well with the orientation of the protein helices, providing useful information for structure validation or refinement (Fig. 6b)22.
Finally, the PPT method can also be used to estimate the lipid bilayer thickness around the TMD. In fact, the rz values in Fig. 6c at which the PREamp start becoming saturated should represent the bicelle boundaries. We note that for accurate determination of the membrane boundaries, the use of solvent PRE is preferred over that of lipophilic PRE, because soluble paramagnetic probes can usually be applied at higher concentration than the lipophilic probes, making it easier to reach PRE saturation. Using this simple PPT analysis, we found that different TMDs can indeed modulate different lipid bilayer thickness around them. For example, the bilayer thickness around the Fas-TMD is 32 Å (Fig. 6c, left)5. The bilayer thickness around the HIV-1 gp41 MPER-TMD is significantly larger, at 46 Å (Fig. 6c, middle)21. However, the HCV p7 caused drastic thinning of the bilayer to 28 Å (Fig. 6c, right)22, which is consistent with the fact that the ER membrane, where the p7 resides, is much thinner than other membranes.
Acknowledgements
This work was supported by a US National Institutes of Health grants GM116898 and AI127193 to J.J.C.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Call ME, Wucherpfennig KW & Chou JJ The structural basis for intramembrane assembly of an activating immunoreceptor complex. Nat Immunol 11, 1023–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Call ME & Wucherpfennig KW Common themes in the assembly and architecture of activating immune receptors. Nat Rev Immunol 7, 841–50 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Endres NF et al. Conformational coupling across the plasma membrane in activation of the EGF receptor. Cell 152, 543–56 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arkhipov A et al. Architecture and membrane interactions of the EGF receptor. Cell 152, 557–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu Q et al. Structural Basis and Functional Role of Intramembrane Trimerization of the Fas/CD95 Death Receptor. Mol Cell 61, 602–613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.MacKenzie KR, Prestegard JH & Engelman DM A transmembrane helix dimer: structure and implications. Science 276, 131–3 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Trenker R, Call ME & Call MJ Crystal Structure of the Glycophorin A Transmembrane Dimer in Lipidic Cubic Phase. J Am Chem Soc 137, 15676–9 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Call ME et al. The structure of the zetazeta transmembrane dimer reveals features essential for its assembly with the T cell receptor. Cell 127, 355–68 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bocharov EV et al. Spatial structure of the dimeric transmembrane domain of the growth factor receptor ErbB2 presumably corresponding to the receptor active state. J Biol Chem 283, 6950–6 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Lau TL, Kim C, Ginsberg MH & Ulmer TS The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J 28, 1351–61 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett PJ et al. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–71 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dev J et al. Structural basis for membrane anchoring of HIV-1 envelope spike. Science 353, 172–175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen W et al. Familial Alzheimer’s mutations within APPTM increase Abeta42 production by enhancing accessibility of epsilon-cleavage site. Nat Commun 5, 3037 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee J et al. Structure of the Ebola virus envelope protein MPER/TM domain and its interaction with the fusion loop explains their fusion activity. Proc Natl Acad Sci U S A 114, E7987–E7996 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kyte J & Doolittle RF A simple method for displaying the hydropathic character of a protein. J Mol Biol 157, 105–32 (1982). [DOI] [PubMed] [Google Scholar]
- 16.Gasteiger E et al. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31, 3784–8 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glover KJ et al. Structural evaluation of phospholipid bicelles for solution-state studies of membrane-associated biomolecules. Biophys J 81, 2163–71 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Piai A, Fu Q, Dev J & Chou JJ Optimal Bicelle Size q for Solution NMR Studies of the Protein Transmembrane Partition. Chemistry 23, 1361–1367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanders CR, Hare BJ, Howard KP & Prestegard JH Magnetically-Oriented Phospholipid Micelles As A Tool For The Study Of Membrane-Associated Molecules. Progress In Nuclear Magnetic Resonance Spectroscopy 26, 421–444 (1994). [Google Scholar]
- 20.Caldwell TA et al. Low-q Bicelles Are Mixed Micelles. The Journal of Physical Chemistry Letters 9, 4469–4473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piai A, Dev J, Fu Q & Chou JJ Stability and Water Accessibility of the Trimeric Membrane Anchors of the HIV-1 Envelope Spikes. J Am Chem Soc 139, 18432–18435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W et al. The Unusual Transmembrane Partition of the Hexameric Channel of the Hepatitis C Virus. Structure 26, 627–634 e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu Q et al. Structure of the membrane proximal external region of HIV-1 envelope glycoprotein. Proc Natl Acad Sci U S A 115, E8892–E8899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knoblich K et al. Transmembrane Complexes of DAP12 Crystallized in Lipid Membranes Provide Insights into Control of Oligomerization in Immunoreceptor Assembly. Cell Rep 11, 1184–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofer N, Aragao D & Caffrey M Crystallizing transmembrane peptides in lipidic mesophases. Biophys J 99, L23–5 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomaston JL & DeGrado WF Crystal structure of the drug-resistant S31N influenza M2 proton channel. Protein Sci 25, 1551–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper RS, Georgieva ER, Borbat PP, Freed JH & Heldwein EE Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat Struct Mol Biol 25, 416–424 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barbet-Massin E et al. Rapid proton-detected NMR assignment for proteins with fast magic angle spinning. J Am Chem Soc 136, 12489–97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andreas LB et al. Structure and Mechanism of the Influenza A M218–60 Dimer of Dimers. J Am Chem Soc 137, 14877–86 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Dam L, Karlsson G & Edwards K Direct observation and characterization of DMPC/DHPC aggregates under conditions relevant for biological solution NMR. Biochim Biophys Acta 1664, 241–56 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Galperin MY Structural classification of bacterial response regulators: diversity of output domains and domain combinations. J Bacteriol 188, 4169–82 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parkinson JS & Kofoid EC Communication modules in bacterial signaling proteins. Annu Rev Genet 26, 71–112 (1992). [DOI] [PubMed] [Google Scholar]
- 33.Stock AM, Robinson VL & Goudreau PN Two-component signal transduction. Annu Rev Biochem 69, 183–215 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Wolanin PM, Thomason PA & Stock JB Histidine protein kinases: key signal transducers outside the animal kingdom. Genome Biol 3, REVIEWS3013 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blacklow SC & Kim PS Protein folding and calcium binding defects arising from familial hypercholesterolemia mutations of the LDL receptor. Nat Struct Biol 3, 758–62 (1996). [DOI] [PubMed] [Google Scholar]
- 36.North CL & Blacklow SC Solution structure of the sixth LDL-A module of the LDL receptor. Biochemistry 39, 2564–71 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Caldwell TA et al. Low- q Bicelles Are Mixed Micelles. J Phys Chem Lett 9, 4469–4473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lata S, Reichel A, Brock R, Tampe R & Piehler J High-affinity adaptors for switchable recognition of histidine-tagged proteins. J Am Chem Soc 127, 10205–15 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Piai A, Shen HB & Chou JJ An Exhaustive Search Algorithm to Aid NMR-Based Structure Determination of Rotationally Symmetric Transmembrane Oligomers. Sci Rep 7, 17373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwieters CD, Kuszewski JJ, Tjandra N & Clore GM The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160, 65–73 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Mitra K, Ubarretxena-Belandia I, Taguchi T, Warren G & Engelman DM Modulation of the bilayer thickness of exocytic pathway membranes by membrane proteins rather than cholesterol. Proc Natl Acad Sci U S A 101, 4083–8 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharpe HJ, Stevens TJ & Munro S A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell 142, 158–69 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMahon HT & Boucrot E Membrane curvature at a glance. J Cell Sci 128, 1065–70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossman JS, Jing X, Leser GP & Lamb RA Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 142, 902–13 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu C et al. Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 135, 702–13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pielak RM, Oxenoid K & Chou JJ Structural investigation of rimantadine inhibition of the AM2-BM2 chimera channel of influenza viruses. Structure 19, 1655–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schnell JR & Chou JJ Structure and mechanism of the M2 proton channel of influenza A virus. Nature 451, 591–5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.OuYang B et al. Unusual architecture of the p7 channel from hepatitis C virus. Nature 498, 521–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang J, Pielak RM, McClintock MA & Chou JJ Solution structure and functional analysis of the influenza B proton channel. Nat Struct Mol Biol 16, 1267–71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Szyperski T, Neri D, Leiting B, Otting G & Wuthrich K Support of 1H NMR assignments in proteins by biosynthetically directed fractional 13C-labeling. J Biomol NMR 2, 323–34 (1992). [DOI] [PubMed] [Google Scholar]
- 51.Shen Y, Delaglio F, Cornilescu G & Bax A TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J Biomol NMR 44, 213–23 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]