

SUMMARY:
Integrin αvβ8 binds with exquisite specificity to latent transforming growth factor-b (L-TGF-β). This binding is essential for activating L-TGF-β presented by a variety of cell types. Inhibiting αvβ8-mediated TGF-β activation blocks immunosuppressive regulatory T-cell differentiation, which is a potential therapeutic strategy in cancer. Using cryo-electron microscopy, structure-guided mutagenesis, and cell-based assays, we reveal the binding interactions between the entire αvβ8 ectodomain and its intact natural ligand, L-TGF-β, as well as two different inhibitory antibody fragments to understand the structural underpinnings of αvβ8 binding specificity and TGF-β activation. Our studies reveal a mechanism of TGF-β activation where mature TGF-β signals within the confines of L-TGF-β and the release and diffusion of TGF-β is not required. The structural details of this mechanism provide a rational basis for therapeutic strategies to inhibit αvβ8-mediated L-TGF-β activation.
Graphical Abstract
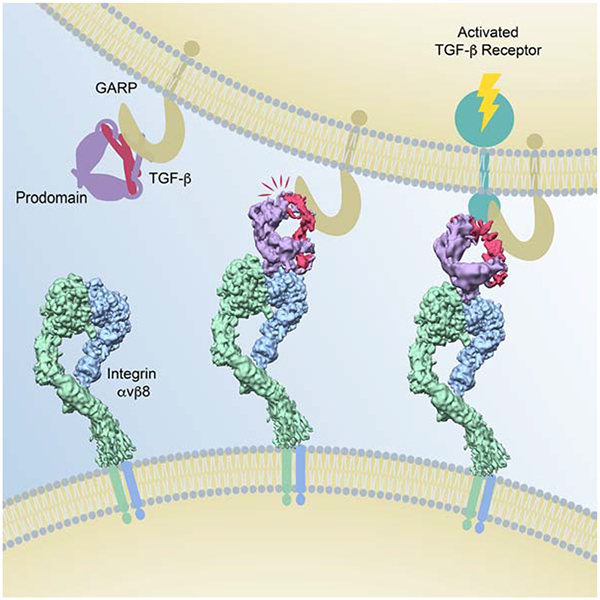
Analysis of intermediate conformations of the interaction between αvβ8 integrin and latent TGF-β suggest an activation mechanism that does not require release and diffusion of mature TGF-β, which has implications for current approaches to targeting TGF-β signaling therapeutically.
INTRODUCTION
Studies in the past three decades have elucidated the diverse and essential functions of TGF-β in immunity and vascular biology (Sanjabi et al., 2017; van Meeteren et al., 2011). While TGF-β and its receptors are ubiquitously expressed (Gordon and Blobe, 2008), TGF-β is always expressed in a latent form (L-TGF-β). During biosynthesis, TGF-β is cleaved by furin from its prodomain, latency associated peptide (LAP), but remains associated with it forming a latent complex, in which LAP forms a ring like structure shielding the active domains of mature TGF-β from its receptors (Fig. S1A)(Shi et al., 2011). Therefore, activation is a major point of regulation of TGF-β function (Miyazono et al., 1990). It is widely presumed that TGF-β activation requires mature TGF-β to be released from LAP to enable receptor binding (Annes et al., 2003; Massagué, 2012; Moses et al., 2016). The majority of therapeutics in development are conceptually tied to inhibiting freely diffusible TGF-β, but have been associated with toxicities (Akhurst, 2017). Targeting TGF-β activation thus has the potential to increase specificity while limiting toxicities associated with such global TGF-β inhibition strategies (Nishimura, 2009).
Integrin αvβ8 is particularly important for TGF-β activation since it is essential for T-cell, myeloid and endothelial cell differentiation during development (Aluwihare et al., 2009; Arnold et al., 2014; Qin et al., 2018; Travis et al., 2007). In post-natal life, αvβ8 plays important roles in fibroinflammatory processes (Kudo et al., 2012; Melton et al., 2010; Minagawa et al., 2014), and anti-tumor immunity (Takasaka et al., 2018). In particular, the immunosuppressive function of regulatory T cells (Tregs), which is a major mechanism of tumor immune evasion, is determined by the activation of L-TGF-β presented by GARP (glycoprotein A repetitions predominant) on their cell surface to αvβ8 (Stockis et al., 2009). How αvβ8 binds to and activates L-TGF-β has not been elucidated.
A structural model of TGF-β activation by a different integrin, αvβ6, suggests that L-TGF-β binding to αvβ6 triggers a global conformational change from extended-closed to extended-open, which allows actin cytoskeletal force to be transmitted through the b-subunit to release mature TGF-β from its latent complex (Fig. S1B)(Dong et al., 2017). However, the αvβ8 integrin has a divergent cytoplasmic domain that does not interact with the actin cytoskeleton and exists only in an extended-closed conformation whether alone or in complex with L-TGF-β (Cormier et al., 2018; Minagawa et al., 2014; Mu et al., 2002; Wang et al., 2017). Therefore, αvβ8-mediated L-TGF-β activation can occur without drastic conformational rearrangements and without cytoskeletal force. Thus, its mechanism of activation must be different from the model proposed for αvβ6 (Fig. S1C).
αvβ8 only binds efficiently to L-TGF-β, which is distinct from all other αv-integrins, which bind to many other ligands in addition to L-TGF-β (Humphries et al., 2006; Mu et al., 2002; Ozawa et al., 2016). Therefore, determining the basis of αvβ8 specificity for L-TGF-β is of general importance for understanding integrin-mediated TGF-β activation, and to guide development of therapeutic antibodies and integrin-specific small molecules, the latter of which has proven difficult (Hatley et al., 2018).
Using cryo-electron microscopy we determined structures of three complexes between the αvβ8 ectodomain and L-TGF-β, or two inhibitory Fabs, one of which is ligand-mimetic. Together with structure-guided mutagenesis, we reveal a binding intermediate and the two key regions that define the specificity of αvβ8 for L-TGF-β, suggesting an alternative strategy to inhibit αvβ8/L-TGF-β binding. The dynamic nature of αvβ8/L-TGF-β interactions suggest an alternative activation mechanism that does not require release and diffusion of mature TGF-β, which we confirmed through cell-based assays. Our findings present a model and conceptual framework to understand the cell-specific regulation of TGF-β activation.
RESULTS
Structure of the αvβ8/L-TGF-β complex
To gain mechanistic insights into TGF-β activation and αvβ8 ligand specificity, we used single particle cryo-EM to determine structures of a stable complex of the human αvβ8 ectodomain with porcine L-TGF-β1, which has 94% sequence identity with human L-TGF-β. This L-TGF-β has two point substitutions: C4S that increases expression and R249A that disrupts the furin cleavage to stabilize the L-TGF-β complex for structural studies. The biochemical relevance of this complex was established in previous studies (Shi et al., 2011). From the entire cryo-EM dataset, we isolated a subset of particles that give a structure with an overall nominal resolution of 3.3 Å, however, the local resolution varies (Figs. 1A–C and S2A). In our map, most of the integrin headpiece has well resolved side chains for reliable atomic model building. In the αv subunit, the resolution of the thigh domain near the headpiece is sufficient to separate β-strands and to trace the backbone, but it becomes less well-resolved towards the lower leg. In the β8 subunit, the resolution of the hybrid domain is also sufficient to trace the backbone of all β-strands with the bulky side chains resolved. However, the resolution of the PSI/EGF1 domain is significantly lower, suggesting that the linkage between the PSI/EGF1 and hybrid domains is flexible. Overall, our reconstruction suggests that there is considerable motion between the headpiece and the legs, which is consistent with our previous studies of the αvβ8 ectodomain (Cormier et al., 2018).
Fig. 1. The αvβ8 integrin ectodomain bound to L-TGF-β1.
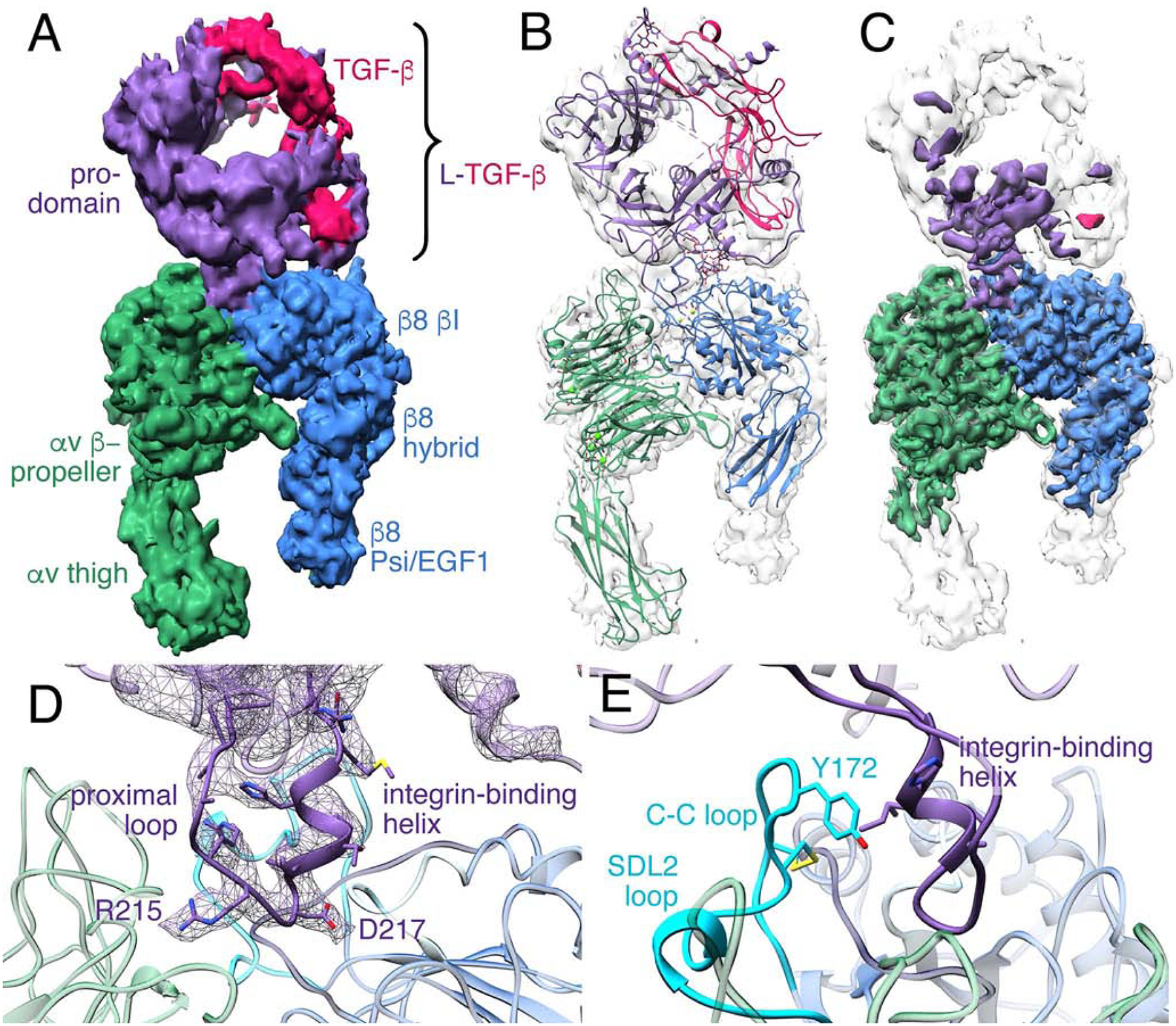
(A) Cryo-EM density map of αvβ8 integrin ectodomain with L-TGF-β1 bound. The map is displayed as unsharpened and at a low-threshold. The color code is as follows: integrin αv-subunit is green, integrin β8-subunit is blue, prodomain of L-TGF-β1 is purple, and mature TGF-β1 is red. (B) Ribbon diagram of αvβ8/L-TGF-β1 displayed within the density map shown in (A). (C) The sharpened cryo-EM map is shown in color, superimposed with the unsharpened map in semi-transparent white. (D) A ribbon diagram shows a close-up view of the binding interface between αvβ8 and L-TGF-β1. The EM density of the integrin-binding motif of L-TGF-β1, including proximal loop, RGD motif, and integrin-binding helix is shown in purple mesh. (E) Ribbon diagram showing the C-C loop within the SDL2 loop of the β8 subunit (cyan) relative to the L-TGF-β1 integrin-binding helix. Related information is in Fig. S1, S3.
The integrin-binding loop of L-TGF-β1 is strikingly well resolved with clear density for the side chains suggesting that this binding interface between integrin αvβ8 and L-TGF-β1 is very stable. In comparison, the rest of L-TGF-β1 is at a lower resolution (5–10Å) suited for rigid body fitting suggesting that bound L-TGF-β1 is flexible (Figs. 1B and 1C). Using the arm domain of L-TGF-β as a mainstay, the crystal structure of L-TGF-β1 (PDB: 3RJR (Shi et al., 2011)) is well accommodated by our map (Fig. 1B).
The L-TGF-β RGD loop is comprised of two main components: the proximal RGD loop, which shows significant variance in length and sequence between TGF-β1 and TGF-β3, and the RGDLXXI/L integrin-binding motif, which is comprised of the tripeptide RGD recognition motif followed by a consensus accessory binding site (218-LATI−221 in L-TGF-β1 and 244-LGRL−247 in L-TGF-β3) (Mu et al., 2002; Ozawa et al., 2016). From here on we refer to the RGDLXXI/L consensus motif as the integrin-binding motif. A crystal structure of L-TGF-β alone shows proximal loop is disordered, and LXXI/L motif forms a loop (Shi et al., 2011). When crystallized in complex with αvβ6, the LXXI/L motif organized into a short one and half turn a-helix (Dong et al., 2014; Dong et al., 2017), which we will refer to as the integrin-binding helix.
In our map, the density of the entire integrin-binding loop is well resolved. The RGD sequence is located in the ligand-binding cleft immediately followed by the integrin binding helix (Fig. 1D). Overall, the integrin-binding motif and helix and the αvβ8 ligand-binding cleft are similar to the crystal structures of the αvβ6 headpiece in complex with L-TGF-β1 or with a L-TGF-β3 RGD-peptide (Fig. S3) (Dong et al., 2014; Dong et al., 2017).
SDL2 is a loop between the α1 and α2 helices of β8 and is located at the back of the ligand binding cleft. A disulfide bond is formed between two conserved residues (C169 and C176) forming the SDL2 C-C loop. The SDL2 C-C loop bends inwards towards the integrin binding helix of L-TGF-β1 and positions its Y172 to form a hydrophobic patch with multiple residues of SDL1–3 (Figs. 1E, S3E). The ArgRGD interacts with the sidechain of αv D218. The AspRGD, together with S114 and S116 of the SDL1 α1-helix, coordinate the MIDAS cation consistent with other αv-integrins in complex with RGD (Fig. S3J–M) (Takagi, 2007).
The αvβ8/C6D4 structure reveals the unliganded integrin binding pocket
In our previous ligand-free structure of αvβ8, we found that SDL2 was flexible, and therefore could not be modeled (Cormier et al., 2018). To define the structure of αvβ8 in the ligand-free state, we used the Fab domain of C6D4 to capture SDL2 in a specific conformation. C6D4 is a potent neutralizing antibody that we previously developed to bind to the αvβ8 headpiece with high affinity and to effectively inhibit binding of L-TGF-β (Takasaka et al., 2018). To further facilitate high-resolution structure determination, we added an additional Fab, 11D12V2, that binds the αv-thigh. Since 11D12V2 is used solely for facilitating alignment and does not impact L-TGF-β binding nor headpiece conformation, for simplicity we refer to this complex as αvβ8/C6D4. We determined a cryo-EM structure of αvβ8/C6D4 in an extended conformation at an overall nominal resolution of 3.9 Å. With further focused alignment on the region of interest, which includes the headpiece and the variable domain of C6D4 Fab, the resolution was improved to 3.5 Å (Figs. 2A, 2B, S3C). The variable domains of C6D4 contact all SDL loops and D218 of the αv-head, thus directly competing with L-TGF-β (Fig. 2B, 3, S3C, Table S1, S3 and S4).
Fig. 2. Mechanisms of action of inhibitory C6D4 and C6-RGD3 Fabs.
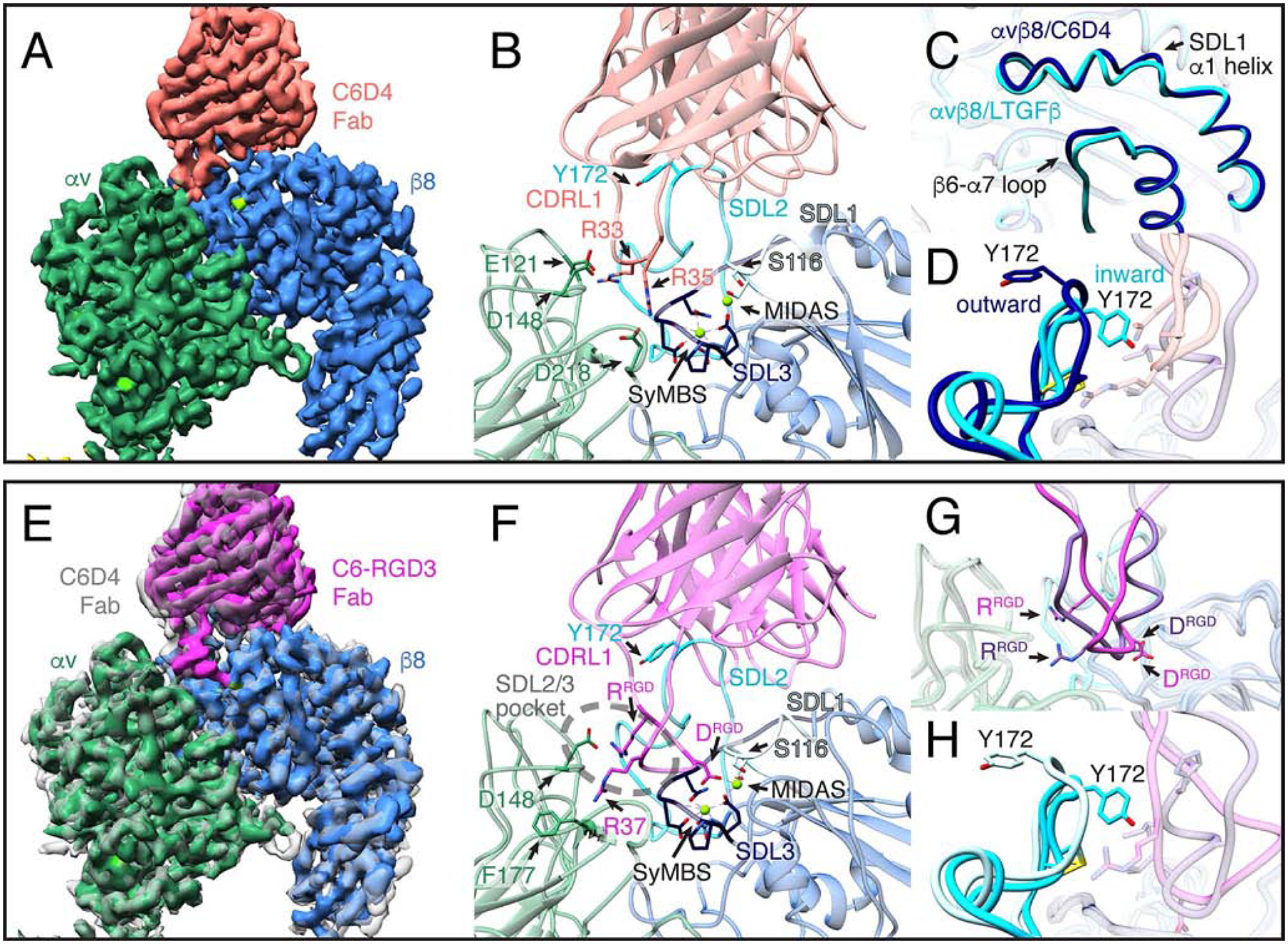
(A) A close-up view of the cryo-EM density map of the αvβ8/C6D4 complex. The full structure is shown in Fig. S2. The αvβ8 ligand-binding cleft is fully occupied by C6D4, demonstrating a steric mode of inhibition. Color code: αv is dark green, β8 is blue, and cations are bright green. (B) Close-up ribbon diagram of the αvβ8/C6D4 interface showing the position of the C6D4 Fab (coral) on the αvβ8 binding-cleft, highlighting interactions between the Fab CDRL1 loop R33 interacting with αv E121 and αv D148, and Fab R35 with αv D218, respectively (for all interactions see Fig. 3, S1, S3 and S4). Additional color code: SyMBS and MIDAS is green, SDL1 is light blue, SDL2 is cyan, SDL3 is dark blue. (C-D) Comparisons are shown between αvβ8/C6D4 and αvβ8/L-TGF-β1 with key changes occurring in β8 upon ligand binding noted. Color code: unliganded C6D4 structure is dark blue, liganded L-TGF-β structure is cyan. There are very subtle ligand-induced changes in the SDL1 α1 helix (~1 Å at the tip) and the β6-α7 loop (C). Y172 of the SDL2 loop is positioned outward relative to the ligand binding site in complex with C6D4 but moves inwards upon ligand binding (D). (E) Cryo-EM density map of αvβ8 (same colors as in A) in complex with the C6-RGD3 ligand-mimetic Fab (magenta) superimposed on the αvβ8/C6D4 map (semi-transparent gray). Aligning the αv-subunit β-propeller domain of the two maps shows a slight shift in the angle of C6-RGD3 Fab binding. (F) Close-up ribbon diagram of the αvβ8/C6-RGD3 binding interface shows the alternative RGD binding mode with C6-RGD3 Fab. Key contacts are: ArgRGD interacting with αv D148 at the edge of the β8 proximal SDL2/3 pocket (dashed ellipse) and C6-RGD3 Fab AspRGD interacting with the MIDAS cation. C6-RGD3 Fab R37 (30-LGRGDLGRL−38) interacts with αv F177 (for all interactions see Fig. 3, S2–4). Integrin color codes are as in B, with C6-RGD3 in magenta. (G) Superimposition of the C6-RGD3 ligand-mimetic RGD loop (magenta) and the native L-TGF-β loop (purple) shows that C6-RGD3 binds in an alternative binding conformation. (H) β8 Y172 in the αvβ8/C6-RGD3 complex (light blue) is in the outward position compared to the αvβ8/LTGFb complex (cyan). Related information is in Fig. S2–4.
Fig. 3. L-TGF-β and inhibitory antibodies contact multiple overlapping residues in β8 SDL loops.
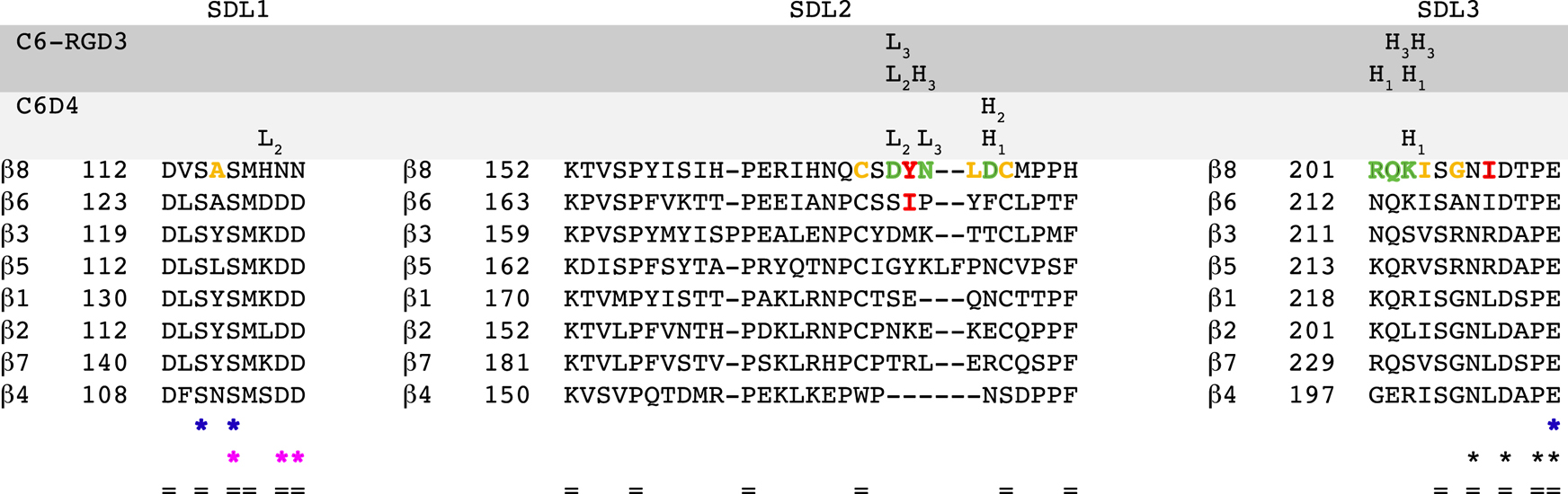
Sequence alignments for integrin b subunits are shown in decreasing order of homology to β8. Each SDL region is indicated above the sequence. Each CDR VH and VL loop of C6-RGD3 and C6D4 is shown above the specific amino acids which it contacts. The β8 SDL residues which contact L-TGF-β as well as C6D4 or C6-RGD3 are shown in green letters. The β8 SDL residues which form the hydrophobic patch that interact with the integrin binding helix of L-TGF-β1 are shown in orange letters. The residues that are mutated in Fig. 5 are shown in red letters. The cation binding sites are indicated below the sequence by colored asterisks with MIDAS in blue, ADMIDAS in magenta, and SyMBS in black. Conserved residues are indicated (=). Related information is in Table S1–4.
Although C6D4 interacts with the ligand-binding pocket, it does not participate in coordination of the MIDAS cation (Fig. 2B). Comparison between the αvβ8/L-TGF-β1 and αvβ8/C6D4 structures shows that upon ligand binding the S116 at the tip of the SDL1 α1-helix position shifts slightly towards the ligand-binding pocket (~1 Å) but the majority of the SDL1 α1-helix itself remains at a similar position (Fig. 2C, S3G, S3R and S3S). This result is surprising because other integrin structures show both larger shifts and straightening of the SDL1 α1-helix upon ligand binding (Fig. S3N–S) (Dong et al., 2014; Dong et al., 2017; Zhu et al., 2013).
The differences in SDL2 positioning are the largest overall conformational differences between the αvβ8/L-TGF-β and αvβ8/C6D4 complexes. C6D4 captures the SDL2 C-C loop in an outward position through interactions with complementary determining regions (CDR) CDRL1,L3 and CDRH1,H3 (Fig. 2D, 3 and Table S1). Notably, the inhibitory Fab Act-1 captures the β7 integrin SDL2 C-C loop in the same position, indicating that SDL2 can assume similar ligand free conformations in other integrins (Fig. S3T and S3U) (Yu et al., 2012). Overall, our structure demonstrates that C6D4 captures αvβ8 in a conformation corresponding to an unliganded state and completely occludes the ligand binding cleft. Thus, it provides a clear mechanism of inhibition and explains the potency of C6D4 in vitro and in vivo (Takasaka et al., 2018).
An intermediate αvβ8 binding state
We hypothesized that the inward position of the SDL2 C-C loop in the αvβ8/L-TGF-β structure is a critical feature that facilitates formation of the integrin-binding helix, which is necessary for positioning of the RGD sequence into the αvβ8 ligand binding pocket. To address this hypothesis, we took advantage of the binding footprint of C6D4, which covers SDL1, 2 and 3 in addition to having its CDRL1 extended into the ligand-binding cleft. It is therefore possible to exchange the CDRL1 with the RGD loop of L-TGF-β while still maintaining binding of the Fab to αvβ8. This would allow visualization of the RGD loop in the ligand binding cleft with SDL2 in an inactive conformation. We designed a chimeric Fab with the CDRL1 loop replaced by the RGD loop of L-TGF-β3. This antibody (C6-RGD3) binds to and blocks function of αvβ8 (Fig. S3V–W). We determined the structure of αvβ8 in complex with C6-RGD3 and 11D12V2 Fabs to a resolution of 3.9Å (Figs. 2E, S2C and S3D). The angle and position at which the C6-RGD3 binds integrin is very similar to C6D4 with the major differences in the engineered CDRL1 loop (Fig. 2E–G, 3, Table S2, S3, and S4). The conformation of this loop is distinct from the canonical position of the RGD in the αvβ8/L-TGF-β1 structure with ArgRGD in a pocket formed by SDL2/3, which we will refer to as the proximal SDL2/3 pocket, where it interacts with D148 of the αv-head (Fig. 2F, S3D and 3SF). This residue is conserved in α8, the only other integrin α-subunit that binds to L-TGF-β, suggesting it may play a key role in L-TGF-β binding (Table S3). In the αvβ8/C6-RGD3 structure, the integrin-binding helix does not form and SDL2 remains in an outward position as in the αvβ8/C6D4 complex (Fig. 2G, 2H, and S3D). In contrast, the position of the SDL1 α1-helix is similar to its position in αvβ8/L-TGF-β and the AspRGD coordinates with the MIDAS cation (Fig. 2F, 4 and S3H, S3R, S3S). These features suggest that this is an intermediate state of L-TGF-β binding and its biological relevance is tested below.
Fig. 4. The ADMIDAS cation is not present in αvβ8.
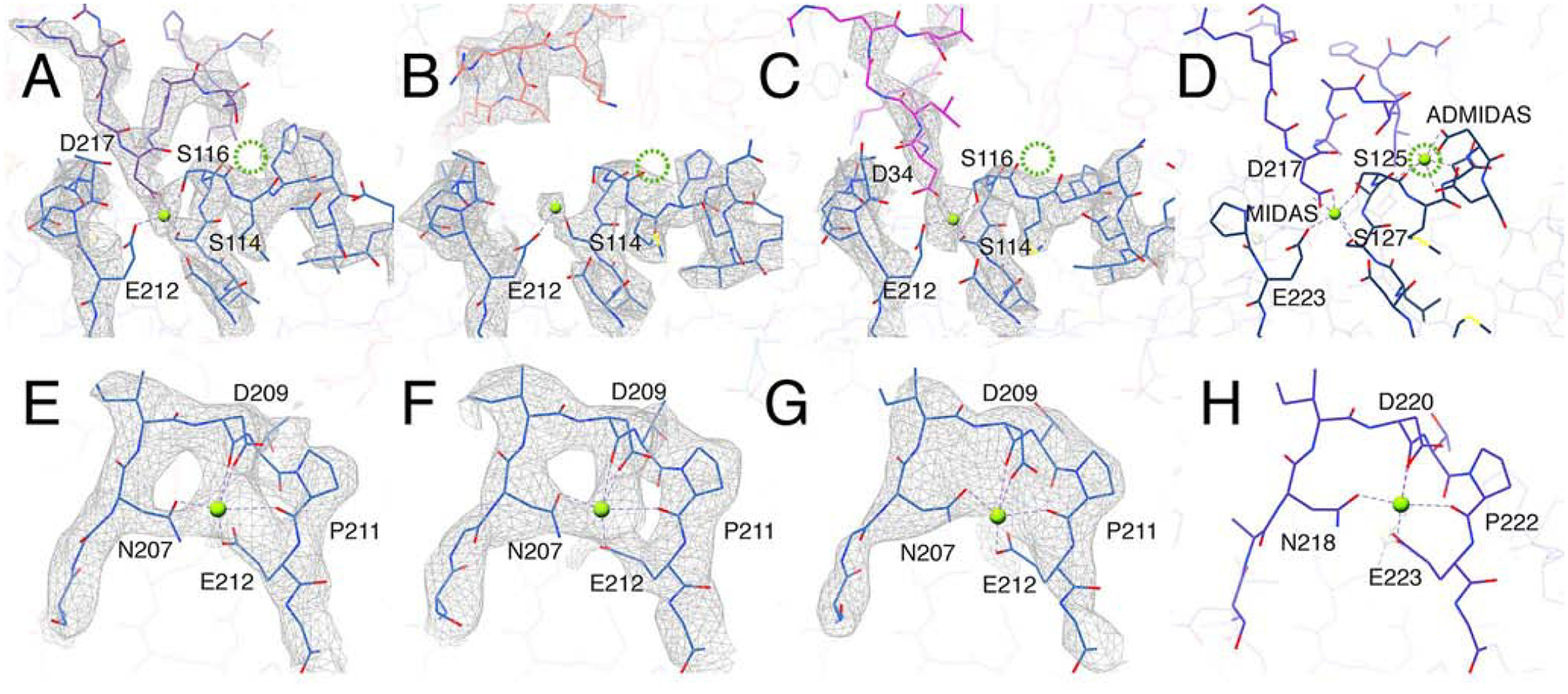
(A-C) The MIDAS cation binding site formed in αvβ8 integrin (blue) bound with: L-TGF-β1 (purple, A) C6D4 (coral, B) and C6-RGD3 (magenta, C). In all three structures there is clear density for the MIDAS cation (bright green). No density for the ADMIDAS cation is observed (the expected position for ADMIDAS is noted by the dotted green circle). Shown are the cation-coordinating residues of the integrin b subunit or the Asp of RGD of L-TGF-β (D217) or C6-RGD3 (D34). (D) The same binding interface in the αvβ6/LTGFβ crystal structure (5FFO (Dong et al., 2017)). (E-H) Maps and models of the SyMBS cation in αvβ8/L-TGF-β (E), αvβ8/C6D4 (F), αvβ8/C6-RGD3 (G), and αvβ6/LTGFβ crystal structure (5FFO) (H). All panels are color coded as in Fig. 2. Related information is in Fig. S3.
C6-RGD3 binds efficiently to αvβ8 and αvβ6, but not αvβ1, αvβ3, or αvβ5, mirroring the binding preference of these integrins to L-TGF-β (Fig. S3W). Since C6D4 does not bind to αvβ6, we assume that C6-RGD3 binding to αvβ6 is entirely through canonical positioning of the integrin-binding loop of CDRL1. We reasoned that the C6-RGD3 CDRL1 loop could also reach a canonical conformation with αvβ8 without a contribution from rest of the Fab binding footprint. Indeed, upon intensive classification, we identified a minor population (~6% of particles) that showed the C6-RGD3 integrin-binding motif in a canonical binding position with helix formation (Fig. S4). These results suggest that the C6-RGD3 Fab is able to bind to αvβ8 in two distinct modes, one reflecting an alternative L-TGF-β RGD position with no integrin-binding helix formation and no contact with the β8 SDL2, and the other, a canonical positioning of L-TGF-β RGD with the integrin-binding helix.
αvβ8 lacks the ADMIDAS cation
When αvβ8 is bound with L-TGF-β the position of the SDL1 α-helix tip is similar to the ligand-bound state of other integrins. In other integrins, the inward motion of the SDL1 α1-helix is accompanied by swinging out of the hybrid domain, a process that is inhibited by Ca2+ and stimulated by Mn2+(Zhu et al., 2013). We previously found that αvβ8 binding to L-TGF-β is not significantly enhanced by Mn2+ over Ca2+ and that hybrid domain swing out is not seen in Mn2+ containing buffers, suggesting that metal-binding properties are different for αvβ8 than other integrins studied so far (Minagawa et al., 2014). Existing crystal structures of integrin ligand binding pockets show that three adjacent conserved cation-binding sites SyMBS, MIDAS and ADMIDAS, are coordinated by residues in SDL1 and SDL3 (Takagi, 2007; Xiong et al., 2001). Such cation coordination is widely thought to be a key determinant of integrin binding affinity and conformational dynamics (Takagi, 2007). Although coordinating residues are conserved across all b-subunits in the MIDAS and SyMBS sites, in the β8 ADMIDAS site, two Asn residues replace otherwise conserved adjacent Asp residues. We find no density for the ADMIDAS cation in any of our structures while densities corresponding to the SyMBS and MIDAS cations are clearly observed (Figs. 4A–H). The absence of an ADMIDAS in αvβ8 provides a structural explanation for the lack of cation preference and movement of SDL1 α1-helix upon L-TGF-β binding, which are expected features in other integrins where conformational coupling between the hybrid domain and ligand binding were observed.
Flexibility of L-TGF-β when bound to αvβ8
To gain further insight into conformational flexibility of L-TGF-β1 bound to αvβ8, we analyzed the complete αvβ8/L-TGF-β1 dataset through comprehensive classification (Fig. S5). We define seven subclasses that represent conformational snapshots of the movements adopted between L-TGF-β1 and the integrin headpiece. Each structure exhibits high resolution in the integrin headpiece and binding interface (3.3–3.6 Å) and a well-defined shape for L-TGF-β1 prodomain. This conformational lineup shows different degrees of rotation between L-TGF-β1 and the αvβ8 headpiece (Fig. 5). In all seven classes the key structural elements that mediate major interactions between αvβ8 and L-TGF-β are similar to those shown in Fig. 1. The proximal RGD loop of L-TGF-β is not as well resolved in most of these structures, and the distal linker that follows the integrin binding helix is resolved but varies considerably from subclass to subclass. This suggests that the proximal RGD loop and the distal sequence act as flexible linkers, which allow the rest of L-TGF-β to tilt around the αvβ8 headpiece in a wide range. This allows the arm domain of L-TGF-β to make contacts with the β8 headpiece in some of the subclasses. The overall density of the prodomain and TGF-β itself varies noticeably indicating a range of plasticity throughout L-TGF-β (Fig. 5).
Fig. 5. L-TGF-β is flexible when bound to αvβ8.
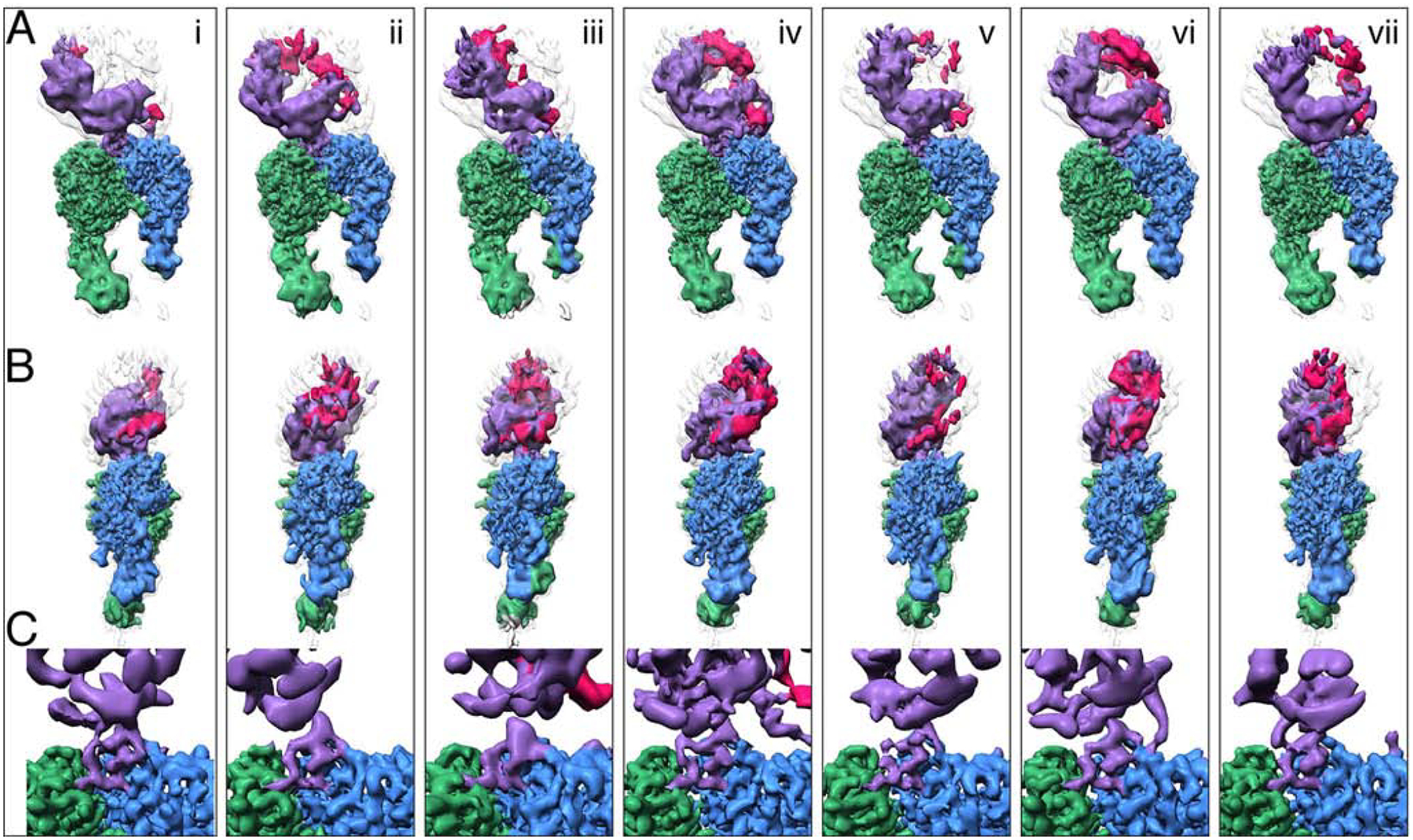
Seven structures (i-vii) illustrate the conformational variability of the αvβ8/LTGFβ complex. All structures are aligned to each other using the αv-subunit b-propeller domain. Each vertical panel shows two views (A, and B) of the same structure rotated 90˚ around the z-axis and a close-up of the integrin-ligand binding interface with the L-TGF-β integrin-binding helix (C), which is formed in all structures. Subclass (iv) is shown in Figs. 1, 2 and 4. Color coding is the same as for Fig. 2. For (A-B) the primary conformation is shown in colors, as defined in Fig. 1A, and all six other conformations are shown as semi-transparent to provide a reference for L-TGF-β’s range of motion. From left to right, the percentage of particles that went into each class are as follows: 14%, 14%, 14%, 19%, 16%, 11%, 12%. Related information is in Fig. S5.
The position of the SDL1 α1-helix is the same and no movement of the β8 hybrid domain is observed in any subclass. The lower leg domains in these subclasses are similarly and consistently at a lower resolution than the rest of the complex. This is in line with what we observed previously and suggests that movement in the lower legs is not correlated with the motion of bound L-TGF-β (Cormier et al., 2018). Such flexibilities in both αvβ8 and L-TGF-β are unlikely to generate any force to liberate the TGF-β from the latent complex. Therefore, our structural data reveal a model where TGF-β can be activated without being released from the latent complex. In such a model, the structural flexibility of L-TGF-β upon binding to αvβ8 would be sufficient to expose the active domain of TGF-β to its receptors.
Dynamic interactions within the ligand binding pocket determine αvβ8 specificity for L-TGF-β
Differences in the proximal SDL2/3 binding pocket and the SDL2 C-C loop define three distinct conformational states captured in our structures. We hypothesized that a series of sequential restructuring steps of the L-TGF-β integrin-binding motif and αvβ8 headpiece lead to stable ligand binding (Fig. 6A). To test this model, we carried out a series of mutations. To ensure that all mutants were properly folded, we confirmed that C6D4, which recognizes a non-linear epitope on the αvβ8 headpiece (Takasaka et al., 2018), binds to these mutants as expected (Fig. S6A).
Fig. 6. Structure based modeling of L-TGF-β binding to αvβ8.
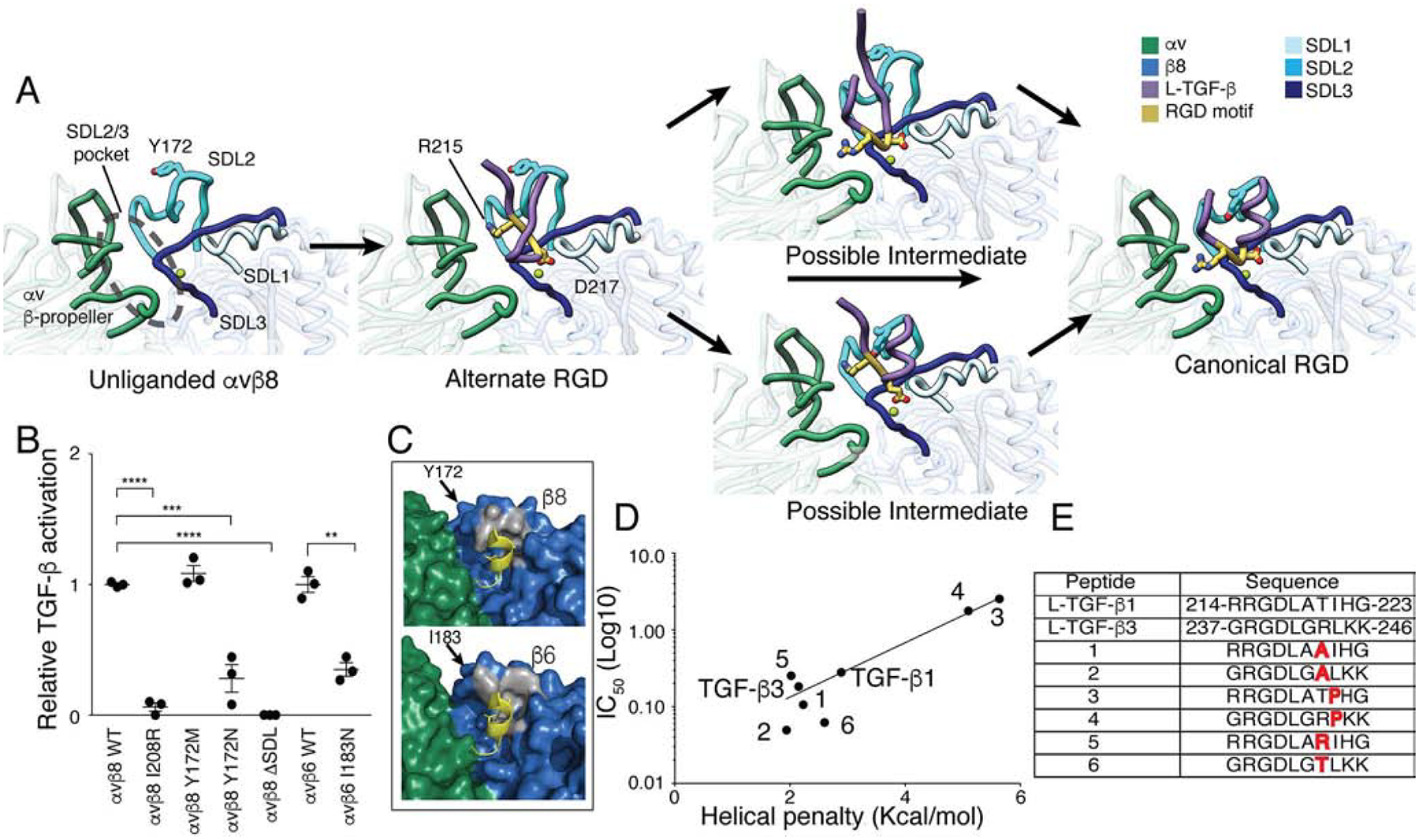
(A) A stepwise model of L-TGF-β binding to αvβ8. Our structures were used to define unliganded (αvβ8/C6D4), alternate (αvβ8/C6-RGD3) and canonical RGD binding modes (αvβ8/L-TGF-β). The hypothesized transitions between these binding states suggest possible intermediates where the integrin binding helix either forms before or after engaging the canonical RGD position (color coding as in Fig. 1). (B) TGF-β activation assays test the function of β8- and β6-subunit SDL mutants. Wild-type (WT) αvβ8 or αvβ6 or mutant receptors (β8: I208R, Y172M, Y182N, entire SDL2 deletion mutant (Δ-SDL), or β6: I183N) were stably expressed in CHO cells, sorted for uniform expression, confirmed for proper folding using C6D4 or anti-β6 antibodies (Fig. S6) and tested for ability to support TGF-β activation with TMLC reporter cells. Activation relative to αvβ8 WT expressing CHO cells is shown. (C) Shown is surface rendering of the b-subunit hydrophobic patch (grey) of αvβ8 (upper) and αvβ6 (lower) and the relationship of β8 Y172 and β6 I183 with the L-TGF-β integrin-binding helix (yellow sticks and cartoon). (D) Inhibition of L-TGF-β1 binding to αvβ8 by TGF-β1 and TGF-β3 integrin-binding motif peptides and Ala and Pro mutants was measured. Shown is the inhibitory concentration (IC50) plotted against helical penalty score (Pace and Scholtz, 1998). (E) Sequences of peptides used in (D) are shown. ** p=0.01, *** p=0.001, **** p=0.0001 by unpaired two-sided Student’s t test. Related information is in Fig. S6.
To rule out the possibility that the alternative binding mode is an artifact of grafting the RGD L-TGF-β loop onto the C6D4 Fab, we mutated the SDL3 reside I208 to its corresponding Arg residue in β3 (I208R). Based on our structure and hypothesis, this substitution would occlude the proximal SDL2/3 binding pocket and prevent the alternative binding mode, while not affecting the placement of RGD in the canonical binding position (Fig. S6E–G). The I208R mutant completely inhibited L-TGF-β binding and activation suggesting that the alternative RGD binding mode is a required initial or intermediate step in the L-TGF-β binding and activation process (Fig. 6B). Mutational validation of the importance of the proximal SDL2/3 pocket including I208 in ligand binding, reinforces the potential of this domain as a therapeutic target.
The differences in position of the SDL2 C-C loop in the αvβ8/C6D4 and αvβ8/RGD3 compared to the αvβ8/L-TGF-β structures suggest a critical role for Y172 in the formation of the L-TGF-β integrin-binding helix (Fig. 6C). The conservation of the hydrophobic patch in β6 suggests a similar role for β6 I183 (Fig. 6C). The positioning of the aromatic ring of Y172 in our structure, along with the knowledge that in αvβ6 the corresponding residue is an Ile (I183) suggest that the hydrophobic interaction is imperative for L-TGF-β integrin-binding helix formation (Fig. 3). In order to test the importance of this residue we made the following substitution mutants: one conservative, Y172M, and one non-conservative, Y172N. As expected, the conservative substitution, Y172M did not affect L-TGF-β activation. Replacing Y172 with the polar Asn significantly reduced L-TGF-β activation to a level similar to when the entire SDL2 region is removed (Fig. 6B) (Dong et al., 2014). A secreted version of αvβ8 expressing Y172A dramatically reduced L-TGF-β binding (Fig. S6C).
The highly flexible SDL2 is poorly conserved between integrin b-subunits and provides the main β8 residues of the hydrophobic patch interacting with the L-TGF-β helix. Despite large sequence variation in SDL2 between the β6 and β8 subunits, the footprint of hydrophobic interactions that promotes stabilization of the L-TGF-β helix appears to be absolutely conserved (Fig. 6C) (Dong et al., 2014). As a test of the generalizability of our model to the αvβ6 integrin, we made the non-conservative mutation of β6 I183N, which corresponds to β8 Y172N (Fig. 3). As expected I183N dramatically inhibits αvβ6-mediated L-TGF-β activation and binding (Fig. 6B, S6D). This suggests that the β6 and β8 SDL2 have similar roles.
Our data reveal two main features that are required for efficient L-TGF-β binding: a conserved hydrophobic residue in the β8 SDL3 I208 position of the proximal SDL2/3 pocket and a bulky hydrophobic residue at the tip of the SDL2 C-C loop. The β3 and β5 integrins contain a charged residue in the β8 SDL3 I208 position, while integrin β1, β2, β4 and β7 subunits do not have a bulky hydrophobic residue in the position of β8 SDL2 Y172 (Fig. 3). Thus, the absence of one or the other features explain the relatively inefficient L-TGF-β binding to these integrins (Dong et al., 2014).
Previous studies showed that peptides derived from the TGF-β3 compared to the TGF-β1 RGDLXXI/L sequence have increased ability to inhibit L-TGF-β binding to αvβ6 (Dong et al., 2014). Because the formation of integrin binding helix is a critical step in L-TGF-β binding to integrin, the ability of the LXXI/L motif to form a helix may underlie this difference. Therefore, we synthesized a series of peptide mutants that were designed to differ in their predicted abilities to form a helix. As expected, the ability to form a helix correlated with the IC50 of each peptide and the peptides with proline substitutions in the terminal hydrophobic position of LXXI/L that were designed to disrupt the helix had the highest IC50. Similarly, the TGF-β3 peptide has a lower IC50 than the TGF-β1 peptide because TGF-β3 peptide has a higher propensity to form a helix (Fig. 6D). Overall, our findings suggest that conformational dynamics within the L-TGF-β integrin-binding helix are critical for binding ability to αvβ8.
Integrin αvβ8-mediated TGF-β activation does not require TGF-β release
In the latent form, the active domain of TGF-β is shielded by a flexible loop of the prodomain known as the latency lasso. Without extensive conformational changes and cytoskeletal linkage, force cannot be transmitted through the β8 subunit to release TGF-β from its latent complex. However, the flexible nature of L-TGF-β we observed in our structure may expose the active domain of mature TGF-β without releasing it.
A recent crystal structure of the L-TGF-β/GARP complex reveals how GARP functions as a stable platform for L-TGF-β presentation, which suggests that a complex of αvβ8 with L-TGF-β/GARP forms between two opposing cells with one presenting αvβ8 and the other presenting L-TGF-β (Lienart et al., 2018). Furthermore, it is already known that L-TGF-β/GARP presenting immune cells also present its receptor, TGF-βR2 (Li et al., 2006). We thus envision a new TGF-β activation model in which L-TGF-β binding to αvβ8 exposes the active domain of mature TGF-β so that it can bind to TGF-βR2, recruit a type I TGF-βR (i.e. ALK5) to form the minimal TGF-β signaling module to initiate the TGF-β signaling cascade (Huang et al., 2011). We generated a structural model using our cryo-EM structure of αvβ8/L-TGF-β with crystal structures of L-TGF-β/GARP (Lienart et al., 2018) and TGF-β in complex with TGF-βR2 (Groppe et al., 2008; Hart et al., 2002; Radaev et al., 2010) (Fig. 7A). In this multicomponent model, TGF-β signaling can occur within the confines of L-TGF-β and release and diffusion of TGF-β is not required.
Fig. 7. A structural model of the αvβ8/L-TGF-β/GARP/TGF-βR2 complex predicts that releasing of mature TGF-β is not required for αvβ8-mediated TGF-β activation.
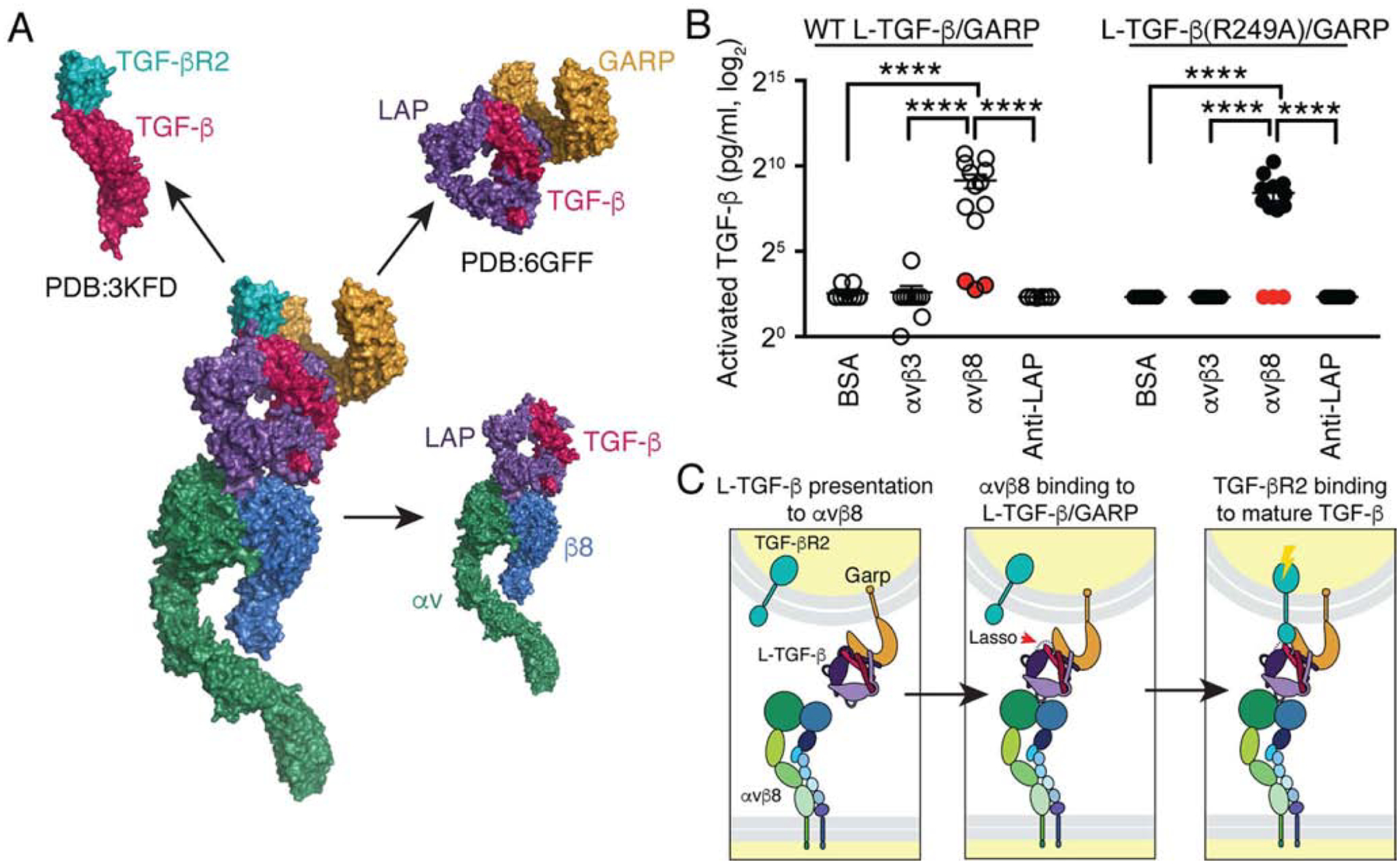
(A) Surface representation of a model of the putative complex derived from our αvβ8/L-TGF-β1 structures combined with the crystal structures of the GARP/L-TGF-β1 complex (PDB: 6GFF (Lienart et al., 2018)), and the TGF-β/TGF-βR2 complex (PDB: 3KFD (Radaev et al., 2010)) is shown. Color code: integrin αvβ8 (αv-green, β8-blue), L-TGF-β1 (LAP-purple, TGF-β-red), GARP-orange and TGF-βR2-teal. (B) WT L-TGF-β1/GARP or L-TGFβ1(R249A)/GARP expressed on the surface of TGF-β reporter cells (TMLC) show efficient TGF-β activation when plated on immobilized αvβ8. TMLC are stably transfected with a highly specific TGF-β responsive promoter driving a truncated plasminogen activator inhibitor type I (PAI-1) promoter, which contains SMAD-binding elements driving a luciferase reporter cassette (Abe, et al, 1994). TGF-β activation is then measured as luciferase activity. A R249A mutation in TGF-β prevents proteolytic cleavage by furin of mature TGF-β from LAP during intracellular processing (Shi, et al, 2011). TMLC expressing L-TGF-β1 (R249A)/GARP cannot produce diffusible active TGF-β and thus report only TGF-β activated within the L-TGF-β1 complex (Fig. S7). Minimal or undetectable levels of TGF-β were released into the supernatants from WT L-TGF-β1/GARP or L-TGF-β1 (R249A)/GARP TMLC cells when plated on αvβ8 or control substrates (supernatants were removed and assayed separately using TMLC cells, filled red circles). Luciferase activities are normalized and converted to amount of active TGF-β (pg/ml), as described (Annes et al., 2003). The y-axis is in log2 scale. ****p<0.0001 by one-way ANOVA and Tukey’s multiple comparisons test. (C) Model of TGF-β mediated TGF-β activation is shown. Activation occurs in three steps: Left panel: Initially, αvβ8 in an extended-closed conformation surveys the environment for cells presenting L-TGF-β on their cell surface by co-expression of adaptor molecules such as GARP. Middle panel: Binding of one cell expressing αvβ8 to a L-TGF-β expressing cell increases the flexibility of the latency lasso of TGF-β (red arrow) which exposes the active domain on one fingertip of the mature TGF-β homodimer. Right panel: TGF-βR2 expressed by the same cell presenting L-TGF-β binds to the exposed active domain of mature TGF-β, which initiates the TGF-β signaling cascade. Related information is in Fig. S7.
To test this hypothesis, we sought to express a form of TGF-β that could not be released from L-TGF-β in a model system that reproduces a physiologic milieu where αvβ8 normally activates TGF-β. Stimulated T-cells present L-TGF-β on their cell surfaces via covalent linkage to transmembrane adaptor proteins such as GARP and demonstrate TGF-β-dependent differentiation into regulatory T-cells (Treg) (Edwards, et al, 2014). These findings indicate that cell surface L-TGF-β can be activated to interact with its own TGF-β receptors in a T cell-intrinsic mechanism of TGF-β activation and signaling (Stockis, et al, 2012). The integrin αvβ8 either provided from contacting cells or Treg cells themselves has been implicated in stimulating such cell-intrinsic T-cell differentiation (Takasaka, et al, 2018; Edwards, et al, 2014). No reporter system currently exists to investigate the mechanistic basis of cell-intrinsic TGF-β activation, whereby the L-TGF-β presenting cell is also the cell that responds to TGF-β signaling. The most widely used in vitro TGF-β activation system relies on co-culturing a highly sensitive and specific TGF-β reporter cell line (TMLC) with integrin-expressing and/or L-TGF-β/GARP presenting cells (Wang, et al 2012). In such systems, TMLC must be in contact with these other cell-types for detection of TGF-β activation (Munger, et al, Cell, 1999). TMLC cells are a stable subclone of mink lung epithelial cells (MLEC) stably transfected with an expression construct containing a TGF-β specific promoter fragment, consisting of a truncated plasminogen activator inhibitor-1 (PAI-1) promoter, fused to the firefly luciferase reporter gene (Abe, et al, 1994). TMLC cells are widely used to measure active TGF-β because of their low background, specificity for TGF-β and abundant expression of TGF-β receptors and downstream signaling molecules that allow measurement of dose-dependent increases in TGF-β concentration in the physiologic range (Abe, et al, 1994). However, TMLC cells cannot report cell-intrinsic TGF-β activation since they do not present L-TGF-β themselves.
To build a cell-intrinsic TGF-β activation system, TMLC cells were stably transfected with wild-type (WT) TGF-β. Without co-transfecting GARP, TMLC do not present L-TGF-β on their cell surface. When co-transfected with TGF-β and GARP, high levels of cell surface expression of L-TGF-β can be detected (Fig S7A, C). To build a cell-intrinsic TGF-β activation system, which express a non-releasable form of TGF-β, we similarly expressed the L-TGF-β (R249A)/GARP complex on the surface of TGF-β reporter cells (TMLC) in which the L-TGF-β carries the same mutation (R249A) as described above that prevents cleavage of TGF-β from its prodomain during biosynthesis (Fig S7B, D). We verified that the transfected L-TGF-β (R249A)/GARP reporter cells expressed the cell surface complex at equivalent levels as the WT L-TGF-β/GARP reporter cells (Fig. S7C, D), and confirmed the absence of cleaved mature TGF-β from LAP in the R249A mutant (Fig. S7G). The resulting cell lines were used to determine cell-intrinsic TGF-β activation and signaling and as well as the role for release of mature TGF-β.
TMLC non-transfected or transfected with WT L-TGF-β, L-TGF-β (R249A), WT L-TGF-β/GARP, or L-TGF-β (R249A)/GARP were plated on the immobilized αvβ8 ectodomain, or control substrates (αvβ3 as a low-affinity TGF-β binding integrin, an antibody to LAP as a high-affinity L-TGF-β binder, or BSA) (Fig. S7E). In this configuration, actin cytoskeletal force cannot be generated or applied from the integrin to cause release of TGF-β. Robust TGF-β activation was observed at similar levels using cells expressing L-TGF-β (R249A)/GARP or WT L-TGF-β/GARP when plated on αvβ8, while no activation was seen when these same cells were plated on control substrates (Fig. 7B). Activation in this system was significantly enhanced by cell surface presentation of L-TGF-β by GARP since minimal TGF-β activation was seen when TMLC transfected with WT L-TGF-β or L-TGF-β (R249A) without GARP were plated on αvβ8 (Fig. S7F). The ability of GARP to present L-TGF-β and to increase efficiency of integrin-mediated TGF-β activation is consistent with previous findings (Wang, et al, 2012). No active TGF-β was detected in the supernatant from reporter cells expressing either WT or L-TGF-β (R249A)/GARP when plated on αvβ8 (Fig. 7B). These results demonstrate that αvβ8-dependent TGF-β activation can occur independently of actin-cytoskeletal force and does not require release of mature TGF-β (Fig. 7C).
DISCUSSION
Our studies reveal an intermediate step of L-TGF-β binding to αvβ8, and the flexible nature of L-TGF-β in complex with αvβ8. Together with mutagenesis and cell-based assays, we reveal an unanticipated mechanism for TGF-β activation, where mature TGF-β signals within the confines of L-TGF-β. Thus, αvβ8-mediated TGF-β activation does not require actin-cytoskeletal force nor release and diffusion of mature TGF-β. This model differs from all other activation models that require release and diffusion of mature TGF-β (Annes et al., 2003; Massagué, 2012; Moses et al., 2016). Instead, αvβ8-mediated TGF-β activation directs TGF-β signaling to the opposing L-TGF-β/GARP expressing cell through formation of a large multicomponent cell-cell protein complex.
Protein inhibitors in clinical development that globally inhibit mature TGF-β or TGF-β receptors have been associated with toxicities (Akhurst, 2017; Tolcher et al., 2017). Furthermore, our model predicts that such global TGF-β inhibition strategies would be ineffective in blocking αvβ8 mediated TGF-β activation due to the inaccessibility of their targeted epitopes within the geometrically constrained cell-cell αvβ8/L-TGF-β/GARP complex. In contrast, targeting αvβ8 prevents the complex from forming, which would increase specificity and efficacy while mitigating the risks of global TGF-β inhibition.
This TGF-β activation mechanism provides the structural basis to explain why conditional deletion of αvβ8 on one cell type primarily affects another (Arnold et al., 2019; Kitamura et al., 2011; Mohammed et al., 2016; Travis et al., 2007). This mechanism also explains why deletion of GARP, or its homologue, on immune cells negatively affects their TGF-β-dependent differentiation state (Qin et al., 2018; Salem et al., 2019; Wallace et al., 2018). In tumors, we have found that αvβ8 expressing tumors cells direct the immunosuppressive differentiation of L-TGF-β/GARP expressing Treg and myeloid cells (Takasaka et al., 2018).
Inhibiting αvβ8 in tumors can specifically target local immunosuppressive mechanisms of tumor immune evasion by blocking αvβ8-mediated contact of tumor cells with immune cells (Takasaka et al., 2018). We have defined the mechanisms of action of two antibody prototypes that efficiently block binding of αvβ8 to L-TGF-β. One of these is ligand-mimetic and defines an alternative RGD binding mode that not only reveals the initial phase of ligand recognition for L-TGF-β, but also reveals the proximal SDL2/3 pocket as a possible therapeutic target for αvβ8. Based on the variation of this pocket between other αv-integrins, it is possible that small molecules designed to bind within this pocket could increase specificity, which has been difficult with small molecules that directly target the conserved RGD binding site (Hatley et al., 2018). After the initial phase of ligand binding, it is clear from our structures that SDL2 plays a prominent role in stable binding of αvβ8 to L-TGF-β, a mechanism that we have experimentally extended to the αvβ6 integrin. We anticipate that the dynamic interactions occurring during binding between αvβ8 and L-TGF-β could provide a roadmap for understanding how flexible ligands conform to the SDL loops deep within the ligand binding clefts of other integrins as well.
In summary, our data provide unprecedented structural insights into the TGF-β activation process that expand the basic concepts of TGF-β activation for improved biologic understanding of TGF-β function in the immune and vascular systems and lay the foundation for structural investigations of other intact integrin ectodomains with their ligands to improve strategies to target those functions for therapeutic benefit.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents may be addressed to the Lead Contact, Stephen 5 Nishimura (stephen.nishimura@ucsf.edu). Antibodies, cell lines and plasmids used in this manuscript will be available from the Lead Contact upon execution of a materials transfer agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
Adult airways were collected from first through fourth order bronchi from lobectomy specimens from resections performed for primary lung cancer. Informed consent was obtained from all surgical participants as part of an approved ongoing research protocol by the University of California San Francisco Committee on Human Research, in full accordance with the declaration of Helsinki principles. The specimens were de-indentified.
Normal bronchial epithelial cells (HBEC)
Normal human bronchial epithelial cells (HBEC) were isolated from human lung specimens, as previously described (Araya et al., 2007). Specifically, airway epithelium was stripped freshly from bronchi from surgical specimens and after washing in PBS with dithiothreitol (5 mM) were digested overnight at 4°C with a protease XIV solution (0.4 mg/ml, Sigma). After washing with bronchial epithelial growth medium (BEGM, Lonza) with 2.5% FCS and tituration, HBEC were plated onto rat-tail Col I-coated (10 μg/ml, Corning, cat. no. 354236) dishes and incubated overnight; then the medium was changed to fresh BEGM. HBEC were passaged under conditional reprogramming conditions with BEGM with ROCK inhibitor (10 mM) and irradiated NIH3T3 fibroblasts, as described (Liu et al., 2012).
Human embryonic kidney cells (HEK293)
HEK293 are an authenticated female line from a commercial vendor (ATCC). HEK293 cells were grown in DMEM + 10% FBS + penicillin-streptomycin + amphotericin B + 0.11mg/mL Sodium Pyruvate + 2mM L-glutamine + MEM nonessential amino acids, cultured at 37°C with 5%C O2. HEK293 and all other cell lines were routinely tested for mycoplasma contamination. For all established cell lines cell culture media and antibiotics were prepared by the University of California, San Francisco Cell Culture Facility using deionized water and analytical grade reagents. Fetal calf serum was obtained from Invitrogen (Carlsbad, CA).
Chinese Hamster Ovary Cells (CHO)
CHO-K1 are an authenticated female line from a commercial vendor (ATCC). CHOLec 3.2.8.1 have four independent mutations in the N- and O-glycosylation pathways producing proteins all of the high mannose glycosylation pattern (Stanley, 1989). CHOLec 3.2.8.1 cells were grown in CHO-S-SFM II medium + 10% FBS + penicillin-streptomycin + amphotericin B, cultured at 37°C with 5%CO 2. CHOLec 3.2.8.1 cells were provided by Pamela Stanley (Stanley, 1989). Authentication of CHOLec 3.2.8.1 cells was performed by changes in glycosylation patterns as estimated by migration of secreted proteins by SDS-PAGE.
Transformed Mink Lung Epithelial Cells (TMLC)
TMLC is a stable mink lung epithelial reporter cell line derived from mixed sex American Mink fetal lung epithelial cells that stably expresses a portion of the plasminogen activator inhibitor 1 promoter (Abe et al., 1994). TMLC cells were grown in DMEM + 10% FBS + penicillin-streptomycin + amphotericin B, cultured at 37°C with 5%CO 2. TMLC are a gift from John Munger (NYU medical center, New York City, New York). Authentication was performed by increased luciferase activity in response to recombinant TGF-β.
METHOD DETAILS
Antibody isolation, characterization and production
The C6D4, 68, 8B8, 3G9, 1D11 antibodies have been previously described (Minagawa et al., 2014; Mu et al., 2002; Takasaka et al., 2018; Weinreb et al., 2004). Mouse anti-human αv, clone 11D12V2 was isolated as described, with the following modifications. Screening of hybridomas produced a polyclonal hybridoma 11D12 with reactivity against cell lines expressing the human αv-subunit (CHO-αvβ8, or SW480-β6). Subcloning produced two clones 11D12V1 and 11D12V2 both with reactivity against the αv-subunit. Variable (V) genes were isolated, sequenced, and the VH/VK genes cloned into mouse IgG2a expression vectors and stably transfected into CHO-K1 cells. C6-RGD3 was created by splice overlap extension (SOE) polymerase chain reaction (PCR) into the VL CDRL1 region of a C6D4 IgG2a expression vector (Minagawa et al., 2014) with the following oligonucleotide primers using the C6D4 IgG2a expression construct as a template: 5’-GATCTGGGGCGCCTCAAGAAGAACGCCTTGGCTTGGTACCAGCAG-3’; 5’-CTTGAGGCGCCCCAGATCTCCACGGCCGAGCAGACTCTGACTGGATTTG-3’. C6-RGD3 was transfected into CHO-K1 cells. Antibodies were produced and purified as described previously (Minagawa et al., 2014). Briefly, stable transfected CHO-K1 cells were grown in spinner cultures in CHO SFMII media with antibiotics. Antibodies were purified from culture media using protein G sepharose columns (HiTrap, GE Healthcare). C6D4, C6-RGD3, and 11D12V2 fragments were generated by papain digestion (Pierce) of the IgG followed by Protein-A Agarose (Millipore) incubation and to remove Fc fragments and intact antibodies and final separation using Mono S ion-exchange chromatography (GE Healthcare).
The 11D12V1 and 11D12V2 binding epitopes were estimated by negative staining electron microscopy (ns-EM), essentially as described below and in previous studies (Minagawa et al., 2014; Takasaka et al., 2018). Clone 11D12V1 bound to the αv-head and 11D12V2 to the αv-thigh. Anti-β6 (MAB4155) was purchased (R&D Systems).
A bioassay to measure cell-intrinsic TGF-β signaling
To develop a cell intrinsic TGF-β activation system we used stable transfection of TMLC (Amaxa) with a vector containing either a WT human TGF-β1 IRES GFP, or a human TGF-β1 (R249A) IRES GFP cassette with puromycin resistance to obtain TMLC cells expressing WT L-TGF-β either capable of dissociating into LAP and mature TGF-β or not, due to the R249A mutation that normally allows furin cleavage of LAP from mature TGF-β (Shi, et al, 2011). Human TGF-β1 IRES GFP or human TGF-β1 (R249A) IRES GFP TMLC cells were sorted for equal expression using GFP fluorescence and did not present any L-TGF-β on their cell surface (Fig. S7). In contrast, stable transfection of these lines with a HA-GARP construct with a blastacidin resistance cassette followed by selection and sorting resulted in high surface expression of TGF-β1/GARP or TGF-β1 (R249A)/GARP, as measured by anti-HA (clone 5E11D8, GenScript, Piscataway, NJ) or anti LAP (R&D Systems, AF426). To confirm lack of releasable of TGF-β from the L-TGF-β R249A/GARP cell surface complex, L-TGF-β (R249A)/GARP expressing or WT L-TGF-β/GARP TMLC cells were surface biotinylated using EZ-link sulfo-NHS biotin (Thermo Fisher Scientific) and immunoprecipitated using anti-HA, resolved by 4–12% gradient SDS-PAGE, under non-reducing conditions, immunoblotted, probed with streptavidin-HRP and detected by chemiluminescence, essentially as described (Mu, et al, 2002). Immunoprecipitations confirmed association of GARP with WT L-TGF-β, L-TGF-β (R249A), and absence of cleavage of LAP from mature TGF-β in the L-TGF-β (R249A)/GARP TMLC cells (Fig. S7).
A TMLC assay to measure integrin-mediated cell-intrinsic TGF-β signaling
The αvβ8 ectodomain was coated along with the controls αvβ3 (R&D Systems), BSA (Sigma-Aldrich) or anti-LAP (R&D AF426, 1 μg/ml) onto ELISA plates in PBS (1mM Ca2+ and 1mM Mg2+) 1 hour at RT. Wells were subsequently washed in PBS and blocked in PBS with 1% BSA WT L-TGF-β1, L-TGF-β1 (R249A), WT L-TGF-β/GARP, L-TGF-β (R249A)/GARP expressing TMLC cells were plated at a density of 1×105 cells/ml in basal media. After 16 hrs, media was removed and applied to wells containing TMLC reporter cells to measure diffusible mature TGF-β, which were incubated overnight prior to lysis and determination of luciferase activity (Promega) as reported (Mu, et al, 2002). To measure cell-intrinsic TGF-β1 activation, the attached L-TGF-β1, L-TGF-β1 (R249A), WT L-TGF-β/GARP, L-TGF-β (R249A)/GARP or parental TMLC cells were lysed and assayed for luciferase activity. To facilitate comparison between different TMLC lines expressing WT L-TGF-β1, L-TGF-β1 (R249A), WT L-TGF-β/GARP, L-TGF-β (R249A)/GARP (or parental TMLC cells) normalized luciferase activity was expressed as activated TGF-β in pg/ml. Normalization was performed by interpolating luciferase activity against standard curves generated using each TMLC line with varying doses of recombinant human TGF-β1 (R&D Systems).
Integrin DNA constructs
Wild-type and mutant recombinant human integrin αvβ8 and αvβ6 truncated at the junction of the ectodomains and transmembrane domains in pcDNA1neo have been described (Nishimura et al., 1994; Weinreb et al., 2004). β8 Δ SDL was prepared as described (Takasaka et al., 2018). To create mutant β8 subunits, SOE was performed using PCR with WT β8 as a template to create mutant constructs all in pcDNA6 V5HisA (Invitrogen) with a stop codon inserted before the V5/His tag. The following mutagenic primers (all in 5 ‘to 3’ orientation) were used: β8 I208R5’-CAGAAGATCTCTGGAAACAGAGATACACC-3’; 5’-GAAGTTTGGTCGACATAATGC-3’ β8 Y172N: 5’-CAATGCAGTGACAACAATTTAGACTGC-3’, 5’-GCAGTCTAAATTGTTGTCACTGCATTG-3’; β8 Y172M: 5’-GATTCATAATCAATGCAGTGACATGAATTTAGACTGCATGCC-3’, 5’-GGCATGCAGTCTAAATTCATGTCACTGCATTGATTATGAATC-3’; β8 Y172A: 5’-GATTCATAATCAATGCAGTGACGCCAATTTAGACTGCATGCC-3’, 5’-GGCATGCAGTCTAAATTGGCGTCACTGCATTGATTATGAATC-3’; β6 I183N: 5’-CCCTTGCAGTAGTAATCCATACTTCTG-3’, 5’-CAGAAGTATGGATTACTACTGCAAGGG-3’.
TGF-β DNA constructs
Porcine L-TGF-β1 with a C4S mutation, to improve secretion and prevent association with L-TGF-β binding proteins, and an N-terminal cleavable (HRV 3C) 7x Histidine-streptavidin binding protein tag to facilitate purification (Shi et al., 2011), were joined by SOE PCR from pcDNA-GS-TGF-β1 (gift from Dr. Sun, National Institutes of Health, Bethesda, MD (Zou and Sun, 2004)) and subcloned into pcDNA6 (Invitrogen). Porcine L-TGF-β1 C4S pcDNA6 was modified using the following mutagenic primers, RGE (a mutation that disrupts the integrin binding recognition sequence on L-TGF-β and therefore minimizes the number of L-TGF-βs that bind two integrins simultaneously): 5’-CCGCCGGGGTGAACTGGCCAC-3’; 5’-GTGGCCAGTTCACCCCGGCGG-3’; R249A: 5’-ACCTGCACAGCTCCCGGCACCGCGCAGCCCTGG-3’. Wild type human TGFβ1 was derived from human TGFβ1_pLX307 (Rosenbluh et al., 2016) by removing the C-terminal V5 tag using PCR with the following primers; 5’-ATGGCCACCCCGCTGG-3’, 5’-CTCTACTAGTCTCGAGTTATCAGCTGCACTTGCAGGAGCGCAC-3’. The WT human TGF-β1 IRES GFP cassette was then subcloned into a version of pcDNA6 (Invitrogen) with a puromycin resistance cassette which was amplified from TGFβ1_pLX307 using 5’-ATCGTTTCAGACCCACCTCCC-3’ and 5’-CTCTGCTTAGCGAATTCGTTAACTGGCACCGGG-3’. A R249A mutant of this construct was produced using SOE PCR employing the following mutagenic primers; 5’-CACCGCGCAGCCCTGGACACCAAC-3’, 5’-CCAGGGCTGCGCGGTGCCGGGAG-3’.
GARP DNA constructs
N-terminal HA tagged human GARP pcDNA3 (Cuende et al., 2015) was provided by Sophie Lucas (de Duve Institute, UCLouvain, Brussels, Belgium) and the entire HA-GARP reading frame transferred into pcDNA6 (Invitrogen). All constructs were verified by sequencing.
Secreted protein expression and purification
Integrin constructs were expressed using stably expressing CHOLec 3.2.8.1 cells grown in spinner cultures in CHO SFMII (Thermo Fisher) with antibiotics, for structural studies, or transiently transfected in HEK293 cells using 293 SFMII (Thermo Fisher) for biochemical studies. Integrin purification was carried out by affinity chromatography using a Protein G-clone 8B8 column followed by size exclusion chromatography (Superdex 200 Increase 10/300 GL, GE Healthcare) in 20mM Tris-HCL pH 7.5, 150 mM NaCl, 1mM CaCl2 and 1mM MgCl2. To produce L-TGF-β for structural studies, 293 cells were transiently transfected with equal amounts of porcine L-TGF-β1 C4S R249A RGD and C4S R249A RGE plasmids, to favor formation of L-TGF-β1 with a single intact RGD binding site to favor 1:1 binding stoichiometry to reduce sample heterogeneity, and purified as described (Shi et al., 2011). Briefly, supernatants were collected from spinner cultures, clarified by centrifugation, filtered though a PES (polyethersulfone) membrane, 0.2 mm pore size (Millipore), concentrated, and purified using Ni-NTA agarose (Qiagen), washed with three column volumes of 0.6 M NaCl, 0.01 M Tris (pH 8.0) and eluted with 0.25 M imidazole in TBS. The was adjusted to pH 7.4 then applied to Strep-tactin agarose (IBA) (1 ml per 1 L of culture supernatant) and washed with TBS (pH 7.4). Tag was cleaved with recombinant His-tagged HRV-3C protease (Novagen, 100 U mg–1, 1 mg ml–1), diluted 20-fold in TBS (pH 7.4) with 10% glycerol, applied to the column, and incubated at 4 °C for 16 h. The flow-through was washed with two column volumes of TBS (pH 7.4), containing untagged proTGF-β1, then concentrated using centrifugal concentrators (Millipore) to about 1 mg ml–1 in 10 mM Tris (pH 7.5), 75 mM NaCl. The homogeneity and purity of all protein preparations were verified by SDS-PAGE stained with Coomassie blue and protein concentrations were measured by bicinchoninic acid assay (Pierce).
Cryo-EM sample preparation
To prepare integrin-Fab or integrin-L-TGF-β complexes, 100 mg of recombinant αvβ8 was incubated in a 2-fold molar excess of each Fab or L-TGF-β, incubated at room temperature for 30 min, subjected to size exclusion chromatography and concentrated to 6 to 9 mg/ml. For cryo-EM grid preparation, 2.5 μL of purified αvβ8 complex were deposited onto Quantifoil grids. For the αvβ8/L-TGF-β-R249A at 0.25 mg/ml, a 400 mesh 1.2/1/3 holey carbon gold grid was used. For the αvβ8/L-TGF-β/RGD-RGE complex at 0.075 mg/mL, a 400 mesh 2/2 holey carbon copper grid that had been covered with a thin layer of graphene oxide was used. For the αvβ8/C6D4/11d12v2 and the αvβ8/C6-RGD3/11d12v2 complexes, both at 6.8 mg/mL, 300 mesh 1.2/1.3 holey carbon gold grids were used. Except for the graphene-coated grid, grids were glow-discharged for 60s at 15 mA prior to sample application and freezing. The αvβ8/L-TGF-β C4S R249A RGD and αvβ8/L-TGF-β C4S R249A RGD/RGE complexes were frozen using a FEI Vitrobot Mark IV using a 4 second blot time. The αvβ8/C6D4/11d12v2 and αvβ8/C6-RGD3/11d12v2 complexes were frozen using a FEI Vitrobot Mark III using a 3–4 second blotting time. All grids were frozen with 100% humidity at 20°C and plunge-frozen in liquid ethane cooled by liquid nitrogen.
Cryo-EM data acquisition
Four datasets were acquired on a FEI Titan Krios transmission electron microscope operated in nano-probe mode at 300 kV equipped with a Gatan Quantum GIF energy filter, operated in zero-loss mode with a slit width of 20 eV and a Gatan K2 Summit direct detector. Automated data collection was carried out using the SerialEM software (Mastronarde, 2005). Movies were recorded in super-resolution mode with a super-resolution pixel size of 0.673 Å/px and a nominal magnification of 105kx at a dose rate of ~8 e−/px/s. For the αvβ8/L-TGF-β-R249A complex and the αvβ8/LTGF-β/RGD-RGE, each 16 second movie contained 80 frames of 200 ms each, which corresponds to a total dose of ~70 e−/Å2. For the αvβ8/C6D4/11d12v2 complex and the αvβ8/C6RGD3/11d12v2 complex, each 12 second movie contained 60 frames of 200 ms, which corresponds to a total dose of ~50 e−/Å2. Each dataset was collected in a single session with a nominal defocus range of 1.0 – 2.5 μm under focus. Total micrographs collected for each dataset are as follows: αvβ8/L-TGF-β-R249A complex: 1684 micrographs; αvβ8/L-TGF-β-RGDRGE complex: 2682 micrographs; αvβ8/C6D4/11d12v2 complex: 1644 micrographs; αvβ8/C6RGD3/11d12v2 complex: 4033 micrographs.
Imaging Processing
Dose fractionated super-resolution image stacks were motion corrected and binned 2 × 2 by Fourier cropping using MotionCor2 (Zheng et al., 2017). Motion corrected sums without dose-weighting were used for contrast transfer function (CTF) determination using GCTF (Zhang, 2016) or CTFFIND4 (Rohou and Grigorieff, 2015). Particles were picked using the reference-free method using Gautomatch (http://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch) and boxed out using Relion 3.0 (Zivanov et al., 2018) with a box size of 300 pixels and binned to 64 pixels. After 2D alignment and classification was carried out using cryoSPARC (Punjani et al., 2017), selected particles were re-extracted in Relion 3.0 and binned to 128 pixels to generate ab initio initial models using cryoSPARC. 3D classification schemes are outlined for the αvβ8/LTGFB-R249A complex and the αvβ8/L-TGF-β-RGD-RGE complex in Fig. S5 and for the αvβ8/C6RGD3/11d12v2 complex in Fig. S4. No 3D classification was used for the αvβ8/C6D4/11d12v2 complex dataset. For all final maps, non-uniform refinement, local resolution refinement, local resolution estimation, sharpening, and local filtering was carried out using cryoSPARC to yield maps with a final pixel size of 1.345Å/px. The number of particles contributing to the final maps are as follows: αvβ8/L-TGF-β (conformation iv): 43,600 particles; αvβ8/C6D4/11d12v: 84,266 particles; αvβ8/C6-RGD3/11d12v2: 221,159 particles. Images were rendered using UCSF Chimera (Pettersen et al., 2004) and PyMol (DeLano, 2002).
Model Building and Refinement
The atomic model of the αv headpiece from the crystal structure of αvβ6 (PDB: 4UM8) with glycans removed was fitted to the cryo-EM map as a rigid body. An atomic model of the β8 headpiece was generated by rigid body fitting of a homology model based on the same crystal structure (4UM8) using Modeller (Webb and Sali, 2014) and adjusted around the SyMBS cation area using the crystal structure of αvβ3 (PDB: 3IJE), then fitted into the cryo-EM density map as a rigid body. Atomic models of Fabs were generated with RosettaAntibody using multiple-template grafting and H3 loop modelization (Lyskov et al., 2013) based on the primary sequence of their Vh/Vk. An atomic model of the arm domain of L-TGF-β1 was generated from the crystal structure of L-TGF-β1 bound to αvβ6 (PDB: 5FFO). The models for Fab C6D4 and Fab C6-RGD3 or L-TGF-β1 were then fitted as a rigid body to the map. Prototypical CHOLec3.2.8.1 glycans were added back to the model at the solvent exposed N-glycosylation consensus sites using GLYCAM (Singh et al., 2016). The sugar base of glycans were trimmed to fit into the corresponding densities and further refined in Rosetta using a dedicated protocol that uses physically realistic geometries based on prior knowledge of saccharide chemical properties (Frenz et al., 2019). After rigid body fitting, all models were manually adjusted to fit the cryo-EM density maps in COOT (Emsley et al., 2010), followed by real space refinement using Phenix (Adams et al., 2010), and Rosetta (Wang et al., 2016). All modeling was aided by using EM maps that were focused on specific regions, as well as sharpened and unsharpened maps. All maps used for modeling have been deposited.
Antibody binding assays
ELISA plates were coated with recombinant αv-integrins (1 mg/ml coating concentration, all from R&D systems) blocked with 5% BSA in PBS for 1 hour, and antibodies allowed to bind for 2 hours at RT and detected with anti-mouse-HRP.
Cell adhesion assays
ELISA plates were coated with integrins (2 mg/ml coating concentration) and blocked with PBS with 5% BSA for 1 hour and then CHO-GARP/L-TGF-β, or CHO mock (5 ×105) transfectants in the presence of various concentrations of C6D4, C6-RGD3 or 3G9 were centrifuged onto integrin coated wells (10 × g) for 5 min allowed to adhere for 30 min at RT after which the plates were inverted and centrifuged (10 × g) for 5 min and then immediated fixed and stained (1% formaldehyde, 20% MeOH, 0.5% crystal violet) and after extensive washing, dye was solubilized in PBS with 1% T-X100 for 1 hr at RT and attached cells estimated by absorbance (A595).
L-TGF-β1 binding assays
ELISA plates were coated with recombinant porcine L-TGF-β1 (0.5 μg/ml coating concentration), blocked with 5% BSA in PBS for 1 hour, and integrins at various concentrations were allowed to bind for 2 hours at RT, and detected with 8B8 antibody and anti-mouse-HRP.
Cell staining, flow cytometry and cell sorting
Cell staining for L-TGF-β1 was confirmed by flow cytometry using anti-LAP (R&D biotinylated anti-LAP BAF246 and streptavidin APC) and GARP cell surface expression was confirmed by anti-HA staining (clone 5E11D8 (Genscript, Piscataway, NJ)). EGFP expression was also used as a surrogate marker for L-TGF-β1 expression. High-expressing pools of expressing cells were established by sorting (BD FACSAria, BD Biosciences, US).
Peptide competition assays
For peptide competition assays, ELISA plates were coated with recombinant porcine L-TGF-β1 (0.5 μg/ml coating concentration), blocked with 5% BSA in PBS for 1 hour, and integrins at 0.5 μg/ml were pre-incubated with peptides at various concentrations for 20 minutes, allowed to bind for 2 hours at RT, and detected with 8B8 antibody and anti-mouse-HRP.
Sequence alignments
Multiple protein sequence alignments for integrins were generated using Clustal Omega (Madeira et al., 2019).
QUANTIFICATION AND STATISTICAL ANALYSIS
ELISA and TMLC assays are reported as means ± s.e.m. All assays were repeated a minimum of 3 times. All statistical analyses were performed using the software package Prism 7 (GraphPad Software, San Diego, CA).
Supplementary Material
Fig. S1 Integrins and TGF-β: nomenclature and models of activation, related to Introduction, Fig. 7 (A) Integrin and L-TGF-β domains and subdomains and their membrane or extracellular matrix localization via coupling to adaptor proteins are shown using common nomenclature. (B) In the global-rearrangement or “switchblade” model of integrin activation, the bent conformation moves through an extended-closed conformation to an extended-open conformation. In this model, high-affinity ligand binding and actin cytoskeletal force transduction occur in the extended-open conformation. In the case of αvβ6 it has been proposed that such force releases TGF-β and allows diffusion to its target receptors (Dong et al., 2017). Pink arrows indicate movements in the headpiece. (C) The αvβ8 integrin assumes a single conformation, extended-closed, that accommodates ligand binding and affinity regulation. The essential immune cell functions of αvβ8 have been tied to binding to L-TGF-β presented by type I transmembrane adaptor proteins such as GARP on immune cell surfaces. In this model, we hypothesize that the “latency lasso” of the straitjacket domain of L-TGF-β loosens upon binding to αvβ8 to allow the active domain of TGF-β to interact with its receptors.
Fig. S2. Cryo-EM micrographs and statistics for αvβ8 complexes, related to Figs. 1 and 2 Details of data collection and processing data for (A) αvβ8/LTGF-β (subclass iv), (B) αvβ8/C6D4 and (C) αvβ8/C6-RGD3. For each complex the following are shown: First column: a representative motion corrected micrograph of the particles suspended in vitreous ice. Second column: the gold standard FSC (top) and the angular distributions of particles (bottom) used in the final map, as estimated by cryoSPARC. Third column: (A) the unsharpened map displayed at a low thresh, (B,C) the unsharpened map before focused refinement displayed at a low threshold. Fourth column: the map after focused alignment displayed at a high threshold sharpened to a b-factor of (A) −71 (B) −83 or (C) −84. Maps are colored on the same scale, as indicated in row A, based on local resolution estimates. Scale bar = 100 nm.
Fig. S3. Comparisons of αvβ8 structures with integrin crystal structures, model quality, and characterization of C6-RGD3, related to Figs. 1, 2 and 4 (A, B) Superimpositions of ribbon models of αvβ8/L-TGF-β1 with published models from crystal structures of liganded αvβ6 (RGD peptide, PDB: 4UM9 (Dong et al., 2014) (A); L-TGF-β1, PDB: 5FFO (Dong et al., 2017) (B)). From αvβ8/L-TGF-β1 αv-subunit, green; β8-subunit, blue; RGD loop, purple; β6 subunit and L-TGF-β3 RGD peptide, salmon (A); β6 subunit and L-TGF-β1 RGD loop, lime green.
(C, D) Close-up of the binding interface of the αvβ8 integrin (light green and blue) and Fab C6D4 (coral, C) or Fab C6-RGD3 (pink, D) with the corresponding sharpened density map (grey volume). (E) Close-up of the binding interface of the β8 integrin subunit SDL2 loop (cyan) and the L-TGF-β1 proximal loop, RGD motif, and ligand-binding helix (purple) superimposed on their respective sharpened density maps (mesh). (F) Close-up of the Fab C6-RGD3 CDRL1 loop (pink) and its sharpened density map (pink mesh). (G, H) Close-up of the integrin β8 SDL1 α1 helix and β6-α7 loop when in complex with L-TGF-β1 (cyan, G) or Fab C6-RGD3 (dark blue, H).
(I) Map to model FSC curves for the αvβ8/L-TGF-β1 (purple), αvβ8/C6D4 (orange), αvβ8/C6-RGD3 (magenta) complexes.
(J-M) View of the metal ions and MIDAS cation coordination in various liganded integrin structures in ribbon models: αvβ8/L-TGF-β1 complex (J), αvβ3/fibronectin 10th domain RGD complex (PDB: 4MMX) (Van Agthoven et al., 2014) (K), αiibβ3/fibrinogen RGD peptide complex (PDB: 2VDR) (Springer et al., 2008)(L), αvβ6/L-TGF-β1 complex (PDB: 5FFO) (M). The coordinating residues are indicated in sticks.
(N-S) Superimpositions of the α1-helix, with the β6-α7 loop and MIDAS cation (dotted circle) as ribbon models with superimpositions from unliganded or liganded αiibβ3 (PDB: 3T3P (Zhu et al., 2012) or 2VDR(Xiong et al., 2009), respectively): (N) liganded (yellow) or unliganded αiibβ3, red; (O) liganded αiibβ3 (red) or αvβ8/C6-RGD3, green; (P) liganded αiibβ3 (yellow) or αvβ8/C6D4, light blue; (Q) liganded αiibβ3 (orange) or αvβ8/L-TGF-β1, pink; (R) αvβ8/C6D4 (light blue), αvβ8/C6-RGD3 (green), or αvβ8/L-TGF-β1, pink. Movement of the tip of the SDL1 α1-helix is highlighted by the S116 (β8)/S123 (β3) residues in sticks. The AspRGD is represented in sticks for liganded structures. (T, U) Superimposition of ribbon models of the αvβ8/C6D4 and α4β7/Act-1 (PDB: 3V4P) (Yu et al., 2012) complexes. Both C6D4 and Act-1 epitopes are located in the SDL2 region of the integrin (front view (T); rotated view, U)). αv-green, β8-blue, C6D4-orange, α4-lime green, β7-light blue, Act-1-magenta.
(V) Ribbon model of the αvβ8/C6D4 complex with the CDRL1 loop highlighted in red that was replaced with the L-TGF-β integrin-binding motif and helix to create C6-RGD3 (αv-green, β8-blue, C6D4-gold). (W) Binding assay to immobilized αv-integrins to show specificity of C6D4 for αvβ8, and C6-RGD3 for αvβ6 and αvβ8. Shown is a representative experiment of three (n=3).
(X, Z) Inhibition of cell adhesion of CHO cells co-expressing GARP and L-TGF-β1 on their cell surface to immobilized (X) αvβ8 or (Z) αvβ6 in the presence of indicated concentrations of C6D4 or C6-RGD3 or anti-β6 (3G9). TGF-β activation assays of (Y) CHO cells stably transfected with αvβ8 or (AA) human bronchial epithelial cells (HBEC), which naturally express high levels of αvβ6 and low levels of αvβ8 (Araya et al., 2007) in the presence of indicated concentrations of C6D4, C6-RGD3 or 3G9 (Weinreb et al., 2004). Activation is shown relative to a pan-TGF-β neutralizing antibody (1D11). n=3, error bars show s.e.m.
Fig. S4. Processing schematic for the αvβ8/C6-RGD3 complex, related to Fig. 2 A schematic flowchart showing the classification scheme of the αvβ8/C6-RGD3 complex. Particle numbers at each step and for each class are indicated. From top to bottom: After 3D classification, the data was separated into three groups: particles with C6-RGD3 bound at the expected angle, particles with C6-RGD3 at a skewed angle, and particles without C6-RGD3. These three groups were processed separately. The map showing C6-RGD3 at the expected angle shows the RGD motif in the alternative, non-helical binding conformation. The map showing C6-RGD3 at the skewed angle shows the RGD motif in a helical conformation. All maps are colored on the same scale, as indicated, based on local resolution estimates.
Fig. S5. Processing schematic for the αvβ8/LTGF-β complex, related to Fig. 5A schematic flowchart showing the classification scheme of the αvβ8/L-TGF-β complex. Due to the preferred orientations adopted by the particles, two types of grids were used and two separate datasets were collected. Upon angular assignment, it was found that although particles on both grids suffered from preferred orientations, the orientations complement each other to form a more complete orientation sampling. Particle numbers at each step and for each class are indicated. 3D reconstructions of seven out of sixteen subclasses showed significant density for L-TGF-β, and reached high resolution after refinement. All maps are colored on the same scale, as indicated, based on local resolution estimates.
Fig. S6. β8 and β6 SDL mutations, and structure-guided mutagenesis of the β8 SDL2/3 binding pocket, related to Fig. 6 (A, B) CHOlec 3.2.8.1 cells were stably transfected with various β8, β6 WT or SDL mutants, as indicated, sorted several times to establish highly expressing pools, and tested for expression using C6D4 (A) compared to clone 68 (B), a βI domain antibody that binds to the tail of the β8 α1-helix (Minagawa et al., 2014). Note that the mutant I208R does not bind C6D4 as well as other mutants, as this mutation would be expected to clash with the position of C6D4 CDRL1. (C, D) Binding of L-TGF-β1 to truncated secreted forms of C) WT and a Y172A αvβ8 mutant, or D) αvβ6 WT and a I183N αvβ6 mutant. n=3, error bars show s.e.m.
(E-G) Ribbon models of the αvβ8 head domain of the CDRL1 loops of (E) C6D4, (F) C6-RGD3 and (G) L-TGF-β (gold sticks), with boxed ligand pocket (upper panels) shown in close up views in middle and lower panels. Color code: αv-subunit (green), β8-SDL1 (light blue), -SDL2 (cyan) and -SDL3 (purple) loops. The positioning of the C6D4 CDRL1; C6-RGD3 CDRL1; or L-TGF-β1 integrin-binding loops are shown positioned in the binding cleft of β8 wild-type (middle panels); and the I208R mutant (lower panels). Middle panel color code: L-TGF-β integrin-binding loop R215 (gold), the β8 SDL2 Y172 (red) and SDL1 S116 (red). Lower panel color code: the SDL3 I208R mutation is indicated in red spheres demonstrating clash with the C6D4 CDRL1; C6-RGD3 CDRL1 but not the canonical position of the L-TGF-β integrin-binding loop. The SyMBS (grey) and MIDAS (green) cations are shown.
Fig. S7. Expression and characterization of WT L-TGF-β, L-TGF-β (R249A), WT L-TGF-β/GARP and L-TGF-β (R249A)/GARP TMLC reporter cells, related to Fig. 7 (A-D) WT L-TGF-β/GARP and L-TGF-β (R249A)/GARP are equally expressed on the surface of TGF-β reporter cells (TMLC). TMLC cells were first stably transfected with either (A) WT L-TGF-β IRES GFP or, (B) L-TGF-β (R249A) IRES GFP and sorted for equal expression of GFP. Histograms show GFP expression on the x-axis and surface localization of L-TGF-β, as assessed by anti-LAP-APC staining, on the y-axis. Stably transfected and sorted cells show no surface staining for L-TGF-β, despite showing ~40–50% GFP bright cells. In contrast, when (C) WT L-TGF-β IRES GFP or (D) L-TGF-β (R249A) IRES GFP expressing TMLC were subsequently stably transfected with a N-terminal HA tagged GARP construct, bright surface expression of L-TGF-β was detected in ~40% of cells. (C) WT L-TGF-β IRES GFP/HA GARP or, (D) L-TGF-β (R249A) IRES GFP/HA GARP TMLC were sorted for equal surface expression of L-TGF-β, as assessed by anti-LAP-APC staining. (E) Cartoon representation of the experiment. Individual wells of 96-well ELISA plates were coated with either αvβ8, αvβ3 (which binds with low affinity to L-TGF-β), polyclonal anti-LAP (which binds to LAP, but does not activate or inhibit activation of L-TGF-β), or BSA as a non-specific binding control. TMLC cells either non-transfected or transfected as in A-D with WT L-TGF-β or L-TGF-β (R249A) with or without GARP were applied to coated wells. (F) Parental TMLC cells, WT L-TGF-β IRES GFP, L-TGF-β (R249A) IRES GFP, WT L-TGF-β IRES GFP/HA GARP or, L-TGF-β (R249A) IRES GFP/HA GARP expressing TMLC cells were added to wells coated as indicated in the legend with each substrate and assayed for luciferase activity after 16–20 hr. Each condition was repeated in triplicate with three independent experiments. *p < 0.05, ** p <0.01, **** p <0.0001, ns = not significant, as determined by unpaired Student’s t-test. Note L-TGF-β (R249A) IRES GFP/HA GARP expressing TMLC show dramatic and significant αvβ8-dependent TGF-β activation similar to that seen with WT L-TGF-β IRES GFP/HA GARP TMLC cells. (G) Characterization of secreted L-TGF-β (R249A) and WT L-TGF-β compared to cell surface expressed L-TGF-β (R249A)/HA GARP and WT L-TGF-β IRES GFP/HA GARP. Left panels: Shown are recombinant secreted L-TGF-β (R249A) and WT L-TGF-β under non-reducing (NR) or reducing (R) conditions as assessed by 4–12% gradient SDS-PAGE and Coomassie Brilliant Blue staining. Shown are molecular weight markers (MWM). Irrelevant lanes have been digitally removed for clarity. The identity of the bands at the expected molecular weights is indicated. Right panel: L-TGF-β (R249A) IRES GFP/HA GARP and WT L-TGF-β IRES GFP/HA GARP expressing TMLC cells were surface labeled with biotin, lysed and immunoprecipitated with anti-HA to capture the cell surface GARP/L-TGF-β complexes. After resolving immunoprecipitated proteins by 4–12% SDS page under reducing conditions, biotinylated proteins were detected by immunoblotting with streptavidin-HRP and results were aligned with MWM on the left panel. Results show that both L-TGF-β (R249A) IRES GFP/HA GARP and WT L-TGF-β IRES GFP/HA GARP form complexes on the surface of TMLC cells and that TGF-β in the R249A mutant does not cleave from the LAP.
HIGHLIGHTS:
Integrin αvβ8 in the extended-closed conformation binds to and activates L-TGF-β
The L-TGF-β RGD loop initiates contact with αvβ8 through an alternate binding mode
L-TGF-β is flexible when bound to αvβ8
Mature TGF-β activates signaling without being released from the latent TGF-β complex
Acknowledgments:
We thank Michael Braunfeld and Drs. Alexander Myasnikov, David Bulkley and Eric Tse for supporting the cryo-EM facility at UCSF; Dr. Shenping Wu for her involvement in the early stages of this project; Drs. Feng Wang and Zanlin Yu for sharing the protocol for the preparation of the graphene oxide grid used in this study, and Daniel Asarnow for sharing scripts in pyEM, Rik Derynck for helpful suggestions. This work is partially supported by grants from National Institute of Health (U54HL119893 and R01HL113032 to S.L.N.; R01HL134183 to S.L.N. and Y.C.; R01GM098672, S10OD020054 and S10OD021741 to Y.C.; and P41CA196276 to J.M.; R01DK093646 to J.L.B., the UCSF Liver Center - P30DK026743 to J.L.B. and S.L.N.). YC is an Investigator of Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: A.C., J.D.M., J.L., J.L.B., Y.C., and S.L.N. are inventors on a patent on anti-αvβ8 antibodies, which have been out-licensed. S.L.N. is on the Scientific Advisory Board of Venn Therapeutics, LLC.
DATA AND CODE AVAILABILITY
All structures and models have been deposited into the EMDB and the PDB. The accession numbers for primary conformation of αvβ8/L-TGF-β (conformation iv) are EMD-20794 and PDB ID 6UJA. For the additional αvβ8/L-TGF-β conformations, i-iii and v-vii the accession numbers are EMD-20797 to EMD-20802. A consensus map of all seven conformations focused at the headpiece region with a nominal resolution of 2.9Å was used for modeling and is deposited with the accession number EMD-21125. For αvβ8/C6D4, the accession numbers are EMD-20795 and PDB ID 6UJB and for αvβ8/C6-RGD3 they are EMD-20796 and PDB ID 6UJC. Three cryo-EM dataset collections containing movies, aligned micrographs, and stacks of αvβ8 bound to L-TGF-β or C6D4 have been deposited into the EMPIAR database: EMPIAR-10343 (αvβ8 bound to L-TGF-β collected on holey carbon grids), EMPIAR-10344 (αvβ8 bound to L-TGF-β collected on graphene oxide grids) and EMPIAR-10345 (αvβ8 bound to C6D4 collected on holey carbon grids).
ADDITIONAL RESOURCES N/A
REFERENCES
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, and Rifkin DB (1994). An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem 216, 276–284. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhurst RJ (2017). Targeting TGF-beta Signaling for Therapeutic Gain. Cold Spring Harb Perspect Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, and Munger JS (2009). Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci 122, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Munger JS, and Rifkin DB (2003). Making sense of latent TGFbeta activation. J Cell Sci 116, 217–224. [DOI] [PubMed] [Google Scholar]
- Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus VC, Sheppard D, et al. (2007). Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest 117, 3551–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold TD, Lizama CO, Cautivo KM, Santander N, Lin L, Qiu H, Huang EJ, Liu C, Mukouyama YS, Reichardt LF, et al. (2019). Impaired αVβ8 and TGFβ signaling lead to microglial dysmaturation and neuromotor dysfunction. J Exp Med 216, 900–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold TD, Niaudet C, Pang MF, Siegenthaler J, Gaengel K, Jung B, Ferrero GM, Mukouyama YS, Fuxe J, Akhurst R, et al. (2014). Excessive vascular sprouting underlies cerebral hemorrhage in mice lacking alphaVbeta8-TGFbeta signaling in the brain. Development 141, 4489–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier A, Campbell MG, Ito S, Wu S, Lou J, Marks J, Baron JL, Nishimura SL, and Cheng Y (2018). Cryo-EM structure of the alphavbeta8 integrin reveals a mechanism for stabilizing integrin extension. Nat Struct Mol Biol 25, 698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuende J, Lienart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J, Huygens C, Colau D, Somja J, Delvenne P, et al. (2015). Monoclonal antibodies against GARP/TGF-beta1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci Transl Med 7, 284ra256. [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002). Pymol: An open-source molecular graphics tool. CCP4 Newsletter On Protein Crystallography 40, 82–92. [Google Scholar]
- Dong X, Hudson NE, Lu C, and Springer TA (2014). Structural determinants of integrin beta-subunit specificity for latent TGF-beta. Nat Struct Mol Biol 21, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, Engen JR, and Springer TA (2017). Force interacts with macromolecular structure in activation of TGF-beta. Nature 542, 55–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenz B, Rämisch S, Borst AJ, Walls AC, Adolf-Bryfogle J, Schief WR, Veesler D, and DiMaio F (2019). Automatically Fixing Errors in Glycoprotein Structures with Rosetta. Structure 27, 134–139.e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon KJ, and Blobe GC (2008). Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta 1782, 197–228. [DOI] [PubMed] [Google Scholar]
- Groppe J, Hinck CS, Samavarchi-Tehrani P, Zubieta C, Schuermann JP, Taylor AB, Schwarz PM, Wrana JL, and Hinck AP (2008). Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol Cell 29, 157–168. [DOI] [PubMed] [Google Scholar]
- Hart PJ, Deep S, Taylor AB, Shu Z, Hinck CS, and Hinck AP (2002). Crystal structure of the human TbetaR2 ectodomain--TGF-beta3 complex. Nat Struct Biol 9, 203–208. [DOI] [PubMed] [Google Scholar]
- Hatley RJD, Macdonald SJF, Slack RJ, Le J, Ludbrook SB, and Lukey PT (2018). An alphav-RGD Integrin Inhibitor Toolbox: Drug Discovery Insight, Challenges and Opportunities. Angew Chem Int Ed Engl 57, 3298–3321. [DOI] [PubMed] [Google Scholar]
- Huang T, David L, Mendoza V, Yang Y, Villarreal M, De K, Sun L, Fang X, Lopez-Casillas F, Wrana JL, et al. (2011). TGF-beta signalling is mediated by two autonomously functioning TbetaRI:TbetaRII pairs. EMBO J 30, 1263–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries JD, Byron A, and Humphries MJ (2006). Integrin ligands at a glance. J Cell Sci 119, 3901–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura H, Cambier S, Somanath S, Barker T, Minagawa S, Markovics J, Goodsell A, Publicover J, Reichardt L, Jablons D, et al. (2011). Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin alphavbeta8-mediated activation of TGF-beta. J Clin Invest 121, 2863–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, Wang Y, Bernstein X, Li JT, Atabai K, et al. (2012). IL-17A produced by αβ T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med 18, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, and Flavell RA (2006). Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25, 455–471. [DOI] [PubMed] [Google Scholar]
- Lienart S, Merceron R, Vanderaa C, Lambert F, Colau D, Stockis J, van der Woning B, De Haard H, Saunders M, Coulie PG, et al. (2018). Structural basis of latent TGF-beta1 presentation and activation by GARP on human regulatory T cells. Science 362, 952–956. [DOI] [PubMed] [Google Scholar]
- Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, et al. (2012). ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 180, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S, Chou FC, Conchuir SO, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P, et al. (2013). Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE). PLoS One 8, e63906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J (2012). TGFβ signalling in context. Nat Rev Mol Cell Biol 13, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Melton AC, Bailey-Bucktrout SL, Travis MA, Fife BT, Bluestone JA, and Sheppard D (2010). Expression of alphavbeta8 integrin on dendritic cells regulates Th17 cell development and experimental autoimmune encephalomyelitis in mice. J Clin Invest 120, 4436–4444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minagawa S, Lou J, Seed RI, Cormier A, Wu S, Cheng Y, Murray L, Tsui P, Connor J, Herbst R, et al. (2014). Selective targeting of TGF-beta activation to treat fibroinflammatory airway disease. Sci Transl Med 6, 241ra279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazono K, Yuki K, Takaku F, Wernstedt C, Kanzaki T, Olofsson A, Hellman U, and Heldin CH (1990). Latent forms of TGF-beta: structure and biology. Ann N Y Acad Sci 593, 51–58. [DOI] [PubMed] [Google Scholar]
- Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, Kashem SW, Welty NE, Igyártó BZ, Wijeyesinghe S, Thompson EA, et al. (2016). Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-β. Nat Immunol 17, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses HL, Roberts AB, and Derynck R (2016). The Discovery and Early Days of TGF-beta: A Historical Perspective. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, and Nishimura SL (2002). The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol 157, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura SL (2009). Integrin-mediated transforming growth factor-beta activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol 175, 1362–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura SL, Sheppard D, and Pytela R (1994). Integrin alpha v beta 8. Interaction with vitronectin and functional divergence of the beta 8 cytoplasmic domain. J Biol Chem 269, 28708–28715. [PubMed] [Google Scholar]
- Ozawa A, Sato Y, Imabayashi T, Uemura T, Takagi J, and Sekiguchi K (2016). Molecular Basis of the Ligand Binding Specificity of alphavbeta8 Integrin. J Biol Chem 291, 11551–11565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace CN, and Scholtz JM (1998). A helix propensity scale based on experimental studies of peptides and proteins. Biophys J 75, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, Mistry M, Bronson RT, Santoro D, Franco C, et al. (2018). A Milieu Molecule for TGF-beta Required for Microglia Function in the Nervous System. Cell 174, 156–171 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radaev S, Zou Z, Huang T, Lafer EM, Hinck AP, and Sun PD (2010). Ternary complex of transforming growth factor-beta1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J Biol Chem 285, 14806–14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J, Mercer J, Shrestha Y, Oliver R, Tamayo P, Doench JG, Tirosh I, Piccioni F, Hartenian E, Horn H, et al. (2016). Genetic and Proteomic Interrogation of Lower Confidence Candidate Genes Reveals Signaling Networks in β-Catenin-Active Cancers. Cell Syst 3, 302–316.e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem M, Wallace C, Velegraki M, Li A, Ansa-Addo E, Metelli A, Kwon H, Riesenberg B, Wu B, Zhang Y, et al. (2019). GARP Dampens Cancer Immunity by Sustaining Function and Accumulation of Regulatory T Cells in the Colon. Cancer Res 79, 1178–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjabi S, Oh SA, and Li MO (2017). Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, and Springer TA (2011). Latent TGF-beta structure and activation. Nature 474, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Tessier MB, Pederson K, Wang X, Venot AP, Boons GJ, Prestegard JH, and Woods RJ (2016). Extension and validation of the GLYCAM force field parameters for modeling glycosaminoglycans. Can J Chem 94, 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer TA, Zhu J, and Xiao T (2008). Structural basis for distinctive recognition of fibrinogen gammaC peptide by the platelet integrin alphaIIbbeta3. J Cell Biol 182, 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley P (1989). Chinese hamster ovary cell mutants with multiple glycosylation defects for production of glycoproteins with minimal carbohydrate heterogeneity. Mol Cell Biol 9, 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockis J, Colau D, Coulie PG, and Lucas S (2009). Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol 39, 3315–3322. [DOI] [PubMed] [Google Scholar]
- Takagi J (2007). Structural basis for ligand recognition by integrins. Curr Opin Cell Biol 19, 557–564. [DOI] [PubMed] [Google Scholar]
- Takasaka N, Seed RI, Cormier A, Bondesson AJ, Lou J, Elattma A, Ito S, Yanagisawa H, Hashimoto M, Ma R, et al. (2018). Integrin alphavbeta8-expressing tumor cells evade host immunity by regulating TGF-beta activation in immune cells. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolcher AW, Berlin JD, Cosaert J, Kauh J, Chan E, Piha-Paul SA, Amaya A, Tang S, Driscoll K, Kimbung R, et al. (2017). A phase 1 study of anti-TGFbeta receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother Pharmacol 79, 673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, Wang Y, Bernstein X, Huang X, Reichardt LF, et al. (2007). Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Agthoven JF, Xiong JP, Alonso JL, Rui X, Adair BD, Goodman SL, and Arnaout MA (2014). Structural basis for pure antagonism of integrin αVβ3 by a high-affinity form of fibronectin. Nat Struct Mol Biol 21, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meeteren LA, Goumans MJ, and ten Dijke P (2011). TGF-beta receptor signaling pathways in angiogenesis; emerging targets for anti-angiogenesis therapy. Curr Pharm Biotechnol 12, 2108–2120. [DOI] [PubMed] [Google Scholar]
- Wallace CH, Wu BX, Salem M, Ansa-Addo EA, Metelli A, Sun S, Gilkeson G, Shlomchik MJ, Liu B, and Li Z (2018). B lymphocytes confer immune tolerance via cell surface GARP-TGF-beta complex. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dong X, Zhao B, Li J, Lu C, and Springer TA (2017). Atypical interactions of integrin alphaVbeta8 with pro-TGF-beta1. Proc Natl Acad Sci U S A 114, E4168–E4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RY, Song Y, Barad BA, Cheng Y, Fraser JS, and DiMaio F (2016). Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb B, and Sali A (2014). Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Bioinformatics 47, 5 6 1–32. [DOI] [PubMed] [Google Scholar]
- Weinreb PH, Simon KJ, Rayhorn P, Yang WJ, Leone DR, Dolinski BM, Pearse BR, Yokota Y, Kawakatsu H, Atakilit A, et al. (2004). Function-blocking integrin alphavbeta6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J Biol Chem 279, 17875–17887. [DOI] [PubMed] [Google Scholar]
- Xiong JP, Mahalingham B, Alonso JL, Borrelli LA, Rui X, Anand S, Hyman BT, Rysiok T, Muller-Pompalla D, Goodman SL, et al. (2009). Crystal structure of the complete integrin alphaVbeta3 ectodomain plus an alpha/beta transmembrane fragment. J Cell Biol 186, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, and Arnaout MA (2001). Crystal structure of the extracellular segment of integrin alpha Vbeta3. Science 294, 339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Zhu J, Mi LZ, Walz T, Sun H, Chen J, and Springer TA (2012). Structural specializations of alpha(4)beta(7), an integrin that mediates rolling adhesion. J Cell Biol 196, 131–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: Real-time CTF determination and correction. J Struct Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Choi WS, McCoy JG, Negri A, Naini S, Li J, Shen M, Huang W, Bougie D, Rasmussen M, et al. (2012). Structure-guided design of a high-affinity platelet integrin αIIbβ3 receptor antagonist that disrupts Mg2+ binding to the MIDAS. Sci Transl Med 4, 125ra132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhu J, and Springer TA (2013). Complete integrin headpiece opening in eight steps. J Cell Biol 201, 1053–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, and Sun PD (2004). Overexpression of human transforming growth factor-beta1 using a recombinant CHO cell expression system. Protein Expr Purif 37, 265–272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Integrins and TGF-β: nomenclature and models of activation, related to Introduction, Fig. 7 (A) Integrin and L-TGF-β domains and subdomains and their membrane or extracellular matrix localization via coupling to adaptor proteins are shown using common nomenclature. (B) In the global-rearrangement or “switchblade” model of integrin activation, the bent conformation moves through an extended-closed conformation to an extended-open conformation. In this model, high-affinity ligand binding and actin cytoskeletal force transduction occur in the extended-open conformation. In the case of αvβ6 it has been proposed that such force releases TGF-β and allows diffusion to its target receptors (Dong et al., 2017). Pink arrows indicate movements in the headpiece. (C) The αvβ8 integrin assumes a single conformation, extended-closed, that accommodates ligand binding and affinity regulation. The essential immune cell functions of αvβ8 have been tied to binding to L-TGF-β presented by type I transmembrane adaptor proteins such as GARP on immune cell surfaces. In this model, we hypothesize that the “latency lasso” of the straitjacket domain of L-TGF-β loosens upon binding to αvβ8 to allow the active domain of TGF-β to interact with its receptors.
Fig. S2. Cryo-EM micrographs and statistics for αvβ8 complexes, related to Figs. 1 and 2 Details of data collection and processing data for (A) αvβ8/LTGF-β (subclass iv), (B) αvβ8/C6D4 and (C) αvβ8/C6-RGD3. For each complex the following are shown: First column: a representative motion corrected micrograph of the particles suspended in vitreous ice. Second column: the gold standard FSC (top) and the angular distributions of particles (bottom) used in the final map, as estimated by cryoSPARC. Third column: (A) the unsharpened map displayed at a low thresh, (B,C) the unsharpened map before focused refinement displayed at a low threshold. Fourth column: the map after focused alignment displayed at a high threshold sharpened to a b-factor of (A) −71 (B) −83 or (C) −84. Maps are colored on the same scale, as indicated in row A, based on local resolution estimates. Scale bar = 100 nm.
Fig. S3. Comparisons of αvβ8 structures with integrin crystal structures, model quality, and characterization of C6-RGD3, related to Figs. 1, 2 and 4 (A, B) Superimpositions of ribbon models of αvβ8/L-TGF-β1 with published models from crystal structures of liganded αvβ6 (RGD peptide, PDB: 4UM9 (Dong et al., 2014) (A); L-TGF-β1, PDB: 5FFO (Dong et al., 2017) (B)). From αvβ8/L-TGF-β1 αv-subunit, green; β8-subunit, blue; RGD loop, purple; β6 subunit and L-TGF-β3 RGD peptide, salmon (A); β6 subunit and L-TGF-β1 RGD loop, lime green.
(C, D) Close-up of the binding interface of the αvβ8 integrin (light green and blue) and Fab C6D4 (coral, C) or Fab C6-RGD3 (pink, D) with the corresponding sharpened density map (grey volume). (E) Close-up of the binding interface of the β8 integrin subunit SDL2 loop (cyan) and the L-TGF-β1 proximal loop, RGD motif, and ligand-binding helix (purple) superimposed on their respective sharpened density maps (mesh). (F) Close-up of the Fab C6-RGD3 CDRL1 loop (pink) and its sharpened density map (pink mesh). (G, H) Close-up of the integrin β8 SDL1 α1 helix and β6-α7 loop when in complex with L-TGF-β1 (cyan, G) or Fab C6-RGD3 (dark blue, H).
(I) Map to model FSC curves for the αvβ8/L-TGF-β1 (purple), αvβ8/C6D4 (orange), αvβ8/C6-RGD3 (magenta) complexes.
(J-M) View of the metal ions and MIDAS cation coordination in various liganded integrin structures in ribbon models: αvβ8/L-TGF-β1 complex (J), αvβ3/fibronectin 10th domain RGD complex (PDB: 4MMX) (Van Agthoven et al., 2014) (K), αiibβ3/fibrinogen RGD peptide complex (PDB: 2VDR) (Springer et al., 2008)(L), αvβ6/L-TGF-β1 complex (PDB: 5FFO) (M). The coordinating residues are indicated in sticks.
(N-S) Superimpositions of the α1-helix, with the β6-α7 loop and MIDAS cation (dotted circle) as ribbon models with superimpositions from unliganded or liganded αiibβ3 (PDB: 3T3P (Zhu et al., 2012) or 2VDR(Xiong et al., 2009), respectively): (N) liganded (yellow) or unliganded αiibβ3, red; (O) liganded αiibβ3 (red) or αvβ8/C6-RGD3, green; (P) liganded αiibβ3 (yellow) or αvβ8/C6D4, light blue; (Q) liganded αiibβ3 (orange) or αvβ8/L-TGF-β1, pink; (R) αvβ8/C6D4 (light blue), αvβ8/C6-RGD3 (green), or αvβ8/L-TGF-β1, pink. Movement of the tip of the SDL1 α1-helix is highlighted by the S116 (β8)/S123 (β3) residues in sticks. The AspRGD is represented in sticks for liganded structures. (T, U) Superimposition of ribbon models of the αvβ8/C6D4 and α4β7/Act-1 (PDB: 3V4P) (Yu et al., 2012) complexes. Both C6D4 and Act-1 epitopes are located in the SDL2 region of the integrin (front view (T); rotated view, U)). αv-green, β8-blue, C6D4-orange, α4-lime green, β7-light blue, Act-1-magenta.
(V) Ribbon model of the αvβ8/C6D4 complex with the CDRL1 loop highlighted in red that was replaced with the L-TGF-β integrin-binding motif and helix to create C6-RGD3 (αv-green, β8-blue, C6D4-gold). (W) Binding assay to immobilized αv-integrins to show specificity of C6D4 for αvβ8, and C6-RGD3 for αvβ6 and αvβ8. Shown is a representative experiment of three (n=3).
(X, Z) Inhibition of cell adhesion of CHO cells co-expressing GARP and L-TGF-β1 on their cell surface to immobilized (X) αvβ8 or (Z) αvβ6 in the presence of indicated concentrations of C6D4 or C6-RGD3 or anti-β6 (3G9). TGF-β activation assays of (Y) CHO cells stably transfected with αvβ8 or (AA) human bronchial epithelial cells (HBEC), which naturally express high levels of αvβ6 and low levels of αvβ8 (Araya et al., 2007) in the presence of indicated concentrations of C6D4, C6-RGD3 or 3G9 (Weinreb et al., 2004). Activation is shown relative to a pan-TGF-β neutralizing antibody (1D11). n=3, error bars show s.e.m.
Fig. S4. Processing schematic for the αvβ8/C6-RGD3 complex, related to Fig. 2 A schematic flowchart showing the classification scheme of the αvβ8/C6-RGD3 complex. Particle numbers at each step and for each class are indicated. From top to bottom: After 3D classification, the data was separated into three groups: particles with C6-RGD3 bound at the expected angle, particles with C6-RGD3 at a skewed angle, and particles without C6-RGD3. These three groups were processed separately. The map showing C6-RGD3 at the expected angle shows the RGD motif in the alternative, non-helical binding conformation. The map showing C6-RGD3 at the skewed angle shows the RGD motif in a helical conformation. All maps are colored on the same scale, as indicated, based on local resolution estimates.
Fig. S5. Processing schematic for the αvβ8/LTGF-β complex, related to Fig. 5A schematic flowchart showing the classification scheme of the αvβ8/L-TGF-β complex. Due to the preferred orientations adopted by the particles, two types of grids were used and two separate datasets were collected. Upon angular assignment, it was found that although particles on both grids suffered from preferred orientations, the orientations complement each other to form a more complete orientation sampling. Particle numbers at each step and for each class are indicated. 3D reconstructions of seven out of sixteen subclasses showed significant density for L-TGF-β, and reached high resolution after refinement. All maps are colored on the same scale, as indicated, based on local resolution estimates.
Fig. S6. β8 and β6 SDL mutations, and structure-guided mutagenesis of the β8 SDL2/3 binding pocket, related to Fig. 6 (A, B) CHOlec 3.2.8.1 cells were stably transfected with various β8, β6 WT or SDL mutants, as indicated, sorted several times to establish highly expressing pools, and tested for expression using C6D4 (A) compared to clone 68 (B), a βI domain antibody that binds to the tail of the β8 α1-helix (Minagawa et al., 2014). Note that the mutant I208R does not bind C6D4 as well as other mutants, as this mutation would be expected to clash with the position of C6D4 CDRL1. (C, D) Binding of L-TGF-β1 to truncated secreted forms of C) WT and a Y172A αvβ8 mutant, or D) αvβ6 WT and a I183N αvβ6 mutant. n=3, error bars show s.e.m.
(E-G) Ribbon models of the αvβ8 head domain of the CDRL1 loops of (E) C6D4, (F) C6-RGD3 and (G) L-TGF-β (gold sticks), with boxed ligand pocket (upper panels) shown in close up views in middle and lower panels. Color code: αv-subunit (green), β8-SDL1 (light blue), -SDL2 (cyan) and -SDL3 (purple) loops. The positioning of the C6D4 CDRL1; C6-RGD3 CDRL1; or L-TGF-β1 integrin-binding loops are shown positioned in the binding cleft of β8 wild-type (middle panels); and the I208R mutant (lower panels). Middle panel color code: L-TGF-β integrin-binding loop R215 (gold), the β8 SDL2 Y172 (red) and SDL1 S116 (red). Lower panel color code: the SDL3 I208R mutation is indicated in red spheres demonstrating clash with the C6D4 CDRL1; C6-RGD3 CDRL1 but not the canonical position of the L-TGF-β integrin-binding loop. The SyMBS (grey) and MIDAS (green) cations are shown.
Fig. S7. Expression and characterization of WT L-TGF-β, L-TGF-β (R249A), WT L-TGF-β/GARP and L-TGF-β (R249A)/GARP TMLC reporter cells, related to Fig. 7 (A-D) WT L-TGF-β/GARP and L-TGF-β (R249A)/GARP are equally expressed on the surface of TGF-β reporter cells (TMLC). TMLC cells were first stably transfected with either (A) WT L-TGF-β IRES GFP or, (B) L-TGF-β (R249A) IRES GFP and sorted for equal expression of GFP. Histograms show GFP expression on the x-axis and surface localization of L-TGF-β, as assessed by anti-LAP-APC staining, on the y-axis. Stably transfected and sorted cells show no surface staining for L-TGF-β, despite showing ~40–50% GFP bright cells. In contrast, when (C) WT L-TGF-β IRES GFP or (D) L-TGF-β (R249A) IRES GFP expressing TMLC were subsequently stably transfected with a N-terminal HA tagged GARP construct, bright surface expression of L-TGF-β was detected in ~40% of cells. (C) WT L-TGF-β IRES GFP/HA GARP or, (D) L-TGF-β (R249A) IRES GFP/HA GARP TMLC were sorted for equal surface expression of L-TGF-β, as assessed by anti-LAP-APC staining. (E) Cartoon representation of the experiment. Individual wells of 96-well ELISA plates were coated with either αvβ8, αvβ3 (which binds with low affinity to L-TGF-β), polyclonal anti-LAP (which binds to LAP, but does not activate or inhibit activation of L-TGF-β), or BSA as a non-specific binding control. TMLC cells either non-transfected or transfected as in A-D with WT L-TGF-β or L-TGF-β (R249A) with or without GARP were applied to coated wells. (F) Parental TMLC cells, WT L-TGF-β IRES GFP, L-TGF-β (R249A) IRES GFP, WT L-TGF-β IRES GFP/HA GARP or, L-TGF-β (R249A) IRES GFP/HA GARP expressing TMLC cells were added to wells coated as indicated in the legend with each substrate and assayed for luciferase activity after 16–20 hr. Each condition was repeated in triplicate with three independent experiments. *p < 0.05, ** p <0.01, **** p <0.0001, ns = not significant, as determined by unpaired Student’s t-test. Note L-TGF-β (R249A) IRES GFP/HA GARP expressing TMLC show dramatic and significant αvβ8-dependent TGF-β activation similar to that seen with WT L-TGF-β IRES GFP/HA GARP TMLC cells. (G) Characterization of secreted L-TGF-β (R249A) and WT L-TGF-β compared to cell surface expressed L-TGF-β (R249A)/HA GARP and WT L-TGF-β IRES GFP/HA GARP. Left panels: Shown are recombinant secreted L-TGF-β (R249A) and WT L-TGF-β under non-reducing (NR) or reducing (R) conditions as assessed by 4–12% gradient SDS-PAGE and Coomassie Brilliant Blue staining. Shown are molecular weight markers (MWM). Irrelevant lanes have been digitally removed for clarity. The identity of the bands at the expected molecular weights is indicated. Right panel: L-TGF-β (R249A) IRES GFP/HA GARP and WT L-TGF-β IRES GFP/HA GARP expressing TMLC cells were surface labeled with biotin, lysed and immunoprecipitated with anti-HA to capture the cell surface GARP/L-TGF-β complexes. After resolving immunoprecipitated proteins by 4–12% SDS page under reducing conditions, biotinylated proteins were detected by immunoblotting with streptavidin-HRP and results were aligned with MWM on the left panel. Results show that both L-TGF-β (R249A) IRES GFP/HA GARP and WT L-TGF-β IRES GFP/HA GARP form complexes on the surface of TMLC cells and that TGF-β in the R249A mutant does not cleave from the LAP.