

Abstract
Hormonal therapy targeting androgen receptor (AR) is initially effective to treat prostate cancer (PCa), but it eventually fails. It has been hypothesized that cellular heterogeneity of PCa, consisting of AR+ luminal tumor cells and AR− neuroendocrine (NE) tumor cells, may contribute to therapy failure. Here, we describe the successful purification of NE cells from primary fresh human prostate adenocarcinoma based on the cell surface receptor C-X-C motif chemokine receptor 2 (CXCR2). Functional studies revealed CXCR2 to be a driver of the NE phenotype, including loss of AR expression, lineage plasticity, and resistance to hormonal therapy. CXCR2-driven NE cells were critical for the tumor microenvironment by providing a survival niche for the AR+ luminal cells. We demonstrate that the combination of CXCR2 inhibition and AR targeting is an effective treatment strategy in mouse xenograft models. Such a strategy has the potential to overcome therapy resistance caused by tumor cell heterogeneity.
INTRODUCTION
Prostate cancer (PCa) is a major cause of cancer-related death (1). Hormonal therapy targeting androgen receptor (AR) is the treatment of choice for advanced PCa. Although the initial response is usually clinically evident, the development of castration-resistant PCa (CRPC) is nearly inevitable. Second-generation hormonal therapy drugs enzalutamide and abiraterone acetate have shown efficacy against CRPC, but resistance still occurs (2,3). Therefore, the disease remains incurable despite near-maximal AR inhibition. In addition, there are cases of AR− PCa that do not respond to AR-targeted therapies. A classic example is small cell neuroendocrine (NE) carcinoma (SCNC) (4), which is composed entirely of NE cells that do not express AR and do not respond to hormonal therapy. Therefore, there is an urgent need to develop AR-independent therapeutic strategies.
Histologically, most primary PCa is classified as adenocarcinoma, composed of bulk luminal-type tumor cells expressing AR and prostate-specific antigen (KLK3) and a minor component (~1%) of NE cells. Unlike the luminal-type tumor cells, the NE cells do not express AR or KLK3 and are quiescent (5). NE tumor cells are enriched in high-grade, high-stage tumors and therapy-resistant PCa (6). However, up to now, immunohistochemistry (IHC) staining of paraffin-embedded tumor tissue has been the only method to study NE cells, because such cells are generally rare in human PCa and are scattered among the more abundant luminal-type tumor cells (7). Molecular characterization of NE tumor cells in primary human PCa tissue has never been reported because of the lack of a specific cell surface marker for this rare cell population and the technical challenge in obtaining fresh human PCa tissue. As a result, little is known about the function of NE tumor cells in therapy resistance and disease progression.
Several markers have been used to identify NE cells in PCa by IHC, among which chromogranin A (CHGA) is considered the most sensitive and specific (8). However, no cell surface marker has been identified that can be used to purify the rare NE tumor cells from fresh PCa. Our previous work demonstrated that the rare NE cells in PCa secrete interleukin 8 (IL-8) and overexpress IL-8 receptor C-X-C motif chemokine receptor 2 (CXCR2), whereas the bulk luminal tumor cells are CXCR2− (7). CXCR2 is a G protein-coupled receptor for angiogenic CXC chemokine family members and is involved in leukocyte chemotaxis and inflammatory responses (9). The CXCL8-CXCR1/2 axis may play an important role in tumor progression and metastasis by regulating cancer stem cell proliferation and self-renewal (10). However, it is unclear whether CXCR2 expressed by NE cells mediates NE cell function and whether it plays a role in therapy resistance and progression of PCa. In this study, we purified NE tumor cells from fresh human PCa tissue and performed RNA sequencing (RNA-seq). We also studied the role of NE cells in PCa therapy resistance and progression. We revealed the sensitivity of aggressive PCa to CXCR2 inhibition in vitro and in vivo, with inhibition of NE cells and suppression of tumor growth. We conclude that targeting NE cells of PCa through CXCR2 inhibition is an AR-independent therapeutic strategy that can improve therapeutic efficacy for the treatment of lethal PCa.
RESULTS
CXCR2 is a cell surface marker for NE cells of human PCa
To determine whether CXCR2 is a specific surface marker for the NE cells of human PCa, we performed immunofluorescence analysis of tissue microarrays (TMAs) from human PCa samples of different grades and stages using antibodies against CXCR2, NE marker CHGA, and luminal cell marker cytokeratin 8 (KRT8). The TMA included 131 cases of low-grade PCa (LG-PCa), 8 cases of high-grade PCa (HG-PCa), 16 cases of CRPC, and 12 cases of SCNC. Scattered or clustered NE cells expressing CHGA were identified (Fig. 1A). The CHGA+ NE cells specifically expressed CXCR2, whereas KRT8+ luminal cells were negative for CXCR2 expression (Fig. 1, B and C, and fig. S1).
Fig. 1. CXCR2 is a surface marker for NE cells in human PCa and is associated with disease progression.
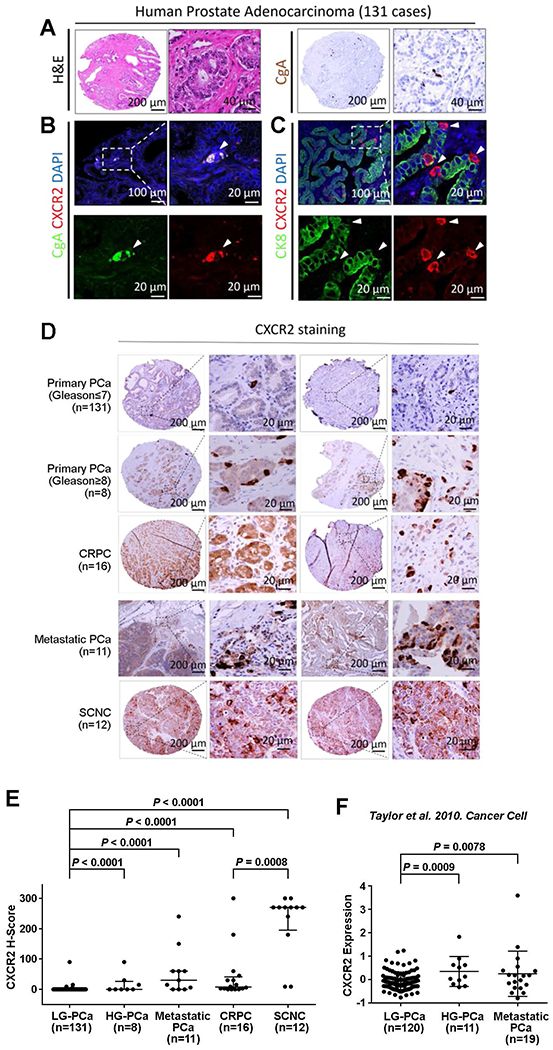
(A) Representative hematoxylin and eosin (H&E) staining (left) and IHC for NE marker CHGA (right) of human primary PCa. Nuclei (DAPI staining) are shown in blue. (B) Representative immunofluorescence images of CXCR2 (red) and NE marker CHGA (green) staining in human primary PCa. White arrowheads point to CXCR2+ CHGA+ NE tumor cells. (C) Immunofluorescence of CXCR2 (red) and luminal marker KRT8 (green) in human primary prostate adenocarcinoma. White arrowheads indicate CXCR2+ NE cells (red), which are negative for KRT8 (green). (D and E) Representative images (D) and quantification (E) of CXCR2 IHC staining on TMAs. Logistic regression analysis was performed using nonparametric Mann-Whitney U test; lines represent median and interquartile range. (F) Analysis of CXCR2 expression among primary and metastatic PCa tumors from the dataset of Taylor et al. (11). Logistic regression analysis was performed using t test; lines represent means ± SD.
CXCR2 expression is associated with Gleason score and PCa progression
Because the NE population in PCa increases with disease progression (6), we sought to determine whether there is a correlation between CXCR2 expression and PCa grade and stage. We examined CXCR2 expression by IHC staining using TMAs of LG-PCa (Gleason score ≤ 7, n = 131), HG-PCa (Gleason score ≥ 8, n = 8), CRPC (n = 16), and SCNC (n = 12), as well as lymph node specimens containing metastatic PCa (n = 11). We found that the CXCR2+ cells were present as a rare population in LG-PCa, comprising <0.5% of the cancer cells, but were significantly enriched in HG-PCa, CRPC, metastatic PC, and SCNC, with an average percentage of 6, 14, 20, and 74%, respectively (P < 0.0001; Fig. 1, D and E). SCNC (74%), a histologic variant of PCa that usually occurs in end-stage disease, was found to harbor more CXCR2+ NE cells in comparison to other tumors (Fig. 1D and fig. S1B). To further validate this finding, we analyzed the publicly available dataset generated by Taylor et al. (11), which includes 35 of 120 cases of LG-PCa, 11 cases of HG-PCa, and 19 cases of metastatic PCa. Similarly, CXCR2 mRNA expression was significantly higher in HG-PCa (P = 0.0009) and metastatic PCa (P = 0.0078) when compared to LG-PCa (Fig. 1F). These findings support the notion that CXCR2+ NE cells are enriched in high-grade and advanced tumors.
CXCR2+ NE tumor cell isolated from primary PCa have stem-like features
We developed a tissue procurement program to isolate pure populations of epithelial cells from fresh prostatectomy specimens immediately after surgery, using cell surface markers TACSTD2 (tumor-associated calcium signal transducer 2, TROP2), PTPRC (protein tyrosine phosphatase receptor type C, CD45), and ITGA6 (integrin subunit α6, CD49f) (12). We have published extensively on the oncogenic mechanisms of PCa using purified basal and luminal cells from fresh human prostates (13, 14). To characterize NE cells in primary PCa, we isolated rare NE cells from fresh prostatectomy specimens by fluorescence-activated cell sorting (FACS) with an anti-CXCR2 antibody (Fig. 2A and fig. S1E). Owing to the rarity of NE cells (< 0.5%) in patients’ fresh prostate tumors (Fig. 1D), we were not able to collect enough cells with good RNA quality for RNA-seq from all 16 samples procured. In total, we acquired qualified RNA samples for NE (TACSTD2+PTPRC−CXCR2+) and luminal (TACSTD2+PTPRC−CXCR2−) tumor cells from PCa of three prostatectomy specimens for RNA-seq. We also isolated basal cells (TACSTD2+PTPRC−ITGA6+) from the benign region of patients’ fresh prostatectomy specimens for gene expression profile analyses.
Fig. 2. Distinct signaling was observed between CXCR2+ NE and CXCR2− luminal cells isolated from human primary PCa.
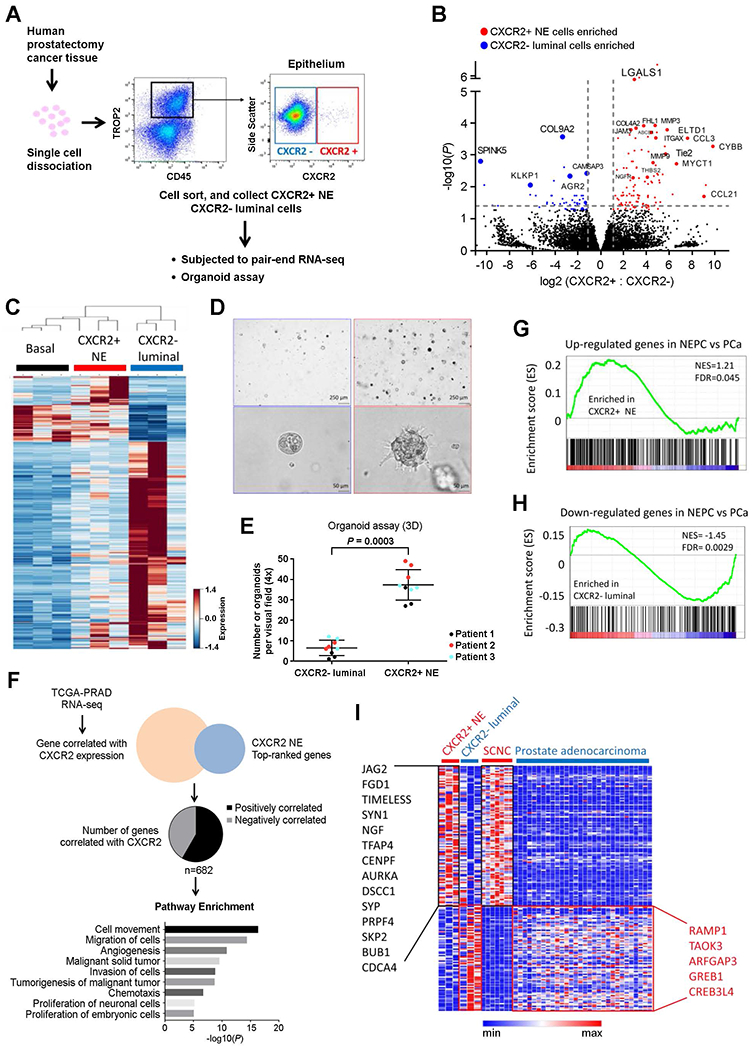
(A) Experimental scheme for gene expression and cellular function analysis of human PCa CXCR2+ NE and CXCR2− luminal populations. (B) Volcano plot of differentially expressed genes between CXCR2+ NE and CXCR2− luminal cells isolated from fresh primary human PCa tissue. (C) Heat map of genes from sorted benign basal cells (ITGA6+), NE tumor cells (CXCR2+), and luminal-type tumor cells (CXCR2−). (D and E) Tumor organoids generated by CXCR2+ NE cells (right) and CXCR2− luminal cells (left). Representative images (D) and quantification (E) of three independent patients’ samples are shown. 3D, three-dimensional. (F) Signaling pathways associated with CXCR2 expression in PCa. Gene expression of 459 primary PCa was obtained from TCGA prostate adenocarcinoma database (TCGA-PRAD). Ingenuity pathway analysis was performed using 682 genes that were correlated with CXCR2 expression (P< 0.05, Pearson’s correlation). (G and H) The enrichment scores of NE gene sets (G) and prostate adenocarcinoma gene sets (H) in CXCR2+ NE cells isolated from primary human PCa tissue (18). FDR, false discovery rate; NES, normalized enrichment score. (I) Heat map of gene expression among CXCR2+ NE and CXCR2− luminal tumor cells from fresh primary human PCa tissue, SCNC/neuroendocrine prostate cancer (NEPC), and prostate adenocarcinoma (18).
To validate whether TACSTD2+PTPRC−CXCR2+ cells are indeed the NE cell population in patients’ fresh PCa tissue, we carried out gene set enrichment analysis (GSEA) using 5917 Gene Ontology (GO) gene set from Molecular Signatures Database (MSigDB) (15). Compared to TACSTD2+PTPRC−CXCR2− cells, TACSTD2+PTPRC−CXCR2+ cells were significantly enriched in the 15 neuron-related GO gene sets (P < 0.05) (fig. S1F). CXCR2+ NE tumor cells were enriched in the gene sets associated with epithelial-mesenchymal transition (EMT), stem cell, extracellular matrix organization, tumorigenesis, angiogenesis, E2F signaling, MYC signaling, and transforming growth factor-β signaling [false discovery rate (FDR) < 0.001] (Fig. 2B, figs. S1G and S2A, and table S1). In contrast, CXCR2− luminal population was enriched for gene sets associated with luminal cells, PCa, response to androgen, and cholesterol biosynthesis (FDR < 0.001) (figs. S1F and S2B and table S2). Together, these findings confirmed that CXCR2 is a surface marker that can be used to purify NE cells from fresh primary human PCa tissue.
The basal cell population within benign human prostates contains stem/progenitor cells that give rise to all epithelial populations, and our prior work has demonstrated that it is a cell of origin for PCa (13). We found that CXCR2+ NE cells had a gene expression profile distinct from CXCR2− luminal-type PCa cells but similar to basal cells, particularly in the gene sets associated with EMT, stem cell, tumorigenesis, and androgen response, and these pathways distinguish CXCR2+ NE cells from the CXCR2− luminal tumor cells (FDR < 0.0001) (Fig. 2C and fig. S2, C to F). To determine whether TACSTD2+PTPRC− CXCR2+ NE cells isolated from PCa tissue were neoplastic (as opposed to contamination by benign NE cells), we compared their expression profiles with those of benign basal cells. We found that 46 of 189 oncogenic gene sets from MSigDB were significantly enriched in CXCR2+ NE cancer cells (Q < 0.05; table S3), including gene sets associated with tumor-specific mutation status, such as KRAS-mutant tumors (16) and phosphatase and tensin homolog (PTEN) inactivation (17), supporting the neoplastic nature of the purified NE cells.
Consistent with the stem cell-enriched gene profiles, freshly purified CXCR2+ NE tumor cells were highly capable of organoid formation, compared to CXCR2− luminal cells (Fig. 2, D and E). Moreover, the organoids formed by CXCR2+ NE tumor cells infiltrated the surrounding Matrigel, whereas the CXCR2− luminal organoids failed to invade (Fig. 2D). These findings further support the intrinsic EMT and invasive properties of prostatic CXCR2+ NE tumor cells.
To further study the relationship between CXCR2 expression and biological function of NE cells, we analyzed the gene expression profiles of 459 primary prostate adenocarcinoma samples in The Cancer Genome Atlas database. Among the genes associated with CXCR2+ NE tumor cells (Fig. 2B), the expression of 682 genes was significantly correlated with CXCR2 expression in primary PCa (P < 0.05, Pearson correlation; Fig. 2F, fig. S2G, and table S4). Ingenuity pathway analysis revealed that these 682 genes were functionally related to cell movement, migration and invasion, angiogenesis, tumorigenesis, chemotaxis, and proliferation of neuronal and embryonic cells (Fig. 2F and fig. S2C).
The CXCR2+ NE population resembles prostatic SCNC
CXCR2+ NE PCa cells were enriched for the gene networks associated with E2F targets and NMYC targets (FDR < 0.001; Fig. 2B), two pathways up-regulated in prostate SCNC, the most lethal histologic variant of PCa, composed of highly aggressive NE tumor cells (18). We therefore applied the CXCR2+ NE gene signature to the Beltran dataset that contains gene expression data for 7 cases of prostate SCNC and 30 cases of prostate adenocarcinomas (18). The CXCR2+ NE population was significantly enriched for the SCNC signature [normalized enrichment score (NES) = 1.21, FDR = 0.045; Fig. 2G], whereas the CXCR2− luminal population was enriched for the gene signature of primary prostate adenocarcinoma (NES = −1.45, FDR = 0.0029; Fig. 2H). The enriched genes in both CXCR2+ PCa NE cells and SCNC were important for tumorigenesis, metastasis, EMT, and neuronal development (Fig. 2I), whereas the genes enriched in both CXCR2− luminal PCa cells and primary adenocarcinoma were involved in AR response (Fig. 2I). These findings demonstrate that CXCR2+ NE cells in hormone-sensitive prostate adenocarcinoma have genomic features of the most lethal PCa (SCNC), which is seen in a large proportion of patients who have failed systemic AR-targeted therapies (19).
Hormonal therapy enriches CXCR2+ NE tumor cells in human PCa
The presence of the rare CXCR2+ NE tumor cell population in early-stage, hormone therapy-naïve PCa that harbors genomics features of advanced and aggressive PCa raises the possibility that these cells represent preexisting drug-resistant clone(s) that survive hormonal therapy and may be enriched by the therapy. To test this hypothesis, we turned to commonly used PCa cell line models. Unexpectedly, similar to human PCa tissue, cellular heterogeneity is also present in PCa cell lines, with bulk CXCR2− luminal-type tumor cells and a minor population of CXCR2+ NE tumor cells. In LNCaP cells, the vast majority of the cells were CXCR2−, while there was a rare population (~3.6%) of CXCR2+ cells (Fig. 3A and fig. S3A). Castration-resistant C4-2B cells and CWRR1 cells similarly harbored such cellular heterogeneity with varying proportions (9.3 and 0.3%, respectively) of CXCR2+ NE tumor cells (Fig. 3B and fig. S3, B and C). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis demonstrated differential expression of NE markers CHGA, SYP, and ENO2 in the sorted CXCR2+ and CXCR2− subpopulations (fig. S3D).
Fig. 3. CXCR2-mediated phenotypic switch drives therapy resistance in PCa.
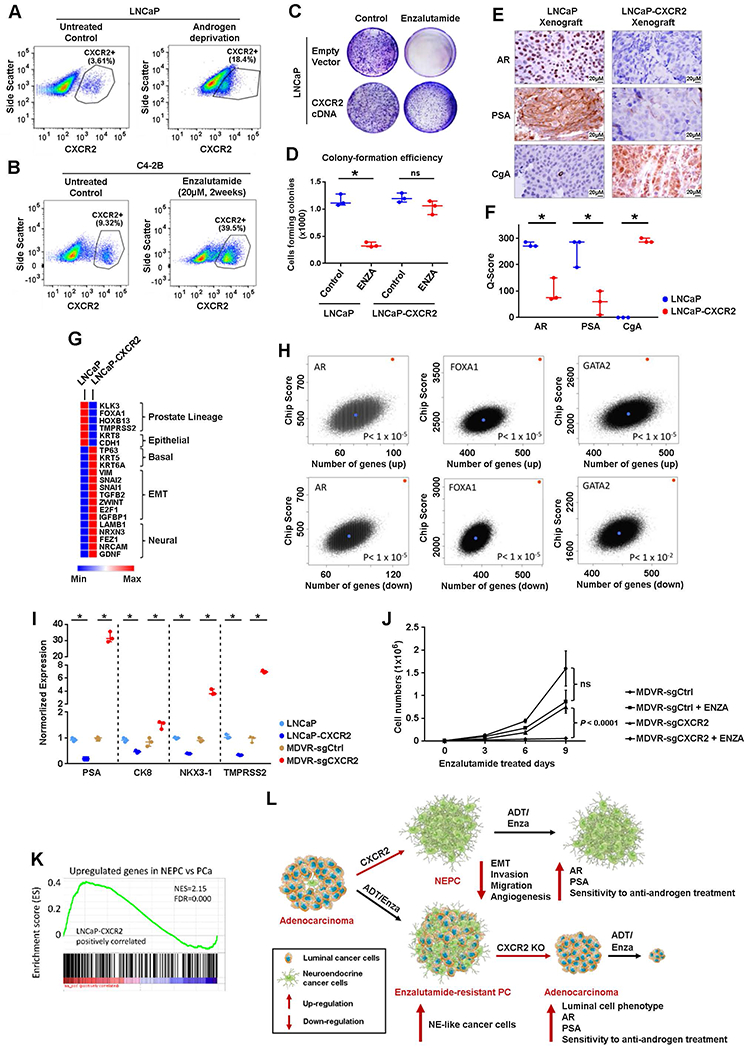
(A) Flow cytometric quantification of CXCR2+ cells in parental LNCaP cell line (left) and LNCaP cells cultured in charcoal-stripped (androgen-deprived) medium for 2 weeks (right). Representative images of three independent experiments are shown.(B) Flow cytometric quantification of CXCR2+ cells in parental C4-2B cell line (left) and C4-2B cells treated with enzalutamide (20 μM) for 2 weeks (right). Representative images of three independent experiments are shown. (C and D) Parental LNCaP cells and LNCaP cells overexpressing CXCR2 (LNCaP-CXCR2) were treated without (left) or with (right) enzalutamide (ENZA) for 2 weeks. Representative image (C) and quantification (D) of colony formation assay are shown. cDNA, complementary DNA. (E and F) CXCR2 expression induces a lineage switch from the luminal phenotype (AR+, KLK3+, and CHGA−) to the NE phenotype (AR−, KLK3−, and CHGA+) in an in vivo xenograft model. Representative images (E) and quantification (F) are shown. (G) Heat map of selected differentially expressed genes between LNCaP cells and LNCaP-CXCR2 cells. (H) Enrichment for binding sites for luminal-defining transcription factors in LNCaP cells measured by ChIP-seq analyses. The red dot represents the query signature of 1000 up-and down-regulated genes between LNCaP and LNCaP-CXCR2 cells. The blue dot represents one of a total of 100,000 randomly sampled gene lists of equal size to the query signature. The rank of the query gene list divided by the total number of resample instances was then used as the P value for the probability of enrichment by chance. (I) mRNA expression of luminal markers in LNCaP cells with/without CXCR2 overexpression or C4-2B cells with/without CRISPR-Cas9 knockout of CXCR2. (J) Growth curves of C4-2B/MDVR-sgCtrl and C4-2B/MDVR-sgCXCR2 (CRISPR-Cas9 knockout of CXCR2) with or without enzalutamide (20 μM) treatment. (K) The enrichment score of NE gene sets (18) in LNCaP-CXCR2 cell. (L) A model demonstrating how CXCR2 drives NE phenotype in PCa cells and renders CXCR2+ NE cells resistant to hormonal therapy [androgen deprivation therapy or enzalutamide treatment (ADT/Enza)]. Logistic regression analysis was performed using nonparametric Mann-Whitney U test; lines represent median and interquartile range. ns, nonsignificant; *P < 0.05.
To determine whether these cell lines molecularly recapitulate the cellular heterogeneity of primary PCa, we sorted CXCR2+ and CXCR2− populations from C4-2B cells by FACS. Consistent with our observations in fresh human PCa tissue, we found that CXCR2+ cells were enriched for stem cell markers (CD44, PROM1, POU5F1, SOX9, and KLF4) (20) (fig. S3E), CRPC-associated genes (IGF1R, REG4, UBE2C, BCL2, E2F3, and SOX2) (21), and EMT marker SNAI1 (fig. S3, F and G). To model the clinically used therapies for hormone-sensitive PCa and CRPC, we cultured LNCaP cells in androgen-deprived medium and treated C4-2B cells with enzalutamide, respectively. Although the treatments inhibited the growth of tumor cells, significant increases in the CXCR2+ cell populations were observed in both cell lines (P < 0.001, Fisher’s exact test; Fig. 3, A and B). These results suggest that CXCR2+ NE tumor cells are resistant to the currently used therapies, and such therapies enrich for NE cells, which may contribute to treatment failure and disease progression, consistent with our observation that CRPC tissue contains more NE tumor cells than primary PCa.
CXCR2 drives NE phenotype and therapeutic resistance
To investigate whether CXCR2 is a driver of the molecular and cellular features observed in NE tumor cells, we overexpressed CXCR2 in LNCaP cells (LNCaP-CXCR2; fig. S3H) and found that LNCaP-CXCR2 cells were resistant to enzalutamide (Fig. 3, C and D). Using GSEA, we found that the gene signatures in LNCaP-CXCR2 cells were concordant with those in CXCR2+ NE cells from patients’ primary PCa samples (figs. S1F and S3I). We further established mouse xenograft tumors of LNCaP and LNCaP-CXCR2 and found that CXCR2 overexpression in LNCaP cells (LNCaP-CXCR2 tumors) shut down the expression of luminal markers AR and KLK3 but turned on the expression of NE marker CHGA (Fig. 3, E and F).
Comparing the gene expression of lineage markers in LNCaP and LNCaP-CXCR2 cells, we discovered lineage plasticity in LNCaP cells after CXCR2 expression, which transforms LNCaP cells from prostate luminal phenotype to basal, EMT, and neural phenotype (Fig. 3G). To further elucidate the lineage switch induced by CXCR2, we assessed whether CXCR2 expression regulated a genome-wide regulatory gene program of luminal-type PCa. Using public datasets from chromatin immunoprecipitation and sequencing (ChIP-seq) analysis of luminal LNCaP cells (22, 23), we analyzed the enrichment of binding sites for the luminal-defining transcription factors AR, FOXA1, and GATA-binding protein 2 (GATA2) in the genes most differentially expressed between LNCaP and LNCaP-CXCR2 cells. The top 1000 overexpressed and 1000 underexpressed genes in the LNCaP cells relative to the LNCaP-CXCR2 cells were enriched for binding sites of AR, FOXA1, and GATA2 (Fig. 3H), suggesting that CXCR2 suppressed the activation of a global gene regulatory program associated with luminal phenotype in PCa.
We next investigated whether inhibition of CXCR2 expression in tumor cells was associated with a luminal phenotype. We generated CXCR2 knockout lines in enzalutamide-resistant C4-2B cells [C4-2B/enzalutamide (MDV3100)-resistant (MDVR)], which display NE features (fig. S3, J to L) (24). CXCR2 knockout caused the up-regulation of luminal and epithelial markers, including KLK3, KRT8, NKX3-1, and TMPRSS2 (Fig. 3I and fig. S3M), and restored sensitivity to enzalutamide treatment (Fig. 3J). Our findings demonstrate that CXCR2 expression is necessary and sufficient to induce lineage plasticity and therapeutic resistance in PCa cells.
LNCaP-CXCR2 cells and lethal PCa SCNC both show up-regulation of MYC targets, E2F targets, and TNF signaling (14, 25), prompting us to further explore their molecular similarities (18). GSEA revealed a strong positive association between LNCaP-CXCR2 cells and SCNC gene signature (FDR < 0.0001; Fig. 3K), suggesting that CXCR2 expression contributes to the clinical phenomenon that prostatic adenocarcinoma frequently transforms to the highly aggressive SCNC after hormonal therapy (19). Together, our findings suggest that CXCR2+ NE cells are enriched by AR-targeted therapies, and the expression of CXCR2 confers tumor cells resistance to hormonal therapy (Fig. 3L) and may directly contribute to the emergence of SCNC in the terminal stage of the disease.
CXCR2+ NE cells play important roles in the remodeling of tumor microenvironment
Because CXCR2+ NE cells were involved in the extracellular matrix organization, angiogenesis, and invasion (fig. S3F), we next sought to determine the role of the rare NE cells in regulating the tissue microenvironment in primary PCa. Using PECAM1 (platelet and endothelial cell adhesion molecule 1, CD31) IHC staining of patients’ PCa tissue samples (n = 77), we observed a positive correlation between the abundance of CXCR2+ NE cells and the density of blood vessels (P = 0.0011, Spearman correlation; Fig. 4, A and B, and fig. S4A). Because GO analysis of LNCaP-CXCR2 gene signature also revealed that the angiogenesis pathway is among the top-ranked gene sets (figs. S3I and S4B), we performed IHC staining of LNCaP and LNCaP-CXCR2 xenograft tumors for the expression of PECAM1 and found that CXCR2 overexpression enhanced blood vessel formation (Fig. 4, C and D).
Fig. 4. CXCR2 expression in NE cells drives the secretion of proangiogenic factors and promotes the formation of premetastatic niche in the tumor environment.
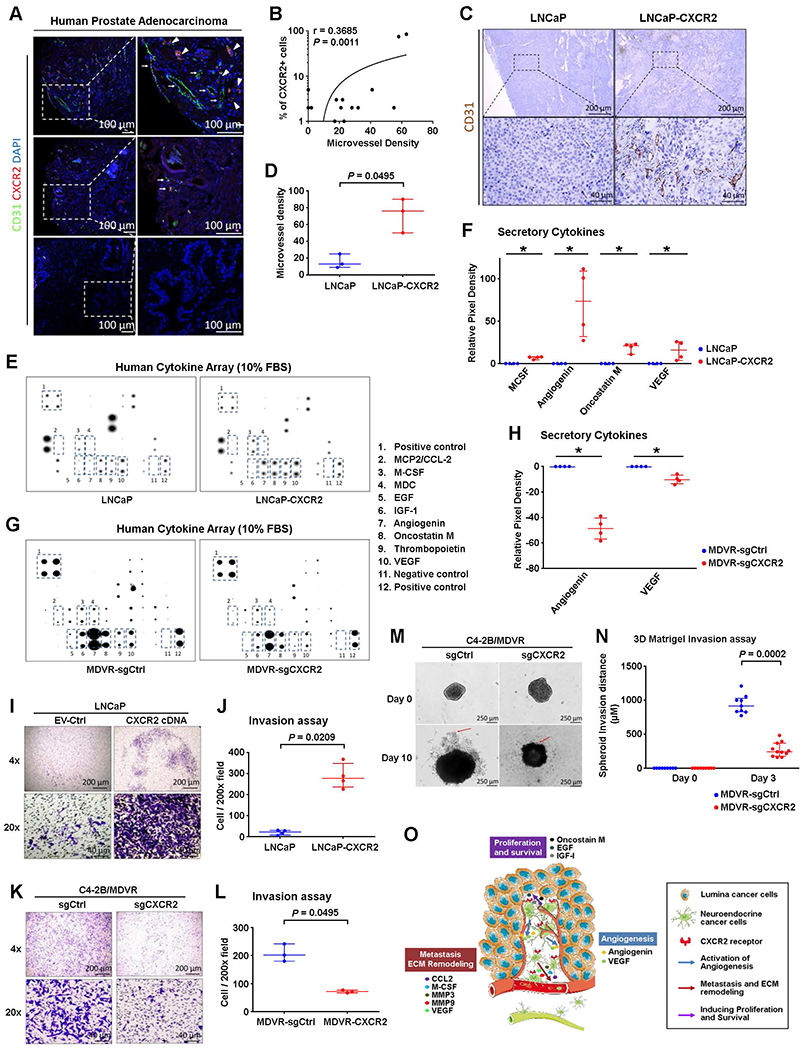
(A and B) Representative images of immunofluorescence for CXCR2 (red) and vascular endothelial cell marker PECAM1 (green) in TMAs of human primary PCa are shown in (A). Nuclei (DAPI staining) are shown in blue. Spearman correlation between CXCR2 expression and blood vessel density in a panel of 77 PCa cases is shown in (B). (C and D) Representative IHC images(A) and quantification (D) of PECAM1 expression in LNCaP and LNCaP-CXCR2 xenograft tumors. (E to H) Representative images (E) and quantification (F) of human cytokine antibody arrays of LNCaP or LNCaP-CXCR2. Representative images (G) and quantification (H) of human cytokine antibody arrays of C4–2B/MDVR-sgCtrl or C4–2B/MDVR-sgCXCR2. FBS, fetal bovine serum. (I to N) Representative images (I) and quantification (J) of transwell Matrigel invasion assay of LNCaP and LNCaP-CXCR2 cells. The data were normalized to average invasion. Representative images (K) and quantification (L) of transwell Matrigel invasion assay of C4–2B/MDVR-sgCtrl and C4–2B/MDVR-sgCXCR2 cells. The data were normalized to average invasion. Representative images (M) and quantification (N) of spheroid invasion of C4–2B/MDVR-sgCtrl and C4–2B/MDVR-sgCXCR2 cells at days 0 and 10, with leading edges indicated by the red arrows. Invasion distance normalized to day 0 distance for each sphere. (O) A model of CXCR2+ NE cells remodeling the tumor microenvironment. MMP, matrix metalloproteinase. VEGF, vascular endothelial growth factor. Logistic regression analysis was performed using nonparametric Mann-Whitney U test; lines represent median and interquartile range. *P < 0.05.
To identify the proangiogenic factors secreted by CXCR2+ NE cells, we conducted human cytokine array analysis of secreted factors in LNCaP and LNCaP-CXCR2 cells. Overexpression of CXCR2 promoted the secretion of a number of proangiogenic factors (Fig. 4, E and F, and fig. S4C). This result was further confirmed by comparing the secreted cytokine concentrations in the parental and CXCR2-knockout enzalutamide-resistant C4-2B/MDVR cells (Fig. 4, G and H). In addition, LNCaP cells cultured in conditioned medium from LNCaP-CXCR2 cells also showed increased invasiveness (fig. S4D). These data suggest that CXCR2 expression may promote angiogenesis and the dissemination of PCa cells. CXCR2 overexpression markedly increased the invasion by LNCaP cells (P = 0.0209, Mann-Whitney U test; Fig. 4, I and J). Furthermore, we found that knocking out CXCR2 in enzalutamide-resistant C4-2B/MDVR cells reduced cell invasion in both transwell Matrigel assay and a three-dimensional Matrigel spheroid assay (Fig. 4, K to N, and fig. S4E). Therefore, CXCR2 promotes angiogenesis [angiogenin and vascular endothelial growth factor (EGF)], metastasis [C-C motif chemokine ligand 2 (CCL2), macrophage colony-stimulating factor (M-CSF), matrix metalloproteinase, etc.], and proliferation and survival of neighboring luminal-type cancer cells (EGF, insulin-like growth factor-I, oncostatin M, etc.) (Fig. 4O).
CXCR2 is a therapeutic target for advanced and therapy-resistant PCa
Because CXCR2+ NE subpopulation was enriched after hormonal therapy or enzalutamide treatment, we set out to determine whether targeting the NE population through CXCR2 inhibition would be an effective therapeutic strategy. We found that CXCR2 knockout significantly (P < 0.01) suppressed the tumorigenesis of enzalutamide-resistant C4-2B/MDVR cells (Fig. 5, A to C) (24). We then tested the therapeutic effect of CXCR2 inhibitor navarixin in xenograft tumors using CRPC models C4-2B and enzalutamide-resistant C4-2B/MDVR xenografts. Both xenograft tumors were highly sensitive to navarixin, with almost complete inhibition of tumor growth after drug treatment (Fig. 5, D to F, and fig. S5, A to C). Histologic analyses showed inhibition of angiogenesis and massive tumor cell apoptosis in navarixin-treated tumors (Fig. 5, G to L, and fig. S5, D and E). As a proof of principle, we also tested a combination of navarixin with enzalutamide, which has the potential to target all PCa cells (luminal tumor cells and NE tumor cells). In C4-2B xenografts, combination therapy induced significantly (P = 0.0006) better tumor inhibition and more apoptosis compared to either treatment alone (Fig. 5, M to O).
Fig. 5. Advanced and therapy-resistant PCa is sensitive to CXCR2 inhibition.
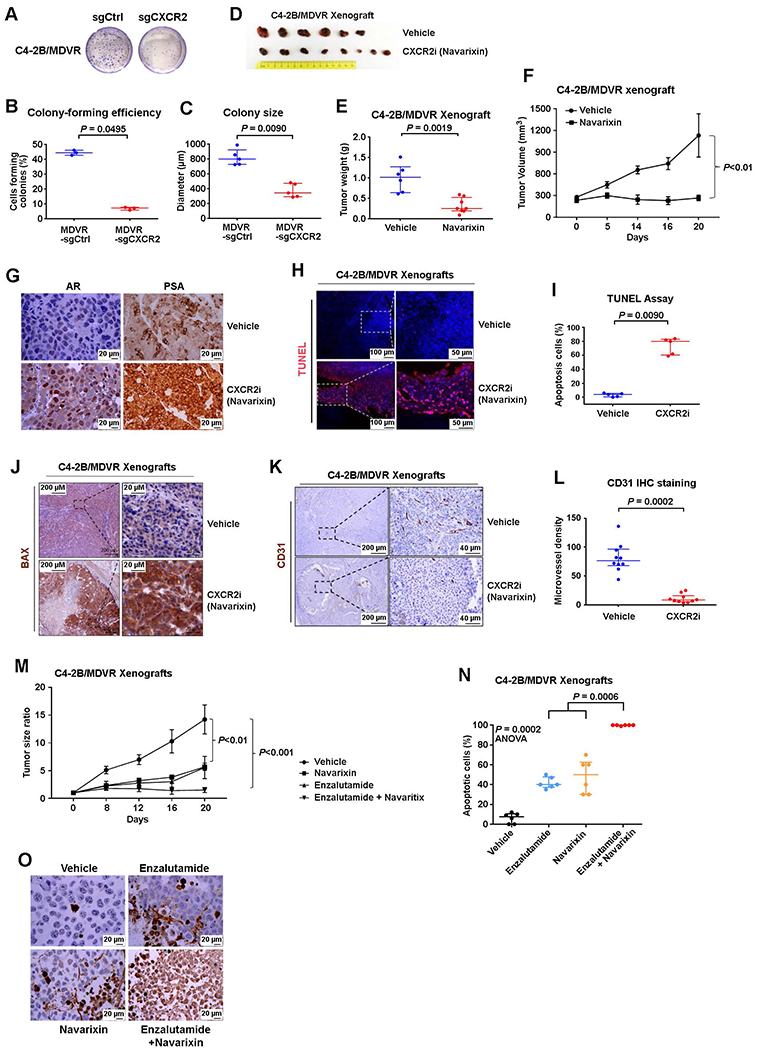
(A to C) Tumorigenesis of C4-2B/MDVR cells with/without CRISPR-Cas9 knockout of CXCR2. Representative images (A), quantification of colony-forming efficiency (B), and colony size (C) of C4-2B/MDVR cells with/without the CXCR2 gene deleted. Cells were cultured for 2 weeks. (D to F) Effect of navarixin on enzalutamide-resistant PCa C4-2B/MDVR cells’growth in vivo. Images (D), weights (E), and volumes (F) of C4-2B/MDVR tumors in mice treated with navarixin (70 mg/kg) or vehicle control. (G) Representative images of IHC staining for AR and prostate-specific antigen in control and navarixin-treated C4-2B/MDVR xenografts. (H to J) Representative images (H) and quantification (I) of immunostaining for terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL) in tumors from mice treated with vehicle or CXCR2 inhibitor navarixin for 3 weeks. Representative images (J) of BCL2 associated X, apoptosis regulator (BAX) immunostaining of xenograft tumors in mice treated with/without navarixin for 3 weeks. (K and L) Representative images (K) and quantification (L) of blood vessel marker CD31 in mice treated with vehicle or navarixin (70 mg/kg) for 3 weeks. (M) Quantification of C4-2B tumor burden in mice treated with vehicle, enzalutamide, navarixin, or both enzalutamide and navarixin. (N and O) Quantification (N) and representative images (O) of immunostaining for TUNEL in mice treated with vehicle, enzalutamide, navarixin, or both enzalutamide and navarixin. ANOVA, analysis of variance. Logistic regression analysis was performed using nonparametric Mann-Whitney U test; lines represent median and interquartile range.
PC3 cells do not express AR and are resistant to AR-targeted therapies. We have shown that PC3 cells have features of SCNC (26). Our study demonstrated that PC3 xenograft tumors were insensitive to enzalutamide treatment (fig. S5, F and G). However, treatment by navarixin, in the absence or presence of enzalutamide, achieved significant (P = 0.0005) tumor-killing effects (fig. S5, F and G). These results suggest that targeting CXCR2 has the potential to treat tumors that are advanced and resistant to current therapies (conventional hormonal therapy and newer drugs such as enzalutamide), including the AR−, treatment-resistant SCNC, as shown schematically in fig. S5I.
DISCUSSION
NE tumor cells, which generally constitute no more than 1% of all tumor cells in primary human PCa, are found in greater numbers in high-grade, high-stage PCa and metastatic CRPC (6). The treatments that are currently in clinical use mostly target AR+ luminal-type tumor cells and leave NE tumor cells untouched. In this study, we demonstrate that hormonal therapy enriches CXCR2+ NE cell population, which likely contributes to therapy failure and disease progression. Unfortunately, there is no effective therapy against the NE tumor cell population.
CXCR2 is specifically expressed by prostate NE cells and not by the bulk luminal-type tumor cells (5). We had developed a tissue procurement method to obtain fresh human PCa tissue from prostatectomy specimens (12). Whereas previous research mostly focused on using purified luminal and basal cells (12, 27), our current work has purified NE tumor cells from fresh primary PCa tissue, which offers an opportunity to comprehensively study the molecular features of NE cells. The results described here demonstrate that (i) NE cells have properties of aggressive cancer cells and therapy resistance; (ii) CXCR2 is a cell surface marker for NE cells in human PCa and can be used to purify NE cells from fresh primary human PCa tissue; (iii) CXCR2 drives NE phenotype and therapeutic resistance in PCa; (iv) CXCR2 expression in NE cells stimulates secretion of proangiogenic factors and promotes angiogenesis in PCa; and (v) CXCR2 is a therapeutic target for advanced and therapy-resistant PCa.
Systemic therapies of PCa mainly target AR, but resistance develops quickly (3). We have approached this problem from the angle of pathology and have established that the presence of cellular heterogeneity in PCa (with bulk luminal-type tumor cells and a minor component of NE tumor cells) has important implications in PCa therapy. We propose that the AR− NE cells are missed by AR-targeted therapies, resulting in therapy failure and disease progression. In addition, a large number of patients eventually develop SCNC composed of pure NE tumor cells, further highlighting the need to target NE cells to achieve better clinical outcome (19).
CXCR2 inhibitor navarixin has been used in clinical trials for the treatment of chronic obstructive pulmonary diseases, with known safety and toxicity profiles (28). On the basis of, in part, the findings of our current study, a Merck-sponsored clinical trial of navarixin has been initiated to evaluate its efficacy in the treatment of advanced PCa (NCT03473925). We believe that targeting NE cells in PCa, in combination with an AR-targeted drug such as enzalutamide, will provide an opportunity to target both the luminal and NE tumor cells to achieve durable therapeutic effect.
In addition, we uncovered a cross-talk between CXCR2-driven NE tumor cells and the tumor microenvironment. We showed that CXCR2+ NE cells provide a source of secreted chemokines and cytokines, which induce angiogenesis, tumor invasion, cancer cell survival, and therapy resistance. Cytokines and factors known to induce MDSC expansion and immunosuppression, including CCL2 and M-CSF, were also up-regulated in CXCR2+ NE tumor cells, suggesting a role of CXCR2-driven NE cells in MDSC recruitment and T cell suppression. Because of the central role of NE cells in PCa tumor biology, therapy resistance, and disease progression, targeting CXCR2 may achieve marked therapeutic efficacy. Because CXCR2 is ubiquitously expressed by PCa of all stages (hormone sensitive, metastatic, CRPC, and SCNC), targeting CXCR2 may particularly benefit patients whose tumors are advanced, recurrent, and resistant to currently available therapies.
Our study used multiple model systems and human tissue, but there are still limitations. For example, the xenograft tumor model lacks the tumor environment provided by human prostate stromal cells, making it difficult to directly observe interactions among NE tumor cells, luminal tumor cells, and the stromal component. In addition, disease progression from hormone-sensitive adenocarcinoma to CRPC or SCNC is a dynamic process that occurs over years. Human tissue samples used in our study only represent snapshots along this process, and longitudinal sampling from the same patients would be ideal but is difficult for many reasons. Therefore, despite the compelling data presented here, the real implications of our findings need to be tested in clinical settings to determine whether patients with advanced PCa benefit from CXCR2 inhibition, alone or in combination with an AR inhibitor.
MATERIALS AND METHODS
Study design
The objective of this study was to determine whether targeting CXCR2 on NE cells in combination with AR inhibition is potentially an effective treatment strategy. Using immunofluorescence analysis of TMAs, we demonstrated that NE cells specifically expressed CXCR2, and CXCR2 expression was associated with PCa progression. We hypothesized CXCR2 to be a driver of the NE phenotype, and targeting NE cells of PCa through CXCR2 inhibition is an AR-independent therapeutic strategy. TMA analysis and isolation of pure populations of epithelial cells from fresh prostatectomy specimens were conducted according to our previous published protocols (12, 29). For mouse experiments, on the basis of previous experience with the model systems, we selected sample sizes to have a power of 0.8 and P < 0.05, using the MATLAB function sampsizepwr (MathWorks Inc.). Mice were randomized to treatment groups after tumor establishment. The number of replicates is indicated in the figure legends. No data, including outlier values, were excluded. All animal work was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by Duke Institutional Animal Care and Use Committee. Primary data are reported in data file S1.
Patients and tissue samples
Several TMA were constructed and have been reported previously (29). A TMA of localized prostate adenocarcinoma was constructed with prostatectomy specimens including PCa and the adjacent benign prostate tissue from 150 patients. Three cores were obtained from benign and cancer areas of each of the prostatectomy specimens and incorporated into TMAs. CRPC TMA was built using CRPC tissue from 16 patients. These patients had primary adenocarcinoma of the prostate but were treated with hormonal therapy instead of surgery. Eventually, the tumor recurred (CRPC) and caused urinary obstruction. Transurethral resection of the prostate was performed to relieve the obstructive symptoms, and tissues were histologically diagnosed as adenocarcinoma and incorporated into a TMA of CRPC. An SCNC TMA was constructed from 12 primary SCNC cases. In addition, 20 cases of metastatic prostate tumor tissue were collected at Duke University Hospital as part of standard-of-care practice (surgery or biopsy collection) between 1 June 2004 and 1 June 2014. For cell sorting and RNA-seq, fresh tumor samples were obtained from 16 patients with localized prostate adenocarcinoma who received prostatectomy at Ronald Reagan UCLA Medical Center and Duke University Hospital between 2015 and 2018. All samples were collected from patients with informed consent, and all related procedures were performed with the approval of the internal review and ethics boards of the indicated hospitals.
Fluorescence-activated cell sorting
The processing of human prostate tissue and acquisition of epithelial subpopulations was performed as previously described (12). Primary PCa cells were resuspended in Advanced Dulbecco’s Modified Eagle Medium (ADMEM)/F12 containing 1× Hepes and 1× GlutaMAX and incubated with antibodies against PTPRC, TACSTD2, ITGA6, and CXCR2 for 30 min at 4°C. Different subsets of PCa cells (TACSTD2+ CXCR2-luminal cells and TACSTD2+CXCR2+ NE cells) were selected by FACS using a flow cytometer (BD FACSDiva).
IHC and immunofluorescence
Sections were deparaffinized, rehydrated, and boiled in a water bath for 40 min in citrate buffer (pH 6.0). Antibodies were incubated for 1 hour at room temperature. Horseradish peroxidase-conjugated secondary antibodies (Dako EnVision+Kit) were applied for 30 min and visualized with diaminobenzidine (DAB) or secondary Alexa Fluor 488 or 594 dye-conjugated antibodies (Thermo Fisher Scientific) applied for 30 min at room temperature. Fluorescent antibody-labeled slides were counterstained with 4’,6-diamidino-2-phenylindole (DAPI), and images were obtained by laser-scanning confocal microscopy (Leica SP5 inverted confocal).
TMAs were constructed as stated above. Four-micrometer sections were stained for CXCR2, and staining was evaluated in a blinded fashion by the pathologists. Scoring was assessed on the basis of staining intensity from 0 (no staining) to 3 (strong) and percentage of tumor cell expression (1 to 100%), creating a composite score from 0 to 300.
Cell line models
To construct CXCR2-overexpressing LNCaP cell line, 1 × 106 LNCaP cells were transfected with plasmid DNA carrying CXCR2 complementary DNA (cDNA) construct for 24 hours at 37°C using X-tremeGENE HP DNA Transfection Reagent. CXCR2 cDNA plasmid was synthesized by OriGene (SC321915). G418 (400 μg/ml) was used to select stably transfected cells. CXCR2-overexpressing LNCaP cells (LNCaP-CXCR2) were identified through qRT-PCR (table S5) and IHC staining.
To construct CXCR2-knockout C4-2B/MDVR cells, we designed CXCR2 single-guide RNA (sgRNA) oligos and inserted them into px458 backbone to generate pSpCas9(BB)-2A-GFP(PX458)-CXCR2-E1.2-sgRNA and pSpCas9(BB)-2A-GFP(PX458)-CXCR2-E3.2-sgRNA plasmid DNAs. The gRNAs were designed to target the first and third exons of CXCR2. A total of 1 × 106 C4-2B/MDVR cells were transfected with the px458 plasmid DNA carrying CXCR2 sgRNA constructs for 24 hours at 37°C using X-tremeGENE HP DNA Transfection Reagent. Green fluorescent protein-positive cells were sorted 3 days after transfection and were plated in 96-well plates for single-colony screening. Primers used for CXCR2-knockout screening are shown in table S6.
Xenograft mouse models
Immunocompromised NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) and nude (nu/nu) mice were from The Jackson Laboratories. A total of 2 × 106 of LNCaP or LNCaP-CXCR2 cells or 1 × 106 of C4-2B/MDVR, C4-2B, or PC3 cells were suspended in 0.1-ml 1× RPMI 1640 and 10% fetal bovine serum (FBS) with 50% Matrigel (Corning), then inoculated subcutaneously into the bilateral flanks of six-week-old male NSG (LNCaP LNCaP-CXCR2, C4-2B, and C4-2B/MDVR) nude (PC3) mice, as previously described (30). For LNCaP and LNCaP-CXCR2 xenografts, mice were euthanized with CO2 12 weeks after implantation, and the xenografts were harvested, fixed in 10% formalin, and embedded in paraffin for subsequent analysis. For C4-2B/MDVR, C4-2B, or PC3 xenografts, tumor size was measured twice a week with a caliper, and the volume was calculated using the formula volume (mm3) = (length × height2)/2. Enzalutamide dissolved in 5% dimethyl sulfoxide and 1% methyl cellulose was administered daily by oral gavage at a dose of 30 mg/kg body mass for 4 weeks of treatment. For in vivo studies of CXCR2 inhibitor, once tumors achieved a volume of 200 mm3, mice were treated with CXCR2 inhibitor navarixin 70 mg/kg body mass or vehicle (1% methyl cellulose) orally daily.
Quantification and statistical analysis
Statistical analyses were performed using R Project for Statistical Computing, Statistica, GraphPad, and MATLAB. The information about statistical details and methods is indicated in the figure legends, results, or materials and methods. IHC staining slides were scanned with an Aperio AT2 microscope (Leica Biosystems) at ×20 magnification and analyzed with Aperio software. Quantification was performed by pathologists blinded to the diagnosis of the tissue cores. Flow cytometric analysis was performed using BD FACSDiva software and analyzed by FlowJo 10.0. The quantification of human cytokine array assay was performed using ImageJ.
Supplementary Material
Data file S1. Primary data.
Fig. S1. CXCR2 is a surface marker for NE cells at all stages of PCa evolution.
Fig. S2. CXCR2+ NE cells from human primary PCa show distinct gene expression signatures from CXCR2− luminal cell subpopulation.
Fig. S3. CXCR2 drives NE phenotype and therapeutic resistance.
Fig. S4. CXCR2 drives the NE secretion of proangiogenic factors and contributes to premetastatic niche formation.
Fig. S5. CXCR2 inhibitor inhibits CRPC growth in preclinical model.
Table S1. Gene set enriched in CXCR2+ NE cells.
Table S2. Gene set enriched in CXCR2− luminal cells.
Table S3. Oncogenic gene set enrichment in CXCR2 NE (CXCR2+ malignant versus basal benign).
Table S4. Six hundred eighty-two-gene signature in CXCR2 NE cells.
Table S5. Primers for qRT-PCR.
Table S6. Targeting sequences for CXCR2 sgRNA.
Acknowledgments:
We thank the Tissue Procurement Core Laboratory at University of California, Los Angeles and Biorepository and Precision Pathology Center at Duke University for assistance on tissue processing and hematoxylin and eosin staining, J. Groth for IHC technical support, Duke Cancer Institute Flow Cytometry Shared Resource for FACS, and Duke Center for Genomic and Computational Biology for processing RNA-seq results. We thank E. Wilson (University of North Carolina, Chapel Hill) for the CWRR1 cell line.
Funding: This study was supported by the Prostate Cancer Foundation and the National Institutes of Health (grant number 1R01CA205001) to J.H.
Footnotes
Competing interests: A.J.A. has consulting roles and/or receives research support from Merck, Astellas/Pfizer, and Janssen and Bayer. M.B.R. is on the speakers’ bureau of Johnson & Johnson and Bayer; received research grants from Novartis, Johnson & Johnson, Medivation, and Astellas; and served as steering committee for clinical trial of Constellation Pharmaceuticals. O.N.W. currently has consulting, equity, and/or board relationships with Trethera Corporation, Kronos Biosciences, Sofie Biosciences, and Allogene Therapeutics. J.H. has consulting roles for MoreHealth, OptraSCAN, KingMed Diagnostics, Genetron, and Gem Flower Healthcare. M.F. is an employee of Foundation Medicine Inc., B.A.S. is an employee of Allogene Therapeutics, and S.F.-K. is an employee of Merck. None of these companies contributed to or directed any of the research reported in this article. The other authors declare that they have no competing interests.
Data and materials availability: The RNA-seq data have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GEO: GSE114326 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114326). All other data associated with this study are present in the paper or Supplementary Materials.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A, Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin 64, 252–271 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Claessens F, Helsen C, Prekovic S, van den Broeck BT, Spans L, Van Poppel PH, Joniau S, Emerging mechanisms of enzalutamide resistance in prostate cancer. Nat. Rev. Urol 11, 712–716 (2014). [DOI] [PubMed] [Google Scholar]
- 3.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OBF Jr., Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI, COU-AA-301 Investigators, Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med 364, 1995–2005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BVSK, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, Van Allen EM, Elemento O, Sboner A, Garraway LA, Rubin MA, Demichelis F, Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang J, Yao JL, di Sant’Agnese PA, Yang Q, Bourne PA, Na Y, Immunohistochemical characterization of neuroendocrine cells in prostate cancer. Prostate 66, 1399–1406 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Terry S, Beltran H, The many faces of neuroendocrine differentiation in prostate cancer progression. Front. Oncol 4, 60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang J, Yao JL, Zhang L, Bourne PA, Quinn AM, di Sant’Agnese PA, Reeder JE, Differential expression of interleukin-8 and its receptors in the neuroendocrine and non-neuroendocrine compartments of prostate cancer. Am. J. Pathol 166, 1807–1815 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abrahamsson PA, Falkmer S, Fält K, Grimelius L, The course of neuroendocrine differentiation in prostatic carcinomas. An immunohistochemical study testing chromogranin A as an “endocrine marker”. Pathol. Res. Pract 185, 373–380 (1989). [DOI] [PubMed] [Google Scholar]
- 9.Chapman RW, Phillips JE, Hipkin RW, Curran AK, Lundell D, Fine JS, CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol. Ther 121, 55–68 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Ha H, Debnath B, Neamati N, Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 7, 1543–1588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, Socci ND, Lash AE, Heguy A, Eastham JA, Scher HI, Reuter VE, Scardino PT, Sander C, Sawyers CL, Gerald WL, Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldstein AS, Drake JM, Burnes DL, Finley DS, Zhang H, Reiter RE, Huang J, Witte ON, Purification and direct transformation of epithelial progenitor cells from primary human prostate. Nat. Protoc 6, 656–667 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON, Identification of a cell of origin for human prostate cancer. Science 329, 568–571 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, Baertsch R, Sokolov A, Meyerowitz JG, Mathis C, Cheng D, Stuart JM, Shokat KM, Gustafson WC, Huang J, Witte ON, N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell 29, 536–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Fröhling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, Weir BA, Reiling JH, Sheng Q, Gupta PB, Wadlow RC, Le H, Hoersch S, Wittner BS, Ramaswamy S, Livingston DM, Sabatini DM, Meyerson M, Thomas RK, Lander ES, Mesirov JP, Root DE, Gilliland DG, Jacks T, Hahn WC, Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vivanco I, Palaskas N, Tran C, Finn SP, Getz G, Kennedy NJ, Jiao J, Rose J, Xie W, Loda M, Golub T, Mellinghoff IK, Davis RJ, Wu H, Sawyers CL, Identification of the JNK signaling pathway as a functional target of the tumor suppressor PTEN. Cancer Cell 11, 555–569 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST, Dhir R, Nelson JB, de la Taille TA, Allory Y, Gerstein MB, Perner S, Pienta KJ, Chinnaiyan AM, Wang Y, Collins CC, Gleave ME, Demichelis F, Nanus DM, Rubin MA, Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 1, 487–495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, Weinstein AS, Friedl V, Zhang C, Witte ON, Lloyd P, Gleave M, Evans CP, Youngren J, Beer TM, Rettig M, Wong CK, True L, Foye A, Playdle D, Ryan CJ, Lara P, Chi KN, Uzunangelov V, Sokolov A, Newton Y, Beltran H, Demichelis F, Rubin MA, Stuart JM, Small EJ, Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: A multi-institutional prospective study. J. Clin. Oncol 36, 2492–2503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks MD, Burness ML, Wicha MS, Therapeutic implications of cellular heterogeneity and plasticity in breast cancer. Cell Stem Cell 17, 260–271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen C-C, Wongvipat J, Ku S-Y, Gao D, Cao Z, Shah N, Adams EJ, Abida W, Watson PA, Prandi D, Huang C-H, De SE, Lowe SW, Ellis L, Beltran H, Rubin MA, Goodrich DW, Demichelis F, Sawyers CL, SOX2 promotes lineage plasticity and antiandrogen resistance in TP53-and RB1-deficient prostate cancer. Science 355, 84–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Lan X, Thomas-Ahner JM, Wu D, Liu X, Ye Z, Wang L, Sunkel B, Grenade C, Chen J, Zynger DL, Yan PS, Huang J, Nephew KP, Huang TH-M, Lin S, Clinton SK, Li W, Jin VX, Wang Q, Agonist and antagonist switch DNA motifs recognized by human androgen receptor in prostate cancer. EMBO J. 34, 502–516 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu D, Sunkel B, Chen Z, Liu X, Ye Z, Li Q, Grenade C, Ke J, Zhang C, Chen H, Nephew KP, Huang TH, Liu Z, Jin VX, Wang Q, Three-tiered role of the pioneer factor GATA2 in promoting androgen-dependent gene expression in prostate cancer. Nucleic Acids Res. 42, 3607–3622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu C, Lou W, Zhu Y, Yang JC, Nadiminty N, Gaikwad NW, Evans CP, Gao AC, Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 75, 1413–1422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith BA, Sokolov A, Uzunangelov V, Baertsch R, Newton Y, Graim K, Mathis C, Cheng D, Stuart JM, Witte ON, A basal stem cell signature identifies aggressive prostate cancer phenotypes. Proc. Natl. Acad. Sci. U.S.A 112, E6544–E6552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tai S, Sun Y, Squires JM, Zhang H, Oh WK, Liang C-Z, Huang J, PC3 is a cell line characteristic of prostatic small cell carcinoma. Prostate 71, 1668–1679 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park JW, Lee JK, Phillips JW, Huang P, Cheng D, Huang J, Witte ON, Prostate epithelial cell of origin determines cancer differentiation state in an organoid transformation assay. Proc. Natl. Acad. Sci. U.S.A 113, 4482–4487 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nair P, Gaga M, Zervas E, Alagha K, Hargreave FE, O’Byrne PM, Stryszak P, Gann L, Sadeh J, Chanez P, Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: A randomized, placebo-controlled clinical trial. Clin. Exp. Allergy 42, 1097–1103 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Park JW, Lee JK, Witte ON, Huang J, FOXA2 is a sensitive and specific marker for small cell neuroendocrine carcinoma of the prostate. Mod. Pathol 30, 1262–1272 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stoyanova T, Riedinger M, Lin S, Faltermeier CM, Smith BA, Zhang KX, Going CC, Goldstein AS, Lee JK, Drake JM, Rice MA, Hsu EC, Nowroozizadeh B, Castor B, Orellana SY, Blum SM, Cheng D, Pienta KJ, Reiter RE, Pitteri SJ, Huang J, Witte ON, Activation of Notch1 synergizes with multiple pathways in promoting castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A 113, E6457–E6466 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drost J, Karthaus WR, Gao D, Driehuis E, Sawyers CL, Chen Y, Clevers H, Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc 11, 347–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vinci M, Box C, Eccles SA, Three-dimensional (3D) tumor spheroid invasion assay. J. Vis. Exp 2015, e52686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roswall P, Bocci M, Bartoschek M, Li H, Kristiansen G, Jansson S, Lehn S, Sjolund J, Reid S, Larsson C, Eriksson P, Anderberg C, Cortez E, Saal LH, Orsmark-Pietras C, Cordero E, Haller BK, Hakkinen J, Burvenich IJG, Lim E, Orimo A, Höglund M, Ryden L, Moch H, Scott AM, Eriksson U, Pietras K, Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat. Med 24, 463–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Primary data.
Fig. S1. CXCR2 is a surface marker for NE cells at all stages of PCa evolution.
Fig. S2. CXCR2+ NE cells from human primary PCa show distinct gene expression signatures from CXCR2− luminal cell subpopulation.
Fig. S3. CXCR2 drives NE phenotype and therapeutic resistance.
Fig. S4. CXCR2 drives the NE secretion of proangiogenic factors and contributes to premetastatic niche formation.
Fig. S5. CXCR2 inhibitor inhibits CRPC growth in preclinical model.
Table S1. Gene set enriched in CXCR2+ NE cells.
Table S2. Gene set enriched in CXCR2− luminal cells.
Table S3. Oncogenic gene set enrichment in CXCR2 NE (CXCR2+ malignant versus basal benign).
Table S4. Six hundred eighty-two-gene signature in CXCR2 NE cells.
Table S5. Primers for qRT-PCR.
Table S6. Targeting sequences for CXCR2 sgRNA.