

Abstract
Among the numerous immune interactions, or lack-thereof, that occur during cancer progression, tumor-associated macrophages (TAMs) – cancer cell interactions have been shown to play an important role in modulating the tumor-microenvironment to an immune suppressive mode, promoting accelerated tumor growth, survival and metastatic spread. TAMs are predominantly polarized to a pro-tumorigenic M2-phenotype through macrophage colony stimulating factor 1 (MCSF) cytokines that bind to the colony-stimulating factor 1 receptor (CSF1R), a class III receptor tyrosine kinase. This MCSF-CSF1R interaction results in autophosphorylation of CSF1R and subsequent phosphorylation and activation of downstream signaling pathways including mitogen-activated protein kinase (MAPK) pathway leading to proliferation, survival and functional activity of M2 TAMs. Therapeutic inhibition of CSF1R and MAPK signaling could effectively re-polarize M2 macrophages to an anti-tumorigenic M1 phenotype; however, this is challenging. In this study, we demonstrate that concurrent and sustained inhibition of the CSF1R and MAPK signaling pathways using dual-kinase inhibitor-loaded supramolecular nanoparticles (DSNs) enhance repolarization of pro-tumorigenic M2 macrophages to the anti-tumorigenic M1 phenotype. The supramolecular nanoparticles exhibited physical stability of over 7 days during storage conditions at 4°C and over 24 hours in human serum, released the inhibitors in a sustained manner and showed significantly higher internalization and accumulation of inhibitors in the M2 macrophages even at longer time points. When tested in a highly aggressive 4T1 breast cancer model, the supramolecular nanoparticles accumulated in TAMs at a significantly higher concentration, increased M1-like phenotype at significantly higher proportion and improved anti-tumor efficacy as compared to combination of single-inhibitor nanoparticles or the small molecule inhibitors. Our data suggests that concurrent, vertical inhibition of multiple intracellular kinase signaling pathways is important for repolarization of M2 macrophages to M1 phenotype, and by utilizing dual-inhibitor loaded supramolecular nanoparticles, further increase the ability to produce more M1 macrophages as compared to M2 macrophages in the tumor microenvironment. This results in enhanced tumor growth inhibition and reduced toxicity. Therefore, vertical, co-inhibition of CSF1R and downstream signaling pathways like MAPK could be a promising macrophage immunotherapy strategy for aggressive cancers.
Keywords: Macrophages, Immunotherapy, Supramolecular Nanoparticles, Cancer, Kinase Signaling
Introduction:
Macrophages are an important part of the mononuclear phagocytic system and play a crucial role in both the innate and the adaptive immune system.[1–3] In addition to the normal immune functions, macrophages also play an important role in tumor progression.[4, 5] Tumor-associated macrophages (TAMs) are a major component of the tumor microenvironment (TME) and account for approximately 50% of tumor mass in some solid tumors. Indeed, recent preclinical and clinical studies show that increased tumor infiltration of macrophages is associated with poor prognosis and increased metastasis.[6–9] Macrophages are inherently plastic cells and exhibit remarkable functional and phenotypic diversity. They can adopt two broadly classified phenotypes, M1 (classically activated, pro-inflammatory) and M2 (alternatively activated, anti-inflammatory), based on their distinct functional states in response to environmental stimuli.[10, 11] M1-polarized macrophages are traditionally anti-tumorigenic while the M2 macrophages are pro-tumorigenic.[12] The tumor microenvironment produces an array of cytokines and chemokines which favors the polarization of the macrophages to an M2 phenotype. These M2 macrophages help in tumor metastasis and angiogenesis, and also play an immunosuppressive role by secreting various immunosuppressive cytokines, in turn aiding with tumor progression.[13] In contrast, M1 macrophages inhibit tumor progression by releasing pro-inflammatory cytokines, activating T cells and directly engulfing cancer cells. This inherent plasticity of macrophages which allows for different functions and phenotypes depending on the environment, can be used to engineer therapeutic strategies to reprogram the macrophages to the desired phenotype, especially for cancer immunotherapy.[14] Several recent studies have shown positive results when repolarizing macrophages in order to decrease tumor progression or increase survival in animal models.[15–17]
The macrophage colony stimulating factor (MCSF) cytokine is frequently produced in the tumor microenvironment by cancer cells. This potent cytokine effectively aids in the recruitment and polarization of macrophages to an M2 phenotype by binding to the colony stimulating factor 1 receptor (CSF1R), and transmembrane type III protein tyrosine kinase receptor, present on macrophages and monocytes.[18, 19] There have been several clinical trials in recent times targeting this MCSF-CSF1R axis for different cancer types including melanoma, prostate cancer, glioblastoma (GBM), classical Hodgkin lymphoma (cHL), neurofibroma, sarcoma, and leukemias.[20] Pexidartinib (PLX3397) is a small molecular CSF1R kinase inhibitor used in clinical and preclinical settings as a monotherapy. In addition to this, several other CSF1R-targeting small molecules, including ARRY-382, PLX7486, BLZ945, and JNJ-40346527 are also being used.[21] Emactuzumab, AMG820, IMC-CS4 are monoclonal antibodies which are also used in clinical trials to target the CSF1R axis.[22, 23] However, clinical trial analysis showed that single agents of the CSF1R pathway including small molecules inhibitors were ineffective.[20, 24] For example, PLX3397 showed no significant progression free survival in recurrent glioblastoma phase II clinical trial, whereas a multicenter phase I/II study with JNJ-40346527 in patients with relapsed or refractory Hodgkin lymphoma showed limited efficacy as monotherapy.[25, 26] Also, several dose-limiting toxicities that are associated with global shutdown of CSF1-CSF1R pathway and global activation of M1 macrophages were observed. For example, serious liver toxicities were observed in patients treated with PLX3397 and facial edema is reported for up to 64% of patients treated with anti-CSF1R monoclonal antibody, emactuzumab.[20] We have recently shown that focal inhibition of CSF1R signaling in M2 macrophages in the tumor microenvironment results in improved efficacy with reduced toxicity.[27] However, we observed that sustained inhibition of CSF1R and downstream signaling pathways is required for efficient repolarization of M2 macrophages to M1 phenotype. While, MAPK pathway has been shown to play important role in cancer cells growth, recent studies have shown that this pathway is also crucial for M2 macrophages growth and survival via CSF1R signaling.[28–30] Indeed, binding of MCSF to CSF1R on macrophages triggers activation of multiple downstream signal transduction pathways including MAPK signaling which are important for survival, growth, differentiation and proper function of M2 macrophages.[31] Recent studies have shown the impact of this vertical inhibition of a single pathway, resulting in increased efficacy of therapeutic systems.[32, 33] However, limited studies exist on the effect of sustained MAPK inhibition on macrophage repolarization as well as combination with CSF1R inhibitors. Here, we rationalized that a synergistic, vertical inhibition of CSF1R and MAPK signaling pathways can repolarize M2 macrophages to the M1 phenotype at a superior efficiency than individual inhibitors and improve anticancer effect. Indeed, recent study has shown that, through dual inhibition of pathways, increased target efficacy can be achieved in a controlled and sustained manner, further promoting that synergistic inhibitor-use is advantageous in the field of immunotherapy.[34] However, deterministic delivery of both CSF1R and MAPK inhibitor to tumor associated macrophages is challenging.
To address these challenges, we utilized our recently developed supramolecular nanotherapeutics platform, where amphiphilic CSF1R and MAPK inhibitors self-assemble into nanostructures facilitated by co-lipids. We proposed that such a dual-inhibitors loaded supramolecular nanoparticles (DSN) that can co-deliver CSF1R and MAPK inhibitors to the tumor-associated macrophages, could simultaneously inhibit these signaling pathways in a sustained manner leading to efficient M2 to M1 repolarization (Fig. 1). Through blocking the phosphorylation of CSF-1R, at the cytoplasmic-side of the cell membrane, and downstream MAPK signaling via the sustained release of the inhibitors from DSN, we hypothesize that an increased repolarization of M2 to M1 macrophages is possible. Here, we demonstrate that DSNs internalize into M2 macrophages and release the inhibitors in a sustained manner resulting in sustained inhibition of phosphorylation of CSF1R and MARK pathways without exhibiting toxicity to the macrophages. This sustained inhibition leads to enhanced repolarization efficiency and improved anticancer effect in aggressive 4T1 breast cancer model. Interestingly, sustained inhibition of MAPK signaling using single MAPK-inhibitor loaded supramolecular nanoparticles repolarized M2 macrophages to M1 phenotype while the free inhibitor resulted in significant cytotoxicity to macrophages at equivalent concentration. Thus, this study shows that vertical blockade of multiple signaling pathways, CSF1R and downstream MAPK signaling, in M2 macrophages could lead to clinically applicable combination strategy for macrophage immunotherapy.
Figure 1|. Design of dual-inhibitor loaded supramolecular nanoparticles (DSN) and its mechanism of action.

(a) Schematic illustrating the immunosuppressive environment caused by increased amounts of MCSF1, increasing the prevalence of M2-phenotypical macrophages. (b) DSNs are synthesized by supramolecular self-assembly of amphiphilic MEK-inhibitor (iMEK) and CSF1R-inhibitor (iCSF1R) in presence of co-lipids, phosphatidylcholine (PC) and DSPE-PEG2000-amine. DSNs accumulate into tumor and deliver dual kinase-inhibiting molecules into the immunosuppressive M2-like tumor associated macrophages (M2-TAMs). Sustained inhibition of CSF-1R and MEK/ERK signaling pathways lead to efficient repolarization of M2 macrophages to pro-inflammatory M1 phenotype, resulting in improved anti-tumor efficacy.
Materials and Methods:
Materials:
All chemical reagents were of analytical grade, used as supplied without further purification unless otherwise specified. All reactions performed under inert conditions unless otherwise specified. Dichloromethane (DCM), methanol, and N,N-dimethyleformamide (DMF) were purchased from Fisher Scientific. 4-dimethylaminopyridine (DMAP), L-α-phosphatidylcholine (PC), and Sephadex G-25 were purchased from Sigma-Aldrich. 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Amino(Polythylene Glycol)2000] (DSPE-PEG2000-amine), cholesterol hemisuccinate, and the mini handheld extruder kit (including 0.4 μm and 0.2 μm Whatman Nucleopore Track-Etch Membrane, Whatman filter supports, and 1 M1 Hamiltonian syringes) were brought from Avanti Polar Lipids. 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) was purchased from Tokyo Chemical Industry Co., LTD. BLZ-945 drug was purchased from Selleck Chem. Selumetinib was purchased from LC Laboratories. Analytical thin-layer chromatography (TLC) was performed using precoated silica gel aluminum sheets 60 F254, purchased from Sigma-Aldrich. Spots on the TLC plates were visualized under UV light. 6 wells and 12 wells, 5 M1, and 10 M1 plates were purchased from Corning. DMEM, FBS, and antibiotic-antimycotic were purchased from Gibco, Life Technologies. Arginase-1, phospho CSF1R, total CSF-1R, phospho ERK1/2, total ERK1/2, phosphor AKT, total AKT, and beta actin antibodies were purchased from Cell Signaling Technology. CD45, CD16/32 (Fc-block), CD80, CD86, CD206, CD11b, Annexin V, and MHCII FACS antibodies were purchased from Biolegend Inc. MTS reagent was obtained from Promega. LysoTracker Red DND-99 was obtained from Invitrogen.
Synthesis of CSF-1R- and MAPK-inhibiting amphiphile:
BLZ-945 (BLZ) or Selumetinib (SEL) was dissolved in 2 M1 anhydrous DCM along with 1.1 molar equivalents of cholesterol hemisuccinate, DMAP, and EDC. The reaction was stirred at room temperature under inert conditions for 24 hours. The progress of the reaction was monitored through thin-layer chromatography (TLC) at intervals over 24 hours. Once the reaction was completed, the corresponding final product was purified through column chromatography by eluting with a 3% methanol:DCM gradient, to get the CSF1R and MAPK-inhibiting amphiphile as an opaque, off-beige color. The final products were analyzed by 1H NMR and Mass spectrometry for purity and structural integrity and confirmed with previously reported values. [27, 35]
Synthesis and characterization of single-inhibitor supramolecular nanoparticles and dual-inhibitor supramolecular nanoparticles (DSNs):
Supramolecular nanoparticles were synthesized using lipid-film hydration method as described previously.[27, 35] Briefly, L-alpha-phosphatidylcholine, DSPE-PEG2000-amine, and kinase inhibiting amphiphiles at 6:3:1 molar ratios were dissolved in 1 M1 DCM. For single-inhibitor supramolecular nanoparticles, 10 mol% of either CSF1R inhibiting amphiphile or MAPK-inhibiting amphiphile was used, and DSNs were comprised of 5 mol% of each inhibitor as compared to 90% of co-lipids. The solvent was evaporated to form a thin film in a round-bottom flask followed by hydration with PBS at 60°C for 2 hours to obtain self-assembled supramolecular nanoparticles of either single or dual-inhibitors. The nanoparticles were then extruded through a 0.4 μm and 0.2 μm polycarbonate membranes using a mini-extruder. The hydrodynamic diameter was measured by Dynamic Light Scattering (DLS) using Malvern Zetasizer Nano ZSP (Malvern, UK). 10 μL of the nanoparticle solution was diluted into 1 M1 using Milli-Q water and 3 sets of 10 measurements were each performed at a 90-degree scattering angle to get the mean particle size. The zeta potential was measured using the Malvern Zetasizer Nano ZSP (Malvern, UK. The inhibitor loading was quantified by measuring the absorbance using Shimadzu UV3600 UV-VIS spectrophotometer. Standard loading curves were made in order to quantify total inhibitor loading.
Synthesis and stability studies of BLZ-945 and Selumetinib loaded lipid nanoparticles:
Free inhibitors loaded lipid nanoparticles were synthesized by using lipid-film hydration method as described previously. Briefly, the ratio of BLZ and SEL was kept as 1:1 while varying the total molar concentration of the inhibitors in the formulations. Compositions of 2 mol%, 6 mol% and 10 mol% inhibitors were used, with the remaining concentration being the co-lipids. The size and stability of the synthesized nanoparticles was analyzed at different time points.
Stability Studies of single inhibitor-loaded and dual-inhibitor loaded supramolecular nanoparticles:
The amount of inhibitor(s) loaded in the single and dual-inhibitor supramolecular nanoparticles was determined using UV-VIS spectroscopy. The loading efficiency was determined as the percentage of inhibitor recovered from the nanoparticle fractions as compared to the initial loading amount. The physical stability of the supramolecular nanoparticles was evaluated by measuring changes in mean particle size and zeta potential during storage condition at 4°C at different time points.
Stability in human serum:
DSNs (0.5 mg/M1) were incubated with equal volumes of human serum. The stability of DSN in serum was calculated by measuring the average hydrodynamic diameter of the nanoparticles using DLS at specified time intervals.
Release kinetics studies:
DSNs (1 mg inhibitor combination/M1, 5 M1) were suspended in PBS buffer (pH 7.4) or RAW274.6 cell lysate (pH ~6.2) and sealed in dialysis tube (MWCO = 3500 Da, Float-A-Lyzer™, Spectrum Labs). Sealed dialysis tubing was suspended in 500 M1 of PBS with gentle stirring to simulate the infinite sink tank condition. 100 μL portions of the aliquot was collected from the incubation medium at predetermined time intervals and replaced by equal amounts of PBS buffer. The portions taken from the dialysis tubing was analyzed using UV-VIS spectrophotometry and was plotted in terms of cumulative inhibitor release over several days.
Cryo-transmission electron microscopy for single-drug and DSNs:
Nanoparticle samples were preserved using a Vitrobot and liquid ethane. Briefly, the sample was prepared on plasma-treated lacey carbon 400-mesh copper grids. 5 μL of nanoparticle suspension was placed onto the plasma treated grids, subsequently blotted with filter paper and processed by vitrification in liquid ethane. Electron microscopy was performed using a Phillips CM 120 Cryo operating at 120 keV using a Gatan Oris 2k by 2k CCD camera system. Vitreous ice grids were transferred into the cryo-electron on a cryostage that maintains the grids at a temperature below −180 C. Images were acquired at 66 kx (0.152 nm pixels) under low-dose conditions at 8–10 electrons Å2.
DSN Internalization Assay:
The FITC-dye tagged dual-inhibitor-loaded supramolecular nanoparticles were synthesized as described previously. Briefly, 9 mol% of both the inhibitors, 1 mol% FITC-cholesterol conjugate, 30mol% DSPE-PEG-Amine2000, and 60mol% L-α-phosphatidylcholine were dissolved in 1 M1 of DCM, followed by formation of the thin film by evaporation of DCM. The lipid film was hydrated to form the supramolecular nanoparticles. The amount of FITC-cholesterol present was determined using a fluorescence spectrometer. RAW264.7 cells were seeded at a density of 50 × 103 cells per well onto an 8-well chamber slide. The cells were incubated with IL-4 (20 ng/M1) for 24h to induce an M2 phenotype. The FITC-tagged DSNs were then added at 10 μM (dye concentration) and incubated for 4h. After 4 hours, cells were washed with PBS to remove the particles that were not internalized. The cells were then surface stained with AlexaFluor 594 SIRP-α (macrophage surface stain) antibody. Cells were then fixed using 4% PFA and stained with DAPI followed by mounting using Invitrogen Glass Antifade reagent. Images were taken at 60x using a Nikon A1R-SIMe confocal microscope and were analyzed using NIS Elements 4.6. DSN-treated macrophages were compared to un-treated cells for quantifying the amount nanoparticles internalized in the macrophages.
DSN Internalization Assay with Lysosome Staining:
The FITC-dye tagged DSNs were synthesized as previously described. RAW264.7 cells were seeded at a density of 50 × 103 cells per well onto an 8-well chamber slide. The cells were incubated for a total time of 2.5 hours with 75 nM LysoTracker Red DND-99, where FITC-tagged DSNs were added either simultaneously as LysoTracker Red or 1 hour after at 10 μM concentration. The cells were then washed with PBS to remove the particles that were not internalized. The cells were then surface stained with AlexaFluor 594 SIRP-α (macrophage surface stain) antibody. Cells were then fixed using 10% Formalin Buffered Solution and stained with DAPI followed by mounting using Invitrogen Glass Antifade reagent. Images were taken at 60x using a Nikon A1R-SIMe confocal microscope and were analyzed using NIS Elements 4.6.
Western Blot Assay to study inhibition of CSF-1R and MAPK pathways at different time points:
5 × 105 RAW264.7 cells were seeded into 5 M1 plates. The cells were incubated with different treatments including either vehicle or 5 μM of BLZ + SEL free inhibitors (1:1 ratio), BLZ-SNP, SEL-SNP, BLZ-SNP+ SEL-SNP (1:1) and the DSNs. After 4 hours of treatment, the cells were washed once with PBS and then replenished with complete media followed by 7 hour-and 36 hour-incubation periods at 5% CO2 and 37°C. Cells were washed after the timepoints with PBS and lysed using RIPA lysis buffer with protease and phosphatase inhibitor (Thermofisher Scientific, Cat.No.: 78442). BCA assays were conducted to measure the amount of protein and equal amount of protein lysates were electrophoresed on a 10% polyacrylamide gel transferred to a PVDF membrane blocked in TBST-T (5% skim milk). Membranes were incubated in 1% BSA in TBST with phospho-CSFIR (1:1000 dilution), CSF1R (1:1000 dilution), phosphor-AKT (1:1000 dilution), AKT (1:1000 dilution), phosphor-ERK (1:1000 dilution), ERK (1:1000 dilution), Arg1 (1:1000 dilution), and beta-actin (1:2000 dilution) antibodies (Cell Signaling Technology) overnight at 4°C. After washing with TBST, membranes were incubated with horseradish peroxidase-conjugated secondary antibody (1:2000) for 1 hour. Detection was done using Biorad’s Clarity ECL (Catalog No.: 1705061) and image processing was done by Image J.
Fluorescent microscopy to assess the extent of NO production of macrophages:
RAW264.7 cells were seeded at a density of 1 × 105 cells per well in an 8-chamber slide and were allowed to reach sub-confluency. The cells were then incubated with 20 ng/M1 of IL-4 in DMEM media for 24 hours. Post stimulation of IL-4, cells were treated with 1 μM BLZ-SNPs, SEL-SNPs, BLZ+SEL, BLZ-SNP+SEL-SNP, Vehicle, and DSNs for 48 hours. Wells were then treated with 1 μM DAF2DA (green) to identify nitric oxide (NO) which is indicative of M1 macrophages, then fixed with 4% PFA, and finally DAPI stained (blue). Images were taken at 20x using a Nikon A1R-SIMe confocal microscope and were analyzed using NTS Elements 4.6.
Flow cytometry assay to quantify toxicity of DSNs on macrophages:
RAW264.7 cells were seeded at a density of 1.5 × 105 cells per well in 6-well plates and were allowed to reach sub-confluency. The cells were then stimulated with 20 ng/M1 of recombinant mouse IL-4 in DMEM media for 24 hours. Post-stimulation of IL-4, cells were treated with BLZ + SEL free inhibitors or the DSNs at varying concentrations (10 nM – 10μM) in 5% FBS DMEM for 72 hours. Following the treatment, the cells were collected in media then washed and centrifuged in Annexin V buffer. The cells were incubated with Annexin V antibody as per manufacturer’s protocol. Post staining, the cells were washed and further incubated with propidium iodide and analyzed using a Novocyte flow cytometer (Acea Biosciences) and the results were processed using NovoExpress 1.2.5. The cells were gated for a live population and isolated singlets in order to reduce autofluorescence from doublets. The singlets were then isolated out into separate populations to quantify the amounts of Annexin V negative cells to signify viability.
Flow cytometry assay to quantify repolarization of M2 macrophages to M1 phenotype after inhibition of CSF-1 and MAPK signaling:
RAW264.7 macrophages were seeded at a density of 7.5 × 104 cells per well in 12-well plates and were allowed to reach sub-confluency. The cells were then stimulated with 20 ng/M1 of recombinant mouse IL-4 in DMEM media for 24 hours. Post stimulation with IL-4, cells were treated with 10 nM, 100nM, 500nM, 1 μM, or 10 μM BLZ945, Selumetinib, BLZ+SEL combination, BLZ-SNP, SEL-SNP or DSNs in 5% FBS DMEM for 48 hours. Following the treatment, the cells were washed with PBS once, split into two tubes per well, and centrifuged. The cells were double-stained for APC CD11b (pan macrophage marker) and either FITC CD80 (M1 narker) or FITC CD206 (M2 marker). Post-staining, the cells were washed and quantified using a Novocyte flow cytometer (Acea Biosciences) and the results were analyzed using NovoExpress 1.2.5. The cells were gated for a live population and then isolated for singlets to reduce autofluorescence due to doublets. The singlets were then isolated out for CD80+/CD11b+ M1 macrophages and CD206+/CD11b+ M2 macrophages, from the double-stained sample. The ratio of M1/M2 macrophages was calculated based on the % M1 positive cells to % M2 positive cells.
Flow cytometry assay to quantify repolarization of bone-marrow derived macrophages (BMDMs) from an M2 to M1 phenotype after inhibition of CSF-1 and MAPK signaling:
Femur and Tibia were harvested from C57BL/6 mice and the bone marrow was flushed out into a petri plate using PBS. The marrow was collected, centrifuged and resuspended in DMEM media. To this 20ng/M1 of MCSF was added and the cells were incubated for 7 days in 37°C while the media was replenished every alternate day. At the end of 7 days, the BMDMs would have adhered and these were then polarized to a M2 phenotype by incubating the with B16F10 tumor conditioned media (TCM) for 24h. Post stimulation with TCM, cells were treated with 10 nM, 100nM, 500nM, 1 μM, or 10 μM BLZ945, Selumetinib, BLZ+SEL combination, BLZ-SNP, SEL-SNP or DSNs in 5% FBS DMEM for 48 hours. Following the treatment, the cells were washed with PBS once and centrifuged. The cells were triple stained for APC CD11b (pan macrophage marker), FITC CD80 (M1 narker) or FITC CD206 (M2 marker). Post-staining, the cells were washed and quantified using a Novocyte flow cytometer (Acea Biosciences) and the results were analyzed using NovoExpress 1.2.5. The cells were gated for a live population and then isolated for singlets to reduce autofluorescence due to doublets. The singlets were then isolated out for CD80+/CD11b+ M1 macrophages and CD206+/CD11b+ M2 macrophages. The ratio of M1/M2 macrophages was calculated based on the % M1 positive cells to % M2 positive cells.
Stability and released kinetics profiles of NIR dye from dye-tagged DSNs when incubated in human serum:
DiR dye-labeled DSNs were synthesized as described previously. DSNs were incubated in equal parts PBS and human serum in dialyzer tube (Slide-A-Lyzer™, Thermo Fisher) over 24 hours at room temperature in 500 M1 PBS with gentle stirring. 100 μL portions of the nanoparticle solution was taken and collected at several time points. The portions taken from the dialysis cassettes were analyzed using a Perkin Elmer IVIS SpectrumCT In Vivo Imaging system for the amount of dye retained in the nanoparticle. Exposure times for acquiring data were kept the same, and the stage settings and fiber optic adjustable illuminator arms were also kept the same for the duration of the imaging. Fluorescence signals were normalized and quantified using Living Image 4.7.2 and was used to conduct spectral unmixing. Spectral unmixing was performed by analyzing only the dye in PBS solution in the field of view and using automatic spectral unmixing for each data set to eliminate autofluorescence due to the stage, leaving fluorescent intensities driven only by the DiR dye itself. The data represents average radiance values at different time points.
In vivo tumor distribution study:
NiR dye-labelled DSNs were synthesized using the methods described above. Briefly, 50 mol% L-α-phosphatidylcholine, 19 mol% combination of the kinase-inhibiting amphiphiles, 1 mol% of DiR dye and 30 mol% DSPE-PEG-Amine were mixed in 1 M1 DCM followed by evaporation of the solvent to form thin film. The film was then hydrated to form DSN which were then extruded. For tumor induction, 4T1 breast cancer cells (1 × 106) were injected into the right flank of female BALB/c mice (4–5 weeks old, ~16–20g). Once the tumor reached volumes of ~500 mm , the NiR dye-labelled DSNs were injected via tail-vein at 2nM concentration (DiR dye concentration, 200 uL). The imaging was performed at 2h, 8h, 12h, and 24h post-injection utilizing IVIS filter set (excitation 745 nm and emission 800 nm). All the images were taken using a Perkin Elmer IVIS SpectrumCT In Vivo Imaging system. Exposure times for acquiring data were kept the same, and the stage settings and fiber optic adjustable illuminator arms were kept the same for the duration of the observation. Fluorescence signals were normalized and quantified using Living Image 4.7.2 and was used to conduct spectral unmixing. Spectral unmixing was performed by analyzing only the mice in the field of view and using automatic spectral unmixing for each data set to eliminate autofluorescence due to the stage and the mice, leaving fluorescent intensities driven only by the DiR dye itself. All animal procedures were approved by the University of Massachusetts Amherst Institutional Use and Care of Animals Committee.
Efficacy study of DSNs in murine 4T1 breast cancer BALB/c model:
4T1 murine breast cancer cells were injected subcutaneously (1 × 106 cells) in the right flanks of 4–5-week-old BALB/c mice (weighing between 16–20g, Charles River Laboratories). The treatments were started on day 7 (now referred to as Day 0 of treatment). After the tumors reached ~50mm volume, animals were randomized in different treatment groups as follows: Vehicle, BLZ+ SEL combination inhibitors in DMSO (15 mg/kg, i.p. injection), DSN (15 mg/kg of each drug equivalent), BLZ-SNP (15 mg/kg, concentration equivalent to free drug), SEL-SNP (15 mg/kg, concentration equivalent to free drug), BLZ-SNP+ SEL-SNP (15 mg/kg each, concentration equivalent to free drugs). The different doses were administered on day 0, 3, and 5. The tumor volumes and body weights were monitored throughout the entire treatment, every other day. In order to calculate the tumor volume, the volume of an ellipsoid was used (L x B2/2, L being the longest diameter of the tumor, B being the shortest) and measured using a Vernier caliper. Mice were sacrificed when tumor volumes reached 2000 mm3 or tumors exhibited necrosis or mice became lethargic. Tumor, lung, liver, kidney, and spleen from each mouse was harvested for further analysis. All animal procedures were approved by the UMass Institutional Animal Care and Use Committee (IACUC).
Treatment with FITC-Cholesterol-incorporated DSNs for TAM internalization:
4T1 murine breast cancer cells were injected subcutaneously (1 × 106 cells) in the right flanks of 4–5-week-old BALB/c mice (weighing between 16–20g, Charles River Laboratories). Once the tumors reached ~500 mm3, mice were injected intra-venously with FITC-cholesterol incorporated DSNs (2.5 mg/kg FITC-Chol loading) and sacked 24 hours following. Cryo-sections were taken of the samples and stained with DAPI (blue) and anti-CD11b (red) in order to signify TME cells or macrophages, respectively. Those same tumors were also processed. Harvested tumors were thoroughly minced with and suspended in individual 4 M1 Type-I Collagenase solution (1 mg/M1 in complete DMEM). Each sample was incubated in collagenase solution for 1 hour at 37°C and 5% CO2 and passed through a 40 μm filter. Washing of impurities and debris was done in DMEM media by centrifuging at 2000 rpm for 5 min. The supernatant was aspirated, and the pellets were stained for CD45+/CD11b+ TAMs. Samples were gated to have the above characterization and then the colocalization with FITC was analyzed.
Western Blot analysis of in vivo tumor samples:
The excised tumors from different treatment groups were taken from BALB/c mice and flash frozen at −80°C. Samples were then resected, homogenized and lysed in ice-cold NP40 cell lysis buffer containing protease and phosphatase inhibitors. Centrifugation at 15,000 rpm was done in order to remove cellular debris and the supernatant was used for the rest of the experiment. BCA protein assay kit was used for protein estimation. 45ug of protein was loaded in each well and probed for phospho-CSFIR, total CSF1R, phospho-ERK, total ERK, phosphor-AKT, AKT, Arg1, and beta-actin. Biorad’s Clarity ECL was used for detection and image analysis was done by ImageJ.
FACS Analysis of in vivo tumor samples (4T1 breast cancer model):
Harvested tissues were thoroughly minced with and suspended in individual 4 M1 Type-I Collagenase solution (1 mg/M1 in complete DMEM). Each sample was incubated in collagenase solution for 1 hour at 37°C and 5% CO2 and passed through a 40 μm filter. Washing of impurities and debris was done in DMEM media by centrifuging at 2000 rpm for 5 min. The supernatant was aspirated, and the pellet was split into the following groups for flow cytometric analysis: CD206+/CD80+/CD11b+, and CD80+/CD11b+. The resulting, stained, tumor cell suspensions were analyzed and quantified using a Novocyte Flow cytometer and analyzed using NovoExpress 1.2.5.
Histopathology and TUNEL Assay:
The harvested tissues from different treatment groups were flash frozen in OCT (Tissue Tek) and sectioned into 5 uM thin slices. Sections were stained with Alexa Fluor 588 Click -iT™ Plus TUNEL Assay Kit (Thermo Fisher) as per the manufacturer’s recommended protocol. Nuclei were stained with DAPI (Blue). Imaging was performed with Nikon A1R-SIMe confocal microscope at 20x and analyzed with NIS Elements 4.6.
H&E staining to analyze inflammation in tissues:
Immunohistochemistry (IHC) was performed on a DakoCytomation autostainer using the Envision HRP Detection system (Dako, Carpinteria, CA). Each tissue block was sectioned at 4 μm on a graded slide, deparaffinized in xylene, rehydrated in graded ethanols, and rinsed in Tris-phosphate-buffered saline (TBS). Heat induced antigen retrieval was performed in a microwave at 98°C in 0.01 M citrate buffer. After cooling for 20 minutes, sections were rinsed in TBS and subjected to the following primary antibodies: Rat anti mouse CD8a l:100(eBioscience) for 30 mins at room temperature and then incubated with Goat anti Rat Biotin (Invitrogen) for 30 mins at room temperature followed by incubation with Streptavidin Horse radish peroxidase (BD Pharmingen) for 30 mins at room temperature. Immunoreactivity was visualized by incubation with chromogen diaminobenzidine (DAB) for 5 minutes. Tissue sections were counterstained with hematoxylin, dehydrated through graded ethanols and xylene, and cover-slipped. Images were captured with an EVOS FL Auto microscope at magnification of 20x.
Fluorescent staining for F4/80+ cells to analyze inflammation in tissues:
Each tissue block was sectioned at 4 μm on a graded slide, deparaffinized in xylene, rehydrated in graded ethanols, and rinsed in Tris-phosphate-buffered saline (TBS). Heat induced antigen retrieval was performed in a microwave at 98°C in 0.01 M citrate buffer. After cooling for 20 minutes, sections were rinsed in TBS and subjected incubated with normal rat serum for 30 mins at room temperature. This was followed by overnight incubation with Alexa Flour 488 F4/80 (eBioscience) overnight. After subsequent washing, the tissues were counter stained with DAPI and mounted and imaged using a Nikon A1R-SIMe confocal microscope at 10X and analyzed with NTS elements 4.6.
Results and Discussion:
Design and synthesis of dual CSF1R and MAPK-inhibiting lipid-based supramolecular nanoparticle:
A nanoparticle system that can co-deliver both CSF1R and MAPK inhibitors to the tumor-associated macrophages (TAMs) at high concentrations would be important for activation of macrophages to anti-tumor phenotype. As a first step we tested if stable liposomal-based delivery systems can be used to load the CSF1R inhibitor, BLZ-945 and MAPK inhibitor, selumetinib in the same liposome. We systematically varied both the inhibitors to co-lipids molar ratio in the liposomes synthesis while maintaining the BLZ-945 to selumetinib ratio as 1:1. As shown in Fig. 2a–b, very unstable liposomes were formed even at lower inhibitor concentrations of (a) 2mol% (with 1% of BLZ-945, 1mol% of selumetinib and 98% co-lipids) and (b) 6mol% (with 3mol% of BLZ-945, 3mol% of selumetinib and 94% co-lipids). At 10 mol% of inhibitors concentration (with 5mol% of BLZ-945, 5mol% of selumetinib and 90% co-lipids), there was significant precipitation observed within few minutes of synthesis (Fig. 2c). Also, the size measurements of different liposomal systems revealed non-uniform size distribution at all the inhibitors concentrations with a higher peak was observed at ~10–20 nm and a second lower peak was observed between 100–1000nm. These studies showed that in the liposomal system, where the drugs are encapsulated in between the spaces of the self-assembled lipid bilayer, the number of drugs with different physicochemical properties that can be stably loaded is limited. This validates our previous observations where we couldn’t load more than 5mol% of free drug in the liposomal system. However, we have previously shown that these challenges can be overcome by designing an amphiphilic inhibitor that is biologically active and additionally can rapidly self-assemble to supramolecular nanostructures of ~100 nm size, thereby significantly improving the loading efficiency and stability. Using this platform, amphiphilic inhibitors were synthesized by conjugating either BLZ-945 (BLZ) or selumetinib (SEL) to cholesterol hemisuccinate to form kinase inhibitor-cholesterol conjugates.[27, 36] Our recent studies have shown that the amphiphilic inhibitors form a stable supramolecular assembly at higher concentrations (>10 mol% total) in the presence of co-lipids, in turn allow for higher loading efficiencies. Consistent with these observations, CSF1R and MAPK amphiphiles (at equimolar concentrations, total 10 mol%) formed a supramolecular self-assembly at nanoscale (dual-inhibitors loaded supramolecular nanoparticles, DSNs), facilitated by co-lipids phosphatidylcholine (PC) and DSPE-PEG (2000) amine (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethyleneglycol)-2000] (ammonium salt)). We choose PC, since it has been widely used as a safer lipid molecule for drug delivery. With the addition of DSPE-PEG, a PEGylated coating will have the ability to “stealth coat” the particle and allow for increased circulation time in vivo, while the DSPE end stably incorporates itself into the lipid bilayer of PC. Cryo-transmission electron microscopy analysis showed spherical nanoparticles of average diameter 98.5±31nm (Fig. 2d). Interestingly, a mixture of unilamellar and multilamellar structures were observed with an average ratio of 0.63:0.32:0.05 unilamellar:bilamellar:multilamellar structures. Dynamic light scattering analysis revealed a hydrodynamic diameter of 190.1± 27 nm with a zeta potential of approximately −17.1 ± 7.3 mV (Fig 2e). No precipitation was observed even after 2 days of the synthesis of nanoparticles. We next tested if the DSNs maintain their structural integrity for longer periods of time by measuring the size and zeta potential differences over several days while stored in PBS (pH 7.4) at 4°C. The system exhibited a stable size and zeta potential over a 6-day period of time (Fig. 2f), which showed that the synthesized DSNs is indeed stable. We also tested the stability of DSNs in physiologically-relevant conditions such as in serum. We incubated the DSNs in human serum and measured the changes in size and zeta potential over time. It is known that protein corona formation can significantly reduce the stability of nanoparticles. The DSNs were stable for over 10h in human serum as shown by minimum changes in diameter and zeta potential (Fig 2g–h). Once morphological stability was studied, we next analyzed the release kinetics profiles of the inhibitors from DSNs. To study this, DSNs were put in either macrophage lysate solution (pH 6.2) or PBS (pH 7.4) in order to show the impact of pH and enzymatic activity on the degradation of the supramolecular system. The hypothesis is that in PBS in comparison to macrophage cell lysate, the extent of release will be lower due to physiologically relevant conditions. The higher stability at physiological pH and serum conditions would mean that the DSN can circulate for longer time leading to higher accumulation in the tumor. Macrophage cell lysate was used in order to replicate the activity inside of the macrophages and how readily it would break down at the site of action, rather than in circulation, demonstrating systemic stability while also ability to rapidly release the drugs only upon internalization by macrophages. As anticipated, when comparing the release of the drugs in either PBS or macrophage cell-lysate, there was an increased release in the macrophage cell lysate, over 80% of the drugs released in 80h, as compared to that of the physiologically relevant pH 7.4, only ~20% of the drugs was released (Fig. 2i). This is consistent with our hypothesis that the DSN are stable under physiologically relevant conditions, however release the drugs rapidly once internalized by macrophages. The synthesis of DSNs was achieved by designing a system that is stable during normal physiological conditions but selectively release the payload in increasingly acidic and enzyme-rich conditions.
Figure 2|. Biophysical characterization of DSNs.

Representative size distribution graphs show that BLZ-945 and Selumetinib combinations don’t form stable lipid nanoparticles at different drug concentrations, (a) 1:1 – BLZ-945: Selumetinib, (b) 3:3 – BLZ-945: Selumetinib and (c) 5:5 – BLZ-945: Selumetinib. Inset shows the representative image of free drugs loaded nanoparticles 24h after synthesis. The nanoparticles are unstable as shown by significant precipitation of drugs. (d) Representative high-resolution cryo-TEM image of DSN, showing a size of ~98.5 nm and exhibit multi-lamellarity and spherical morphology. (e) Representative graph of the size distribution (hydrodynamic diameter) of DSNs as measured by dynamic light scattering. Inset shows the representative image of DSN 72h after synthesis. No visible precipitation is observed which shows that DSN are very stable. (f) Graph shows stability of DSNs based on a function of change in size distribution and zeta potential over time during storage condition at 4°C. (g-h) Graphs show stability of DSNs when incubated with human serum as a function of the change in size distribution and zeta potential, measured by Dynamic Light Scattering. (i) Drug release profiles of iMEK and iCSF1R from the DSNs incubated in macrophage cell lysate (pH = 6.2) and PBS (pH 7.4).
In vitro characterization of DSNs:
We next tested if DSNs can internalize and deliver both the CSF1R and MAPK inhibitors into M2 macrophages. Fig. 3a shows the schematic of internalization assay. RAW 264.7 cells were treated with IL-4 cytokine for 24h, which is known to polarize macrophages to M2 phenotype. We then added either combination of free inhibitors (BLZ+SEL) or DSN and incubated for 4h. After the incubation period, the cells were washed to remove the drugs or nanoparticles that were not internalized. The cells were then harvested and the amount of inhibitors internalized in the cells was quantified. As shown in Fig. 3b, significantly higher accumulation of inhibitors was observed in M2 macrophages when added as DSNs as compared to combination of free inhibitors. This was validated by imaging the internalization of FITC dye-tagged DSNs after incubation with M2 macrophages for 4h (Fig. 3c). Indeed, we observed significantly higher internalization of FITC-tagged DSNs in M2 macrophages as compared to untreated group (Supplemental Fig. 2). Since DSNs were found to be internalized within 4 hours, we decided to track the colocalization of dye-tagged DSNs and lysosomes using LysoTracker Red DND-99 (Fig 3d). It was observed that the dye-tagged DSNs were co-localized with lysosomes at when imaged at 1.5 hours. We next tested if higher internalization of DSNs and sustained release of inhibitors in M2 macrophages have any toxicity to cells. M2-polarized macrophages were treated with either free inhibitor combination or DSN at different concentrations (10nM - 10μM) for 72h in order to evaluate if the DSNs or free inhibitor combination treatments result in depletion of macrophages. Interestingly, Annexin V and propidium iodide staining of treated macrophages (Fig. 3e–f) showed that free inhibitor combination showed a dose dependent toxicity to M2 macrophages with an IC50 of 66 nM, whereas the DSN did not exhibit any toxicity to macrophages. Finally, to ensure that there is no other cytotoxicity involved with the DSN system, D4M melanoma and Jurkat T-cells were exposed to varying concentrations of DSNs (Supplemental Fig. 3). As shown in Supplemental Fig. 3, there was no significant reduction in cell viability was observed, showing that the effective concentrations have a low probability of creating off-target cytotoxicity. These results demonstrate that although DSNs internalize in M2 macrophages, they have minimal toxicity to macrophages due to extended release mechanisms inherent to supramolecular nanoparticles that allow sustained release of drugs in sub-cytotoxic amounts.
Figure 3|. DSNs internalize into M2 macrophages.
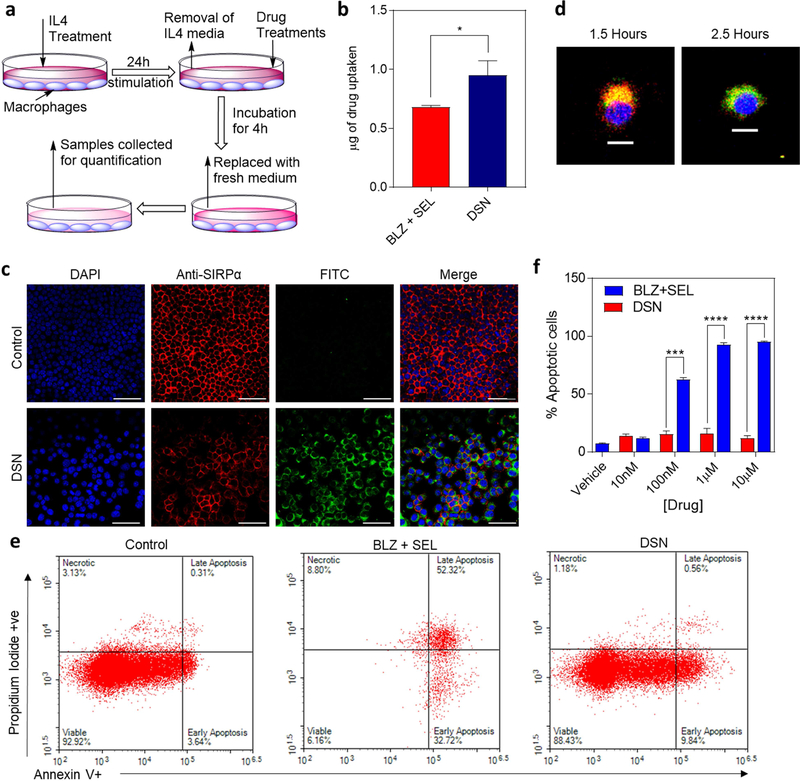
(a) Schematic representation of the internalization assay. RAW264.7 macrophages were polarized to M2 phenotype by stimulation with IL4 for 24h. Then the treatments were added in fresh medium and incubated for 4h followed by washing and sample collection for quantification. (b) Graph shows the quantification of drug internalized in M2 macrophages after 4 hours as measured by UV-Vis spectrophotometry. Data shown are mean ± s.e.m. (n =3). Statistical analysis was performed with student t-test. *p< 0.05. (c) Representative confocal images showing internalization of FITC-tagged DSNs in M2 macrophages after 4h. The cells nuclei are stained with DAPI (blue). Scalebar is 100 μm. (d) Representative confocal microscopy images demonstrating FITC-Chol labeled DSNs co-localized in lysosomes at 1.5 hours of incubation, and not co-localizing at 2.5 hours (blue, DAPI; red, LysoTracker Red DND-99; green, FITC-Chol DSNs) (scalebar 10 um). (e) Representative flow cytometry plots of apoptosis and necrosis of macrophages following treatment with increasing concentration of free drugs combination and DSN. DSN induced minimal apoptosis and no significant necrosis. (f) Quantification of Annexin V/Propidium iodide assay showing the relationship between concentration of DSNs and free inhibitor concentration with cell viability. Data shown are mean ± s.e.m. (n =3). Statistical analysis was performed with student t-test. ***p< 0.001, ****p< 0.0001. DSNs did not exhibit any toxicity to M2 macrophages whereas free drugs combination showed significant toxicity at higher drug concentrations.
DSNs enable sustained inhibition of CSF1R and MAPK (MEK/ERK) signaling:
We next tested if the significant internalization of both CSF1R and MAPK inhibitors in macrophages and sustained release of the inhibitors result in sustained inhibition of kinase signaling pathways. RAW 264.7 macrophages were treated with IL-4 that can activate CSF1R and MAPK signaling independent of MCSF cytokine. After IL-4 treatment, the macrophages were exposed to 5μM of either free inhibitor combination, CSF1R-inhibiting supramolecular nanoparticles (BLZ-SNP), MAPK-inhibiting supramolecular nanoparticles (SEL-SNP) or DSNs (Fig. 4a). While there is limited inhibition for DSNs in comparison to the vehicle in the first 7 hours, 36 hours exhibits comparable inhibition of pERK1/2 and better pCSF-1R reduction than other groups present (Fig 4b–d). This inhibition of kinase signaling at longer timepoints shows that the DSN system exhibits sustained inhibition of TME-polarizing mechanisms. To further investigate polarization to an M1 phenotype, nitric oxide staining was performed, which is indicative of this inflammatory response (Fig 4e). In comparison to free inhibitor and vehicle treatment, there is significant increase in NO signal, further reiterating the DSN system’s capability to repolarize macrophages to an anti-tumorigenic phenotype.
Figure 4|. DSNs effectively repolarize M2 macrophages to M1 phenotype.

(a) Schematic showing the repolarization assay. RAW264.7 macrophages were polarized to M2 phenotype by stimulation with IL4 for 24h. Then the treatments were added in fresh medium and incubated for 48h followed by washing and sample collection for Western blot assay and flow cytometry analysis, (b) Western blots showing expressions of phospho-CSFIR and phospho-ERKl/2 in macrophages treated with different treatments. DSN exhibited significant inhibition of CSF1R and MAPK signaling, (c-d) Quantification of the western blot results in (b) for the ratio of (c) phopho-ERK1/2 and (d) phospho–CSFIR vs their respective total protein expression. Data plotted is mean ± s.e.m, (n = 2). (e) Representative confocal images showing the expression of Nitric Oxide (green) after different treatments on M2 macrophages. Higher NO expression was observed with DSN treatment as compared to other treatment groups. (Scalebar 100 μm). (f) Quantification of flow cytometry plots showing ratio of expression of M1 (CD11b+, CD80+) to M2 (CD11b+, CD206+) macrophages treated with different concentrations of drugs for 48 hours post-M2 polarization (against equivalent of each free drugs). Data plotted is mean ± s.e.m. (n= 2–3), n.s. – not significant, ****p <0.0001 (one-way ANOVA). Increase in concentration of drug treatments results in significantly higher repolarization efficiency by DSN. (g-i) Quantification of flow cytometry data demonstrating a time-dependent relationship between the relative expression of M1 (CD11b+, CD80+) compared to M2 (CD11b+, CD206+) macrophages at 1 μM concentration of different treatments. Time points are 4 h (g), 24 h (h), and 48 h (i). Data shown are mean ± s. e. m. (n = 2–3). *p< 0.05, **p < 0.01, and ***p< 0.001 (one-way ANOVA). DSNs show significant repolarization efficacy after 24 hours through 48 hours in comparison to other groups.
In vitro re-education of M2 macrophages to M1 phenotype post-DSN treatment:
Activated CSF1R and downstream signaling pathways such as MAPK signaling are important for M2 macrophages differentiation, proliferation and activity. Recent studies have shown that small molecular inhibitors of CSF1R signaling can repolarize M2 macrophages to M1 phenotype. But the effect of MAPK inhibition on macrophage repolarization has been underexplored. In order to establish an optimal concentration to quantify the repolarization of M2 macrophages from both inhibitors, we analyzed re-polarization efficacy of DSNs at different concentrations. Briefly, RAW264.7 macrophages were plated and exposed to IL-4 in order to polarize them to an M2 phenotype. After 24 hours, the cells were incubated with different concentrations (10 nM- 10μM) of either DSNs or BLZ+SEL. Following 48h of incubation, the macrophages were triple-stained with a pan-macrophage marker antibody (CD11b), an M1-specific antibody (CD80), and an M2-specific antibody (CD206). The M1 to M2 ratio was calculated by measuring the %M1 positive cells (CD11b+CD80+) to %M2 positive cells (CD11b+CD206+). As shown in Fig. 4f and Supplemental Fig. 4, M2 macrophages were significantly re-educated to an M1 phenotype (p < 0.0001) in comparison to their free inhibitor analogs at 1μM and 10 μM. This shows that the sustained release characteristics of the supramolecular structures allow for sufficient manipulation of macrophage phenotype over a 48h time period. It is interesting to note that the maximum M1:M2 ratio was achieved after the treatment with 1 μM of DSNs. Hence, all further repolarization experiments were performed using 1 μM of DSNs.
After analyzing the concentration dependent effect on repolarization, we next wanted to evaluate the kinetics of the repolarization over time. IL-4 treated RAW264.7 macrophages were treated with either 1 μM of DSNs, BNP-SNP, SEL-SNP, a combination of both supramolecular single systems (BLZ-SNP+SEL-SNP), free inhibitors, or vehicle. Repolarization experiments was performed, and the repolarization efficacy was analyzed at 4h, 24h, and 48h respectively (Fig 4g–i). In the first 4 hours, it was observed that DSNs, BLZ-SNPs and BLZ-SNP+SEL-SNPs show significantly better repolarization efficacy than the free inhibitors and vehicle. This points to the fact that supramolecular systems have the ability to be uptaken by macrophages more effectively and induce repolarization. At 24 hours, the DSN system reached a peak M1 to M2 ratio of 6-fold higher than 4 hours, whereas the vehicle and free inhibitors exhibit a 1.5 ratio. This significance shows that this system allows for strong, sustained interactions with macrophages allowing for a robust repolarizing platform. The other single inhibitor supramolecular systems show better repolarization, however not as robust as that of the dual-inhibitor system. Finally, 48 hours post-treatment show that DSNs continue to have an enhanced macrophage repolarization effect for longer time periods, showing significance in comparison to all other treatment groups. We have confirmed these findings in bone marrow derived macrophages where DSN showed significantly higher M1 repolarization as compared to other treatment groups (Supplemental Fig. 5)
DSNs exert enhanced tumor growth inhibition in breast cancer model:
In order to evaluate if the in vitro efficacy exerted by DSNs in terms of sustained inhibition of CSF1R and MAPK signaling and repolarization of macrophages from M2 to M1 phenotype, can be translated into in vivo tumor growth inhibition. We used an aggressive and highly metastatic 4T1 murine breast cancer tumor model. We first wanted to quantify how much of the DSNs is able to accumulate into the tumor microenvironment, we conducted biodistribution studies by measuring the accumulation of NIR-dye tagged DSNs in different organs at different time points. Recent studies have indicated that the NIR dye from the nanoparticles can get prematurely released while in circulation [37, 38]. This could have impact on the quantification of nanoparticle accumulation in different organs as the results might not be representative of the actual distribution of the nanoparticles. We performed a release kinetics study of NIR dye from the DSNs over different time periods when incubated in human serum. Briefly, NIR-dye tagged DSNs were synthesized in PBS and incubated with equal volume of human serum. The resulting nanoparticle solution was dialyzed against 500M1 PBS buffer and the amount of dye retained in the nanoparticle solution was calculated by imaging the aliquots of the nanoparticles solution using IVIS imaging system and the data was represented as average radiance over time. As shown in Fig. 5a, no significant changes in the NIR florescence was observed for 24h. This is consistent with the stability and release kinetics profiles observed with DSNs.
Figure 5|. In vivo biodistribution study of NIR-dye tagged DSNs in 4T1 breast cancer model.

(a) Graph shows release kinetics profile of NIR dye from the DSN when incubated in presence of human serum or PBS buffer. Minimum release of the dye shows that it was stably loaded in DSN. (b) Representative NIR fluorescence images of 4T1 tumor bearing mice at different time points after NIR-dye tagged DSN injection. The tumor location in shown with dotted circle. (c) Graph shows quantification of DSN accumulation in tumors at different time points after injection. Data shows mean ± s. e. m. (n = 3). (d) Ex-vivo NIR imaging of the excised organs after 24h. (e) Graph shows the quantification of the NIR-dye accumulation in different organs. Data shown are mean ± s. e. m. (n = 3). *p < 0.05 (one-way ANOVA).
We next tested the distribution of NIR-dye tagged nanoparticles in 4T1 tumor bearing mice. Briefly, the tumor bearing mice were injected with 2nM NiR-tagged DSNs (dye concentration, 200μL volume) and the imaging was performed at different time points. After 24h of the injection, the mice were euthanized, and the harvested organs were imaged. As shown in Fig. 5b–c, a time dependent increase in tumor accumulation was observed as measured by increase in NIR dye fluorescence in tumor, consistent with our previous results. Ex vivo fluorescence analysis showed that the nanoparticles were accumulated in tumors at significantly higher concentration than spleen, kidneys, lung and heart (Fig. 5 d–e). However, we did observe accumulation in liver, consistent with a RES uptake. Recent studies have demonstrated that majority of the nanoparticles accumulated in tumor are phagocyted by tumor associated macrophages. [39] This is advantageous for nanoparticles targeted to tumor associated macrophages since we can get significantly higher drug accumulation in tumor associated macrophages as compared to other cells in the tumor including cancer cells.
We next tested if the higher nanoparticle accumulation in the tumor can result in better antitumor efficacy. When the tumors reached ~ 50mm3 volume, the mice were randoM1y assigned into six groups, with three doses of the following treatments at 15 mg/kg doses (relative to total kinase-inhibitor concentration): Vehicle, BLZ-SNP (CSF1R inhibiting supramolecular nanoparticles), SEL-SNP (MAPK-inhibiting supramolecular nanoparticles), BLZ-SNP+SEL-SNP, BLZ-945+SEL free drug and DSNs. We choose sub-optimal concentrations for the kinase inhibitors so as to dissect out the combination effect. It was shown in our previous work [27] that 45 mg/kg of CSF1R-inhibitor delivered using nanoparticles exhibited significant repolarization of macrophages, and in another study 100 mg/kg of BLZ945 was used p.o. to show survival benefit and different levels of cellular expression in mice models [40]. Selumetinib has also been administered at 50 mg/kg in a variety of animal models as described in recent studies.[41, 42] By utilizing sub-optimal doses for these therapeutics, the synergistic effect of both the inhibitors could be dissected out when delivered using DSN as compared to combination of individual inhibitors or single inhibitor loaded nanoparticles. All the doses were administered via tail-vein injection, excluding the free drug. Free inhibitors were dissolved in dimethyl sulfoxide and given intraperitoneally because of their low solubility in PBS. Indeed, we observed significant precipitation of the inhibitors at the 15mg/kg dose concentration in PBS within few minutes. Seven days after the injection of cancer cells, injections were started (now to be considered day 0), and the animals were euthanized upon observable necrosis or if the tumors reach 2 cm in diameter. The animals in all the groups were euthanized at the same point to evaluate the effect of treatments on tumor immune microenvironment. As shown in Fig. 6a, there were significant inhibition of tumor volume in DSN treated mice as compared to other treatment groups. It is also shown that the combination of single-inhibitor nanoparticles and SEL-SNPs have still a significant impact on tumor regression as compared to the control. Body weight was monitored over the entirety of treatment and is within reasonable limits which shows that the treatments did not exhibit significant toxicity (Fig. 6b). To perform mechanistic analysis of in vivo efficacy, we isolated the cells from excised tumors and stained with M1 and M2 markers. Flow cytometric analysis of TAM expression and the effects of treatment were analyzed, showing a higher M1 (CD11b+ CD80+) expression in macrophages treated with DSNs and SEL-SNPs in comparison to other treatment groups, which explains the observed in vivo effect (Fig. 6c–e). Interestingly, it was observed that the DSN system outperformed the combination of individual inhibitor-loaded nanoparticles in terms of repolarization efficacy and tumor regression. This could be due to the fact that suboptimal doses were used to dissect out the effect of the combination therapy using DSN over monotherapies involving single inhibitors as well as individual inhibitors or nanoparticles combination. Moreover, this is consistent with recent reports and our previous studies where concurrent delivery of two different drugs in the single nanoparticle was more effective in inhibiting the tumor growth than delivery of the individual drugs in two different nanoparticles[27, 43, 44]. Deterministic delivery of two kinase inhibitors using a single nanoparticle results in spatially constrained distribution of both the kinase inhibiting amphiphiles into the same cellular compartment as compared to stochastic delivery of the kinase inhibitors using combination of individual nanoparticles. This ensures that both kinase pathways are inhibited simultaneously, thus validating our rationale for simultaneous delivery and inhibition of both CSF1R and MAPK signaling pathways in the same cell. Furthermore, since both of these pathways are activated in M2 macrophages and important for its functions, necessitates the need for concurrent inhibition of these pathways in the same macrophage.
Figure 6|. In vivo efficacy study of DSNs in 4T1 breast cancer model.

(a) Graph shows relative tumor growth profiles in 4T1 tumor bearing mice after different multi-dose treatments. The tumor bearing animals were injected with 3 doses of either vehicle (control), sub-optimal doses of BLZ-945+ Selumetinib combination (15 mg/kg each drug), BZL-SNP (15 mg/kg equivalent dose of BLZ-945), SEL-SNP (15 mg/kg equivalent dose of Selumetinib) or DSN (15 mg/kg equivalent of each drug) at day 0, 3 and 5. Data shown are mean ± s. e. m. (n = 4–5). *p < 0.05 (one-way ANOVA). (b) Graph shows the body weight measurements of 4T1 tumor bearing mice after different treatments. No significant changes in body weight was observed which shows that the treatments did not exhibit toxicity. Data shown are mean ± s. e. m. (n = 3). (c-e) Graphs shows quantification of M1 (CD11b+CD80+, CD11b+CD86+) and M2 (CD11b+CD206+) markers in the different tumor samples. Data shown are mean ± s. e. m. (n = 3). *p< 0.05 (one-way ANOVA). (f) Representative Confocal microscopy images to demonstrate the internalization of FITC-Chol loaded DSNs into TAMs (blue, DAPI; red, CD11b+). (Scalebar 100 μm). (g) Flow cytometry analysis showing internalization of FITC-cholesterol-loaded DSNs (right) into CD11b+ TAMs in comparison to untreated control (left).
Next, we wanted to confirm that the nanoparticles were internalized by macrophages. Tumor bearing mice were injected with FITC-incorporated DSNs. After 24 hours, which was shown through IVIS biodistribution to have a higher concentration of nanoparticles at the site of the tumor (Fig. 5b–c), the mice were euthanized, and their tumors were cryo-sectioned and stained to identify TAMs. The tissue sections were stained with DAPI (blue) and CD11b (red) and imaged to identify whether or not there is colocalization of treatment in TAMs versus the surrounding cells (Fig 6f). We show that DSNs were indeed internalized by TAMs at higher amount. This was also confirmed by making a single cell suspension of the tumor samples and processing them through flow cytometry double staining for CD11b+ cells that also have FITC signal (Fig 6g).
Western blot analysis of the tumor samples revealed a significant inhibition of phospho-CSF-1R and phospho-ERK1/2 in DSN treatment groups as compared to other treatments (Fig. 7a–c). Interestingly, we also observed inhibition of phospho-AKT signaling which is downstream of phospho-CSF1R. Consistent with previous results, sustained inhibition of CSF-1R signaling results in inhibition of AKT signaling pathway. [27] We next tested if the inhibition of CSF1R and MAPK signaling pathways and repolarization of M2 macrophages to M1 phenotype result in enhanced tumor cell death. We performed a TUNEL staining of the tumor sections from different treatment groups. As shown in Fig. 7d–e, significantly higher tumor cells death was observed in DSN treated groups where as other treatment groups didn’t show significant tumor cells death, consistent with tumor growth progression. Since we observed significant accumulation of DSN in liver, we wanted to test if that leads to any toxicity to the liver cells. We performed the TUNEL staining of liver tissue sections from different treatment groups. As shown in Supplemental Fig. 7 no treatment resulted in apoptosis in liver cells which shows that the treatments are not toxic to liver. Finally, we wanted to assess the inflammatory effects of the treatments on the microenvironments of the most-highly trafficked areas by the nanoparticles: tumor and liver. This was conducted by H&E staining of paraffin-embedded tissue samples for CD8a+ T-cells as well as fluorescent labeling of F4/80+ cells in the tumor microenvironment. In the tumor tissue sections, we found similar expression levels of F4/80+ cells across all treatment groups, however increased levels of CD8a+ T-cells in both the DSN and BNP+SNP treatments were observed (Supplemental Figure 8). Also, we observed that the livers show similar levels of CD8a+ T-cells as well as F4/80+ cells regardless of the treatment regimen given (Supplemental Figure 9). These studies demonstrate that the treatments did not show off-target effects. Taken together, these results indicate that concurrent inhibition of CSF-1R and MAPK signaling pathways in TAMs using dual-inhibitors loaded supramolecular nanoparticles result in enhanced antitumor efficacy without any off-target effects.
Figure 7|. DSN inhibits CSF1R and MAPK signaling in TAMs at a significantly higher proportion and significantly enhances tumor cells death in 4T1 breast cancer model.

(a) Western blot showing expression of phospho-AKT, total-AKT, phospho-ERK1/2, total-ERK1/2, phospho-CSFIR and total-CSFIR in tumor samples from different treatment groups, (b-c) Quantification of the western blot results in (a) for the ratio of (d) phospho–CSFIR and (c) phopho-ERKl/2 vs their respective total expression. Data plotted is mean +- s.e.m, (n = 3). *p < 0.05 (one-way ANOVA). (d) Representative confocal images of tumor sections from animals treated with different treatment group as described in Fig. 6. The sections were labeled for apoptosis using TUNEL (red) stain and counterstained with DAPI (blue). (Scalebar 100 μm). (e) Graph shows the quantification of apoptosis from the labeled tumor sections as a percentage of TUNEL+ve cells as a function of total nuclei. Data shown are mean ± s. e. m. (n = 3). *p < 0.05 (one-way ANOVA).
Conclusions:
In summary, we have demonstrated that a stable lipid-based supramolecular nanoparticle that can carry two kinase inhibitors of different structural properties, can be designed by synthesizing the amphiphilic kinase-inhibiting building blocks. These building blocks can then self-assemble in the presence of co-lipids to form stable supramolecular nanoparticles with active inhibitors at higher concentrations. Sustained release of the inhibitors from the nanoparticles along with higher internalization in tumor associated M2 macrophages results in sustained inhibition of both CSF1R and MAPK signaling pathways and enhanced repolarization of M2 macrophages to M1 phenotype. MAPK-inhibiting supramolecular nanoparticles induce repolarization of M2 macrophages which could also be additional way current MAPK inhibitors be used to improve the effect in clinics. Tumor-homing abilities of the supramolecular nanoparticles resulting from increased circulation, could result in better tumor accumulation of the inhibitors and deterministic delivery of both inhibitors to the same tumor associated macrophages. Improved delivery of the inhibitors coupled with inhibition of CSF1R and MAPK signaling pathways in tumor associated macrophages, results in enhanced anti-tumor efficacy in an aggressive 4T1 tumor model with reduced systemic toxicity. In the future, to better improve the efficacy of repolarization of this immunotherapeutic system, higher doses can be employed to potentially ensure decrease in the prevalence of M2 TAMs while continuing to re-educate them to an anti-tumorigenic, M1 phenotype. This novel combination strategy could provide a new way to treat aggressive cancers and can be combined with current standard of care treatments including chemotherapy, immune checkpoint blockade for improved response.
Supplementary Material
Acknowledgements:
We are thankful for the support of a National Cancer Institute of the National Institutes of Health (P50CA168504) and Hearst Foundation/Brigham & Women’s Hospital Young Investigator Award, Melanoma Research Alliance Young Investigator Award (510283) and Cancer Research Institute Technology Impact Award (118–1501) to A. K. We would like to thank Biophysical Characterization Core Facility and Animal Imaging Facility at the Institute for Applied Life Sciences (IALS), University of Massachusetts Amherst for their consulting and assistance in the characterization experiments. We would like to acknowledge the support of the Electron Microscopy Core facility at the university of Massachusetts Medical School for Cryo-TEM imaging experiments. The project described was supported in part by Award Number S10 0D025113–01 from the National Center For Research Resources. We would like to acknowledge the Biospecimen Resource and Molecular Analysis Facility at the Pioneer Valley Life Sciences Institute for their histology work.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability:
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
References:
- 1.Dunkelberger JR and Song WC, Complement and its role in innate and adaptive immune responses. Cell Res, 2010. 20(1): p. 34–50. [DOI] [PubMed] [Google Scholar]
- 2.Elhelu MA, The role of macrophages in immunology. J Natl Med Assoc, 1983. 75(3): p. 314–7. [PMC free article] [PubMed] [Google Scholar]
- 3.Mogensen TH, Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev, 2009. 22(2): p. 240–73, Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quail DF and Joyce JA, Microenvironmental regulation of tumor progression and metastasis. Nat Med, 2013. 19(11): p. 1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mantovani A and Sica A, Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol, 2010. 22(2): p. 231–7. [DOI] [PubMed] [Google Scholar]
- 6.Bostrom MM, et al. , Tumor-Associated Macrophages Provide Significant Prognostic Information in Urothelial Bladder Cancer. PLoS One, 2015. 10(7): p. e0133552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noy R and Pollard JW, Tumor-associated macrophages: from mechanisms to therapy. Immunity, 2014. 41(1): p. 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruffell B and Coussens LM, Macrophages and therapeutic resistance in cancer. Cancer Cell, 2015. 27(4): p. 462–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aras S and Zaidi MR, TAMeless traitors: macrophages in cancer progression and metastasis. Br J Cancer, 2017. 117(11): p. 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poh AR and Ernst M, Targeting Macrophages in Cancer: From Bench to Bedside. Front Oncol, 2018. 8: p. 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bronte V and Murray PJ, Understanding local macrophage phenotypes in disease: modulating macrophage function to treat cancer. Nat Med, 2015. 21(2): p. 117–9. [DOI] [PubMed] [Google Scholar]
- 12.Martinez FO and Gordon S, The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep, 2014. 6: p. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coffelt SB, Hughes R, and Lewis CE, Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochim Biophys Acta, 2009. 1796(1): p. 11–8. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence T and Natoli G, Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol, 2011. 11(11): p. 750–61. [DOI] [PubMed] [Google Scholar]
- 15.Pyonteck SM, et al. , CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med, 2013. 19(10): p. 1264–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng X, et al. , Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget, 2017. 8(29): p. 48436–48452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lizotte PH, et al. , Attenuated Listeria monocytogenes reprograms M2-polarized tumor-associated macrophages in ovarian cancer leading to iNOS-mediated tumor cell lysis. Oncoimmunology, 2014. 3: p. e28926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chanmee T, et al. , Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel), 2014. 6(3): p. 1670–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chitu V and Stanley ER, Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol, 2006. 18(1): p. 39–48. [DOI] [PubMed] [Google Scholar]
- 20.Cannarile MA, et al. , Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer, 2017. 5(1): p. 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peyraud F, Cousin S, and Italiano A, CSF-1R Inhibitor Development: Current Clinical Status. Curr Oncol Rep, 2017. 19(11): p. 70. [DOI] [PubMed] [Google Scholar]
- 22.Ries CH, et al. , Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell, 2014. 25(6): p. 846–59. [DOI] [PubMed] [Google Scholar]
- 23.Papadopoulos KP, et al. , First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin Cancer Res, 2017. 23(19): p. 5703–5710. [DOI] [PubMed] [Google Scholar]
- 24.Moskowitz CH, et al. , CSF1R Inhibition by PLX3397 in Patients with Relapsed or Refractory Hodgkin Lymphoma: Results From a Phase 2 Single Agent Clinical Trial. Blood, 2012. 120(21): p. 1638–1638. [Google Scholar]
- 25.Butowski N, et al. , Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase IIstudy. Neuro Oncol, 2016. 18(4): p. 557–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Tresckow B, et al. , An Open-Label, Multicenter, Phase I/IIStudy of JNJ-40346527, a CSF-1R Inhibitor, in Patients with Relapsed or Refractory Hodgkin Lymphoma. Clin Cancer Res, 2015. 21(8): p. 1843–50. [DOI] [PubMed] [Google Scholar]
- 27.Kulkarni A, et al. , A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nature Biomedical Engineering, 2018. 2(8): p. 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou D, et al. , Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal, 2014. 26(2): p. 192–7. [DOI] [PubMed] [Google Scholar]
- 29.Caescu CI, et al. , Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood, 2015. 125(8): p. e1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richardson ET, et al. , ERK Signaling Is Essential for Macrophage Development. PLoS One, 2015. 10(10): p. e0140064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boulakirba S, et al. , IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci Rep, 2018. 8(1): p. 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molnar E, et al. , Pan-RAF and MEK vertical inhibition enhances therapeutic response in non-V600BRAFmutant cells. BMC Cancer, 2018. 18(1): p. 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woo SU, et al. , Vertical inhibition of the PI3K/Akt/mTOR pathway is synergistic in breast cancer. Oncogenesis, 2017. 6(10): p. e385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramesh A, et al. , Dual Inhibitors-Loaded Nanotherapeutics that Target Kinase Signaling Pathways Synergize with Immune Checkpoint Inhibitor. Cellular and Molecular Bioengineering, 2019. 12(5): p. 357–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kulkarni A, et al. , Combining Immune Checkpoint Inhibitors and Kinase-Inhibiting Supramolecular Therapeutics for Enhanced Anticancer Efficacy. ACS Nano, 2016. 10(10): p. 9227–9242. [DOI] [PubMed] [Google Scholar]
- 36.Kulkarni A, et al. , Rationally Designed 2-in-1 Nanoparticles Can Overcome Adaptive Resistance in Cancer. ACS Nano, 2016. 10(6): p. 5823–5834. [DOI] [PubMed] [Google Scholar]
- 37.Cohen S and Margel S, Engineering of near IR fluorescent albumin nanoparticles for in vivo detection of colon cancer. Journal of Nanobiotechnology, 2012. 10(1): p. 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng F, et al. , Quantitative Assessment of Nanoparticle Biodistribution by Fluorescence Imaging, Revisited. ACS Nano, 2018. 12(7): p. 6458–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dai Q, et al. , Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano, 2018. 12(8): p. 8423–8435. [DOI] [PubMed] [Google Scholar]
- 40.Quail DF, et al. , The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science, 2016. 352(6288): p. aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Decaudin D, et al. , Selumetinib-based therapy in uveal melanoma patient-derived xenografts. Oncotarget, 2018. 9(31): p. 21674–21686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loi S, et al. , RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res, 2016. 22(6): p. 1499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldman A, et al. , Rationally Designed 2-in-1 Nanoparticles Can Overcome Adaptive Resistance in Cancer. ACS Nano, 2016. 10(6): p. 5823–34. [DOI] [PubMed] [Google Scholar]
- 44.Miao L, et al. , Nanoparticles with Precise Ratiometric Co-Loading and Co-Delivery of Gemcitabine Monophosphate and Cisplatin for Treatment of Bladder Cancer. Advanced Functional Materials, 2014. 24(42): p. 6601–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.