

Abstract
The Lennox–Gastaut syndrome is a devastating early-onset epileptic encephalopathy, associated with severe behavioural abnormalities. Its pathophysiology, however, is largely unknown. A de novo mutation (c.G358A, p.D120N) in the human GABA type-A receptor β3 subunit gene (GABRB3) has been identified in a patient with Lennox–Gastaut syndrome. To determine whether the mutation causes Lennox–Gastaut syndrome in vivo in mice and to elucidate its mechanistic effects, we generated the heterozygous Gabrb3+/D120N knock-in mouse and found that it had frequent spontaneous atypical absence seizures, as well as less frequent tonic, myoclonic, atonic and generalized tonic–clonic seizures. Each of these seizure types had a unique and characteristic ictal EEG. In addition, knock-in mice displayed abnormal behaviours seen in patients with Lennox–Gastaut syndrome including impaired learning and memory, hyperactivity, impaired social interactions and increased anxiety. This Gabrb3 mutation did not alter GABA type-A receptor trafficking or expression in knock-in mice. However, cortical neurons in thalamocortical slices from knock-in mice had reduced miniature inhibitory post-synaptic current amplitude and prolonged spontaneous thalamocortical oscillations. Thus, the Gabrb3+/D120N knock-in mouse recapitulated human Lennox–Gastaut syndrome seizure types and behavioural abnormalities and was caused by impaired inhibitory GABAergic signalling in the thalamocortical loop. In addition, treatment with antiepileptic drugs and cannabinoids ameliorated atypical absence seizures in knock-in mice. This congenic knock-in mouse demonstrates that a single-point mutation in a single gene can cause development of multiple types of seizures and multiple behavioural abnormalities. The knock-in mouse will be useful for further investigation of the mechanisms of Lennox–Gastaut syndrome development and for the development of new antiepileptic drugs and treatments.
Keywords: genetic epilepsies, GABRB3, Lennox–Gastaut syndrome, early-onset epileptic encephalopathy, seizures
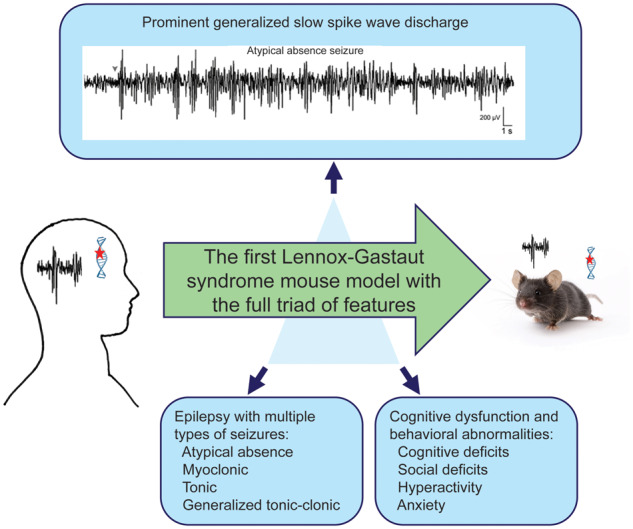
We generated the Gabrb3+/D120N knock-in mouse harbouring a mutation in the GABAAR β3 subunit identified in an epileptic patient with Lennox–Gastaut syndrome. This mouse displays the full triad of features that characterize Lennox–Gastaut syndrome: multiple seizure semiologies, generalized slow spike-and-wave discharge on EEG and cognitive dysfunction and behavioural abnormalities.
Graphical Abstract
Graphical Abstract.
Introduction
The Lennox–Gastaut syndrome (LGS) is a severe early-onset epileptic encephalopathy of infancy and childhood (McTague et al., 2016), accounting for 3–10% of childhood epilepsies (Abu Saleh and Stephen, 2008). LGS can be genetic or secondary to an underlying brain disorder and is characterized by a triad of features including epilepsy with multiple seizure semiologies, a generalized spike wave discharge (SWD) pattern on EEG and cognitive and behavioural abnormalities (Trevathan, 2002). The most common seizure types include atypical absence, atonic and tonic seizures, and less common types include clonic, myoclonic and generalized tonic–clonic seizures (GTCSs). LGS onset usually occurs between 1 and 8 years and frequently persists and worsens through adolescence and adulthood (McTague et al., 2016). Most patients with LGS experience behavioural abnormalities that can include varying degrees of developmental delay, intellectual impairment and other behavioural problems such as hyperactivity and autism spectrum disorder (ASD). There is no reliably effective treatment for LGS, and up to 10% of patients with LGS die before the age of 11. LGS prognosis is generally poor as seizures are generally refractory to antiepileptic drugs (AEDs) (Trevathan, 2002). It is difficult to address the cause and underlying mechanisms of genetic LGS in human studies, and lack of animal genetic LGS models has hindered progress in understanding the pathophysiology and development of treatment for this devastating disease.
While the aetiopathology of LGS is largely unknown at cellular and molecular levels, next-generation DNA sequencing has identified mutations in genes implicated in its pathogenesis (EuroEPINOMICS-RES Consortium, 2014). The de novo mutation (c.G358A, p.D120N) was one of the several GABRB3 mutations found in patients with LGS by whole-exome sequencing of familial triads (child affected, parents unaffected) (Epi et al., 2013). GABRB3 encodes the human β3 subunit of the heteropentameric GABA receptor type-A (GABAA) receptors that mediate fast inhibitory synaptic neurotransmission. Dysfunction of GABAA receptors can lead to a variety of seizure types (Macdonald et al., 2010), and in addition, to LGS. GABRB3 mutations are implicated in several other childhood epilepsies including childhood absence epilepsy, infantile spasms (IS) and other early-onset epileptic encephalopathies (Homanics et al., 1997; McTague et al., 2016; Shen et al., 2017). The β3 subunit plays an important role during brain development, with the expression of β3 subunits being widespread and abundant in embryonic and neonatal brains, especially in the cerebral cortex, thalamus, hippocampus and other regions involved in generating seizures (Laurie et al., 1992; Hortnagl et al., 2013).
Amino acid sequence alignment analysis showed that an aspartic acid residue at Position 120 was invariant across all GABAA receptor subunits, and structural modelling revealed that the D120N (aspartic acid residue at position 120 is substituted with an asparagine) amino acid substitution was in a conserved domain of the GABAA receptor β3 subunit near the GABA binding pocket. We previously demonstrated in vitro in transfected HEK293T cells that the mutant β3 subunit not only significantly reduced GABAA receptor currents but also altered their kinetic properties without affecting receptor assembly or trafficking (Janve et al., 2016). However, a major question remains: does the single-point mutation in the mutant β3(D120N) subunit cause the multiple LGS seizure types and behavioural abnormalities, and if so, how? Although these in vitro results strongly suggest that the mutant β3(D120N) subunit contributes to the generation of LGS seizure types and behavioural abnormalities, its causal role can only be established directly by determining its effect on behaviour and brain excitability in vivo. To do this, we generated and characterized a mouse model of LGS [the heterozygous Gabrb3+/D120N knock-in (KI) mouse], which has the human GABRB3 mutation, c.G358A, p.D120N, that was identified in a patient with LGS. Currently, there are no reports of mouse models exhibiting the triad features of LGS and no mouse models carrying LGS-associated human mutations. Here, we show that heterozygous Gabrb3+/D120N KI mice recapitulated both the seizure and behavioural abnormalities associated with LGS. While expression and trafficking were unaltered, cortical miniature inhibitory post-synaptic currents (mIPSCs) in coronal slices had reduced amplitude and spontaneous prolonged thalamocortical oscillations in horizontal thalamocortical slices were recorded. Finally, AEDs commonly used to treat LGS were similarly effective at reducing seizures in the KI mice. Therefore, Gabrb3+/D120N KI mice are an excellent model for LGS and should be useful for further characterization of LGS pathophysiology and for the development of novel therapies. Importantly, Gabrb3+/D120N KI mice show that it is possible to fully recapitulate a complex human epilepsy syndrome in rodents with a single-point mutation, indicating overlap of the seizure syndrome and behavioural comorbidity circuitry.
Materials and methods
All animals were cared for and used in accordance with the policies of the Vanderbilt University Medical Center IACUC and to the National Institutes of Health Guide for Care and Use of Laboratory Mice. Mice were maintained in cages of up to five adults, except after EEG headmount surgery when mice were single-housed with enrichment. Mice were housed in either the VU Level 5 (not specific-pathogen-free) breeding facility or the Level 7 Neurobehavioral Core, both of which are maintained by the Department of Animal Care. Facilities use a 12-h light/dark cycle, corn-cob bedding and ad libitum food and water. Power analyses were conducted with preliminary data to determine final sample sizes. Mice used for data were discrete groups in every case except EEG surgeries were performed on some mice after behavioural testing, and some animals used in the initial v-EEG analysis were used in drug studies. Where possible, the observer was blinded to the genotype until after analysis was complete.
Generation of the Gabrb3+/D120N KI mouse
The Gabrb3 G358A mutation changes a GAC codon to AAC in Exon 4, thus converting an aspartate to an asparagine (D120N) in the β3 subunit. The KI mouse was generated in collaboration with the Vanderbilt Transgenic Mouse/ES Cell Shared Resource by using conventional homologous recombination methods. The replacement targeting vector was constructed by cloning the upstream 5.4-kb 5′ and downstream 4.6-kb 3′ homology arms into an Flippase recognition site (FRT)-flanked PGK-Neo cassette, in a vector with the negative selection marker MC1-thymidine kinase cassette. Embryonic stem cells (C57BL/6N-PRX-B6N) with a correctly targeted locus, identified first by long-range polymerase chain reaction (PCR) with one of the primers located outside of homology arms and followed by PCR/sequencing, were micro-injected into the C57BL/6J albino blastocysts to produce chimeric mice. Chimeric mice were bred with Actin-Flpe C57BL/6J transgenic mice (Rodriguez et al., 2000) to screen for germline transmission and to excise the PGK-Neo selection cassette. The KI mice were genotyped by using a pair of primers (forward 5′-GTGCACTGGCATTGAGTTTCTCAC-3′ and backward 5′- CTTCATAGAGCACCAATGAGACTGTACC-3′). Mice were further backcrossed to C57BL/6J, which differs from C57BL/6N by only 0.8% (Mekada et al., 2009), for a minimum of six generations.
Video-EEG recordings and analysis
Synchronized video-EEGs were recorded from 4.5- to 6.5-month-old wild type littermate and KI mice after electrode implantation (# 8201; Pinnacle Technology) with a video-EEG monitoring system from Pinnacle Technology (Arain et al., 2012), and following published protocols (Warner et al., 2017). Prior to surgery, mice were weighed and anesthetized using 2–4% isoflurane and placed into a stereotactic frame. The headmount was placed on the skull with the front edge 3.0 to 3.5 mm anterior of the bregma and secured with four screws (# 8209; Pinnacle Technology) such that all four screws rest in the cerebral cortex, providing electrical contact between the brain surface and the headmount. Two EMG leads were inserted into the nuchal muscles. Mice were allowed to recover for a minimum of 7 days before testing. Video-EEG and EMG data were analysed offline with Sirenia® Seizure software using a widely used approach of visual inspection. The entire 24-h recording was analysed. Mice were maintained for several months post-operative with headmounts in place and were checked daily for the signs of infection or irritation. Power spectra were generated using EDF browser.
Postnatal spasm observations
Pups from P1 to P6 were observed in their home cage with their mother to reduce separation-induced stress and rejection. P6–P11 and P12–P18 pups were separated from the home cage for 5 and 10 min, respectively, and observed and videotaped individually for general behaviours and spontaneous seizures. Pups were genotyped at weaning.
Mouse behavioural testing
The mouse behaviour experiments were performed in the Vanderbilt University Mouse Neurobehavioral Laboratory. Each cohort was subjected to the full suite of behaviour tests, and they were performed in the order in which they are described below. If mice displayed obvious seizure activity, they were allowed to recover to normal behaviour before further testing. In addition, behaviour tests were conducted between 9 am and 5 pm to minimize seizure occurrence, as KI mice have increased seizure incidence during the light-to-dark transition. Extensive handling was done prior to behavioural testing to avoid unnecessary anxiety responses.
The elevated zero maze test was used for assessing anxiety-related behaviours, following a previously described protocol (Kang et al., 2015). The elevated circular platform is divided into equal fourths, with two enclosed arenas opposite each other and two open arenas between the enclosed arenas. Each mouse explored the maze for 300 s. Activity was monitored via an overhead camera using ANY-maze software (Stoelting, Wood Dale, IL, USA).
The locomotor activity tests were performed as previously described (Fukada et al., 2012; McLaughlin et al., 2012; Kang et al., 2015) using the standard protocol in the Vanderbilt University Mouse Neurobehavioral Laboratory. Each mouse was placed for 60 min in an open field activity chamber (Med Associates, 27 cm × 27 cm × 20.3 cm). Location and movement were detected by the interruption of infrared beams by the mouse (in each horizontal direction and elevated by 4 cm to measure rearing/jumping) and were measured by the Med Associates Activity Monitoring programme.
The three-chamber socialization test (3CST) protocol follows that of a previous study (Carter et al., 2011)—habituation: subject mouse freely explores empty apparatus for 10 min; sociability: subject mouse freely explores a stranger mouse (novel mouse) and empty pencil cup (novel object) for 10 min; and social novelty: subject mouse freely explores a novel mouse and familiar mouse for 10 min. Mice, excluding the subject mouse, were contained in inverted pencil cups. Time subject mouse spent actively investigating each stimulus was recorded by hand using ANY-maze.
The Barnes maze test was based on a protocol described in detail in a previous study (Harrison et al., 2006). The procedure includes three components: pretraining, training and a memory probe. All data were collected using ANY-maze. In addition, search strategies were manually scored as either a direct path (the mouse travels either directly to the target hole or to a hole on either side before immediately going to the target hole, worth zero points), a serial path (the mouse visits two or more non-target holes in a sequential order before entering the target hole, also known as a thigmotaxic strategy, worth one point) or a random path (the mouse uses a non-direct or non-sequential method, worth two points), based on the strategies outlined in Harrison, et al. (2006).
Behavioural data were analysed with GraphPad Prism software. Independent-sample Student’s t-tests were used in the analyses of locomotor activity, elevated zero maze and the probe trials of the Barnes maze. A two-way ANOVA was used for the three-chamber socialization test for both sociability and social novelty. A repeated-measures ANOVA was used to analyse the training trials of the Barnes maze. Post hoc and a priori Bonferroni comparisons were conducted to evaluate individual mean comparisons where appropriate.
Reverse transcriptase PCR
Total RNAs were extracted from brain tissues of P10–P12 mice by using Direct-zol™ RNA MiniPrep Plus kit (Zymo Research, Irvine, CA, USA). Reverse transcriptase PCR was carried out by using QIAGEN OneStep reverse transcriptase PCR kit. Forward primer was 5′-GTAGAGTGGAGCAGTTCCCACTCAG-3′ (upstream of the start codon), and backward primer was 5′-GACAGGCAGGGTAATATTTCACTCAGTG-3′ (downstream of the stop codon). Glyceraldehyde 3-phosphate dehydrogenase primers were 5′-GTGAAGGTCGGTGTGAACGGATTTG-3′ and 5′-GATGGCATGGACTGTGGTCATGAG-3′.
Western blots
Cerebellum, cortex, hippocampus and thalamus were dissected from 1- to 3-month-old mouse brains and homogenized with radio immunoprecipitation assay (RIPA) buffer. Proteins were fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with following antibodies: ATPase (1:2000, a6F; DSHB), GABAA receptor α1 subunit (1:500, 75-136; NeuroMab), GABAA receptor β3 subunit (1:500, B300-199; Novus Biologicals), GABAA receptor γ2 subunit (1:500, AB 5559; Millipore), gephyrin (1:1000, 3B11; Cedarlane) and glyceraldehyde 3-phosphate dehydrogenase (1:6000, MAB374; Millipore) antibodies. Surface expression of GABAA receptor subunits was evaluated by the extraction of plasma membrane proteins from brain using the 101Bio kit (101bio). Proteins were fractionated by SDS-PAGE and subjected to immunoblot analyses. Synaptosomes were isolated following a previous study (Kang et al., 2015). Full gel images are shown in Supplementary Fig. 1.
Whole-cell slice recording
Brain slices were prepared according to previously published methods (Huang et al., 2017). Coronal brain slices (300 μm) taken from 30-day-old mice (males and females) were cut using a LEICA VT-1200S vibratome (Leica Inc.) with oxygenated dissection solution (mM: 2.5 KCl, 0.5 CaCl2, 10 MgSO4, 1.25 NaH2PO4, 24 NaHCO3, 11 glucose, 214 sucrose). The brain slices were transferred into a glass chamber with oxygenated artificial cerebrospinal fluid (mM: 126 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 26 NaHCO3, 10 glucose, pH 7.4) and then recovered at room temperature for 1 h before experiments.
With an upright Eclipse FN-1IR-DIC microscope (Nikon) and a MultiClamp 700B amplifier and Digidata 1440A (Molecular Devices), whole-cell recordings of layer V/VI pyramidal neurons (with typical apical dendrites within somatosensory cortex) were obtained according to previously published methods (Huang et al., 2017). Layer V pyramidal neurons were distinguished by their large soma size with apical dendrites, and layer VI pyramidal neurons were distinguished by their position right above white matter within coronal section brain slices. We did not notice any significant morphological difference (IR-DIC imaging) in pyramidal neurons from wild type or KI mice. The mIPSCs were isolated pharmacologically by including 10–20 µM NBQX and 1 µM tetrodotoxin in the artificial cerebrospinal fluid. The chloride ion reversal potential with the external artificial cerebrospinal fluid and internal solution used [mM: 135 CsCl, 10 HEPES, 10 EGTA, 5 QX-314, 5 ATP-Mg (290–295 mOsm, pH = 7.3)] was 0 mV, and neurons were voltage-clamped at −60 mV. Access resistances during recording were continuously monitored and confirmed to be <25 MΩ. Data were collected using the Clampex programme 10.2 (Molecular Devices) (Zhou et al., 2011; Huang et al., 2017). Histograms and cumulative graphs were constructed using Clampfit (Molecular Devices).
Thalamocortical oscillations
Horizontal thalamocortical brain slices (350–400 µm) were prepared (Huguenard and Prince, 1994) from P30 mice to maintain thalamocortical and corticothalamic circuit projections. Slices were placed on a self-made nylon mesh interface (only one side of the slice contacted the artificial cerebrospinal fluid solution flowing below). One unipolar tungsten electrode (MultiClamp 700B, current-clamp mode) was placed in the thalamic ventrobasal nucleus of the thalamus to record spontaneous multi-unit activity, which was band-filtered between 100 Hz and 3 kHz. Experiments were performed at 31–32°C for 1–2 h to collect sufficient spontaneous multi-unit activity. Data were analysed using both Clampfit and MATLAB. Oscillation indices and duration for the spontaneous multi-unit activity in thalamocortical slices from wild type and KI mice were calculated and compared (Sugihara et al., 1995; Sohal et al., 2003).
AED treatment of LGS KI mice
Head mounts were surgically implanted on the skull surface of adult KI mice (6–8 months old). The following classic AEDs were tested: ethosuximide (200 mg/kg in saline), clobazam (2.5 mg/kg in 30% PEG 400 solution) and topiramate (30 mg/kg in saline). Experiments began at 10 am, with AEDs given at 12 pm. Two hours prior to drug injection, animals were injected with the AED diluent to serve as control observations. A single dose of AED was injected intraperitoneally in a solution volume of ∼200 μl. Mice were monitored by synchronized video-EEG recording for 2 h after vehicle administration and for 2 h after AED injection. WIN 55,212-2, a cannabinoid receptor 1 agonist, was given daily for 7 days at a dose of 0.1 mg/kg dissolved in dimethyl sulfoxide. Video-EEGs were recorded from 12 pm to 2 pm on the first day for baseline (prior to the first injection) and from 12 pm to 2 pm on the eighth day also. For drugs with published mouse data, dosing was based on available information. For drugs without published mouse data, dosing was based on either the mid-range of human dosing or previous experimental knowledge of the laboratory. Mice were sacrificed after drug treatment.
Statistical analysis
Statistical analyses were performed, and graphs were generated using GraphPad Prism. Tests performed on each data set are outlined in the corresponding figure legend. Where n < 10, all data points were displayed with mean and SEM. Where n > 10, violin plots were utilized showing the median, quartiles (25% and 75%) and range of the data. All analyses used an alpha level of 0.05 to determine statistical significance.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Material. These data are available from the corresponding author upon reasonable request.
Special note: the naïve EEG profile was scored and analysed by V.J., while R.K.H. scored and analysed the drug data. There may be subtle differences between their scoring strategies, but their data were not cross-analysed to avoid any observer-specific effects.
Results
Gabrb3+/D120N mice
To establish an LGS mouse model with the specific mutation in GABRB3 (c.G358A, p.D120N) reported in a human with LGS, we generated mice with a targeted G358A mutation at the endogenous Gabrb3 locus using conventional homologous recombination in B6N embryonic stem cells (Primogenix, Inc.) derived from the C57BL/6N mouse (Fig. 1). The G358A mutation located in the 5′ homologous arm was generated by site-directed mutagenesis, resulting in the substitution of asparagine for aspartic acid at Residue 120. The correctly targeted embryonic stem cell clones were identified by long-range PCR with one of the primers (A or D) located outside the homologous arms to exclude the clones with random vector insertion (Fig. 1A). PCR fragments were digested with the HindIII (H3) enzyme to confirm further correctly targeted recombination (Fig. 1B). Chimeric mice were bred with Flpe transgenic mice to screen for germline transmission and to excise the PGK-Neo selection cassette. The leftover FRT site allowed convenient genotyping (Fig. 1C).
Figure 1.

Creation of the Gabrb3+/D120N KI mouse. (A) Scheme for targeted KI of the Gabrb3 locus. A vector was constructed to replace Gabrb3 Exon 4 genomic DNA with an Exon 4 containing the G358A mutation and the positive selection cassette (PGK-Neo). (B) Long-range PCR was used to identify the correctly targeted clones, G3 and F5. The 6- and 4.6-kb fragments were amplified from correctly targeted clones using primers A/B and C/D, respectively. The PCR fragments were digested with the HindIII restriction enzyme (to generate the predicted 4.2- and 1.8-kb fragments for the 5′ end and 3.6- and 1-kb fragments for the 3′ end) to further confirm correct homologous recombination. (C) The KI mice were genotyped. PCR amplified a 392-bp fragment from the wild type β3 subunit allele and a 480-bp fragment from the β3 subunit KI allele due to the insertion of FRT sequences and other modifications. (D) Sequencing chromatogram of RT-PCR derived from total brain RNA showed the presence of the G358A mutation and corresponding equal level of wild type and mutant nucleotide expression. (E1) Semi-quantitative RT-PCR showed that the KI allele did not affect Gabrb3 gene expression, using GAPDH as a loading control. (E2) Band intensities of the RT-PCR products were first normalized to GAPDH for quantification and then to wild type levels. (F1) Immunoblotting showed equal β3 subunit expression in KI and wild type littermate mouse brain whole-cell lysates. GAPDH served as a loading control. (F2) Protein levels were normalized first to internal GAPDH and then to wild type levels. (G) The body weight of KI and their littermate mice at weaning were plotted (n = 22 wild type, 18 KI). GAPDH: glyceraldehyde 3-phosphate dehydrogenase; RT-PCR: reverse transcriptase PCR.
The presence of the G358A mutation was further confirmed by PCR/sequencing. The sequencing chromatogram of the reverse transcriptase PCR products of total RNA isolated from the heterozygous KI mice showed similar intensity of peak G (wild type mRNA) and peak A (mutant mRNA) nucleotides (Fig. 1D), indicating that both alleles were transcribed identically. In addition, to determine if the gene editing affected Gabrb3 expression at the transcriptional level, semi-quantitative reverse transcriptase PCR was carried out with total RNAs isolated from whole brain (Fig. 1E1). The results revealed that expression levels of Gabrb3 mRNA from total brain were equivalent in KI and wild type littermate mice (Fig. 1E2). Sequence analysis using reverse transcriptase PCR that included the entire coding region of the Gabrb3 transcript from Gabrb3+/D120N mouse brain confirmed that there were no DNA sequence changes other than the intended G358A mutation. At the translation level, western blots with whole brain lysates (Fig. 1F1) showed that the expression of β3 subunits in brain of Gabrb3+/D120N mice was the same as that of wild type littermate mice (Fig. 1F2).
Gabrb3+/D120N mice had increased mortality, reduced body weight and other abnormalities
P18.5–P19.5 homozygous Gabrb3D120N/D120N embryos collected from heterozygous KI intercrossed timed-pregnant mice were viable, and there were no apparent differences at this embryonic age among homozygous KI, heterozygous KI and wild type littermates. However, no homozygous pups survived to the 3-week weaning and genotyping age, indicating that homozygous D120N mutations were neonatal lethal.
While heterozygous KI mice achieved normal body weight by adulthood, at P21 KI pups weighed significantly less than wild type pups (Fig. 1G). No major differences were observed between wild type and KI pups in the neonatal (P0–P5) period. From P6 to P11, KI pups had delays in the development of skin pigmentation and in righting when they were placed on their backs. Adult male KI mice did not fight when combined from different cages. Males had a limited reproductive time window that was restricted to when they were relatively young, so breeding was done primarily between female KI and male wild-type mice. In addition, first-time KI dams failed to nurture their offspring irrespective of pup genotype; the nurturing behaviour improved for subsequent litters after KI dams were housed with wild type foster dams.
Mating of female KI mice with male wild-type mice revealed a ratio of KI to wild type offspring at weaning that was inconsistent with Mendel’s law. Out of 525 weanlings genotyped, 294 were wild type and 231 were KI, suggesting that KI mice had increased mortality. A traditional large survival analysis was not performed due to experimental use and breeding challenges; however, surviving KI mice had a lifespan similar to that of wild-type mice. Using Nissl stain, there were no apparent differences in brain cross sectional anatomy between KI mice and their wild type littermates (data not shown), suggesting that the increased death rate of heterozygous mice was likely related to seizures.
Gabrb3+/D120N mice had seizure semiologies and ictal EEG patterns consistent with LGS
We next determined if the KI mice had LGS seizure semiologies and ictal EEGs by home-cage observation and synchronized video-EEG analyses. In adult mice, EEG head mounts were implanted ∼4 months of age in both wild type and KI mice, and after 1 week of recovery, the mice were monitored by synchronized video-EEG across a range of ages. We observed multiple types of spontaneous seizures in the KI mice (see Video 1), but not in wild type littermates [except for infrequent, brief, typical absence-like seizures, as is expected in C57BL6 mice (Letts et al., 2014)].
Typical absence seizures in humans are classified by (i) generalized, symmetrical SWD with a peak ictal spectral frequency >2.5 Hz, usually ∼3 Hz; (ii) duration average of 9.4 s (range 1–44 s); and (iii) abrupt onset and offset of loss of consciousness that is time-locked with the SWD. In addition, they are common in different epilepsies, can occur at any age (though typically onset is at 6 or 12 years old) and are generally considered benign, and there is a high remission rate (57–74% for childhood absence epilepsy) (Tenney and Glauser, 2013). wild type littermates had rare, brief absence seizures (Fig. 2A and B) characterized by (i) generalized SWD with a high peak frequency (peak 7.6 Hz, range 6–8 Hz), (ii) a short duration (mean 9.9 ± 0.1 s, range 1.3–30.7 s) and (iii) behavioural arrest with sudden onset and offset time-locked to the SWD. These seizures reflect typical absence seizures in humans and those reported in absence epilepsy mouse models where duration is 0.3–10 s and the predominant frequency range is 6–8 Hz (Letts et al., 2014; Jarre et al., 2017).
Figure 2.

Spontaneous seizures were observed in adult KI mice. Representative EEG traces for typical absence seizures seen in wild-type mice, and atypical absence, myoclonic, tonic and GTCS seen in KI mice. The arrowheads indicate the start and end of the seizure. Note that the atypical absence seizure extends beyond the selected time frame, so no endpoint is shown. (A) Typical absence seizures in wild-type mice were brief and behavioural arrest was time-locked to SWD onset and offset. (B) Power spectral density for typical absence seizure EEGs in wild-type mice with peak frequency at 7.6 Hz. (C) Atypical absence seizure EEG trace; atypical absence seizures were not always time-locked with the behavioural onset and/or had brief movements during the seizure (see Video 1 for full seizure). (D) Power spectral density of atypical absence EEGs often started with (i) higher-frequency components that (ii) decreased over time; however, the frequency over the entire seizure (iii) was low. (E) Myoclonic seizure EEG discharges were brief (∼300 µs), and both EEG and EMG traces had prominent spikes. (F) EEG during tonic seizures did not stand out from the baseline in amplitude but showed an increased frequency compared with the baseline. (G) GTCS EEGs were striking with the largest increase in EEG amplitude. The tonic phases during GTCS had slightly lower amplitude but higher waveform frequencies compared with the clonic phase, which was followed by electrodecrement (see Video 1 for examples of KI video-EEG recordings for each seizure type). (H) Bar graph showing average number seizures in wild type and KI mice in a 24-h period (note: y-axis is log scale). Atypical absence seizures and myoclonic seizures were the dominant seizures in KI mice. (I, J) Most atypical absence seizures in KI mice occurred during the dark (active) period with a peak incidence at the light-to-dark period transition (paired t-test, n = 7, light mean = 142, dark mean = 297.7, *P = 0.011). As the light cycle does not change with daylight savings, all times are set to the appropriate non-daylight savings time (6:00–18:00 light, 18:00–6:00 dark). Video-EEG recordings (for panels A–J) were done in n = 4 wild type (two males, two females) and 7 KI (three males, four females) mice at age 4.5–6.5 months. (K) Spontaneous spasms in wild type and KI mice were observed in pups between P12 and P18. Shown are the percentages of wild type and KI pups that exhibited spasms at each age. Observers were blind (five litters, with n = 16 wild type and n = 19 KI mice).
Atypical absence seizures in humans are classified by (i) generalized, diffuse, irregular slow SWD (SSWD) with a frequency of <2.5 Hz with or without irregular diffuse fast activity; (ii) a longer duration than typical absence; and (iii) impairments of consciousness that are variable in severity and may not be time-locked to the SSWD. In addition, they are more rare and more difficult to control pharmacologically than typical absence seizures, and while they are often considered minor seizures as they are not directly life-threatening, they are believed to contribute to the severe learning disabilities associated with disorders like LGS (Tenney and Glauser, 2013; Vrielynck, 2013). KI mice expressed frequent atypical absence seizures (Fig. 2C and D) that were characterized by (i) an ictal EEG containing SSWDs that were primarily poorly formed high-voltage, low-frequency events (3–7 Hz), which is closer in range to atypical absence seizures observed in the rat AY-9944 induction model and the GABABR1a overexpression mouse model (5–6 Hz; Rodriguez et al., 2000; Cortez et al., 2001); (ii) a longer duration than typical absence (mean 20.3 ± 10.2 s; range 0.5–363.5 s); and (iii) behavioural arrest and loss of consciousness that were not necessarily tightly time-locked to seizure onset or offset and behavioural arrest that was not always complete. These events were only identified as seizures if the mouse was clearly awake (visibly open eyes or a sudden interruption of a high level of activity at event onset). It was possible to distinguish atypical absence seizures from non-REM sleep based on behaviour and spectral density (Supplementary Fig. 2). Peak SSWD frequency was often higher at the beginning of the seizure (Fig. 2Di; 4.0 Hz peak, strong 8.0 Hz contribution) and developed a lower frequency as the seizures progressed and became less organized (Fig. 2Dii; 3.6 Hz peak, strong 4.8 Hz contribution). Because of the characteristics of these seizures, including appropriate peak spectral frequency, time and behavioural dynamics, we classified them as atypical absence seizures, the hallmark seizures of LGS.
Myoclonic seizures in humans are characterized by sudden, brief (<350 ms) shock-like movements with a high-amplitude spike on EEG (Striano and Belcastro, 2012). Myoclonic seizures in KI mice were moderately frequent and brief (∼300 ms), with sudden muscle contraction involving whole-body extension and flexion associated with a single, high-amplitude sharp wave (∼200–400 µV) on EEG and EMG channels (Fig. 2E).
Tonic seizures in humans are characterized by either unilateral or bilateral contraction of one or more muscle groups. This contraction is sustained, lasting for a few seconds to up to a minute and may be asymmetrical resulting in turning to one side (Werhahn et al., 2000; Daroff, 2012). Tonic seizures in KI mice involved a sudden and prolonged (up to several seconds) contraction of the limbs, with or without tail stiffening, that was associated with either no change in EEG amplitude or low-amplitude, high-frequency activity, as previously described (Gohma et al., 2007) (Fig. 2F). Tonic seizures often resulted in the ‘rolling’ of the animal, similar to how humans may turn to one side. Tonic seizure incidence may be underestimated because the KI mice did not have an obvious or characteristic EEG pattern. Thus, we were conservative in identifying tonic seizures since the EEG pattern was variable, and unless the mice were moving prior to the seizure, it was difficult to confirm a behavioural event.
GTCSs in humans are characterized by an abrupt seizure onset with the loss of consciousness, a high-amplitude high-frequency tonic phase and finally a lower frequency clonic phase characterized by myoclonic and clonic jerking. The EEG may also show background suppression interrupted by polyspike-wave discharges (Daroff, 2012; Seneviratne et al., 2017). The postictal state encompasses a gradual recovery of the background EEG, consciousness and physical movement. In KI mice, GTCSs started with limb stiffening and extension, followed by rapid clonic movements of the limbs, and proceeded to strong tonic–clonic activity with abrupt jumps and Straub tail, ending in postictal behavioural arrest (Fig. 2G). The GTCSs were mostly observed in older mice. The ictal EEG showed a high-amplitude, high-frequency discharge followed by a low-amplitude baseline rhythm, followed by a low-activity postictal state lasting up to several minutes.
In adult KI mice, atypical absence and myoclonic seizures were observed most frequently, while tonic and GTCSs were observed much less frequently (Fig. 2H). Typical (wild type) and atypical (KI) absence seizure occurrences were graphed together for visual comparison; however, the absence seizures in wild-type mice were 100% typical absence seizures (6–8 Hz), while the absence seizures in the KI mice were ∼97% low-frequency atypical absence seizures (3–5.5 Hz). Because a random sampling of absence seizures in KI mice yielded an average peak frequency of 4.4 Hz, despite the presence of a small percentage of higher-frequency (possibly typical) absence seizures, all absence seizures in KI mice have been grouped together as ‘atypical’ due to the overwhelming majority of these seizures, yielding a range in peak spectral frequency of 3–7 Hz. Atypical absence seizures in KI mice peaked at the light-to-dark transition (Fig. 2I), and KI mice had more atypical absence seizures overall during the dark hours (Fig. 2J). Finally, atonic seizures were observed extremely rarely and were difficult to verify, so they were not quantified; however, an example is available in Video 1.
In addition, while we could not implant our EEG head mounts on pups, blinded observations of young mice, aged P1–P21, showed epileptic spasms in KI mice from age P13 to P17 (Fig. 2K). Approximately 20% of patients with LGS had a diagnosis of IS prior to developing LGS (Shields, 2004), including the D120N patient. Spasms in humans last 0.5–2 s, with neck and trunk flexion and arm abduction in a jack-knife pattern, and they often occur in clusters (Daroff, 2012). In KI mice, spasms were brief, ∼1 s, with rapid flexion–extension movements accompanied by behavioural arrest, staring and loss of responsiveness. While a fraction of wild type pups were scored to have spasms (<30%) from age P13 to P17, all of the KI mice had spasms at that age range. In addition, we observed more seizures during pregnancy and nursing, suggesting that female hormones may affect the epilepsy phenotype (Tauboll et al., 2015). Taken together, the seizure semiologies and ictal EEGs were consistent with a diagnosis of LGS, representing that the first time the LGS seizure semiology has been faithfully replicated in a genetic model system.
Gabrb3+/D120N mice had impaired learning and memory, impaired social interaction, hyperactivity and elevated anxiety
Patients with LGS have behavioural abnormalities, which can include mildly to severely impaired cognition, attention-deficit hyperactivity disorder, ASD and general mood instability and conduct problems (Kerr et al., 2011). These conditions are commonly seen in other epilepsy syndromes (Hamiwka and Wirrell, 2009; Ettinger et al., 2015; Holmes, 2015) but are particularly impactful to patients with LGS and often dictate poor outcomes (Vignoli et al., 2017). We conducted a series of behavioural studies to assess whether the behaviour in KI mice recapitulated the abnormalities seen in humans with LGS (Figs 3 and 4). These tests were conducted in both male and female mice around two ages, juvenile to young adulthood (P30–P55, called P49) and mature adulthood (P180–P220, called P200).
Figure 3.

KI mice have spatial learning and memory deficits. The Barnes maze test demonstrated a delay in spatial learning and a deficit in spatial memory in the KI mice at both P49 and P200. Eight days of learning trials depict spatial learning abilities in P49 (A–C) and P200 (D–F) wild type and KI mice. Each day represents the average across all mice of each genotype with the data point of each mouse being the average of the four trials conducted that day. (A, D) The time it takes each animal (latency) to find the target hole for each day was plotted. (B, E) The number of non-target hole zones entered (errors) was plotted for each day. (C, F) Search strategy scores for the 8 days of learning trials were plotted for each day. Mice earned zero points for a direct path to the target hole, one point for a serial path and two points for a random path. Higher scores represented less efficient searching. (G–J) Five minute probe trial for spatial memory performed on Day 8 after the learning trials were plotted for (G, H) P49 and (I, J) P200 mice. The target hole was then covered and appeared identical to the other 11 holes. (G, I) Time spent near the target hole and (P49 wild type 26.25 ± 2.27 s, KI 18.44 ± 1.45 s; P200 wild type 26.50 ± 2.28 s, KI 22.27 ± 2.02 s). (H, J) Number of non-target hole zones entered (errors) (P49 wild type 64.14 ± 5.74, KI 87.10 ± 8.06; P200 wild type 66.36 ± 3.29, KI 102.1 ± 5.88). (A–F) Values expressed as mean ± SEM. (A–F) Two-way ANOVA for repeated measures: genotype effect #P < 0.05, ###P < 0.001, ####P < 0.0001. Bonferroni post-tests: *P < 0.05, **P < 0.01, ***P < 0.001. (G–J) Graphed values are expressed as median ± SEM. Unpaired two-tailed Student’s t-test, *P < 0.05, ***P < 0.001. n = 21–22 for all panels.
Impaired cognition is present in 80–90% of patients with LGS (Widdess-Walsh et al., 2013; McTague et al., 2016), and the patient in which the GABRB3 c. G358A mutation was identified had an adaptive test score of <20 (Epi et al., 2013). To assess cognitive deficiencies of KI mice, spatial learning and memory were assayed using the Barnes maze at P49 (Fig. 3A–C) and P200 (Fig. 3D–F) mice. In the first cohort, it was determined that for the KI mice to reach the same level of learning as their wild type littermates, 8 days (more than the standard of 5 days; Harrison et al., 2006) of learning trials were necessary. At P49, KI mice displayed a spatial learning deficit in the Barnes maze as measured by the latency to find the target hole (Fig. 3A) and the number of errors committed (non-target holes visited) before entering the target hole (Fig. 3B). In addition, P49 KI mice displayed a less effective search strategy overall (Fig. 3C), as well as having a persistently worse search strategy on the final day. P200 KI mice displayed a spatial learning deficit similar to that of the P49 cohort, with notable spatial learning delays (Fig. 3D–F). In the memory trial, P49 KI mice spent less time investigating the target hole area (Fig. 3G) and made an increased number of errors (Fig. 3H) showing that young KI mice have a spatial memory deficit. P200 KI mice showed a similar phenotype, with a trend towards a decrease in the amount of time spent investigating the target hole area (Fig. 3I), and a significant increase in the number of errors made (Fig. 3J). Together these data showed that both spatial learning and spatial memory deficits in KI mice began early in life and persisted across their lifespan.
Hyperactivity, one of the hallmark features of attention-deficit hyperactivity disorder (American Psychiatric Association Publishing, 2013) is one of the most common comorbidities diagnosed in patients with LGS (Hamiwka and Wirrell, 2009; Hancock and Cross, 2013). We assessed hyperactivity using locomotor activity chambers. At P49, KI mice had a mild hyperactive phenotype with an increased number of vertical counts (jumps in which the feet leave the floor, a phenotype often observed in the home cage, which we sought to quantify) (Fig. 4A), and a trend towards an increase in the total distance travelled in the chamber (a more traditional measure of hyperactivity) compared to wild-type mice (Fig. 4B). P200 mice displayed a more robust hyperactive phenotype, with significant increases in both parameters (Fig. 4C and D). Together, these data demonstrate that KI mice had hyperactivity that worsened with age.
Figure 4.

Gabrb3+/D120N KI mice exhibited hyperactivity, social deficits and anxiety. (A–D) KI mice had a mild hyperactive phenotype at P49, which became more pronounced at P200. Data shown were collected from 60 min in the locomotor activity chambers. (A) Vertical counts, or the number of rearings, detected at P49 were plotted (group means: wild type 293.6 ± 29.10, KI 417.0 ± 44.42). (B) Total distance travelled (centre and surround included) in the locomotor activity chambers at P49 (group means: wild type 49.03 ± 3.22 m, KI 58.71 ± 4.38 m). (C) Vertical counts at P200 (group means: wild type 373.3 ± 32.00, KI 527.5 ± 63.17). (D) Total distance travelled at P200 (group means: wild type 52.68 ± 3.97 m, KI 70.71 ± 7.09 m). Graphed values are expressed as median ± SEM. Unpaired two-tailed Student’s t-test, *P < 0.05. n = 20–25. (E–H) The 3CST showed social deficits in KI mice that begin at P49 and become more pronounced at P200. Not shown is Stage 1, a 10-min familiarization stage for the test mouse to acclimate to the set-up. (E, F) Stage 2 of the 3CST was a 10-min trial in which subject mice had two socialization options: (i) an empty inverted pencil cup in one side chamber or (ii) an inverted pencil cup containing a novel age-matched female mouse in the other side chamber. Two-way ANOVA was not significant for a genotype effect at P49 in E, and genotype effect was significant at P200 at P < 0.05 in F (group means: P49, novel mouse: wild type 1.89 ± 0.13 min, KI 1.86 ± 0.18 min; novel object: wild type 1.19 ± 0.08 min, KI 1.06 ± 0.14 min; total exploration: wild type 3.00 ± 0.24 min, KI 2.64 ± 0.18 min. P200, novel mouse: wild type 1.60 ± 0.17 min, KI 1.00 ± 0.14 min; novel object: wild type 0.79 ± 0.08 min, KI 0.76 ± 0.11 min; total exploration: wild type 2.39 ± 0.21 min, KI 1.73 ± 0.22 min). (G, H) Stage 3 of the 3CST was a 10-min trial in which test mice had two socialization options: (i) familiar socialization, in which the novel mouse from Stage 2 remained where she was or (ii) novel socialization, in which a new novel mouse was placed under the previously empty pencil cup. Two-way ANOVA showed a significant genotype effect P < 0.01 at both (G) P49 (group means: familiar mouse: wild type 1.11 ± 0.14 min, KI 0.72 ± 0.07 min; novel mouse: wild type 1.54 ± 0.12 min, KI 1.27 ± 0.14 min; total exploration: wild type 2.65 ± 0.21 min, KI 2.00 ± 0.17 min) and (H) p200 (group means: familiar mouse: wild type 0.89 ± 0.08 min, KI 0.82 ± 0.14 min; novel mouse: wild type 1.49 ± 0.19 min, KI 0.72 ± 0.09 min; total exploration: wild type 2.38 ± 0.21 min, KI 1.58 ± 0.17 min). Graphed values expressed as median ± SEM. Statistical tests shown on graphs are a priori Bonferroni post-tests after two-way ANOVAs. Four comparisons were made in each graph; however, only significant or otherwise relevant results were shown. Bonferroni multiple-comparison correction significance levels for E–H were: *P < 0.05, **P < 0.01, ***P < 0.001. n = 21–23. (I–L) KI mice displayed elevated anxiety in both the locomotor activity chambers and the elevated zero maze, with a mild anxiety phenotype at P49 and more pronounced anxiety at P200. (I, J) Each subject spent 60 min in the locomotor activity chamber. The chamber was divided into two 50% sections by area: the centre and the surround. Distance travelled within the surround (edge) area as a percentage of total distance travelled was shown for (I) P49 (group means: wild type 44.80 ± 1.92, KI 37.84 ± 1.67) and (J) P200 (group means: wild type 53.63 ± 2.49, KI 44.96 ± 2.08). (K, L) Each subject spent 5 min in the elevated zero maze. The distance travelled in the open arms at (K) P49 (group means: wild type 6.87 ± 0.43, KI 6.39 ± 0.33) and (L) P200 (group means: 5.82 ± 0.39, KI 4.386 ± 0.58). Data not shown: total distance travelled was not changed for the KI mice at either age in the zero maze. Graphed values are expressed as median ± SEM. Unpaired two-tailed Student’s t-test, for I–L, *P < 0.05, **P < 0.01. n = 22–28. 3CST: three-chamber socialization test.
While the percentage of patients with both LGS and ASD is not specifically known, the coincidence is often referenced (Hamiwka and Wirrell, 2009; Epi et al., 2013; Hancock and Cross, 2013), with 5–37% of patients with primary epilepsy also having an ASD diagnosis (Jeste and Tuchman, 2015) and 30–40% of patients with ASD having an epilepsy diagnosis (Pickett et al., 2011; Jacob, 2016). Therefore, an ASD-like phenotype may be an important attribute when phenotyping the KI mouse. One of the principal diagnostic criteria for ASD is altered social interaction (American Psychiatric Association Publishing, 2013), and so we tested socialization using the three-chamber socialization test. Independent of age, wild type littermate mice showed a preference for novel socialization over exploration of a novel object (Fig. 4E and F). P49 KI mice showed this preference as well (Fig. 4E); however, by P200, KI mice had reduced exploratory behaviour overall (total time spent investigating targets) and the preference for novel socialization over a novel object was lost (Fig. 4F). A wild type littermate mouse at both ages preferred socializing with the novel mouse over the familiar mouse (Fig. 4G and H). At P49, KI mice did show this preference, although there was a significant reduction in overall exploratory behaviour (Fig. 4G). P200 KI mice lost the preference for novel over familiar socialization and also had a reduction in overall exploration (Fig. 4H). Together, the three-chamber socialization test indicated that P49 mice had reduced socialization, which evolved into completely aberrant socialization by P200, at which time KI mice showed none of the social preferences of their wild type littermates.
The final behavioural comorbidity of LGS that we assessed was elevated anxiety (Fisher and Noble, 2017). To test for anxiety, we took data from two tests: the locomotor activity chambers and the elevated zero maze. When in the locomotor activity chambers, a more anxious mouse will spend more time on the perimeter of the chamber, avoiding the centre (called thigmotaxis) (Simon et al., 1994). At both P49 and P200, KI mice travelled less through the centre of the chamber (Fig. 4I and J). On the elevated zero maze, a more anxious mouse will spend more of its time in the closed arms, avoiding the open arms (Shepherd et al., 1994). At P49, KI mice did not have a significant change in the time spent in the open arms of the maze (Fig. 4K), while at P200, KI mice spent significantly less time in the open arms of the maze (Fig. 4L). Together, these data indicated that young KI mice had a mild anxiety phenotype that became worse with age.
Expression and trafficking of GABAA receptor α1, β3 and γ2 subunits were unchanged in brains of Gabrb3+/D120N mice
Having confirmed that KI mice had the seizure and behavioural phenotypes of LGS, we next examined the underlying mechanisms for the seizures. The majority of GABR epilepsy mutations produce subunits with impaired biogenesis, leading to altered cell surface expression (Macdonald and Kang, 2012). To determine if the effects of the mutant β3(D120N) subunits on GABAA receptor currents were related to its expression and trafficking in vivo, we assayed total expression in cerebellum (Ce), cortex (Co), hippocampus (Hi) and thalamus (Th) (Fig. 5A and B), surface expression in whole brain (Fig. 5C and D) and synaptosomal content (Fig. 5E and F) of GABAA receptor subunits but found no differences between KI and wild type littermate mice (see Methods section). These results demonstrated that mutant β3(D120N) subunits did not affect GABAA receptor biogenesis, trafficking or synaptic distribution and suggested that the mutant β3 subunits can effectively compete with wild type subunits to form intact, but functionally impaired, GABAA receptors in brain, which leads to decreased GABAergic inhibition and seizures.
Figure 5.

Biogenesis and trafficking of receptors containing mutant β3(D120N) subunits were unaltered in Gabrb3+/D120N KI mice. The mutant β3(D120N) subunits did not affect expression, surface trafficking or distribution in synaptosomes of either their own or other GABAA receptor subunits. (A) Whole-cell lysates from different brain regions [cortex (Co), cerebellum (Ce), hippocampus (Hi) and thalamus (Th)] were collected from KI and wild type littermate mice and subjected to SDS–PAGE and immunoblotted with anti-α1, -β3 and -γ2 subunit antibodies. GAPDH served as a loading control. (B) Band intensities of α1, β3 and γ2 subunits were normalized to GAPDH signal and, then, KI was compared with wild type (n = 3 KI: wild type pairs). One-way ANOVA followed by the Dunnett’s multiple-comparison test was used to determine significance; however, no changes were identified. (C) Plasma membrane proteins from mouse brain were isolated, analysed by SDS–PAGE and immunoblotted with anti-α1, -β3 and -γ2 subunit antibodies. Na+/K+ ATPase served as a loading control, and GAPDH served as cytosolic protein contamination control. (D) Band intensities of α1, β3 and γ2 subunits were normalized to ATPase signal and, then, KI bands were compared with wild type bands (n = 3 KI: wild type pairs). (E) Proteins from synaptosomes were analysed by SDS–PAGE and immunoblotted with anti-α1, -β3 and -γ2 subunit and gephyrin antibodies. Na+/K+ ATPase served as a loading control. The distribution of GABAA receptor subunits was not changed in KI or wild type littermate mice. (F) Band intensities of α1, β3 and γ2 subunits and gephyrin were normalized to the ATPase signal and, then, KI bands were compared with wild type bands (n = 3 KI: wild type pairs). Student’s t-tests were used in D and F with a significance level of α = 0.05. GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
mIPSCs recorded from SS cortex layer V/VI pyramidal neurons of Gabrb3+/D120N mice had reduced amplitude
During brain development, β3 subunits are a major constituent of GABAA receptors and are widely expressed in the brain, especially highly in the cerebral cortex, amygdala, olfactory bulb, nucleus reticularis of the thalamus and hippocampus (Hortnagl et al., 2013). Our previous in vitro results showed that the mutant subunits significantly reduced peak amplitude and altered kinetic properties of α1β3(D120N)γ2 GABAA receptor currents in transfected HEK293T cells (Janve et al., 2016). We determined whether the D120N amino acid substitution affected the functional properties of GABAergic inhibitory interneuron synapses on cortical layer V/VI pyramidal neurons. We targeted somatosensory cortex layer V/VI pyramidal neurons as they participate in the thalamocortical feedback loop (Ledergerber and Larkum, 2010). Using the whole-cell patch-clamp technique and acute coronal thalamocortical slices, we recorded mIPSCs in the pyramidal neurons from wild type littermate and KI mice (Fig. 6A and B). In KI mice, we observed decreased GABAA receptor-mediated mIPSC amplitude (Fig. 6C–E) and increased mIPSC decay time constant (Fig. 6F), with no change to mIPSC frequency. Consistent with the decrease in mIPSC amplitudes, mIPSC charge transfer was also decreased in neurons from het KI mice compared with that from wild-type mice (wild type 617.12 ± 89.89 pA ms, n = 5 mice; KI 363.14 ± 38.04 pA ms, n = 5 mice; Student’s t-test P = 0.040). The reduced current amplitudes and charge transfer of mutant GABAA receptors containing β3(D120N) subunits in layer V/VI somatosensory cortical pyramidal neurons would be predicted to decrease interneuron phasic GABAergic inhibition and lower the seizure threshold by promoting neuronal hyperexcitability, resulting in epilepsy.
Figure 6.

mIPSCs recorded from KI mouse SS cortex layer V/VI neurons were altered. mIPSCs were recorded from SS cortex layer V/VI neurons (voltage-clamped at −60 mV with equal chloride concentration inside and outside cells) in thalamocortical slices from littermate (A) wild type and (B) KI mice. (C) Normalized mIPSC events for wild type and KI mice showed the frequency of mIPSCs in each amplitude bin. (D) Normalized cumulative distribution was plotted for wild type and KI mouse mIPSCs. Compared with mIPSCs recorded from wild type littermate mice, those from SS cortex layer V/VI neurons in KI mice had (E) significantly reduced amplitudes (wild type −49.67 ± 2.71 pA; KI −31.48 ± 1.76 pA) and (F) slowed mIPSC decay (wild type 25.05 ± 2.87 ms; KI 39.13 ± 2.72 ms). wild type, n = 6 mice; KI, n = 5 mice. Student’s t-test, **P < 0.01, ***P < 0.001. SS: somatosensory.
Thalamocorticothalamic oscillations recorded from VBn neurons of Gabrb3+/D120N mice had prolonged duration
Excitatory–inhibitory balance critically regulates cortical network function and activity, and an imbalance is the basis for pathogenesis for seizures and other neuropsychiatric disorders (McCormick and Contreras, 2001; Magloczky and Freund, 2005; Marin, 2012; Penzes et al., 2013). In addition, as atypical absence seizures with SSWD are the predominant seizure type in LGS KI mice, and thalamocortical hypersynchrony is known to be the primary cause of SWD (Schofield et al., 2009), we asked whether mutant β3(D120N) subunits affected neural network activity at the cellular level in the thalamocortical circuit. Spontaneous thalamocortical network oscillatory activity was measured in the thalamic ventrobasal nucleus in horizontal slices from wild type littermate and KI mice that retained the thalamocortical circuitry (Fig. 7A and B). Multi-unit recordings from thalamic ventrobasal nucleus in slices from KI mice exhibited prolonged and high amplitude, spontaneous network oscillations compared with the very short, spontaneous network bursts in thalamocortical slices from wild type littermate mice (Fig. 7C), consistent with the notion that excessive thalamocortical synchronization and hyperexcitability occur during seizures, particularly absence seizures. However, these results do not exclude a role for other brain areas such as hippocampus from participating in the spontaneous oscillations.
Figure 7.

Spontaneous thalamocortical network oscillations from KI mice were longer and more frequent than those from wild type littermate mice. Representative extracellular multiple unit recordings from VBn in slices showing (A) spontaneous brief thalamocortical bursts in wild-type mice and (B) spontaneous prolonged thalamocortical oscillations in KI mice. One short burst from a wild type and one oscillation from a KI mouse were expanded in each panel to show the multiple spikes in the burst or oscillation. Scale bars are as indicated. (C) Average duration of spontaneous bursts for P42 wild-type mice (0.78 ± 0.71 s, n = 6 mice) and spontaneous oscillations from P42 KI mice (6.88 ± 1.39 s, n = 9 mice) were plotted (**P < 0.01, Student’s t-test). VBn: thalamic ventrobasal nucleus.
AED treatment in Gabrb3+/D120N mice reflects efficacy in patients with LGS
To further assess the validity of the Gabrb3+/D120N mouse as an LGS model, we evaluated the effectiveness of several AEDs used to treat LGS in the KI mice, ethosuximide, clobazam and topiramate. Ethosuximide is the primary drug of choice for the treatment of absence seizures (Vrielynck, 2013) and is also used in LGS treatment when other treatments are ineffective, or as an adjunct to other AEDs to increase efficacy (Gomez and Klass, 1983; Schmidt and Bourgeois, 2000). Ethosuximide can also reduce atypical absence seizure occurrence, and in some patients, it may reduce tonic and myoclonic seizures as well (Schmidt and Bourgeois, 2000; van Rijckevorsel, 2008). Clobazam and topiramate are commonly used for the treatment of LGS (Schmidt and Bourgeois, 2000). In addition, as cannabidiol has been shown recently to be effective in treatment-resistant epilepsies such as LGS (Devinsky et al., 2016; O’Connell et al., 2017), we assessed the effectiveness of the cannabinoid receptor 1-specific agonist WIN 55,212-2 for the treatment of LGS seizures. While these drugs are used in the treatment of LGS, the AEDs sometimes have severe side effects. These AEDs are often not efficacious in every patient or may require a specific combination of several drugs unique to each patient to achieve efficacy (Schmidt and Bourgeois, 2000), making drug therapy difficult. We sought to determine whether any of these drugs were efficacious in the KI mice.
For the evaluation of AED efficacy (Fig. 8A and B), KI mice were injected with solvent and video-EEG was recorded for 2 h to establish seizure baseline. Seizure time and count were found to be stable during these 2 h, so an average of ‘per hour’ was used for baseline for each animal. Mice were then injected with the AED dissolved in solvent, and another 2 h of video-EEG were recorded. These 2 h were split into 1 h bins as AEDs have a plasma half-life that is shorter in mice than that in humans, often as short as 1 h. Here, we quantified only atypical absence seizure frequency and duration as other seizures were less frequent and may not provide useful information for the short experimental protocol used here such as this. Two AEDs, ethosuximide and clobazam, showed main effects by one-way ANOVA. Bonferroni post-tests indicated a significant decrease in cumulative atypical absence seizure duration in the hour after injection for both drugs (Fig. 8A). Ethosuximide resulted in a decrease in atypical absence seizure time of 94%, and clobazam resulted in a decrease of 73%. Clobazam showed a full return to baseline by the second hour after injection, while ethosuximide showed a partial return to baseline. Ethosuximide had a similar effect on seizure count as it did on cumulative duration whereas clobazam had no effect on count (Fig. 8B), indicating that mice had shorter seizures after dosing with clobazam. Topiramate was not effective in reducing atypical absence seizure cumulative time or count. Cannabinoids may require a long-term dosing strategy to achieve an effect; therefore, WIN 55,212-2 was given to mice daily for 7 days and cumulative atypical absence seizure duration and count within a 2-h window on Day 8 were compared with the baseline state for each mouse recorded on Day 1 (Fig. 8C and D). Administration of WIN 55,212-2 produced a decrease by 75% in the time spent in atypical absence seizures as well as a reduction in the number of these seizures.
Figure 8.

Ethosuximide, clobazam and a cannabinoid, but not topiramate, reduced atypical absence seizures in KI mice. (A, B) Three AEDs were administered individually to KI mice. Total observation period by video-EEG lasted 4 h. At Hour 0, an injection of each drug’s respective vehicle was given as a control observation. Vehicle and vehicle washout were found to be stable and were therefore averaged as a baseline. Drug + vehicle were administered at Hour 2. The 2 h after the drugs were administered were split into single hour observations: the first hour after administration and the second hour after administration. A one-way ANOVA was performed on each drug individually (shown above each drug set), and Tukey post-tests were conducted comparing all three pairs of time points (shown on bar graphs where significant). (A) Both ethosuximide and clobazam, but not topiramate, significantly reduced the cumulative time spent in atypical absence seizures. (B) Ethosuximide resulted in a significant reduction in the number of atypical absence seizures, while clobazam and topiramate did not. n = 8–9, 6–8 months old, males and females. Repeated-measures one-way ANOVA #P < 0.05; Tukey post-tests adjusted P-value *P < 0.05 compares drug to baseline, @P < 0.05 compares drug to drug washout. (C, D) Win 55,212-2 was administered daily for 1 week. KI mice had a significant reduction in both (C) cumulative time spent in atypical absence seizures and (D) atypical absence seizure count after 1 week of dosing. n = 6, paired Student’s t-test, 5–8 months old, males and females, *P < 0.05, **P < 0.01.
Discussion
The Gabrb3+/D120N KI mouse is a model of human LGS
LGS is a severe form of early-onset epileptic encephalopathy, which has multiple genetic and symptomatic structural causes. LGS is characterized by seizures with multiple semiologies, a generalized SSWD pattern on EEG, and cognitive and behavioural abnormalities (Trevathan, 2002). In a given case of genetic LGS, it is unclear whether the multiple seizures and behavioural abnormalities are caused by mutations of several genes and varying genetic background or are the result of a single mutation in a key gene. To answer this question, it would be useful to have an animal model of LGS caused by a single-gene mutation and with a uniform genetic background and to determine if the animal model develops the multiple seizures and behavioural comorbidities.
The GABRB3 (c.G358A, p.D120N) mutation was shown to be associated with LGS by its presence only in the proband of an LGS triad, but its causal role in the development of LGS can be established directly by generating a mouse model with the corresponding G358A KI allele. Generation of a genetic mouse model with a human LGS mutation displaying the LGS phenotype is an important advance for understanding the pathophysiology of LGS and for the potential development of new treatments for LGS and associated behavioural abnormalities. The Gabrb3+/D120N KI mouse generated for this study recapitulated both the occurrence and semiologies of LGS seizures (spontaneous atypical absence, generalized tonic-clonic, myoclonic and tonic seizures with appropriate ictal EEG correlates) and the behavioural phenotypes (cognitive disability, hyperactivity, social deficits and anxiety) seen in both the proband and broadly in patients with LGS. Together, these results show that we have generated a novel mouse model for LGS based on a human genetic mutation.
Gabrb3+/D120N mice had seizure semiologies like those of patients with LGS
Home-cage monitoring revealed that KI mice had modest developmental delay in the infantile period, and video-EEG monitoring in adult KI mice showed atypical absence and myoclonic seizures as the most prominent seizure types, with tonic seizures being observed less frequently, although all of these seizures were observed throughout adulthood. GTCSs were observed infrequently and were seen more often in older mice (>6 months of age). In addition, we observed spontaneous epileptic spasms in young KI mice between P13 and P17. These flexion–extension spasms, accompanied by behavioural arrest, staring and loss of responsiveness, were similar to spasms reported in models of IS (Dulla, 2018), a disorder whose diagnosis often precedes the diagnosis of LGS, as it did in the patient bearing the D120N mutation. Seizure semiologies observed at all ages in the KI mice were consistent with those of LGS and demonstrated a worsening of seizure semiology from an early-life onset to late-life persistence. It is worth noting that while tonic seizures are observed in the majority of the patients with LGS (Crumrine, 2011), they were infrequently observed in the KI mice. This could be the result of scoring difficulties, or due to different underlying genetics or genetic modifiers in patients with idiopathic LGS compared with our inbred KI mice. Nonetheless, the KI mice recapitulated LGS-associated seizures.
Patients with LGS are reported to have atypical absence seizures (1–2.5 Hz SSWDs) (Gastaut et al., 1966). There are several rodent models of typical absence seizures (6–8 Hz), but atypical absence seizures have been reported only after administration of AY-9944 (a cholesterol biosynthesis inhibitor) to young rats (Cortez et al., 2001) or by overexpressing the GABABR1a receptor in mice (Stewart et al., 2009), neither of which reflect human genetics or result in a complete LGS phenotype. Atypical absence seizures are a hallmark symptom of LGS and are thought to contribute significant cognitive impairment (Gastaut et al., 1966) that is more severe than that seen in patients with typical absence seizures. This KI mouse offers an opportunity to identify neural circuits underpinning atypical absence seizures and to determine if worsening of seizures with age can be prevented by blocking them as soon as they are observed.
Gabrb3+/D120N mice exhibited behavioural comorbidities like those of patients with LGS
LGS is known to have several comorbidities, including cognitive disability, attention-deficit hyperactivity disorder, ASD and anxiety. Treatment of LGS necessitates not only a reduction in seizures but also management of the complex behavioural abnormalities that accompany the disorder (Kerr et al., 2011; Archer et al., 2014). Thus, the KI mice were assessed for comorbid conditions seen in patients with LGS. Because onset of LGS occurs early in life, but often persists throughout adulthood, mice were studied at young adult (P49) and older adult (P200) ages. While the Barnes maze test is traditionally thought to require hippocampal processing, evidence suggests that the cortex is also required for spatial navigation and spatial memory formation (Negron-Oyarzo et al., 2018). In addition, although we have largely studied the thalamocortical circuit in this study, which is the primary circuit involved in typical absence seizures (Bomben et al., 2016; Barad et al., 2017), at least one model of atypical absence seizures indicated that the hippocampus is also involved (Vrielynck, 2013). We decided that the Barnes maze test would be an effective low-stress method of assessing cognitive ability in this model. Both adolescent and adult KI mice displayed deficits in spatial learning and spatial memory as assessed in the Barnes maze test, consistent with the involvement of hippocampus in atypical absence seizures.
Social impairment is a hallmark symptom of ASD, a condition commonly associated with epilepsy (Hamiwka and Wirrell, 2009; Epi et al., 2013; Hancock and Cross, 2013), and in the three-chamber socialization test, KI mice were shown to have mild social impairment in adolescence that evolved into a profound social deficit by adulthood. Because Gabrb3 is highly expressed in the olfactory bulb (Hortnagl et al., 2013) and because olfactory cues are important for socialization in mice (Arakawa et al., 2008), we tested for differences in olfaction between wild type littermate and KI mice. We did not find any olfactory deficits in KI mice (data not shown).
KI mice were also more hyperactive than their wild type littermates, a phenotype that reflects human attention-deficit hyperactivity disorder and which began in adolescence and worsened with age. Finally, while adolescent KI mice showed a mild anxiety phenotype, adult KI mice were substantially more anxious. While we assessed motor deficiencies using Rotorod and TreadScan tests and for depression using the tail suspension test, we did not find significant effects in any of these tests (data not shown). Together, these comorbidities recapitulate the symptoms observed in many patients with LGS. Of note, while it is possible that some of the comorbid conditions may impact each other, there is no evidence that any one of the conditions explains any other condition. The data suggest that each abnormality is a distinct condition associated with the LGS mutation.
Cortical layer V/VI neurons in thalamocortical slices from Gabrb3+/D120N mice showed decreased inhibition
After establishing that the KI mice recapitulated the salient features of LGS, the molecular mechanism of the mutation was assessed. Our previous work in vitro demonstrated that there was no change in assembly or trafficking of α1β3(D120N)γ2 GABAA receptors in transfected HEK293T cells (Janve et al., 2016). Here, using western blots of wild type littermate and KI mouse brain tissue, the expression and trafficking of β3(D120N) subunits were confirmed to be normal in brain in vivo. We saw no evidence for a decrease in the presence of β3 subunits in assembled receptors or in the overall expression of β3 subunits and β2 subunits have been shown to be unable to substitute for β3 subunits as their expression is generally discrete (Homanics et al., 1997), so we do not predict a change in receptor composition, quantity or localization. Previously, whole-cell recordings from HEK293T cells transfected with α1β3(D120N)γ2 GABAA receptor subunits showed a decrease in current amplitude with 1 mM GABA applied and, single-channel recordings from transfected HEK293T cells showed decreased open probability, opening frequency and burst duration with 1 mM GABA applied (Janve et al., 2016). In adult mice, β3 subunits are highly expressed in cortical layers V/VI, which plays a role in thalamocortical oscillations. We recorded mIPSCs from somatosensory cortical layer V/VI pyramidal cells that result from the release of GABAergic vesicles from local interneurons. In KI mice, the amplitudes of mIPSCs were reduced 37% and the decay times were increased 53% with no change in frequency, and an overall reduction in net charge transfer of 44%. These results suggest a change in post-synaptic function without a change in presynaptic neurotransmitter release (Choi and Lovinger, 1997), which is consistent with our finding that surface levels and synaptic quantity of β3(D120N) subunits were unchanged by the mutation.
Network disturbances in Gabrb3+/D120N mice provide opportunities for discovery
The above data suggested that network wiring resulted in similar patterns of GABAergic synaptic inputs in KI as in wild type littermate mice, and the changes we report were due instead to dysfunction of post-synaptic GABAA receptors. Because the GABAA receptor β3 subunit is highly expressed in selective locations in the brain such as the cortex where we observed substantial mIPSC changes, the β3 subunit D120N substitution had profound effects on brain function. These data indicate that a reduction in interneuronal inhibition likely is the cause of the LGS. Generation of absence seizures is known to involve both cortex and thalamus; however, there is conflicting evidence about whether absence seizures occur due to an increase or a decrease in activity in thalamocortical activity (Blumenfeld, 2005). Our data suggest that for atypical absence seizures, this is due to an increase in network activity in the LGS mouse. GTCS also are associated with increases in cortical activity (Blumenfeld, 2005), among other regions, but the networks involved in tonic, atonic, clonic and myoclonic seizures are more complicated and less well understood. This mouse offers the opportunity to further study and understand the networks involved in these types of seizures. Of particular value, a single-point mutation observed in a human with LGS was able to recapitulate both the seizure and behavioural comorbidity profiles seen in this complex disorder when expressed in this rodent model. This fact also indicates that the circuitry involved in the LGS seizure profile overlaps with that which is involved in the observed comorbidities. Therefore, future work may elucidate the connections between seizure circuitry and behavioural circuitry.
Gabrb3+/D120N mice fill a gap in animal models and will be useful for the development of novel therapies for the treatment of LGS
The Epi4k consortium also discovered two de novo mutations in dynamin 1 (DNM1) thought to be causative of LGS in patients (Epi et al., 2013), and the fitful mouse containing a spontaneous murine Dnm1 mutation has been reported to have seizures. While this mouse is certainly a useful model, the fitful mouse was not derived from a human mutation and exhibits only a partial set of the seizure profile observed in patients (Asinof et al., 2015). In addition, the fitful mouse has a limited expression of human comorbidities, with no observed social deficits, and cognitive tests showed either no deficit or were confounded by severe hyperactivity. Separately, Gabrb3 knock-out mice were shown to recapitulate some of the features of Angelman syndrome, including a subset of the common seizure and comorbid phenotypes of Angelman syndrome patients (DeLorey et al., 1998; Fiumara et al., 2010). This does not appear to be an LGS-like phenotype, suggesting that LGS in the KI mouse is not caused by haploinsufficiency only.
In addition to GABRB3, Epi4k identified nine mutations in the sodium channel gene SCN1A alone and mutations in 16 other genes thought to be causative of LGS (Epi et al., 2013). These genes include five of the seven genes aside from GABRB3 that the National Institutes of Health lists as having mutations associated with LGS (2020). Our KI mice mirror the seizure types and behavioural comorbid phenotypes associated with LGS. Mechanistically, LGS was shown to occur at least in part via a reduction in mIPSC amplitude in cortical layer V/VI pyramidal neurons and development of thalamocortical oscillations. LGS is a disorder that is notoriously difficult to treat, and seizures are poorly controlled with AEDs. The need for novel therapeutic strategies is great. In the KI mice, we found that ethosuximide, clobazam and a cannabinoid receptor 1 agonist were effective at reducing atypical absence seizures, the most-frequent seizure type observed in KI mice. However, topiramate was not effective and none of the drugs were effective at stopping all seizure activity. While the KI mouse represents one mutation in one gene that is causative for LGS, we believe that the strong similarity between the mouse and the human seizure types and behavioural phenotypes will enable future studies of the mechanisms of and treatments for LGS. Of further interest, 20% of patients with LGS have a history of prior IS (Shields, 2004). The proband patient in which the GABRB3 G358A mutation was discovered had a diagnosis of IS prior to the LGS diagnosis (Epi et al., 2013), and here, we report epileptic spasms in young LGS KI mice. The LGS KI mice will also serve as a tool to elucidate the changes that occur between infancy and the juvenile period, on to adulthood, which may reflect this progression from IS to LGS and the age-related worsening of LGS, offering another tool for future therapeutic exploration.
Supplementary Material
Acknowledgements
We would like to thank the Vanderbilt University Mouse Neurobehavioral Lab, particularly Dr John Allison, for assistance with mouse video-EEG and behavioural studies. We would also like to thank Laurel G. Jackson for her generous help finalizing this manuscript.
Funding
This work was funded by the National Institutes of Health NS33300 and NS096483.
Competing interests
The authors report no competing interests.
Glossary
- AEDs =
antiepileptic drugs
- ASD =
autism spectrum disorder
- D120N =
aspartic acid residue at Position 120 is substituted with an asparagine
- GABAA =
GABA receptor type A
- GTCSs =
generalized tonic–clonic seizures
- IS =
infantile spasms
- KI =
knock-in
- LGS =
Lennox–Gastaut syndrome
- mIPSCs =
miniature inhibitory post-synaptic currents
- SSWD =
slow spike wave discharge
- SWD =
spike wave discharge
References
- Abu Saleh T, Stephen L.. Lennox Gastaut syndrome, review of the literature and a case report. Head Face Med 2008; 4: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arain FM, Boyd KL, Gallagher MJ.. Decreased viability and absence-like epilepsy in mice lacking or deficient in the GABAA receptor alpha1 subunit. Epilepsia 2012; 53: e161–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa H, Arakawa K, Blanchard DC, Blanchard RJ.. A new test paradigm for social recognition evidenced by urinary scent marking behavior in C57BL/6J mice. Behav Brain Res 2008; 190: 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer JS, Warren AE, Jackson GD, Abbott DF.. Conceptualizing Lennox-Gastaut syndrome as a secondary network epilepsy. Front Neurol 2014; 5: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asinof SK, Sukoff Rizzo SJ, Buckley AR, Beyer BJ, Letts VA, Frankel WN, et al. Independent neuronal origin of seizures and behavioral comorbidities in an animal model of a severe childhood genetic epileptic encephalopathy. PLoS Genet 2015; 11: e1005347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association Publishing. Diagnostic and statistical manual of mental disorders, 5th edn.Arlington, VA: American Psychiatric Publishing; 2013. [Google Scholar]
- Barad Z, Grattan DR, Leitch B.. NMDA receptor expression in the thalamus of the stargazer model of absence epilepsy. Sci Rep 2017; 7: 42926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H. Consciousness and epilepsy: why are patients with absence seizures absent? Prog Brain Res 2005; 150: 271–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomben VC, Aiba I, Qian J, Mark MD, Herlitze S, Noebels JL.. Isolated P/Q calcium channel deletion in layer VI corticothalamic neurons generates absence epilepsy. J Neurosci 2016; 36: 405–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter MD, Shah CR, Muller CL, Crawley JN, Carneiro AM, Veenstra-VanderWeele J.. Absence of preference for social novelty and increased grooming in integrin beta3 knockout mice: initial studies and future directions. Autism Res 2011; 4: 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Lovinger DM.. Decreased frequency but not amplitude of quantal synaptic responses associated with expression of corticostriatal long-term depression. J Neurosci 1997; 17: 8613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez MA, McKerlie C, Snead OC 3rd. A model of atypical absence seizures: EEG, pharmacology, and developmental characterization. Neurology 2001; 56: 341–9. [DOI] [PubMed] [Google Scholar]
- Crumrine PK. Management of seizures in Lennox-Gastaut syndrome. Paediatr Drugs 2011; 13: 107–18. [DOI] [PubMed] [Google Scholar]
- Daroff RB. Bradley’s neurology in clinical practice , 6th edn In: Jankovic J, Mazziotta JC, Pomeroy SL, editors. Philadelphia, PA: Elsevier/Saunders; 2012. [Google Scholar]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, et al. Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci 1998; 18: 8505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Marsh E, Friedman D, Thiele E, Laux L, Sullivan J, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol 2016; 15: 270–8. [DOI] [PubMed] [Google Scholar]
- Dulla CG. Utilizing animal models of infantile spasms. Epilepsy Curr 2018; 18: 107–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi KCEpilepsy Phenome/Genome ProjectAllen AS, Berkovic SF, Cossette P, Delanty N, et al. De novo mutations in epileptic encephalopathies. Nature 2013; 501: 217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger AB, Ottman R, Lipton RB, Cramer JA, Fanning KM, Reed ML.. Attention-deficit/hyperactivity disorder symptoms in adults with self-reported epilepsy: results from a national epidemiologic survey of epilepsy. Epilepsia 2015; 56: 218–24. [DOI] [PubMed] [Google Scholar]
- EuroEPINOMICS-RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium. De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 2014; 95: 360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher PL, Noble AJ.. Anxiety and depression in people with epilepsy: the contribution of metacognitive beliefs. Seizure 2017; 50: 153–9. [DOI] [PubMed] [Google Scholar]
- Fiumara A, Pittala A, Cocuzza M, Sorge G.. Epilepsy in patients with Angelman syndrome. Ital J Pediatr 2010; 36: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada M, Hanai A, Nakayama A, Suzuki T, Miyata N, Rodriguiz RM, et al. Loss of deacetylation activity of Hdac6 affects emotional behavior in mice. PLoS One 2012; 7: e30924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastaut H, Roger J, Soulayrol R, Saint-Jean M, Tassinari CA, Regis H, et al. Epileptic encephalopathy of children with diffuse slow spikes and waves (alias “petit mal variant”) or Lennox syndrome. Ann Pediatr (Paris) 1966; 13: 489–99. [PubMed] [Google Scholar]
- Gohma H, Kuramoto T, Matalon R, Surendran S, Tyring S, Kitada K, et al. Absence-like and tonic seizures in aspartoacylase/attractin double-mutant mice. Exp Anim 2007; 56: 161–5. [DOI] [PubMed] [Google Scholar]
- Gomez MR, Klass DW.. Epilepsies of infancy and childhood. Ann Neurol 1983; 13: 113–24. [DOI] [PubMed] [Google Scholar]
- Hamiwka LD, Wirrell EC.. Comorbidities in pediatric epilepsy: beyond “just” treating the seizures. J Child Neurol 2009; 24: 734–42. [DOI] [PubMed] [Google Scholar]
- Hancock EC, Cross JH.. Treatment of Lennox-Gastaut syndrome. Cochrane Database Syst Rev, Issue 2, 2013; CD003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison FE, Reiserer RS, Tomarken AJ, McDonald MP.. Spatial and nonspatial escape strategies in the Barnes maze. Learn Mem 2006; 13: 809–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GL. Cognitive impairment in epilepsy: the role of network abnormalities. Epileptic Disord 2015; 17: 101–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, et al. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci USA 1997; 94: 4143–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortnagl H, Tasan RO, Wieselthaler A, Kirchmair E, Sieghart W, Sperk G.. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience 2013; 236: 345–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Zhou C, Tian M, Kang JQ, Shen W, Verdier K, et al. Overexpressing wild-type gamma2 subunits rescued the seizure phenotype in Gabrg2+/Q390X Dravet syndrome mice. Epilepsia 2017; 58: 1451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA.. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J Neurosci 1994; 14: 5485–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J. Cortical interneuron dysfunction in epilepsy associated with autism spectrum disorders. Epilepsia 2016; 57: 182–93. [DOI] [PubMed] [Google Scholar]
- Janve VS, Hernandez CC, Verdier KM, Hu N, Macdonald RL.. Epileptic encephalopathy de novo GABRB mutations impair GABAA receptor function. Ann Neurol 2016; 79: 806–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarre G, Guillemain I, Deransart C, Depaulis A.. Genetic models of absence epilepsy in rats and mice In: Models of seizures and epilepsy. 2nd edn. Elsevier; 2006. pp. 455–71. [Google Scholar]
- Jeste SS, Tuchman R.. Autism spectrum disorder and epilepsy: two sides of the same coin? J Child Neurol 2015; 30: 1963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JQ, Shen W, Zhou C, Xu D, Macdonald RL.. The human epilepsy mutation GABRG2(Q390X) causes chronic subunit accumulation and neurodegeneration. Nat Neurosci 2015; 18: 988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr M, Kluger G, Philip S.. Evolution and management of Lennox-Gastaut syndrome through adolescence and into adulthood: are seizures always the primary issue? Epileptic Disord 2011; 13 (Suppl 1): S15–26. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH.. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. J Neurosci 1992; 12: 4151–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledergerber D, Larkum ME.. Properties of layer 6 pyramidal neuron apical dendrites. J Neurosci 2010; 30: 13031–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennox–Gastaut Syndrome. 2020. https://ghr.nlm.nih.gov/condition/lennox-gastaut-syndrome (24 March 2020, date last accessed).
- Letts VA, Beyer BJ, Frankel WN.. Hidden in plain sight: spike-wave discharges in mouse inbred strains. Genes Brain Behav 2014; 13: 519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ.. mRNA surveillance and endoplasmic reticulum quality control processes alter biogenesis of mutant GABAA receptor subunits associated with genetic epilepsies. Epilepsia 2012; 53 (Suppl 9): 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Kang JQ, Gallagher MJ.. Mutations in GABAA receptor subunits associated with genetic epilepsies. J Physiol 2010; 588: 1861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magloczky Z, Freund TF.. Impaired and repaired inhibitory circuits in the epileptic human hippocampus. Trends Neurosci 2005; 28: 334–40. [DOI] [PubMed] [Google Scholar]
- Marin O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci 2012; 13: 107–20. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D.. On the cellular and network bases of epileptic seizures. Annu Rev Physiol 2001; 63: 815–46. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Buendia MA, Saborido TP, Palubinsky AM, Stankowski JN, Stanwood GD.. Haploinsufficiency of the E3 ubiquitin ligase C-terminus of heat shock cognate 70 interacting protein (CHIP) produces specific behavioral impairments. PLoS One 2012; 7: e36340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTague A, Howell KB, Cross JH, Kurian MA, Scheffer IE.. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol 2016; 15: 304–16. [DOI] [PubMed] [Google Scholar]
- Mekada K, Abe K, Murakami A, Nakamura S, Nakata H, Moriwaki K, et al. Genetic differences among C57BL/6 substrains. Exp Anim 2009; 58: 141–9. [DOI] [PubMed] [Google Scholar]
- Negron-Oyarzo I, Espinosa N, Aguilar-Rivera M, Fuenzalida M, Aboitiz F, Fuentealba P.. Coordinated prefrontal-hippocampal activity and navigation strategy-related prefrontal firing during spatial memory formation. Proc Natl Acad Sci USA 2018; 115: 7123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell BK, Gloss D, Devinsky O.. Cannabinoids in treatment-resistant epilepsy: a review. Epilepsy Behav 2017; 70: 341–8. [DOI] [PubMed] [Google Scholar]
- Penzes P, Buonanno A, Passafaro M, Sala C, Sweet RA.. Developmental vulnerability of synapses and circuits associated with neuropsychiatric disorders. J Neurochem 2013; 126: 165–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickett J, Xiu E, Tuchman R, Dawson G, Lajonchere C.. Mortality in individuals with autism, with and without epilepsy. J Child Neurol 2011; 26: 932–9. [DOI] [PubMed] [Google Scholar]
- Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, et al. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet 2000; 25: 139–40. [DOI] [PubMed] [Google Scholar]
- Schmidt D, Bourgeois B.. A risk-benefit assessment of therapies for Lennox-Gastaut syndrome. Drug Saf 2000; 22: 467–77. [DOI] [PubMed] [Google Scholar]
- Schofield CM, Kleiman-Weiner M, Rudolph U, Huguenard JR.. A gain in GABAA receptor synaptic strength in thalamus reduces oscillatory activity and absence seizures. Proc Natl Acad Sci USA 2009; 106: 7630–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seneviratne U, Cook MJ, D’Souza WJ.. Electroencephalography in the diagnosis of genetic generalized epilepsy syndromes. Front Neurol 2017; 8: 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen D, Hernandez CC, Shen W, Hu N, Poduri A, Shiedley B, et al. De novo GABRG2 mutations associated with epileptic encephalopathies. Brain 2017; 140: 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JK, Grewal SS, Fletcher A, Bill DJ, Dourish CT.. Behavioural and pharmacological characterisation of the elevated “zero-maze” as an animal model of anxiety. Psychopharmacology 1994; 116: 56–64. [DOI] [PubMed] [Google Scholar]
- Shields WD. Diagnosis of infantile spasms, Lennox-Gastaut syndrome, and progressive myoclonic epilepsy. Epilepsia 2004; 45 (Suppl 5): 2–4. [DOI] [PubMed] [Google Scholar]
- Simon P, Dupuis R, Costentin J.. Thigmotaxis as an index of anxiety in mice. Influence of dopaminergic transmissions. Behav Brain Res 1994; 61: 59–64. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Keist R, Rudolph U, Huguenard JR.. Dynamic GABA(A) receptor subtype-specific modulation of the synchrony and duration of thalamic oscillations. J Neurosci 2003; 23: 3649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart LS, Wu Y, Eubanks JH, Han H, Leschenko Y, Perez Velazquez JL, et al. Severity of atypical absence phenotype in GABAB transgenic mice is subunit specific. Epilepsy Behav 2009; 14: 577–81. [DOI] [PubMed] [Google Scholar]
- Striano P, Belcastro V.. Treatment of myoclonic seizures. Expert Rev Neurother 2012; 12: 1411–7; quiz 8. [DOI] [PubMed] [Google Scholar]
- Sugihara I, Lang EJ, Llinas R.. Serotonin modulation of inferior olivary oscillations and synchronicity: a multiple-electrode study in the rat cerebellum. Eur J Neurosci 1995; 7: 521–34. [DOI] [PubMed] [Google Scholar]
- Tauboll E, Sveberg L, Svalheim S.. Interactions between hormones and epilepsy. Seizure 2015; 28: 3–11. [DOI] [PubMed] [Google Scholar]
- Tenney JR, Glauser TA.. The current state of absence epilepsy: can we have your attention? Epilepsy Curr 2013; 13: 135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevathan E. Infantile spasms and Lennox-Gastaut syndrome. J Child Neurol 2002; 17 (Suppl 2): 2S9–22. [DOI] [PubMed] [Google Scholar]
- van Rijckevorsel K. Treatment of Lennox-Gastaut syndrome: overview and recent findings. Neuropsychiatr Dis Treat 2008; 4: 1001–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignoli A, Oggioni G, De Maria G, Peron A, Savini MN, Zambrelli E, et al. Lennox-Gastaut syndrome in adulthood: long-term clinical follow-up of 38 patients and analysis of their recorded seizures. Epilepsy Behav 2017; 77: 73–8. [DOI] [PubMed] [Google Scholar]
- Vrielynck P. Current and emerging treatments for absence seizures in young patients. Neuropsychiatr Dis Treat 2013; 9: 963–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner TA, Liu Z, Macdonald RL, Kang JQ.. Heat induced temperature dysregulation and seizures in Dravet Syndrome/GEFS+ Gabrg2(+/Q390X) mice. Epilepsy Res 2017; 134: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werhahn KJ, Noachtar S, Arnold S, Pfander M, Henkel A, Winkler PA, et al. Tonic seizures: their significance for lateralization and frequency in different focal epileptic syndromes. Epilepsia 2000; 41: 1153–61. [DOI] [PubMed] [Google Scholar]
- Widdess-Walsh P, Dlugos D, Fahlstrom R, Joshi S, Shellhaas R, Boro A, et al. ; the EPGP Investigators. Lennox-Gastaut syndrome of unknown cause: phenotypic characteristics of patients in the Epilepsy Phenome/Genome Project. Epilepsia 2013; 54: 1898–904. [DOI] [PubMed] [Google Scholar]
- Zhou C, Lippman JJ, Sun H, Jensen FE.. Hypoxia-induced neonatal seizures diminish silent synapses and long-term potentiation in hippocampal CA1 neurons. J Neurosci 2011; 31: 18211–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary Material. These data are available from the corresponding author upon reasonable request.
Special note: the naïve EEG profile was scored and analysed by V.J., while R.K.H. scored and analysed the drug data. There may be subtle differences between their scoring strategies, but their data were not cross-analysed to avoid any observer-specific effects.