

Abstract
Medial arterial calcification (MAC) is a major complication of chronic kidney disease (CKD) and an indicator of poor prognosis. Aortic overexpression of tissue-nonspecific alkaline phosphatase (TNAP) accelerates MAC formation. The present study aimed to assess whether a TNAP inhibitor, SBI-425, protects against MAC and improves survival probability in a CKD- mineral and bone disorder (MBD) mouse model. CKD-MBD mice were divided in three groups: vehicle, SBI-10, and SBI-30. They were fed a 0.2% adenine and 0.8% phosphorus diet from 14 to 20 weeks of age to induce CKD, followed by a high-phosphorus (0.2% adenine and 1.8% phosphorus) diet for another 6 weeks. At 14–20 weeks of age, mice in the SBI-10 and SBI-30 groups were given 10 and 30 mg/kg SBI-425 by gavage once a day, respectively, vehicle-group mice were given distilled water. Control mice were fed a standard chow (0.8% phosphorus) between the ages of 8–20 weeks. Computed tomography imaging, histology, and aortic tissue calcium content revealed that, compared to vehicle animals, SBI-425 nearly halted the formation of MAC. Mice in the Control, SBI-10 and SBI-30 groups exhibited 100% survival, which was significantly better than vehicle-treated mice (57.1%). Aortic mRNA expression of Alpl, encoding TNAP, as well as plasma and aortic tissue TNAP activity, were suppressed by SBI-425 administration, whereas plasma pyrophosphate increased. We conclude that a TNAP inhibitor successfully protected the vasculature from MAC and improved survival in a mouse CKD-MBD model, without causing any adverse effects on normal skeletal formation and residual renal function.
Keywords: tissue-nonspecific alkaline phosphatase, medial arterial calcification, phosphorus, chronic kidney disease, mineral bone disorder
Introduction
Chronic kidney disease (CKD)-mineral bone disorder (MBD), which includes ectopic medial arterial calcification (MAC) and renal osteodystrophy, is a principal complication of CKD and a risk factor for CKD-related death [1,2]. MAC, also known as Mönckeberg’s arteriosclerosis, is commonly associated with diabetes [3], CKD [4], and aging [5], and is a risk factor for heart failure, cardiovascular diseases, and poor prognosis [3,4]. Among reported risk factors [4,6,7], hyperphosphatemia is directly associated with MAC [7] and is a strong predictor of morbidity and cardiovascular mortality in dialysis patients [4]. Hyperphosphatemia reportedly stimulates the transformation of vascular smooth muscle cells (VSMCs) into cells with osteoblast-like characteristics [7–10]. VSMC trans-differentiation is stimulated by increased intracellular uptake of phosphorus through type III sodium-dependent phosphate co-transporters Pit-1 (Slc20a1) and Pit-2 (Slc20a2) [7,11,12], and is mediated by upregulation of runt-related transcription factor 2 (Runx2) [7–10]. This is followed by upregulation of ossification markers, such as osteopontin (Spp1), bone morphogenetic protein 2 (Bmp2), Sp7 transcription factor 7 (Sp7, encoding Osterix), and tissue-nonspecific alkaline phosphatase (Alpl, encoding TNAP), indicative of acquiring osteoblast-like characteristics [7–10,13]. TNAP is crucial for bone matrix mineralization as it hydrolyzes extracellular pyrophosphate (PPi), a potent mineralization inhibitor, to phosphate. Through controlled hydrolysis of PPi, TNAP maintains the correct PPi:phosphate ratio, enabling normal skeletal and dental calcification [14–16]. TNAP overexpression in soft tissue causes ectopic calcification [17,18], including MAC of aortic tissue [8,14,19–22]. Although the mechanism of MAC formation has been extensively investigated, no effective therapeutic strategies to stop MAC formation in patients with CKD are available. The main approach against MAC is based on correcting hyperphosphatemia, achieved by increasing phosphorus removal in hemodialysis [23], prescribing phosphate binders [24], and a phosphorus-restricted diet [25,26]. Nevertheless, the life expectancy of patients with advanced CKD remains limited [3,4,27,28].
TNAP inhibition to raise PPi and prevent soft tissue calcification has been suggested for MAC treatment [14,20,29]. Numerous TNAP inhibitors have been identified, but most display very weak binding and/or are not selective against other alkaline phosphatases [14], limiting their applicability in clinical practice. Pinkerton et al. applied a structure-activity-relationship to optimize a lead molecule identified during a high-throughput screening for specific TNAP inhibitors and generated a potent derivative with drug-like properties, 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide [14]. The compound, SBI-425, displays very high oral exposure (AUC of >800 μg.h/ml after a 10 mg/kg oral dose), high selectivity against other alkaline phosphatases, and limited cross-reactivity [14]. A single 10 mg/kg oral dose of SBI-425 inhibited TNAP activity in plasma by >75% after 8 h and by ~50% after 24 h [14], suggesting that SBI-425 is a potent, selective, and orally bioavailable compound that robustly inhibits TNAP in vivo. Millán et al. reported that SBI-425 inhibited ectopic calcification in various animal models [17–19,29–31]. SBI-425 administration to a transgenic mouse model overexpressing TNAP in VSMCs with massive MAC significantly reduced cardiac hypertrophy and extended lifespan [19]. In another transgenic model overexpressing TNAP in endothelial cells, SBI-425 reduced coronary calcification in the vascular endothelium [18]. SBI-425 attenuated the development and progression of calcification in Abcc6−/− mice, a model of pseudoxanthoma elasticum, a rare disease characterized by mutations in ABCC6 and ectopic calcification [30,31]. Given the inhibitory effect of SBI-425 on ectopic calcification [18,19,30,31], and the intrinsic association between TNAP and MAC formation [7–10,13], SBI-425 might have therapeutic and/or prophylactic effects on MAC in patients with advanced CKD. Also, as MAC is a risk factor for heart failure, cardiovascular diseases, and poor prognosis [1–4], we hypothesized that MAC inhibition by SBI-425 might improve life expectancy in patients. To test these hypotheses, we sought to evaluate the inhibitory effect of SBI-425 on MAC, and whether reduced MAC improves survival probability in an animal model that simulated CKD-MBD in clinical practice [32].
We recently established a novel CKD-MBD mouse model displaying hyperphosphatemia and secondary hyperparathyroidism, mimicking CKD-related complications of MAC and renal osteodystrophy in patients with CKD [32]. This model allows induction of CKD-MBD through an adenine and high-phosphate diet, avoiding the need for drugs. This induction modality is key, given that C57BL/6J mice tolerate well ectopic calcification [32,33]. In this study, the CKD-MBD mouse model was employed to evaluate whether the favorable effects of a TNAP inhibitor on MAC formation and mortality rate could be achieved. As TNAP is important for normal bone mineralization [14], the study examined whether TNAP inhibition affected normal skeletal formation.
MATERIALS AND METHODS
Detailed descriptions of experimental materials and methods are presented in supplementary material, Supplementary materials and methods.
Animal studies
Thirty-eight 8-week-old C57/BL6J male mice were randomly allocated to the Control, Vehicle, SBI-10 and SBI-30 groups (Figure 1A). Mice in the control groups (n=8) were fed a standard chow (MF; Oriental yeast Co., Tokyo, Japan) containing 0.8% phosphorus (Pi) for 12 weeks. Mice in the Vehicle (n=14), SBI-10 (n=8) and SBI-30 (n=8) groups were fed a MF-based special chow (Oriental yeast Co.) containing 0.2% adenine (Wako pure chemical industries Co., Osaka, Japan) and 0.8% Pi diet for 6 weeks to induce CKD. Mice were then randomly assigned to the following groups, Vehicle, SBI-10, and SBI-30 (CKD-MBD groups) and were fed a MF-based special chow (Oriental yeast Co.) containing 0.2% adenine and 1.8% phosphorus (Pi) for 6 weeks. Mice in SBI-10 and SBI-30 groups were also given 10 mg/kg and 30 mg/kg respectively of SBI-425 by gavage once per day. One day prior to sacrifice, mice underwent a 24 h urine collection in individual metabolic cages. At 14, 16, 18, and 20 weeks of age, mice were anaesthetized and scanned by micro-computed tomography (CT) scans. At sacrifice, blood samples, aorta, heart and right femoral bone were harvested from each animal and were stored for further analysis.
Figure 1. Protocols for the induction of CKD and phosphate loading, and survival curve by the Kaplan–Meier method.
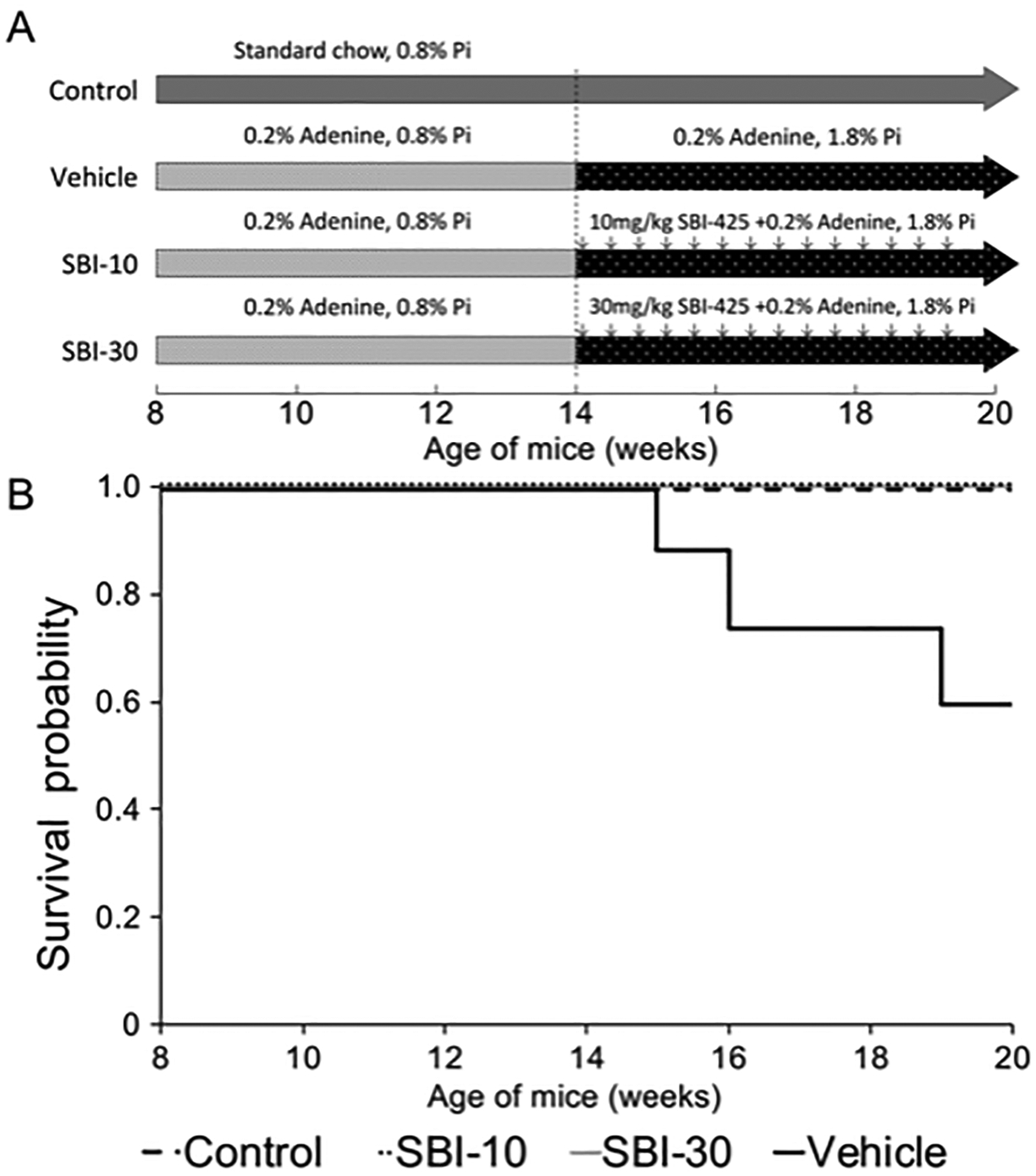
(A) Thirty-eight 8-week-old C57BL/6J mice were randomly assigned to the following experimental groups: Control (n=8), Vehicle (n=14), SBI-10 (n=8), and SBI-30 (n=8). Mice in the Control group were fed a standard MF pellet chow containing 0.8% phosphorus for 12 weeks. Mice in the CKD-MBD groups, namely Vehicle, SBI-10, and SBI-30 group, were first fed a MF-based special chow containing 0.2% adenine and normal (0.8%) levels of phosphorus for 6 weeks to induce CKD. They were then fed a MF-based diet containing 0.2% adenine and 1.8% phosphorus for 6 weeks. From 14 to 20 weeks of age, mice in the SBI-10 and SBI-30 groups were administered 10 mg/kg and 30 mg/kg of SBI-425 dissolved in distilled water by gavage once a day, respectively. Mice in the Vehicle group were administered equivalent volume of distilled water as vehicle solution instead. (B) Cumulative survival probability started to decrease in vehicle-treated mice at 15 weeks of age and dropped to 57.1% by the end of the study. No animals in the Control, SBI-10, and SBI-30 groups died prior to the end of the study.
Serum and urine biochemistry
Plasma and urine samples were analyzed as reported previously [32]. Plasma and urine phosphorus, creatinine, plasma calcium, and urea levels were measured using an automated analyzer as explained in supplementary material, Supplementary materials and methods. Intact parathyroid hormone and fibroblast growth factor 23 were measured by enzyme-linked immunosorbent assay. Plasma PPi levels were determined as described previously with some modifications as detailed in supplementary material, Supplementary materials and methods [30,34].
Alkaline phosphatase (ALP) activity assay
Plasma and aortic tissue alkaline phosphatase activity were determined by the rate of hydrolysis of p-NPP as described previously [35]. The ALP activity of an aorta was assayed in supernatants of homogenized organ standardized by milligrams of protein.
Histology and quantitative analysis of calcified lesions
Hematoxylin and eosin (H&E), Elastica Masson-Goldner (EMG), von Kossa staining, as well as subsequent histological analyses were carried out as reported previously [32]. Identification of fibrosis-positive area of kidney and calcification-positive areas of aortic rings and heart were performed on tissue sections using Meta Morph version 7.10 (Molecular Devices Co., Sunnyvale, CA, USA).
In vivo quantitative CT (qCT) analysis
qCT analysis was performed using a LaTheta LCT-200 CT scanner (Hitachi Aloka Medical Ltd., Tokyo, Japan) with the scan resolution of 24–48 μm, x-ray energy of 50 kV without using any filters as reported previously [32] with modifications as detailed in supplementary material, Supplementary materials and methods. Quantification of abdominal aortic calcification was performed to encompass the entire region of pathological calcification of an abdominal aorta from celiac artery branch section to the common iliac artery branch section, and a threshold was determined at the CT value of 350 to calculate the total area of ectopic calcification by using the scanner’s standard image analysis software as previously described [36]. Three-dimensional (3D) images of the calcified aorta were obtained using the Amira 5 3D visualizer application (FEI Co., Hillsboro, OR, USA).
Morphometric parameters of the distal metaphysis of the femur bones (n=8 per each group) were measured as described previously [32]; each region of interest was identified as total length of 1.2 mm; starting from 1.6 mm proximal to the distal end of the growth plate, toward the distal end of the bone. The manufacturer’s software was used to calculate the following parameters: cortical bone mineral density, bone mineral density, cortical bone mineral content, bone mineral content, cortical bone thickness.
Bone histomorphometry
In brief, the right femoral bone samples fixed with 70% ethanol were stained with Villanueva Osteochrome Bone Stain for 7 days. The specimens were then dehydrated and defatted and were cut at a thickness of 5 μm on a microtome for static and dynamic histomorphometric analyses.
Quantification of aortic calcium
Aortic calcium content was quantified by the o-cresolphthalein complexone (OCPC) method as reported previously [32].
Gene expression analysis
Reverse transcription - quantitative real-time polymerase chain reaction was carried out as reported previously [32]. In brief, RT-PCR amplification was carried out in a Step One Plus Real-Time PCR System (Life Technologies Co., Carlsbad, CA, USA), using the TaqMan gene expression assay probes (Life Technologies Co.). Gene expression was normalized to the reference Rn18s (18S rRNA) and expressed as fold-increase using the ΔΔCt method.
Statistical analysis
All statistical analyses were performed using SPSS version 25.0 (IBM, Chicago, IL, USA). All values are expressed as the mean ± standard error of the mean (SEM), unless otherwise stated. Non-repeated analysis of variance (ANOVA) with Games-Howell’s honest significance difference post hoc analysis was performed to assess any major effects and determine whether there were differences between multiple groups. A P-value <0.05 was considered significant.
Results
SBI-425 administration to CKD-MBD mice improves cumulative survival
To investigate whether administration of SBI-425 improved the survival rate, we used the Kaplan–Meier method to estimate the survival function and plotted a survival curve for each group of mice (Figure 1) Cumulative survival probability began to decrease in vehicle-treated mice after induction of phosphorus loading, and dropped to 57.1% that of the control animals (eight-live animals) by the end of the study. Mice in the Control, SBI-10, and SBI-30 groups maintained cumulative survival probability of 100% at p-values of 0.025, 0.024 and 0.037, respectively, as determined by a chi-square test using log rank against the Vehicle group. In the following experiments, eight samples per group were analyzed at sacrifice, unless otherwise stated.
Pathological and biochemical parameters, except plasma TNAP activity and PPi, are comparable between CKD-MBD groups
Intergroup comparisons of pathological and serum/urine mineral metabolism parameters are summarized in Table 1. Mice in the Vehicle, SBI-10, and SBI-30 groups (collectively referred to as CKD-MBD groups) were associated with increased renal and heart weight per body weight, oral phosphate, and water intake, but significantly decreased body weight compared to the Control group. Changes in body weight displayed by CKD-MBD groups were not associated with lower food intake (g/day) relative to the Control group. Plasma calcium levels were lower, while blood urea nitrogen (BUN) and creatinine (Cre) levels were significantly higher in the CKD-MBD groups than in control animals. CKD-MBD mice also exhibited a 3.51–3.54, 37.8–41.4, and 25.3–27.0-fold increase in plasma phosphate, intact parathyroid hormone, and fibroblast growth factor 23 levels, respectively, compared to the Control group. Notably, all these values were similar among CKD-MBD groups. Serum TNAP activity was 3.85-fold higher in the Vehicle group than in the Control but was lower than the latter in SBI-10 and SBI-30 groups. Plasma PPi was 2.10 and 2.54 nmol/ml in SBI-10 and SBI-30 groups, respectively, significantly more than the 1.36 nmol/ml of the Vehicle group.
Table 1.
Serum, urine biochemistries and pathological parameters in CKD-MBD and control mice at 20 weeks old age.
| Control (n = 8) | Vehicle (n = 8) | SBI-10 (n = 8) | SBI-30 (n = 8) | |
|---|---|---|---|---|
| Percent body weight gain/loss (%) | 25.9 ± 2.3 | −33.6 ± 2.8** | −33.9 ± 0.9** | −30.3 ± 0.6** |
| Food consumption (g/day) | 2.39 ± 0.13 | 3.70 ± 0.32* | 3.38 ± 0.25* | 3.62 ± 0.47 |
| Water intake (ml/day) | 3.75 ± 0.31 | 11.26 ± 1.54* | 10.58 ± 0.48** | 12.88 ± 0.32** |
| Phosphate intake (mg/day) | 19.1 ± 1.0 | 61.4 ± 7.3** | 60.8 ± 4.6** | 65.1 ± 8.5** |
| Lt. Renal weight (mg/g body weight) | 5.86 ± 0.22 | 6.76 ± 0.15* | 6.88 ± 0.08* | 6.86 ± 0.29* |
| Heart weight (mg/g body weight) | 4.70 ± 0.09 | 7.88 ± 0.44* | 7.21 ± 0.25** | 6.93 ± 0.33** |
| BUN (mg/dl) | 28.9 ± 0.9 | 108.8 ± 13.6** | 110.5 ± 6.3** | 108.2 ± 3.1** |
| Creatinine (mg/dl) | 0.12 ± 0.01 | 0.43 ± 0.02** | 0.42 ± 0.02** | 0.41 ± 0.02** |
| 24 h CCr (ml/min/kg) | 9.28 ± 1.46 | 2.70 ± 0.52* | 2.03 ± 0.20** | 2.74 ± 0.21* |
| Calcium (mg/dl) | 8.47 ± 0.18 | 5.41 ± 0.22** | 5.79 ± 0.33** | 6.22 ± 0.07** |
| Phosphorus (mg/dl) | 7.66 ± 0.58 | 27.2 ± 2.3** | 26.9 ± 1.5** | 27.1 ± 3.1** |
| Fephos | 0.05 ± 0.01 | 0.79 ± 0.02** | 0.77 ± 0.03** | 0.73 ± 0.04** |
| 24 h PiCr (ml/min/kg) | 0.52 ± 0.04 | 2.18 ± 0.39* | 1.65 ± 0.16** | 2.11 ± 0.20* |
| ALP activity (nMol/min/ml) | 9.30 ± 0.69 | 35.78 ± 1.57**##δδ | 7.78 ± 0.44δ | 5.35 ± 0.45**# |
| PPi (nMol/ml) | 1.58 ± 0.17 | 1.36 ± 0.13#δ | 2.10 ± 0.26* | 2.54 ± 0.45* |
| intact PTH (pg/ml) | 164 ± 27 | 6194 ± 928** | 6660 ± 860** | 6785 ± 1930** |
| FGF-23 (pg/ml) | 115 ± 12 | 3074 ± 323** | 2920 ± 232** | 3118 ± 243** |
Percent body weight loss, body weight at 20 week old age/ body weight at 14 weeks of age*100; BUN, blood urea nitrogen; 24 h CCr, 24 h creatinine clearance; FEphos, fractional excretion of phosphorus; 24 h PiCr, 24 h phosphorus clearance; ALP, alkaline phosphatase; PPi, pyrophosphate; PTH, parathyroid hormone; FGF-23, fibroblast growth factor 23. Mean ± SEM, n = 8.
P < 0.05 versus Control,
P < 0.01 versus Control,
P < 0.05 versus SBI-10,
P < 0.01 versus SBI-10,
P < 0.05 versus SBI-30,
P < 0.01 versus SBI-30 by non-repeated measures ANOVA with Games-Howell’s honest significance difference post hoc analysis.
Tubular impairment and fibrotic changes in the kidneys are similar among CKD-MBD mice
Intergroup histological differences are shown in Figure 2. Compared to the Control group (Figure 2A, E, I, M), the kidneys of mice in the CKD-MBD groups exhibited an irregular surface, indicating atrophy (Figure 2B–D); more dropout tubular epithelial cells, principally in distal tubules, associated with significant invasion of inflammatory cells (Figure 2F–H); and tubular luminal expansion (Figure 2F–H, J–L, N–P; white arrowheads). We observed EMG-stained cross-sections to quantify renal fibrosis; we found comparable fibrosed areas (%) in the Vehicle, SBI-10 and SBI-30 groups (15.15±0.58, 14.57± 0.60 and 14.12±0.63, respectively); these values were significantly higher than the 2.05±0.04 in the Control group (mean ± SEM; n=4 each).
Figure 2. Representative histopathological findings in renal tissues.
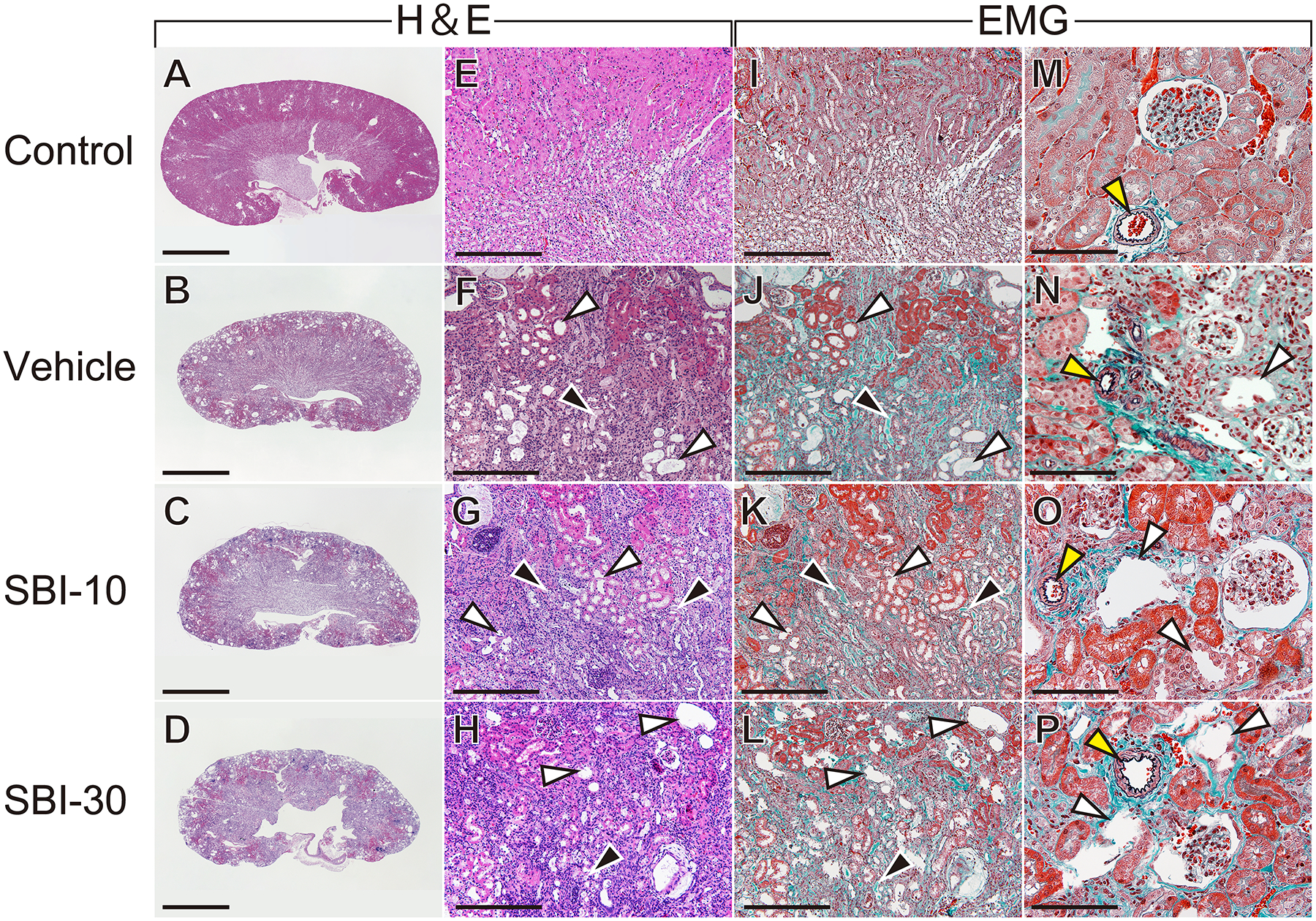
CKD mice in the Vehicle, SBI-10 and SBI-30 group exhibited irregular surfaces, indicative of atrophy (B–D), they also exhibited a higher prevalence of dropout of tubular epithelial cells and significant invasion of inflammatory cells (F–H) and tubular luminal expansion (F–H, J–L, N–P; white arrowheads) compared to control animals (A, E, I, M). Fibrotic changes were identified by EMG staining in the CKD groups (J–L), and cropped tubular epithelium and casts was also evident in renal tubules (F–H, J–L; black arrowheads). These abnormal histological findings were comparable among CKD groups. The glomerular structure remained normal, and degenerative changes was not observed in the small artery wall in CKD mice and control (N–P, yellow arrowheads). Scale bar, 2 mm for (A–D), 300 μm for (E–I) and 100 μm for (M–P); EMG, elastica Masson-Goldner staining.
TNAP inhibitor suppresses the time-dependent development of MAC in CKD-MBD mice
CT imaging was repeated for the same mouse every other week between 14 and 20 weeks of age, to detect aortic calcification as early as two weeks after initiation of phosphorus loading. Vascular calcification became more severe as phosphorus loading periods increased (Figure 3A) In the Vehicle group, the volume of aortic calcification at 20 weeks of age was 1.47 and 4.97-fold higher than at 18 or 16 weeks, respectively. No remarkable aortic calcification was detected in the other groups. A 3D reconstruction of CT images of 20-week-old animals confirmed vascular calcification mainly in the ascending aorta and the abdominal aorta, from the celiac artery branch section to the aortic bifurcation (Figure 3B-ii; for a 3D rotating movie, see Supplementary Video S1–S4). The Control, SBI-10, and SBI-30 groups maintained an intact aorta (Figure 3B-i, iii, iv); only one animal out of ten in the SBI-10 group displayed micro-aortic calcification. Histological analysis of the abdominal aorta at sacrifice (Figure 4) revealed clear evidence of MAC by von Kossa staining in vehicle-treated mice (Figure 4J,N) but not in control, SBI-10, or SBI-30 animals (Figure 4I,K,L,M,O,P). Elastin layers in these calcified areas appeared disorganized and disrupted (Figure 4F); no evidence of inflammation or atherosclerotic lesions was observed (Figure 4B,F). Calcification of the heart, mainly in the left ventricle, was detected in all CKD-MBD groups (supplementary material, Figure S1).
Figure 3. Calcified volumes of abdominal aorta and three-dimensional (3D) reconstructions of ectopic calcifications by CT imaging.
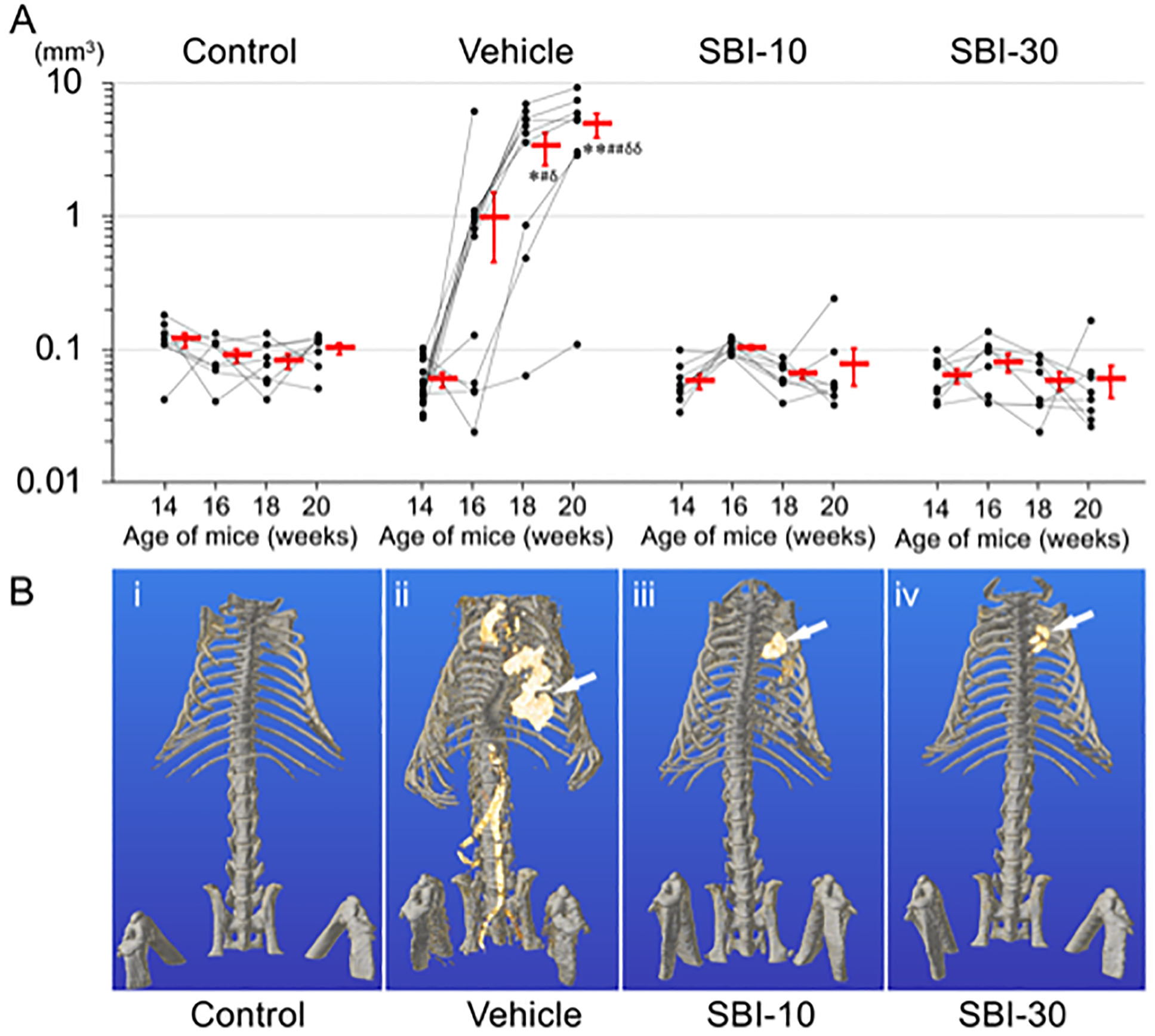
A) Computed tomography (CT) imaging was performed repeatedly to measure calcification volume of abdominal aorta (mm3) for each animal at 14, 16, 18 and 20 weeks of age (Line charts with black dots). The missing plots in some of the Vehicle groups’ charts represent mortality of the corresponding mouse and unavailability of data. The mean values of aortic calcification volumes for each group at 14, 16, 18 and 20 weeks of age are presented as the mean ± SEM (Red bars). Calcification of abdominal aorta was identified in Vehicle group at 16, 18 and 20 weeks of age with the volume of 0.98±0.53, 3.32±0.91 and 4.89±1.00 mm3, respectively. The y-axis of graph is expressed as log scale. *P < 0.05 versus Control, **P < 0.01 versus Control, #P < 0.05 versus SBI-10, ##P < 0.01 versus SBI-10, δP <0.05 versus SBI-30, δδP <0.01 versus SBI-30 by non-repeated measures ANOVA with Games-Howell’s honest significance difference post hoc analysis. (B) Three-dimensional (3D) reconstruction of CT images confirmed vascular calcification mainly in the ascending aorta, and the abdominal aorta from celiac artery branch section to the aortic bifurcation (B-ii). Mice in the control, SBI-10 and the SBI-30 groups were free from vascular calcification (B-i, iii, iv). Calcification of the heart, mainly in the left ventricle, was also identified in mice with CKD (B-ii, iii, iv white arrows).
Figure 4. Micrographs of H&E, EMG, Von Kossa and Alizarin Red stained sections of the abdominal aorta.
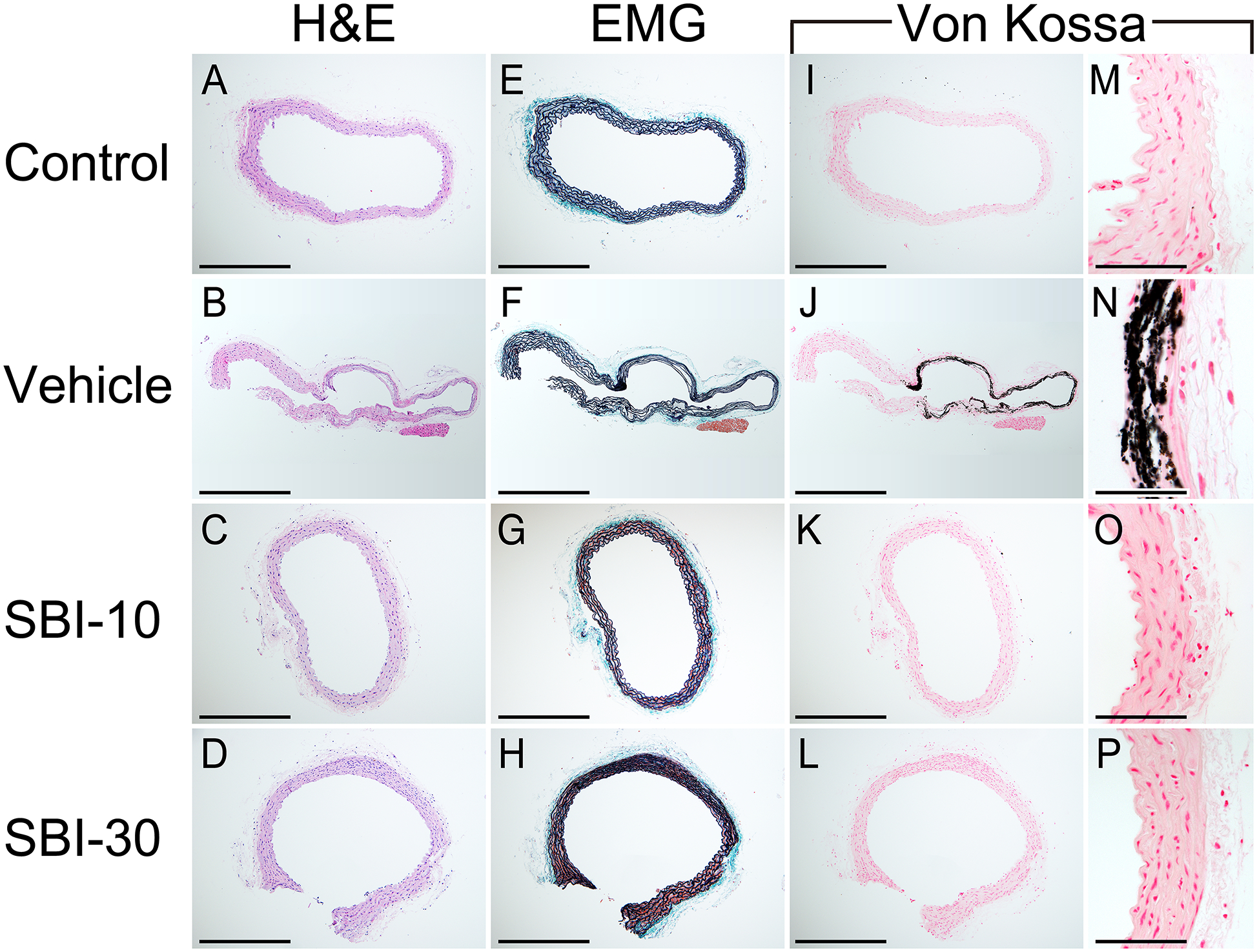
Sections of the abdominal aorta for the Control, Vehicle, SBI-10 and SBI-30 groups are shown. Medial arterial calcification (MAC) were observed in vehicle mice as von-Kossa staining positive legions (J, N). MAC was barely observed among mice in the Control, SBI-10, and SBI-30 groups (I, K, L, M, O, P). Elastin layers in these calcified areas grossly appeared to be disorganized and disrupted (F), while no evidence of cartilaginous metaplasia, inflammation or atherosclerotic lesions were observed (B, F), in line with control samples (A, E). Scale bar, 500 μm for (A–L) and 50 μm for (M–P); EMG, elastica Masson-Goldner staining.
To quantitatively verify whether SBI-425 treatment inhibited ectopic calcification observed in the Vehicle group, calcification in the abdominal aorta and heart was measured by either/both CT imaging and histology (von Kossa-positive areas) at sacrifice (20 weeks of age). In accordance with representative CT and histology images (Figure 3B and Figure 4), aortic calcification was significantly inhibited in SBI-10 and SBI-30 groups, dropping to a level comparable to the Control (Table 2, supplementary material, Figure S2A,B). Similarly, the OCPC assay revealed that calcium in aortic tissue extracts was abundant in the Vehicle group but reduced to nearly control levels in SBI-10 and SBI-30 groups (Table 2, supplementary material, Figure S2C). TNAP activity in aortic tissue was high in the Vehicle group but suppressed in SBI-10 and SBI-30 animals compared to both Control and Vehicle groups (Table 2, supplementary material, Figure S2D). Calcification volume of the heart, assessed by CT imaging (Table 2, supplementary material, Figure S2E) and histology (Table 2, supplementary material, Figure S2F), was comparable among all CKD-MBD groups.
Table 2.
Quantification of ectopic calcification by multiple analytical methods.
| Organ | Control (n = 8) | Vehicle (n = 8) | SBI-10 (n = 8) | SBI-30 (n = 8) | |
|---|---|---|---|---|---|
| Calcified volume by qCT analysis (mm3) | Aorta | 0.10 ± 0.01 | 4.89 ± 1.00**##δδ | 0.08 ± 0.02 | 0.06 ± 0.02 |
| Heart | 0.10 ± 0.03 | 8.10 ± 2.55** | 6.00 ± 0.48* | 3.772 ± 1.11 | |
| Percentage calcified surface (%) | Aorta | N.D. | 22.9 ± 6.2*#δ | 0.02 ± 0.02 | N.D. |
| Heart | N.D. | 3.47 ± 1.28 | 3.92 ± 1.04 | 1.89 ± 1.14 | |
| OCPC assay [Ca2+] (μmol/mg dry weight) | Aorta | 0.010 ± 0.001 | 7.26 ± 0.95**##δδ | 0.066 ± 0.004 | 0.073 ± 0.003 |
| TNAP activity (nmol/min/mg protein) | Aorta | 4.83 ± 0.10 | 11.05 ± 0.41**##δδ | 4.01 ± 0.21 **δ | 2.32 ± 0.12**# |
Quantification of medial arterial calcification (MAC) by multiple analytical methods. qCT, quantitative computed tomography; OCPC, o-cresolphthalein complexone; Ca, calcium. Mean ± SEM, (n = 8). N.D.: not detected.
P < 0.05 versus Control,
P < 0.01 versus Control,
P < 0.05 versus SBI-10,
P < 0.01 versus SBI-10,
P < 0.05 versus SBI-30,
P < 0.01 versus SBI-30 by non-repeated measures ANOVA with Games-Howell’s honest significance difference post hoc analysis.
Osteoblastic trans-differentiation-related genes are upregulated in VSMCs of the Vehicle group
Relative mRNA levels of vascular trans-differentiation-related genes were quantified by RT-qPCR analysis of aortic tissue. Runx2, ALPL, Bmp2, and Sp7 were found significantly more abundant in the Vehicle group by, 4.39, 2.98, 4.93, and 2.59-fold, respectively (Figure 5A–D). The mRNA level of Spp1 was 452-fold higher in the Vehicle group than in control animals, whereas that of Slc20a1 and Slc20a2 was 2.98 and 2.52-fold higher, respectively (Figure 5E–G). Transgelin mRNA (Tagln, encoding SM22-α) was significantly lower in all CKD-MBD groups (Figure 5H). Expression of phosphatase, orphan 1 (Phospho1) was comparable between all groups (Figure 5I); expression of ectonucleotide pyrophosphatase/phosphodiesterase 1 (Enpp1) was 2.94-fold higher in the Vehicle group but significantly lower (by about 90%) in SBI-10 and SBI30 groups than in the Control group (Figure 5J).
Figure 5. RT-qPCR analysis of aortic tissue.
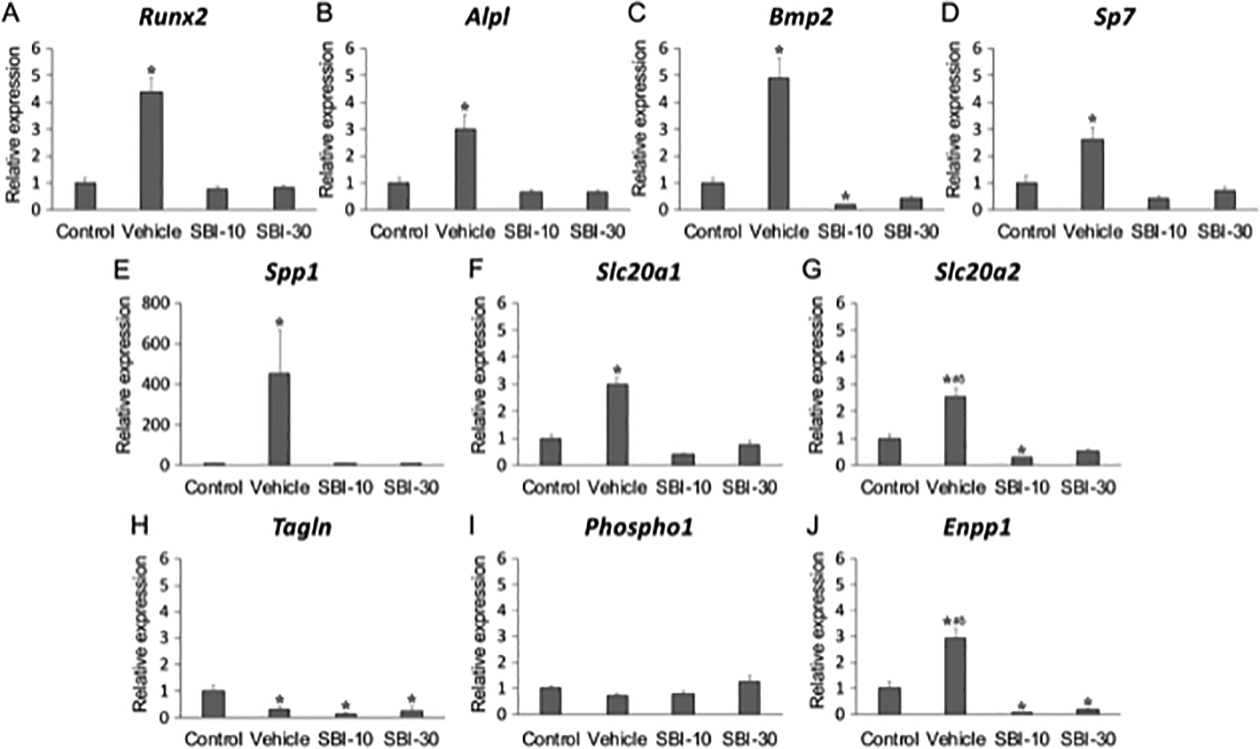
Relative expression levels of runt-related transcription factor 2 (Runx2) (A), tissue-nonspecific alkaline phosphatase (Alpl) (B), bone morphogenetic protein 2 (Bmp2) (C), Sp7 transcription factor 7 (Sp7) (D), osteopontin (Spp1) (E), secreted phosphoprotein 1 (Slc20a1) (F), secreted phosphoprotein 2 (Slc20a2) (G), transgelin (Tagln) (H), phosphatase, orphan 1 (Phospho1) (I) and ectonucleotide pyrophosphatase/phosphodiesterase 1 (Enpp1) (J) Levels of mRNA were determined by RT-qPCR in triplicate and normalized to 18S rRNA (Rn18s) levels. Gene expression of each group was relative to the control referred to as unity. Mean ± SEM (n=8); *P < 0.05 versus Control, **P < 0.01 versus Control, ##P < 0.01 versus SBI-10, δδP <0.01 versus SBI-30 by non-repeated measures ANOVA with Games-Howell’s honest significance difference post hoc analysis.
A TNAP inhibitor does not exacerbate renal osteodystrophy in CKD-MBD mice
Intergroup differences in bone density and femur structure were evaluated by CT imaging and Villanueva Osteochrome Bone Staining. Compared to the thick and homogeneous appearance of wild-type murine cortical bone in the Control group (Figure 6A, E, I), the femur bone cortex of CKD-MBD groups was thin and irregular (Figure 6B–D, F–H, J–L). CT images of the distal femoral metaphysis revealed a significant decrease in cortical bone mineral density (Ct. BMD), cortical bone mineral content (Ct. BMC), BMD, BMC and cortical bone thickness (Ct. B. Th) in CKD-MBD groups compared to the Control (Table 3, supplementary material, Figure S3A–E). Static and dynamic histomorphometric parameters revealed that, bone volume per tissue volume (BV/TV), and trabecular number (Tb. N) were lower in CKD-MBD animals than in the Control (Table 3, supplementary material, Figure S3E, F, H). The bone resorption parameters, eroded surface per bone surface (ES/BS) and osteoclast surface per bone surface (Oc. S/BS), were drastically increased in CKD-MBD groups compared to the Control (Table 3, supplementary material, Figure S3I,J). Although high-turnover bone metabolism disorders, such as renal osteodystrophy, predominantly in the bone cortex, were clearly demonstrated in CKD-MBD groups, disease severity was comparable between SBI-425 treated and non-treated animals, suggesting no influence of TNAP inhibitor on bone formation.
Figure 6. Representative histological and CT images of the distal femur bone.
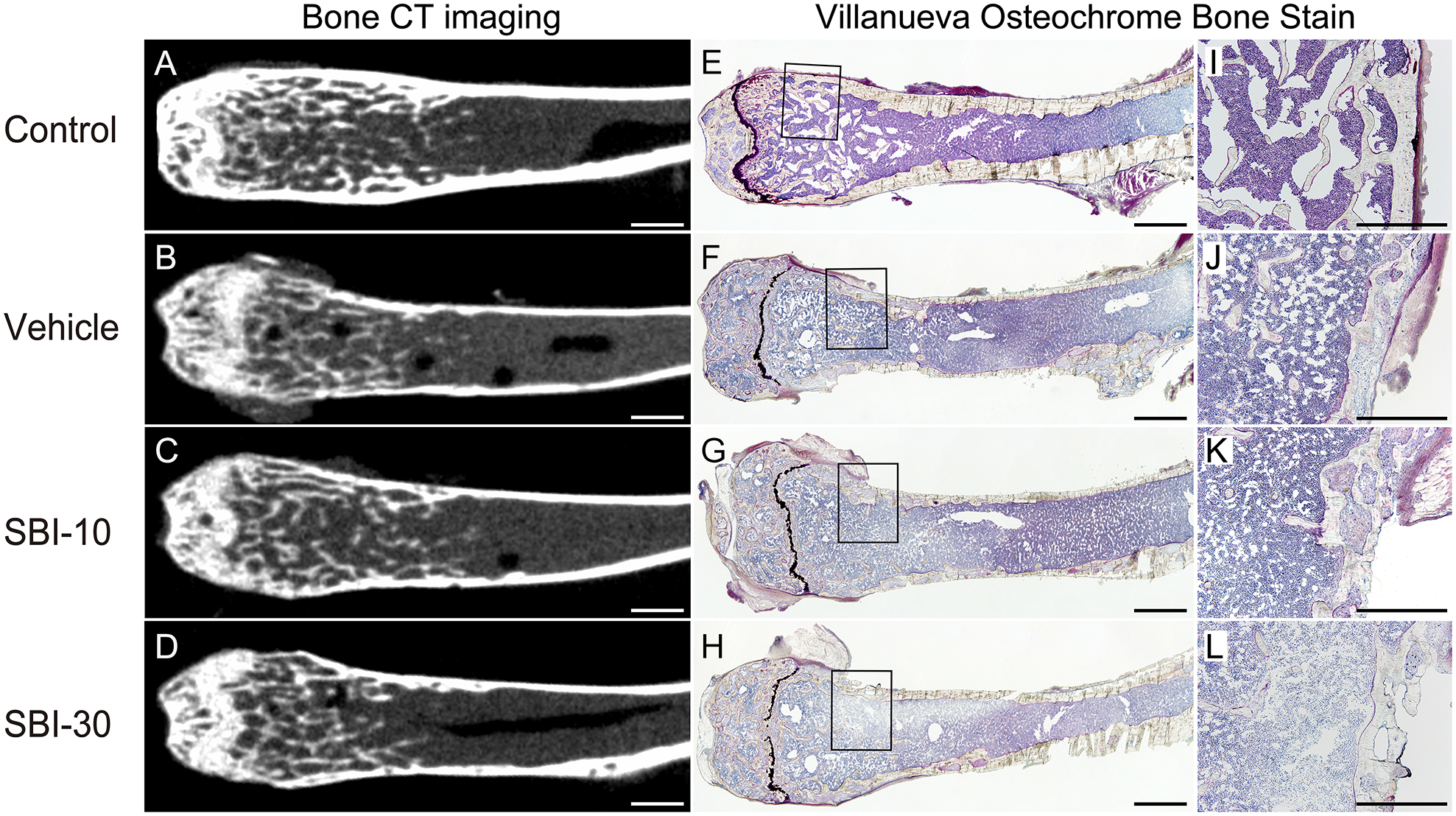
Histological and CT images from the Control (A, E, I), Vehicle (B, F, J), SBI-10 (C, G, K), and SBI-30 (D, H, L) groups are shown. The enlarged figure (I–L) represents black-squared area of the corresponding entire histological image (E-H). The thinning of the femur cortex bone was commonly observed in the CKD-MBD group (B–D, F–H, J–L), compared to the thick and homogeneous appearance of the wild-type cortical bone in the Control group (A, E, I). Histological changes in the morphology of the diaphysis and irregularity of cortical bones was evident in all CKD-MBD mice (F–H, J–L). Scale bar, 1000 μm for (A–H) and 500 μm for (I-L).
Table 3.
Parameters of the distal femoral metaphysis by CT imaging and histomorphometric analysis.
| Control (n = 8) | Vehicle (n = 8) | SBI-10 (n = 8) | SBI-30 (n = 8) | |
|---|---|---|---|---|
| Ct. BMD (mg/cm3) | 1077.5 ± 10.4 | 577.4 ± 24.9** | 590.4 ± 18.5** | 629.9 ± 12.9** |
| BMD (mg/cm3) | 452.2 ± 20.7 | 322.8 ± 22.4** | 285.9 ± 8.9** | 299.3 ± 13.1** |
| Ct. BMC (mg) | 0.95 ± 0.03 | 0.43 ± 0.02** | 0.48 ± 0.01** | 0.49 ± 0.01** |
| BMC (mg) | 1.77 ± 0.15 | 1.06 ± 0.10** | 1.19 ± 0.06** | 1.22 ± 0.08** |
| Ct. B. Th (mm x10−1) | 10.65 ± 0.16 | 9.08 ± 0.10** | 8.78 ± 0.05** | 8.83 ± 0.02** |
| BV/TV (%) | 12.14 ± 1.11 | 8.57 ± 1.29** | 7.74 ± 1.41** | 7.57 ± 1.14** |
| Tb.Th (μm) | 36.45 ± 1.92 | 35.92 ± 1.01 | 35.81 ± 1.52 | 37.47 ± 1.50 |
| Tb.N (N/mm) | 3.32 ± 0.16 | 2.39 ± 0.34** | 2.13 ± 0.30** | 2.01 ± 0.28** |
| ES/BS (%) | 2.06 ± 0.46 | 14.31 ± 3.89** | 13.19 ± 2.34** | 15.87 ± 1.14** |
| Oc.S/BS (%) | 0.83 ± 0.22 | 8.44 ± 2.67** | 8.03 ± 2.16** | 9.88 ± 1.32** |
| MAR (μm/day) | 1.00 ± 0.22 | 0.81 ± 0.10 | 1.26 ± 0.03 | 1.17 ± 0.13 |
Ct. BMD, cortical bone mineral density; BMD, bone mineral density; Ct. BMC, cortical bone mineral content; BMC, bone mineral content; Ct. B. Th, Cortical bone thickness; BV/TV, bone volume per tissue volume; Tb.Th, trabecular thickness; Tb.N, trabecular number; ES/BS, eroded surface per bone surface; Oc.S/BS, osteoclast surface per bone surface; MAR, mineral apposition rate. Mean ± SEM, n = 8.
P < 0.05 versus Control,
P < 0.01 versus Control by non-repeated measures ANOVA with Games-Howell’s honest significance difference post hoc analysis.
DISCUSSION
Here, we have evaluated the potential efficacy of pharmacological TNAP inhibition in preventing MAC. TNAP inhibitor significantly reduced MAC formation at 10 mg/kg and 30 mg/kg, and improved cumulative survival probability. Changes in pathological/biochemical parameters of CKD-MBD mice were in accordance with our previous report [32] and, except for plasma TNAP activity and PPi concentration, they were comparable between CKD-MBD groups. Accordingly, these parameters, which potentially affect MAC formation [6], played no role in suppressing MAC in SBI-10 and SBI-30 groups. The comparable histological findings, quantitative assessment of renal histopathological findings by adenine administration, and renal dysfunction observed among CKD-MBD groups were also consistent with previous findings [32] and confirmed no influence of this dosage of SBI-425 on kidneys.
CKD-MBD mice closely mimic MAC formation in patients with advanced CKD [37]. The time-dependent increase of MAC formation in vehicle-treated mice observed using CT imaging confirmed a strong association between MAC and phosphorus loading [6,7,32,38–40]. Here, we based SBI-425 dosage upon previous reports on inhibition of TNAP activity and ectopic calcification [18,19,30]. An oral SBI-425 dose of 10 or 30 mg/kg/day inhibited TNAP activity in plasma and aortic tissue by 78.3–85.0% and 63.7–79.0%, respectively, 2 h after administration. As administration of SBI-425 significantly suppressed TNAP activities of aortic tissues compared to not only the Vehicle group but also to the Control, in a dose-dependent manner, the inhibitory effect of SBI-425 on aortic TNAP activity was probably achieved by the TNAP inhibitor itself, not by smaller volumes of aortic calcification than the vehicle-treated mice. The potency of the TNAP inhibitor to suppress MAC was confirmed by established quantitative methods [32]. Although significant reduction of MAC by SBI-425 was also suggested by tissue calcium content and histological analysis, microcalcification smaller than the scan resolution of CT (48 μm) was not detected in the qCT analysis; this is a technical limitation. Importantly, SBI-425 is thought to inhibit MAC formation independently of plasma concentrations of uremic toxins such as phosphorus [6,7,38–40]. Because 10 mg/kg/day of SBI-425 almost completely blocked MAC formation, a smaller dose of 3–5 mg/kg/day may be attempted in CKD-MBD mice in further studies, as a means to identify the minimum required dosage of the drug to inhibit MAC formation in patients with advanced CKD.
MAC leads to reduced compliance of arterial walls and altered mechanical and hemodynamic properties, causing left ventricular hypertrophy and decreased coronary perfusion. These structural and functional changes result in heart failure, cardiovascular diseases, and poor prognosis [1–4,27]. As TNAP inhibition by SBI-425 significantly reduced formation of MAC in the abdominal aorta, we believe that this reduction directly contributed to better outcomes in SBI-10 and SBI-30 group mice. This hypothesis is supported by comparable levels of biochemical parameters that may influence mortality rates and indicate renal dysfunction, and levels of uremic toxins (e.g. BUN, serum creatinine, serum phosphorus, PTH, and FGF-23) between CKD-MBD groups. However, as blood concentrations of vasoprotective proteins, such as matrix gla protein and fetuin A, were not measured, we cannot exclude that SBI-425 administration caused their plasma and/or tissue concentrations to fluctuate and thus inhibited MAC formation. Also, as we did not examine blood pressure levels and cardiac ultrasonography, this study lacks objective physiological data to reflect the degree of cardiac hypertrophy. Although further investigation is required, it is probable that SBI-425-mediated inhibition of MAC formation directly contributed to the greater survival of SBI-10 and SBI-30 groups compared with the Vehicle group.
Upregulation of Slc20a1, Slc20a1, Runx2, Bmp2, Sp7, OPN, and Alpl mRNA expression in the Vehicle group demonstrates that formation of MAC in this CKD-MBD model presumably originates from trans-differentiation of VSMCs into osteoblast-like cells, and is promoted by elevated serum phosphate levels, as reported previously [8–13,32]. Based on previous reports [18,19] and our study, suppression of locally overexpressed TNAP in VSMCs and TNAP activity in plasma and arterial extracts may contribute to inhibition of MAC formation. One possibility is that SBI-425 raises PPi levels by inhibiting the strong PPiase activity of TNAP [17–19,30,41]. Given the documented high inhibitory action of PPi on mineralization [14,20–22], it is likely that in this study, elevated plasma PPi is sufficient to halt MAC formation. Changes in PPi concentration may also have influenced mRNA expression of Enpp1, which was suppressed in SBI-10 and SBI-30 groups but overexpressed in the vehicle group. ENPP1 is a phosphatase that hydrolyzes adenosine triphosphate (ATP) into PPi and adenosine monophosphate (AMP) [15,16]. Therefore, TNAP inhibition by SBI-425 may have suppressed ENPP1 activity through elevated in loco PPi levels [42]. Conversely, overexpressed TNAP in the Vehicle group may have promoted ENPP1 activity via a lower PPi:phosphate ratio [42]. Additional direct/indirect mechanisms may explain the contribution of elevated PPi to inhibition of MAC formation. According to one hypothesis, PPi can prevent ectopic calcification by directly stimulating OPN production in osteoblasts via the MAPK signaling pathway, which then limits hydroxyapatite formation [42,43]. Alternatively, the presence of a second substrate such as β-glycerophosphate allows PPi to directly alter TNAP conformation, inhibit its enzymatic activity, and reduce PPi hydrolysis [43].
Overexpression/suppression of TNAP in the vascular wall may have influenced MAC formation by affecting the signaling factors required for VSMC trans-differentiation. Besides being an efficient PPiase, TNAP functions as a potent ATPase and ADPase, whereas ENPP1 acts as a weak ADPase and PPiase in addition to ATPase [15,16,41,44]. Thus, suppression of TNAP activity could change extracellular purine nucleotide and purine base concentrations, affecting MAC formation in a number of ways. For example, locally released ATP, which is hydrolyzed by ENPP1, is the major source of extracellular PPi and can act locally to regulate calcification [44]. Alternatively, ATP and related-purine nucleotides act via purinergic P2 receptors to regulate cell proliferation and differentiation [44], and VSMCs express multiple P2 receptor subtypes. ATP and uridine triphosphate (UTP) reportedly prevented calcification in VSMCs, at least in part, by reducing VSMC apoptosis [44]. Thus, changes in concentrations of extracellular purine nucleotides following TNAP inhibition can affect cell signaling, differentiation, and apoptosis, and consequently inhibit MAC formation [16]. This hypothesis is also supported by the effectiveness of SBI-425 in a mouse model of pseudoxanthoma elasticum, whose etiology is supposed to derive from a deficiency in ATP metabolism owing to ABCC6 mutations [30,31]. Therefore, SBI-425-mediated inhibition of upstream trans-differentiation signals to TNAP may be attributed to changes in extracellular purine metabolism. As the precise nature of alterations to the microenvironment remains speculative, further investigation is required to demonstrate how the TNAP inhibitor suppresses MAC formation.
In the CKD-MBD group, ectopic calcification of the heart was not affected by TNAP inhibition, which may be explained by the pharmacokinetics of SBI-425 in the body. Pinkerton et al. reported that the simulated volume of distribution of SBI-425 was 1.03 L/kg, indicating some level of intracellular penetration but not significant concentration in tissues [14]. Therefore, the drug’s concentration in the heart is expected to be lower than that in the blood. This explains the selective effect of SBI-425 in blood vessels.
The morphological changes to the bone of CKD-MBD groups resemble typical secondary hyperparathyroidism observed in patients with advanced CKD [2,32], and indicate renal osteodystrophy. No difference was observed in bone-related histological and parametric changes between CKD-MBD groups, suggesting no adverse effects on bone formation by the TNAP inhibitor, consistent with previous reports [19,30]. Two reasons could explain this phenomenon: pharmacokinetics and loading of the drug. First, as in the heart, the drug concentration was lower in the bone than in blood, and not sufficient to fully inhibit bone TNAP activity. Second, the overall duration of inhibition was insufficient to halt strong TNAP activity in the bone compared with that of an aortic wall. For future clinical applications, SBI-425 might effectively inhibit MAC formation without adversely affecting normal bone formation and ectopic calcification in other organs.
In summary, the novel and potent TNAP inhibitor SBI-425 protected the vasculature from MAC formation, and significantly improved survival in a CKD-MBD mouse model characterized by uremic conditions with hyperphosphatemia, without causing adverse effects on normal skeletal formation and residual renal function. As MAC is a risk factor for poor prognosis [3,4], and hyperphosphatemia strongly correlates with complication of MAC in patients with advanced CKD [6,7,38–40], SBI-425 is expected to benefit clinical practice by inhibiting MAC formation and improving prognosis. However, because many other factors contribute to MAC formation in patients with advanced CKD [6], further studies, such as challenging suppressive effects of MAC formation in an animal model under uremic conditions without hyperphosphatemia, are desired. Nevertheless, our data strongly suggest that TNAP inhibition is a promising therapeutic strategy for MAC and can contribute to better clinical outcomes in patients with advanced CKD.
Supplementary Material
Figure S1. Representative histopathological findings in heart tissues
Figure S2. Quantification of medial arterial calcification (MAC) and heart calcification by multiple analytical methods at 20 weeks of age
Figure S3. Parameters of the distal femoral metaphysis by CT imaging and histomorphometric analysis
Video S1. 3D reconstruction of CT images of 20-week-old animals (Vehicle group)
Video S2. 3D reconstruction of CT images of 20-week-old animals (SBI-10 group)
Video S3. 3D reconstruction of CT images of 20-week-old animals (SBI-30 group)
Video S4. 3D reconstruction of CT images of 20-week-old animals (Control group)
Acknowledgements
We wish to express our gratitude to Dr. Masanori Izumi and Dr. Koutaro Ishibashi (Daiichi Sankyo Co., Ltd.) for technical assistance with plasma pyrophosphate measurements.
Footnotes
Conflict of Interest Statement
ABP and JLM are co-inventors on a patent application covering SBI-425 (PCT WO 2013126608). All other authors declare no competing financial interests.
References
- 1.Gorriz JL, Molina P, Cerveron MJ, et al. Vascular calcification in patients with nondialysis CKD over 3 years. Clin J Am Soc Nephrol 2015; 10: 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alem AM, Sherrard DJ, Gillen DL, et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58: 396–399. [DOI] [PubMed] [Google Scholar]
- 3.Lehto S, Niskanen L, Suhonen M, et al. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler Thromb Vasc Biol 1996; 16: 978–983. [DOI] [PubMed] [Google Scholar]
- 4.London GM, Guerin AP, Marchais SJ, et al. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 2003; 18: 1731–1740. [DOI] [PubMed] [Google Scholar]
- 5.McEniery CM, McDonnell BJ, So A, et al. Aortic calcification is associated with aortic stiffness and isolated systolic hypertension in healthy individuals. Hypertension 2009; 53: 524–531. [DOI] [PubMed] [Google Scholar]
- 6.Shroff R, Long DA, Shanahan C. Mechanistic insights into vascular calcification in CKD. J Am Soc Nephrol 2013; 24: 179–189. [DOI] [PubMed] [Google Scholar]
- 7.Yamada S, Giachelli CM. Vascular calcification in CKD-MBD: Roles for phosphate, FGF23, and Klotho. Bone 2017; 100: 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lomashvili KA, Garg P, Narisawa S, et al. Upregulation of alkaline phosphatase and pyrophosphate hydrolysis: potential mechanism for uremic vascular calcification. Kidney Int 2008; 73: 1024–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giachelli CM. Vascular calcification: in vitro evidence for the role of inorganic phosphate. J Am Soc Nephrol 2003; 14: 300S–304. [DOI] [PubMed] [Google Scholar]
- 10.Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol 2004; 15: 2857–2867. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res 2006; 98: 905–912. [DOI] [PubMed] [Google Scholar]
- 12.Crouthamel MH, Lau WL, Leaf EM, et al. Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: redundant roles for PiT-1 and PiT-2. Arterioscler Thromb Vasc Biol 2013; 33: 2625–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lomashvili KA, Cobbs S, Hennigar RA, et al. Phosphate-induced vascular calcification: role of pyrophosphate and osteopontin. J Am Soc Nephrol 2004; 15: 1392–1401. [DOI] [PubMed] [Google Scholar]
- 14.Pinkerton AB, Sergienko E, Bravo Y, et al. Discovery of 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorg Med Chem Lett 2018; 28: 31–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Millan JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int 2013; 93: 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buchet R, Millan JL, Magne D. Multisystemic functions of alkaline phosphatases. Methods Mol Biol 2013; 1053: 27–51. [DOI] [PubMed] [Google Scholar]
- 17.Savinov AY, Salehi M, Yadav MC, et al. Transgenic overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in vascular endothelium results in generalized arterial calcification. J Am Heart Assoc 2015; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romanelli F, Corbo A, Salehi M, et al. Overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in endothelial cells accelerates coronary artery disease in a mouse model of familial hypercholesterolemia. PLoS One 2017; 12: e0186426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheen CR, Kuss P, Narisawa S, et al. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J Bone Miner Res 2015; 30: 824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Neill WC. Treatment of vascular calcification. Kidney Int 2008; 74: 1376–1378. [DOI] [PubMed] [Google Scholar]
- 21.Dedinszki D, Szeri F, Kozak E, et al. Oral administration of pyrophosphate inhibits connective tissue calcification. EMBO Mol Med 2017; 9: 1463–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Neill WC, Lomashvili KA, Malluche HH, et al. Treatment with pyrophosphate inhibits uremic vascular calcification. Kidney Int 2011; 79: 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daugirdas JT. Removal of phosphorus by hemodialysis. Semin Dial 2015; 28: 620–623. [DOI] [PubMed] [Google Scholar]
- 24.Kovesdy CP, Kuchmak O, Lu JL, et al. Outcomes associated with phosphorus binders in men with non-dialysis-dependent CKD. Am J Kidney Dis 2010; 56: 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wald R, Sarnak MJ, Tighiouart H, et al. Disordered mineral metabolism in hemodialysis patients: an analysis of cumulative effects in the Hemodialysis (HEMO) Study. Am J Kidney Dis 2008; 52: 531–540. [DOI] [PubMed] [Google Scholar]
- 26.Lynch KE, Lynch R, Curhan GC, et al. Prescribed dietary phosphate restriction and survival among hemodialysis patients. Clin J Am Soc Nephrol 2011; 6: 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blacher J, Guerin AP, Pannier B, et al. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 2001; 38: 938–942. [DOI] [PubMed] [Google Scholar]
- 28.Niskanen L, Siitonen O, Suhonen M, et al. Medial artery calcification predicts cardiovascular mortality in patients with NIDDM. Diabetes Care 1994; 17: 1252–1256. [DOI] [PubMed] [Google Scholar]
- 29.Narisawa S, Harmey D, Yadav MC, et al. Novel inhibitors of alkaline phosphatase suppress vascular smooth muscle cell calcification. J Bone Miner Res 2007; 22: 1700–1710. [DOI] [PubMed] [Google Scholar]
- 30.Ziegler SG, Ferreira CR, MacFarlane EG, et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci Transl Med 2017; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Q, Huang J, Pinkerton AB, et al. Inhibition of tissue-nonspecific alkaline phosphatase attenuates ectopic mineralization in the Abcc6(−/−) mouse model of PXE but not in the Enpp1 mutant mouse models of GACI. J Invest Dermatol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tani T, Orimo H, Shimizu A, et al. Development of a novel chronic kidney disease mouse model to evaluate the progression of hyperphosphatemia and associated mineral bone disease. Sci Rep 2017; 7: 2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ivandic BT, Utz HF, Kaczmarek PM, et al. New Dyscalc loci for myocardial cell necrosis and calcification (dystrophic cardiac calcinosis) in mice. Physiol Genomics 2001; 6: 137–144. [DOI] [PubMed] [Google Scholar]
- 34.Jansen RS, Duijst S, Mahakena S, et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler Thromb Vasc Biol 2014; 34: 1985–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sogabe N, Oda K, Nakamura H, et al. Molecular effects of the tissue-nonspecific alkaline phosphatase gene polymorphism (787T > C) associated with bone mineral density. Biomed Res 2008; 29: 213–219. [DOI] [PubMed] [Google Scholar]
- 36.Wang JH, Eguchi K, Matsumoto S, et al. The omega-3 polyunsaturated fatty acid, eicosapentaenoic acid, attenuates abdominal aortic aneurysm development via suppression of tissue remodeling. PLoS One 2014; 9: e96286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moe SM, Chen NX. Pathophysiology of vascular calcification in chronic kidney disease. Circ Res 2004; 95: 560–567. [DOI] [PubMed] [Google Scholar]
- 38.Mazhar AR, Johnson RJ, Gillen D, et al. Risk factors and mortality associated with calciphylaxis in end-stage renal disease. Kidney Int 2001; 60: 324–332. [DOI] [PubMed] [Google Scholar]
- 39.Shigematsu T, Kono T, Satoh K, et al. Phosphate overload accelerates vascular calcium deposition in end-stage renal disease patients. Nephrol Dial Transplant 2003; 18 Suppl 3: iii86–89. [DOI] [PubMed] [Google Scholar]
- 40.Adeney KL, Siscovick DS, Ix JH, et al. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J Am Soc Nephrol 2009; 20: 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciancaglini P, Yadav MC, Simao AM, et al. Kinetic analysis of substrate utilization by native and TNAP-, NPP1-, or PHOSPHO1-deficient matrix vesicles. J Bone Miner Res 2010; 25: 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harmey D, Hessle L, Narisawa S, et al. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol 2004; 164: 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orriss IR, Arnett TR, Russell RG. Pyrophosphate: a key inhibitor of mineralisation. Curr Opin Pharmacol 2016; 28: 57–68. [DOI] [PubMed] [Google Scholar]
- 44.Patel JJ, Zhu D, Opdebeeck B, et al. Inhibition of arterial medial calcification and bone mineralization by extracellular nucleotides: The same functional effect mediated by different cellular mechanisms. J Cell Physiol 2018; 233: 3230–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dahl R, Sergienko EA, Su Y, et al. Discovery and validation of a series of aryl sulfonamides as selective inhibitors of tissue-nonspecific alkaline phosphatase (TNAP). J Med Chem 2009; 52: 6919–6925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El-Abbadi MM, Pai AS, Leaf EM, et al. Phosphate feeding induces arterial medial calcification in uremic mice: role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int 2009; 75: 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bouxsein ML, Boyd SK, Christiansen BA, et al. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 2010; 25: 1468–1486. [DOI] [PubMed] [Google Scholar]
- 48.Dempster DW, Compston JE, Drezner MK, et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2013; 28: 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Representative histopathological findings in heart tissues
Figure S2. Quantification of medial arterial calcification (MAC) and heart calcification by multiple analytical methods at 20 weeks of age
Figure S3. Parameters of the distal femoral metaphysis by CT imaging and histomorphometric analysis
Video S1. 3D reconstruction of CT images of 20-week-old animals (Vehicle group)
Video S2. 3D reconstruction of CT images of 20-week-old animals (SBI-10 group)
Video S3. 3D reconstruction of CT images of 20-week-old animals (SBI-30 group)
Video S4. 3D reconstruction of CT images of 20-week-old animals (Control group)