

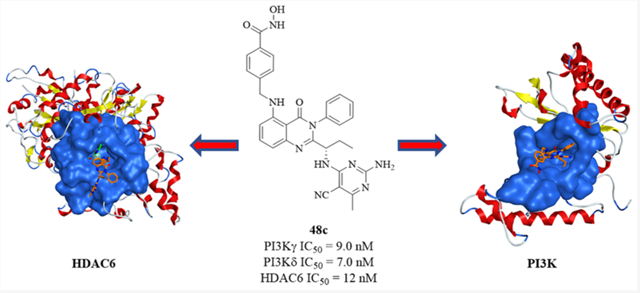
Abstract
A series of quinazolin-4-one based hydroxamic acids was rationally designed and synthesized as novel dual PI3K/HDAC inhibitors by incorporating an HDAC pharmacophore into a PI3K inhibitor (Idelalisib) via an optimized linker. Several of these dual inhibitors were highly potent (IC50 < 10 nM) and selective against PI3Kγ, δ and HDAC6 enzymes and exhibited good antiproliferative activity against multiple cancer cell lines. The lead compound 48c, induced necrosis in several mutant and FLT3-resistant AML cell lines and primary blasts from AML patients, while showing no cytotoxicity against normal PBMCs, NIH3T3, and HEK293 cells. Target engagement of PI3Kδ and HDAC6 by 48c was demonstrated in MV411 cells using the cellular thermal shift assay (CETSA). Compound 48c showed good pharmacokinetics properties in mice via intraperitoneal (ip) administration and provides a means to examine the biological effects of inhibiting these two important enzymes with a single molecule, either in vitro or in vivo.
Graphical Abstract
INTRODUCTION
Most of the currently approved drugs based on the “single-target single drug” model are becoming less effective in the treatment of complex, heterogeneous, multigenic diseases, like cancer.1,2 While drug combination therapies are employed as an alternative approach to achieve efficacy, their benefits are often negated by adverse drug–drug interactions, unpredictable pharmacokinetic (PK) and safety profiles, and poor patient compliance.3,4 In recent years, there has been an increasing inclination toward discovering “multitarget drugs” to address the limited efficacy and resistance or toxicity associated with many single-target or combination-based therapies.5–12
Class I phosphoinositide 3-kinases (PI3Ks) are lipid kinase enzymes that transduce signals from cell surface receptors (RTK, GPCR, etc.) to downstream effectors (Akt, mTOR, etc.), leading to a variety of cellular processes, including cell proliferation, survival, differentiation, metabolism, and angiogenesis.13,14 Specifically, class I PI3Ks phosphorylate phosphatidylinositol (4,5)-bisphosphate (PI(4,5)P2 or PIP2) in vivo to form the secondary messenger phosphatidylinositol (3,4,5)-trisphosphate (PI(3,4,5)P3 or PIP3), which activates the pleckstrin homology (PH)-domain of protein kinases (Akt, BTK, etc.), leading to downstream signaling.15,16 The four isoforms of Class I PI3Ks (PI3Kα, PI3Kβ, PI3Kγ, and PI3Kδ) exist as heterodimers, consisting of a catalytic p110 (α, β, δ, γ) subunit and a regulatory p85 (α, β, δ) or p101/p84 (γ) subunit.14 The PI3Kα and PI3Kβ isoforms are ubiquitously expressed, whereas PI3Kγ and PI3Kδ expression is limited to leukocytes.16 Aberrant regulation of class I PI3Ks has been implicated in several cancer types, and their inhibition continues to be an active cancer therapeutic area with four recently approved PI3K inhibitor drugs (Idelalisib, Copanlisib, Duvelisib, and Alpelisib) (Figure 1) and several others in ongoing clinical trials.17–20 Despite tremendous progress in the discovery of PI3K inhibitors (PI3Ki) over the past decade, clinical trials with PI3Ki as a monotherapy have shown poor efficacy,21 prompting their evaluation in combination therapies and/or developing PI3K-based multitarget drugs.19
Figure 1.
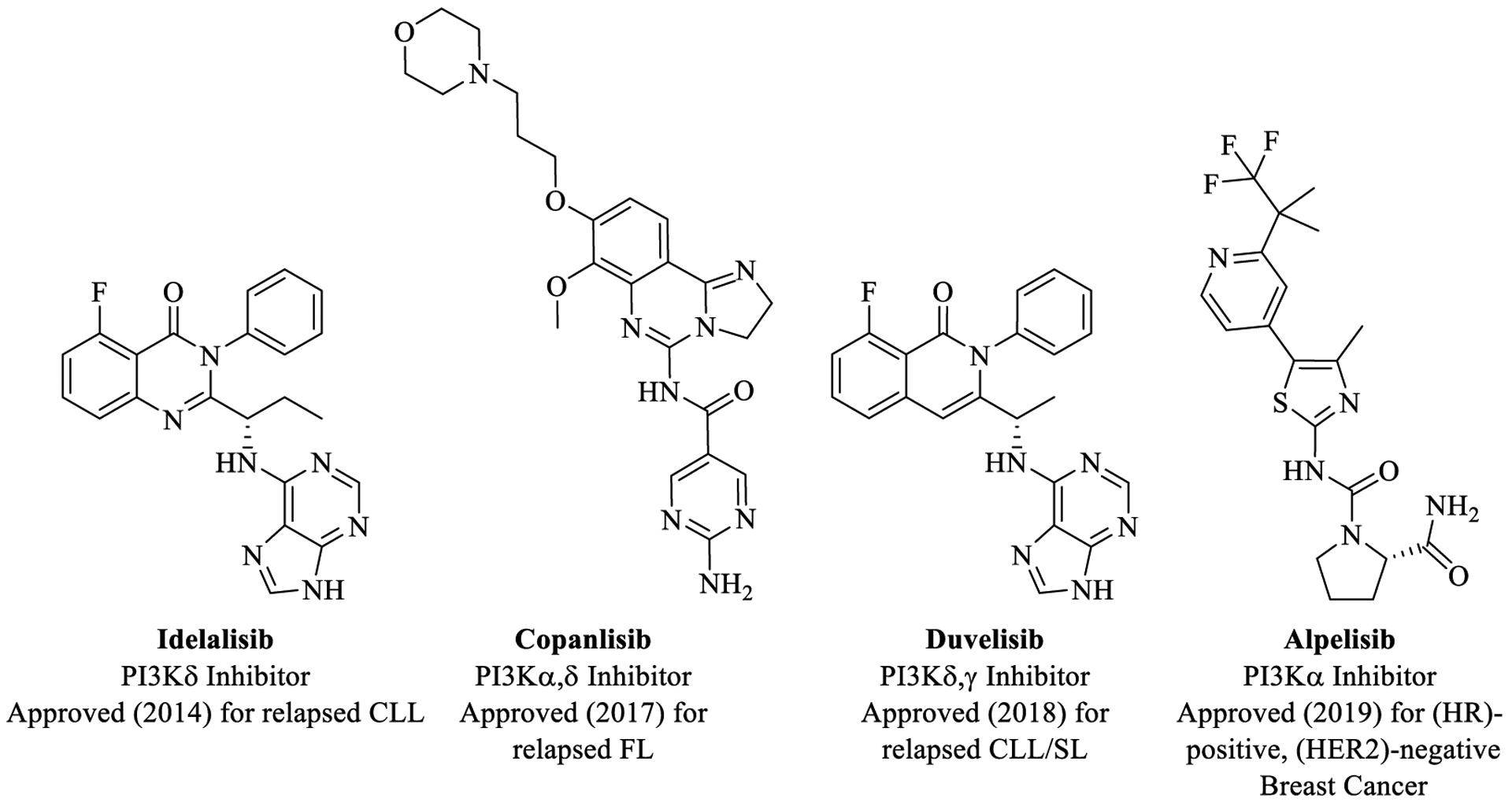
FDA Approved PI3K inhibitor drugs for cancer (CLL = chronic lymphocytic leukemia; FL = follicular lymphoma; SLL = small lymphocytic lymphoma).
Histone deacetylases (HDACs) are an important class of epigenetic enzymes that regulate gene expression by removing acetyl groups from ε-amino lysine residues on histones.22 HDACs play an important role in regulating expression of various proteins, including tumor suppressors (p53, p21, etc.) and transcription factors (e.g., TFIIE, TCF, SF1, etc.).23 Dysregulation of HDACs is involved in cancer initiation and proliferation.24 HDAC inhibition has emerged as a therapeutic approach for cancer and several inhibitors, including Vorinostat (SAHA), Romidepsin, Belinostat, Panobinostat, and Chidamide, have been approved in recent years (Figure 2A).25 Pan-HDAC inhibitors, such as Panobinostat and Vorinostat (SAHA), modulate the acetylation status of a wide range of protein targets leading to a therapeutic response; however, these molecules also have undesirable effects, including hematological, gastrointestinal, and cardiac toxicity.25–27 SAHA monotherapy was approved by the U.S. Food and Drug Administration (FDA) in 2006 for the treatment of cutaneous T-cell lymphoma (CTCL), however, it has been demonstrated to have little efficacy.28
Figure 2.
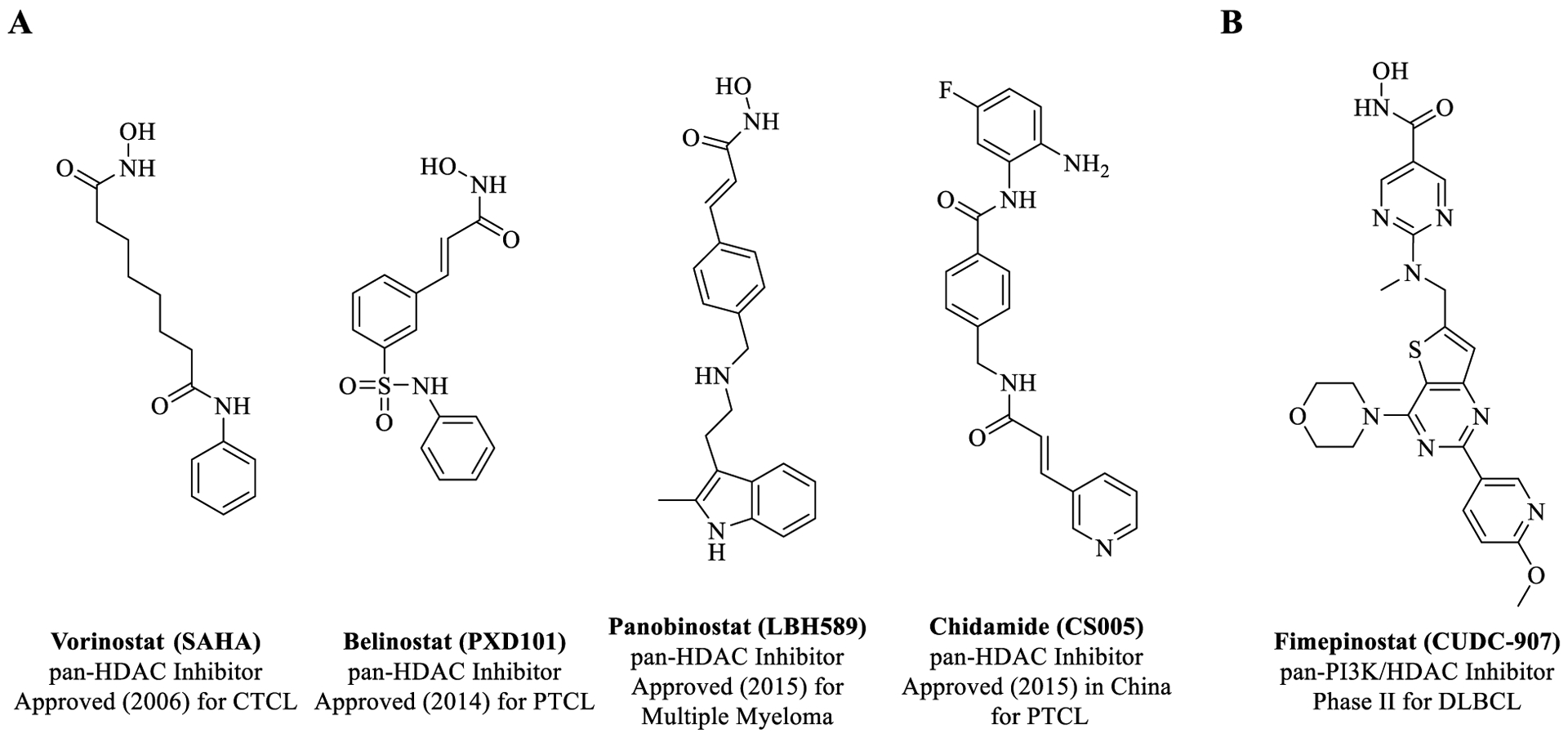
(A) Selected approved HDAC inhibitor drugs for cancer. (B) Chemical structure of dual PI3K/HDAC inhibitor, Fimepinostat (CTCL = cutaneous T-cell lymphoma; PTCL = peripheral T-cell lymphoma; DLBCL = diffuse large B-cell lymphoma).
Although both PI3K and HDAC inhibitor drugs are limited by insufficient efficacy and resistance mutations, there is strong evidence that simultaneous inhibition of both PI3K and HDAC can synergistically inhibit tumor growth and address these limitations by improving efficacy, limiting resistance, and providing a better therapeutic window than single inhibitors.28–30 Selective inhibitors of specific isoforms of HDAC and PI3K potentially would have a better toxicity profile and therefore bigger therapeutic window.31 In this context, several dual PI3K/HDAC inhibitors have been recently reported in literature,32–34 with Curis Inc.’s Fimepinostat (CUDC-907) currently being evaluated in phase 2 clinical trials for treatment of diffuse large B-cell lymphoma (DLBCL) [Figure 2B].19,35,36 Dual inhibitors, such as CUDC-907, may thus offer improved therapeutic benefit through synergistic effects in inhibiting cancer cell proliferation and their compensatory pathways for survival.19,35 However, CUDC-907 exhibits pan-HDAC and pan-PI3K inhibition, which might lead to toxicity and low tolerability.37,38 Thus, there remains an unmet need for new dual inhibitors having high potency and selectivity.
Herein we report the design, synthesis, and biological evaluation of potent and selective dual PI3K/HDAC inhibitors.
RESULTS AND DISCUSSION
Design of Dual Inhibitors.
The rational design of our PI3K/HDAC dual inhibitors commenced by analyzing the crystal structures of Idelalisib (1) bound to PI3Kδ [PDB 4XE0]39 (Figure 3A) and Vorinostat (2) bound to HDAC2 [PDB 4LXZ]40 (Figure 3B), respectively. As shown in Figure 3A, the purine moiety in Idelalisib binds to the hinge (Glu826, Val828) and the quinazolinone core extends up into a pocket formed by the G loop residues, Trp760 and Met752. The carbonyl and fluoro group in the quinazolinone core at fourth and fifth positions, respectively, extend into the solvent exposed area, and therefore, serve as desired locations to attach the HDAC pharmacophore. The phenyl ring on the nitrogen in the quinazolinone core is largely solvent exposed (on top) but makes hydrophobic contacts underneath to methyl groups on Thr833 and Met900. In the Vorinostat-HDAC2 structure (Figure 3B), Zn2+ is penta-coordinated (Asp181, Asp260, His183, and both oxygens on hydroxamic acid) and the hydroxamic acid group forms hydrogen bonds to two charge relay histidines (His145 and His146) and a nearby Tyr308. The Vorinostat aliphatic chain traverses a hydrophobic portion of the pocket (Leu144, Phe155). The phenyl group forms a hydrogen bond to the rim of the pocket at Asp104 via the amino nitrogen.
Figure 3.
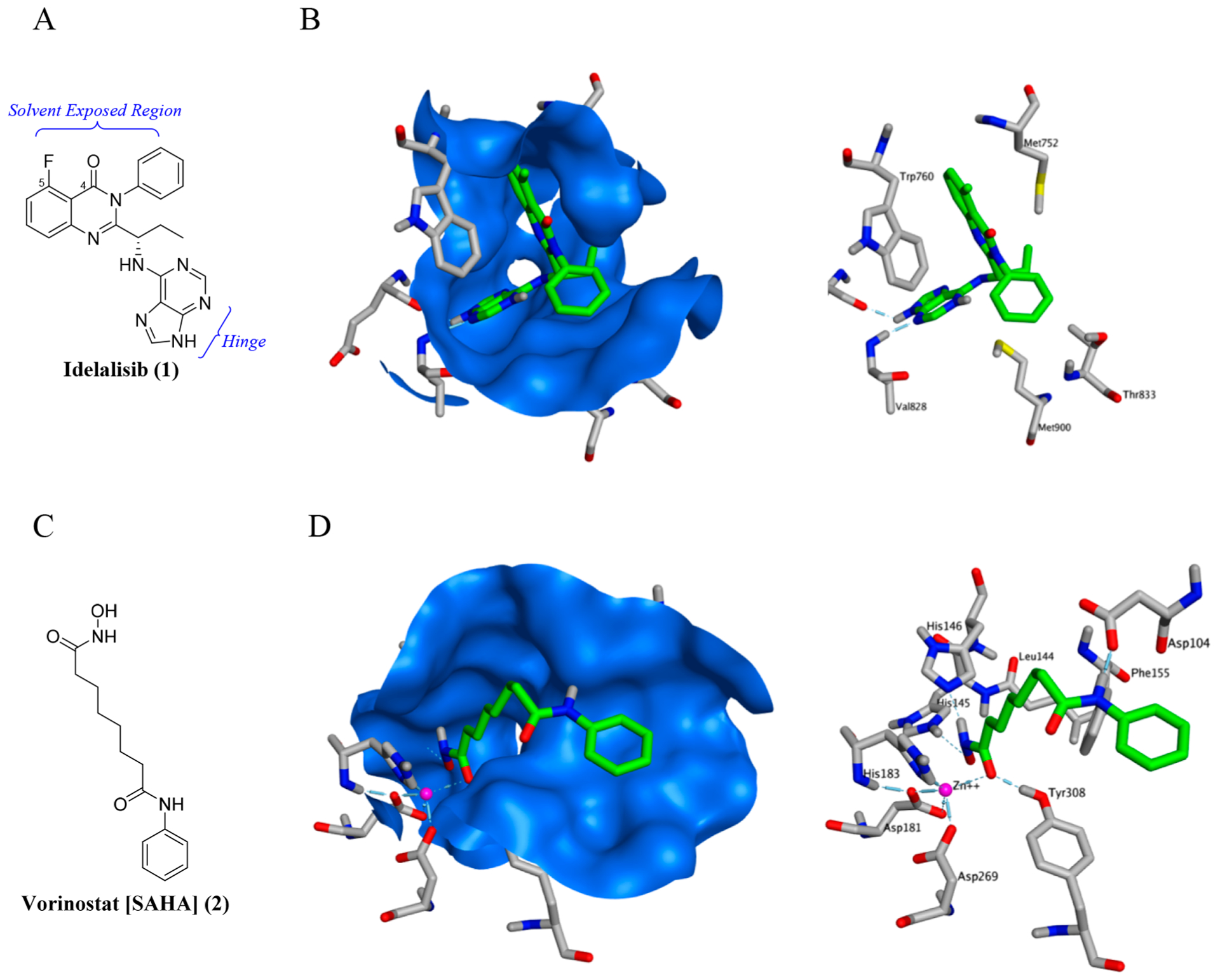
(A) Chemical structure of the PI3Kδ inhibitor, Idelalisib (1). (B) Crystal structures of Idelalisib (1) [top view] bound to PI3Kδ (PDB 4XE0) with protein surface (blue) and without protein surface, respectively. (C) Chemical structure of the HDAC inhibitor, Vorinostat (2). (D) Crystal structures of Vorinostat (2) bound to HDAC2 (PDB 4LXZ) with protein surface (blue) and without protein surface, respectively.
In general, most of the HDAC inhibitors, including Vorinostat, share these three common structural components: a zinc binding group (e.g., hydroxamic acid), a linker occupying the hydrophobic tunnel in the active site, and a cap group that sits outside the hydrophobic tunnel and interacts with the HDAC surface residues (Figure 4).41 More importantly, the HDAC inhibitors can accommodate a wide variety of cap groups, which allows for replacement of the cap with a second target pharmacophore to design HDAC inhibitor based multitarget drugs.32,33,42–50 Thus, by assessing the binding features of Idelalisib and Vorinostat in PI3Kδ and HDAC2 enzymes, respectively, we designed quinazoline- and quinazolinone-based dual PI3K/HDAC inhibitors (3 and 4, Figure 4) by incorporating the PI3Kδ inhibitor as the cap group of an HDAC pharmacophore and optimizing for the appropriate linker-hydroxamic acid combination at quinazo-line’s C-4 position (3) or quinazolinone’s C-5 position (4) to achieve potencies against both the targets.
Figure 4.
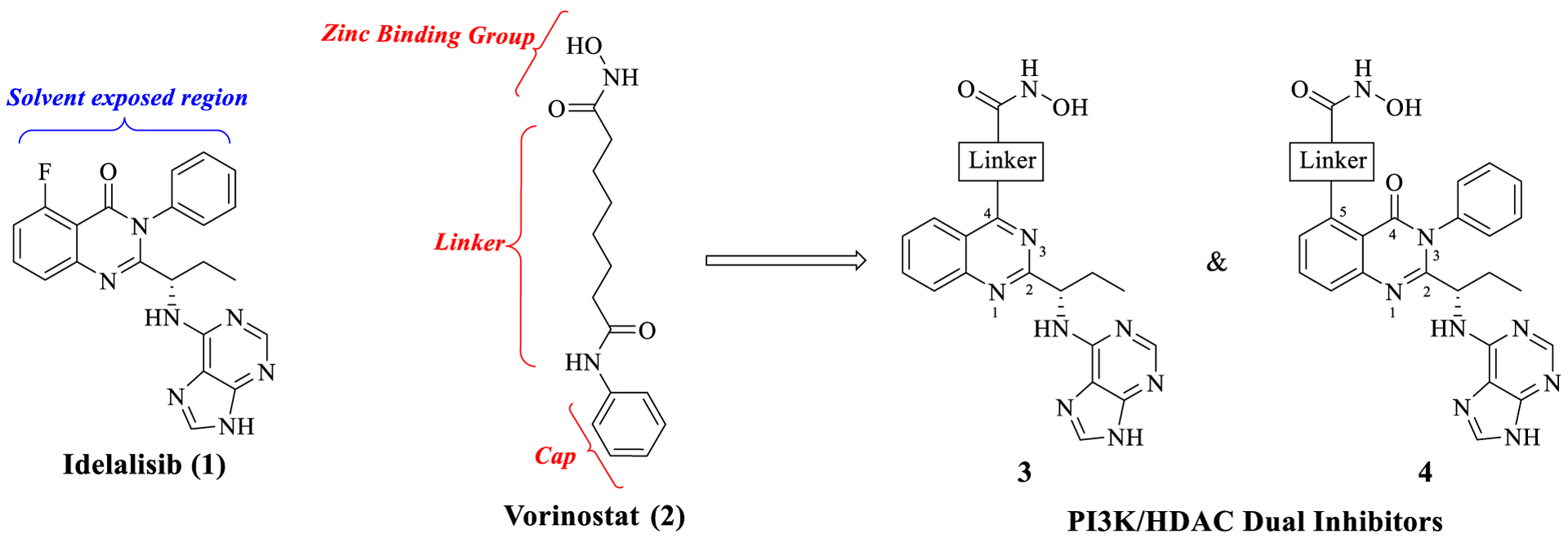
Design strategy for dual PI3K/HDAC inhibitor types 3 and 4.
Chemistry.
The C-4 substituted quinazoline compounds (10a,b and 14a,b) were synthesized from tert-butyl (S)-(1-(4-chloroquinazolin-2-yl)propyl)carbamate 551 in 6–7 steps as outlined in Scheme 1. The compound 5 was subjected to Sonogashira cross-coupling reactions with alkynes 6a and 6b, using a copper-free Pd/PtBu3-catalytic system to afford 7a,b. Reduction of alkyne group to alkane in 7a,b using Pd/C hydrogenation, followed by boc-deprotection and nucleophilic aromatic substitution of 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine 8 with the resulting free amine, yielded compounds 9a,b. Hydrolysis of the alkyl ester in 9a,b to the respective carboxylic acid, followed by coupling with NH2OTHP (THP = tetrahydropyran) and THP-deprotection using trifluoroacetic acid formed the target compounds 10a,b.
Scheme 1.
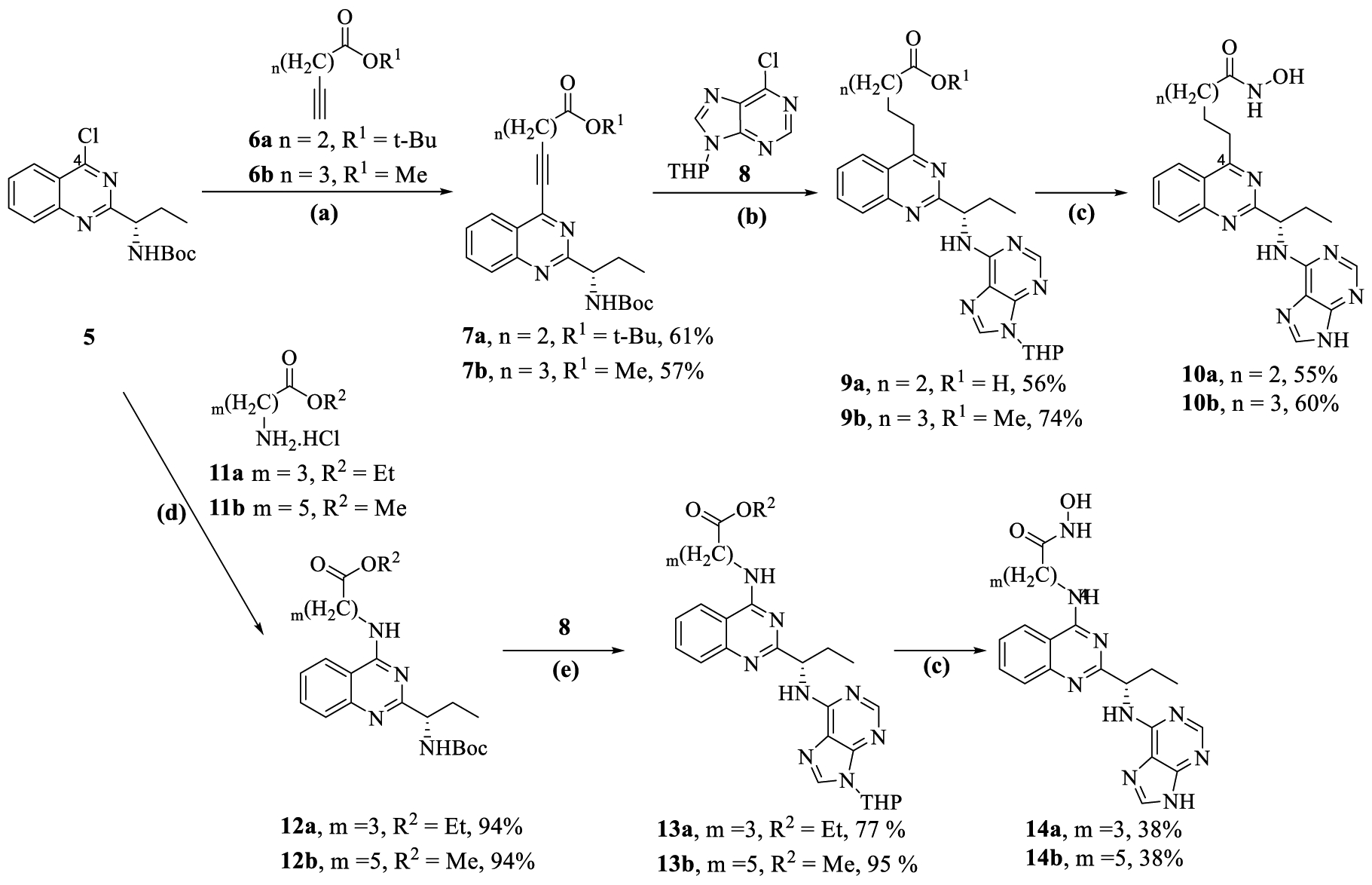
Synthesis of C-4 Alkyl/Amino-alkyl Substituted Quinazolines (10a,b and 14a,b)a
aReagents and conditions: (a) 6a, 6b (1.2 equiv), [PdCl(allyl)]2 (0.05 equiv), PtBu3HBF4 (0.20 equiv), 1,4-dioxane, 16 h, rt; (b) (i) 10 wt % Pd/C, H2 balloon, EtOAc, 20 h, rt, (ii) CF3CO2H (20.0 equiv), DCM, 3 h, rt, (iii) 8 (2.0 equiv), triethyamine (4.0 equiv), EtOH, 1 h, MW, 100 1/2C; (c) (i) LiOH·H2O (2.0 equiv), MeOH/H2O, 10 h, rt, (ii) NH2OTHP (3.1 equiv), N-methyl morpholine (3.0 equiv) 3-(((ethylimino)-methylene)amino)-N,N-dimethylpropan-1-amine hydrochloride [EDC·HCl] (1.4 equiv), 1H-[1,2,3]triazolo[4,5-b]pyridin-1-ol [HOAT] (1.2 equiv), DMF, 16 h rt; (iii) CF3CO2H (20.0 equiv), DCM, 20 h, rt (For 9a, only c(ii) and c(iii) were used); (d) 11a,b (2.0 equiv), triethyamine (4.0 equiv), EtOH, 1 h, 100 1/2C; (e) 8 (2.0 equiv), triethyamine (4.0 equiv), EtOH, 1 h, 100 1/2C.
The synthesis of C-4 amino-alkyl substituted compounds (14a,b) began by subjecting compound 5 to nucleophilic aromatic substitution with amines 11a,b to yield compounds 12a,b. Boc-deprotection in 12a,b, followed by the installation of a purine kinase hinge binding moiety using nucleophilic aromatic substitution (SNAr) on 8, afforded compounds 13a,b. Target compounds 14ab,b were synthesized from 13a,b by converting alkyl ester to hydroxamic acid, in a fashion similar to that described above for compounds 10a,b.
The procedure for syntheses of C-5 alkyl/aryl substituted quinazolinone-based compounds is depicted in Scheme 2. The quinazolinone compound 15a52 was subjected to Sonogashira cross-coupling reactions with alkynes 16a–c to yield compounds 17a–c. Hydrogenation of alkyne in 17a–c followed by Boc-deprotection and nucleophilic aromatic substitution on purine 8 afforded compounds 18a–c. Target compounds 19a–c were derived from 18a–c by either: (a) hydrolyzing the alkyl ester to carboxylic acid, coupling with NH2OTHP followed by deprotection of THP-protecting groups using TFA, or (b) converting the alkyl ester directly to hydroxamic acid using aq NH2OH/LiOH and subsequent removal of the THP-protecting group using TFA. The latter method also formed carboxylic acid as the minor side product due to ester hydrolysis (see Supporting Information).
Scheme 2.
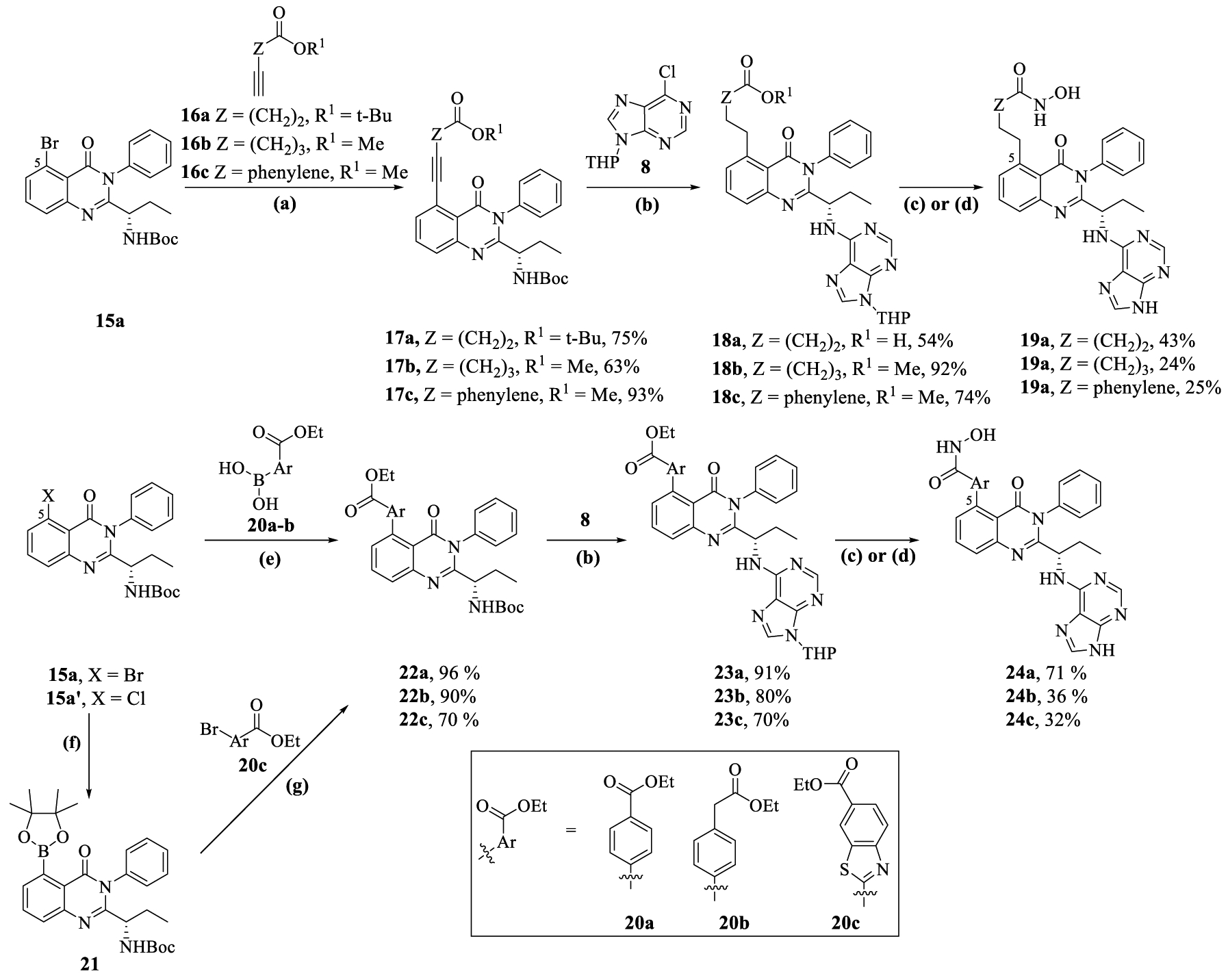
Synthesis of C-5 Alkyl/Aryl Substituted Quinazolinones (19a–c and 24a–c)a
aReagents and conditions: (a) 16a, 16b, 16c (1.2 equiv), [PdCl(allyl)]2 (0.05 equiv), PtBu3HBF4 (0.20 equiv), 1,4-dioxane, 16 h, rt; (b) (i) 10 wt % Pd/C, H2 balloon, EtOAc, 20 h, rt, (ii) CF3CO2H (20.0 equiv), DCM, 3 h, rt, (iii) 8 (1.5–2.5 equiv), triethyamine (3.0–5.0 equiv), EtOH, 4 h, MW, 100 1/2C; (c) (i) LiOH·H2O (2.0 equiv), MeOH/H2O, 10 h, rt, (ii) NH2OTHP (3.1 equiv), N-methyl morpholine (3.0 equiv) 3-(((ethylimino)methylene)amino)-N,N-dimethylpropan-1-amine hydrochloride [EDC·HCl] (1.4 equiv), 1H-[1,2,3]triazolo[4,5-b]pyridin-1-ol [HOAT] (1.2 equiv), DMF, 16 h rt, (iii) CF3CO2H (20.0 equiv), DCM/MeOH, 20 h, rt, (For17a, only c(ii) and c(iii) were used); (d) (i) aq 50 wt % NH2OH (30.0 equiv), LiOH·H2O (1.1 equiv), MeOH/H2O, 20 h, 0 1/2C to rt, (ii) CF3CO2H (20.0 equiv), DCM/MeOH, 20 h, rt; (e) boronic acid/ester 20a,b (1.2 equiv), [XPhos Pd(crotyl)Cl] (0.05 equiv), K3PO4 (3.0 equiv), 1,4-dioxane/water, 1–2 h MW, 100 1/2C; (f) [Bpin]2 (1.2 equiv), [Pd(dppf)Cl2] (0.05 equiv), KOAc (3.0 equiv) 1,4-dioxane, 16 h, 100 1/2C; (g) 20c (0.85 equiv), [XPhos Pd(crotyl)Cl] (0.05 equiv), K3PO4 (3.0 equiv), 1,4-dioxane/water, 10 h, 100 1/2C; (h) (i) CF3CO2H (20.0 equiv), DCM, 3 h, rt, (ii) 8 (1.5–2.5 equiv), triethyamine (3.0–5.0 equiv), EtOH, 4 h, 100 1/2C.
For C-5 aryl substituted quinazolinone-based compounds, 5-bromo or 5-chloro-quinazolinone (15a or 15a′) was either subjected to a Suzuki–Miyaura cross-coupling reaction with boronic acids 20a,b or first converted to boronate ester 21 using Pd(dppf)Cl2/Bpin2, followed by Suzuki–Miyaura coupling with aryl bromide 20c, to form compounds 22a–c. These C-5 arylated molecules 22a–c were converted to target compounds 24a–c in a sequence of steps similar to that previously described for compounds 19a–c.
The synthesis of C-5 amino-alkyl/aryl substituted quinazolinones involved the Pd/XantPhos-catalyzed amination of 5-bromo-quinazolinone 15a,b with amines 25a–h, yielding compounds 26a–i (Scheme 3). Subsequent attachment of purine 8 and conversion of alkyl esters to hydroxamic acids afforded the target compounds 28a–h. For compound 28i, the methyl ester in 27d was converted to N-methyl hydroxamic acid via hydrolysis to carboxylic acid, followed by its coupling with N-methyl hydroxylamine using HATU/DIPEA.
Scheme 3.
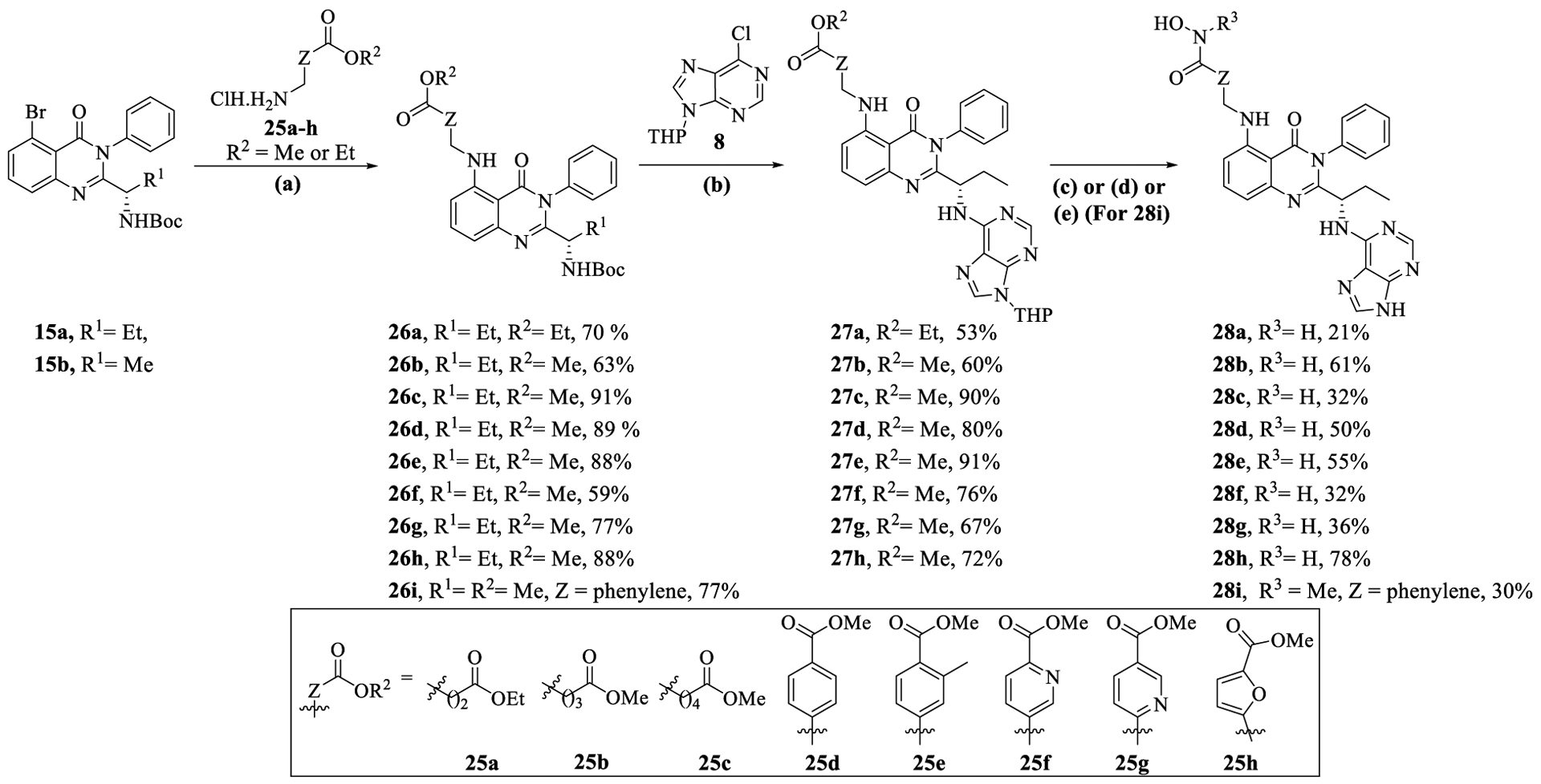
Synthesis of C-5 Amino-alkyl/Aryl Substituted Quinazolinones (28a–i)a
aReagents and conditions: (a) 25a–h (1.3 equiv), [Xantphos Palladacycle Gen. 4] (0.03 equiv), Cs2CO3 (3.0 equiv), toluene or 1,4-dioxane, 16 h, rt; (b) (i) CF3CO2H (20.0 equiv), DCM, 3 h, rt, (ii) 8 (1.5–2.5 equiv), triethyamine (3.0–5.0 equiv), EtOH, 4 h, MW, 100 1/2C; (c) (i) LiOH·H2O (2.0 equiv), MeOH/H2O, 10 h, rt, (ii) NH2OTHP (3.1 equiv), N-methyl morpholine (3.0 equiv) 3-(((ethylimino)methylene)amino)-N,N-dimethylpropan-1-amine hydrochloride [EDC·HCl] (1.4 equiv), 1H-[1,2,3]triazolo[4,5-b]pyridin-1-ol [HOAT] (1.2 equiv), DMF, 16 h rt, (iii) CF3CO2H (20.0 equiv), DCM/MeOH, 20 h, rt; (d) (i) aq 50 wt % NH2OH (30.0 equiv), LiOH·H2O (1.1 equiv), MeOH/H2O, 20 h, 0 1/2C to rt, (ii) CF3CO2H (20.0 equiv), DCM/MeOH, 20 h, rt; (e) (i) LiOH·H2O (1.1 equiv), MeOH/H2O, 20 h, 0 1/2C to rt, (ii) N-methylhydroxylamine hydrochloride (1.5 equiv), HATU (1.5), DIPEA (2.5 equiv), DMF, rt, (iii) CF3CO2H (20.0 equiv), DCM/MeOH, 20 h, rt.
The procedure for synthesizing C-5 alkyl-amino substituted quinazolinones commenced from the Pd-catalyzed cyanation of C-5 bromide in 15a,b and subsequent hydrogenation of the nitrile group to form compounds 30a,b (Scheme 4). The free amine in 30a was reacted with ethyl 2-bromothiazole-4-carboxylate, 31 to yield 32a, which was converted to final compound 33 using a similar sequence of steps as described in previous schemes. For target compounds 37, 40a,b, and 43, compound 29a was subjected to Boc-deprotection and nucleophilic aromatic substitution on purine 8 to afford 34 (Scheme 4). Hydrogenation of the nitrile group in 34 to free amine was utilized in synthesizing a variety of linkers using (a) amide-coupling with 35, (b) reductive amination with 38a,b, and (c) substitution with 41, to form compounds 36, 39a,b, and 42. Subsequent conversion of alkyl esters to hydroxamic acids and removal of the THP protecting groups afforded the target compounds, 37, 40a,b, and 43.
Scheme 4.
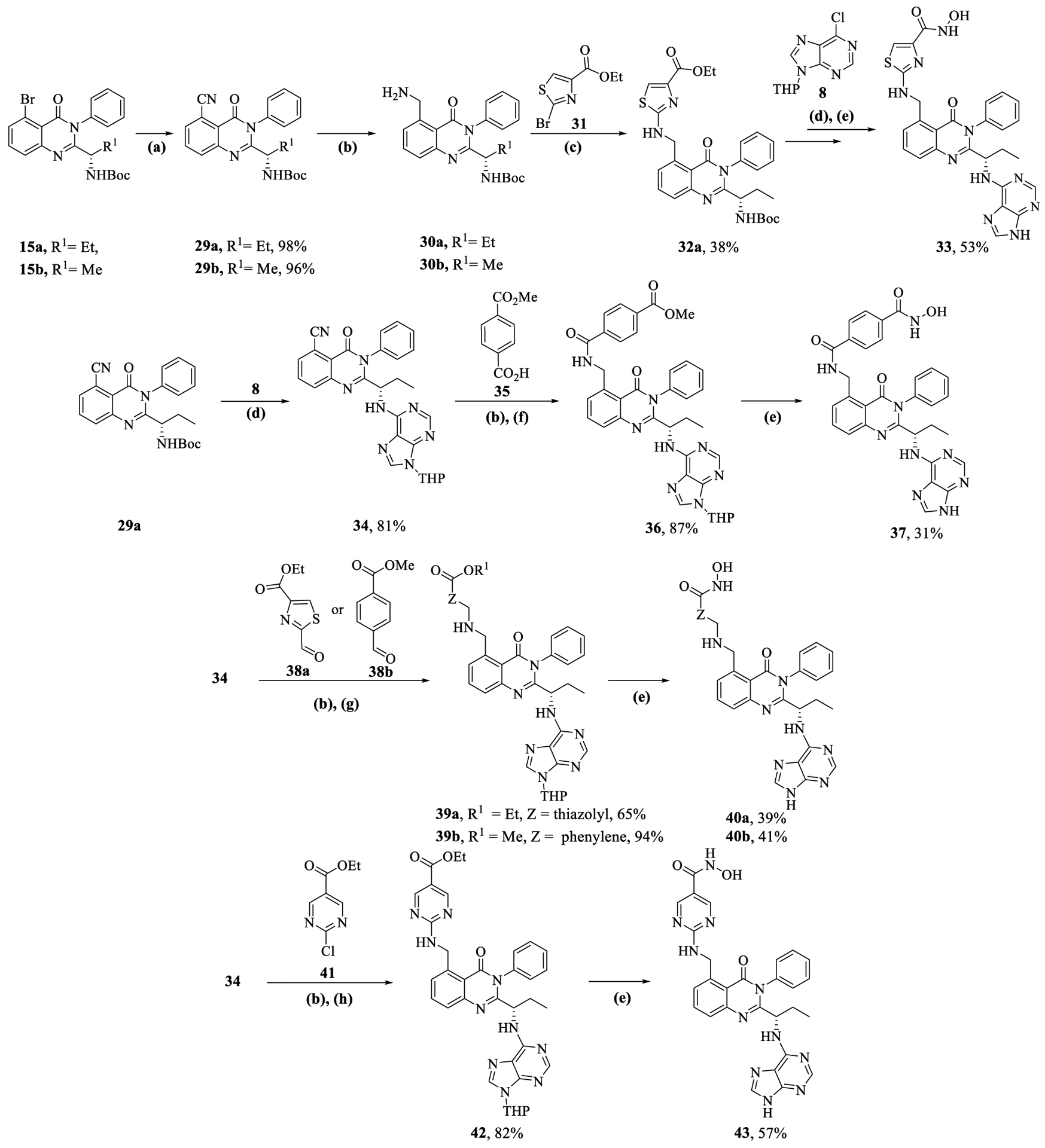
Synthesis of C-5 Alkyl-amino Substituted Quinazolinones (33, 37, 40, and 43)a
aReagents and conditions: (a) Pd(PPh3)4 (0.05 equiv), Zn(CN)2 (1.2 equiv), DMF, 100 1/2C, 16 h; (b) Raney Ni, H2, MeOH, 20 h; (c) 31 (2.0 equiv) DIPEA (4.0 equiv), DMF, 180 1/2C, MW, 30 min; (d) (i) CF3CO2H (20.0 equiv), DCM, 3 h, rt, (ii) 8 (1.5–2.5 equiv), triethyamine (3.0–5.0 equiv), EtOH, 4 h, MW, 100 1/2C; (e) (i) LiOH·H2O (2.0 equiv), MeOH/H2O, 10 h, rt, (ii) NH2OTHP (3.1 equiv), N-methyl morpholine (3.0 equiv) 3-(((ethylimino)methylene)amino)-N,N-dimethylpropan-1-amine hydrochloride [EDC·HCl] (1.4 equiv), 1H-[1,2,3]triazolo[4,5-b]pyridin-1-ol [HOAT] (1.2 equiv), DMF, 16 h rt, (iii) CF3CO2H (20.0 equiv), DCM/MeOH, 20 h, rt; (f) 35 (1.5 equiv), HATU (1.2 equiv), DIPEA (3.0 equiv), DMF, 16 h, rt; (g) 38 (1 equiv), cat. AcOH, DCE, 2 h, then NaBH(OAC)3 (3.0 equiv), 2 h; (h) 41 (2.0 equiv), triethylamine (4.0 equiv), EtOH, 100 1/2C, MW, 3 h.
C-5 alkyl-substituted quinazolinones with pyrimidine kinase hinge binding groups (46a–d) were readily synthesized from 17b,c in four steps as shown in Scheme 5. The alkyne group in 17b,c was hydrogenated using Raney-Ni followed by Bocdeprotection and SNAr on chloropyrimidines 44a–c to form compounds 45a–d. Subsequent conversion of methyl esters in 45a–d to hydroxamic acids using hydroxylamine afforded the target compounds 46a–d.
Scheme 5.
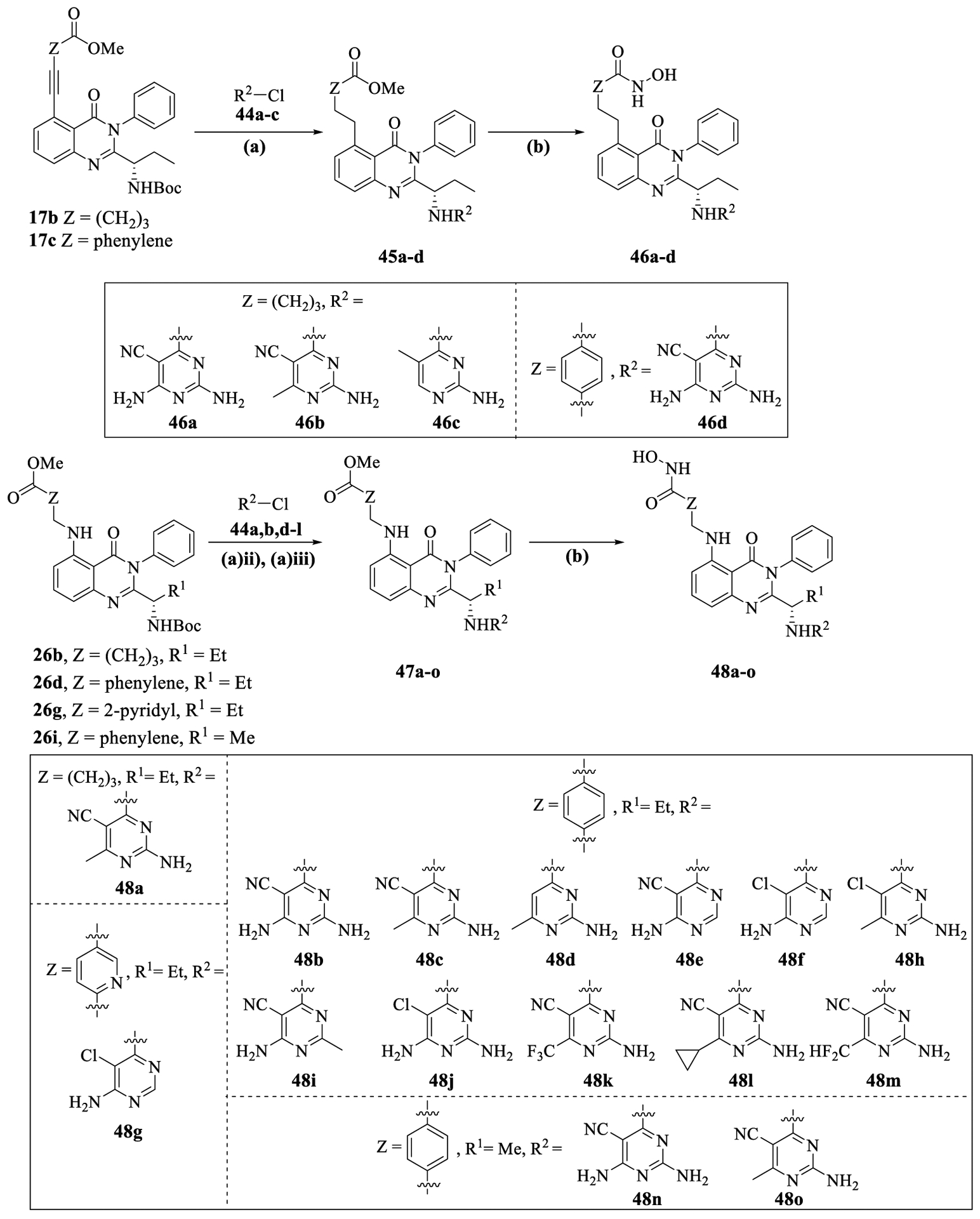
Synthesis of C-5 Substituted Quinazolinones with Pyrimidine Hinge Binding Groups (46a–d and 48a–o)a
aReagents and conditions: (a) (i) 10 wt % Pd/C, H2 balloon, EtOAc, 20 h, rt, (ii) CF3CO2H (20.0 equiv), DCM, 3 h, rt, (iii) 44 (1.5–2.0 equiv), DIPEA (3.0–4.0 equiv), n-butanol, 2–3 h, MW, 130 1/2C, 38–88% (3 steps); (b) aq 50 wt % NH2OH (30.0 equiv), LiOH·H2O (1.1 equiv), MeOH/H2O, 20 h, 0 1/2C to rt; 36–82%.
C-5 amino-alkyl substituted quinazolinones (48a–o) were synthesized in a similar fashion from compounds 26b,d,g,i by attaching the pyrimidine kinase binding moieties using SNAr reactions on 44a,b,d–l and converting methyl esters in 47a–o to hydroxamic acids.
For synthesis of quinazolinones 51 and 54, the nitrile group in compound 29b was hydrogenated using Raney Ni/H2 to generate free amine, which was either coupled to 4-(methoxycarbonyl)benzoic acid 35 using HATU/DIPEA or subjected to SNAr reaction with ethyl 2-chloropyrimidine-5-carboxylate 41 to form compounds 49 and 52, respectively (Scheme 6). Subsequent attachment of the pyrimidine kinase hinge binding group and transformation of alkyl esters to hydroxamic acids yielded the target compounds 51 and 54.
Scheme 6.
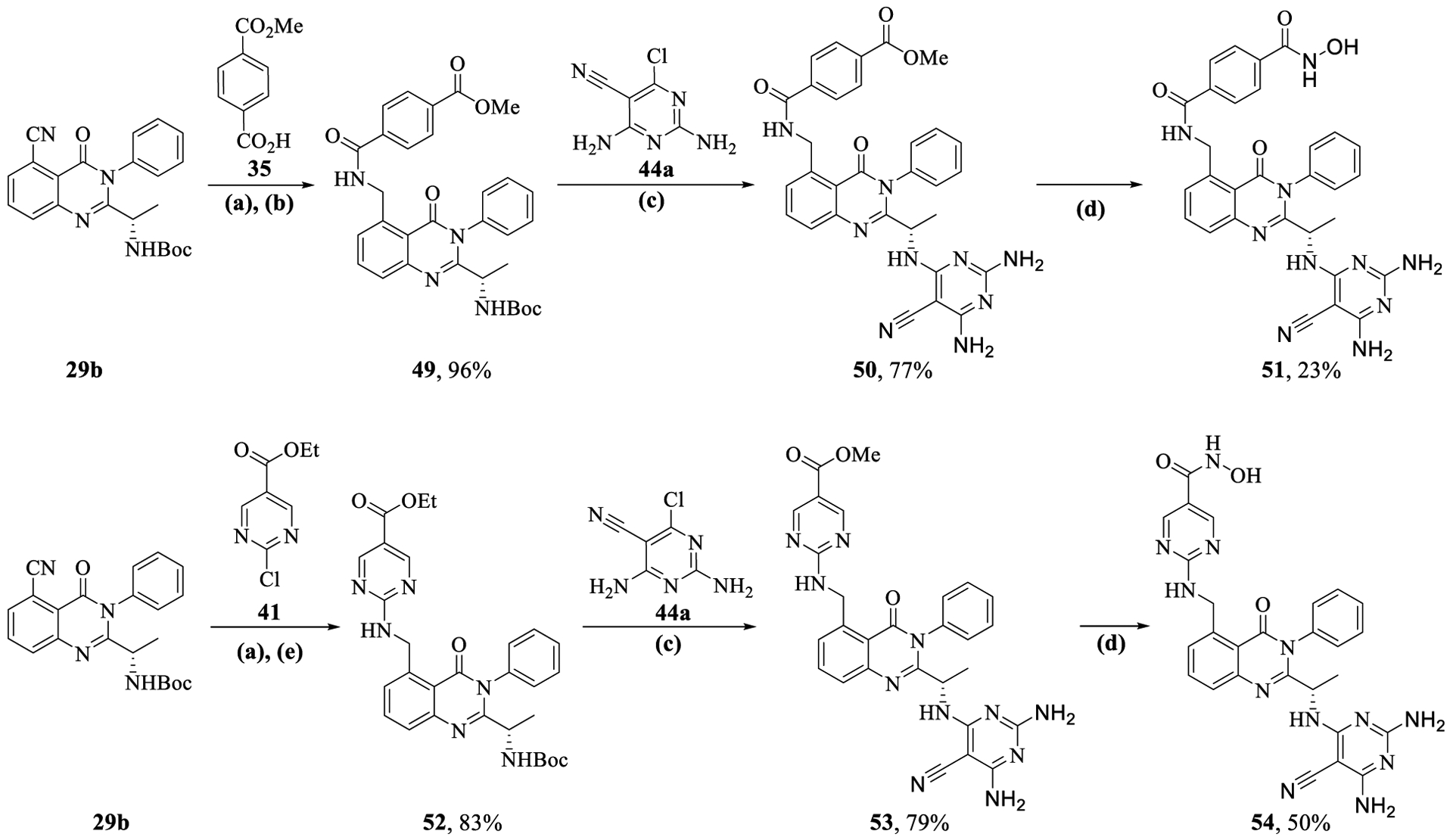
Synthesis of C-5 Alkyl-amino Substituted Quinazolinones (51 and 54)a
aReagents and conditions: (a) Raney Ni, H2, MeOH, 20 h; (b) 35 (1.5 equiv), HATU (1.2 equiv), DIPEA (3.0 equiv), DMF, 16 h; (c) (i) CF3CO2H (20.0 equiv), DCM, 3 h, rt, (ii) 44a (1.5 equiv), DIPEA (3.0 equiv), n-butanol, 2–5 h, MW, 130–150 1/2C; (d) aq 50 wt % NH2OH (30.0 equiv), LiOH·H2O (1.1 equiv), MeOH/H2O, 20 h, 0 1/2C to rt; (e) 41 (1.5 equiv), DIPEA (3.0 equiv), n-butanol, 2 h, MW, 130 1/2C.
PI3K and HDAC: Enzyme Inhibition Activity and SAR.
The dual inhibitors were screened against PI3K and HDAC enzymes at Reaction Biology Corporation, Malvern, PA, USA. Initially, we compared the enzyme inhibitory activities of 4-substituted quinazolines vs 5-substituted quinazolinones by screening against PI3Kδ and HDAC1–11 enzymes (Table 1). In general, 5-substituted quinazolinones were consistently found to be more potent than the 4-substituted quinazolines for PI3K and HDAC enzyme inhibition. As shown for a few examples in Table 1, 5-substituted quinazolinones having 4- and 5-carbon alkylene linkers (19a, 19b) and amino-propylene linker (28a), showed more potent PI3K/HDAC activities than their 4-substituted quinazoline counterparts (10a, 10b, 14a). Additionally, the 5-substituted compounds showed high potency and selectivity for HDAC6 inhibition, leading us to focus only on 5-substituted quinazolinones.
Table 1.
Enzyme Inhibitory Activities of 4-Substituted Quinazolines (10a,b, 14a) vs 5-Substituted Quinazolinones (19a,b, 28a)a
 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | ||||||||||||
| HDACs | ||||||||||||
| compd | PI3Kδ | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| 10a | 49 | 5846 | 18190 | 7075 | 2169 | 1937 | 33730 | 23030 | ||||
| 19a | <5 | 429 | 1231 | 427 | 70910 | 23610 | 36 | 90810 | 385 | >105 | 2073 | 2089 |
| 10b | 68 | 5390 | 14100 | 10600 | >105 | 43700 | 39 | 353 | 16900 | 9819 | ||
| 19b | 0.2 | 388 | 11290 | 8106 | 5 | 24400 | 253 | 34850 | 1502 | 2155 | ||
| 14a | 520 | >105 | >105 | >105 | >105 | 431 | 6194 | 74140 | ||||
| 28a | 46 | 1575 | 3247 | 1031 | 9087 | 37 | 1758 | NT | 3362 | |||
| PI-103 | 3 | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| TSA | NT | 9 | 25 | 14 | NT | NT | 3 | NT | 371 | NT | 61 | 3470 |
| TMP 269 | NT | NT | NT | NT | 281 | 333 | NT | 108 | NT | 49 | NT | NT |
Compounds were tested in singlet 10-dose IC50 mode with 3-fold serial dilution starting at 1 μM for PI3Kδ and 10 μM for HDAC1–11 enzymes. Empty cells indicate no inhibition or compound activity that could not be fit to an IC50 curve. NT = Compound not tested against enzyme. PI-103 was used as a control compound for PI3Kδ, whereas trichostatin A (TSA) and TMP 269 were used as control compounds for HDAC enzymes.
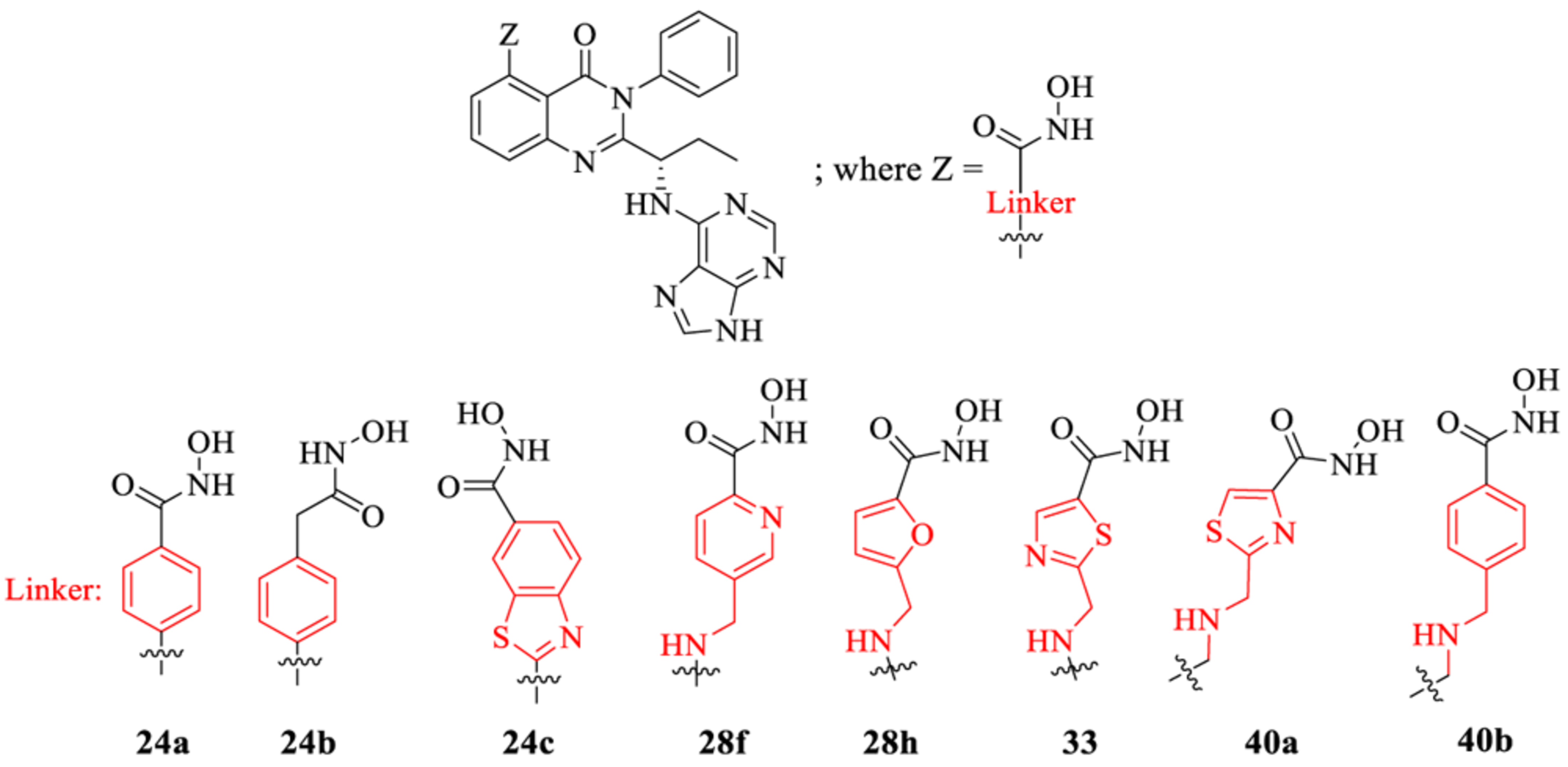
Next, we explored the linker region on 5-substituted quinazolinones by incorporating a variety of aryl, heteroaryl, and amino-aryl groups on the PI3K core. Initially we screened against PI3K isoforms (α, β, γ, δ) and HDAC isoforms (1, 2, 3, 6, 8, and 10) at a single dose (1 μM for PI3Ks and 10 μM for HDAC enzymes). As shown in Table 2, dual inhibitors with a variety of linkers were synthesized and tested to establish SAR. Dual inhibitors with aryl (24a) and heteroaryl linkers such as benzothiazole (24c) showed high potency for PI3Kδ and HDAC6, 8, whereas benzyl linker (24b) only showed high PI3Kδ activity. The use of amino-alkyl heterocycle linkers such as 3-(methylamino) pyridine (28f) and 2-(methylamino) thiazole (33) showed good inhibition for PI3K and HDAC6, but the corresponding alkyl-amino furan (28h) lost its potency against both enzymes. Linkers containing basic amines (40a,b) exhibited modest to high HDAC6 activity (80% for 40a, 99% for 40b) but were inactive against PI3Ks. Select dual inhibitors that showed high single-point potency (% inhibition) in both PI3Kδ and HDAC6 were subsequently tested in 10-dose IC50 titration against PI3Ks (α, β, γ, δ) and HDAC 1–11 isozymes.
Table 2.
PI3K/HDAC Enzyme Inhibition (%) Activities of 5-Substituted Quinazolinones with Purine as the Hinge Binding groupa
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| inhibition (%) at 1 μM | inhibition (%) at 10 μM | |||||||||
| PI3K | HDAC | |||||||||
| compd | α | β | γ | δ | 1 | 2 | 3 | 6 | 8 | 10 |
| 24a | 84.5 ± 0.3 | 44.0 ± 0.6 | 81.6 ± 0.2 | 94.7 ± 0.02 | 63.3 ± 3.2 | 52.3 ± 0.4 | 48.2 ± 0.9 | 93.3 ± 0.3 | 99.1 ± 0.1 | 54.0 ± 0.3 |
| 24b | 64.1 ± 1.0 | 37.5 ± 2.4 | 79.1 ± 0.02 | 95.8 ± 1.3 | −0.9 ± 2.8 | 11.5 ± 0.1 | −13.4 ± 0.3 | 70.7 ± 2.9 | 70.1 ± 0.6 | 20.0 ± 0.6 |
| 24c | 84.7 ± 0.2 | 38.6 ± 1.4 | 88.0 ± 0.6 | 92.6 ± 0.6 | 50.0 ± 2.3 | 40.5 ± 1.7 | 20.9 ± 7.2 | 95.6 ± 0.4 | 98.2 ± 0.3 | 33.1 ± 0.1 |
| 28f | 45.8 ± 2.7 | 4.4 ± 4.3 | 39.9 ± 5.0 | 88.3 ± 0.5 | 20.5 ± 0.3 | 16.3 ± 0.9 | 14.3 ± 1.6 | 90.5 ± 0.7 | 87.2 ± 0.1 | −7.0 ± 1.6 |
| 28h | 18.1 ± 0.4 | 3.8 ± 1.3 | 48.0 ± 1.3 | 61.2 ± 1.0 | 18.3 ± 6.7 | 28.0 ± 3.6 | −6.3 ± 3.2 | 72.3 ± 0.03 | 68.4 ± 3.3 | 17.0 ± 2.8 |
| 33 | 45.6 ± 1.0 | 6.8 ± 0.7 | 59.9 ± 0.4 | 95.2 ± 0.1 | 5.2 ± 3.9 | 4.1 ± 1.1 | 2.0 ± 3.5 | 89.1 ± 0.1 | 58.4 ± 0.8 | −4.6 ± 4.7 |
| 40a | 1.8 ± 0.7 | 5.3 ± 0.3 | −12.2 ± 0.7 | 56.0 ± 1.0 | 24.3 ± 3.3 | 30.4 ± 3.8 | −4.3 ± 1.1 | 79.9 ± 0.7 | 54.5 ± 0.3 | 26.4 ± 0.7 |
| 40b | −6.8 ± 0.2 | −6.0 ± 0.3 | −9.1 ± 3.4 | 20.5 ± 0.7 | 65.6 ± 2.0 | 57.2 ± 5.7 | 19.1 ± 1.4 | 99.2 ± 0.7 | 93.5 ± 0.3 | 57.2 ± 1.2 |
| PI-103 (IC50) | 5 | 13 | 63 | 7 | NT | NT | NT | NT | NT | NT |
| TSA (IC50) | NT | NT | NT | NT | 18 | 48 | 40 | 4 | 519 | 63 |
Compounds were tested in single dose duplicate at 1 μM for PI3Ks and 10 μM for HDAC enzymes. NT = Compound not tested against enzyme. PI-103 was used as a control compound for PI3Kδ, whereas trichostatin A (TSA) was used as control compounds for HDAC enzymes.
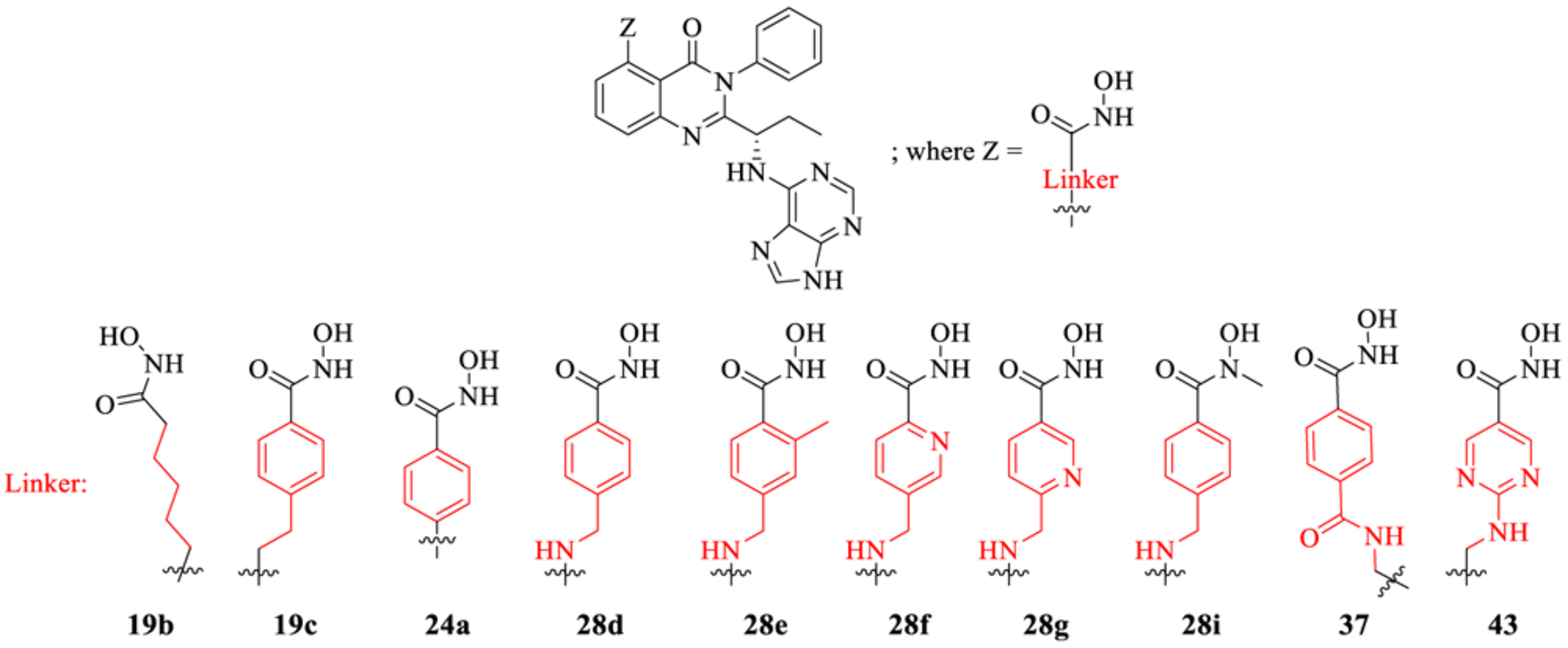
Table 3 shows the PI3K and HDAC enzyme potencies of the 5-substituted quinazolinone with purine hinge binding group. The dual inhibitor containing the pentyl group as linker (19b) exhibited high activity and selectivity for both PI3Kδ (IC50 = 0.2 nM, >210-fold selectivity over PI3Kα,β,γ) and HDAC6 (IC50 = 13 nM, >65 fold-selectivity over HDAC1–5,7–11). Substitution with a homobenzyl (19c) or phenyl linker (24a) showed high PI3Kδ inhibition but poor activity against HDAC6. Swapping the homobenzylic linker with 1-(methylamino) benzyl group (28d) restored HDAC6 activity and selectivity along with good PI3Kδ activity. Interestingly, substituting the 1-(methylamino) benzyl linker in 28d with a 1-(methylamino)-3-methylbenzyl group in 28e maintained the PI3Kδ activity but completely abolished the HDAC activity. A similar trend in PI3Kδ/HDAC6 potency was observed with (3-methylamino)pyridyl linker in 28f, where HDAC6 activity was lost, presumably due to hydrogen-bonding interaction of the pyridyl N atom with O–H group on hydroxamic acid, thereby impacting the chelation of hydroxamic acid with Zn in the HDAC6 enzyme (Figure 5). To further support this hydrogen bonding rationale, we designed and synthesized compound 28g with a (2-methylamino)pyridyl linker, where the possibility of H-bonding interaction with the pyridyl N atom and −OH group of hydroxamic acid was eliminated. Gratifyingly, the HDAC6 potency was restored in 28g (IC50 = 9 nM) when compared with 28f (IC50 = 169 nM) (Figure 5).
Table 3.
PI3K/HDAC Enzyme Inhibitory Activities of 5-Substituted Quinazolinones with Purine Hinge Binding Groupa
 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | |||||||||||||||
| PI3K | HDAC | ||||||||||||||
| compd | α | β | γ | δ | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
| 19b | 122 | 286 | 43 | 0.2 | 1390 | 3030 | 2400 | 8060 | 13 | 856 | 1240 | 1840 | |||
| 19c | NT | NT | NT | 5 | >105 | 889 | 3840 | 9000 | |||||||
| 24a | NT | NT | NT | 0.7 | NT | NT | NT | NT | NT | 362 | NT | 71 | NT | NT | NT |
| 28d | 254 | 103 | 47 | 3925 | 7173 | 6699 | 1751 | 2144 | 5 | 229 | 250 | 3335 | 14700 | 324 | |
| 28e | 517 | 203 | 41 | NT | NT | 2699 | 6922 | NT | NT | ||||||
| 28f | NT | NT | NT | 43 | NT | NT | NT | NT | NT | 169 | NT | NT | NT | NT | NT |
| 28g | 392 | 135 | 26 | 6067 | 10990 | 381 | 439 | 9 | 104 | 204 | 922 | 9996 | 2255 | ||
| 28i | 387 | 162 | 81 | ||||||||||||
| 37 | 266 | 234 | 0.6 | 8434 | 10 | 23120 | 138 | 19890 | 1778 | ||||||
| 43 | 422 | 995 | 49 | 67870 | 25480 | 1428 | 3827 | 6 | 60 | 320 | 5349 | ||||
| PI-103 | 4 | 7 | 81 | 8 | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| TSA | NT | NT | NT | NT | 10 | 34 | 19 | NT | NT | 2 | NT | 445 | NT | 45 | 4826 |
| TMP 269 | NT | NT | NT | NT | NT | NT | NT | 244 | 278 | NT | 62 | NT | 19 | NT | NT |
Compounds were tested in singlet 10-dose IC50 mode with 3-fold serial dilution starting at 1 μM for PI3Kα, β, γ, δ, and 10 μM for HDAC1–11 enzymes. Empty cells indicate no inhibition or compound activity that could not be fit to an IC50 curve. NT = Compound not tested against enzyme. PI-103 was used as a control compound for PI3Ks, whereas trichostatin A (TSA) and TMP 269 were used as control compounds for HDAC enzymes.
Figure 5.
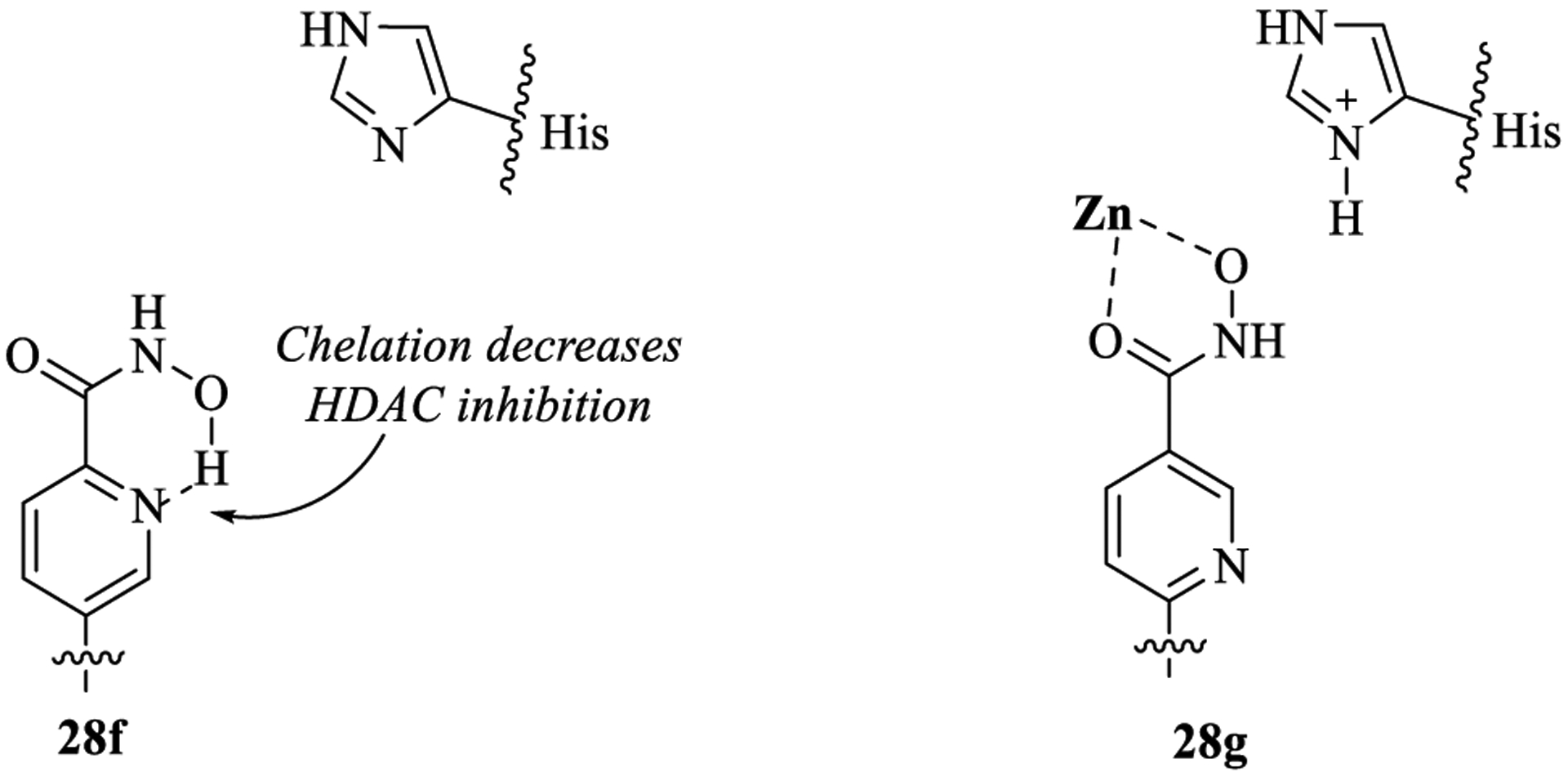
Hydrogen bonding rationale for HDAC activity comparison in 28f and 28g.
Substituting the hydroxamic acid in 28d to N-methyl hydroxamic acid as zinc-binding group in 28i led to a 2-fold drop in PI3Kδ potency and complete loss in activity against all HDACs tested. The dual inhibitor containing N-methylbenzamide (37) as the linker resulted in high activity and selectivity in both PI3Kδ and HDAC6. Compound 43 with a 2-(aminomethyl)-pyrimidine linker also showed good potency and selectivity against PI3Kδ and HDAC6 inhibition.
Despite high activity and selectivity observed with several 5-substituted quinazolinones against PI3Kδ/HDAC6, most did not show expected antiproliferative/cell kill activities when tested against the NCI-60 panel (see section titled, “NCI-60 Human Tumor Cell Lines Screen”). We hypothesized that their poor cellular potency might be due to low permeability (predicted by PAMPA permeability)53 generally observed with these dual inhibitors. Table 4 shows the PAMPA permeability values of selected 5-substituted quinazolinones with a purine hinge binding group.
Table 4.
PAMPA Permeability Data of Selected 5-Substituted Quinazolinones with Purine Hinge Binding Groupa
| compd | PAMPA permeabilitya,b |
|---|---|
| 19b | <1 |
| 24a | 1.2 ± 0.2 |
| 24c | 2.4 ± 0.1 |
| 28d | 6.4 ± 0.8 |
| 28e | 2.6 ± 0.1 |
| 33 | 1.3 ± 0.4 |
| 37 | <1 |
| 40b | <1 |
| 43 | <1 |
Average of two runs ± standard deviation (SD). PAMPA permeability values were determined at pH 7.4 (10−6 cm/s).
No SD values provided if all PAMPA permeability values are less than 1.
To improve upon the cellular potencies of 5-substituted quinazolinones, we investigated a new series of dual inhibitors by substituting the purine kinase hinge binding moiety with amino-pyrimidine groups.52,54–56 Table 5 shows the PI3K and HDAC enzyme inhibition data for selected 5-substituted quinazolinones with amino-pyrimidine hinge binding groups. Compounds 46a,b with 2,4-diamino-6-pyrimidine-5-carbon-itrile and 2-amino-6-methylpyrimidine-5-carbonitrile hinge binding groups, respectively, showed excellent PI3Kδ activity (IC50 = 0.2–0.7 nM), but a weaker HDAC6 potency (IC50 = 28–69 nM), when compared with compound 19b from the purine series (PI3Kδ IC50 = 0.2 nM, HDAC6 IC50 = 13 nM). Compound 46d with a homobenzyl linker and 2,4-diamino-6-pyrimidine-5-carbonitrile hinge binding group was highly active against PI3Kδ (IC50 = 0.2 nM) but showed a modest potency for HDAC6 (IC50 = 77 nM). Dual inhibitors with amino-benzyl/amino-methylpyridyl linkers were explored with a variety of amino-pyrimidine kinase hinge binders to establish SAR. Replacement of the pentyl linker in 46b with aminobutyl linker in 48a with the same hinge binding group showed high potencies for PI3Kγ,δ but no activity against HDACs. Compound 48b with 2,4-diamino-6-pyrimidine-5-carbonitrile hinge binder showed moderate potency for PI3Kγ,δ but high potency and selectivity for HDCA6. The PI3K potency was restored in 48c using a 2-amino-6-methylpyrimidine-5-carbon-itrile hinge binding group while maintaining good potency for HDAC6. Removing the nitrile group in 48c gave compound 48d, which was inactive against PI3K. Dual inhibitors with amino-pyrimidines lacking the 2-amino group in the hinge region (48e,f,g) showed high HDAC6 activity (IC50 = 3–4 nM) but poor PI3Kδ potency (PI3Kδ IC50 = 60 nM for 48e, 133 nM for 48f, 189 nM for 48g). Similar detrimental effects on PI3K potencies were observed when the C-5 nitrile group on the aminopyrimidine was changed to Cl (48h, 48j) and the C-2 amino group was changed to Me (48i). Replacement of the C-6 methyl/amino group in the kinase hinge to CF3, cyclopropyl, and CF2H, respectively, were also not good for PI3K activity (48k, 48l, 48m). Overall, only 2-amino-6-methylpyrimidine-5-carbonitrile and 2,4-diamino-6-pyrimi-dine-5-carbonitrile hinge binding groups were revealed as optimal hinge binders for achieving high potencies for both PI3K and HDAC enzymes. While HDAC6 activity and selectivity obtained by using these amino-pyrimidine hinge binders were similar to their purine hinge binder counterparts, less PI3K selectivity was observed with aminopyrimidine series, with almost equipotent activities against PI3Kγ and PI3Kδ in many examples. Swapping the ethyl group at R1 with a methyl group was tolerated well to obtain compounds (48n, 48o) with single-digit nM activities against PI3Kγ,δ and HDAC6. Compounds having 2,4-diamino-6-pyrimidine-5-carbonitrile as hinge binder with N-methylbenzamide and 2-(amino-methyl)-pyrimidine linkers (51 and 54) also showed high PI3K/HDAC6 activity. In general, dual inhibitors with aminopyrimidine hinge binders showed comparatively better PAMPA permeability (Table 6) than their purine hinge binding counterparts. Additionally, 5-substituted quinazolinones with amino-pyrimidine hinge binders showed potent cell activities when tested in the NCI-60 panel (see section NCI-60 assays and Supporting Information).
Table 5.
PI3K/HDAC Enzyme Inhibitory Activities of 5-Substituted Quinazolinones with Amino-pyrimidine Hinge Bindersa
| IC50 (nM) | HDAC6 isoform selectivityb | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | L | R1 | R2 | PI3K | HDAC6 | ||||
| α | β | γ | δ | ||||||
| 46a |  |
Et | 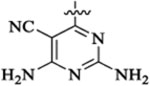 |
52 | 33 | 3 | 0.7 | 69 | >13 |
| 46b | 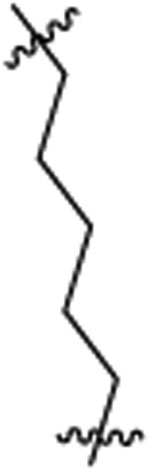 |
Et | 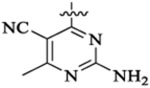 |
9 | 12 | 2 | 0.2 | 28 | >36 |
| 46c | 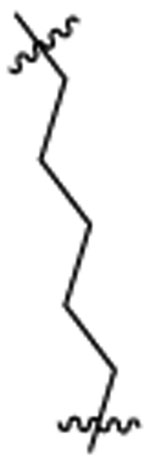 |
Et | 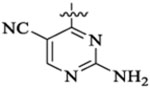 |
5210 | 1769 | 96 | 45 | >22 | |
| 46d | 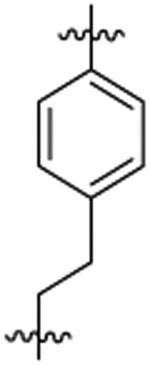 |
Et | 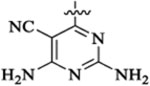 |
13 | 26 | 2 | 0.2 | 77 | >7 |
| 48a | 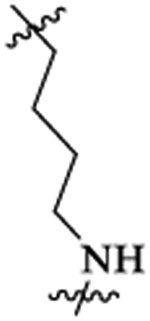 |
Et | 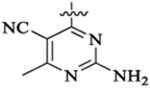 |
186 | 279 | 8 | 5 | 1238 | >2 |
| 48b | 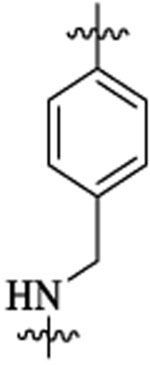 |
Et | 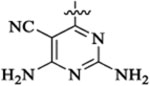 |
50 | 842 | 45 | 32 | 4 | >73 |
| 48c | 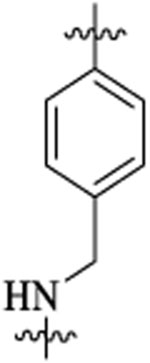 |
Et | 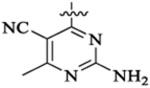 |
47 | 9 | 7.0 | 12 | >39 | |
| 48d |  |
Et | 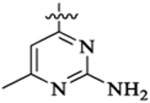 |
4 | >58 | ||||
| 48e |  |
Et | 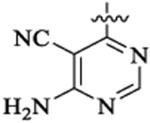 |
444 | 752 | 60 | 4 | >113 | |
| 48f | 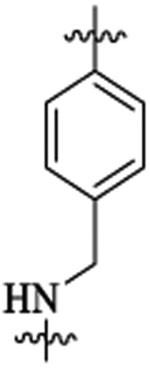 |
Et | 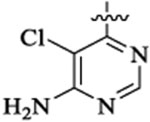 |
133 | 4 | >5 | |||
| 48g | 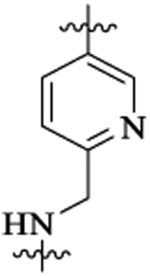 |
Et | 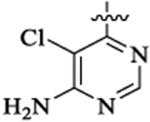 |
189 | 3 | >81 | |||
| 48h | 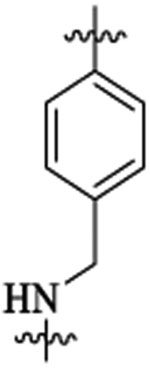 |
Et | 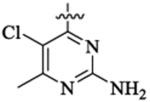 |
663 | 22 | >15 | |||
| 48i | 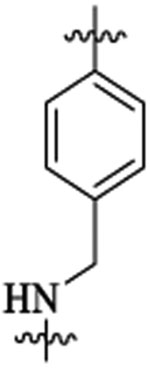 |
Et | 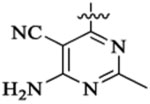 |
491 | 14 | >20 | |||
| 48j | 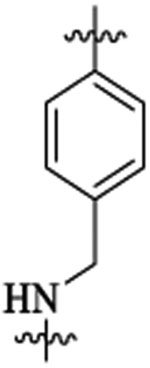 |
Et | 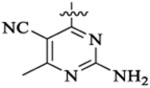 |
269 | 9 | >24 | |||
| 48k | 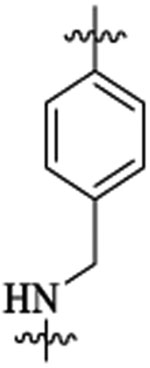 |
Et | 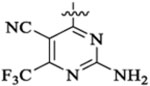 |
52 | >13 | ||||
| 481 |  |
Et | 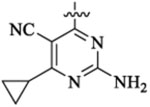 |
47 | >9 | ||||
| 48m |  |
Et | 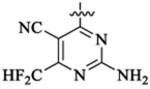 |
222 | 390 | 27 | >6 | ||
| 48n | 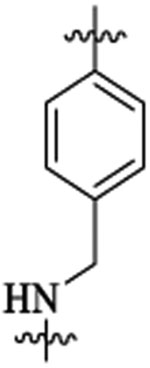 |
Me | 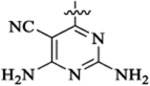 |
42 | 159 | 4 | 4 | 6 | >31 |
| 48o | 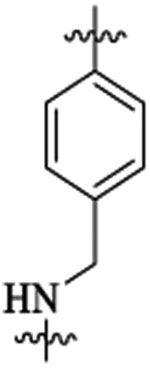 |
Me | 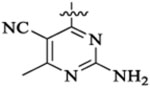 |
75 | 188 | 2 | 4 | 8 | >15 |
| 51 | 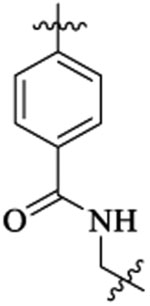 |
Me | 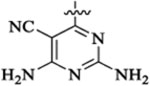 |
18 | 379 | 10 | 0.7 | 16 | >6 |
| 54 | 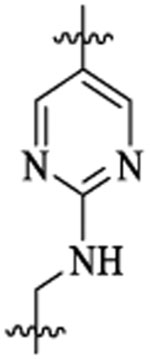 |
Me | 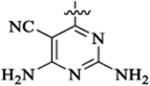 |
42 | 293 | 15 | 3 | 3 | >25 |
| PI-103 | - | - | - | 4 | 7 | 55 | 5 | NT | - |
| TSA | - | - | - | NT | NT | NT | NT | 6 | - |
| TMP269 | - | - | - | NT | NT | NT | NT | NT | - |
Table 6.
PAMPA Permeability Data of Selected 5-Substituted Quinazolinones with Amino-pyrimidine Hinge Binding Groups
| compd | PAMPA permeabilitya,b |
|---|---|
| 46b | 25.3 ± 0.8 |
| 46c | 22.7 ± 7.6 |
| 48b | 4.9 ± 0.1 |
| 48c | 12.0 ± 0.7 |
| 48d | 15.7 ± 5.6 |
| 48f | 50.5 ± 12.6 |
| 48g | 77.5 ± 11.7 |
| 48h | 30.2 ± 3.2 |
| 48i | 12.4 ± 1.2 |
| 48j | 149.1 ± 2.1 |
Compounds were tested in singlet 10-dose IC50 mode with 3-fold serial dilution starting at 1 μM for PI3Kα, β, γ, δ, and 10 μM for HDAC1–11 enzymes. Empty cells indicate no inhibition or compound activity that could not be fit to an IC50 curve. NT = Compound not tested against enzyme. PI-103 was used as a control compound for PI3Ks, whereas trichostatin A (TSA) and TMP 269 were used as control compounds for HDAC enzymes.
Selectivity of HDAC6 inhibition over other HDAC isoforms (1, 2, 3, 4, 5, 7, 8, 9, 10, and 11). Please see Table S1 in Supporting Information for full IC50 data against HDAC1–11.
Average of two runs ± standard deviation (SD). PAMPA permeability values were determined at pH 7.4 (10−6 cm/s).
No SD values provided if all PAMPA permbeability values are less than 1.
Molecular Modeling: HDAC6 Binding and Selectivity.
The dual inhibitor 19b was docked into the solved crystal structure of hHDAC6 (PDB 5EDU.pdb) (Figure 6A).57 The geometry of the protein around the 19b hydroxamic acid headgroup mirrors that of Vorinostat (see Figure 3). The aliphatic linker chain, in similar fashion, traverses a hydro-phobic region of the pocket. The capping group, which is composed of a quinazolinone ring system, on the other hand, forms a hydrogen bond to Ser568 via the quinazolinone carbonyl. This hydrogen bonding interaction on the rim of the pocket is distinctly different than that made by Vorinostat.
Figure 6.
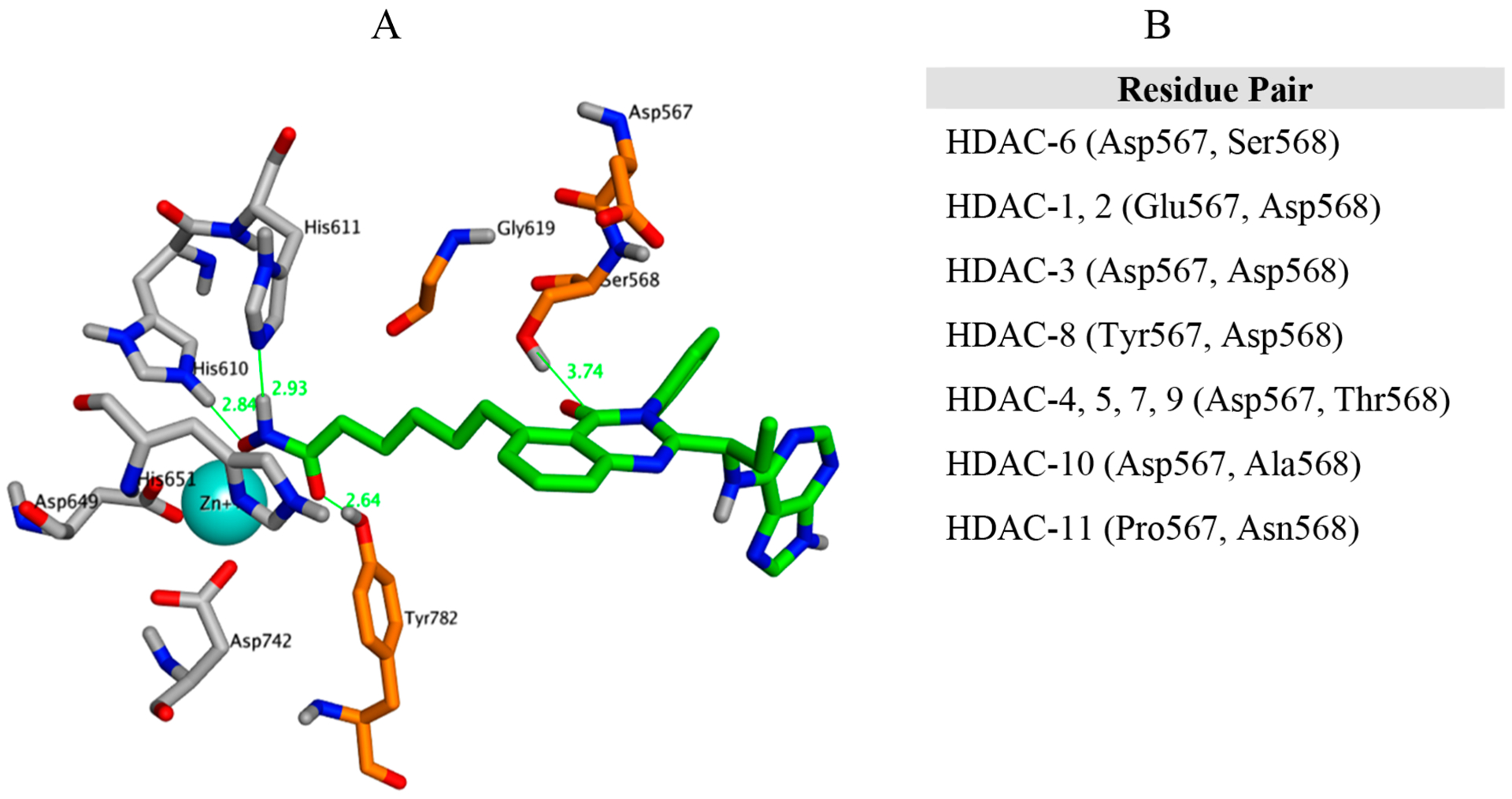
(A) Compound 19b bound to HDAC6 (PDB 5EDU.pdb). (B) Variation in amino acid residues at positions 567 and 568 across different HDACs.
HDAC6 Selectivity.
Using the procedure described in Methods, we identified Ser568 as the only residue on HDAC6 that could be responsible for the HDAC selectivity profile of 19b. As shown in Figure 6A, Ser568 in HDAC6 forms a hydrogen bond with the quinazolinone carbonyl on 19b. In the case of HDACs-1, 2, 3, and 8, Ser568 becomes Asp568 (Figure 6B). The negatively charged carboxyl group on Asp568 is a hydrogen bond acceptor and does not form a hydrogen bond with the quinazolinone carbonyl on 19b (Figure S1–S3, Supporting Information). Rather, the Asp568 carboxyl repulses the quinazolinone carbonyl and swings away from the ligand. The nearby presence of Glu567 (HDAC1,2, Figure S1, Supporting Information) and Asp567 (HDAC3, Figure S2, Supporting Information) contributes to this process as these residues have negatively charged carboxyl side chains.
In the case of HDAC8, the residue adjacent to Asp568 is tyrosine, which contains a neutral phenyl side chain (Figure S3, Supporting Information). As a result, the conformational shift in the carboxyl group on position 568 is less significant than for HDACs-1,2,3. Nevertheless, the H bond to the quinazolinone carbonyl is still broken. In the case of HDACs-4,5,7, and 9, threonine occupies position 568. Thr568 contains a hydrogen bond donor (hydroxyl), but the hydroxyl group does not interact with the quinazolinone carbonyl on the ligand. That is because the extra methyl group on the Thr568 side chain clashes with nearby Gly619 and prevents the OH group from obtaining a favorable hydrogen bonding position with respect to the quinazolinone carbonyl. In fact, it is easier for the Thr568 OH group to hydrogen bond with the carboxyl side chain of nearby Asp567 (Figure S4, Supporting Information). In the case of HDAC10, Ser568 becomes Ala568 resulting in an obvious loss of hydrogen bonding to the quinazolinone carbonyl on the ligand (Figure S5, Supporting Information). In HDAC11, Ser568 becomes Asn568. Although asparagine has a hydrogen bond donating nitrogen group, its side chain is one carbon too long to adopt a reasonable orientation for hydrogen bonding to the quinazolinone carbonyl on the ligand (Figure S6, Supporting Information). The 19b-HDAC interaction energies relative to 19b-HDAC6 were computed for each of the scenarios described above. These interaction energies are completely consistent with the idea that there is one less hydrogen bond between 19b and HDACs-1, 2, 3, 4, 5, 7, 8, 9, 10,11 than there is between 19b and HDAC6 hence, explaining the selectivity of 19b to HDAC6.
Kinase Selectivity.
The kinase selectivity of 48c was investigated at 1 μM concentration against a panel of 412 kinases, including 24 atypical kinases and 17 lipid kinases at Reaction Biology Corporation, PA. As shown in Figure 7A, the dual inhibitor 48c showed very high selectivity across the diverse kinase families, including atypical kinases, with less than 50% inhibition among all the kinases tested (Table S2, Supporting Information). In the case of the highly homo-logated lipid kinases (Figure 7B), compound 48c showed between 74 and 83% inhibition for PI3Kα, β, and other PI3K mutants while maintaining the high selectivity for PI3Kγ and PI3Kδ (≥99% inhibition), as previously observed.
Figure 7.
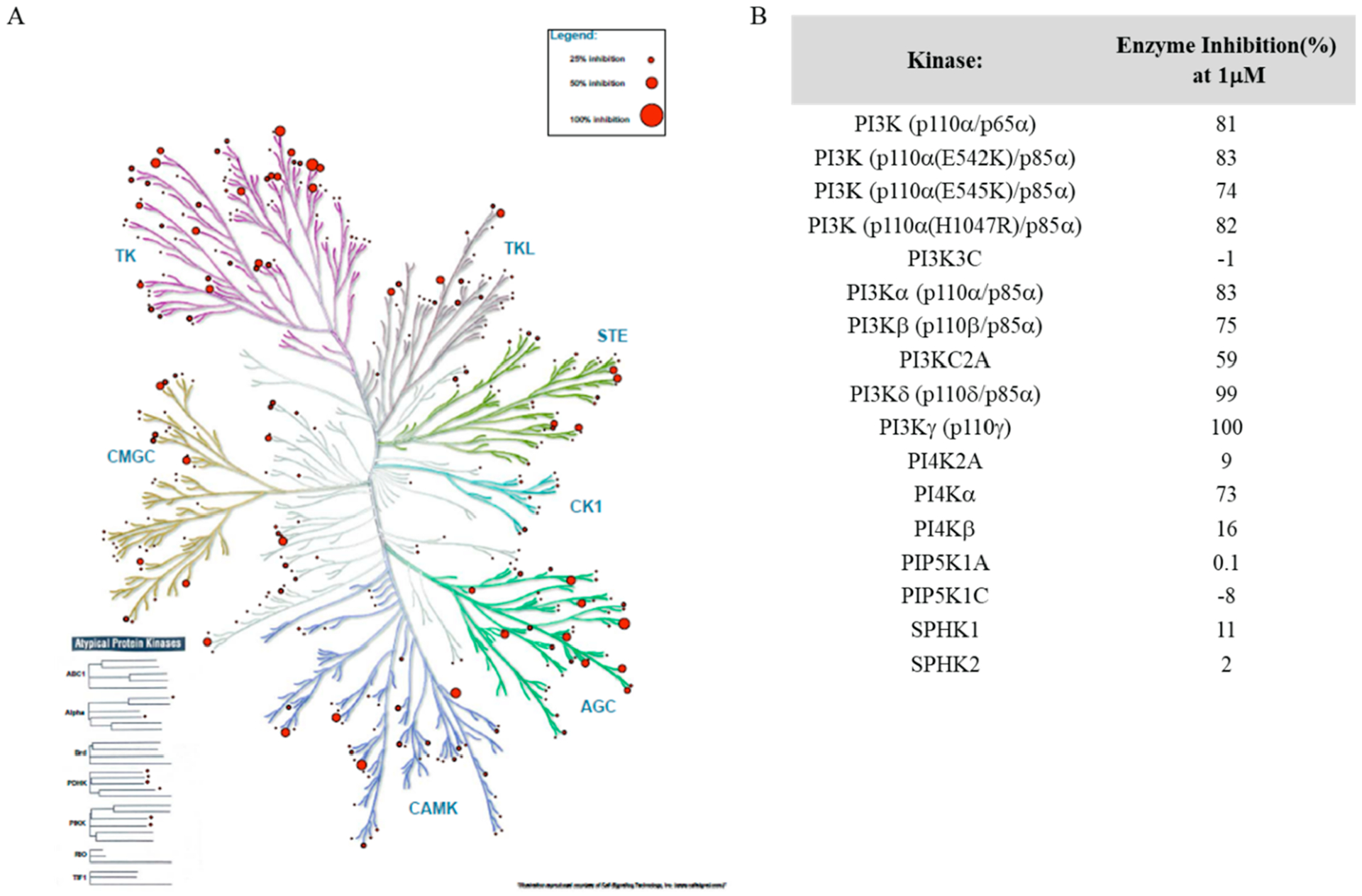
(A) Kinase selectivity profile of compound 48c (1 μM concentration) against 412 kinases. (B) Selectivity data (% Inhibition at 1 μM) of compound 48c against lipid kinases. Kinase profiling was done at Reaction Biology, Malvern, PA, USA.
NCI-60 Human Tumor Cell Lines Screen.
The dual inhibitors were tested against 60 human cancer cell lines at the National Cancer Institute (NCI-60 panel).58 The NCI-60 employs an initial singe-dose (10 μM compound concentration) growth inhibition screen against 60 cancer cell lines and selects only the most potent compounds for subsequent 5-point dose–response studies. As previously mentioned, most of the dual inhibitors (except 28d) with the purine hinge binding group showed a poor growth inhibition profile (average growth inhibition (%) = 0–33%) across all the 60 cell lines tested and therefore were not evaluated in dose–response IC50. Compound 28d with the amino-benzyl linker and purine hinge binder was the only dual inhibitor (from the purine series) selected for NCI-60–cell line dose–response and showed potent antiproliferative activities against multiple human cancer cell line types (Table 7). In contrast to the purine series, several dual inhibitors bearing amino-pyrimidine kinase hinge binding groups were evaluated for dose–response, with a few of them even showing better GI50 than Idelalisib and SAHA.
Table 7.
Antiproliferative Activities of Selected Dual Inhibitors against Various Human Cancer Cell Lines at NCI-60a
| GI50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| cancer type | cell line | 28d | 46b | 48b | 48c | 48o | Idelalisib | Vorinostat (SAHA) |
| leukemia | CCRF-CEM | 3.1 | 0.4 | 1.6 | 0.7 | 0.05 | 22.3 | 0.7 |
| SR | 2.5 | 1.2 | 1.4 | 0.7 | 0.6 | 0.4 | ||
| non-small cell lung cancer | HOP-62 | 11.8 | 2.7 | 1.9 | 2.4 | 1.4 | >100.0 | 1.6 |
| HOP-92 | 2.2 | 0.2 | 1.1 | 0.9 | 0.3 | 14.1 | 2.9 | |
| colon cancer | HCT-2998 | 3.0 | 2.2 | 1.7 | 2.1 | 1.8 | 38.5 | 1.9 |
| KM-12 | 2.3 | 4.0 | 1.9 | 1.7 | 0.4 | 1.2 | 0.8 | |
| CNS cancer | SNB-75 | 3.0 | 0.3 | 1.3 | 0.7 | 0.4 | 1.2 | 0.8 |
| U251 | 2.5 | 1.5 | 1.7 | 1.7 | 1.4 | 53.2 | 1.6 | |
| melanoma | LOX IMVI | 1.8 | 1.4 | 1.8 | 1.6 | 1.5 | 33.5 | 1.2 |
| M14 | 5.5 | 1.8 | 2.3 | 1.8 | 1.5 | 37.8 | 1.3 | |
| ovarian cancer | IGROV1 | 2.3 | 0.5 | 1.9 | 1.5 | 0.9 | 4.8 | 1.1 |
| OVCAR3 | 3.4 | 1.2 | 2.2 | 1.2 | 1.3 | 17.7 | 1.4 | |
| renal cancer | A498 | 6.1 | 0.2 | 1.4 | 1.0 | 0.7 | 1.1 | 1.4 |
| CAKI-1 | 14.6 | 1.3 | 3.2 | 1.3 | 0.8 | 20.5 | 1.2 | |
| RXF 393 | 3.2 | 0.2 | 1.4 | 1.2 | 0.5 | 1.4 | 1.3 | |
| breast cancer | MDA-MB-231/ATCC | 2.8 | 1.8 | 2.3 | 1.8 | 1.5 | 42.3 | 2.5 |
| HS 578T | 4.5 | 0.4 | 2.5 | 2.0 | 0.9 | 6.0 | 3.6 | |
| T-47D | 1.7 | 0.3 | 1.6 | 1.2 | 0.3 | 5.2 | 0.5 | |
Compounds were tested at NCI-60 panel in singlet 5-dose IC50 mode with 10-fold serial dilution starting at a concentration of 100 μM.
Dual Inhibitors Decrease Survival and Induce Cell Death in Diverse Types of AML Cells.
Dual inhibitor 48c was evaluated in cell viability assays against several myeloma and leukemia cell lines as shown in Figure 8A,B. Cell-viability was determined using the CellTiter-Glo (Promega) assay59 or by the trypan blue exclusion assay.60 Compound 48c showed good inhibitory activity against FLT3-ITD mutant AML cell lines, MOLM-14 and MV411. Further studies were performed with 48c in other types of AML cell lines. THP-1 is a spontaneously immortalized monocyte-like cell line derived from the peripheral blood of a childhood case of acute monocytic leukemia (M5 subtype).61 HL-60 is a human promyelocytic leukemia cell line with p53-null mutation.62 Compound 48c demonstrated antiproliferative activity against these cell lines with IC50 values in the 2–5 μM range (Figure 8A). Like MOLM-14 cells, MOLM-13 AML cells have one wild-type FLT3 and one FLT3-ITD allele.63 Treatment of MOLM-13 cells with 48c resulted in inhibition of growth that was concentration-dependent [Figure 8B(2)]. As shown previously, loss of MOLM-13 cell survival was observed in response to treatment with the type II ATP competitive inhibitor HG-7-85-01 (HG) (data not shown).60 Interestingly, MOLM-13 cells selected for resistance to HG were also sensitive to compound 48c treatment [Figure 8B(3)], indicating that targeting PI3Kδ/HDAC6 is effective against MOLM-13 cells resistant to FLT3 inhibitors. Dual inhibitors 48c, 48n, and 48o, when screened alongside Idelalisib,64 SAHA65 and CUDC-90766 (at their IC50 range) against MV411 cell line displayed better potency than Idelalisib and SAHA but were less potent than the pan PI3K/HDAC inhibitor, CUDC-907, as expected (Figure 8C). Importantly, dual inhibitor 48c showed no cytotoxicity against normal human peripheral blood mononuclear cells (PBMCs) [Figure 8D], demonstrating its high selectivity in targeting leukemia cells over PBMCs. Additionally, dual inhibitors 48c, 48n, and 48o, were nontoxic against NIH3T3 normal fibroblasts, whereas significant toxicity was observed with the pan-inhibitor CUDC-907, even at a low concentration of 50 nM (Figure 8E). These studies clearly indicate that our isoform-selective dual inhibitors display potent antitumor activity with no toxicity in normal cells.
Figure 8.
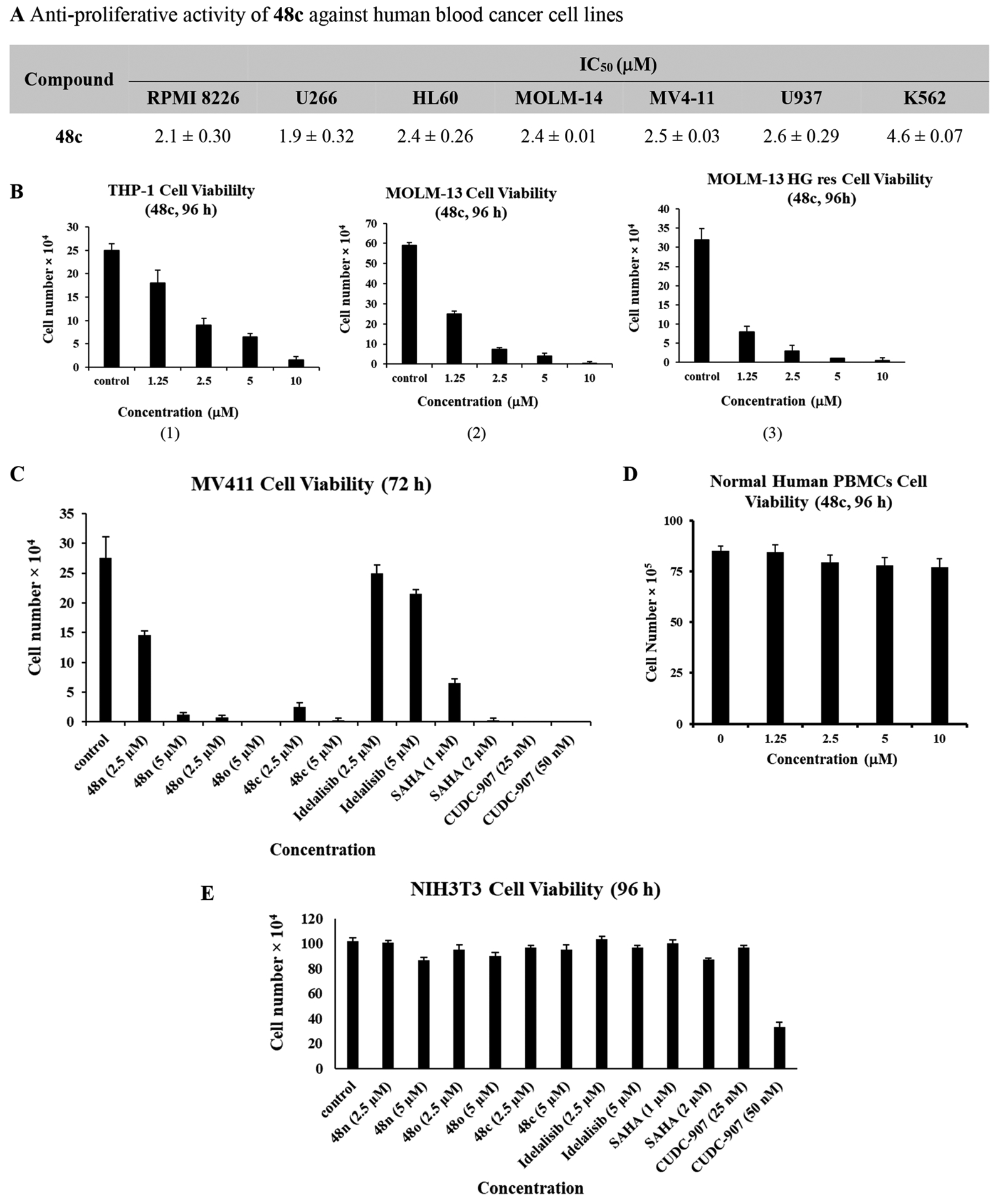
(A) Cell viability assays were performed in duplicate with IC50 values expressed as mean ± SD of two independent runs. (B) In vitro cell-kill activity of 48c. (C) In vitro cell-kill activity of compounds 48n, 48o, 48c, Idelalisib, SAHA, and CUDC-907 against MV411 cells. (D) PBMCs viability with 48c treatment. (E) NIH3T3 cell viability upon treatment with compounds 48n, 48o, 48c, Idelalisib, SAHA, and CUDC-907.
To further define the activity of these dual inhibitors on AML cell lines, we determined potential induction of cell death as measured by propidium iodide (PI) alone or PI/Annexin V staining followed by flow cytometry.67 Dual inhibitors 48c, 48n, and 48o induced cell death via necrosis in multiple AML cell lines as shown in Figure 9A–C. Both compounds 48n and 48o were able to induce >50% necrosis at 5 μM concentration in MOLM-14, MV4–11, and HL-60 tumor cells (Figure 9A,B). Similarly, dual inhibitor 48c was highly active for inducing necrosis across several AML cell lines, including >60% necrosis in MOLM-13 FLT3 inhibitor-resistant cell line (MOLM-13 HG Res) (Figure 9C). To determine whether primary AML cells also respond to compound 48c, blasts from a patient with AML were grown in short-term culture. The AML primary cells were then treated with 5 μM compound 48c for 72 h. The results demonstrate induction of substantial necrosis in these primary cells (Figure 9D). Notably, dual inhibitors 48c, 48n, and 48o displayed no significant necrotic activity against normal human HEK293 cells, whereas pan-inhibitors SAHA and CUDC-907, induced necrosis in these cells, again demonstrating the selective targeting of tumor cells over normal cells by our compounds.
Figure 9.
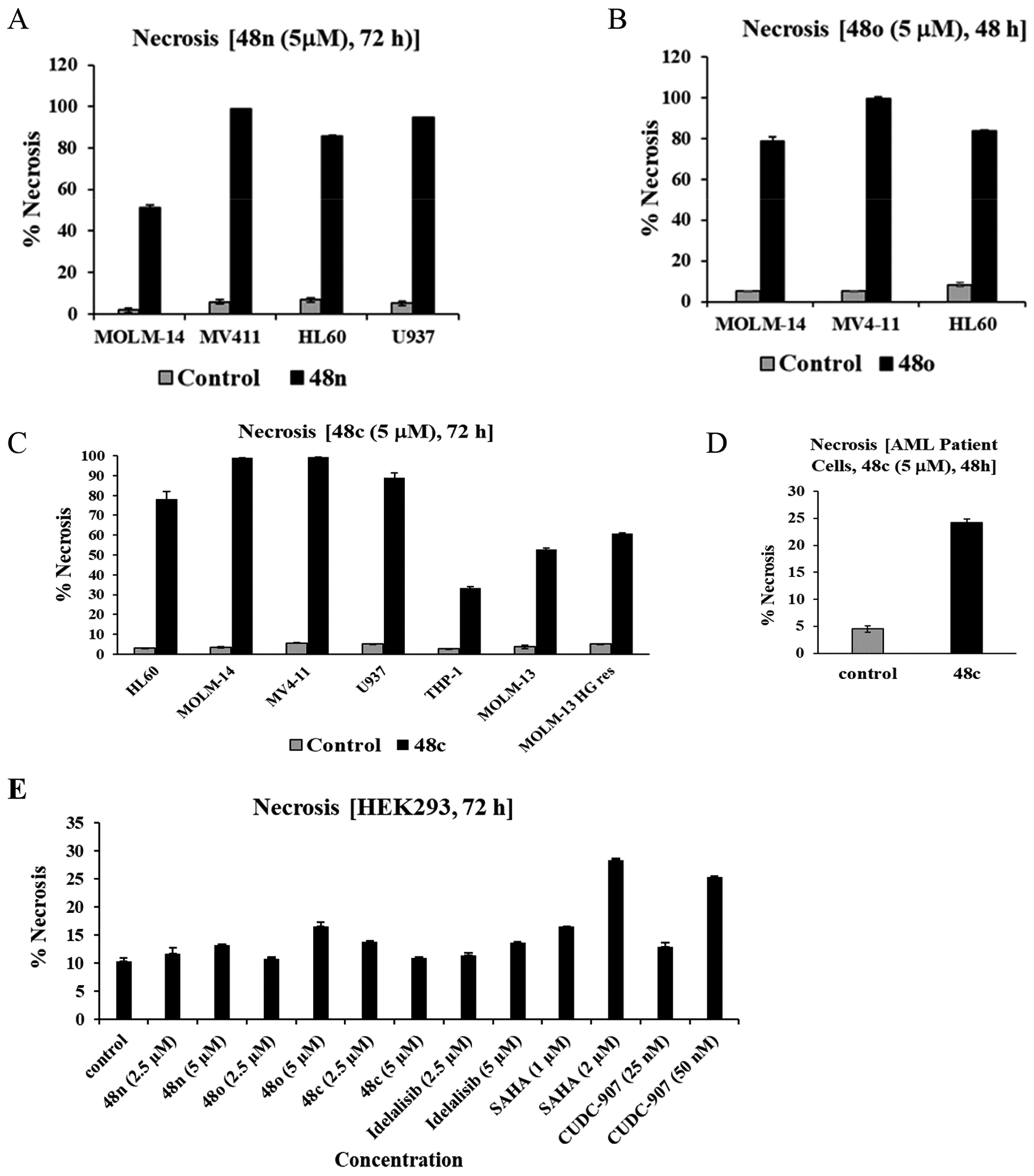
(A) Necrosis rate (%) of 48n at 5 μM concentration. (B) Necrosis rate (%) of 48o at 5 μM concentration. (C) Necrosis rate (%) of 48c at 5 μM concentration. (D) Necrosis rate (%) of 48c at 5 μM concentration against primary AML blasts. (E) Necrosis rate (%) of compounds 48n, 48o, 48c, Idelalisib, SAHA, and CUDC-907 against HEK293 cells. Necrosis assays were performed in duplicate and % necrosis values are expressed as mean ± SD of two independent runs.
Clonogenic Survival Assay.
Encouraged by their growth inhibitory activities against multiple AML cell lines, dual inhibitors 48c and 48o were tested in an in vitro clonogenic assay for inhibition against MOLM-14 colony formation (Figure 10). MOLM-14 cells were seeded in 6-well plates for 24 h and then left untreated or treated with either 2.5 or 5 μM of 48c and 48o, respectively. After 48 h, the cells were washed and seeded in soft agar for the generation of colonies. After 10 days, colonies of >50 cells were counted. Plating efficiency of untreated MOLM-14 cells as colony formation is very high as multiple colonies (>50 cells) were seen. Gratifyingly, treatment of MOLM-14 cells with our dual inhibitors 48c and 48o resulted in significant decrease in clonogenic survival as shown below in Figure 10.
Figure 10.
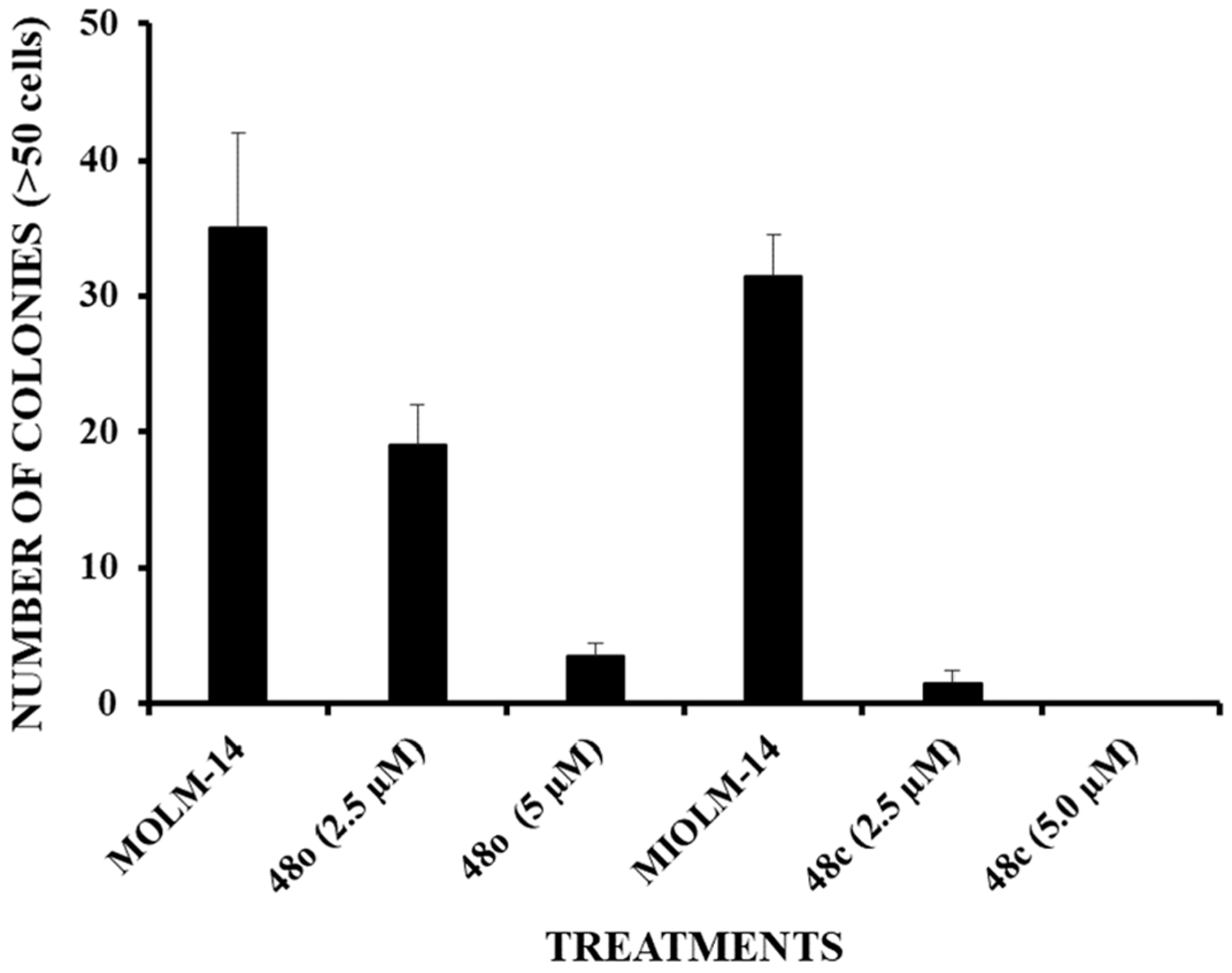
Inhibition of MOLM-14 colony formation by 48c and 48o.
PI3Kδ Inhibition Decreases Activation of Downstream Effectors.
PI3K activates the AKT pathway. MOLM-14 cells were treated for 1, 6, and 24 h, with dual inhibitor 48c (5.0 μM) and analyzed for p-Akt and total Akt levels by immunoblotting. In concert with the demonstration that dual inhibitors downregulate PI3K activation, treatment of MOLM-14 cells with compound 48c was associated with complete inhibition of p-AKT with no change in the total AKT protein levels as shown in Figure 11.
Figure 11.
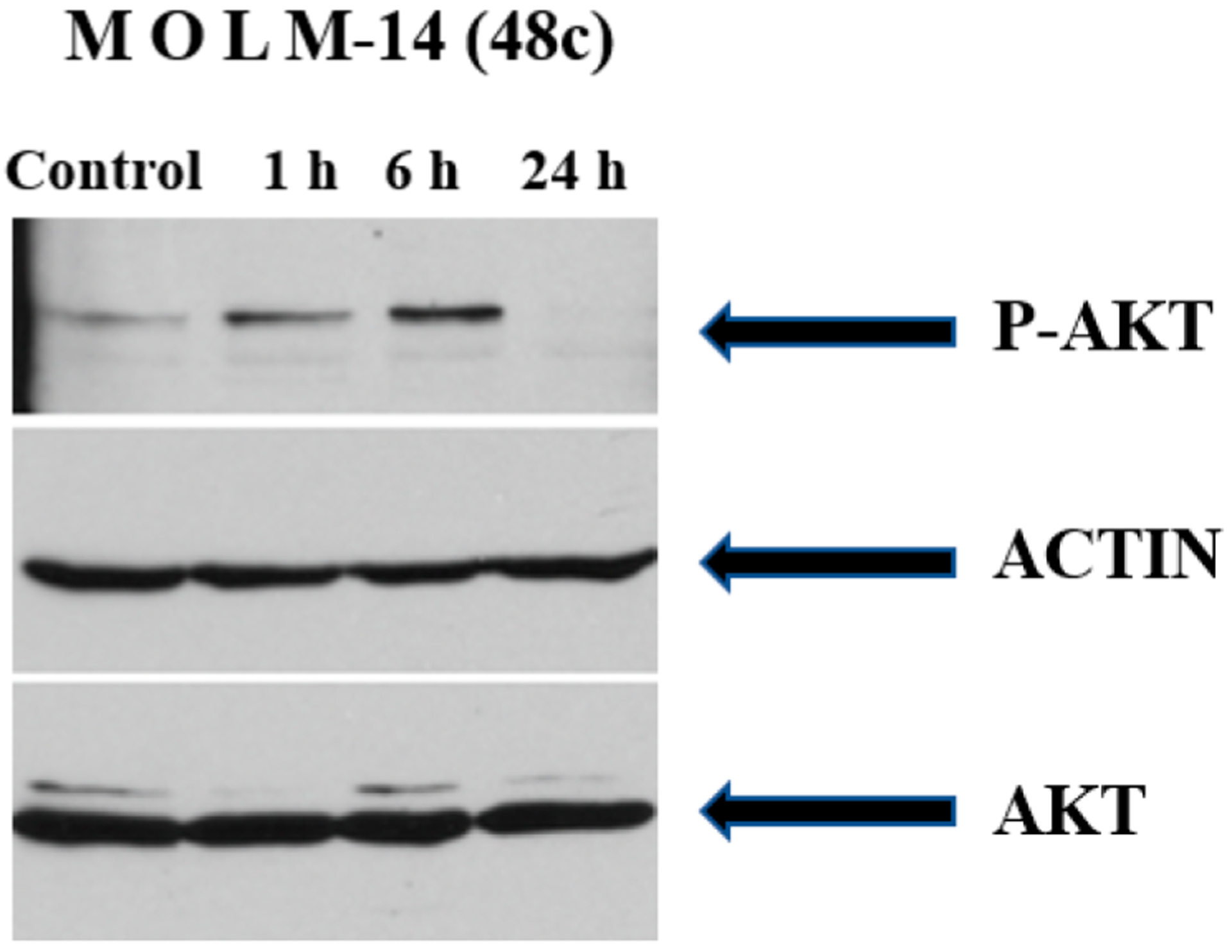
Immunoblot analysis for PI3K inhibition in MOLM-14 cells.
CETSA.
Next, we examined target engagement using the cellular thermal shift assay (CETSA).68 Three lymphocytic cell lines were profiled by immunoblot for HDAC6 and PI3Kδ expression, and MV411 was selected for subsequent experiments based on detection of both targets (Figure S7, Supporting Information). The CETSA melt profile (Tagg) of each protein was then measured (Figure 12A,B), and 58 1/2C (HDAC6) and 52 1/2C (PI3Kδ) were selected for isothermal dose–response experiments. The control compounds showed the anticipated responses, with CAY10603 (HDAC6 inhibitor) stabilizing HDAC6 and Idelalisib (PI3Kδ inhibitor) stabilizing PI3Kδ. For the dual inhibitors, robust HDAC6 stabilization was observed for 48c, with less stabilization observed for 46b and HDAC6 only inhibitor 48d (Figure 12C). For PI3Kδ, PI3K inhibitor 45b, and dual inhibitor, 48c stabilized the protein, whereas 46b and 48d did not (Figure 12D). These results indicate compound 48c can engage both HDAC6 and PI3Kδ in the complex cellular environment.
Figure 12.
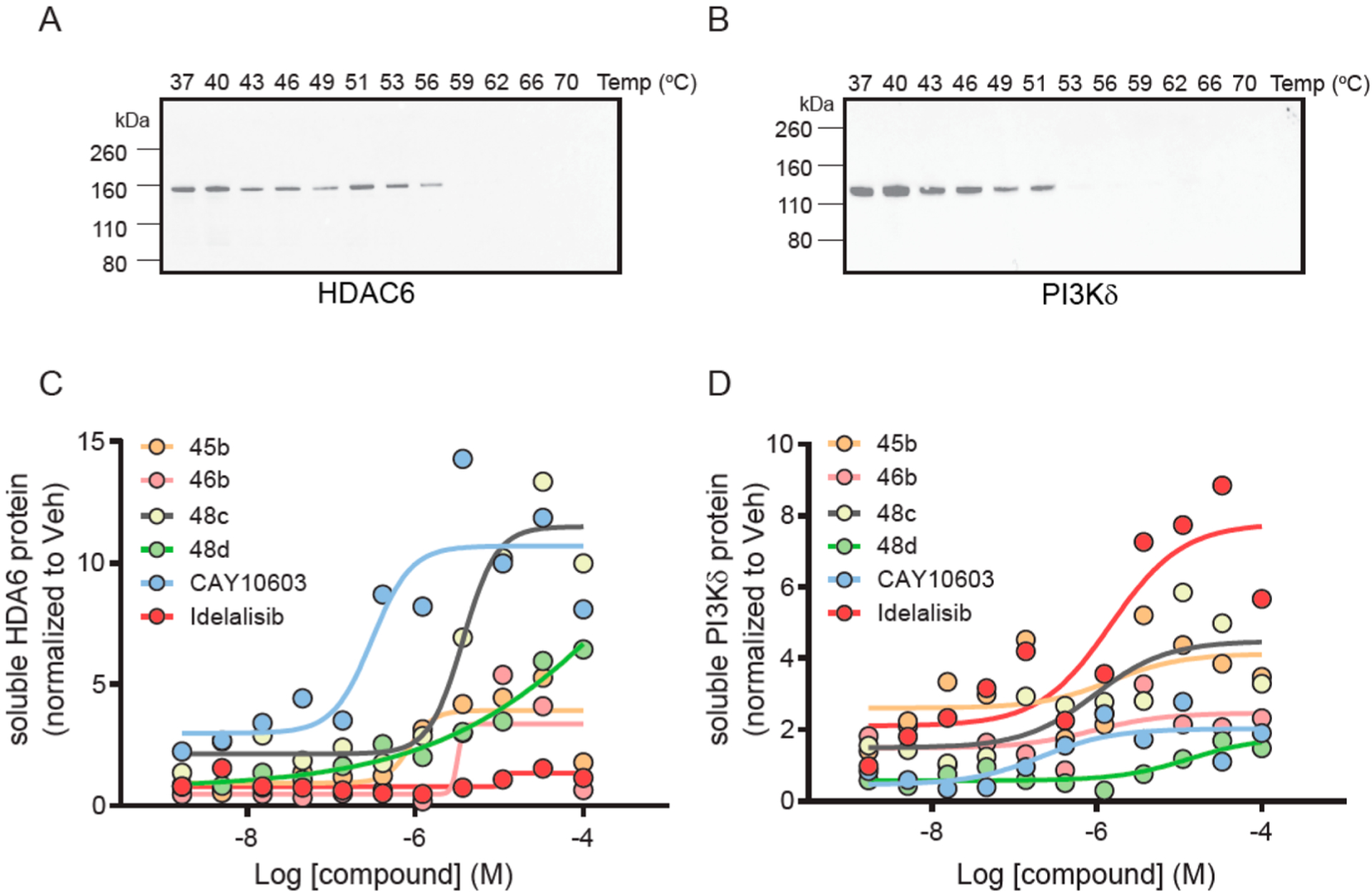
CETSA analysis. (A) Tagg measurement for HDAC6 in MV411 cells, (B) Tagg measurement for PI3Kδ in MV411 cells, (C) melt profile for HDAC6, (D) melt profile for PI3Kδ.
In Vitro ADME Assays.
Selected dual inhibitors were evaluated in multispecies (human, mouse, and rat) liver microsome/cytosol stability and Caco-2 permeability and efflux assays (Table 8). Compound 19b showed good stability in human liver microsomes (HLM) but was poor in mouse (MLM) and rat liver microsomes (RLM). 19b displayed low (t1/2 < 30 min) to moderate (30 < t1/2 < 60 min) metabolic stability in liver cytosol from the three species. Additionally, it was found to have low permeability in Caco-2 cell monolayer assay and an efflux ratio of 11, indicating the involvement of efflux transporters. Dual inhibitor 46b, the pyrimidine hinge binding analogue of 19b, showed similar stability in HLM and MLM assays as 19b but was significantly better in RLM and cytosol assays. In contrast to compounds 19b and46b, the dual inhibitors bearing amino-benzyl linker (48b, 48c, 48n, 48o) displayed moderate to high stability in MLM and RLM assays but the HLM stability varied from low to high with compound 48n, showing high stability across the three species tested. Additionally, these compounds show high cytosolic stability in human, mouse, and rat assays. The lead compound 48c had low cell permeability (1.9 × 10−6 cm/s), however, its efflux ratio of 3.5 was found to be on the lower side.
Table 8.
In Vitro Metabolic Stability (Liver Microsome and Cytosol) and Caco-2 Permeability of Selected Dual PI3K/HDAC Inhibitorsa
| microsomal stability (t1/2, min)b,d | cytosolic stability (t1/2, min)c,d | ||||||
|---|---|---|---|---|---|---|---|
| compd | HLM | MLM | RLM | HLC | MLC | RLC | Caco-2 efflux ratio (BA/AB)e |
| 19b | 93.1 ± 4.9 | 9.0 ± 0.03 | 19.1 ± 0.1 | 40.6 ± 7.5 | 27.7 ± 1.5 | 19.1 ± 0.1 | 9.5/0.9 |
| 46b | 91.5 ± 2.2 | 9.0 ± 0.4 | 49.8 ± 3.6 | >120 | >120 | >120 | ND |
| 48b | 45.7 ± 15.1 | 60.4 ± 1.4 | 95.4 ± 4.5 | >120 | 52.1 ± 4.7 | >120 | 7.5/1.4 |
| 48c | 22.4 ± 2.2 | 51.7 ± 0.02 | 100.6 ± 14.6 | 116.5 ± 4.9 | 78.4 ± 13.5 | >120 | 6.5/1.9 |
| 48n | 95.1 ± 30.6 | 92.8 ± 17.3 | >120 | >120 | >120 | >120 | ND |
| 48o | 32.2 ± 2.7 | 74.4 ± 5.3 | 98.6 ± 12.5 | 105.7 ± 20.2 | >120 | >120 | ND |
Average of two runs ± standard deviation (SD).
Multispecies liver microsomal stability in human (HLM), mouse (MLM) and rat (RLM).
Multispecies liver cytosolic stability in human (HLC), mouse (MLC) and rat (RLC).
No SD values provided if all replicate half-life values exceeded 120 min.
Caco-2 permeability [apical to basolateral (AB); basolateral to apical (BA); 10−6 cm/s). ND = not determined.
Pharmacokinetics.
Because of its potent PI3K/HDAC inhibitory and antitumor activity, compound 48c was progressed to an in vivo pharmacokinetic (PK) study in female Balb/c mice (Table 9 and Figure 13). The plasma PK of 48c via intravenous (iv) route at 3 mg/kg dose revealed its high clearance at 57 mL/min/kg with terminal half-life (t1/2) of 1 h (Figure 13A). Unfortunately, po administration of 48c at 10 mg/kg dose exhibited very low exposure with an oral bioavailability (F) of only 0.7% (Figure 13A). When administered intraperitoneally (ip) at 50 and 150 mg/kg dose, respectively, compound 48c displayed better PK properties with significantly improved in vivo exposure in terms of Cmax and AUC when compared to po administration (Figure 13B). This preliminary PK assessment suggested that ip administration would be the preferred dosing route for the proof of concept (POC) purpose in the in vivo antitumor efficacy study in mouse model.
Table 9.
PK Profile of Compound 48ca
| dosing route | ||||
|---|---|---|---|---|
| iv | po | ip | ip | |
| dose (mg/kg) | 3 | 10 | 50 | 150 |
| t1/2 (h) | 1.01 | 1.4 | 2.7b | 1.7b |
| Tmax (h) | 0.167 | 1 | 2 | |
| Cmax (ng/mL) | 18.4 | 1443 | 7783 | |
| AUC0-t (h·ng/mL) | 869 | 20.1 | 8493 | 40100 |
| AUC0-t/D (h·mg/mL) | 291 | 2.1 | 170 | 267 |
| Vdss (L/kg) | 3.6 | |||
| CLp (mL/min/kg) | 57 | |||
| F (%) | 0.7 | NCc | NCc | |
Formulation: IV (3 mg/kg), EtOH/PEG-300/sterile water (10:60:30); PO (10 mg/kg), EtOH/PEG-300/sterile water (10:60:30); IP (50 mg/kg), DMSO/PEG-300/sterile water (10:40:50); IP (150 mg/kg), DMSO/PEG-300/”20% HP-β-CD in water (1:4)” (10:40:50), adjusted to pH 3.0 with 1 M HCl.
The 24 h data point was excluded for terminal half-life (t1/2) calculation.
NC = not calculated.
Figure 13.
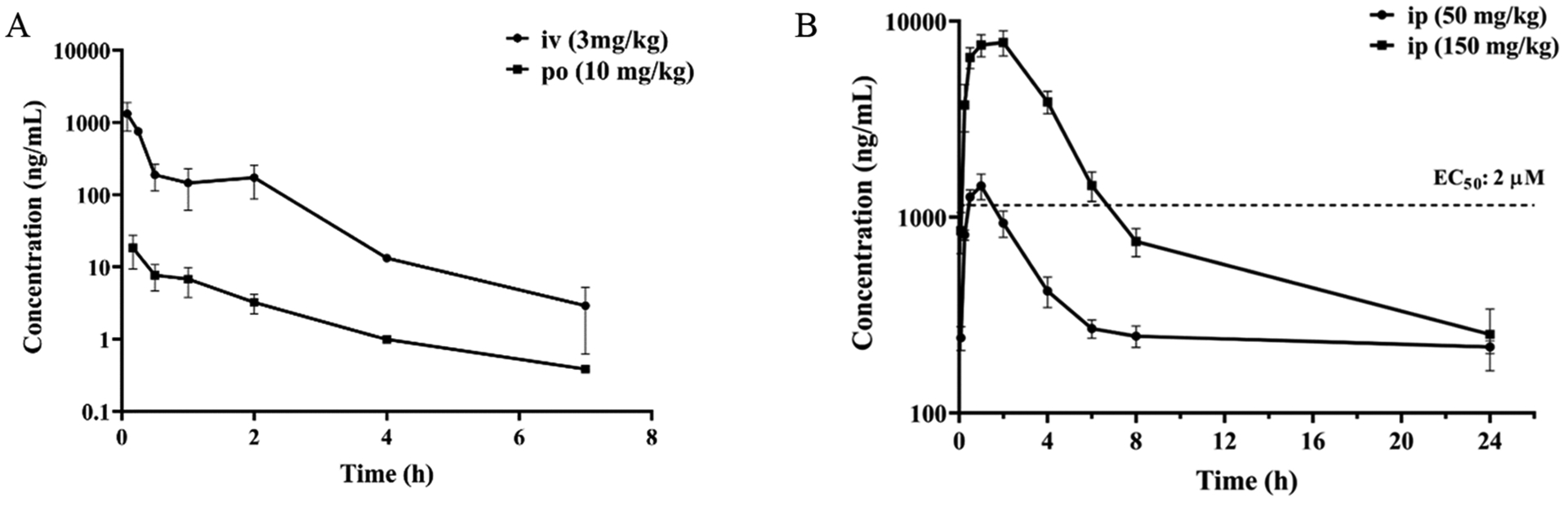
Plasma concentration vs time profiles for 48c in female Balb/c mice via: (A) iv (3 mg/kg) and po (10 mg/kg) dosing, and (B) ip (50 mg/kg and 150 mg/kg) dosing.
CONCLUSIONS
In summary, we have rationally designed and synthesized a series of quinazolin-4-one-based hydroxamic acids for the dual inhibition of PI3K and HDAC enzymes. Structure–activity relationship (SAR) exploration revealed several compounds as potent and selective PI3Kγ, δ/HDAC6 inhibitors with good cellular potencies against a panel of 60 different cancer cell lines at National Cancer Institute (NCI-60). Particularly, compound 48c was identified as potent dual inhibitor with high kinome selectivity and potent antiproliferative activity against various cancer cell lines, including leukemia, melanoma, renal, nonsmall cell lung cancer, central nervous system (CNS) cancer, and breast cancer. Dual inhibitor 48c also showed good potency in inducing cell death via necrosis in multiple AML cell lines, including the FLT3-ITD mutant and FLT3-inhibitor resistant cell lines, and AML patient’s primary cells. The dual PI3Kδ/HDAC6 activity of 48c was further supported by the target engagement studies in MV411 cell line using the cellular thermal shift assay (CETSA). Compound 48c showed decent microsomal stability and in vivo PK profile and warrants further exploration in antitumor efficacy study in mice. Further lead optimization efforts to achieve new potent dual inhibitors with favorable in vivo PK profiles are currently underway in our laboratory.
EXPERIMENTAL SECTION
Chemistry.
All air or moisture sensitive reactions were performed under positive pressure of nitrogen with oven-dried glassware. Chemical reagents and anhydrous solvents were obtained from commercial sources and used as is. Preparative purification was performed on a Waters semipreparative HPLC instrument. The column used was a Phenomenex Luna C18 (5 μm, 30 mm × 75 mm) at a flow rate of 45 mL/min. The mobile phase consisted of acetonitrile and water (each containing 0.1% trifluoroacetic acid). A gradient from 10% to 50% acetonitrile over 8 min was used during the purification. Fraction collection was triggered by UV detection (220 nm). Alternately, flash chromatography on silica gel was performed using forced flow (liquid) of the indicated solvent system on Biotage KP-Sil prepacked cartridges and using the Biotage SP-1 automated chromatography system. Analytical analysis for purity was determined by two different methods denoted as final QC methods 1 and 2. Method 1: Analysis was performed on an Agilent 1290 Infinity series HPLC instrument. UHPLC long gradient equivalent from 4% to 100% acetonitrile (0.05% trifluoroacetic acid) in water over 3 min run time of 4.5 min with a flow rate of 0.8 mL/min. A Phenomenex Luna C18 column (3 μm, 3 mm × 75 mm) was used at a temperature of 50 1/2C. Method 2: Analysis was performed on an Agilent 1260 with a 7 min gradient from 4% to 100% acetonitrile (containing 0.025% trifluoroacetic acid) in water (containing 0.05% trifluoroacetic acid) over 8 min run time at a flow rate of 1 mL/min. A Phenomenex Luna C18 column (3 μm, 3 mm × 75 mm) was used at a temperature of 50 1/2C. Purity determination was performed using an Agilent diode array detector for both method 1 and method 2. Mass determination was performed using an Agilent 6130 mass spectrometer with electrospray ionization in the positive mode. All assay compounds had >95% purity based on both analytical methods. 1H NMR spectra were recorded on Varian 400 MHz spectrometers. All proton spectra are referenced relative to the deuterated solvent peak: 7.27 ppm for CDCl3, 2.50 ppm (center line signal) for DMSO-d6. High resolution mass spectrometry results were recorded on Agilent 6210 time-of-flight LC/MS system.
General Procedure for Sonogashira Cross-Coupling Reaction (GP1).
Aryl bromide (1.0 equiv), allylpalladium(II) chloride dimer (0.05 equiv), tri-tert-butylphosphonium tetrafluoroborate (0.20 equiv), and alkyne (1.2 equiv) (if solid at room temperature) were weighed and added to a MW vial equipped with a stir bar. The vial was covered with a rubber septum and placed under nitrogen atmosphere. In a separate scintillation vial, DABCO was weighed and dissolved in dry 1,4-dioxane (5 mL/mmol of aryl bromide). This DABCO solution and alkyne (if liquid at room temperature) were added to the MW vial via syringe, and the resulting mixture was bubbled with nitrogen for 5 min followed by stirring for 16 h at room temperature under nitrogen atmosphere. After 16 h, the crude reaction mixture was filtered through a short pad of Celite and concentrated in vacuo. The remaining residue was purified by flash chromatography on silica using forced flow of ethyl acetate/hexanes system on Biotage KP-Sil prepacked cartridges and using the Biotage SP-1 automated chromatography system to afford the cross-coupled product.
General Procedure for Pd/C-Catalyzed Hydrogenation (GP2).
The substituted alkyne (1.0 equiv) and 10 wt % Pd/C were added to a 20 mL scintillation vial fitted with a rubber septum. The reaction vial was evacuated followed by the addition of dry EtOAc (0.1M). The vacuum was removed, and the reaction vial was stirred for 20 h under an atmosphere of hydrogen using a balloon. After completion of reaction (determined by LC-MS), the crude reaction mixture was filtered using Celite, concentrated in vacuo, and carried over to the next step without purification.
General Procedure for Attachment of Purine Hinge Binding Group (GP3).
The Boc-protected amine (1.0 equiv) was dissolved in DCM (0.1 M) in a vial, and trifluoroacetic acid (20 equiv) was added dropwise to it. The resulting mixture was stirred at room temperature for 3 h. After completion of reaction (by LC-MS), the reaction mixture was worked-up by either of the following two methods. Method A: The crude reaction was quenched with aqueous saturated NaHCO3 solution and extracted three times with DCM. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated in vacuo to afford the free amine. Method B: The crude reaction mixture was concentrated in vacuo, redissolved in 1–2 mL of DCM, and passed through preconditioned PL-HCO3 MP SPE device (bicarbonate resin solid phase extraction) and washed with 2 mL of DCM. The filtrate was concentrated in vacuo to afford the free amine. The resulting free amine was dissolved in ethanol (0.4 M) in a microwave vial equipped with a stir bar followed by the addition of 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine (1.5–2.0 equiv) and triethylamine (3.0–4.0 equiv) to it. The vial was sealed and heated for 4 h at 100 1/2C in a microwave. After completion of reaction (by LCMS), the reaction mixture was concentrated in vacuo and the remaining residue was purified by flash chromatography on silica gel using forced flow of indicated solvent system on Biotage KP-Sil prepacked cartridges and using the Biotage SP-1 automated chromatography system to afford the product.
General Procedure for Conversion of Alkyl Ester to Hydroxamic Acid (GP4).
The alkyl ester was converted to hydroxamic acid in the final product by either of the following two methods. Method A: The alkyl ester was dissolved in THF/water (0.1 M, 1:1 by vol) in a vial equipped with a stir bar, and LiOH·H2O (2.0 equiv) was added to it. The resulting mixture was stirred at room temperature for 10 h and concentrated in vacuo to afford the carboxylic acid. The crude carboxylic acid was dissolved in DMF (0.1 M) in a vial, and O-(tetrahydro-2H-pyran-2-yl)hydroxylamine [NH2OTHP] (3.1 equiv), N-methyl morpholine (3.0 equiv), 3-(((ethylimino)methylene)-amino)-N,N-dimethylpropan-1-amine hydrochloride [EDC·HCl] (1.4 equiv), and 1H-[1,2,3]triazolo[4,5-b]pyridin-1-ol [HOAT] (1.2 equiv) were added to it. The resulting mixture was stirred at room temperature for 16 h, concentrated in vacuo, and purified by flash chromatography on silica using forced flow of 0–10% MeOH/DCM system on Biotage KP-Sil prepacked cartridges and using the Biotage SP-1 automated chromatography system. The purified compound was dissolved in DCM (0.1 M), and TFA (20.0 equiv) was added to it. The resulting mixture was stirred for 20 h. After completion of reaction (by LC-MS), the reaction mixture was concentrated in vacuo and purified on preparative HPLC to afford the final compound. Method B: The alkyl ester was dissolved in MeOH (0.1M) or THF/MeOH (0.1 M, 1:1 by vol) in a MW vial equipped with a stir bar and LiOH·H2O (1.1 equiv), and 50% hydroxylamine in water solution (30.0 equiv) were added at to it 0 1/2C. The MW vial was sealed, and the resulting solution was stirred at 0 1/2C for 2 h, then allowed to warmup to room temperature overnight. After completion of reaction by LC-MS, the reaction mixture was concentrated in vacuo, dissolved in DCM/MeOH (0.1M, 1:1 by vol), and TFA (20.0 equiv) was added to it. The resulting mixture was stirred at room temperature for 20 h. After completion of reaction (by LC-MS), the reaction mixture was concentrated in vacuo and purified on preparative HPLC to afford the final compound.
Procedure for Synthesis of C4-Subsitiuted Quinazolines (10a,b and 14a,b, Scheme 1).
Sonogashira Cross-Coupling of Compound 5 with Alkynes 6a,b. (S)-tert-Butyl 5-(2-(1-((tert-Butoxycarbonyl)-amino)propyl)quinazolin-4-yl)pent-4-ynoate (7a). The general procedure GP1 was used with (S)-tert-butyl (1-(4-chloroquinazolin-2-yl)propyl)carbamate 5 (180.0 mg, 0.56 mmol), allylpalladium(II) chloride dimer (10.1 mg, 0.028 mmol), tri-tert-butylphosphonium tetrafluoroborate (16.0 mg, 0.06 mmol), DABCO (125.0 mg, 1.12 mmol), and tert-butyl pent-4-ynoate 6a (104.0 mg, 0.67 mmol) in dry 1,4-dioxane (2.8 mL). The remaining residue was purified by flash chromatography on silica using 0–25% ethyl acetate/hexanes system to afford (S)-tert-butyl 5-(2-(1-((tert-butoxycarbonyl)amino)propyl)-quinazolin-4-yl)pent-4-ynoate 7a (150.0 mg, 0.34 mmol) as a yellow oil in 61% yield. LC–MS (method 1): tR = 3.85 min, m/z (M + H)+ = 440.3. 1H NMR (400 MHz, CDCl3) δ 8.29–8.24 (m, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.88 (ddd, J = 8.5, 6.9, 1.4 Hz, 1H), 7.62 (ddd, J = 8.3, 6.9, 1.2 Hz, 1H), 5.90 (d, J = 8.2 Hz, 1H), 5.02 (d, J = 7.3 Hz, 1H), 2.91 (t, J = 7.4 Hz, 2H), 2.69 (t, J = 7.1 Hz, 2H), 2.15–2.02 (m, 1H), 1.89 (dt, J = 14.0, 7.3 Hz, 1H), 1.49 (s, 9H), 1.47 (s, 9H) 0.90 (t, J = 7.4 Hz, 3H).
Methyl (S)-6-(2-(1-((tert-Butoxycarbonyl)amino)propyl)-quinazolin-4-yl)hex-5-ynoate (7b). The general procedure GP1 was used with (S)-tert-butyl (1-(4-chloroquinazolin-2-yl)propyl)-carbamate 5 (225.0 mg, 0.70 mmol), allylpalladium(II) chloride dimer (13.0 mg, 0.04 mmol), tri-tert-butylphosphonium tetrafluoroborate (20.0 mg, 0.07 mmol), DABCO (157.0 mg, 1.4 mmol), and methyl hex-5-ynoate 6b (115.0 mg, 0.91 mmol) in dry 1,4-dioxane (3.5 mL). The remaining residue was purified by flash chromatography on silica using 0–30% ethyl acetate/hexanes system to afford the product, methyl (S)-6-(2-(1-((tert-butoxycarbonyl)amino)-propyl)quinazolin-4-yl)hex-5-ynoate 7b (163.6 mg, 0.398 mmol) as a yellow oil in 57% yield. LC–MS (method 1): tR = 3.51 min, m/z (M + H)+ = 412.3. 1H NMR (400 MHz, CDCl3) δ 8.26 (ddd, J = 8.3, 1.5, 0.7 Hz, 1H), 8.03–7.97 (m, 1H), 7.90 (ddd, J = 8.4, 6.9, 1.4 Hz, 1H), 7.65 (ddd, J = 8.2, 6.8, 1.2 Hz, 1H), 5.91 (d, J = 8.2 Hz, 1H), 5.03 (d, J = 6.9 Hz, 1H), 3.72 (s, 3H), 2.74 (t, J = 7.1 Hz, 2H), 2.60 (t, J = 7.3 Hz, 2H), 2.10 (p, J = 7.2 Hz, 2H), 1.90 (dt, J = 13.6, 7.2 Hz, 2H), 1.46 (s, 9H), 0.91 (dd, J = 8.2, 7.0 Hz, 3H).
(S)-5-(2-(1-((9H-Purin-6-yl)amino)propyl)quinazolin-4-yl)-N-hydroxypentanamide (10a). Step 1. (i) Alkyne hydrogenation. The general procedure GP2 was used with compound 7a (45.0 mg, 0.102 mmol) and 10 wt % Pd/C (4.5 mg). After completion of reaction (determined by LC-MS), the crude reaction mixture was filtered using Celite, concentrated in vacuo, and carried over to the next step without purification. (ii) Attachment of purine hinge binding group. The general procedure GP3 was used on the hydrogenated product (formed above) with trifluoroacetic acid (233.0 mg, 2.04 mmol, 0.16 mL) in DCM (1.0 mL). The crude reaction was worked-up (method B) and reacted with 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine 8 (48.7 mg, 0.20 mmol) and triethylamine (41.3 mg, 0.41 mmol, 57 μL) in ethanol (0.3 mL). The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH (0.5% AcOH)/ DCM to afford the product 5-(2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)quinazolin-4-yl)pentanoic acid 9a (27.7 mg, 0.057 mmol) in 56% yield. LC–MS (method 1): tR = 2.92 min, m/z (M + H)+ = 490.3. Step 2. Conversion of carboxylic acid to hydroxamic acid. The general procedure GP4 (method A, without the LiOH hydrolysis step) was used with carboxylic acid, 9a (16.3 mg, 0.033 mmol) to afford the final product, (S)-5-(2-(1-((9H-purin-6-yl)amino)propyl)quinazolin-4-yl)-N-hydroxypentanamide, TFA (10a) (7.5 mg, 0.014 mmol) in 55% yield. LC–MS (method 2): tR = 3.40 min, m/z (M + H)+ = 421.2. 1H NMR (400 MHz, DMSO-d6) δ 10.50 (s, 1H), 8.40–8.28 (m, 3H), 8.03–7.94 (m, 2H), 7.76–7.67 (m, 1H), 3.32 (h, J = 7.9 Hz, 2H), 2.23–2.14 (m, 1H), 2.07 (dt, J = 19.8, 6.9 Hz, 3H), 1.85–1.76 (m, 2H), 1.76–1.66 (m, 2H), 1.62–1.57 (m, 1H), 0.93 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M+ H]+ calcd for C21H25N8O2 421.2095, found 421.2087.
Compound 9b was synthesized using a similar procedure, as described above for 9a.
(S)-6-(2-(1-((9H-Purin-6-yl)amino)propyl)quinazolin-4-yl)-N-hydroxyhexanamide (10b). The general procedure GP4 (method A) was used with compound 9b (70.4 mg, 0.14 mmol) to afford the final product, (S)-6-(2-(1-((9H-purin-6-yl)amino)propyl)quinazolin-4-yl)-N-hydroxyhexanamide, TFA (10b) (45.0 mg, 0.082 mmol) in 60% yield. LC–MS (method 2): tR = 3.53 min, m/z (M + H)+ = 435.3. 1H NMR (400 MHz, DMSO-d6) δ 10.35 (s, 1H), 8.39 (d, J = 8.8 Hz, 2H), 8.32 (dt, J = 8.4, 1.1 Hz, 1H), 7.99–7.95 (m, 2H), 7.76–7.70 (m, 1H), 5.59 (s, 1H), 3.29 (h, J = 7.1 Hz, 2H), 2.20 (dd, J = 14.6, 7.7 Hz, 1H), 2.08 (dt, J = 13.6, 7.4 Hz, 1H), 1.93 (t, J = 7.3 Hz, 2H), 1.79 (p, J = 7.5 Hz, 2H), 1.53 (p, J = 7.3 Hz, 2H), 1.34 (p, J = 7.7 Hz, 2H), 0.94 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M+ H]+ calcd for C22H27N8O2 435.2251, found 435.2250.
(S)-4-((2-(1-((9H-Purin-6-yl)amino)propyl)quinazolin-4-yl)-amino)-N-hydroxybutanamide (14a). Step 1. N-Alkylation. Compound 5 (103.0 mg, 0.32 mmol) was dissolved in ethanol (0.7 mL) in a 5 mL microwave vial equipped with a stir bar, and ethyl 4-aminobutyrate hydrochloride (107.0 mg, 0.64 mmol) and triethylamine (130.0 mg, 1.28 mmol, 0.18 mL) were added to it. The vial was sealed and heated at 100 1/2C for 1 h in a microwave. After completion of reaction (by LC-MS), the reaction mixture was concentrated in vacuo and the remaining residue was purified on silica using 0–30% EtOAc/hexanes to afford (S)-ethyl 4-((2-(1-((tert-butoxycarbonyl)-amino)propyl)quinazolin-4-yl)amino)butanoate 12a (125.0 mg, 0.30 mmol) in 94% yield. LC–MS (method 1): tR = 2.85 min, m/z (M + H)+ = 417.3. 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 8.4 Hz, 1H), 7.75–7.63 (m, 2H), 7.40 (t, J = 7.6 Hz, 1H), 6.51 (s, 1H), 6.04 (d, J = 7.9 Hz, 1H), 4.75 (q, J = 6.6 Hz, 1H), 4.12 (q, J = 7.2 Hz, 2H), 3.71 (q, J = 6.2 Hz, 2H), 2.50 (t, J = 6.7 Hz, 2H), 2.07 (p, J = 6.8 Hz, 2H), 2.03–1.97 (m, 1H), 1.88 (dt, J = 13.8, 7.0 Hz, 1H), 1.47 (s, 9H), 1.22 (t, J = 7.1 Hz, 3H), 0.89 (t, J = 7.5 Hz, 3H). Step 2. Attachment of purine hinge binding group. The general procedure GP3 was used compound 12a (120.0 mg, 0.29 mmol) and with trifluoroacetic acid (661.0 mg, 5.8 mmol, 0.44 mL) in DCM (3.0 mL). The crude Bocdeprotected amine was worked-up (method A) and reacted with 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine 8 (140.4 mg, 0.58 mmol) and triethylamine (118.0 mg, 1.16 mmol, 0.16 mL) in ethanol (0.7 mL). The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH/DCM to afford ethyl 4-((2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)-propyl)quinazolin-4-yl)amino)butanoate 13a (113.0 mg, 0.22 mmol) in 77% yield. LC–MS (method 1): tR = 2.78 min, m/z (M + H)+ = 519.3. Step 3. Conversion of alkyl ester to hydroxamic acid. The general procedure GP4 (method A) was used with compound 13a (100.0 mg, 0.193 mmol) to afford the final product, (S)-4-((2-(1-((9H-purin-6-yl)amino)propyl)quinazolin-4-yl)amino)-N-hydroxybutanamide, TFA (14a) (39.0 mg, 0.073 mmol) in 38% yield. LC–MS (method 2): tR = 2.70 min, m/z (M + H)+ = 422.1. 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.38 (d, J = 8.1 Hz, 1H), 8.28 (d, J = 16.0 Hz, 2H), 8.05–7.96 (m, 1H), 7.88–7.81 (m, 1H), 7.74 (t, J = 7.7 Hz, 1H), 3.63 (ddd, J = 35.6, 13.3, 6.7 Hz, 3H), 2.20–2.02 (m, 4H), 2.02–1.88 (m, 1H), 1.78 (dq, J = 13.4, 6.7 Hz, 1H), 1.03 (t, J = 7.4 Hz, 3H). HRMS (ESI) m/z: [M+ H]+ calcd for C20H24N9O2 422.2047, found 422.2055.
Compound 13b was synthesized from 5 and 11b, using the similar procedure as described for 13a.
(S)-6-((2-(1-((9H-Purin-6-yl)amino)propyl)quinazolin-4-yl)-amino)-N-hydroxyhexanamide (14b). The general procedure GP4 (method A) was used with compound 13b (280.0 mg, 0.526 mmol) to afford the final product, (S)-6-((2-(1-((9H-purin-6-yl)amino)-propyl)quinazolin-4-yl)amino)-N-hydroxyhexanamide, TFA 14b (112.5 mg, 0.200 mmol) in 38% yield. LC–MS (method 2): tR = 2.77 min, m/z (M + H)+ = 450.3. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.38 (dd, J = 8.5, 1.2 Hz, 2H), 8.31 (s, 1H), 8.22 (s, 1H), 8.01 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.85 (dd, J = 8.5, 1.1 Hz, 1H), 7.74 (ddd, J = 8.3, 7.2, 1.2 Hz, 1H), 5.24 (s, 1H), 3.69 (dq, J = 13.2, 6.6 Hz, 1H), 3.55 (dt, J = 13.1, 6.6 Hz, 1H), 2.14 (q, J = 7.2 Hz, 2H), 1.88 (t, J = 7.3 Hz, 2H), 1.56–1.41 (m, 2H), 1.41–1.34 (m, 2H), 1.17 (q, J = 7.7 Hz, 2H), 1.06 (t, J = 7.4 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C22H28N9O2 450.2360, found 450.2377.
Procedure for Synthesis of C5-Subsitiuted Quinazolinones (19a–c and 24a–c, Scheme 2).
Sonogashira Cross-Coupling of Compound 15a with Alkynes 16a–c. tert-Butyl (S)-5-(2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)pent-4-ynoate (17a). The general procedure GP1 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (150.0 mg, 0.33 mmol), allylpalladium(II) chloride dimer (5.9 mg, 0.02 mmol), tri-tert-butylphosphonium tetrafluoroborate (9.5 mg, 0.03 mmol), tert-butyl pent-4-yn-oate 16a (60.6 mg, 0.39 mmol), and DABCO (73.4 mg, 0.66 mmol) in 2.5 mL of dry 1,4-dioxane. The remaining residue was purified by flash chromatography on silica using 0–25% ethyl acetate/ hexanes to afford tert-butyl (S)-5-(2-(1-((tert-butoxycarbonyl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)pent-4-ynoate 17a (130.0 mg, 0.245 mmol) as a yellow oil in 75% yield. LC– MS (method 1): tR = 3.90 min, m/z (M + H)+ = 532.4. 1H NMR (400 MHz, CDCl3) δ 7.67–7.48 (m, 6H), 7.37 (d, J = 8.0 Hz, 1H), 7.32–7.28 (m, 1H), 5.55 (s, 1H), 4.38 (s, 1H), 2.74 (dd, J = 8.4, 6.7 Hz, 2H), 2.55 (dd, J = 8.3, 6.7 Hz, 2H), 1.47–1.40 (m, 18H), 0.76 (t, J = 7.4 Hz, 3H).
Methyl (S)-6-(2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)hex-5-ynoate (17b). The general procedure GP1 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (234.0 mg, 0.51 mmol), allylpalladium(II) chloride dimer (9.3 mg, 0.03 mmol), tri-tert-butylphosphonium tetrafluoroborate (15.0 mg, 0.05 mmol), methyl hex-5-ynoate 16b (77.0 mg, 0613 mmol), and DABCO (115.0 mg, 1.02 mmol) in 2.5 mL of dry 1,4-dioxane. The remaining residue was purified by flash chromatography on silica using 0–30% ethyl acetate/hexanes to afford methyl (S)-6-(2-(1-((tert-butoxycarbonyl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)hex-5-ynoate 17b (162.0 mg, 0.322 mmol) as a yellow oil in 63% yield. LC–MS (method 1): tR = 3.60 min, m/z (M + H)+ = 504.3. 1H NMR (400 MHz, CDCl3) δ 7.67–7.47 (m, 6H), 7.39–7.28 (m, 2H), 5.49 (d, J = 9.0 Hz, 1H), 4.38 (s, 1H), 3.65 (s, 3H), 2.53 (dt, J = 15.0, 7.2 Hz, 4H), 1.94 (p, J = 7.2 Hz, 2H), 1.79–1.66 (m, 1H), 1.57–1.45 (m, 2H), 1.43 (s, 9H), 0.75 (t, J = 7.4 Hz, 3H).
Methyl (S)-4-((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)ethynyl)benzoate (17c). The general procedure GP1 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (183.0 mg, 0.40 mmol), allylpalladium(II) chloride dimer (7.2 mg, 0.02 mmol), tri-tert-butylphosphonium tetrafluoroborate (12.0 mg, 0.04 mmol), methyl 4-ethynylbenzoate 17c (77.0 mg, 0.48 mmol), and DABCO (90.0 mg, 0.80 mmol) in 2.0 mL of dry 1,4-dioxane. The resulting mixture was stirred at room temperature for 16 h and concentrated in vacuo. The remaining residue was purified by flash chromatography on silica using 0–25% ethyl acetate/hexanes to afford methyl (S)-4-((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)ethynyl)benzoate 17c (200.0 mg, 0.37 mmol) as a yellow solid in 93% yield. LC–MS (method 1): tR = 3.78 min, m/z (M + H)+ = 538.3. 1H NMR (400 MHz, CDCl3) δ 8.00–7.95 (m, 2H), 7.74–7.69 (m, 3H), 7.66–7.49 (m, 5H), 7.40 (d, J = 7.9 Hz, 1H), 7.36–7.30 (m, 1H), 5.49 (d, J = 9.0 Hz, 1H), 4.40 (s, 1H), 3.91 (s, 3H), 1.75 (ddd, J = 13.9, 7.3, 4.6 Hz, 1H), 1.55–1.48 (m, 1H), 1.43 (s, 9H), 0.77 (t, J = 7.4 Hz, 3H).
(S)-5-(2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxypentanamide (19a). Step 1. (i) Alkyne hydrogenation. The general procedure GP2 was used with compound 17a (102.0 mg, 0.19 mmol) and 10 wt % Pd/C (10.0 mg) in dry ethyl acetate (1.9 mL). After completion of reaction (determined by LC-MS), the crude reaction mixture was filtered using Celite, concentrated in vacuo, and carried over to the next step without purification. (ii) Attachment of purine hinge binding group. The general procedure GP3 was used on the hydrogenated product (formed above) with trifluoroacetic acid (438.0 mg, 3.84 mmol, 0.29 mL) in DCM (1.9 mL). The crude Boc-deprotected amine was worked-up (method B) and reacted with 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine 8 (92.0 mg, 0.38 mmol) and triethylamine (78.0 mg, 0.77 mmol, 0.1 mL) in ethanol (0.5 mL). The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc (0.5% AcOH by vol)/hexanes to afford 5-(4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)-amino)propyl)-3,4-dihydroquinazolin-5-yl)pentanoic acid 5.2n (60.0 mg, 0.103 mmol) in 54% yield. LC–MS (method 1): tR = 3.17 min, m/z (M + H)+ = 582.4. Step 2. Conversion of carboxylic acid to hydroxamic acid. The general procedure GP4 (method A, without the LiOH hydolysis step) was used with carboxylic acid, 18a (17.0 mg, 0.03 mmol), to afford the final product, (S)-5-(2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxypentanamide, TFA 19a (8.0 mg, 0.013 mmol) in 43% yield. LC–MS (method 2): tR = 3.74 min, m/z (M + H)+ = 513.2. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.26 (s, 1H), 7.68 (t, J = 7.7 Hz, 1H), 7.56 (d, J = 7.3 Hz, 3H), 7.50 (d, J = 1.2 Hz, 1H), 7.48 (s, 1H), 7.28 (dd, J = 7.6, 1.2 Hz, 1H), 7.20 (s, 1H), 7.07 (s, 1H), 6.95 (s, 1H), 4.74 (s, 1H), 3.81 (s, 1H), 3.14 (s, 2H), 1.97–1.90 (m, 2H), 1.90–1.79 (m, 1H), 1.53–1.48 (m, 3H), 0.76 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C27H29N8O3 513.2357, found 513.2346.
Compounds 19b and 19c were synthesized using the similar procedure as described for 19a.
(S)-6-(2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxyhexanamide (19b). The general procedure GP4 (method B) was used with compound 18b (60.0 mg, 0.098 mmol) to afford the final product (S)-6-(2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxyhexanamide, TFA 19b (15.0 mg, 0.023 mmol) in 24% yield. LC–MS (method 2): tR = 3.75 min, m/z (M + H)+ = 527.3. 1H NMR (400 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.32 (s, 2H), 7.73–7.64 (m, 1H), 7.59–7.46 (m, 5H), 7.29 (dd, J = 7.5, 1.3 Hz, 1H), 4.76 (s, 1H), 3.13 (t, J = 7.7 Hz, 2H), 2.04–1.95 (m, 1H), 1.94–1.78 (m, 3H), 1.47 (p, J = 7.6 Hz, 4H), 1.27 (dt, J = 14.4, 7.6 Hz, 2H), 0.77 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C28H31N8O3 527.2514, found 527.2524.
(S)-4-(2-(2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)ethyl)-N-hydroxybenzamide (19c). The general procedure GP4 (method A) was used with compound 18c (12.0 mg, 0.019 mmol) to afford the final product (S)-4-(2-(2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)ethyl)-N-hydroxybenzamide, TFA 19c (3.0 mg, 0.005 mmol) in 25% yield. LC–MS (method 2): tR = 3.94 min, m/z (M + H)+ = 561.3. HRMS (ESI) m/z: [M + H]+ calcd for C31H29N8O3 561.2357, found 561.2357.
(S)-4-(2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxybenzamide (24a). Step 1. Suzuki cross-coupling reaction. ((S)-tert-butyl (1-(5-chloro-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a′ (261.0 mg, 0.63 mmol), chloro(crotyl)(2-dicyclohexylphosphino-2′,4′,6′-triisopropybiphenyl)palladium(II) [Pd-170] (21.0 mg, 0.03 mmol), and (4-(ethoxycarbonyl)phenyl)boronic acid 20a (147.0 mg, 0.76 mmol) were suspended in 1,4-dioxane/water (2.0 mL, 4:1 by vol) in a MW vial equipped with a stir bar under N2 atmosphere, and potassium phosphate (402.0 mg, 1.89 mmol) was added to it. The MW vial was sealed and heated at 100 1/2C for 1 h in a MW reactor. The reaction mixture was allowed to cool to RT, quenched with water, and then extracted 3 times with ethyl acetate. The combined organic fractions were dried over MgSO4 and then concentrated in vacuo. The remaining residue was purified by flash chromatography on silica using 0–35% ethyl acetate/hexanes to afford ethyl (S)-4-(2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)benzoate 22a (320.0 mg, 0.61 mmol) as a colorless solid in 96% yield. LC–MS (method 1): tR = 3.82 min, m/z (M + H)+ = 528.3. 1H NMR (400 MHz, CDCl3) δ 8.05–8.00 (m, 2H), 7.80–7.76 (m, 2H), 7.56–7.40 (m, 3H), 7.39–7.30 (m, 3H), 7.27 (t, J = 4.4 Hz, 1H), 7.22–7.16 (m, 1H), 5.57 (d, J = 9.1 Hz, 1H), 4.41 (d, J = 8.6 Hz, 1H), 4.36 (q, J = 7.1 Hz, 2H), 1.76 (ddd, J = 14.2, 7.4, 4.6 Hz, 1H), 1.62–1.53 (m, 1H), 1.45 (s, 9H), 0.78 (t, J = 7.4 Hz, 3H). Step 2. Installation of purine hinge binding group. The general procedure GP3 was used with 22a (320.0 mg, 0.61 mmol) and trifluoroacetic acid (1.38 g, 12.13 mmol, 0.93 mL) in DCM (6.0 mL). The crude Boc-deprotected amine was worked-up (method A) and reacted with 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine (289.0 mg, 1.21 mmol) and triethylamine (245 mg, 2.42 mmol, 0.34 mL) in ethanol (1.2 mL). The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH/DCM to afford ethyl 4-(4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazolin-5-yl)benzoate 23a (348.0 mg, 0.55 mmol) in 91% yield. LC–MS (method 1): tR = 3.56 min, m/z (M + H)+ = 630.3. Step 3. Conversion of alkyl ester to hydroxamic acid. The general procedure GP4 (method A) was used with compound 23a (73.2 mg, 0.116 mmol) to afford the final product, (S)-4-(2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxybenzamide, TFA 24a (53.0 mg, 0.082 mmol) in 71% yield. LC–MS (method 2): tR = 3.56 min, m/z (M + H)+ = 533.2. 1H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H), 8.69 (s, 1H), 8.42 (s, 2H), 7.84 (dd, J = 8.2, 7.4 Hz, 1H), 7.74–7.64 (m, 3H), 7.57–7.37 (m, 5H), 7.34–7.24 (m, 3H), 5.75 (s, 1H), 4.80 (s, 1H), 2.03 (ddd, J = 11.6, 7.4, 3.6 Hz, 1H), 1.87 (dt, J = 14.7, 7.6 Hz, 1H), 0.79 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C29H25N8O3 533.2044, found 533.2044.
Compound 24b was synthesized using the similar procedure as described for 24a.
(S)-2-(4-(2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)phenyl)-N-hydroxyacetamide (24b). The general procedure GP4 (method A) was used with compound 23b (33.4 mg, 0.052 mmol) to afford the final product, (S)-2-(4-(2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)phenyl)-N-hydroxyacetamide, TFA 24b (12.7 mg, 0.019 mmol) in 36% yield. LC–MS (method 2): tR = 3.66 min, m/z (M + H)+ = 546.2. 1H NMR (400 MHz, DMSO-d6) δ 10.64 (s, 1H), 8.67 (s, 1H), 8.42 (s, 2H), 7.81 (t, J = 7.8 Hz, 1H), 7.67 (dd, J = 8.1, 1.3 Hz, 1H), 7.57–7.39 (m, 5H), 7.25 (dd, J = 7.4, 1.3 Hz, 1H), 7.21–7.14 (m, 4H), 5.75 (s, 2H), 4.81 (s, 1H), 2.03 (dd, J = 12.1, 6.8 Hz, 1H), 1.86 (dt, J = 14.7, 7.7 Hz, 1H), 0.79 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C30H27N8O3 547.2201, found 547.2203.
Ethyl (S)-2-(2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)benzo[d]thiazole-6-carboxylate (22c). To a mixture of ((S)-tert-butyl (1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (116.0 mg, 0.25 mmol) and 4,4,4′,4′,5,5,5′,5′-octamethyl-2,2′-bi(1,3,2-dioxaborolane) [BPin]2 (77.0 mg, 0.30 mmol) in 1,4-dioxane (1.0 mL) in a sealed tube, Pd(dppf)Cl2 (9.3 mg, 13.0 μmol) and potassium acetate (74.5 mg, 0.76 mmol) were added under N2 bubbling through the solvent. The resulting mixture was stirred at 100 1/2C for 16 h. After completion of the reaction, the crude reaction mixture was filtered into a MW vial equipped with a stir bar, and ethyl 2-bromobenzo[d]thiazole-6-carboxylate 20c (60.0 mg, 0.21 mmol) and 0.1 mL of water were added to it. [Pd-170] (10.6 mg, 7.1 μmol) and potassium phosphate (134.0 mg, 0.63 mmol) were added to this mixture under nitrogen atmosphere. The MW vial was sealed and heated at 100 1/2C for 10 h. The reaction mixture was allowed to cool to room temperature, quenched with water, and then extracted 3 times with ethyl acetate. The combined organic fractions were dried over MgSO4 and then concentrated in vacuo. The remaining residue was purified by flash chromatography on silica using 0–45% EtOAc/hexanes to afford the coupled product, ethyl (S)-2-(2-(1-((tert-butoxycarbonyl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)benzo[d]thiazole-6-carboxylate (87.0 mg, 0.15 mmol) 22c in 70% yield. LC–MS (method 1): tR = 3.80 min, m/z (M + H)+ = 585.2. 1H NMR (400 MHz, CDCl3) δ 8.59 (d, J = 1.6 Hz, 1H), 8.14 (dd, J = 8.6, 1.7 Hz, 1H), 8.05 (d, J = 8.6 Hz, 1H), 7.94 (d, J = 8.3 Hz, 1H), 7.90–7.82 (m, 1H), 7.63–7.40 (m, 4H), 7.36 (d, J = 8.0 Hz, 1H), 7.23 (d, J = 7.3 Hz, 1H), 5.50 (d, J = 9.1 Hz, 1H), 4.42 (q, J = 7.1 Hz, 3H), 1.75 (dt, J = 12.6, 6.9 Hz, 1H), 1.58–1.52 (m, 1H), 1.44 (m, 9H), 0.78 (t, J = 7.4 Hz, 3H).
Compound 23c was synthesized from 22c using the similar procedure as described for 23a.
(S)-2-(2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxybenzo[d]thiazole-6-carboxamide (24c). The general procedure GP4 (method B) was used with compound 23c (16.4 mg, 0.024 mmol) to afford the final product, (S)-2-(2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxybenzo[d]thiazole-6-carboxamide, TFA 24c (5.0 mg, 0.007 mmol) in 32% yield. LC–MS (method 2): tR = 3.64 min, m/z (M + H)+ = 590.2. 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 8.47 (d, J = 1.7 Hz, 1H), 8.27 (s, 2H), 8.02 (d, J = 8.5 Hz, 1H), 7.97–7.83 (m, 4H), 7.67–7.60 (m, 2H), 7.58–7.47 (m, 4H), 7.42 (s, 3H), 4.73 (s, 1H), 2.01 (s, 1H), 1.89 (s, 1H), 0.78 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C30H24N9O3S 590.1717, found 590.1708.
General Procedure for Pd-Catalyzed Amination Reaction (GP5). Aryl bromide (1 equiv), methanesulfonato[9,9-dimethyl-4,5-bis-(diphenylphosphino)xanthene](2′-methylamino-1,1′-biphenyl-2-yl)-palladium(II) XantPhos Palladacycle (methanesulfonato[9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene](2′-methylamino-1,1′-biphenyl-2-yl)palladium(II), Strem Chemicals Inc.) (0.025–0.05 equiv), and amine [if solid] (1.3 equiv) were weighed and added to a microwave vial equipped with a stir bar. The vial was covered with a rubber septum, evacuated, and then filled with nitrogen. Dry toluene or 1,4-dioxane (0.2 M) and amine [if oil at room temperature] (1.3 equiv) were added to the vial, followed by the addition of Cs2CO3 (3.0 equiv) under nitrogen bubbling through the solvent. The microwave vial was sealed and heated at 110 1/2C for 20 h. After 20 h, the crude reaction mixture was filtered through a short pad of Celite and concentrated in vacuo. The remaining residue was purified by flash chromatography on silica using forced flow of ethyl acetate/ hexanes system on Biotage KP-Sil prepacked cartridges and using the Biotage SP-1 automated chromatography system to afford the coupled product.
Procedure for Synthesis of C5-Subsitiuted Quinazolinones (28a–h, Scheme 3).
Pd-Catalyzed Amination of Compounds 15a or 15b with Amines 25a–g. Ethyl (S)-4-((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)butanoate (26a). The general procedure GP5 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (75.2 mg, 0.16 mmol), [XantPhos Palladacycle] (3.9 mg, 4.10 μmol), Cs2CO3 (160.0 mg, 0.49 mmol), and 4-ethoxy-4-oxobutan-1-aminium chloride 25a (35.8 mg, 0.21 mmol) were combined in dry toluene (0.8 mL). The resulting mixture was heated at 110 1/2C for 20 h and concentrated in vacuo. The remaining residue was purified by flash chromatography on silica using 0–30% ethyl acetate/hexanes to afford the coupled product, ethyl (S)-4-((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)butanoate 26a (58.4 mg, 0.115 mmol) in 70% yield. LC–MS (method 1): tR = 3.77 min, m/z (M + H)+ = 509.4. 1H NMR (400 MHz, CDCl3) δ 8.49 (s, 1H), 7.62 (d, J = 7.1 Hz, 1H), 7.55 (q, J = 8.0, 7.6 Hz, 3H), 7.40 (s, 1H), 6.93 (d, J = 27.4 Hz, 1H), 6.56 (d, J = 8.4 Hz, 1H), 5.57 (s, 1H), 4.39–4.32 (m, 1H), 4.12 (q, J = 7.1 Hz, 2H), 3.25 (q, J = 6.5 Hz, 2H), 2.41 (t, J = 7.3 Hz, 2H), 1.97 (p, J = 7.2 Hz, 2H), 1.73 (s, 2H), 1.43 (s, 9H), 1.24 (t, J = 7.1 Hz, 3H), 0.77 (t, J = 7.4 Hz, 3H).
Methyl (S)-5-((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)pentanoate (26b). The general procedure GP5 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (218.4 mg, 0.48 mmol), [XantPhos Palladacycle] (13.7 mg, 14.0 μmol), Cs2CO3 (466.0 mg, 1.43 mmol), and 5-methoxy-5-oxopentan-1-aminium chloride 25b (96.0 mg, 0.57 mmol) in dry toluene (2.4 mL). The remaining residue was purified by flash chromatography on silica using 0–50% ethyl acetate/hexanes to afford the coupled product, methyl (S)-5-((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)pentanoate 26b (153.0 mg, 0.30 mmol) in 63% yield. LC–MS (method 1): tR = 3.75 min, m/z (M + H)+ = 509.3. 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 7.57 (td, J = 18.2, 14.9, 7.7 Hz, 4H), 7.41 (s, 1H), 7.26 (s, 1H), 6.53 (d, J = 8.4 Hz, 1H), 4.35 (s, 1H), 3.66 (s, 3H), 3.20 (q, J = 6.2 Hz, 2H), 2.34 (t, J = 6.9 Hz, 2H), 1.79–1.64 (m, 6H), 1.43 (s, 9H), 0.77 (t, J = 7.3 Hz, 3H).
Methyl (S)-6-((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)hexanoate (26c). The general procedure GP5 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (121.2 mg, 0.26 mmol), [XantPhos Palladacycle] (6.3 mg, 6.61 μmol), Cs2CO3 (258.0 mg, 0.79 mmol), and 6-methoxy-6-oxohexan-1-aminium chloride 25c (62.4 mg, 0.34 mmol) in dry toluene (1.3 mL). The remaining residue was purified by flash chromatography on silica using 0–20% ethyl acetate/hexanes to afford the coupled product, methyl (S)-6-((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)hexanoate 26c (126.0 mg, 0.24 mmol) in 91% yield. LC–MS (method 1): tR = 3.83 min, m/z (M + H)+ = 523.3. 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 5.3 Hz, 1H), 7.66–7.48 (m, 4H), 7.37 (d, J = 7.9 Hz, 1H), 6.84 (d, J = 7.8 Hz, 1H), 6.50 (d, J = 8.4 Hz, 1H), 4.40–4.30 (m, 1H), 3.65 (s, 3H), 3.17 (td, J = 6.9, 5.1 Hz, 2H), 2.31 (t, J = 7.5 Hz, 2H), 1.67 (ddt, J = 17.5, 15.2, 7.6 Hz, 6H), 1.58–1.45 (m, 2H), 1.43 (s, 9H), 0.76 (t, J = 7.4 Hz, 3H).
Methyl (S)-4-(((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate (26d). The general procedure GP5 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (95.2 mg, 0.21 mmol), [XantPhos Palladacycle] (4.9 mg, 5.19 μmol), Cs2CO3 (203.0 mg, 0.62 mmol), and methyl 4-(aminomethyl)benzoate hydrochloride 25d (54.3 mg, 1.3 mmol) in dry toluene (1.0 mL). The remaining residue was purified by flash chromatography on silica using 0–30% ethyl acetate/hexanes to afford the coupled product, methyl (S)-4-(((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 26d (100.0 mg, 0.184 mmol) as an off-white solid in 89% yield. LC–MS (method 1): tR = 3.82 min, m/z (M + H)+ = 542.3. 1H NMR (400 MHz, CDCl3) δ 9.03 (t, J = 5.6 Hz, 1H), 8.02–7.95 (m, 2H), 7.57 (td, J = 17.2, 14.9, 8.1 Hz, 3H), 7.49–7.37 (m, 4H), 7.31–7.28 (m, 1H), 6.91 (d, J = 7.8 Hz, 1H), 6.40 (d, J = 8.4 Hz, 1H), 5.54 (s, 1H), 4.49 (d, J = 5.8 Hz, 2H), 4.43–4.33 (m, 1H), 3.90 (s, 3H), 1.79–1.70 (m, 1H), 1.64–1.56 (m, 1H), 1.43 (s, 9H), 0.77 (t, J = 7.4 Hz, 3H).
Methyl (S)-4-(((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-2-methylbenzoate (26e). The general procedure GP5 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)-carbamate 15a (170.0 mg, 0.411 mmol), [XantPhos Palladacycle] (20.0 mg, 0.02 mmol), Cs2CO3 (401.0 mg, 1.23 mmol), and methyl 5-(aminomethyl)picolinate dihydrochloride 25e (88.0 mg, 0.493 mmol) in dry toluene (2.0 mL). The remaining residue was purified by flash chromatography on silica using 0–50% ethyl acetate/hexanes to afford the coupled product, methyl (S)-4-(((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-2-methylbenzoate 26e (175.0 mg, 0.31 mmol) in 88% yield. LC–MS (method 1): tR = 3.88 min, m/z (M + H)+ = 557.3.
Methyl (S)-5-(((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)picolinate (26f). The general procedure GP5 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (96.5 mg, 0.21 mmol), [XantPhos Palladacycle] (5.0 mg, 5.26 μmol), Cs2CO3 (206.0 mg, 0.63 mmol), and methyl 5-(aminomethyl)picolinate dihydrochloride 25f (54.3 mg, 1.3 mmol) in dry toluene (1.0 mL). The remaining residue was purified by flash chromatography on silica using 0–80% ethyl acetate/hexanes to afford the coupled product, methyl (S)-5-(((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl) picolinate 26f (67.5 mg, 0.124 mmol) in 59% yield. LC–MS (method 1): tR = 3.56 min, m/z (M + H)+ = 544.3. 1H NMR (400 MHz, CDCl3) δ 9.07 (s, 1H), 8.73 (d, J = 2.1 Hz, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.82 (dd, J = 8.1, 2.2 Hz, 1H), 7.67–7.51 (m, 3H), 7.47 (t, J = 8.1 Hz, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.29 (s, 1H), 6.97 (s, 1H), 6.38 (d, J = 8.4 Hz, 1H), 4.54 (d, J = 5.8 Hz, 2H), 4.38 (s, 1H), 4.01 (d, J = 1.2 Hz, 3H), 1.78–1.72 (m, 1H), 1.63–1.53 (m, 1H) 1.43 (s, 9H), 0.78 (t, J = 7.3 Hz, 3H).
Methyl (S)-6-(((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)nicotinate (26g). The general procedure GP5 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 15a (109.0 mg, 0.24 mmol), [XantPhos Palladacycle] (6.8 mg, 0.007 mmol), Cs2CO3 (232.0 mg, 0.71 mmol), and methyl 5-(aminomethyl)picolinate dihydrochloride 25g (58.0 mg, 0.29 mmol) in dry toluene (1.2 mL). The remaining residue was purified by flash chromatography on silica using 0–50% ethyl acetate/hexanes to afford the coupled product, methyl (S)-6-(((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)nicotinate 26g (100.0 mg, 0.18 mmol) in 77% yield. LC–MS (method 1): tR = 3.58 min, m/z (M + H)+ = 544.3. 1H NMR (400 MHz, CDCl3) δ 9.28–9.15 (m, 2H), 8.20 (dd, J = 8.2, 2.2 Hz, 1H), 7.67–7.50 (m, 3H), 7.44 (dt, J = 13.8, 9.0 Hz, 3H), 7.33–7.28 (m, 1H), 6.91 (d, J = 7.9 Hz, 1H), 6.37 (d, J = 8.3 Hz, 1H), 5.56–5.51 (m, 1H), 4.64 (d, J = 6.0 Hz, 2H), 4.39 (s, 1H), 3.94 (d, J = 1.6 Hz, 3H), 1.78–1.70 (m, 1H), 1.60–1.50 (m, 1H), 1.40 (s, 9H), 0.77 (t, J = 7.4 Hz, 3H).
Methyl (S)-5-(((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)furan-2-carboxylate (26h). The general procedure GP5 was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)-carbamate 15a (56.6 mg, 0.12 mmol) [XantPhos Palladacycle] (2.9 mg, 3.09 μmol), Cs2CO3 (121.0 mg, 0.37 mmol), and methyl 5-(aminomethyl)furan-2-carboxylate hydrochloride 25h (35.5 mg, 0.18 mmol) in dry toluene (0.6 mL). The remaining residue was purified by flash chromatography on silica using 0–55% ethyl acetate/hexanes to afford the coupled product, methyl (S)-5-(((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)furan-2-carboxylate 26h (58.0 mg, 0.11 mmol) in 88% yield. LC–MS (method 1): tR = 3.71 min, m/z (M + H)+ = 533.3. 1H NMR (400 MHz, CDCl3) δ 8.94 (t, J = 5.9 Hz, 1H), 7.66–7.48 (m, 4H), 7.38 (d, J = 7.9 Hz, 1H), 7.09 (d, J = 3.5 Hz, 1H), 6.94 (d, J = 7.9 Hz, 1H), 6.51 (d, J = 8.4 Hz, 1H), 6.34 (d, J = 3.3 Hz, 1H), 4.48 (d, J = 5.8 Hz, 2H), 4.39 (d, J = 11.2 Hz, 1H), 3.88 (s, 3H), 1.74 (ddd, J = 14.3, 7.4, 4.8 Hz, 1H), 1.55 (dt, J = 14.0, 7.1 Hz, 1H), 1.43 (s, 9H), 0.77 (t, J = 7.4 Hz, 3H).
Methyl (S)-4-(((2-(1-((tert-Butoxycarbonyl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate (26i). The procedure mentioned in Scheme 1(3) was used with tert-butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)ethyl)-carbamate 15a (300.0 mg, 0.675 mmol) [XantPhos Palladacycle] (19.0 mg, 0.02 mmol), Cs2CO3 (660.0 mg, 2.03 mmol), and methyl 4-(aminomethyl)benzoate hydrochloride (177.0 mg, 0.88 mmol) in dry toluene (3.4 mL). The remaining residue was purified by flash chromatography on silica using 0–50% ethyl acetate/hexanes to afford the coupled product, methyl (S)-4-(((2-(1-((tert-butoxycarbonyl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 26i (274.0 mg, 0.52 mmol) as an off-white solid in 77% yield. LC–MS (method 1): tR = 3.53 min, m/z (M + H)+ = 529.3. 1H NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 8.02–7.96 (m, 2H), 7.69–7.37 (m, 8H), 7.30 (d, J = 7.3 Hz, 1H), 6.42 (d, J = 8.4 Hz, 1H), 4.52 (m, 1H), 4.49 (d, J = 5.9 Hz, 2H), 3.91 (d, J = 1.2 Hz, 3H), 1.43 (s, 9H), 1.32 (m, 3H). (S)-4-((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-N-hydroxybutanamide (28a). Step 1. Installation of purine hinge binding group. The general procedure GP3 was used with 26a (83.0 mg, 0.16 mmol) and trifluoroacetic acid (374.0 mg, 3.28 mmol, 0.25 mL) in DCM (1.6 mL). The crude Bocdeprotected amine was worked-up (method A) and reacted with 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine (78.0 mg, 0.33 mmol) and triethylamine (66.0 mg, 0.66 mmol, 91.0 μL) in ethanol (0.5 mL). The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH/DCM to afford ethyl 4-((4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazolin-5-yl)amino)butanoate 27a (53.0 mg, 0.09 mmol) in 53% yield. LC–MS (method 1): tR = 3.46 min, m/z (M + H)+ = 611.4. Step 2. Conversion of alkyl ester to hydroxamic acid. The general procedure GP4 (method B) was used with compound 27a (50.0 mg, 0.08 mmol) to afford the final product, (S)-4-((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-N-hydroxybutanamide, TFA 28a (8.8 mg, 0.02 mmol) in 21% yield. LC–MS (method 2): tR = 3.50 min, m/z (M + H)+ = 514.2. 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.49 (s, 1H), 8.38 (s, 2H), 7.59–7.47 (m, 5H), 6.70 (d, J = 7.9 Hz, 1H), 6.57 (d, J = 8.5 Hz, 1H), 5.75 (s, 1H), 4.74 (s, 1H), 3.16 (d, J = 4.9 Hz, 3H), 2.08–1.92 (m, 3H), 1.79 (h, J = 7.3, 6.9 Hz, 3H), 0.76 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C26H28N9O3 514.2310, found 514.2285 [Note: In addition to compound 28a, a side product originating from the hydrolysis of ethyl ester in 27a to carboxylic acid was also isolated after TFA deprotection step, in 7% yield].
Compounds 26b–g were converted to 27b–g using the similar procedure, as described above for 27a.
(S)-5-((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-N-hydroxypentanamide (28b). The procedure mentioned in Scheme 3 (method B) was used with compound 27b (68.5 mg, 0.112 mmol) to afford the product, (S)-5-((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-N-hydroxypentanamide, TFA 28b (42.0 mg, 0.065 mmol) in 61% yield. LC–MS (method 2): tR = 3.69 min, m/z (M + H)+ = 528.3. 1H NMR (400 MHz, DMSO-d6) δ 10.33 (s, 1H), 8.71 (s, 1H), 8.44 (s, 2H), 7.52 (td, J = 13.5, 5.2 Hz, 5H), 6.70 (d, J = 7.9 Hz, 1H), 6.56 (d, J = 8.4 Hz, 1H), 4.76 (s, 1H), 3.14 (d, J = 5.7 Hz, 2H), 2.10–1.93 (m, 3H), 1.82 (dt, J = 15.1, 7.6 Hz, 1H), 1.56 (dt, J = 7.0, 3.5 Hz, 4H), 0.77 (t, J = 7.3 Hz, 3H).
(S)-6-((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-N-hydroxyhexanamide (28c). The general procedure GP4 (method B) was used with compound 27c (135.0 mg, 0.216 mmol) to afford the final product, (S)-6-((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-N-hydroxyhexanamide, TFA 28c (30.2 mg, 0.056 mmol) in 32% yield. LC–MS (method 2): tR = 4.02 min, m/z (M + H)+ = 542.3. 1H NMR (400 MHz, DMSO-d6) δ 10.33–10.28 (m, 1H), 8.89 (s, 1H), 8.48 (s, 2H), 7.53 (dq, J = 13.3, 8.0, 4.8 Hz, 5H), 6.70 (d, J = 7.9 Hz, 1H), 6.56 (d, J = 8.5 Hz, 1H), 4.77 (s, 1H), 3.13 (t, J = 6.9 Hz, 2H), 2.01 (ddd, J = 14.2, 7.5, 4.5 Hz, 1H), 1.93 (t, J = 7.4 Hz, 2H), 1.83 (h, J = 7.4 Hz, 1H), 1.53 (dp, J = 22.8, 7.3 Hz, 4H), 1.37–1.25 (m, 2H), 0.77 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C28H32N9O3 542.2623, found 542.2647 [Note: In addition to compound 28c, a side product originating from the hydrolysis of methyl ester in 27c to carboxylic acid was also isolated after TFA deprotection step, in 14% yield].
(S)-4-(((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide (28d). The general procedure GP4 (method A) was used with compound 27d (78.1 mg, 0.121 mmol) to afford the final product, (S)-4-(((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 28d (35.5 mg, 0.106 mmol) in 50% yield. LC–MS (method 2): tR = 3.88 min, m/z (M + H)+ = 562.2. 1H NMR (400 MHz, DMSO-d6) δ 11.15 (s, 1H), 8.97 (s, 1H), 8.77 (s, 1H), 8.45 (s, 2H), 7.73–7.66 (m, 2H), 7.58 (s, 2H), 7.55 (t, J = 1.6 Hz, 1H), 7.52–7.36 (m, 5H), 6.73 (d, J = 7.9 Hz, 1H), 6.48 (d, J = 8.4 Hz, 1H), 4.77 (s, 1H), 4.52–4.46 (m, 2H), 2.01 (ddd, J = 14.2, 7.6, 4.6 Hz, 1H), 1.83 (dt, J = 14.4, 7.5 Hz, 1H), 0.77 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C30H28N9O3 562.2310, found 562.2322.
(S)-4-(((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxy-2-methylbenzamide (28e). The general procedure GP4 (method B) was used with compound 27e (105.0 mg, 0.159 mmol) to afford the final product (S)-4-(((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxy-2-methylbenzamide, TFA 28e (60.0 mg, 0.087 mmol) in 55% yield. LC–MS (method 2): tR = 4.18 min, m/z (M + H)+ = 576.2. 1H NMR (400 MHz, DMSO-d6) δ 10.76 (s, 1H), 8.88 (s, 1H), 8.74 (s, 1H), 8.44 (s, 2H), 7.61–7.52 (m, 3H), 7.56–7.43 (m, 4H), 7.26–7.13 (m, 3H), 6.73 (d, J = 7.9 Hz, 1H), 6.51 (dd, J = 8.5, 1.0 Hz, 1H), 4.78 (s, 1H), 4.41 (s, 2H), 2.30 (s, 3H), 2.01 (ddd, J = 14.0, 7.4, 4.4 Hz, 1H), 1.83 (dt, J = 14.8, 8.0 Hz, 1H), 0.77 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C31H30N9O3; 576.2466, found 576.2471 [Note: In addition to compound 28e, a side product originating from the hydrolysis of methyl ester in 27e to carboxylic acid was also isolated after TFA deprotection step, in 13% yield].
(S)-5-(((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxypicolinamide (28f). The general procedure GP4 (method A) was used with compound 28f (60.5 mg, 0.094 mmol) to afford the final product, (S)-5-(((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxypicolinamide, TFA 28f (20.0 mg, 0.03 mmol) in 32% yield. LC–MS (method 2): tR = 3.77 min, m/z (M + H)+ = 563.2. 1H NMR (400 MHz, DMSO-d6) δ 11.35 (s, 1H), 9.02 (s, 1H), 8.85 (s, 1H), 8.58 (d, J = 2.0 Hz, 1H), 8.47 (s, 2H), 7.96–7.83 (m, 2H), 7.66–7.40 (m, 6H), 6.75 (d, J = 7.9 Hz, 1H), 6.53 (d, J = 8.4 Hz, 1H), 4.78 (s, 1H), 4.58 (d, J = 3.4 Hz, 2H), 2.02 (ddd, J = 14.4, 7.5, 4.5 Hz, 1H), 1.82 (dt, J = 14.7, 7.7 Hz, 1H), 0.77 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C29H27N10O3 563.2262, found 563.2273.
(S)-6-(((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxynicotinamide (28g). The general procedure GP4 (method B) was used with compound 28g (79.1 mg, 0.122 mmol) to afford the final product, (S)-6-(((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxynicotinamide, TFA 28g (30.0 mg, 0.044 mmol) in 36% yield. LC–MS (method 2): tR = 3.54 min, m/z (M + H)+ = 563.3. 1H NMR (400 MHz, DMSO-d6) δ 11.32 (s, 1H), 9.22 (s, 1H), 8.96 (s, 1H), 8.85 (dd, J = 2.2, 0.8 Hz, 1H), 8.51 (s, 2H), 8.06 (dd, J = 8.1, 2.3 Hz, 1H), 7.63–7.42 (m, 6H), 6.76 (d, J = 7.9 Hz, 1H), 6.54–6.49 (m, 1H), 4.81 (s, 1H), 4.59 (s, 2H), 2.08–1.98 (m, 1H), 1.83 (dt, J = 14.7, 7.8 Hz, 1H), 0.79 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C29H27N10O3 563.2262, found 563.2283.
(S)-5-(((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxyfuran-2-carbox-amide (28h). The general procedure GP4 (method A) was used with compound 28h (61.5 mg, 0.097 mmol) to afford the final product, (S)-5-(((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxyfuran-2-carboxamide, TFA 28h (41.0 mg, 0.076 mmol) in 78% yield. LC–MS (method 2): tR = 3.73 min, m/z (M + H)+ = 552.2. 1H NMR (400 MHz, DMSO-d6) δ 11.09 (s, 2H), 8.83 (s, 3H), 8.46 (d, J = 2.9 Hz, 2H), 7.65–7.42 (m, 6H), 6.95 (d, J = 3.4 Hz, 1H), 6.77 (d, J = 7.9 Hz, 1H), 6.69 (d, J = 8.4 Hz, 1H), 6.45 (d, J = 3.4 Hz, 1H), 4.76 (dd, J = 10.5, 4.9 Hz, 1H), 4.48 (d, J = 3.7 Hz, 2H), 2.00 (ddd, J = 14.3, 7.4, 4.3 Hz, 1H), 1.82 (dt, J = 14.5, 7.6 Hz, 1H), 0.76 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C28H26N9O4 552.2102, found 552.2106.
(S)-4-(((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxy-N-methylbenzamide (28i). Compound 27d (40.6 mg, 0.063) was suspended in MeOH/water (1.2 mL, 1:1) in a vial equipped with a stir bar, and LiOH·H2O (5.3 mg, 0.13 mmol) was added to it. The resulting mixture was stirred at room temperature for 10 h and concentrated in vacuo. This crude product was dissolved in DMF (1.2 mL), and N-methylhydroxylamine hydrochloride (8.0 mg, 0.95 mmol), DIPEA (18.0 mg, 0.16 mmol), and 2-(3H-[1,2,3]triazolo[4,5-b]pyridin-3-yl)-1,1,3,3-tetramethylisouronium hexafluorophosphate(V) [HATU] (36.0 mg, 0.095 mmol) were added to it. The resulting mixture was stirred at room temperature for 16 h, concentrated in vacuo, and redissolved in DCM (1.2 mL) in a vial followed by dropwise addition of TFA (144.0 mg, 1.26 mmol, 0.10 mL) into it. The resulting mixture was stirred for 10 h, concentrated in vacuo, and purified on preparative HPLC to afford the final compound, (S)-4-(((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxy-N-methylbenzamide, TFA 28i (13.0 mg, 0.019 mmol) in 30% yield. LC–MS (method 2): tR = 4.15 min, m/z (M + H)+ = 576.3. 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 8.96 (s, 1H), 8.54 (s, 1H), 8.39 (s, 1H), 7.57 (dt, J = 6.6, 1.9 Hz, 4H), 7.47 (dd, J = 10.6, 5.7 Hz, 3H), 7.40–7.32 (m, 2H), 6.73 (d, J = 7.8 Hz, 1H), 6.50 (dd, J = 8.5, 1.0 Hz, 1H), 4.77 (s, 1H), 4.48 (s, 2H), 3.22 (s, 3H), 1.99 (ddd, J = 11.7, 7.4, 3.7 Hz, 1H), 1.87–1.78 (m, 1H), 0.77 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C31H30N9O3 576.2466, found 576.2494.
Procedure for Synthesis of C5-Substituted Quinazolinones (33, 37, 40a,b, 47, Scheme 4).
tert-Butyl (S)-(1-(5-Cyano-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate (29a). tert-Butyl (S)-(1-(5-bromo-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)-ethyl)carbamate 15a (181.0 mg, 0.40 mmol), zinc cyanide (58.0 mg, 0.49 mmol), and tetrakis(triphenylphosphine)Pd(0) (23.0 mg, 0.02 mmol) were suspended in dry DMF (2.0 mL) in a MW vial equipped with a stir bar under nitrogen atmosphere. The mixture was bubbled with N2 gas for 2 min, sealed and heated at 100 1/2C for 16 h. After 16 h, the crude reaction mixture was filtered through a short pad of Celite and concentrated in vacuo. The remaining residue was purified by flash chromatography on silica using 0–45% ethyl acetate/hexanes to afford tert-butyl (S)-(1-(5-cyano-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 29a (154.0 mg, 0.38 mmol) as a colorless solid in 96% yield. LC–MS (method 1): tR = 3.64 min, m/z (M + H)+ = 405.2. 1H NMR (400 MHz, CDCl3) δ 7.99–7.92 (m, 1H), 7.91–7.79 (m, 2H), 7.65–7.50 (m, 3H), 7.42 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 7.1 Hz, 1H), 5.39 (d, J = 9.0 Hz, 1H), 4.46 (s, 1H), 1.75 (ddd, J = 12.2, 7.3, 4.6 Hz, 1H), 1.55–1.49 (m, 1H), 1.43 (s, 9H), 0.78 (t, J = 7.4 Hz, 3H).
tert-Butyl (S)-(1-(5-(Aminomethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate (30a). Compound 29a (180.0 mg, 0.44 mmol) was dissolved in methanolic ammonia (2.2 mL, 7N in MeOH) in a 20 mL scintillation vial, and Raney Ni (20.0 mg (approx)) was added to it. The reaction vial was evacuated and then kept under hydrogen atmosphere using a balloon. The resulting suspension was stirred at room temperature for 20 h. After completion of reaction (by LC-MS), the crude reaction mixture was carefully filtered under nitrogen and concentrated in vacuo to afford tert-butyl (S)-(1-(5-(aminomethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)propyl)carbamate 30a. LC–MS (method 1): tR = 2.85 min, m/z (M + H)+ = 409.3, that was carried to the next step without purification.
Compound 30b was synthesized from 15a′ using the similar procedure as described for 30a.
Ethyl (S)-2-(((2-(1-((tert-Butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)thiazole-4-carboxylate (32a). Compound 30a (102.0 mg, 0.25 mmol) was dissolved in dry DMF (0.6 mL) in a microwave vial, and ethyl 2-bromothiazole-4-carboxylate 31 (118.0 mg, 0.5 mmol) and N-ethyl-N-isopropylpropan-2-amine [DIPEA] (129.0 mg, 1.00 mmol) were added to it. The microwave vial was sealed and heated at 180 1/2C for 30 min in a microwave. After completion of the reaction, the reaction mixture was concentrated in vacuo and the remaining residue was purified using 0–5% MeOH/DCM to afford ethyl (S)-2-(((2-(1-((tert-butoxycarbonyl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)thiazole-4-carboxylate 32a (52.8 mg, 0.094 mmol) in 38% yield. LC–MS (method 1): tR = 3.55 min, m/z (M + H)+ = 564.3.
(S)-2-(((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)-N-hydroxythiazole-4-carboxamide (33). The general procedure GP3 was used with compound 32a (78.8 mg, 0.14 mmol) and trifluoroacetic acid (319.0 mg, 2.80 mmol, 0.21 mL) in DCM (1.4 mL). The crude Bocdeprotected amine was worked-up (method A) and reacted with 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine 8 (66.8 mg, 0.28 mmol) and triethylamine (56.7 mg, 0.56 mmol, 78.0 μL) in ethanol (0.3 mL). The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH/DCM to afford ethyl 2-(((4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazolin-5-yl)methyl)amino)thiazole-4-carboxylate (68.1 mg, 0.102 mmol) in 73% yield. LC–MS (method 1): tR = 3.35 min, m/z (M + H)+ = 666.3.
The general procedure GP4 (method A) was used with compound 32a (62.4 mg, 0.09 mmol) to afford the final product, (S)-2-(((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)-N-hydroxythiazole-4-carboxamide, TFA 33 (17.6 mg, 0.03 mmol) in 33% yield. LC–MS (method 2): tR = 3.71 min, m/z (M + H)+ = 569.2. 1H NMR (400 MHz, DMSO-d6) δ 10.62 (s, 1H), 8.50 (s, 1H), 8.37 (s, 2H), 8.05 (t, J = 6.2 Hz, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.65–7.55 (m, 5H), 7.52 (s, 2H), 7.17 (s, 1H), 5.03 (d, J = 6.0 Hz, 2H), 4.79 (s, 1H), 2.00 (d, J = 11.7 Hz, 1H), 1.90–1.82 (m, 1H), 0.78 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C27H25N10O3S 569.1826, found 569.1805.
4-Oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazoline-5-carbonitrile (34). The general procedure GP3 was used with compound 29a (250.0 mg, 0.63 mmol) and trifluoroacetic acid (1.44 g, 12.7 mmol, 0.97 mL) in DCM (6.3 mL). The crude Boc-deprotected amine was worked-up (method B) and reacted with 6-chloro-9-(tetrahydro-2H-pyran-2-yl)-9H-purine 8 (300.0 mg, 1.26 mmol) and triethylamine (256.0 mg, 2.52 mmol, 0.35 mL) in ethanol (1.6 mL). The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH/DCM to afford 4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazo-line-5-carbonitrile 34 (258.4 mg, 0.51 mmol) in 81% yield. LC–MS (method 1): tR = 3.18 min, m/z (M + H)+ = 507.3.
Methyl 4-(((4-Oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazolin-5-yl)-methyl)carbamoyl)benzoate (36). Compound 34 (136.7 mg, 0.27 mmol) was dissolved in ammonia (1.5 mL, 7N in MeOH) in a 20 mL scintillation vial, and Raney Ni (20.0 mg (approx)) was added to it. The reaction vial was evacuated and then kept under hydrogen atmosphere using a balloon. The resulting suspension was stirred at room temperature for 20 h. After completion of reaction (by LC-MS), the crude reaction mixture was carefully filtered under nitrogen, concentrated in vacuo to afford the hydrogenated product, 5-(aminomethyl)-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)quinazolin-4(3H)-one [LC-MS (method 1): tR = 2.67 min, m/z (M + H)+ = 511.3)]. This hydrogenated product was dissolved in DMF (1.3 mL) in a 20 mL scintillation vial, and 4-(methoxycarbonyl)benzoic acid 35 (68.0 mg, 0.41 mmol), DIPEA (106.0 mg, 0.82 mmol, 0.14 mL), and HATU (125.0 mg, 0.33 mmol) were added to it. The resulting mixture was stirred at room temperature for 16 h and concentrated in vacuo. The remaining residue was purified using 0–10% MeOH/DCM to afford methyl 4-(((4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazolin-5-yl)methyl)-carbamoyl)benzoate 36 (160.0 mg, 0.24 mmol) in 87% yield. LC–MS (method 1): tR = 3.32 min, m/z (M + H)+ = 673.3.
(S)-N1-((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)-N4-hydroxyterephthalamide (37). The general procedure GP4 (method A) was used with compound 36 (80.0 mg, 0.119 mmol) to afford the final product, (S)-N1-((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)-N4-hydroxyterephthalamide, TFA 37 (26.0 mg, 0.037 mmol) in 31% yield. LC–MS (method 2): tR = 3.54 min, m/z (M + H)+ = 590.3. 1H NMR (400 MHz, DMSO-d6) δ 11.35 (s, 1H), 9.04 (t, J = 6.0 Hz, 1H), 8.56 (s, 1H), 8.39 (s, 2H), 7.98–7.94 (m, 2H), 7.86–7.82 (m, 2H), 7.76 (t, J = 7.9 Hz, 1H), 7.65–7.48 (m, 6H), 7.41 (d, J = 7.6 Hz, 1H), 5.02 (d, J = 5.8 Hz, 2H), 4.79 (s, 1H), 2.03 (ddd, J = 14.3, 7.5, 4.5 Hz, 1H), 1.89–1.82 (m, 1H), 0.78 (t, J = 7.3 Hz, 3H)). HRMS (ESI) m/z: [M+ H]+ calcd for C31H28N9O4 590.2259, found 590.2265.
(S)-2-((((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)methyl)-N-hydroxythiazole-4-carboxamide (40a). 5-(Aminomethyl)-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)quinazolin-4(3H)-one (20.0 mg, 0.04 mmol) was dissolved in DCE (1 mL), and ethyl 2-formylthiazole-4-carboxylate 38a (7.3 mg, 0.04 mmol) and a drop of acetic acid were added to it. The resulting mixture was stirred at room temperature for 2 h. After 2 h, sodium triacetoxy borohydride (24.9 mg, 0.12 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. After completion of the reaction (by LCMS), a saturated aqueous solution of sodium bicarbonate was added. The product was extracted three times with DCM. The combined organic layers were washed, dried over MgSO4, filtered, and concentrated in vacuo. The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH (with 1% triethylamine)/DCM to afford ethyl 2-((((4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazolin-5-yl)methyl)amino)methyl)thiazole-4-carboxylate 39a (17.4 mg, 0.026 mmol) in 65% yield. LC–MS (method 1): tR = 2.96 min, m/z (M + H)+ = 680.3. The general procedure GP4 (method B) was used with compound 39a (11.0 mg, 0.018 mmol) to afford the final product, (S)-2-((((2-(1-((9H-purin-6-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)-methyl)-N-hydroxythiazole-4-carboxamide, TFA 40a (5.0 mg, 0.007 mmol) in 39% yield. LC–MS (method 2): tR = 3.14 min, m/z (M + H)+ = 583.2. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 10.08 (s, 1H), 9.22 (s, 2H), 8.30 (s, 1H), 8.21 (s, 2H), 7.87 (dd, J = 8.3, 7.3 Hz, 1H), 7.78–7.73 (m, 1H), 7.68 (dt, J = 6.6, 2.2 Hz, 1H), 7.62 (dd, J = 7.4, 1.3 Hz, 2H), 7.55 (d, J = 7.4 Hz, 2H), 4.76 (d, J = 15.8 Hz, 4H), 4.64 (s, 2H), 1.96–1.89 (m, 2H), 0.77 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + 2H]2+ (M + z/z, z = 2) calcd for C28H28N10O3S 292.1028, found 292.1042.
(S)-4-((((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)methyl)-N-hydroxybenzamide (40b). 5-(Aminomethyl)-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)quinazolin-4(3H)-one (44.1 mg, 0.09 mmol) was dissolved in DCE (2 mL), and ethyl 2-formylthiazole-4-carboxylate 38b (14.1 mg, 0.09 mmol) and a drop of acetic acid were added to it. The resulting mixture was stirred at room temperature for 2 h. After 2 h of stirring, sodium triacetoxy borohydride (54.9 mg, 0.26 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. After completion of the reaction (by LC-MS), a saturated aqueous solution of sodium bicarbonate was added. The product was extracted three times with DCM. The combined organic layers were washed, dried over MgSO4, filtered, and concentrated in vacuo. The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH (with 1% triethylamine)/DCM to afford methyl 4-((((4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)-amino)propyl)-3,4-dihydroquinazolin-5-yl)methyl)amino)methyl)-benzoate 39b (53.3 mg, 0.08 mmol) in 94% yield. LC–MS (method 1): tR = 2.98 min, m/z (M + H)+ = 659.3. The general procedure GP4 (method A) was used with compound 39b (53.3 mg, 0.081 mmol) to afford the final product, (S)-4-((((2-(1-((9H-purin-6-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)-methyl)-N-hydroxybenzamide, TFA 40b (33.5 mg, 0.049 mmol) in 41% yield. LC–MS (method 2): tR = 3.09 min, m/z (M + H)+ = 576.3. 1H NMR (400 MHz, DMSO-d6) δ 11.27 (s, 1H), 8.99 (s, 2H), 8.26 (d, J = 5.8 Hz, 2H), 8.18 (s, 1H), 7.85 (dd, J = 8.3, 7.3 Hz, 1H), 7.81–7.71 (m, 3H), 7.67 (qt, J = 6.2, 3.5 Hz, 2H), 7.59–7.50 (m, 5H), 4.78 (s, 1H), 4.58 (dd, J = 11.5, 5.8 Hz, 2H), 4.29 (t, J = 5.8 Hz, 2H), 1.97–1.85 (m, 2H), 0.77 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + 2H]2+ (M + z/z; z = 2) calcd for C31H31N9O3 288.6269, found 288.6270.
(S)-2-(((2-(1-((9H-Purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)-N-hydroxypyrimidine-5-carboxamide (43). 5-(Aminomethyl)-3-phenyl-2-((1S)-1-((9-(tetra-hydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)quinazolin-4(3H)-one (160.0 mg, 0.31 mmol) was dissolved in ethanol (0.7 mL) in a microwave vial equipped with a sir bar, and ethyl 2-chloropyrimidine-5-carboxylate 41 (117.0 mg, 0.63 mmol) and triethylamine (127.0 mg, 1.25 mmol, 0.18 mL) were added to it. The microwave vial was sealed, and the resulting mixture was heated at 90 1/2C for 3 h in a microwave. After completion of the reaction, the reaction mixture was concentrated in vacuo, and the remaining residue was purified using 0–5% MeOH/DCM to afford ethyl 2-(((4-oxo-3-phenyl-2-((1S)-1-((9-(tetrahydro-2H-pyran-2-yl)-9H-purin-6-yl)amino)propyl)-3,4-dihydroquinazolin-5-yl)methyl)amino)-pyrimidine-5-carboxylate 42 (170.0 mg, 0.26 mmol) in 82% yield. LC–MS (method 1): tR = 3.41 min, m/z (M + H)+ = 661.3.
The general procedure GP4 (method A) was used with compound 42 (72.0 mg, 0.11 mmol) to afford the final product, (S)-2-(((2-(1-((9H-purin-6-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)-N-hydroxypyrimidine-5-carboxamide, TFA 43 (42.0 mg, 0.062 mmol) in 57% yield. LC–MS (method 2): tR = 3.84 min, m/z (M + H)+ = 564.3. 1H NMR (400 MHz, DMSO-d6) δ 11.05 (s, 1H), 10.01 (d, J = 6.8 Hz, 1H), 8.73–8.53 (m, 4H), 8.17 (d, J = 6.6 Hz, 1H), 7.72 (t, J = 7.9 Hz, 1H), 7.66–7.45 (m, 6H), 7.37 (d, J = 7.6 Hz, 1H), 5.08 (d, J = 4.8 Hz, 2H), 4.77 (ddd, J = 10.7, 6.9, 3.8 Hz, 2H), 2.15–2.02 (m, 1H), 1.97–1.82 (m, 1H), 0.86 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C28H26N11O3 564.2215, found 564.2207.
General Procedure for Attachment of Pyrimidine Hinge Binding Group (GP6).
The Boc-protected amine (1.0 equiv) was dissolved in DCM (0.1 M) in a vial, and trifluoroacetic acid (20 equiv) was added dropwise to it. The resulting mixture was stirred at room temperature for 3 h. After completion of reaction (by LC-MS), the reaction mixture was worked-up by either of the following two methods. Method A: The crude reaction was quenched with aqueous saturated NaHCO3 solution and extracted three times with DCM. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated in vacuo to afford the free amine. Method B: The crude reaction mixture was concentrated in vacuo, redissolved in 1–2 mL of DCM, and passed through preconditioned PL-HCO3 MP SPE device (bicarbonate resin solid phase extraction) and washed with 2 mL of DCM. The filtrate was concentrated in vacuo to afford the free amine. The free amine was dissolved in n-butanol (0.4 M) in a microwave vial equipped with a stir bar followed by the addition of substituted chloro-pyrimidine (1.5–2.0 equiv) and DIPEA (3.0–4.0 equiv) to it. The vial was sealed and heated for 1–5 h at 130 1/2C in a microwave. After completion of reaction (by LC-MS), the reaction mixture was concentrated in vacuo and the remaining residue was purified by flash chromatography on silica gel using forced flow of indicated solvent system on Biotage KP-Sil prepacked cartridges and using the Biotage SP-1 automated chromatography system to afford the coupled product.
Procedure for Synthesis of C5-Substituted quinazolinones (46a–d and 48a–o, Scheme 5).
Methyl (S)-6-(2-(1-((2,6-Diamino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)hexanoate (45a). The general procedure GP2 was used with compound 17b (96.0 mg, 0.19 mmol) and 10 wt % Pd/C (9.6 mg). After completion of reaction (determined by LC-MS), the crude reaction mixture was filtered using Celite and concentrated in vacuo. The general procedure GP6 was used on the hydrogenated product (formed above) with trifluoroacetic acid (440.0 mg, 3.86 mmol, 0.30 mL) in DCM (2.0 mL). The crude reaction was worked-up (method B) and heated with 2,4-diamino-6-chloropyrimidine-5-carbonitrile 44a (49.0 mg, 0.29 mmol) and DIPEA (75.0 mg, 0.58 mmol, 0.10 mL) in n-butanol (0.5 mL) at 130 1/2C for 5 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–70% EtOAc/hexanes to afford methyl (S)-6-(2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)hexanoate 45a (66.0 mg, 0.12 mmol) in 63% yield. LC–MS (method 1): tR = 3.13 min, m/z (M + H)+ = 541.3.
Methyl (S)-6-(2-(1-((2-Amino-5-cyano-6-methylpyrimidin-4-yl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-hexanoate (45b). The general procedure GP2 was used with compound 17b (141.0 mg, 0.28 mmol) and 10 wt % Pd/C (9.6 mg). After completion of reaction (determined by LC-MS), the crude reaction mixture was filtered using Celite and concentrated in vacuo. The general procedure GP6 was used on the hydrogenated product (formed above) with trifluoroacetic acid (643.0 mg, 5.64 mmol, 0.43 mL) in DCM (2.8 mL). The crude reaction was worked-up (method B) and heated with 2-amino-4-chloro-6-methylpyrimidine-5-carbon-itrile 44b (71.0 mg, 0.42 mmol) and DIPEA (109.0 mg, 0.85 mmol, 0.15 mL) in n-butanol (0.7 mL) at 130 1/2C for 1 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–70% EtOAc/hexanes to afford methyl (S)-6-(2-(1-((2-amino-5-cyano-6-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)hexanoate 45b (76.0 mg, 0.14 mmol) in 50% yield. LC–MS (method 1): tR = 3.19 min, m/z (M + H)+ = 540.3.
Methyl (S)-6-(2-(1-((2-Amino-5-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)hexanoate (45c). The general procedure GP2 was used with compound 17b (96.0 mg, 0.19 mmol) and 10 wt % Pd/C (9.6 mg). After completion of reaction (determined by LC-MS), the crude reaction mixture was filtered using Celite and concentrated in vacuo. The general procedure GP6 was used on the hydrogenated product (formed above) with trifluoroacetic acid (440.0 mg, 3.86 mmol, 0.30 mL) in DCM (2.0 mL). The crude reaction was worked-up (method B) and heated with 4-chloro-5-methylpyrimidin-2-amine 45c (42.0 mg, 0.29 mmol) and DIPEA (75.0 mg, 0.58 mmol, 0.10 mL) in n-butanol (0.5 mL) at 130 1/2C for 5 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–10% MeOH/DCM to afford the product methyl (S)-6-(2-(1-((2-amino-5-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroqui nazolin-5-yl)hexanoate 45c (38.0 mg, 0.07 mmol) in 38% yield. LC– MS (method 1): tR = 3.2 min, m/z (M + H)+ = 515.3.
Methyl (S)-4-(2-(2-(1-((2,6-Diamino-5-cyanopyrimidin-4-yl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)ethyl)-benzoate (45d). The general procedure GP2 was used with compound 17c (81.2 mg, 0.15 mmol) and 10 wt % Pd/C (8.0 mg). After completion of reaction (determined by LC-MS), the crude reaction mixture was filtered using Celite and concentrated in vacuo. The general procedure GP6 was used on the hydrogenated product (formed above) with trifluoroacetic acid (333.0 mg, 2.92 mmol, 0.22 mL) in DCM (1.5 mL). The crude reaction was worked-up (method B) and heated with 2,4-diamino-6-chloropyrimidine-5-carbonitrile 44a (50.0 mg, 0.29 mmol) and DIPEA (75.0 mg, 0.58 mmol, 0.1 mL) in n-butanol (0.4 mL) at 130 1/2C for 5 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(2-(2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)ethyl)benzoate 45d (64.0 mg, 0.11 mmol) in 76% yield. LC–MS (method 1): tR = 3.23 min, m/z (M + H)+ = 575.3.
(S)-6-(2-(1-((2,6-Diamino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxyhexanamide (46a). The general procedure GP4 (method B) was used with compound 45a (54.0 mg, 0.10 mmol) to afford the final product, (S)-6-(2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxyhexanamide, TFA 46a (40.0 mg, 0.061 mmol) in 61% yield. LC–MS (method 2): tR = 3.91 min, m/z (M + H)+ = 542.3. 1H NMR (400 MHz, DMSO-d6) δ 10.29 (s, 1H), 8.05 (s, 3H), 7.74 (t, J = 7.8 Hz, 2H), 7.57–7.42 (m, 7H), 7.33 (dd, J = 7.6, 1.3 Hz, 1H), 4.77 (td, J = 7.6, 4.9 Hz, 1H), 3.14 (ddq, J = 12.4, 8.0, 5.5, 4.2 Hz, 2H), 1.90 (t, J = 7.2 Hz, 3H), 1.82–1.71 (m, 1H), 1.49 (dp, J = 15.0, 7.4 Hz, 4H), 1.28 (q, J = 8.1 Hz, 2H), 0.71 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M+ H]+ calcd for C28H32N9O3 542.2623, found 542.2635.
(S)-6-(2-(1-((2-Amino-5-cyano-6-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxyhexanamide (46b). The general procedure GP4 (method B) was used with compound 45b (54.0 mg, 0.10 mmol) to afford the final product, (S)-6-(2-(1-((2-amino-5-cyano-6-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxyhexanamide, TFA 46b (45.0 mg, 0.069 mmol) in 69% yield. LC–MS (method 2): tR = 4.04 min, m/z (M + H)+ = 541.3. 1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 8.09 (s, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.59–7.45 (m, 6H), 7.45–7.36 (m, 2H), 7.32 (dd, J = 7.5, 1.3 Hz, 1H), 4.77 (td, J = 7.6, 4.8 Hz, 1H), 3.14 (t, J = 7.7 Hz, 2H), 2.54 (s, 1H), 2.34 (s, 3H), 2.00–1.86 (m, 3H), 1.79 (dp, J = 14.7, 7.4 Hz, 1H), 1.49 (dq, J = 15.1, 7.6 Hz, 4H), 1.28 (p, J = 7.7 Hz, 2H), 0.72 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C29H33N8O3 541.2670, found 541.2680.
(S)-6-(2-(1-((2-Amino-5-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxyhexanamide (46c). The general procedure GP4 (method B) was used with compound 45c (35.0 mg, 0.068 mmol) to afford the final product, (S)-6-(2-(1-((2-amino-5-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-N-hydroxyhexanamide, TFA 46c (30.0 mg, 0.048 mmol) in 70% yield. LC–MS (method 2): tR = 4.06 min, m/z (M + H)+ = 516.3. 1H NMR (400 MHz, DMSO-d6) δ 11.72 (s, 1H), 10.29 (s, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.57–7.50 (m, 4H), 7.42–7.31 (m, 5H), 4.85 (td, J = 7.9, 5.2 Hz, 1H), 3.14 (t, J = 7.7 Hz, 2H), 2.08–1.78 (m, 8H), 1.47 (dt, J = 14.9, 7.0 Hz, 4H), 1.27 (p, J = 7.5, 7.1 Hz, 2H), 0.76 (t, J = 7.3 Hz, 3H)). HRMS (ESI) m/z: [M + H]+ calcd for C28H34N7O3 516.2718, found 516.2717.
(S)-4-(2-(2-(1-((2,6-Diamino-5-cyanopyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)ethyl)-N-hydroxybenzamide (46d). The general procedure GP4 (method B) was used with compound 45d (56.0 mg, 0.097 mmol) to afford the final product, (S)-4-(2-(2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)ethyl)-N-hydroxybenzamide, TFA 46d (27.2 mg, 0.039 mmol) in 40% yield. LC–MS (method 2): tR = 4.34 min, m/z (M + H)+ = 576.2. 1H NMR (400 MHz, DMSO-d6) δ 11.10 (s, 1H), 7.77–7.71 (m, 1H), 7.67– 7.61 (m, 3H), 7.60–7.44 (m, 6H), 7.36–7.25 (m, 4H), 4.76 (q, J = 7.3 Hz, 1H), 2.91–2.82 (m, 2H), 1.94–1.85 (m, 1H), 1.75 (dt, J = 14.5, 7.4 Hz, 1H), 0.74–0.65 (m, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C31H30N9O3 576.2466, found 576.2475 [Note: In addition to compound 46d, a side product originating from the hydrolysis of methyl ester in 45d to carboxylic acid was also isolated in 7% yield].
(S)-5-((2-(1-((2-Amino-5-cyano-6-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-N-hydroxypentanamide (48a). The general procedure GP6 was used with compound 26b (98.0 mg, 0.193 mmol) and trifluoroacetic acid (439.0 mg, 3.85 mmol, 0.3 mL) in DCM (2.0 mL). The crude reaction was worked-up (method B) and heated with 2-amino-4-chloro-6-methylpyrimidine-5-carbonitrile 44b (49.0 mg, 0.29 mmol) and DIPEA (75.0 mg, 0.56 mmol, 0.1 mL) in n-butanol (0.5 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-5-((2-(1-((2-amino-5-cyano-6-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-pentanoate 47a (89.0 mg, 0.164 mmol) in 85% yield. LC–MS (method 1): tR = 3.18 min, m/z (M + H)+ = 541.3.
The general procedure GP4 (method B) was used with compound 47a (74.0 mg, 0.14 mmol) to afford the final product, (S)-5-((2-(1-((2-amino-5-cyano-6-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-N-hydroxypentanamide, TFA 48a (73.5 mg, 0.112 mmol) in 82% yield. LC–MS (method 2): tR = 4.02 min, m/z (M + H)+ = 542.3. 1H NMR (400 MHz, DMSO-d6) δ 10.30 (s, 1H), 8.03 (s, 1H), 7.63–7.51 (m, 3H), 7.51–7.46 (m, 3H), 7.46–7.37 (m, 2H), 6.74 (dd, J = 7.9, 1.0 Hz, 1H), 6.59 (d, J = 8.5 Hz, 1H), 4.74 (td, J = 7.6, 4.8 Hz, 1H), 3.18 (d, J = 6.1 Hz, 2H), 2.54 (s, 1H), 2.35 (s, 3H), 2.02–1.85 (m, 3H), 1.76 (dp, J = 14.6, 7.4 Hz, 1H), 1.57 (q, J = 4.1 Hz, 4H), 0.70 (t, J = 7.3 Hz, 3H)). HRMS (ESI) m/z: [M + H]+ calcd for C28H32N9O3 542.2623, found 542.2626 [Note: In addition to compound 48a, a side product originating from the hydrolysis of methyl ester in 47a to carboxylic acid was also isolated in 7% yield].
(S)-4-(((2-(1-((2,6-Diamino-5-cyanopyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)-N-hydroxybenzamide (48b). The general procedure GP6 was used with compound 26d (80.0 mg, 0.15 mmol) and trifluoroacetic acid (336.0 mg, 2.95 mmol, 0.23 mL) in DCM (1.7 mL). The crude reaction was worked-up (method B) and heated with 2,4-diamino-6-chloropyrimidine-5-carbonitrile 44a (50.0 mg, 0.29 mmol) and DIPEA (76.0 mg, 0.59 mmol, 0.1 mL) in 2-propanol (0.7 mL) at 130 1/2C for 1 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/ hexanes to afford the product methyl (S)-4-(((2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 47b (70.0 mg, 0.10 mmol) in 69% yield. LC–MS (method 1): tR = 3.18 min, m/z (M + H)+ = 576.3. The general procedure GP4 (method B) was used with compound 47b (41.7 mg, 0.072 mmol) to afford the final product, (S)-4-(((2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48b (21.2 mg, 0.031 mmol) in 42% yield. LC–MS (method 2): tR = 3.98 min, m/z (M + H)+ = 577.3. 1H NMR (400 MHz, DMSO-d6) δ 11.12 (s, 1H), 8.96 (s, 1H), 7.70 (dd, J = 8.2, 1.7 Hz, 2H), 7.55 (t, J = 7.3 Hz, 2H), 7.52–7.43 (m, 5H), 7.40 (dd, J = 8.2, 1.7 Hz, 2H), 6.76 (d, J = 7.8 Hz, 1H), 6.51 (d, J = 8.4 Hz, 1H), 4.72 (q, J = 7.1 Hz, 1H), 4.50 (d, J = 4.4 Hz, 2H), 1.85 (q, J = 6.4, 5.8 Hz, 1H), 1.71 (dt, J = 14.5, 7.4 Hz, 1H), 0.72–0.64 (m, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C30H29N10O3 577.2419, found 577.2407.
(S)-4-(((2-(1-((2-Amino-5-cyano-6-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)-N-hydroxybenzamide (48c). The general procedure GP6 was used with compound 26d (68.0 mg, 0.125 mmol) and trifluoroacetic acid (286.0 mg, 2.51 mmol, 0.19 mL) in DCM (1.2 mL). The crude reaction was worked-up (method B) and heated with 2-amino-4-chloro-6-methylpyrimidine-5-carbonitrile 44b (32.0 mg, 0.19 mmol) and DIPEA (48.0 mg, 0.38 mmol, 65.0 μL) in n-butanol (0.3 mL) at 130 1/2C for 1 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–70% EtOAc/hexanes to afford the product methyl (S)-4-(((2-(1-((2-amino-5-cyano-6-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 47c (60.0 mg, 0.10 mmol) in 84% yield. LC–MS (method 1): tR = 3.24 min, m/z (M + H)+ = 575.3. The general procedure GP4 (method B) was used with compound 47c (54.0 mg, 0.094 mmol) to afford the final product, (S)-4-(((2-(1-((2-amino-5-cyano-6-methylpyrimidin-4-yl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)-N-hydroxybenzamide, TFA 48c (34.0 mg, 0.05 mmol) in 52% yield. LC–MS (method 2): tR = 4.18 min, m/z (M + H)+ = 576.3. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.97 (s, 1H), 8.00 (s, 1H), 7.73–7.66 (m, 2H), 7.59–7.45 (m, 5H), 7.45–7.36 (m, 4H), 6.76 (dd, J = 7.9, 0.8 Hz, 1H), 6.51 (dd, J = 8.5, 0.9 Hz, 1H), 4.74 (td, J = 7.7, 4.7 Hz, 1H), 4.50 (s, 2H), 2.33 (s, 3H), 1.90 (dtd, J = 14.2, 7.2, 4.7 Hz, 1H), 1.82–1.71 (m, 1H), 0.70 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C31H30N9O3 576.2466, found 576.2481 [Note: For PK studies, 48c was converted to a free base].
(S)-4-(((2-(1-((2-Amino-6-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide (48d). The general procedure GP6 was used with compound 26d (52.0 mg, 0.096 mmol) and trifluoroacetic acid (219.0 mg, 1.92 mmol, 0.15 mL) in DCM (1.0 mL). The crude reaction was worked-up (method B) and heated with 4-chloro-6-methylpyrimidin-2-amine 44d (21.0 mg, 0.14 mmol) and DIPEA (37.0 mg, 0.29 mmol, 50.0 μL) in n-butanol (0.3 mL) at 130 1/2C for 5 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–10% MeOH/DCM to afford methyl (S)-4-(((2-(1-((2-amino-6-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-benzoate 47d (29.0 mg, 0.053 mmol) in 55% yield. LC–MS (method 1): tR = 2.97 min, m/z (M + H)+ = 550.3. The general procedure GP4 (method B) was used with compound 47d (35.0 mg, 0.064 mmol) to afford the final product, (S)-4-(((2-(1-((2-amino-6-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-amino)methyl)-N-hydroxybenzamide, TFA 48d (18.0 mg, 0.027 mmol) in 42% yield. LC–MS (method 2): tR = 4.23 min, m/z (M + H)+ = 551.2. 1H NMR (400 MHz, DMSO-d6) δ 12.08 (s, 1H), 11.14 (s, 1H), 8.96 (d, J = 7.2 Hz, 2H), 7.72–7.68 (m, 2H), 7.60–7.45 (m, 6H), 7.42–7.37 (m, 3H), 6.73 (d, J = 7.8 Hz, 1H), 6.51 (d, J = 8.4 Hz, 1H), 5.99 (s, 1H), 4.51 (ddd, J = 11.5, 5.5, 2.7 Hz, 3H), 2.20 (s, 3H), 1.88 (ddd, J = 14.2, 7.4, 4.1 Hz, 1H), 1.64 (ddd, J = 13.9, 9.0, 7.0 Hz, 1H), 0.67 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C30H31N8O3 551.2514, found 551.2532 [Note: In addition to compound 48d, a side product originating from the hydrolysis of methyl ester in 47d to carboxylic acid was also isolated in 7% yield].
(S)-4-(((2-(1-((6-Amino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide (48e). The general procedure GP6 was used with compound 26d (48.3 mg, 0.09 mmol) and trifluoroacetic acid (203.0 mg, 1.78 mmol, 0.14 mL) in DCM (1.0 mL). The crude reaction was worked-up (method B) and heated with 4-amino-6-chloropyrimidine-5-carbonitrile 44e (27.5 mg, 0.18 mmol) and DIPEA (46.0 mg, 0.36 mmol, 62.0 μL) in n-butanol (0.3 mL) at 130 1/2C for 1 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((6-amino-5-cyanopyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 47e (25.0 mg, 0.045 mmol) in 50% yield. LC–MS (method 1): tR = 3.39 min, m/z (M + H)+ = 561.3. The general procedure GP4 (method B) was used with compound 47e (72.0 mg, 0.13 mmol) to afford the final product, (S)-4-(((2-(1-((6-amino-5-cyanopyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48e (34.0 mg, 0.05 mmol) in 39% yield. LC–MS (method 2): tR = 4.36 min, m/z (M + H)+ = 562.2. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.96 (s, 1H), 7.93 (s, 1H), 7.73–7.66 (m, 2H), 7.60–7.45 (m, 6H), 7.42–7.32 (m, 3H), 6.78–6.70 (m, 1H), 6.49 (d, J = 8.4 Hz, 1H), 4.65 (td, J = 7.9, 4.8 Hz, 1H), 4.49 (s, 2H), 2.54 (s, 1H), 1.86 (tq, J = 12.2, 7.4, 6.2 Hz, 1H), 1.80–1.72 (m, 1H), 0.70 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C30H28N9O3 562.2310, found 562.2330. [Note: In addition to compounds 48e, a side product originating from the hydrolysis of methyl ester in 47e to carboxylic acid was also isolated in 2% yield].
(S)-4-(((2-(1-((6-Amino-5-chloropyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide (48f). The general procedure GP6 was used with compound 26d (61.8 mg, 0.114 mmol) and trifluoroacetic acid (260.0 mg, 2.28 mmol, 0.17 mL) in DCM (1.2 mL). The crude reaction was worked-up (method B) and heated with 5,6-dichloropyrimidin-4-amine 44f (37.0 mg, 0.23 mmol) and DIPEA (59.0 mg, 0.46 mmol, 79.0 μL) in n-butanol (0.3 mL) at 130 1/2C for 5 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((6-amino-5-chloropyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-benzoate 47f (55.0 mg, 0.096 mmol) in 85% yield. LC–MS (method 1): tR = 3.25 min, m/z (M + H)+ = 570.2. The general procedure GP4 (method B) was used with compound 47f (55.0 mg, 0.096 mmol) to afford the final product, (S)-4-(((2-(1-((6-amino-5-chloropyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-amino)methyl)-N-hydroxybenzamide, TFA 48f (53.0 mg, 0.077 mmol) in 80% yield. LC–MS (method 2): tR = 4.50 min, m/z (M + H)+ = 571.1. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.95 (s, 1H), 8.03 (s, 1H), 7.73–7.66 (m, 2H), 7.60–7.51 (m, 2H), 7.51–7.44 (m, 5H), 7.40 (d, J = 8.1 Hz, 3H), 6.75 (d, J = 7.8 Hz, 1H), 6.49 (d, J = 8.4 Hz, 1H), 4.63 (td, J = 8.1, 4.2 Hz, 1H), 4.49 (s, 2H), 1.93–1.75 (m, 2H), 0.70 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C29H28ClN8O3 571.1967, found 571.1972 [Note: In addition to compound 48f, a side product originating from the hydrolysis of methyl ester in 47f to carboxylic acid was also isolated in 9% yield].
(S)-6-(((2-(1-((6-Amino-5-chloropyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxynicotinamide (48g). The general procedure GP6 was used with compound 26g (66.0 mg, 0.12 mmol) and trifluoroacetic acid (277.0 mg, 2.45 mmol, 0.19 mL) in DCM (1.2 mL). The crude reaction was worked-up (method B) and heated with 5,6-dichloropyrimidin-4-amine 44f (40.0 mg, 0.24 mmol) and DIPEA (63.0 mg, 0.48 mmol, 83.0 μL) in n-butanol (0.3 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-6-(((2-(1-((6-amino-5-chloropyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)nicotinate 47g (30.0 mg, 0.053 mmol) in 43% yield. LC–MS (method 1): tR = 3.06 min, m/z (M + H)+ = 571.2. The general procedure GP4 (method B) was used with compound 47g (30.0 mg, 0.053 mmol) to afford the final product, (S)-6-(((2-(1-((6-amino-5-chloropyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxynicotinamide, TFA 48g (13.0 mg, 0.019 mmol) in 36% yield. LC–MS (method 2): tR = 3.89 min, m/z (M + H)+ = 572.3. 1H NMR (400 MHz, DMSO-d6) δ 11.31 (s, 1H), 9.21 (s, 1H), 8.84 (d, J = 2.1 Hz, 1H), 8.05 (dd, J = 8.1, 2.2 Hz, 1H), 7.89 (s, 1H), 7.60–7.42 (m, 6H), 6.93 (d, J = 22.9 Hz, 2H), 6.76 (d, J = 7.6 Hz, 1H), 6.50 (d, J = 8.5 Hz, 1H), 5.75 (s, 1H), 4.64–4.55 (m, 2H), 3.10 (qd, J = 7.3, 4.8 Hz, 1H), 1.90–1.72 (m, 2H), 0.70 (t, J = 7.2 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C28H27ClN9O3 572.1920, found 572.1936.
(S)-4-(((2-(1-((2-Amino-5-chloro-6-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)-N-hydroxybenzamide (48f). The general procedure GP6 was used with compound 26d (72.0 mg, 0.133 mmol) and trifluoroacetic acid (303.0 mg, 2.65 mmol, 0.20 mL) in DCM (1.3 mL). The crude reaction was worked-up (method B) and heated with 4,5-dichloro-6-methylpyrimidin-2-amine 44g (36.0 mg, 0.20 mmol) and DIPEA (52.0 mg, 0.40 mmol, 70.0 μL) in n-butanol (0.3 mL) at 130 1/2C for 5 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford the product methyl (S)-4-(((2-(1-((2-amino-5-chloro-6-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 47h (68.0 mg, 0.116 mmol) in 88% yield. LC–MS (method 1): tR = 3.15 min, m/z (M + H)+ = 584.2. The general procedure GP4 (method B) was used with compound 47h (68.0 mg, 0.116 mmol) to afford the final product, (S)-4-(((2-(1-((2-amino-5-chloro-6-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48h (62.0 mg, 0.089 mmol) in 76% yield. LC–MS (method 2): tR = 4.19 min, m/z (M + H)+ = 585.2. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.97 (t, J = 5.8 Hz, 1H), 8.39 (d, J = 7.4 Hz, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.61–7.46 (m, 4H), 7.46–7.36 (m, 5H), 6.78 (d, J = 7.8 Hz, 1H), 6.53 (d, J = 8.4 Hz, 1H), 4.78 (td, J = 7.7, 4.8 Hz, 1H), 4.51 (d, J = 4.8 Hz, 2H), 2.33 (s, 3H), 2.03–1.91 (m, 1H), 1.83 (dq, J = 14.4, 7.4 Hz, 1H), 0.72 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C30H30ClN8O3 585.2124, found 585.2151 [Note: In addition to compound 48h, a side product originating from the hydrolysis of methyl ester in 47h to carboxylic acid, was also isolated in 4% yield].
(S)-4-(((2-(1-((6-Amino-5-cyano-2-methylpyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)-N-hydroxybenzamide (48i). The general procedure GP6 was used with compound 26d (86.0 mg, 0.158 mmol) and trifluoroacetic acid (361.0 mg, 3.17 mmol, 0.24 mL) in DCM (1.6 mL). The crude reaction was worked-up (method B) and heated with 4-amino-6-chloro-2-methylpyrimidine-5-carbonitrile 44h (40.0 mg, 0.24 mmol) and DIPEA (61.0 mg, 0.47 mmol, 84.0 μL) in n-butanol (0.3 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((6-amino-5-cyano-2-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 47i (75.0 mg, 0.131 mmol) in 83% yield. LC–MS (method 1): tR = 3.33 min, m/z (M + H)+ = 575.3. The general procedure GP4 (method B) was used with compound 47i (73.0 mg, 0.13 mmol) to afford the final product, (S)-4-(((2-(1-((6-amino-5-cyano-2-methylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48i (56.0 mg, 0.08 mmol) in 64% yield. LC–MS (method 2): tR = 4.17 min, m/z (M + H)+ = 576.3. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.96 (s, 1H), 7.69 (d, J = 8.1 Hz, 2H), 7.59–7.42 (m, 8H), 7.40 (d, J = 8.0 Hz, 2H), 6.76 (d, J = 7.9 Hz, 1H), 6.49 (d, J = 8.4 Hz, 1H), 4.78 (td, J = 8.3, 4.3 Hz, 1H), 4.50 (s, 2H), 2.17 (s, 3H), 1.92–1.75 (m, 2H), 0.68 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C31H30CN9O3 576.2466, found 576.2470 [Note: In addition to compound 48i, a side product originating from the hydrolysis of methyl ester in 47i to carboxylic acid, was also isolated in 3% yield].
(S)-4-(((2-(1-((2,6-Diamino-5-chloropyrimidin-4-yl)amino)-propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)-N-hydroxybenzamide (48j). The general procedure GP6 was used with compound 26d (64.0 mg, 0.12 mmol) and trifluoroacetic acid (269.0 mg, 2.36 mmol, 0.18 mL) in DCM (1.2 mL). The crude reaction was worked-up (method B) and heated with 5,6-dichloropyrimidine-2,4-diamine 44i (32.0 mg, 0.18 mmol) and DIPEA (46.0 mg, 0.35 mmol, 63.0 μL) in n-butanol (0.5 mL) at 130 1/2C for 3 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((2,6-diamino-5-chloropyrimidin-4-yl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)benzoate 47j (32.0 mg, 0.06 mmol) in 46% yield. LC–MS (method 1): tR = 3.78 min, m/z (M + H)+ = 585.2. The general procedure GP4 (method B) was used with compound 47j (31.0 mg, 0.05 mmol) to afford the final product, (S)-4-(((2-(1-((2,6-diamino-5-chloropyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48j (15.0 mg, 0.02 mmol) in 40% yield. LC–MS (method 2): tR = 4.29 min, m/z (M + H)+ = 586.3. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.96 (d, J = 6.4 Hz, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.61–7.46 (m, 7H), 7.46–7.36 (m, 3H), 7.27 (s, 1H), 6.77 (d, J = 7.9 Hz, 1H), 6.51 (d, J = 8.4 Hz, 1H), 4.69 (td, J = 7.7, 4.4 Hz, 1H), 4.50 (d, J = 4.7 Hz, 2H), 1.88 (ddd, J = 12.9, 7.4, 4.9 Hz, 1H), 1.76 (p, J = 7.4 Hz, 1H), 0.68 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C29H29CN9O3 586.2076, found 586.2087.
(S)-4-(((2-(1-((2-Amino-5-cyano-6-(trifluoromethyl)pyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-amino)methyl)-N-hydroxybenzamide (48k). The general procedure GP6 was used with compound 26d (85.0 mg, 0.16 mmol) and trifluoroacetic acid (361.0 mg, 3.17 mmol, 0.24 mL) in DCM (1.6 mL). The crude reaction was worked-up (method B) and heated with 2-amino-4-chloro-6-(trifluoromethyl)pyrimidine-5-carbonitrile 44j (52.0 mg, 0.24 mmol) and DIPEA (61.0 mg, 0.47 mmol, 82.0 μL) in n-butanol (0.5 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((2-amino-5-cyano-6-(trifluoromethyl)pyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 47k (94.0 mg, 0.15 mmol) in 95% yield. LC–MS (method 1): tR = 3.71 min, m/z (M + H)+ = 629.3. The general procedure GP4 (method B) was used with compound 47k (94.0 mg, 0.15 mmol) to afford the final product, (S)-4-(((2-(1-((2-amino-5-cyano-6-(trifluoromethyl)pyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48k (45.0 mg, 0.06 mmol) in 41% yield. LC–MS (method 2): tR = 5.39 min, m/z (M + H)+ = 630.3. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.97 (s, 1H), 7.83 (d, J = 7.3 Hz, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.55–7.45 (m, 3H), 7.40 (d, J = 7.4 Hz, 3H), 7.29 (s, 1H), 6.76 (d, J = 7.9 Hz, 1H), 6.51 (d, J = 8.4 Hz, 1H), 4.80 (td, J = 7.3, 4.8 Hz, 1H), 4.50 (s, 2H), 1.92 (dq, J = 13.3, 7.0 Hz, 1H), 1.76 (hept, J = 7.4 Hz, 1H), 0.72 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C31H27F3N9O3 630.2183, found 630.2205.
(S)-4-(((2-(1-((2-Amino-5-cyano-6-cyclopropylpyrimidin-4-yl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)-N-hydroxybenzamide (48l). The general procedure GP6 was used with compound 26d (92.0 mg, 0.17 mmol) and trifluoroacetic acid (387.0 mg, 3.4 mmol, 0.26 mL) in DCM (1.7 mL). The crude reaction was worked-up (method B) and heated with 2-amino-4-chloro-6-cyclopropylpyrimidine-5-carbonitrile 44k (50.0 mg, 0.26 mmol) and DIPEA (66.0 mg, 0.51 mmol, 89.0 μL) in n-butanol (0.5 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((2-amino-5-cyano-6-cyclopropylpyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate 47l (40.0 mg, 0.07 mmol) in 39% yield. LC–MS (method 1): tR = 3.51 min, m/z (M + H)+ = 601.3. The general procedure GP4 (method B) was used with compound 47l (40.0 mg, 0.07 mmol) to afford the final product, (S)-4-(((2-(1-((2-amino-5-cyano-6-cyclopropylpyrimidin-4-yl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)-N-hydroxybenzamide, TFA 48l (28.0 mg, 0.04 mmol) in 59% yield. LC–MS (method 2): tR = 4.74 min, m/z (M + H)+ = 602.3. 1H NMR (400 MHz, DMSO-d6) δ 11.18–11.06 (m, 1H), 8.96 (s, 1H), 8.01–7.90 (m, 1H), 7.72–7.66 (m, 2H), 7.58–7.44 (m, 6H), 7.40 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 7.1 Hz, 1H), 6.75 (d, J = 7.9 Hz, 2H), 6.50 (d, J = 8.4 Hz, 1H), 4.66 (td, J = 7.7, 4.2 Hz, 1H), 4.50 (s, 2H), 2.05 (p, J = 6.3 Hz, 1H), 1.84 (ddd, J = 14.1, 7.3, 4.6 Hz, 1H), 1.72 (dt, J = 14.4, 7.2 Hz, 1H), 1.00 (d, J = 6.0 Hz, 4H), 0.66 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C33H32N9O3 602.2623, found 602.2617.
(S)-4-(((2-(1-((2-Amino-5-cyano-6-(difluoromethyl)pyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-amino)methyl)-N-hydroxybenzamide (48m). The general procedure GP6 was used with compound 26d (50.5 mg, 0.114 mmol) and trifluoroacetic acid (260.0 mg, 2.28 mmol, 0.17 mL) in DCM (1.1 mL). The crude reaction was worked-up (method B) and heated with 5-bromo-4-chloro-6-(difluoromethyl)pyrimidin-2-amine 44l (44.0 mg, 0.17 mmol) and DIPEA (44.0 mg, 0.34 mmol, 60.0 μL) in nbutanol (0.5 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((2-amino-5-bromo-6-(difluoromethyl)pyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate (65.0 mg, 0.098 mmol) in 86% yield. LC–MS (method 1): tR = 3.70 min, m/z (M + H)+ = 664.2. Methyl (S)-4-(((2-(1-((2-amino-5-bromo-6-(difluoromethyl)pyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)benzoate (65.0 mg, 0.098 mmol) and zinc cyanide (14.0 mg, 0.12 mmol) were suepended in DMF (1.0 mL) under nitrogen atmosphere in a MW vial, and tris(dibenzylideneacetone)dipalladium(0) (9.0 mg, 9.8 μmol) and 1,1′-Bis(diphenylphosphino)ferrocene [dppf] (10.9 mg, 0.02 mmol) were added to it. The resulting mixture was bubbled with nitrogen for 5 min, followed by sealing the MW vial and heating at 1/2C in microwave for 2 h. After completion of reaction, the crude reaction mixture was concentrated in vacuo and purified by silica gel chromatography using 0–60% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((2-amino-5-cyano-6-(difluoromethyl)pyrimidin-4-yl)-amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)benzoate 47m (50.0 mg, 0.08 mmol) in 84% yield. LC–MS (method 1): tR = 3.42 min, m/z (M + H)+ = 611.2. The general procedure GP4 (method B) was used with compound 47m (50.0 mg, 0.08 mmol) to afford the final product, (S)-4-(((2-(1-((2-amino-5-cyano-6-(difluoromethyl)pyrimidin-4-yl)amino)propyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48m (18.0 mg, 0.0258 mmol) in 30% yield. LC–MS (method 2): tR = 4.82 min, m/z (M + H)+ 612.2. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.97 (s, 1H), 7.69 (d, J = 7.9 Hz, 2H), 7.64 (d, J = 7.3 Hz, 1H), 7.55–7.38 (m, 6H), 7.08 (s, 1H), 6.81–6.73 (m, 1H), 6.66 (s, 1H), 6.54–6.48 (m, 1H), 4.75 (q, J = 6.7 Hz, 1H), 4.50 (s, 2H), 1.94–1.84 (m, 1H), 1.75 (dt, J = 14.1, 7.2 Hz, 1H), 0.70 (t, J = 7.3 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C31H28F2N9O3 612.2278, found 612.2262 [Note: In addition to compound 48m, a side product originating from the hydrolysis of methyl ester in 47m to carboxylic acid was also isolated in 4% yield].
(S)-4-(((2-(1-((2,6-Diamino-5-cyanopyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide (48n). The general procedure GP6 was used with compound 26i (103.0 mg, 0.195 mmol) and trifluoroacetic acid (444.0 mg, 3.9 mmol, 0.29 mL) in DCM (2.0 mL). The crude reaction was worked-up (method B) and heated with 2,4-diamino-6-chloropyrimidine-5-carbonitrile 44a (50.0 mg, 0.29 mmol) and DIPEA (76.0 mg, 0.59 mmol, 102.0 μL) in n-butanol (0.5 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)-methyl)benzoate 47n (88.0 mg, 0.16 mmol) in 80% yield. LC–MS (method 1): tR = 3.11 min, m/z (M + H)+ = 562.2. The general procedure GP4 (method B) was used with compound 47n (88.0 mg, 0.16 mmol) to afford the final product, (S)-4-(((2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48n (38.0 mg, 0.06 mmol) in 64% yield. LC–MS (method 2): tR = 3.90 min, m/z (M + H)+ = 563.2. 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 8.97 (s, 1H), 8.09 (d, J = 6.5 Hz, 1H), 7.96 (s, 2H), 7.73–7.68 (m, 2H), 7.54–7.48 (m, 3H), 7.46–7.38 (m, 5H), 6.78 (dd, J = 7.8, 0.9 Hz, 1H), 6.52 (dd, J = 8.6, 1.1 Hz, 1H), 5.75 (s, 2H), 4.89 (p, J = 6.6 Hz, 1H), 4.51 (s, 2H), 1.34 (d, J = 6.6 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C29H27N10O3 563.2262, found 563.2257 [Note: In addition to compound 48n, a side product originating from the hydrolysis of methyl ester in 47n to carboxylic acid was also isolated in 5% yield].
(S)-4-(((2-(1-((2-Amino-5-cyano-6-methylpyrimidin-4-yl)amino)-ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide (48o). The general procedure GP6 was used with compound 26i (134.0 mg, 0.25 mmol) and trifluoroacetic acid (579.0 mg, 5.1 mmol, 0.39 mL) in DCM (2.5 mL). The crude reaction was worked-up (method B) and heated with 2-amino-4-chloro-6-methylpyrimidine-5-carbonitrile 44b (64.2 mg, 0.38 mmol) and DIPEA (98.0 mg, 0.76 mmol, 133.0 μL) in n-butanol (0.5 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–100% EtOAc/hexanes to afford methyl (S)-4-(((2-(1-((2-amino-5-cyano-6-methylpyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-amino)methyl)benzoate 47o (90.0 mg, 0.16 mmol) in 63% yield. LC–MS (method 1): tR = 3.12 min, m/z (M + H)+ = 561.2. The general procedure GP4 (method B) was used with compound 47o (84.0 mg, 0.15 mmol) to afford the final product, (S)-4-(((2-(1-((2-amino-5-cyano-6-methylpyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)amino)methyl)-N-hydroxybenzamide, TFA 48o (53.0 mg, 0.08 mmol) in 52% yield. LC–MS (method 2): tR = 3.99 min, m/z (M + H)+ = 562.1. 1H NMR (400 MHz, DMSO-d6) δ 11.14 (s, 1H), 8.98 (s, 1H), 8.37 (s, 1H), 7.70 (d, J = 7.9 Hz, 2H), 7.54–7.48 (m, 3H), 7.45–7.35 (m, 5H), 6.79 (d, J = 7.9 Hz, 1H), 6.52 (d, J = 8.4 Hz, 1H), 4.91 (p, J = 5.8, 4.9 Hz, 1H), 4.51 (s, 2H), 2.35 (s, 3H), 1.36 (d, J = 6.5 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C29H28N9O3 562.2310, found 562.2301 [Note: In addition to compound 480, a side product originating from the hydrolysis of methyl ester in 47o to carboxylic acid was also isolated in 3% yield].
Procedure for Synthesis of C5-Substituted Quinazolinones (51 and 54, Scheme 6).
(S)-N1-((2-(1-((2,6-Diamino-5-cyanopyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-methyl)-N4-hydroxyterephthalamide (51). Compound 29b (36.0 mg, 0.09 mmol) was dissolved in ammonia (1.0 mL, 7N in MeOH) in a 20 mL scintillation vial and Raney Ni (10.0 mg (approx.)) was added to it. The reaction vial was evacuated and then kept under hydrogen atmosphere using a balloon. The resulting suspension was stirred at room temperature for 20 h. After completion of reaction (by LC-MS), the crude reaction mixture was carefully filtered under nitrogen and concentrated in vacuo. This hydrogenated product was dissolved in DMF (1.0 mL) in a 20 mL scintillation vial, and 4-(methoxycarbonyl)benzoic acid 35 (18.2 mg, 0.10 mmol), DIPEA (24.0 mg, 0.18 mmol, 32.0 μL), and HATU (38.0 mg, 0.10 mmol) were added to it. The resulting mixture was stirred at room temperature for 16 h and concentrated in vacuo. The remaining residue was purified using 0–10% MeOH/DCM to afford methyl (S)-4-(((2-(1-((tert-butoxycarbonyl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)carbamoyl)benzoate 49 (49.0 mg, 0.088 mmol) in 96% yield. LC–MS (method 1): tR = 3.45 min, m/z (M + H)+ = 557.1. The general procedure GP6 was used with compound 49 (49.0 mg, 0.09 mmol) and trifluoroacetic acid (201.0 mg, 1.76 mmol, 0.14 mL) in DCM (1.0 mL). The crude reaction was worked-up (method B) and heated with 2,4-diamino-6-chloropyrimidine-5-carbonitrile 44a (22.3 mg, 0.13 mmol) and DIPEA (34.1 mg, 0.26 mmol, 46.0 μL) in n-butanol (0.5 mL) at 130 1/2C for 5 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH/DCM to afford methyl (S)-4-(((2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)carbamoyl)benzoate 50 (40.0 mg, 0.07 mmol) in 77% yield. LC–MS (method 1): tR = 2.85 min, m/z (M + H)+ = 590.0. The general procedure GP4 (method B) was used with compound 50 (40.0 mg, 0.07 mmol) to afford the final product, (S)-N1-((2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)-amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)-N4-hydroxyterephthalamide, TFA 51 (11.0 mg, 0.016 mmol) in 23% yield. LC–MS (method 2): tR = 3.55 min, m/z (M + H)+ = 591.3. 1H NMR (400 MHz, DMSO-d6) δ 11.36 (s, 1H), 9.07 (t, J = 6.0 Hz, 1H), 8.01–7.94 (m, 2H), 7.89–7.76 (m, 4H), 7.64–7.53 (m, 4H), 7.53–7.40 (m, 5H), 5.03 (d, J = 5.9 Hz, 2H), 4.88 (t, J = 6.8 Hz, 1H), 1.35 (d, J = 6.7 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C30H27N10O4 591.2211, found 591.2210.
(S)-2-(((2-(1-((2,6-Diamino-5-cyanopyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)-N-hydroxypyrimidine-5-carboxamide (54). Compound 29b (125.0 mg, 0.32 mmol) was dissolved in ammonia (2.0 mL, 7N in MeOH) in a 20 mL scintillation vial, and Raney Ni (20.0 mg (approx) was added to it. The reaction vial was evacuated and then kept under hydrogen atmosphere using a balloon. The resulting suspension was stirred at room temperature for 20 h. After completion of reaction (by LC-MS), the crude reaction mixture was carefully filtered under nitrogen, concentrated in vacuo, and redissolved in n-BuOH (0.5 mL) in a MW vial. Ethyl 2-chloropyrimidine-5-carboxylate 41 (85.0 mg, 0.46 mmol) and DIPEA (118.0 mg, 0.91 mmol, 0.16 mL) were added to the MW vial, and the vial was sealed and heated at 130 1/2C for 1 h in a MW reactor. After completion of the reaction, the reaction mixture was concentrated in vacuo, and the remaining residue was purified using 0–100% EtOAc/hexanes to afford ethyl (S)-2-(((2-(1-((tert-butoxycarbonyl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)pyrimidine-5-carboxylate 52 (138.0 mg, 0.25 mmol) in 83% yield. LC–MS (method 1): tR = 3.75 min, m/z (M + H)+ = 545.3. The general procedure GP6 was used with compound 52 (138.0 mg, 0.25 mmol) and trifluoroacetic acid (579.0 mg, 5.1 mmol, 0.39 mL) in DCM (2.5 mL). The crude reaction was worked-up (method B) and heated with 2,4-diamino-6-chloropyrimidine-5-carbonitrile 44a (64.0 mg, 0.38 mmol) and DIPEA (98.0 mg, 0.76 mmol, 133.0 μL) in n-butanol (0.5 mL) at 130 1/2C for 2 h in a MW reactor. The remaining residue was purified by flash chromatography on silica gel using 0–5% MeOH/DCM to afford ethyl (S)-2-(((2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)methyl)amino)pyrimidine-5-carboxylate 53 (116.0 mg, 0.20 mmol) in 79% yield. LC–MS (method 1): tR = 3.11 min, m/z (M + H)+ = 578.1. The general procedure GP4 (method B) was used with compound 53 (116.0 mg, 0.20 mmol) to afford the final product, (S)-2-(((2-(1-((2,6-diamino-5-cyanopyrimidin-4-yl)amino)ethyl)-4-oxo-3-phenyl-3,4-dihydroquinazolin-5-yl)-methyl)amino)-N-hydroxypyrimidine-5-carboxamide, TFA 54 (68.0 mg, 0.10 mmol) in 50% yield. LC–MS (method 2): tR = 3.38 min, m/z (M + H)+ = 565.0. 1H NMR (400 MHz, DMSO-d6) δ 11.36 (s, 1H), 9.07 (t, J = 6.0 Hz, 1H), 8.01–7.94 (m, 2H), 7.89–7.76 (m, 4H), 7.64–7.53 (m, 4H), 7.53–7.40 (m, 5H), 5.03 (d, J = 5.9 Hz, 2H), 4.88 (t, J = 6.8 Hz, 1H), 2.54 (s, 2H), 1.35 (d, J = 6.7 Hz, 3H). HRMS (ESI) m/z: [M + H]+ calcd for C27H25N12O3 565.2167, found 565.2164.
Molecular Modeling.
All structure visualizations and computations were performed using molecular operating environment (MOE).69 The images of Idelalisib bound to PI3Kδ (Figure 3B) and SAHA (Vorinostat) bound to HDAC-2 (Figure 3D) are based on the protein data bank entries 4LXZ.pdb39 and 5XE0.pdb40 respectively. The structure of HDAC-6 bound to 19b (Figure 4) was derived from 5EDU.pdb (Trichostatin A bound to HDAC-6).57 In this derivation, 5EDU.pdb was loaded into MOE, protein prep was run, and hydrogens were added. The structure was then minimized, using the MMFF94 potential,70 to a local minimum associated with the crystal structure coordinates. Then Trichostatin A was truncated all the way to the hydroxamic acid headgroup. 19b was grown out from the hydroxamic acid headgroup one atom at a time. If the placement of an atom resulted in a new torsion angle, the torsion was searched to find a local minimum before the next atom was added. Once 19b was completely grown into the HDAC-6 binding pocket, the whole structure was minimized to 0.001 RMS.
HDAC-6 Selectivity Carboxamide Compounds.
The sequences of HDACs-1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and 11 were downloaded from UNIPROT71 and aligned. Using the structure that we modeled of 19b bound to HDAC-6 we determined all HDAC-6 residues within 4.5 Å of 19b. These residue positions were then marked on the HDAC-6 sequence, and their counterpart residues on the other aligned HDACs were determined. All marked HDAC-6 residue positions that were not conserved across the other kinases were identified. Then one at a time each nonconserved residue position in HDAC-6 was mutated to its value in each of the other HDACs, and the effect of the mutation on 19b interactions with the protein was determined.
Biological Assay Protocols.
PI3K Assay Protocol (HTRF Assay Platform). (1) Assay Principle. The PIP3 product is detected by displacement of biotin-PIP3 from an energy transfer complex consisting of europium-labeled anti-GST monoclonal antibody, a GST-tagged pleckstrin homology (PH) domain, biotinylated PIP3 and streptavidin–allophycocyanin (APC). Excitation of europium in the complex results in an energy transfer to the APC and a fluorescent emission at 665 nm. The PIP3 product formed by PI3-kinase(h) activity displaces biotin-PIP3 from the complex, resulting in a loss of energy transfer, and thus, a decrease in signal. This is a 3-step reaction: First, the kinase reaction with PIP2 substrate is carried out in the presence of ATP, and the reaction is quenched with stop solution, and then, finally detect by adding detection mixture followed by incubation. (2) Reaction Conditions. Assay Buffer: HEPES 50 mM (pH7.0), NaN3 0.02%, BSA 0.01%, orthovanadate 0.1 mM, 1% DMSO. Detection Buffer: HEPES 10 mM (pH7.0), BSA 0.02%, KF 0.16 M, EDTA 4 mM. Substrate: 10 μM PIP2 substrate (PI(4,5)P2). ATP: 10 μM ATP under standard conditions. Control Inhibitor: PI-103. (3) Assay Procedure. (A) Prepare substrate in freshly prepared reaction buffer. (B) Deliver kinase into the substrate solution and gently mix. (C) Deliver compounds in 100% DMSO into the kinase reaction mixture by Acoustic Technology (Echo550; nanoliter range), incubate for 10 min at room temperature. (D) Deliver ATP into the reaction mixture to initiate the reaction. (E) Incubate for 30 min at 30 1/2C. (F) Quench the reaction with stop solution. (G) Add detection mixture and incubate for overnight. H) Measure HTRF: Ex = 320 nm, ratio of Em = 615 nm and Em = 665 nm. (4) Data Analysis. The emission ratio is converted into μM PIP3 production based on PIP3 standard curves. The nonlinear regression to obtain the standard curve and IC50 values are performed using Graphpad Prism software.
HDAC Fluorescent Activity Assay.
Assay Description: The HDAC fluorescent activity assay is based on the unique fluorogenic substrate and developer combination. This assay is a highly sensitive and validated. The assay procedure has two steps (Figure 1, Howitz, 2015 Drug Discovery Today: Technologies). First, the fluorogenic substrate, which comprises an acetylated lysine side chain, is incubated with a purified HDAC enzyme. Deacetylation of the substrate sensitizes the substrate so that, in the second step, treatment with the Developer produces a fluorophore. Compound Handling: Testing compounds were dissolved in 100% DMSO to specific concentration. The serial dilution was conducted by epMotion 5070 in DMSO. Materials and Reagents: HDAC reaction buffer: 50 mM Tris-HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, and 1 mM MgCl2, add fresh 1 mg/mL BSA, 1% DMSO. Substrate: HDAC1,2,3,6,10: Fluorogenic peptide from p53 residues 379–382 (RHKK(Ac)AMC). HDAC4,5,7, 9 and 11: Fluorogenic HDAC class2a substrate (trifluoroacetyl lysine). HDAC 8: fluorogenic peptide from p53 residues 379–382 (RHK(Ac)K(Ac)AMC). General Reaction Procedure: standard IC50 determination. Deacetylation Step: (A) Deliver 2X enzyme in wells of reaction plate except no enzyme control wells. Add buffer in No En wells. (B) Deliver compounds in 100% DMSO into the enzyme mixture by Acoustic technology (Echo550; nanoliter range). Spin down and preincubation. (C) Deliver 2× substrate mixture (fluorogenic HDAC substrate and co-factor if applicable) in all reaction wells to initiate the reaction. Spin and shake. (D) Incubate for 1–2 h at 30 1/2C with seal. Development Step: (E) Add developer with Trichostatin A (or TMP269) to stop the reaction and to generate fluorescent color. (F) Fluorescence was read (excitatory, 360; emission, 460) using the EnVision Multilabel Plate Reader (PerkinElmer). (G) Take end point reading for analysis after the development reaches plateau. Data Analysis: The percentages of enzyme activity (relative to DMSO controls) and IC50 values were calculated using the GraphPad Prism 4 program based on a sigmoidal dose–response equation.
NCI-60 Cellular Assays.
Assay Protocols and Data Analysis procedures are available at: https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm
CETSA Protocol. K-562 (chronic myelogenous leukemia), MV-4–11 (B-myelomonocytic leukemia) and THP-1 (acute monocytic leukemia) were cultured in RPMI 1640 (ThermoFisher, cat. no, 11835) supplemented with 10% FBS (Hyclone) and 0.5× penicillin/streptomycin (ThermoFisher). Then 2 ×106 cells were rinsed with PBS and resuspended in 200 μL of DPBS (+Ca, +Mg) + 1 g/L D-glucose + 1× Halt protease inhibitor cocktail (ThermoFisher). Four freeze–thaw cycles (dry ice/EtOH, 37 1/2C water bath) were performed, and lysates were centrifuged at 17 000g for 15 min (4 1/2C). The soluble fraction was separated on a 4–12% Bis-Tris gel using MOPS running buffer and transferred to a nitrocellulose membrane. Primary antibodies were anti-HDAC6 (ab133493, 1:10 000), anti-HDAC6 (ab82557, 1:1000), and anti-PI3kD (ab109006, 1:1000). Secondary antibodies were HRP conjugated, and SuperSignal West Pico chemiluminescent substrate was used for detection. Membranes were imaged on a ChemiDoc imaging system (BioRad). CETSA melt curves were determined by collecting MV-4–11 cells in DPBS (+Ca, +Mg) supplemented 1 g/L D-glucose, 1× protease inhibitors, and 0.5% DMSO at 15 × 106 cells/mL. Cell suspensions were distributed to 0.2 mL PCR tubes, and samples were heated for 3.5 min to various temperatures. After allowing samples to equilibrate to room temperature, samples were processed by freeze– thaw for immunoblot as outlined above. Isothermal dose–response CETSA was performed by collecting MV-4–11 cells in DPBS (+Ca, +Mg) supplemented 1 g/L D-glucose, 0.5× protease inhibitors, 10 × 106 cells/mL. Then 1 μL of compound (at 100× final concentration; 1% DMSO final) was added to 99 μL of cell suspension and cells were incubated at 37 1/2C for 1 h. Samples were then heated for 3.5 min at either 52 1/2C (PI3Kδ) or 58 1/2C (HDAC6). Cells were lysed by freeze–thaw cycles and processed for immunoblot as outlined above. Densitometry analysis was performed using ImageQuant TL and a rolling ball radius 80 for background subtraction.
Cell Culture and Reagents.
Human MOLM-14 AML, Mv4–11, HL-60, THP-1, MOLM-13 wt, MOLM-13 FLT3 inhibitor resistant, and U-937 cells were maintained in RPMI1640 medium containing 10% heat-inactivated fetal bovine serum (FBS), 100 units/mL penecillin, 100 ug/mL streptomycin, and 2 mM L-glutamine. MOLM-13 cells were selected for resistance to FLT3 inhibitor HG-7-85-01 as described (Weisberg, 2011). Cells were treated with different dual inhibitor compounds (NCATS, NIH).
Cell Viability Assays. RPMI 8226 and U266B1 cells obtained from ATCC and were cultured in RPMI1640 (Gibco, cat. no. 11875093) supplemented with 10% FBS (HyClone), 50 U/mL penicillin, and 50 μg/mL streptomycin (Life Technologies). For cell viability assays, RPMI 8226 and U226B1 cells were seeded at 5 × 104 cells/mL (50 μL volume) into white, tissue culture-treated 384-well plates (Corning, cat. no. 3570BC). T63 nL of compound solutions in DMSO were immediately added using a Wako Pin-tool. Plates were covered with BreatheEasy seals (Diversified Biotech) and incubated at 37 1/2C, 5% CO2 for 5 days. Then 25 μL of CellTiter-Glo (Promega) was then added to each well, and plates were incubated at RT for 10 min. Luminescence was measured on a ViewLux plate reader.
Human MOLM-14, MOLM-13 wt, MOLM-13 FLT3 inhibitor resistant, MV4–11, HL-60, THP-, and U-937 cell lines were treated with different concentrations of dual inhibitors for the indicated times. Cell viability was determined by trypan blue exclusion. For studies with primary AML blasts, cells were obtained pretherapy from AML patients under an approved protocol (DFCI 01–206). Normal PBMCs were obtained from normal donors under consent. Cells were left untreated or treated with test compound for the indicated times. Cell death was determined by staining with propidium iodide (PI) alone or PI/Annexin V and then analyzed by flow cytometry.
Necrosis Assays.
For analysis of cell death by necrosis, cells were stained with propidium iodide (PI) alone or PI/Annexin V, and then analyzed by flow cytometry as described.67
Immunoblot Analysis.
Cells were harvested and rinsed with ice-cold phosphate buffered saline (PBS). Ice-cold lysis buffer [0.5 mL; 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mg/mL leupeptin, 1 mM PMSF] was added to 1 × 107 cells and sonicated on ice four times for 5 s each followed by microcentrifugation for 10 min at 4 1/2C. Soluble proteins were separated on gels and transferred to nitrocellulose filters. The lysates were immunoblotted with antiphospho AKT, total AK,T and b-actin antibodies (Cell Signaling Technology), Antigen–antibody complexes were visualized by enhanced chemiluminescence (GE Healthcare).
Pharmacokinetic Analyses.
Parallel Artificial Membrane Permeability Assay (PAMPA).
Stirring double-sink PAMPA method (patented by pION Inc.) was employed to determine the permeability of compounds via PAMPA. The PAMPA lipid membrane consisted of an artificial membrane of a proprietary lipid mixture and dodecane (Pion Inc.), optimized to predict gastrointestinal tract (GIT) passive permeability. The lipid was immobilized on a plastic matrix of a 96-well “donor” filter plate placed above a 96-well “acceptor” plate. pH 7.4 solution was used in both donor and acceptor wells. The test articles, stocked in 10 mM DMSO solutions, were diluted to 0.05 mM in aqueous buffer (pH 7.4), and the concentration of DMSO was 0.5% in the final solution. During the 30 min permeation period at room temperature, the test samples in the donor compartment were stirred using the Gutbox technology (Pion Inc.) to reduce the aqueous boundary layer. The test article concentrations in the donor and acceptor compartments were measured using a UV plate reader (Nano Quant, Infinite 200 PRO, Tecan Inc., Männedorf, Switzerland). Permeability calculations were performed using Pion Inc. software and were expressed in units of 10−6cm/s. Compounds with low or weak UV signal we analyzed using high resolution LC/MS (Thermo QExactive). The three controls used were ranitidine (low permeability), dexamethasone (moderate permeability), and verapamil (high permeability). Method has been previously published.72
Multispecies Multitime Point Assays.
Microsome Stability Assay: Briefly, each reaction mixture (110 μL) consisted of a test article (1 μM), human, SD rat, or CD-1 mouse microsomal fractions (0.5 mg/mL), and NADPH regenerating system (1 μM) in phosphate buffer at pH 7.4. Cytosol Stability Assay: Briefly, each reaction mixture (110 μL) consisted of a test article (1 μM) and either human, SD rat, or CD-1 mouse cytosol fractions (2 mg/mL) in phosphate buffer (100 mM) at pH 7.4. Samples were incubated in 384-well plates at 37 1/2C for 0, 5, 10, 15, 30, and 60 min. Sample analysis and half-life calculations were performed using a previously described method.73 The three controls used were Buspirone (short half-life), Loperamide (moderate half-life), and Antipyrine (long half-life).
Pharmacokinetics (PK) [NCATS].
Adult female Balb/c mice (n = 3/sampling time point) were obtained from Charles River Laboratory (Frederick, MD). All experimental procedures were approved by the Animal Care and Use Committee (ACUC) of the NIH Division of Veterinary Resources (DVR). Pharmacokinetics (PK) was evaluated after a single dose of 3 mg/kg intravenous (IV) and 10 mg/kg oral gavage (PO) administration. Dosing solutions were freshly prepared on the day of administration in 70% PEG-300/30% H2O/10% ethanol. The blood samples (~80 μL) were collected in K2EDTA tubes after drug administration based on the PK study protocols, and plasma (~30 μL) was harvested after centrifugation at 3000 rpm for 10 min. All plasma samples were stored at −80 1/2C until analysis. Ultraperformance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS) methods were developed to determine compounds’ concentrations in mouse plasma samples. Mass spectrometric analysis was performed on a Waters Xevo TQ-S triple quadrupole instrument using electrospray ionization in positive mode with the selected reaction monitoring. The separation of test compounds from endogenous components was performed on an Acquity BEH C18 column (50 mm × 2.1 mm, 1.7 μ) using a Waters Acquity UPLC system with 0.6 mL/min flow rate and gradient elution. The mobile phases were 0.1% formic acid in water and 0.1% formic acid in acetonitrile. The calibration standards and quality control samples were prepared in the blank mouse plasma. Aliquots of 10 μL plasma samples were mixed with 200 μL of internal standard in acetonitrile to precipitate proteins in a 96-well plate. Then 1 μL of supernatant was injected for the UPLC-MS/MS analysis. Data were analyzed using TargetLynx V4.1 (Waters Corp., Milford, MA). The pharmacokinetic parameters, e.g., plasma clearance (CLp) and volume of distribution at steady-state (Vdss), were calculated using the noncompartmental approach (models 200 and 201) of the pharmacokinetic software Phoenix WinNonlin, version 8.0 (Certara, St. Louis, MO). The area under the plasma concentration versus time curve (AUC) was calculated using the linear trapezoidal method. The slope of the apparent terminal phase was estimated by log linear regression using at least 3 data points, and the terminal rate constant (λ) was derived from the slope. AUC0–∞ was estimated as the sum of the AUC0–t (where “t” is the time of the last measurable concentration) and Ct/λ. The apparent terminal half-life (t1/2) was calculated as 0.693/λ.
Pharmacokinetics (PK) [Pharmaron].
Female Balb/c mice (sourced from Si Bei Fu Laboratory Animal Technology Co. Ltd.), approximately 6–8 weeks of age, and a weight of approximately 20–30 g, were dosed with 48c at 50 and 150 mg/kg via IP injection. The formulations, [DMSO/PEG300/sterile water (10:40:50)] and (DMSO/PEG300/”20% HP-β-CD in water (1:4)” (10:40:50), adjusted pH 3–4 with 1 M HCl) was prepared on the day of dosing. Each cohort had three mice, and plasma was collected at pre, 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, and 24 h post dose for IP. Approximately 0.025 mL of blood was collected by the dorsal metatarsal vein at each time point. All blood samples were transferred into plastic microcentrifuge tubes containing Heparin-Na as anticoagulant. Collection tubes with blood samples and anticoagulant were inverted several times for proper mixing of the tube contents and then placed on wet ice prior to centrifugation for plasma. Blood samples will be centrifuged at 4000g for 5 min at 4 1/2C to obtain plasma. Plasma samples will be stored in polypropylene tubes, quickly frozen in a freezer, and kept at −75 ± 15 1/2C until analyzed by LCMS/MS. The following pharmacokinetic parameters were measured: t1/2, Cmax, Tmax, and AUC0–t.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the funding from Intramural Research Program, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH). We thank Paul Shinn and his team for the assistance with compound management and Christopher Leclair and his team for analytical chemistry and purification support. We also thank Reaction Biology Corporation for screening compounds in enzymatic assays and Pharmaron Inc. for conducting the pharmacokinetic studies.
ABBREVIATIONS USED
- PI3K
phosphoinositide 3-kinase
- PIP2
phosphatidylinositol 4,5-diphosphate
- PIP3
phosphatidylinositol 3,4,5-triphosphate
- RTK
receptor tyrosine kinase
- GPCR
G protein-coupled receptor
- HDAC
histone deacetylase
- SAHA
suberanilohydroxamic acid
- Akt
protein kinase B
- mTOR
mammalian target of rapamycin
- AML
acute monocytic leukemia
- CNS
central nervous system
- SAR
structure– activity relationship
- DCM
dichloromethane
- DMF
N, N-dimethylformamide
- DCE
1,2-dichloroethane
- DMSO
dimethyl sulfoxide
- DABCO
(1,4-diazabicyclo[2.2.2]octane)
- THF
tetrahydrofuran
- TFA
trifluoroacetic acid
- DIPEA
N, N-diisopropylethylamine
- Boc
tert-butyloxycarbonyl
- rt
room temperature
- HATU
N-[(dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexa-fluorophosphate N-oxide
- PAMPA
parallel artificial membrane permeability assay
- CETSA
cellular thermal shift assay
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00193.
PI3K/HDAC enzyme inhibitory activities of selected 5-substituted quinazolinones; HPLC-MS chromatogram of compounds 19b, 46b, 48b, 48c, 48n, and 48o, respectively; figures showing compound 19b bound to HDAC1, 2 (Glu567, Asp568), HDAC3 (Asp567, Asp568), HDAC8 (Tyr567, Asp568), HDAC4, 5, 7, 9 (Asp567, Thr568), HDAC10 (Asp567, Ala568), and HDAC11 (Pro567, Asn568), respectively; kinome scan of compound 48c, and immunoblot analysis for PI3Kδ and HDAC6 expression (PDF)
Molecular strings for all final compounds (CSV)
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
The authors declare the following competing financial interest(s): Ashish Thakur, Gurmit Grewal, Gregory J. Tawa and Anton Simeonov are inventors of a patent application associated with this work.
Contributor Information
Ashish Thakur, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States;.
Gregory J. Tawa, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States
Mark J. Henderson, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States;.
Carina Danchik, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Suiyang Liu, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Pranav Shah, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Amy Q. Wang, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States
Garrett Dunn, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Md Kabir, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Elias C. Padilha, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States
Xin Xu, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Anton Simeonov, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States.
Surender Kharbanda, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Richard Stone, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, United States.
Gurmit Grewal, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, Maryland 20850, United States;.
REFERENCES
- (1).Kamb A; Wee S; Lengauer C Why Is Cancer Drug Discovery So Difficult? Nat. Rev. Drug Discovery 2007, 6, 115–120. [DOI] [PubMed] [Google Scholar]
- (2).Hanahan D; Weinberg RA Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- (3).Anighoro A; Bajorath J; Rastelli G Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem 2014, 57, 7874–7887. [DOI] [PubMed] [Google Scholar]
- (4).Shang E; Yuan Y; Chen X; Liu Y; Pei J; Lai L De Novo Design of Multitarget Ligands with an Iterative Fragment-Growing Strategy. J. Chem. Inf. Model 2014, 54, 1235–1241. [DOI] [PubMed] [Google Scholar]
- (5).Morphy R; Rankovic Z Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem 2005, 48, 6523–6543. [DOI] [PubMed] [Google Scholar]
- (6).O’Boyle NM; Meegan MJ Designed Multiple Ligands for Cancer Therapy. Curr. Med. Chem 2011, 18, 4722–4737. [DOI] [PubMed] [Google Scholar]
- (7).Costantino L; Barlocco D Designed Multiple Ligands: Basic Research vs Clinical Outcomes. Curr. Med. Chem 2012, 19, 3353–3387. [DOI] [PubMed] [Google Scholar]
- (8).Talevi A Multi-Target Pharmacology: Possibilities and Limitations of the “Skeleton Key Approach” from a Medicinal Chemist Perspective. Front. Pharmacol 2015, 6, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Fu RG; Sun Y; Sheng WB; Liao DF Designing Multi-Targeted Agents: An Emerging Anticancer Drug Discovery Paradigm. Eur. J. Med. Chem 2017, 136, 195–211. [DOI] [PubMed] [Google Scholar]
- (10).Anastasio TJ Editorial: Computational and Experimental Approaches in Multi-Target Pharmacology. Front. Pharmacol 2017, 8, 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Proschak E; Stark H; Merk D Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem 2019, 62, 420–444. [DOI] [PubMed] [Google Scholar]
- (12).Bolognesi ML Harnessing Polypharmacology with Medicinal Chemistry. ACS Med. Chem. Lett 2019, 10, 273–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Toker A; Cantley LC Signalling through the Lipid Products of Phosphoinositide-3-OH Kinase. Nature 1997, 387, 673–676. [DOI] [PubMed] [Google Scholar]
- (14).Vanhaesebroeck B; Guillermet-Guibert J; Graupera M; Bilanges B The Emerging Mechanisms of Isoform-Specific PI3K Signalling. Nat. Rev. Mol. Cell Biol 2010, 11, 329–341. [DOI] [PubMed] [Google Scholar]
- (15).Liu P; Cheng H; Roberts TM; Zhao JJ Targeting the Phosphoinositide 3-Kinase Pathway in Cancer. Nat. Rev. Drug Discovery 2009, 8, 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fruman DA; Rommel C PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discovery 2014, 13, 140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Stark AK; Sriskantharajah S; Hessel EM; Okkenhaug K PI3K Inhibitors in Inflammation, Autoimmunity and Cancer. Curr. Opin. Pharmacol 2015, 23, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Garces AE; Stocks MJ Class 1 PI3K Clinical Candidates and Recent Inhibitor Design Strategies: A Medicinal Chemistry Perspective. J. Med. Chem 2019, 62, 4815–4850. [DOI] [PubMed] [Google Scholar]
- (19).Yang J; Nie J; Ma X; Wei Y; Peng Y; Wei X Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 2019, 18, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Markham A Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [DOI] [PubMed] [Google Scholar]
- (21).Pons-Tostivint E; Thibault B; Guillermet-Guibert J Targeting PI3K Signaling in Combination Cancer Therapy. Trends Cancer 2017, 3, 454–469. [DOI] [PubMed] [Google Scholar]
- (22).West AC; Johnstone RW New and Emerging HDAC Inhibitors for Cancer Treatment. J. Clin. Invest 2014, 124, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Zhang L; Han Y; Jiang Q; Wang C; Chen X; Li X; Xu F; Jiang Y; Wang Q; Xu W Trend of Histone Deacetylase Inhibitors in Cancer Therapy: Isoform Selectivity or Multitargeted Strategy. Med. Res. Rev 2015, 35, 63–84. [DOI] [PubMed] [Google Scholar]
- (24).Eckschlager T; Plch J; Stiborova M; Hrabeta J Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci 2017, 18, 1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Suraweera A; O’Byrne KJ; Richard DJ Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol 2018, 8, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Subramanian S; Bates SE; Wright JJ; Espinoza-Delgado I; Piekarz RL Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yee AJ; Raje NS Panobinostat and Multiple Myeloma in 2018. Oncologist 2018, 23, 516–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Thurn KT; Thomas S; Moore A; Munster PN Rational Therapeutic Combinations with Histone Deacetylase Inhibitors for the Treatment of Cancer. Future Oncol. 2011, 7, 263–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Bodo J; Zhao X; Sharma A; Hill BT; Portell CA; Lannutti BJ; Almasan A; Hsi ED The Phosphatidylinositol 3-Kinases (PI3K) Inhibitor GS-1101 Synergistically Potentiates Histone Deacetylase Inhibitor-Induced Proliferation Inhibition and Apoptosis through the Inactivation of PI3K and Extracellular Signal-Regulated Kinase Pathways. Br. J. Haematol 2013, 163, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pei Y; Liu KW; Wang J; Garancher A; Tao R; Esparza LA; Maier DL; Udaka YT; Murad N; Morrissy S; Seker-Cin H; Brabetz S; Qi L; Kogiso M; Schubert S; Olson JM; Cho YJ; Li XN; Crawford JR; Levy ML; Kool M; Pfister SM; Taylor MD; Wechsler-Reya RJ HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Luan Y; Li J; Bernatchez JA; Li R Kinase and Histone Deacetylase Hybrid Inhibitors for Cancer Therapy. J. Med. Chem 2019, 62, 3171–3183. [DOI] [PubMed] [Google Scholar]
- (32).Wu Y; Dai W; Chen X; Geng A; Chen Y; Lu T; Zhu Y Design, Synthesis and Biological Evaluation of 2,3-Dihydroimidazo-[1,2-C]Quinazoline Derivatives as Novel Phosphatidylinositol 3-Kinase and Histone Deacetylase Dual Inhibitors. RSC Adv. 2017, 7, 52180–52186. [Google Scholar]
- (33).Chen D; Soh CK; Goh WH; Wang H Design, Synthesis, and Preclinical Evaluation of Fused Pyrimidine-Based Hydroxamates for the Treatment of Hepatocellular Carcinoma. J. Med. Chem 2018, 61, 1552–1575. [DOI] [PubMed] [Google Scholar]
- (34).Zhang K; Lai F; Lin S; Ji M; Zhang J; Zhang Y; Jin J; Fu R; Wu D; Tian H; Xue N; Sheng L; Zou X; Li Y; Chen X; Xu H Design, Synthesis, and Biological Evaluation of 4-Methyl Quinazoline Derivatives as Anticancer Agents Simultaneously Targeting Phosphoinositide 3-Kinases and Histone Deacetylases. J. Med. Chem 2019, 62, 6992–7014. [DOI] [PubMed] [Google Scholar]
- (35).Sun K; Atoyan R; Borek MA; Dellarocca S; Samson ME; Ma AW; Xu GX; Patterson T; Tuck DP; Viner JL; Fattaey A; Wang J Dual HDAC and PI3K Inhibitor CUDC-907 Downregulates MYC and Suppresses Growth of MYC-Dependent Cancers. Mol. Cancer Ther 2017, 16, 285–299. [DOI] [PubMed] [Google Scholar]
- (36).Sermer D; Pasqualucci L; Wendel HG; Melnick A; Younes A Emerging Epigenetic-Modulating Therapies in Lymphoma. Nat. Rev. Clin. Oncol 2019, 16, 494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Younes A; Berdeja JG; Patel MR; Flinn I; Gerecitano JF; Neelapu SS; Kelly KR; Copeland AR; Akins A; Clancy MS; Gong L; Wang J; Ma A; Viner JL; Oki Y Safety, Tolerability, and Preliminary Activity of CUDC-907, a First-in-Class, Oral, Dual Inhibitor of HDAC and PI3K, in Patients with Relapsed or Refractory Lymphoma or Multiple Myeloma: An Open-Label, Dose-Escalation, Phase 1 Trial. Lancet Oncol. 2016, 17, 622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Shah RR Safety and Tolerability of Histone Deacetylase (HDAC) Inhibitors in Oncology. Drug Saf. 2019, 42, 235–245. [DOI] [PubMed] [Google Scholar]
- (39).Somoza JR; Koditek D; Villasenor AG; Novikov N; Wong MH; Liclican A; Xing W; Lagpacan L; Wang R; Schultz BE; Papalia GA; Samuel D; Lad L; McGrath ME Structural, Biochemical, and Biophysical Characterization of Idelalisib Binding to Phosphoinositide 3-Kinase Delta. J. Biol. Chem 2015, 290, 8439–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lauffer BE; Mintzer R; Fong R; Mukund S; Tam C; Zilberleyb I; Flicke B; Ritscher A; Fedorowicz G; Vallero R; Ortwine DF; Gunzner J; Modrusan Z; Neumann L; Koth CM; Lupardus PJ; Kaminker JS; Heise CE; Steiner P Histone Deacetylase (HDAC) Inhibitor Kinetic Rate Constants Correlate with Cellular Histone Acetylation but Not Transcription and Cell Viability. J. Biol. Chem 2013, 288, 26926–26943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhang L; Zhang J; Jiang Q; Zhang L; Song W Zinc Binding Groups for Histone Deacetylase Inhibitors. J. Enzyme Inhib. Med. Chem 2018, 33, 714–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhang K; Lai F; Lin S; Ji M; Zhang J; Zhang Y; Jin J; Fu R; Wu D; Tian H; Xue N; Sheng L; Zou X; Li Y; Chen X; Xu H Design, Synthesis, and Biological Evaluation of 4-Methyl Quinazoline Derivatives as Anticancer Agents Simultaneously Targeting Phosphoinositide 3-Kinases and Histone Deacetylases. J. Med. Chem 2019, 62, 6992–7014. [DOI] [PubMed] [Google Scholar]
- (43).Yao L; Mustafa N; Tan EC; Poulsen A; Singh P; Duong-Thi MD; Lee JXT; Ramanujulu PM; Chng WJ; Yen JJY; Ohlson S; Dymock BW Design and Synthesis of Ligand Efficient Dual Inhibitors of Janus Kinase (JAK) and Histone Deacetylase (HDAC) Based on Ruxolitinib and Vorinostat. J. Med. Chem 2017, 60, 8336–8357. [DOI] [PubMed] [Google Scholar]
- (44).Huang Y; Dong G; Li H; Liu N; Zhang W; Sheng C Discovery of Janus Kinase 2 (JAK2) and Histone Deacetylase (HDAC) Dual Inhibitors as a Novel Strategy for the Combinational Treatment of Leukemia and Invasive Fungal Infections. J. Med. Chem 2018, 61, 6056–6074. [DOI] [PubMed] [Google Scholar]
- (45).Bhatia S; Krieger V; Groll M; Osko JD; Ressing N; Ahlert H; Borkhardt A; Kurz T; Christianson DW; Hauer J; Hansen FK Discovery of the First-in-Class Dual Histone Deacetylase-Proteasome Inhibitor. J. Med. Chem 2018, 61, 10299–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).He S; Dong G; Wu S; Fang K; Miao Z; Wang W; Sheng C Small Molecules Simultaneously Inhibiting P53-Murine Double Minute 2 (MDM2) Interaction and Histone Deacetylases (HDACs): Discovery of Novel Multitargeting Antitumor Agents. J. Med. Chem 2018, 61, 7245–7260. [DOI] [PubMed] [Google Scholar]
- (47).Zang J; Liang X; Huang Y; Jia Y; Li X; Xu W; Chou CJ; Zhang Y Discovery of Novel Pazopanib-Based HDAC and VEGFR Dual Inhibitors Targeting Cancer Epigenetics and Angio-genesis Simultaneously. J. Med. Chem 2018, 61, 5304–5322. [DOI] [PubMed] [Google Scholar]
- (48).Li Y; Luo X; Guo Q; Nie Y; Wang T; Zhang C; Huang Z; Wang X; Liu Y; Chen Y; Zheng J; Yang S; Fan Y; Xiang R Discovery of N1-(4-((7-Cyclopentyl-6-(Dimethylcarbamoyl)-7H-Pyrrolo[2,3-d]Pyrimidin-2-yl)Amino)Phenyl)-N8-Hydroxyoctanediamide as a Novel Inhibitor Targeting Cyclin-Dependent Kinase 4/9 (CDK4/9) and Histone Deacetlyase1 (HDAC1) against Malignant Cancer. J. Med. Chem 2018, 61, 3166–3192. [DOI] [PubMed] [Google Scholar]
- (49).Chen Y; Yuan X; Zhang W; Tang M; Zheng L; Wang F; Yan W; Yang S; Wei Y; He J; Chen L Discovery of Novel Dual Histone Deacetylase and Mammalian Target of Rapamycin Target Inhibitors as a Promising Strategy for Cancer Therapy. J. Med. Chem 2019, 62, 1577–1592. [DOI] [PubMed] [Google Scholar]
- (50).Liang X; Zang J; Li X; Tang S; Huang M; Geng M; Chou CJ; Li C; Cao Y; Xu W; Liu H; Zhang Y Discovery of Novel Janus Kinase (JAK) and Histone Deacetylase (HDAC) Dual Inhibitors for the Treatment of Hematological Malignancies. J. Med. Chem 2019, 62, 3898–3923. [DOI] [PubMed] [Google Scholar]
- (51).Cakici M; Catir M; Karabuga S; Ulukanli S; Kilic H Synthesis and Asymmetric Catalytic Activity of (1S,1′S)-4,4′-Biquinazoline-Based Primary Amines. Tetrahedron: Asymmetry 2011, 22, 300–308. [Google Scholar]
- (52).Wei M; Zhang X; Wang X; Song Z; Ding J; Meng LH; Zhang A Sar Study of 5-Alkynyl Substituted Quinazolin-4(3H)-ones as Phosphoinositide 3-Kinase Delta (PI3Kδ) Inhibitors. Eur. J. Med. Chem 2017, 125, 1156–1171. [DOI] [PubMed] [Google Scholar]
- (53).Adveef A Permeability—PAMPA In Absorption and Drug Development; John Wiley & Sons, Inc: New York, 2012; pp 319–498. [Google Scholar]
- (54).Patel L; Chandrasekhar J; Evarts J; Forseth K; Haran AC; Ip C; Kashishian A; Kim M; Koditek D; Koppenol S; Lad L; Lepist EI; McGrath ME; Perreault S; Puri KD; Villasenor AG; Somoza JR; Steiner BH; Therrien J; Treiberg J; Phillips G Discovery of Orally Efficacious Phosphoinositide 3-Kinase Delta Inhibitors with Improved Metabolic Stability. J. Med. Chem 2016, 59, 9228–9242. [DOI] [PubMed] [Google Scholar]
- (55).Patel L; Chandrasekhar J; Evarts J; Haran AC; Ip C; Kaplan JA; Kim M; Koditek D; Lad L; Lepist EI; McGrath ME; Novikov N; Perreault S; Puri KD; Somoza JR; Steiner BH; Stevens KL; Therrien J; Treiberg J; Villasenor AG; Yeung A; Phillips G 2,4,6-Triaminopyrimidine as a Novel Hinge Binder in a Series of PI3Kδ Selective Inhibitors. J. Med. Chem 2016, 59, 3532–3548. [DOI] [PubMed] [Google Scholar]
- (56).Perreault S; Chandrasekhar J; Cui ZH; Evarts J; Hao J; Kaplan JA; Kashishian A; Keegan KS; Kenney T; Koditek D; Lad L; Lepist EI; McGrath ME; Patel L; Phillips B; Therrien J; Treiberg J; Yahiaoui A; Phillips G Discovery of a Phosphoinositide 3-Kinase (PI3K) Beta/Delta Inhibitor for the Treatment of Phosphatase and Tensin Homolog (PTEN) Deficient Tumors: Building PI3Kβ Potency in a PI3Kδ-Selective Template by Targeting Nonconserved Asp856. J. Med. Chem 2017, 60, 1555–1567. [DOI] [PubMed] [Google Scholar]
- (57).Hai Y; Christianson DW Histone Deacetylase 6 Structure and Molecular Basis of Catalysis and Inhibition. Nat. Chem. Biol 2016, 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).NCI-60 Human Tumor Cell Lines Screen; National Cancer Institute, 2015; https://dtp.cancer.gov/discovery_development/nci-60/ (accessed 2019-10-10). [Google Scholar]
- (59).Celltiter-Glo Luminescent Cell Viability Assay; Promega, 2015; https://www.promega.com/-/media/files/resources/protocols/technical-bulletins/0/celltiter-glo-luminescent-cell-viability-assay-protocol.pdf?la=en (accessed 2019-10-15). [Google Scholar]
- (60).Weisberg E; Ray A; Nelson E; Adamia S; Barrett R; Sattler M; Zhang C; Daley JF; Frank D; Fox E; Griffin JD Reversible Resistance Induced by FLT3 Inhibition: A Novel Resistance Mechanism in Mutant FLT3-Expressing Cells. PLoS One 2011, 6, e25351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Bosshart H; Heinzelmann M THP-1 Cells as a Model for Human Monocytes. Ann. Transl Med 2016, 4, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Ramachandran RP; Madhivanan S; Sundararajan R; Wan-Ying Lin C; Sankaranarayanan K An in Vitro Study of Electro-poration of Leukemia and Cervical Cancer Cells In Electroporation-Based Therapies for Cancer; Sundararajan R, Ed.; Woodhead Publishing, Cambridge UK, 2014; Chapter 7, pp 161–183. [Google Scholar]
- (63).Matsuo Y; MacLeod RA; Uphoff CC; Drexler HG; Nishizaki C; Katayama Y; Kimura G; Fujii N; Omoto E; Harada M; Orita K Two Acute Monocytic Leukemia (AML-M5a) Cell Lines (MOLM-13 and MOLM-14) with Interclonal Phenotypic Heterogeneity Showing MLL-AF9 Fusion Resulting from an Occult Chromosome Insertion, Ins(11;9)(Q23;P22p23). Leukemia 1997, 11, 1469–1477. [DOI] [PubMed] [Google Scholar]
- (64).Liu X; Wang A; Liang X; Chen C; Liu J; Zhao Z; Wu H; Deng Y; Wang L; Wang B; Wu J; Liu F; Fernandes SM; Adamia S; Stone RM; Galinsky IA; Brown JR; Griffin JD; Zhang S; Loh T; Zhang X; Wang W; Weisberg EL; Liu J; Liu Q Characterization of Selective and Potent PI3Kδ Inhibitor (PI3KDIN-015) for B-Cell Malignances. Oncotarget 2016, 7, 32641–32651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Novotny-Diermayr V; Sangthongpitag K; Hu CY; Wu X; Sausgruber N; Yeo P; Greicius G; Pettersson S; Liang AL; Loh YK; Bonday Z; Goh KC; Hentze H; Hart S; Wang H; Ethirajulu K; Wood JM SB939, a Novel Potent and Orally Active Histone Deacetylase Inhibitor with High Tumor Exposure and Efficacy in Mouse Models of Colorectal Cancer. Mol. Cancer Ther 2010, 9, 642–652. [DOI] [PubMed] [Google Scholar]
- (66).Li X; Su Y; Madlambayan G; Edwards H; Polin L; Kushner J; Dzinic SH; White K; Ma J; Knight T; Wang G; Wang Y; Yang J; Taub JW; Lin H; Ge Y Antileukemic Activity and Mechanism of Action of the Novel PI3K and Histone Deacetylase Dual Inhibitor CUDC-907 in Acute Myeloid Leukemia. Haematologica 2019, 104, 2225–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Yin L; Wu Z; Avigan D; Rosenblatt J; Stone R; Kharbanda S; Kufe D MUC1-C Oncoprotein Suppresses Reactive Oxygen Species-Induced Terminal Differentiation of Acute Myelogenous Leukemia Cells. Blood 2011, 117, 4863–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Martinez NJ; Asawa RR; Cyr MG; Zakharov A; Urban DJ; Roth JS; Wallgren E; Klumpp-Thomas C; Coussens NP; Rai G; Yang S-M; Hall MD; Marugan JJ; Simeonov A; Henderson MJ A Widely-Applicable High-Throughput Cellular Thermal Shift Assay (CETSA) Using Split Nano Luciferase. Sci. Rep 2018, 8, 9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC, 2013; https://www.chemcomp.com/Products.htm (accessed 2019-03-02).
- (70).Halgren TA Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem 1996, 17, 490–519. [Google Scholar]
- (71).The UniProt Consortium. Uniprot: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Sun H; Nguyen K; Kerns E; Yan Z; Yu KR; Shah P; Jadhav A; Xu X Highly Predictive and Interpretable Models for PAMPA Permeability. Bioorg. Med. Chem 2017, 25, 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Shah P; Kerns E; Nguyen D-T; Obach RS; Wang AQ; Zakharov A; McKew J; Simeonov A; Hop CECA; Xu X An Automated High-Throughput Metabolic Stability Assay Using an Integrated High-Resolution Accurate Mass Method and Automated Data Analysis Software. Drug Metab. Dispos 2016, 44, 1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.