

ABSTRACT
Cell surface nucleotide-metabolizing enzyme, ectonucleotidase-CD73, has emerged as a central component of the cellular homeostatic-machinery that counterbalances the danger-molecule (extracellular-ATP)-driven proinflammatory response in immune cells. While the importance of CD73 in microbial host fitness and symbiosis is gradually being unraveled, there remains a significant gap in knowledge of CD73 and its putative role in epithelial cells. Here, we depict a novel host-pathogen adaptation mechanism where CD73 takes a center role in the intracellular persistence of Porphyromonas gingivalis, a major colonizer of oral mucosa, using human primary gingival epithelial cell (GEC) system. Temporal analyses revealed, upon invasion into the GECs, P. gingivalis can significantly elevate the host-surface CD73 activity and expression. The enhanced and active CD73 significantly increases P. gingivalis intracellular growth in the presence of substrate-AMP and simultaneously acts as a negative regulator of reactive oxygen species (ROS) generation upon eATP treatment. The inhibition of CD73 by siRNA or by a specific inhibitor markedly increases ROS production. Moreover, CD73 and P. gingivalis cross-signaling significantly modulates pro-inflammatory interleukin-6 (IL-6) in the GECs. Conversely, exogenous treatment of the infected GECs with IL-6 suppresses the intracellular bacteria via amplified ROS generation. However, the decreased bacterial levels can be restored by overexpressing functionally active CD73. Together, these findings illuminate how the local extracellular-purine-metabolism, in which CD73 serves as a core molecular switch, can alter intracellular microbial colonization resistance. Further, host-adaptive pathogens such as P. gingivalis can target host ectonucleotidases to disarm specific innate defenses for successful intracellular persistence in mucosal epithelia.
KEYWORDS: Ectonucleotidase-CD73, purinergic signaling, opportunistic oral bacteria, epithelial cells, intracellular infection, persistence
Introduction
Epithelial cells are the first-line innate defense cells that invading microbes encounter, therefore how this interaction develops can determine the colonization potential of a microorganism and its severity and duration in the host[1, 2]. In the oral cavity, gingival epithelial cells are centrally essential for initial proper induction of mucosal immune response since these cells often can recognize and respond to pathogens without causing serious damage to the host [1,3–5]. The mucosal epithelia actively participate in the immune defense via transfer of intracellular molecules to the extracellular space, which signals probable danger to the immune cells [6,7]. With starting infection or cellular stress, the host cells release adenosine 5ʹ-triphosphate (ATP) through pannexin-1-hemichannel to the extracellular milieu, thereby modulating and possibly disrupting the homeostatic purinergic signaling [8,9]. The host-derived small danger molecules then can act on ionotropic purinergic receptors, particularly P2X7 receptor, to elicit specific pro-inflammatory signal transduction [10–13]. To counteract the increase in the pro-inflammatory effector extracellular ATP (eATP), a series of host ectonucleotidases dynamically regulate rapid phosphohydrolysis of these nucleotides. Most notably, the production of extracellular adenosine occurs primarily through CD73 (ecto-5ʹ-nucleotidase), a glycosylphosphatidylinositol (GPI)-linked, membrane-bound enzyme that hydrolyzes extracellular nucleoside di/monophosphates into bioactive intermediates [14]. CD73-generated adenosine subsequently can lead to activation of G-protein-coupled adenosine receptors, which later regulate diverse physiologic effects including anti-inflammatory actions [15]. CD73 is known as a ubiquitous enzyme [16]. Expression of CD73 may change dynamically in cancer and chronic inflammatory conditions mostly examined in professional immune cells for its potent immunosuppressive functions [17–23]. However, the presence of CD73 has not been well characterized in epithelial cells and never been studied in the oral epithelia. Only recently is CD73 noted as a putative modulator in regulation of specific immune responses against microbial colonization [9,24] whilst CD73-mediated generation of extracellular adenosine signaling in epithelial cells remains largely unexplored [9].
The oral mucosa is a dynamic ecosystem with more than 700 species of intricately connected microorganisms [25,26]. Among those, Porphyromonas gingivalis, a Gram-negative bacterium and facultative intracellular pathogen, has been named a keystone organism for its major contribution to progression of periodontal disease and dysbiotic microbiota [27,28]. Recently, P. gingivalis has been proposed as an etiologic factor in various other chronic diseases, including orodigestive cancers and Alzheimer’s disease [29–31]. In gingival epithelial cells (GECs), P. gingivalis can establish its intracellular replication niche/reservoir [32–34] and later spread to adjacent cells intercellularly as a means of evading host antimicrobial immune detection [35] during disseminating deeper within the tissue [3,35–39]. Upon invasion into GECs, P. gingivalis can facilitate a long-term survival by altering host danger signal eATP-induced pathways that result in specific intracellular events such as modulation of reactive oxygen species (ROS) generation and pro-inflammatory cytokine Interleukin-1β (IL-1β) secretion [3,37,39–42]. Further, P. gingivalis inhibits GEC cell death induced by various pro-inflammatory or pro-apoptotic molecules [1,32,37,39,43,44]. By remaining viable in these host cells without being cleared, P. gingivalis forms a chronic infection in the oral mucosa, which can subsequently drive microorganismal proliferation/survival as well as dysbiosis in the oral microbiota [45]. Despite the past and ongoing endeavors, it is unclear under what microenvironmental deviations and molecular signals P. gingivalis gains supremacy over innate cellular defenses for a successful chronic microbial establishment in the oral mucosa. The significance of the purinergic signaling, which involves danger signals eATP and adenosine, has lately grown strong for colonization of opportunistic pathogens such as P. gingivalis in the epithelial mucosa [46–48]. Increasing evidence also supports the role of adenosine for progression of chronic inflammatory diseases [49]. Recent reports have investigated involvement of adenosine signaling in periodontal disease [50–52]. A study using rat models showed adenosine-dependent reduction in oral inflammation [52,53]. Moreover, we have previously shown that the purine signaling is critical for P. gingivalis in modulation of IL-1β [41] and that primary GECs express all types of adenosine (Aa) receptors including A2a with anti-inflammatory downstream effects including cAMP generation [54]. Addition of A2a receptor-specific agonist to P. gingivalis-infected GECs resulted in more intracellular P. gingivalis, whereas decreased bacterial growth was found with A2a receptor-specific antagonist, suggesting the pathogen may usurp the danger signal adenosine-coupled A2a signaling for intracellular life [54].
In the present work, using human primary GECs as a biologically relevant reductionist model [55–57], we report critically novel facets of CD73 in modulating epithelial-cell antimicrobial defenses via extracellular-purine-nucleotides. Specifically, we demonstrate for the first time the ectonucleotidase-CD73 expression and activity in the human primary epithelial cells, both of which are significantly increased during the opportunistic pathogen, P. gingivalis infection. We further show that the enhanced CD73 activity also coupled by extracellular AMP availability during the infection can be vital for the intracellular bacterial growth in epithelial cells. Interestingly, CD73 can play a crucial role for cross-modulation of select epithelial innate responses by P. gingivalis, including the danger signal eATP-generated reactive oxygen species (ROS) and dysregulated interleukin-6 (IL-6, one of the leading inflammatory mediators in a chronic inflammatory human disease, periodontitis). Lastly, we reveal that the intracellular level of P. gingivalis can be significantly reduced by exogenous treatment of IL-6, which can be largely restored by overexpressing CD73 in GECs. These findings together allude a novel host-pathogen adaptation mechanism specifically mediated by the host homeostatic CD73 and P. gingivalis interaction in oral mucosal cells. The targeting of CD73 by P. gingivalis can aid the microorganism forming a strategic growth-favorable cellular niche with the weakened actions of innate antibacterial molecules (e.g. ROS and IL-6). The described complex interaction may have a direct bearing on the dysbiotic presence of this keystone pathogen in human mucosa and could be an important mechanism used by other successful persistent pathogens.
Results
Examining the expression of ectonucleotidase-CD73 in GECs and its induction by P. gingivalis infection
We initially examined via qRT-PCR and Western blotting the expression of ectonucleotidase CD73 in P. gingivalis infected GECs over 24 h post-infection and compared the levels with uninfected GECs. Our results showed that both mRNA (Figure 1(a)) and protein (Figure 1(b)) expression of CD73 was significantly increased at 6 h post-bacterial invasion and remained gradually increased over 24 h of P. gingivalis infection. Further examination using confocal microscopy with specifically immuno-stained GECs also depicted significantly increased CD73 expression during infection (Figure 1(c), S1A). We demonstrated the exclusive external localization of CD73 on the cell membranes through immuno-staining (red) by orthogonal views of GECs (Figure 1(d), S1B). These data together indicate that P. gingivalis induces gradual and significantly higher CD73 expression in GECs during infection.
Figure 1.
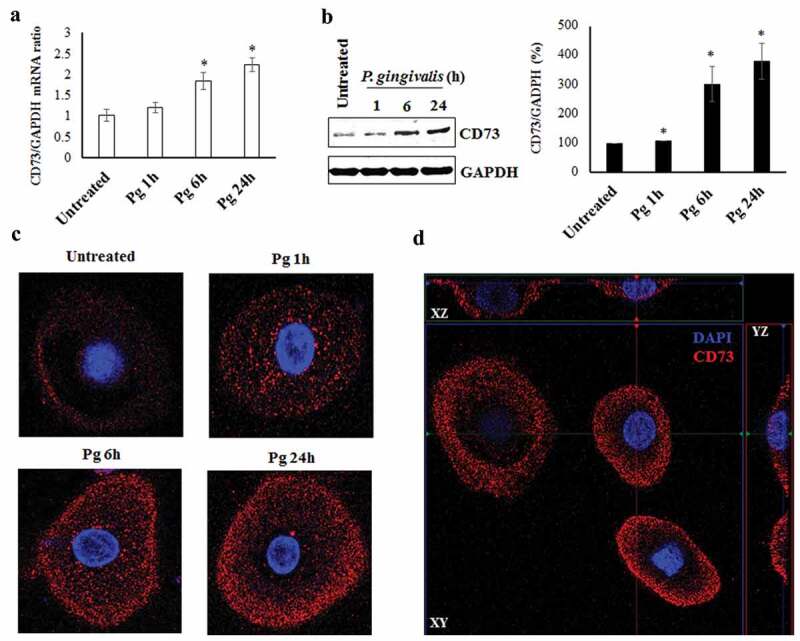
Ectonucleotidase-CD73 expression in primary GECs significantly increases during P. gingivalis infection. a) P. gingivalis strain ATCC 33277 was added at MOI 100 to cultured GECs over 24 h. GAPDH was used as a normalization control. SybrGreen detection of mRNA expression levels of CD73 (nt5e) using qRT-PCR. N = 3, *p < 0.05 as compared to untreated; Mean ± SEM. b) Infection was performed in the same manner as a). Cell lysates were extracted and immunoblotted with a CD73 antibody. Densitometric analysis was performed using NIH ImageJ software. N = 3, *p < 0.05 as compared to untreated. Mean ± SEM. c) Cells with P. gingivalis infection over 24 h were fixed, stained with primary antibody against CD73 and then secondary antibody (Alexa fluor 468; red), and mounted with DAPI (blue) to visualize nuclei. Representative images were obtained via confocal microscopy at 63x objective with oil immersion. d) Confocal micrographs showing orthogonal views of surface CD73 expression in GECs.
Examining the biological activity of ectonucleotidase-CD73 in GECs with or without P. gingivalis infection
To further validate the functional presence of CD73 in human gingival epithelial cells, we first assessed the potency of α,β-methylene adenosine-5′-diphosphate (APCP; a CD73-specific inhibitor) [59] as well as the effect of adenosine monophosphate (AMP; specific substrate for CD73) [60] treatment on CD73 activity in human primary GECs. Cells were incubated in phosphate-free buffer in the presence or absence of APCP and AMP and the amount of free phosphate released was quantified via the malachite green assay method.
The inhibitory effect of APCP on human CD73 in GECs was first determined. The concentration–inhibition curve exhibited an I50 value of 2.95 μM for the inhibitor in GECs pre-treated with AMP to initiate the enzymatic reaction (Figure 2(a)), showing comparable potency of the inhibitor with published data [61,62]. This suggests that ectonucleotidase-CD73 is fully functional in human primary GECs.
Figure 2.
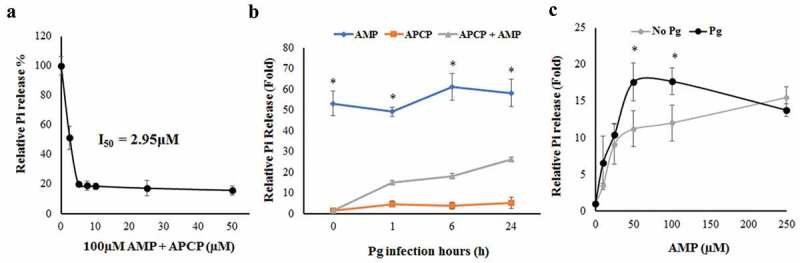
Confirmation of functional presence of the CD73 and the effect of P. gingivalis infection on CD73 activity in GECs. GECs were incubated in phosphate-free buffer and treated with either AMP, APCP, and/or P. gingivalis. All graphs (a-c) were obtained using Malachite Green Phosphate assay kit following the manufacturer’s instructions (Bioassay Systems). a) Inhibition–concentration curve showing CD73 activity of cells pre-treated with AMP [100 μM] and further incubated with varying concentrations of APCP. b) CD73 activity in untreated and P. gingivalis infected GECs (1, 6, and 24 h) in the presence of AMP [100 µM] and/or APCP [10 µM] N = 6, *p < 0.05 as compared for AMP vs. AMP + APCP. Mean ± SEM. c) CD73 activity with/without P. gingivalis infection at varying AMP concentrations. Cells were infected with P. gingivalis for 6 h and stimulated with AMP for 30 min post infection. N = 6, *p < 0.05 as compared to AMP 0 μM; Mean ± SEM.
By the malachite green assay method, we next examined CD73 enzymatic activity in the presence of P. gingivalis infection using APCP and AMP treatment (Figure 2(b,c)). When GECs were infected with P. gingivalis to mimic the inflammatory environment, the AMP treatment alone rapidly exhibited significantly increased phosphate release as expected at ~53-fold, which confirms that AMP is a CD73-specific substrate in GECs and stimulates high conversion to adenosine resulting in increased phosphate release (Figure 2(b)). Cells treated with both APCP and AMP showed significant decrease in phosphate release compared to those treated with AMP only, confirming that APCP functions as a robust CD73 inhibitor in GECs during the infection. While uninfected GECs showed an approximately 10-fold increase in CD73 activity with 50 and 100 μM of AMP pre-treatment, this increase was further augmented by P. gingivalis infection showing enhanced CD73 enzymatic activity by ~17-fold when pre-treated with the same concentrations of AMP. These data collectively suggest that the epithelial cells possess functional CD73 and its activity levels can be significantly enhanced by the infection.
Increased activity of ectonucleotidase-CD73 enhances intracellular growth of P. gingivalis in GECs
We next evaluated whether CD73 can play a specific role in P. gingivalis survival in GECs. Accordingly, we examined the intracellular survival of P. gingivalis in GECs in the presence of CD73 substrate molecule AMP or the CD73 inhibitor APCP using our in-situ antibiotic protection assay [63]. Cells were first incubated with or without 50 μM APCP followed by 100 μM AMP stimulation and 24 h infection with P. gingivalis. We then quantified live, intracellular bacteria by qPCR using 16s rRNA primers (Figure 3(a)). The results showed more than a 3.5-fold increase in the intracellular P. gingivalis levels in GECs treated with AMP compared to the AMP untreated cells only infected with P. gingivalis. Infected cells treated with APCP in addition to AMP stimulation showed significantly reduced bacterial survival than the infected cells treated with AMP only. Interestingly, APCP treatment alone exhibited a significant decrease only when compared to the infected cells treated with both APCP and AMP (Figure 3(a)). Compared to the control cells with only infection, APCP treatment did not have a notable impact on bacterial levels, which points to the importance of AMP as an initiator of the enzymatic reaction. To further confirm our findings via visualization, we performed immunostaining on P. gingivalis infected GECs and analyzed the levels of intracellular bacteria through confocal microscopy (Figure 3(b)). P. gingivalis specifically immuno-stained in green fluorescence is significantly increased in cells treated with AMP compared to the control cells with infection only as well as the host cells further treated with APCP (Figure 3(b)). These findings suggest that high CD73 activity, which was experimentally mimicked by exogenous addition of AMP to cell culture milieu, can be essential for P. gingivalis to establish an optimal level of intracellular growth and survival in the epithelial cells of oral mucosa.
Figure 3.
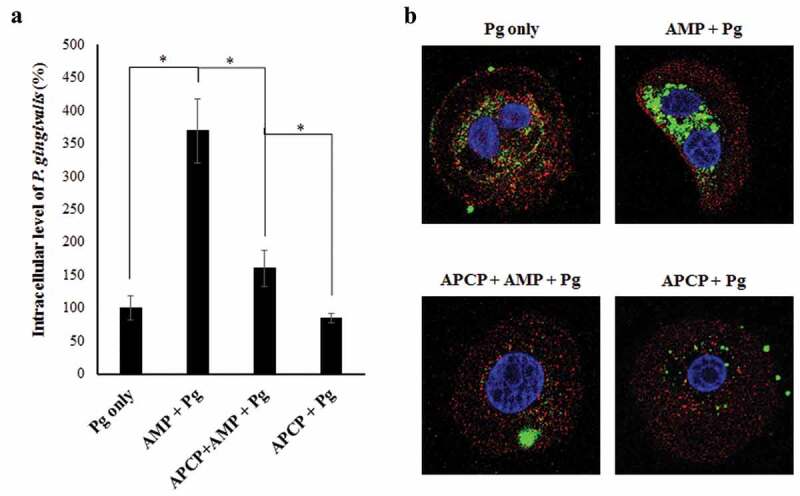
The active CD73 significantly increases intracellular P. gingivalis levels in GECs. a) Antibiotic protection assay – GECs were infected with P. gingivalis (MOI 100) for 24 h. APCP [50 μM] and AMP [100 μM] were added to cells 1 h and 30 min prior to infection, respectively. Cells were then incubated with metronidazole [200 μg/mL] and gentamicin [300 μg/mL] for an additional 1 h. RNAs from each sample were isolated using Trizol and subsequently obtained cDNAs were subjected to qPCR analysis using P. gingivalis 16s rRNA primers. N = 3, *p < 0.05; Mean ± SEM. b) Representative confocal microscopy images of fixed, infected GECs with P. gingivalis (MOI 100) for 24 h with or without AMP and APCP as described in a). CD73 (red) and P. gingivalis (green) were visualized by immunostaining. 63x magnification with oil immersion. The range of z-stacks was kept consistent.
Ectonucleotidase-CD73 is a negative regulator of a danger signal eATP induced ROS production in GECs
Our previous studies showed that P. gingivalis can attenuate eATP-induced biocidal ROS production [3,39] and CD73 may regulate ROS levels in cells or tissues [64,65]. Therefore, we assessed whether CD73 specifically involves in modulation of eATP coupled ROS production in GECs using a ROS-detecting fluorescent probe, DCFDA, for in-situ quantitative and qualitative ROS analysis. Diphenyleneiodonium (DPI; a broad-spectrum NADPH oxidase inhibitor) and hydrogen peroxide (exogenous ROS) were used as controls and underwent the probe incubation in the same manner. Treatment of eATP-stimulated GECs with the CD73 inhibitor APCP significantly increased eATP-induced ROS as early as 30 min after initial ATP treatment (Figure 4(a)). This large increase in ROS levels in the presence of APCP was vigorous and sustained, suggesting the involvement of CD73 in decreasing of cellular ROS produced by the GECs in response to eATP. Live cell imaging for cellular ROS showed that inhibiting CD73 in GECs by APCP also resulted in notably greater ROS production than the cells stimulated with eATP only (Figure 4(b)). Taken together, these results suggest that CD73 perhaps is an important negative regulator of danger signal eATP-induced antimicrobial ROS in GECs.
Figure 4.
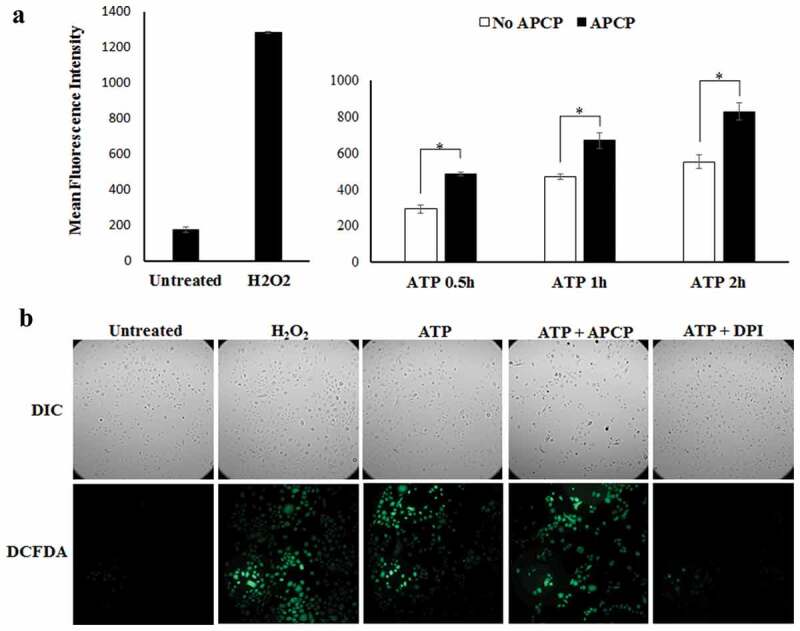
The CD73 significantly diminishes eATP induced reactive oxygen species (ROS) generation in GECs. a) ROS generation in GECs after 30 min pre-treatment with ATP [3 mM] and incubation over 2 h with APCP [50 μM] or vehicle measured as relative fluorescence using a Biotek H1 M monochromatic plate reader at 525 nm. N = 3, *p < 0.05; Mean ± SEM. b) Representative micrographs of CM-H2DCFDA, ROS probe, live fluorescence imaging with Differential Interference Contrast (DIC) microscopy was used for qualitative assessment of ROS generation shown in a). 10x magnification on Leica inverted microscope. Diphenyleneiodonium (DPI), an NAD(p)H oxidase inhibitor, was used as a positive control at 50 μM.
Activation of ectonucleotidase-CD73 by P. gingivalis decreases host cell-mediated induction of antimicrobial interleukin-6 (IL-6) in GECs
Recent studies have shown modulation of IL-6 by ROS accumulation [66,67]. Hence, we next investigated the levels of IL-6, during P. gingivalis infection and whether CD73 plays a role in regulating the expression and/or secretion of IL-6 in GECs. We first measured the effect of P. gingivalis infection on levels of IL-6 secretion over 24 h (Figure 5(a), S2). The infection by P. gingivalis markedly modulated IL-6 secretion with a steady and significant increase at 6 h post-infection, which also resulted in a large decline returning into baseline IL-6 levels at 24 h post-infection. In addition to our published studies which had already demonstrated that P. gingivalis infection does not induce neither apoptosis nor necrosis in primary GECs and the cells are viable at least up to 120 h post-infection [1,3,35,37,40,63,68], we further confirmed that the marked decrease in IL-6 production after 6 h post-infection is not due to GEC cell death (Figure S3).
Figure 5.
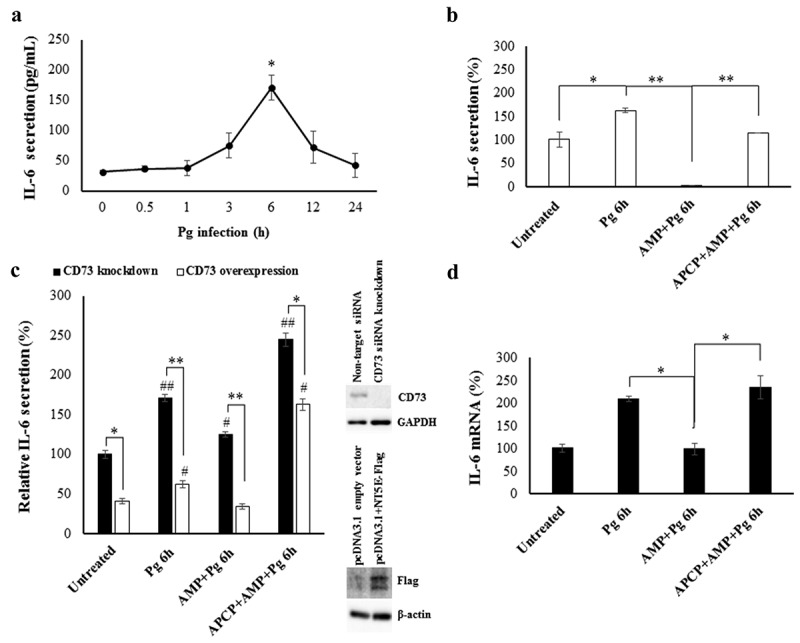
The active and enhanced CD73 attenuates human IL-6 expression and secretion induced by GECs during P. gingivalis infection. a) Quantitative ELISA measurement of human IL-6 expression in GECs with P. gingivalis infection (MOI 100) over 24 h displayed in μM. N = 3, *p < 0.05 as compared to untreated; Mean ± SEM. b,c) Quantitative ELISA measurement of human IL-6 secretion in untreated GECs, CD73 knockdown, and overexpressing GECs incubated with P. gingivalis (MOI 100) for 6 h. Further treatment with APCP [50 μM] and/or AMP [100 μM] was also performed. CD73 depletion/overexpression studies were conducted using non-target siRNA control and empty vector as controls and confirmed by Western blotting. N = 3, #,*p < 0.05, ##,**p < 0.001, # denotes statistical comparison to corresponding untreated; Mean ± SEM. d) GECs were pre-infected with P. gingivalis (MOI 100) for 6 h, treated with APCP [50 μM] and/or AMP [100 μM], and analyzed by qPCR using SYBR Green for IL-6 mRNA levels. N = 3, *p < 0.05; Mean ± SEM.
Subsequently, we examined whether CD73 has any regulatory effect on IL-6 secretion during P. gingivalis infection. ELISA analysis showed that the initial IL-6 secretion produced in GECs by P. gingivalis is significantly weakened upon CD73 stimulation by exogenous AMP. Further, inhibition of CD73 activity by APCP significantly diminished the suppressive effect of AMP (Figure 5(b)). As additional investigations, CD73-specific gene depletion via siRNA or overexpression in GECs was performed (Figure 5(c)). CD73-depleted GECs showed significantly increased IL-6 secretion for corresponding conditions tested compared to CD73-overexpressing cells, indicating that levels of CD73 are also important for attenuating the pro-inflammatory IL-6 production (Figure 5(c)). These results collectively highlight a dynamically specific molecular dialogue between the host CD73 and the microorganism that culminates in reduction of IL-6 production. Interestingly, qRT-PCR analysis for IL-6 gene expression at 6 h of P. gingivalis infection in GECs displayed IL-6 mRNA expression is significantly downregulated by CD73 stimulation with AMP treatment, which is fully rescued by CD73 inhibition with APCP (Figure 5(d)), a similar trend observed in the protein level (Figure 5(b)). This finding further points to a tight regulation of IL-6 by CD73 pathway that may also occur at the IL-6 gene level.
Ectonucleotidase-CD73-mediated inhibition of eATP-coupled ROS generation is critical for IL-6 production in GECs
We next examined the plausible role of CD73-mediated ROS for IL-6 secretion in GECs during P. gingivalis infection. Besides the untreated cells, all GECs were pre-stimulated with eATP for ROS induction and infected with P. gingivalis for 6 h. Some cells were further incubated with either DPI or N-acetyl-l-cysteine (NAC; potent antioxidant) to suppress ROS formation and accumulation, respectively. For both control and CD73-overexpressing GECs, eATP treatment resulted in significantly higher IL-6 secretion levels compared to those when further treated with the ROS inhibitors (Figure 6). More critically, overexpression of CD73 in GECs led to significantly reduced amount of secreted IL-6 when cells were either treated with ATP only or ATP and DPI together. Interestingly, NAC treatment seemed to induce a more robust inhibition of IL-6 secretion than DPI, which suggests the putative link between intracellular ROS and IL-6 secretion may not be entirely limited to membrane-associated NADPH oxidase derived ROS. Overall, these findings support that CD73 can be a key determinant for IL-6 secretion stimulated by the danger signal eATP-mediated ROS production in GECs upon infection.
Figure 6.
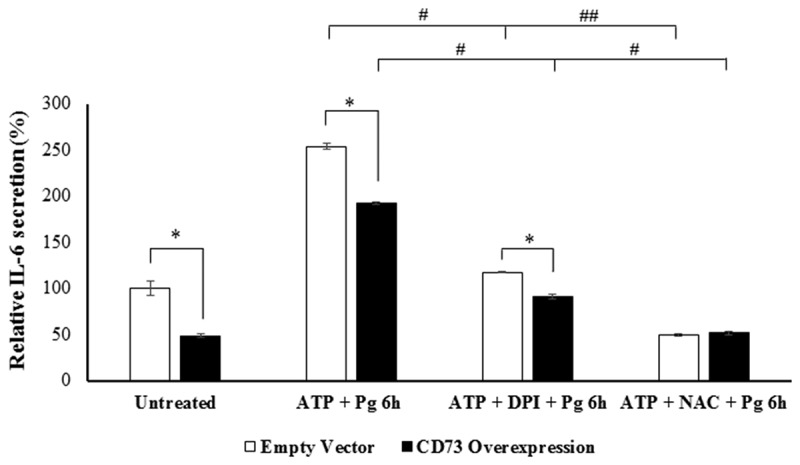
ATP-mediated ROS generation regulates IL-6 secretion in a CD73-dependent manner during P. gingivalis infection. Quantitative ELISA measurement of human IL-6 secretion in control GECs treated with the empty vector, and overexpressing GECs incubated with P. gingivalis (MOI 100) for 6 h. Further treatment with ATP [3 mM], a broad-spectrum NADPH oxidase inhibitor DPI [50 μM] and/or ROS scavenger NAC [50 μM] was also performed. N = 6, *p < 0.05, #p < 0.05, ##p < 0.001; Mean ± SEM.
Exogenous IL-6 treatment reduces the intracellular level of P. gingivalis and the Ectonucleotidase-CD73 overexpression can restore the IL-6-mediated decrease in P. gingivalis survival
Given the putative role of CD73 for promoting intracellular P. gingivalis growth and inhibiting eATP-mediated antimicrobial IL-6 secretion during infection in GECs, we studied the potential direct effect of IL-6 on intracellular P. gingivalis survival. Accordingly, we tried to emulate the pro-inflammatory environment by treating the host cells exogenously with human IL-6 in addition to the CD73-specific substrate (AMP). We first established that extracellular addition of human IL-6 does not induce host cell toxicity in GECs via time-course MTT assay and the bacterial decrease would not be the result of host cell death (Figure 7(a)). We next examined the effect of exogenous IL-6 treatment at varying concentrations on intracellular P. gingivalis levels in GECs at 6 h post-infection where we observed the highest IL-6 production during the infection (Figure 7(b)). Our results showed that exogenous IL-6 treatment substantially decreased the bacterial survival in a dose-dependent manner, exhibiting a significant decrease at 25 and 50 ng/mL.
Figure 7.
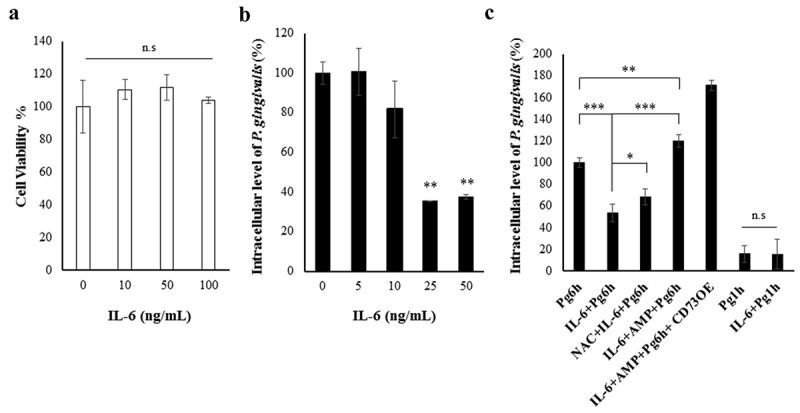
CD73 overexpression reverses the decreasing P. gingivalis intracellular level in GECs caused by exogenous treatment of human IL-6. a) Cells were incubated with multiple doses of IL-6 for 24 h. GEC cell viability was assessed using MTT assay. n.s = no statistically significant difference, N = 3, * p < 0.05. For B and C, GECs were infected with P. gingivalis (MOI 100) for 6 h. b) Human recombinant active IL-6 [25 ng/mL] was added to cells 30 min prior to infection at varying concentrations. IL-6 treatment [25 ng/mL] was performed similarly in B and C. c) Overexpression of CD73 (OE) was implemented in some conditions as described in Material & Methods. For B and C, cells were later incubated with metronidazole [200 μg/mL] and gentamicin [300 μg/mL] for an additional 1 h to kill extracellular bacteria. RNAs from each sample were isolated using Trizol and subsequently obtained cDNAs were subjected to qPCR analysis using P. gingivalis 16s rRNA primers. N = 6, *p < 0.05, **p < 0.001, *** <0.0001; Mean ± SEM. CD73OE = CD73 overexpression, NAC = N-acetyl-l-cysteine (ROS scavenger; 50 µM).
Further, we re-performed the antibiotic protection assay, using P. gingivalis-infected GECs pre-treated with IL-6 as previously done in Figure 7(b). However, we this time amplified the activity of CD73 in two ways: AMP treatment alone and AMP treatment plus CD73 overexpression (Figure 7(c)). Results showed that AMP treatment alone not only significantly restored the IL-6 induced reduction in the intracellular P. gingivalis survival but also presented unexpected synergy by inducing significantly more bacteria compared to the baseline P. gingivalis 6 h levels in GECs. Additional CD73 overexpression accompanied by AMP treatment significantly rescued and further enhanced the bacterial replication, yielding highest (more than a 70%) increase compared to the GECs incubated with P. gingivalis only. Interestingly, additional treatment with a potent ROS scavenger NAC showed a substantial reversion on the antibacterial property of IL-6 compared to that GECs treated with IL-6 only (the NADPH oxidase inhibitor DPI showed a similar reversing effect; data not shown). This result further supports that IL-6 may exert an inhibitory effect on intracellular P. gingivalis survival through ROS induction. We also confirmed no interference of initial bacterial invasion by IL-6 treatment (Figure 7(c), invasion percentage with and without IL-6 treatments at 1 h post-infection is comparable). These findings together describe a new molecular circuitry where the opportunistic P. gingivalis can reprogram the host anti-inflammatory CD73 signaling to disarm the antibacterial effect of high IL-6 levels initially induced by the GECs as a response to growing infection.
Discussion
Increasing studies have highlighted the importance of host-derived small molecules, such as ATP and adenosine, which function as a danger signal in host and alert the immune system to properly respond to threat via purinergic signaling [69–71]. Extracellular ATP accumulation has been strongly associated with upregulation of pro-inflammatory immune responses including cytokine and cellular ROS levels [72]. On the contrary, adenosine is a well-established inflammatory mediator with immunosuppressive actions including inhibition of tumor growth and regulation of Treg cells [73,74]. As a major generator of adenosine, CD73 in various specialized immune cells performs several innate protective functions important for various severe inflammatory diseases [19,21,74]. Growing evidence suggests that CD73 may modulate opportunistic bacterial colonization in host tissues [9,18,21,75]. CD73 also appears to be an important determinant of the extracellular homeostasis for maintaining the physiological state of host cells in the face of potent danger signaling molecules. This distinct ability of CD73 may present a greater degree of importance to host-adapted chronic pathogens such as P. gingivalis for promoting cellular growth and survival, which may later create dysbiotic microbiota. Several lines of in vivo studies have shown that CD73 can impact on various types of microbial infection in the intestinal mucosa through the modulation of adaptive immune responses [18,76,77]. Salmonella-treated murine splenocytes incubated with a CD73-specific inhibitor APCP showed significantly augmented IL17A and IFN-γ expression [21]. Liver tissue from CD73-deficient mice also had attenuated bacterial load of Salmonella, which collectively suggests CD73 expression can exacerbate the outcome of Salmonella infection by regulating host inflammatory responses [21]. Furthermore, these recent studies merely utilized CD73 knockout mice to phenotypically describe and establish the initial importance of this enzyme for mucosal immunity during acute and chronic models of microbial infection [18,21,76,78,79].
Here, we present a newly identified expression of functional CD73 in the human primary epithelial cells and CD73’s novel well-orchestrated molecular actions in regulating a cellular chronic infection by the opportunistic pathogen, P. gingivalis. Both mRNA and protein expression of CD73 is markedly increased by P. gingivalis infection, indicating that the infection acts as a robust danger signal for initiation of the CD73 enzymatic reaction. Furthermore, the immunostaining of CD73 visually confirmed that P. gingivalis infection increases the expression of surface-bound CD73 in GECs. Our results from these expression studies provide a first direct visualization of the interaction between an opportunistic bacterium and CD73 in host epithelial cells.
To date, the primary known action of CD73 is to metabolize AMP to produce adenosine by hydrolyzing nucleotide monophosphates while releasing inorganic phosphate, which have been shown in various immune cells [14,76]. However, no prior studies have confirmed this function in human epithelial cells. While making sure that the concentration of AMP used in the experiments was within the biologically analogous range (i.e. 50–100 μM; AMP higher than 250 μM is considered physiologically high) [80], our novel results showed further increased CD73 activity by P. gingivalis infection in GECs. This finding was consistent with our hypothesis that P. gingivalis favors active CD73 signaling to promote intracellular persistence in the host. Additionally, stimulating P. gingivalis-infected GECs with AMP, a direct substrate that increases CD73 enzymatic activity, induced higher amounts of intracellular P. gingivalis determined by the antibiotic protection assay. This result indicates that more P. gingivalis are present within the host cells upon presence of extracellular AMP and the bacteria are replicating at a higher rate compared to the P. gingivalis without AMP treatment. The observed increase in quantity of intracellular bacteria was markedly abolished when P. gingivalis-infected GECs were further treated with the CD73-specific inhibitor, APCP, signifying CD73 as a key modulator to the shown elevation in bacterial intracellular survival.
We have previously demonstrated that P. gingivalis effector enzyme, nucleoside-diphosphate-kinase (ndk), is extracellularly secreted from P. gingivalis-infected GECs [8]. This enzyme homolog that converts the nucleotides can hydrolyze eATP, thereby inhibiting eATP/P2X7 receptor-mediated ROS production [3,37,40]. Additionally, we have recently published that P. gingivalis in GECs not only modulates antibacterial NADPH oxidase-mediated ROS generation but also induces robust host-antioxidant glutathione synthesis to avoid bacterial clearance [39]. These host-adaptive features of P. gingivalis in the previous publications suggested that eATP-mediated adenosine signaling might regulate the amount of cellular ROS during the infection. Our results from this study also revealed a novel cellular mechanism where CD73 inhibition results in an increase in ROS production, implicating that active CD73 induced by P. gingivalis can be a critical switch of dampening the initial host innate antimicrobial response.
Another key inflammatory mediator that P. gingivalis needs to overcome for successful survival is potent pro-inflammatory cytokine production upon microbial infection [55]. We observed that CD73 inhibition promotes robust and sustained cellular ROS accumulation in GECs (Figure 4). Recent studies demonstrated that ROS can activate IL-6 signaling in nerve cells [66] and inhibition of ROS can specifically block IL-6 release in skeletal muscle cells [67]. IL-6 is a pleiotropic pro-inflammatory cytokine that is dysregulated in initial periodontal inflammation and may contribute to dysbiotic host environment [81–84]. As shown in the present study, P. gingivalis infection alone can lead to robust pro-inflammatory IL-6 secretion by the GECs in earlier stages of infection. However, active and enhanced CD73 was able to abolish this host-mediated IL-6 production against P. gingivalis in GECs, which indicates that the pathogen likely targets anti-inflammatory CD73 signaling to suppress IL-6 levels in the host cell. In addition, our novel results unexpectedly revealed that exogenous treatment of IL-6 can markedly decrease intracellular P. gingivalis in GECs, which could be reversed by stimulating CD73 activity by exogenous AMP treatment and additional CD73 overexpression implemented for a greater reversal effect. There have been some studies showing pro-inflammatory cytokines in clinical isolates can alter growth of pathogens such as Escherichia coli and Staphylococcus aureus [85–87]; however, the studies were performed solely in bacterial growth culture without providing molecular mechanism. The present study for the first time shows that external treatment of a pro-inflammatory cytokine-like IL-6 can alter the intracellular bacterial survival like P. gingivalis in human host cells. Additionally, the concentrations of IL-6 that led to the significant decrease in P. gingivalis intracellular survival in GECs were similar to the published range during systemic inflammation [88]. Interestingly, a recent report showed that CD73 inhibition (with the same CD73-specific inhibitor, APCP) up-regulates macrophage production of both pro-inflammatory cytokine and nitric oxide, which are required host immune responses for bacterial clearance during salmonellosis [79]. Therefore, it is tempting to speculate that other opportunistic bacteria causing intracellular mucosal infections like P. gingivalis may usurp CD73 as described in this study. However, further investigations are needed to fully determine the generality of the current findings.
In summary, our novel findings showed that CD73, a host-protective surface molecule that majorly contributes to eATP danger-molecule enzymatic cascade, can be targeted by P. gingivalis for the enhanced host intracellular symbiosis resulting in robust CD73 expression and activity in epithelial cells. Moreover, we identified potentially a new homeostatic function of the ectonucleotidase-CD73 in host cells for attenuating potent antimicrobial responses such as ROS inhibition and IL-6 secretion, which ultimately supports opportunistic organisms such as P. gingivalis to sustain chronic presence in the epithelial cells (Figure 8). By identifying IL-6 as an important cytokine for P. gingivalis infection in the epithelial cells, along with a newly discovered role of CD73 in epithelial IL-6 biology, we propose the described interactions can present a significant advantage for P. gingivalis’ intracellular persistence in the epithelial cells. Thus, the findings here display a distinct host-microbe adaptation strategy that may contribute to microbial dysbiosis in oral mucosa and might be operational for other chronic bacteria.
Figure 8.
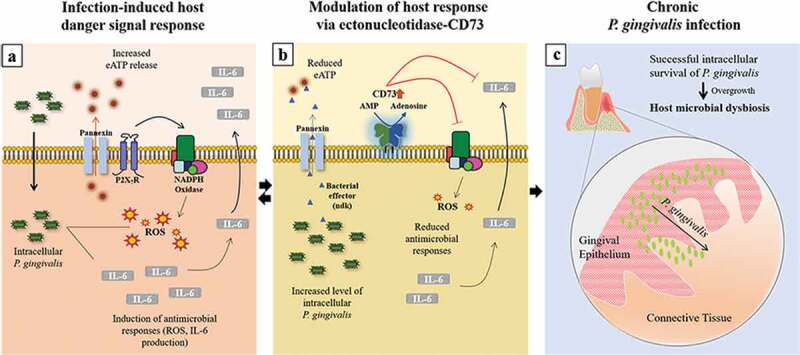
Sequential modulation of the host-danger signaling by the interplay of homeostatic host-enzyme, CD73, and host-adaptive pathogen, P. gingivalis, disarms major cellular innate molecules, ROS and IL-6, in human gingival epithelium. a) Upon P. gingivalis invasion, extracellular ATP (eATP)-induced antimicrobial Reactive-Oxygen-Species (ROS) formation and Interleukin-6 (IL-6) secretion are induced. b) Extracellular secretion of P. gingivalis-nucleoside diphosphate kinase (ndk), acting as an autocrine effector to the host, modulates the P2X7-receptor/pannexin hemichannel (3,8); in parallel, homeostatic CD73 expression and activity is modified by P. gingivalis to counteract the innate host responses resulting in inhibition of epithelial ROS and IL-6 generations. c) Thus, these dysregulated molecular events create a favorable intracellular environment for P. gingivalis in successful evasion of the epithelial antibacterial intracellular immunity, thereby establishing long-term microbial survival. The eventual overgrowth of P. gingivalis and its chronic infection in GECs later might lead to host microbial dysbiosis in oral mucosa.
Materials and methods
Bacterial and eukaryotic cell culture
P. gingivalis ATCC strain 33277, which has been shown to be more invasive in the intracellular infection models compared to other relevant strains (i.e. W83 and 381) [89–92], was used for this study. P. gingivalis ATCC strain 33277 was anaerobically cultured at 37°C in Trypticase soy broth (TSB) supplemented with yeast extract (1 mg/ml), hemin (5 μg/ml) and menadione (1 μg/ml). Bacteria were harvested by centrifugation at 6000 g and 4°C for 10 min and re-suspended in Dulbecco’s phosphate-buffered saline (PBS), pH 7.3. The bacteria were quantified at mid-log phase using a Klett-Summerson photometer and used at a multiplicity of infection (MOI) of 100 for all assays described below.
Primary human GECs were obtained and cultured as described previously [32,37,39,43,93,94]. Briefly, gingival tissue was collected from healthy adult individuals who were selected anonymously and randomly from those presenting at the University of Florida Dental Clinics for tooth crown lengthening or impacted third molar extraction. No patient information was collected. Gingival tissue that would otherwise be discarded was collected after informed consent was obtained by all patients under the approved guidance of the University of Florida Health Science Center Institutional Review Board (IRB, human subjects assurance number FWA 00005790). Cells were cultured in serum‐free keratinocyte growth medium (KGM, Lonza) at 37°C in 5% CO2. To assure consistency, only the early passage numbers of the primary GECs were utilized for experimentation, which were repeated using cells derived from multiple patient samples.
Western blotting analysis for CD73 expression
CD73 protein expression in GECs was evaluated via Western blotting as previously performed [43,63,68]. Cell lysates of GECs infected with P. gingivalis for 1, 6, and 24 h were collected in 1x RIPA buffer with protease inhibitor cocktail (Thermo Fisher) and protein levels were determined by Bradford assay. The cell lysates in 1x Laemmli Sample Buffer were loaded onto a 10% SDS-Page gel at 140 V for 1 h. The separated proteins were transferred to a nitrocellulose membrane via wet transfer at 80 V for 1 h and blocked with 5% nonfat dry milk in Tris-buffered saline and 0.1% Tween-20. The membrane was incubated overnight at 4°C in 1:1,000 mouse CD73 (Abcam) and mouse GAPDH (Abcam) antibodies, washed, and incubated with secondary mouse HRP conjugated antibody for 1 h. Protein bands were visualized using enhanced chemiluminescence (GE Healthcare) and analyzed using NIH ImageJ software.
Confocal microscopy
Cells were infected with P. gingivalis 33277 for 1, 6, or 24 h, fixed using 10% neutral buffered formalin, blocked with 3% BSA, and stained for 1 h with a 1:50 mouse CD73 antibody (Abcam) and a custom-made 1:1,000 rabbit P. gingivalis 33277 antibody (Pacific Immunology, Ramona, CA). The stained cells were washed and incubated for another 1 h with Alexa Fluor 568 conjugated secondary goat anti-mouse antibody and Alexa Fluor 488 conjugated secondary goat anti-rabbit antibody (1:1,000; Invitrogen). Fixed cells were mounted using VectaShield mounting medium containing DAPI (Vector Laboratories). Images were acquired and processed at 63x objective with oil using Zeiss LSM 880 Quasar NLO multiphoton/confocal system with Zeiss Zen Microscope Software.
CD73 activity assay
The enzymatic activity of CD73 was determined by the release of inorganic phosphate (Pi) as previously described [95,96]. Briefly, GECs pre-incubated in phosphate-free buffer (2 mM MgCl2, 125 mM NaCl, 1 mM KCl, 10 mM glucose, 10 mM Hepes pH 7.2, diluted in ddH2O) were treated with α,β-methylene adenosine-5′-diphosphate (APCP) (Sigma), a CD73-specific inhibitor, diluted in phosphate-free buffer [50 μM; unless indicated otherwise]. The cells were incubated with AMP [100 μM, unless indicated otherwise] (Sigma) diluted in phosphate-free buffer for 30 min and the enzymatic reaction was terminated on ice for 10 min. Cell culture supernatant was collected to study the Pi release using Malachite Green Phosphate assay kit (BioAssay Systems) according to the manufacturer’s protocol with a BioTek H1 M monochromatic plate reader at 620 nm. Non-enzymatic hydrolysis was measured by substrate solution without cells. The baseline Pi release for each condition was determined by adding blank buffer. For the determination of kinetic parameters, the AMP and APCP concentrations were selected based on previously used range [97] as well as our results showing no cytotoxic effect on GECs with the chosen concentrations.
IL-6 qRT-PCR
Transcription of IL-6 was measured by qRT-PCR, using the SybrGreen detection system using the following pairs of primers: human-specific IL-6 (Forward: 5ʹ- GTAGCCGCCCCACACAGA-3ʹ; Reverse: 5ʹ-CATGTCTCCTTTCTCAGGGCTG-3ʹ) and human-specific GAPDPH (Forward: 5ʹ-GAAATCCCATCACCATCTTCCAGG-3ʹ; Reverse: 5′-GAGCCCCAGCCTTCTCCATG-3′) [98]. Total RNA was extracted with Trizol (Invitrogen) and 1 μg of the total RNA per sample was reverse-transcribed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was conducted at 95°C for 3 min, followed by 45 cycles at 95°C for 30 s and either 55°C (for IL-6 and GAPDH) for 30 s. Gene expression analysis was performed using the CFX Manager Software (BioRad). The measured expression of GADPH was used as a reference gene. For each condition, uninfected GECs were assigned as control.
IL-6 ELISA
GECs were incubated with AMP [100 μM] and/or APCP [50 μM] 1 h and 30 min, respectively, and infected with P. gingivalis. For CD73 depleted and overexpressing GECs, the transfection described below was performed first for at least 24 h. Cell culture supernatants were collected after selected timepoints of P. gingivalis infection. Uninfected and/or untreated GECs were used as controls. The quantitative measurement of human IL-6 by ELISA was performed following the manufacturer’s protocol (R&D systems). The optical density was read at absorbance of 450 nm using a Biotek H1 M monochromatic plate reader with wavelength correction of 540 nm. A standard curve was obtained from 600 to 9.38 pg/mL. No impact on the initial bacterial invasion by AMP and APCP treatment was confirmed prior to this assay.
Antibiotic protection assay
P. gingivalis intracellular survival in GECs was determined as described previously [63]. Briefly, GECs were incubated with AMP [100 μM] and/or APCP [50 μM] 1 h and 30 min, respectively, and infected with P. gingivalis for 6 or 24 h. GECs were washed with PBS and treated with gentamicin (300 µg/mL) and metronidazole (200 µg/mL) for 1 h. Total RNA was isolated using Trizol Reagent (Invitrogen). Genomic DNA contamination was removed by DNase digestion (Ambion). cDNA was synthesized from 1 μg Total RNA using High Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems). A 1:10 cDNA was used to detect P. gingivalis 16s rRNA by SYBR Green Real-time qPCR (Forward: 5ʹ-TGTAGATGACTGATGGTGAAAACC-3ʹ; Reverse: 5ʹ-ACGTCATCCCCACCTTCCTC-3ʹ) [99]. qPCR was performed in CFX96 real-time system (Bio-Rad) with an initial cycle of 98°C for 3 min followed by 40 cycles of 9 5°C for 15 s, 60.7°C for 30 s, and 72°C for 30 s. The CFU of P. gingivalis in unknown sample was calculated from the standard curve prepared according to the published method. In brief, P. gingivalis was grown to early log phase (OD600 = 0.2) and 1 ml of culture was used for CFU count by 10-fold serial dilution followed by plating on TSB supplemented with yeast extract (5 mg/ml), hemin (5 μg/ml) and menadione (1 μg/ml), agar (1.5%) and sheep blood (5%). Another 1 ml was used to isolate genomic DNA and 10-fold serial dilution of DNA was used for qPCR using the primers and conditions mentioned here. A standard curve was prepared with the Ct (Threshold cycle) value obtained from qPCR and CFU counted on agar plates (correlation coefficient = 0.999). This standard curve was then used to calculate CFU of corresponding Ct value obtained from qPCR performed with cDNA of each infected-GEC condition (as we published previously [63]).
Silencing of CD73 (NT5E) by small interfering RNA
GECs were transfected with ON-TARGETplus Human NT5E siRNA (Dharmcon) or Control siRNA (Life Technologies) using Lipofectamine™ RNAiMax Protocol (Invitrogen) for 24 or 48 h. Cell lysates were collected and the silencing was confirmed by Western blotting as described previously in this study.
Transient transfection with pcDNA‑NT5E plasmid for CD73 overexpression
pcDNA3.0+ expression vector with CD73 coding gene (NT5E) insert (GenScript) was transfected into human primary GECs using Lipofectamine™ 3000 reagent (Invitrogen) following the manufacturer’s protocol. pcDNA3.0+ vector without NT5E insert (empty vector) was used as a control. Confirmation of CD73 overexpression in GECs was done by Western blotting as described previously in this study using Flag antibody (1,1:000; Cell Signaling).
Host cell ROS production measurement
Cellular ROS production in GECs was determined as described previously [3,39]. Briefly, GECs were pre-stimulated with ATP [3 mM] and further incubated with 50 μM APCP. GECs were labeled with the fluorescent probe 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) (Invitrogen) which was solubilized in dimethyl sulfoxide (Sigma) and diluted to 5 μM in Hanks balanced salt solution (HBSS) containing MgCl2 and CaCl2 (Invitrogen). The CM-H2DCFDA (green) fluorescence intensity was measured using a Biotek H1 M monochromatic bottom fluorescence plate reader at excitation 495 nm and emission 525 nm. The representative live images of CM-H2DCFDA fluorescence intensity were taken using epifluorescence (DM IRE2 HC inverted scope, Leica Microsystems GmbH) microscopy equipped with DIC. The single exposure images were collected sequentially in fluorescence and DIC using a Retiga 4000 r CCD camera (Qimaging). Diphenyleneiodonium (DPI), a broad-spectrum NADPH oxidase inhibitor was used as a positive control.
Statistical analysis
All experiments were performed in at least three separate occasions with technical duplicates or triplicates. The results were initially analyzed by one-way ANOVA and then by two-tailed Student’s t-test. P-values determined by the t-test are reported while values of 0.05 or less were considered statistically significant.
Funding Statement
This work was supported by funding from the NIH grants, R01DE016593, R56DE016593, T32DE017551, F30DE029103, and F31DE026065.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Yilmaz O, Jungas T, Verbeke P, et al. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kolenbrander PE. American society for microbiology. Oral microbial communities: genomic inquiry and interspecies communication. Washington (DC): ASM Press; 2011. [Google Scholar]
- [3].Choi CH, Spooner R, DeGuzman J, et al. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell Microbiol. 2013;15:961–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yilmaz O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology. 2008;154:2897–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dale BA. Periodontal epithelium: a newly recognized role in health and disease. Periodontol 2000. 2002;30:70–78. [DOI] [PubMed] [Google Scholar]
- [6].Burnstock G. Purinergic signalling in the gastrointestinal tract and related organs in health and disease. Purinergic Signal. 2014;10:3–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ohman J, Erlinge D. The touching story of purinergic signaling in epithelial and endothelial cells. Purinergic Signal. 2012;8:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Atanasova K, Lee J, Roberts J, et al. Nucleoside-diphosphate-kinase of P. gingivalis is secreted from epithelial cells in the absence of a leader sequence through a pannexin-1 interactome. Sci Rep. 2016;6:37643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee JS, Yilmaz O. Unfolding role of a danger molecule adenosine signaling in modulation of microbial infection and host cell response. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miller CM, Boulter NR, Fuller SJ, et al. The role of the P2X(7) receptor in infectious diseases. PLoS Pathog. 2011;7:e1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Spooner R, Yilmaz O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int J Mol Sci. 2011;12:334–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Guerra AN, Gavala ML, Chung HS, et al. Nucleotide receptor signalling and the generation of reactive oxygen species. Purinergic Signal. 2007;3:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yilmaz O, Lee KL. The inflammasome and danger molecule signaling: at the crossroads of inflammation and pathogen persistence in the oral cavity. Periodontol 2000. 2015;69:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zimmermann H. 5ʹ-Nucleotidase: molecular structure and functional aspects. Biochem J. 1992;285(Pt 2):345–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–787. [DOI] [PubMed] [Google Scholar]
- [16].Jalkanen J, Hollmen M, Jalkanen S, et al. Regulation of CD73 in the development of lower limb atherosclerosis. Purinergic Signal. 2017;13:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Eckle T, Fullbier L, Wehrmann M, et al. Identification of ectonucleotidases CD39 and CD73 in innate protection during acute lung injury. J Immunol. 2007;178:8127–8137. [DOI] [PubMed] [Google Scholar]
- [18].Alam MS, Kurtz CC, Rowlett RM, et al. CD73 is expressed by human regulatory T helper cells and suppresses proinflammatory cytokine production and Helicobacter felis-induced gastritis in mice. J Infect Dis. 2009;199:494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gao ZW, Wang HP, Lin F, et al. CD73 promotes proliferation and migration of human cervical cancer cells independent of its enzyme activity. BMC Cancer. 2017;17:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Velasquez S, Eugenin EA. Role of Pannexin-1 hemichannels and purinergic receptors in the pathogenesis of human diseases. Front Physiol. 2014;5:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Alam MS, Kuo JL, Ernst PB, et al. Ecto-5ʹ-nucleotidase (CD73) regulates host inflammatory responses and exacerbates murine salmonellosis. Sci Rep. 2014;4:4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Francois V, Shehade H, Acolty V, et al. Intestinal immunopathology is associated with decreased CD73-generated adenosine during lethal infection. Mucosal Immunol. 2014;8:773. [DOI] [PubMed] [Google Scholar]
- [23].Wang Y, Telesford KM, Ochoa-Reparaz J, et al. An intestinal commensal symbiosis factor controls neuroinflammation via TLR2-mediated CD39 signalling. Nat Commun. 2014;5:4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bou Ghanem EN. mSphere of influence: adenosine in host defense against bacterial pneumonia-friend or foe? mSphere. 2019;4. DOI: 10.1128/mSphere.00326-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Atanasova KR, Yilmaz O. Prelude to oral microbes and chronic diseases: past, present and future. Microbes Infect. 2015;17:473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Aas JA, Paster BJ, Stokes LN, et al. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10:717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Spooner R, Weigel KM, Harrison PL, et al. In situ anabolic activity of periodontal pathogens Porphyromonas gingivalis and Filifactor alocis in chronic periodontitis. Sci Rep. 2016;6:33638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Olsen I, Yilmaz O. Possible role of Porphyromonas gingivalis in orodigestive cancers. J Oral Microbiol. 2019;11:1563410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Atanasova KR, Yilmaz O. Looking in the Porphyromonas gingivalis cabinet of curiosities: the microbium, the host and cancer association. Mol Oral Microbiol. 2014;29:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5:eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lee K, Roberts JS, Choi CH, et al. Porphyromonas gingivalis traffics into endoplasmic reticulum-rich-autophagosomes for successful survival in human gingival epithelial cells. Virulence. 2018;9:845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Colombo AV, Silva CM, Haffajee A, et al. Identification of oral bacteria associated with crevicular epithelial cells from chronic periodontitis lesions. J Med Microbiol. 2006;55:609–615. [DOI] [PubMed] [Google Scholar]
- [34].Dzink JL, Gibbons RJ, Childs WC 3rd, et al. The predominant cultivable microbiota of crevicular epithelial cells. Oral Microbiol Immunol. 1989;4:1–5. [DOI] [PubMed] [Google Scholar]
- [35].Yilmaz O, Verbeke P, Lamont RJ, et al. Intercellular spreading of Porphyromonas gingivalis infection in primary gingival epithelial cells. Infect Immun. 2006;74:703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dorn BR, Dunn WA Jr., Progulske-Fox A. Bacterial interactions with the autophagic pathway. Cell Microbiol. 2002;4:1–10. [DOI] [PubMed] [Google Scholar]
- [37].Yilmaz O, Yao L, Maeda K, et al. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cell Microbiol. 2008;10:863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hajishengallis G. Immune evasion strategies of Porphyromonas gingivalis. J Oral Biosci. 2011;53:233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Roberts JS, Atanasova KR, Lee J, et al. Opportunistic pathogen Porphyromonas gingivalis modulates danger signal ATP-mediated antibacterial NOX2 pathways in primary epithelial cells. Front Cell Infect Microbiol. 2017;7:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hung SC, Choi CH, Said-Sadier N, et al. P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PloS One. 2013;8:e70210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yilmaz O, Sater AA, Yao L, et al. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol. 2010;12:188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Johnson L, Atanasova KR, Bui PQ, et al. Porphyromonas gingivalis attenuates ATP-mediated inflammasome activation and HMGB1 release through expression of a nucleoside-diphosphate kinase. Microbes Infect. 2015;17:369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lee J, Roberts JS, Atanasova KR, et al. A novel kinase function of a nucleoside-diphosphate-kinase homologue in Porphyromonas gingivalis is critical in subversion of host cell apoptosis by targeting heat-shock protein 27. Cell Microbiol. 2018;20:e12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mao S, Park Y, Hasegawa Y, et al. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell Microbiol. 2007;9:1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. 2018;16:745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Jahngen EG, Brecx M, Rossomando EF. High-performance liquid chromatography analysis of purine nucleosides in human gingival crevicular fluid. Arch Oral Biol. 1984;29:607–610. [DOI] [PubMed] [Google Scholar]
- [47].Barnes VM, Teles R, Trivedi HM, et al. Acceleration of purine degradation by periodontal diseases. J Dent Res. 2009;88:851–855. [DOI] [PubMed] [Google Scholar]
- [48].Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J. 1986;233:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Haskó G, Linden J, Cronstein B, et al. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Murakami S, Hashikawa T, Saho T, et al. Adenosine regulates the IL-1 beta-induced cellular functions of human gingival fibroblasts. Int Immunol. 2001;13:1533–1540. [DOI] [PubMed] [Google Scholar]
- [51].Hashikawa T, Takedachi M, Terakura M, et al. Involvement of CD73 (ecto-5ʹ-nucleotidase) in adenosine generation by human gingival fibroblasts. J Dent Res. 2003;82:888–892. [DOI] [PubMed] [Google Scholar]
- [52].Bitto A, Oteri G, Pisano M, et al. Adenosine receptor stimulation by polynucleotides (PDRN) reduces inflammation in experimental periodontitis. J Clin Periodontol. 2013;40:26–32. [DOI] [PubMed] [Google Scholar]
- [53].Sun CX, Wall NR, Angelov N, et al. Changes in mRNA expression of adenosine receptors in human chronic periodontitis. Chin J Dent Res. 2011;14:113–120. [PubMed] [Google Scholar]
- [54].Spooner R, DeGuzman J, Lee KL, et al. Danger signal adenosine via adenosine 2a receptor stimulates growth of Porphyromonas gingivalis in primary gingival epithelial cells. Mol Oral Microbiol. 2014;29:67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sandros J, Karlsson C, Lappin DF, et al. Cytokine responses of oral epithelial cells to Porphyromonas gingivalis infection. J Dent Res. 2000;79:1808–1814. [DOI] [PubMed] [Google Scholar]
- [56].Sugawara S, Uehara A, Tamai R, et al. Innate immune responses in oral mucosa. J Endotoxin Res. 2002;8:465–468. [DOI] [PubMed] [Google Scholar]
- [57].Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411–420. [DOI] [PubMed] [Google Scholar]
- [58].Knapp K, Zebisch M, Pippel J, et al. Crystal structure of the human ecto-5ʹ-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure. 2012;20:2161–2173. [DOI] [PubMed] [Google Scholar]
- [59].Rahimova R, Fontanel S, Lionne C, et al. Identification of allosteric inhibitors of the ecto-5ʹ-nucleotidase (CD73) targeting the dimer interface. PLoS Comput Biol. 2018;14:e1005943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Freundlieb M, Zimmermann H, Muller CE. A new, sensitive ecto-5ʹ-nucleotidase assay for compound screening. Anal Biochem. 2014;446:53–58. [DOI] [PubMed] [Google Scholar]
- [61].Iqbal J, Jirovsky D, Lee SY, et al. Capillary electrophoresis-based nanoscale assays for monitoring ecto-5ʹ-nucleotidase activity and inhibition in preparations of recombinant enzyme and melanoma cell membranes. Anal Biochem. 2008;373:129–140. [DOI] [PubMed] [Google Scholar]
- [62].Lee J, Roberts JS, Atanasova KR, et al. Human primary epithelial cells acquire an epithelial-mesenchymal-transition phenotype during long-term infection by the oral opportunistic pathogen, Porphyromonas gingivalis. Front Cell Infect Microbiol. 2017;7:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Jian R, Sun Y, Wang Y, et al. CD73 protects kidney from ischemia-reperfusion injury through reduction of free radicals. APMIS. 2012;120:130–138. [DOI] [PubMed] [Google Scholar]
- [64].Chisci E, De Giorgi M, Zanfrini E, et al. Simultaneous overexpression of human E5NT and ENTPD1 protects porcine endothelial cells against H2O2-induced oxidative stress and cytotoxicity in vitro. Free Radic Biol Med. 2017;108:320–333. [DOI] [PubMed] [Google Scholar]
- [65].Liu J, Liu Y, Chen J, et al. The ROS-mediated activation of IL-6/STAT3 signaling pathway is involved in the 27-hydroxycholesterol-induced cellular senescence in nerve cells. Toxicol In Vitro. 2017;45:10–18. [DOI] [PubMed] [Google Scholar]
- [66].Kosmidou I, Vassilakopoulos T, Xagorari A, et al. Production of interleukin-6 by skeletal myotubes: role of reactive oxygen species. Am J Respir Cell Mol Biol. 2002;26:587–593. [DOI] [PubMed] [Google Scholar]
- [67].Yao L, Jermanus C, Barbetta B, et al. Porphyromonas gingivalis infection sequesters pro-apoptotic bad through Akt in primary gingival epithelial cells. Mol Oral Microbiol. 2010;25:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sitkovsky MV, Ohta A. The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol. 2005;26:299–304. [DOI] [PubMed] [Google Scholar]
- [69].Bours MJ, Swennen EL, Di Virgilio F, et al. Adenosine 5ʹ-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. [DOI] [PubMed] [Google Scholar]
- [70].Ishii KJ, Akira S. Potential link between the immune system and metabolism of nucleic acids. Curr Opin Immunol. 2008;20:524–529. [DOI] [PubMed] [Google Scholar]
- [71].Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:177–192. [DOI] [PubMed] [Google Scholar]
- [72].Allard D, Allard B, Gaudreau PO, et al. CD73-adenosine: a next-generation target in immuno-oncology. Immunotherapy. 2016;8:145–163. [DOI] [PubMed] [Google Scholar]
- [73].Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol. 2014;5:304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bou Ghanem EN, Clark S, Roggensack SE, et al. Extracellular Adenosine Protects against Streptococcus pneumoniae Lung Infection by Regulating Pulmonary Neutrophil Recruitment. PLoS Pathog. 2015;11:e1005126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kao DJ, Saeedi BJ, Kitzenberg D, et al. Intestinal epithelial ecto-5ʹ-nucleotidase (CD73) regulates intestinal colonization and infection by nontyphoidal salmonella. Infect Immun. 2017;85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Alam MS, Kurtz CC, Wilson JM, et al. A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4+ helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal Immunol. 2009;2:232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Petit-Jentreau L, Jouvion G, Charles P, et al. Ecto-5ʹ-nucleotidase (CD73) deficiency in Mycobacterium tuberculosis-infected mice enhances neutrophil recruitment. Infect Immun. 2015;83:3666–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Costales MG, Alam MS, Cavanaugh C, et al. Extracellular adenosine produced by ecto-5ʹ-nucleotidase (CD73) regulates macrophage pro-inflammatory responses, nitric oxide production, and favors Salmonella persistence. Nitric Oxide. 2018;72:7–15. [DOI] [PubMed] [Google Scholar]
- [79].Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. [DOI] [PubMed] [Google Scholar]
- [80].Takashiba S, Naruishi K, Murayama Y. Perspective of cytokine regulation for periodontal treatment: fibroblast biology. J Periodontol. 2003;74:103–110. [DOI] [PubMed] [Google Scholar]
- [81].Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer. 2005;41:2502–2512. [DOI] [PubMed] [Google Scholar]
- [82].Dutzan N, Kajikawa T, Abusleme L, et al. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yost S, Duran-Pinedo AE, Krishnan K, et al. Potassium is a key signal in host-microbiome dysbiosis in periodontitis. PLoS Pathog. 2017;13:e1006457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Meduri GU, Kanangat S, Stefan J, et al. Cytokines IL-1beta, IL-6, and TNF-alpha enhance in vitro growth of bacteria. Am J Respir Crit Care Med. 1999;160:961–967. [DOI] [PubMed] [Google Scholar]
- [85].Porat R, Clark BD, Wolff SM, et al. Enhancement of growth of virulent strains of Escherichia coli by interleukin-1. Science. 1991;254:430–432. [DOI] [PubMed] [Google Scholar]
- [86].Denis M, Gregg EO. Recombinant tumour necrosis factor-alpha decreases whereas recombinant interleukin-6 increases growth of a virulent strain of Mycobacterium avium in human macrophages. Immunology. 1990;71:139–141. [PMC free article] [PubMed] [Google Scholar]
- [87].Baran P, Hansen S, Waetzig GH, et al. The balance of interleukin (IL)-6, IL-6.soluble IL-6 receptor (sIL-6R), and IL-6.sIL-6R.sgp130 complexes allows simultaneous classic and trans-signaling. J Biol Chem. 2018;293:6762–6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Saito A, Kokubu E, Inagaki S, et al. Porphyromonas gingivalis entry into gingival epithelial cells modulated by Fusobacterium nucleatum is dependent on lipid rafts. Microb Pathog. 2012;53:234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lamont RJ, Chan A, Belton CM, et al. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63:3878–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Belton CM, Izutsu KT, Goodwin PC, et al. Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol. 1999;1:215–223. [DOI] [PubMed] [Google Scholar]
- [91].Baker PJ, Dixon M, Evans RT, et al. Heterogeneity of Porphyromonas gingivalis strains in the induction of alveolar bone loss in mice. Oral Microbiol Immunol. 2000;15:27–32. [DOI] [PubMed] [Google Scholar]
- [92].Oda D, Watson E. Human oral epithelial cell culture I. Improved conditions for reproducible culture in serum-free medium. In Vitro Cell Dev Biol. 1990;26:589–595. [DOI] [PubMed] [Google Scholar]
- [93].Yilmaz O, Watanabe K, Lamont RJ. Involvement of integrins in fimbriae-mediated binding and invasion by Porphyromonas gingivalis. Cell Microbiol. 2002;4:305–314. [DOI] [PubMed] [Google Scholar]
- [94].Garcia-Hernandez MH, Portales-Cervantes L, Cortez-Espinosa N, et al. Expression and function of P2X(7) receptor and CD39/Entpd1 in patients with type 2 diabetes and their association with biochemical parameters. Cell Immunol. 2011;269:135–143. [DOI] [PubMed] [Google Scholar]
- [95].Xu S, Shao QQ, Sun JT, et al. Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro Oncol. 2013;15:1160–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Adzic M, Nedeljkovic N. Unveiling the role of Ecto-5ʹ-nucleotidase/CD73 in astrocyte migration by using pharmacological tools. Front Pharmacol. 2018;9:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Nhu QM, Shirey K, Teijaro JR, et al. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol. 2010;3:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Tran SD, Rudney JD. Multiplex PCR using conserved and species-specific 16S rRNA gene primers for simultaneous detection of Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. J Clin Microbiol. 1996;34:2674–2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lyons SR, Griffen AL, Leys EJ. Quantitative real-time PCR for Porphyromonas gingivalis and total bacteria. J Clin Microbiol. 2000;38:2362–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.