

Abstract
An outbreak of the novel coronavirus SARS-CoV-2, the causative agent of COVID-19 respiratory disease, has infected over 290,000 people since the end of 2019, killed over 12,000, and caused worldwide social and economic disruption1,2. There are currently no antiviral drugs with proven efficacy nor are there vaccines for its prevention. Unfortunately, the scientific community has little knowledge of the molecular details of SARS-CoV-2 infection. To illuminate this, we cloned, tagged and expressed 26 of the 29 viral proteins in human cells and identified the human proteins physically associated with each using affinity-purification mass spectrometry (AP-MS), which identified 332 high confidence SARS-CoV-2-human protein-protein interactions (PPIs). Among these, we identify 67 druggable human proteins or host factors targeted by 69 existing FDA-approved drugs, drugs in clinical trials and/or preclinical compounds, that we are currently evaluating for efficacy in live SARS-CoV-2 infection assays. The identification of host dependency factors mediating virus infection may provide key insights into effective molecular targets for developing broadly acting antiviral therapeutics against SARS-CoV-2 and other deadly coronavirus strains.
The current pandemic of COVID-19 (Coronavirus Disease-2019), a respiratory disease that has led to over 290,000 confirmed cases and 12,000 fatalities in over 100 countries since its emergence in late 20193,4, is caused by a novel virus strain, SARS-CoV-2, an enveloped, positive-sense, single-stranded RNA betacoronavirus of the family Coronaviridae. Coronaviruses infecting humans historically included several mild common cold viruses e.g. hCoV-OC43, HKU, 229E5. However, over the past two decades, highly pathogenic human coronaviruses have emerged, including SARS-CoV in 2002 and 2003 with 8,000 cases worldwide and a death rate of ~10%, and MERS-CoV in 2012, which caused 2,500 confirmed cases and a fatality rate of 36%6. Infection with these highly pathogenic coronaviruses can result in Acute Respiratory Distress Syndrome (ARDS), which may lead to long-term reduction in lung function, arrhythmia, and death. Compared to MERS or SARS7,8, SARS-CoV-2 appears to spread more efficiently, making it difficult to contain and increasing its pandemic potential. To devise therapeutic strategies to counteract SARS-CoV-2 infection, it is crucial to develop a comprehensive understanding of how this coronavirus hijacks the host during the course of infection, and to apply this knowledge towards developing both new drugs and repurposing existing ones.
So far, no clinically available antiviral drugs have been developed for SARS-CoV, SARS-CoV-2 or MERS-CoV. Clinical trials are ongoing for treatment of COVID-19 with the nucleotide analog RNA-dependent RNA Polymerase (RdRP) inhibitor remdesivir9–11, and recent data suggests a new nucleotide analog may be effective against SARS-CoV-2 infection in laboratory animals12. Clinical trials on several vaccine candidates are also underway13, as well as trials of repurposed host-directed compounds inhibiting the human protease TMPRSS214. We believe there is great potential in systematically exploring the host dependencies of the SARS-CoV-2 virus to identify other host proteins already targeted with existing drugs. Therapies targeting the host-virus interface, where mutational resistance is arguably less likely, could potentially present durable, broad-spectrum treatment modalities15. Unfortunately, our minimal knowledge of the molecular details of SARS-CoV-2 infection precludes a comprehensive evaluation of small molecule candidates for host-directed therapies. We sought to address this knowledge gap by systematically mapping the interaction landscape between SARS-CoV-2 proteins and human proteins.
Cloning and expression of predicted SARS-CoV-2 proteins
Sequence analysis of SARS-CoV-2 isolates suggests that the 30kb genome encodes as many as 14 open reading frames (Orfs). The 5’ Orf1a / Orf1ab encodes polyproteins, which are auto-proteolytically processed into 16 non-structural proteins (Nsp1–16) which form the replicase / transcriptase complex (RTC). The RTC consists of multiple enzymes, including the papain-like protease (Nsp3), the main protease (Nsp5), the Nsp7-Nsp8 primase complex, the primary RNA-dependent RNA polymerase (Nsp12), a helicase/triphosphatase (Nsp13), an exoribonuclease (Nsp14), an endonuclease (Nsp15), and N7- and 2’O-methyltransferases (Nsp10/Nsp16)1,16,17. At the 3’ end of the viral genome, as many as 13 Orfs are expressed from nine predicted sub-genomic RNAs. These include four structural proteins: Spike (S), Envelope (E), Membrane (M) and Nucleocapsid (N)17, and nine putative accessory factors (Fig. 1a)1,16. In genetic composition, the SARS-CoV-2 genome is very similar to SARS-CoV: each has an Orf1ab encoding 16 predicted Nsps and each has the four typical coronavirus structural proteins. However, they differ in their complement of 3’ open reading frames: SARS-CoV-2 possesses an Orf3b and Orf10 with limited detectable protein homology to SARS-CoV16, and its Orf8 is intact while SARS-CoV encodes Orf8a and Orf8b (Fig. 1b)1,16,18.
Figure 1: AP-MS Workflow for Identification of SARS-CoV-2 Host Protein-Protein Interactions.

(a) SARS-CoV-2 genome annotation. (b) Table of the SARS-CoV-2 proteins, including molecular weight, sequence similarity with the SARS-CoV homolog, and inferred function based on the SARS-CoV homolog. (c) Immunoblot detection of 2xStrep tag demonstrates expression of each bait in input samples, as indicated by red arrowhead. (d) Experimental workflow for expressing each 2xStrep tagged SARS-CoV-2 fusion protein in biological triplicate in HEK293T cells, followed by affinity purification-mass spectrometry, and PPI scoring to identify 332 high confidence protein-protein interactions.
Mature Nsps and all predicted proteins expressed from other SARS-CoV-2 Orfs (27 proteins plus one mutant) were codon optimized and cloned into a mammalian expression vector with a 2xStrep tag fused to either the N- or C-terminus19. Protein expression plasmids were transfected into human HEK293T cells, which were incubated for 40 hours before lysis in a mild detergent buffer. Viral proteins were affinity purified using MagStrep beads. Beads were washed, on-bead digestion was performed overnight, and peptides were desalted and analyzed by protein mass spectrometry. High confidence interactors were identified using SAINTexpress and the MiST algorithm19,20.
To verify viral protein expression, we performed an anti-Strep western blot on input cell lysate, and with the exception of Nsp4, Nsp6, Nsp11, and Orf3b, we observed bands consistent with predicted protein sizes (24 of 28 constructs). Despite the lack of detection via western blot we were able to detect expression of viral peptides Nsp4, Nsp6, and Orf3b in the proteomic analysis. The fourth construct not confirmed by western blot, the small peptide Nsp11, had a predicted molecular mass of 4.8 kDa (including tag) but an apparent mass of approximately 30 kDa (Fig. 1c). SARS-CoV-2 Orf3b has limited homology to SARS-CoV Orf3b16, and its restricted expression as measured by both western blot and AP-MS may indicate that it is either not a bonafide protein coding gene or that it requires co-expression of other SARS-CoV-2 proteins for robust expression. Ultimately we proceeded with analysis of protein interaction data from 27 baits, and excluded Orf7b, which had high prey detection background (Fig. 1d).
Global analysis of SARS-CoV-2 host interacting proteins
Our affinity purification-mass spectrometry analysis identified 332 protein interactions between SARS-CoV-2 proteins and human proteins (Extended Data Fig. 1, Supplementary Tables 1, 2; also see Fig. 3). We studied the interacting human proteins in regards to their cell biology, anatomical expression patterns, expression changes during SARS-CoV-2 infection21 and in relation to other maps of pathogen interacting proteins19,22–30(Fig. 2a). For each of the viral proteins, we performed Gene Ontology enrichment analysis (Fig. 2b, Extended Data Fig. 2), identifying the major cell biological processes of the interacting proteins, including lipoprotein metabolism (S), nuclear transport (Nsp7), and biogenesis of ribonucleoprotein (Nsp8). To discover potential binding interfaces, we performed an enrichment for domain families within the interacting proteins of each viral bait (Extended Data Fig. 3). As examples, we note the enrichment of DNA polymerase domains in the interactors of Nsp1 and the enrichment of bromodomains and extra-terminal domain (BET) family for interactors of E (see also Figs. 3, 4). In line with the latter, the interactors of E are also enriched in genes annotated for binding to acetylated histones (Fig. 2b).
Figure 3: SARS-CoV-2 Protein-Protein Interaction Network.

In total, 332 high confidence interactions are represented between 26 SARS-CoV-2 proteins and their human interactors. Red diamonds represent a SARS-CoV-2 viral protein, interacting human host proteins are represented with circles, with drug targets in orange. Edge color is proportional to MiST score and edge thickness proportional to spectral counts. Physical interactions among host proteins are noted as thin black lines, protein complexes are highlighted in yellow, and proteins sharing the same biological process are highlighted in blue.
Figure 2: Global Analysis of SARS-CoV-2 Protein Interactions.

(a) Overview of global analyses performed. (b) Gene Ontology (GO) enrichment analysis performed on the human interacting proteins of each viral protein (Methods). The top GO term of each viral protein was selected for visualization. (c) Degree of differential protein expression for the human interacting proteins across human tissues. We obtained protein abundance values for the proteome in 29 human tissues and calculated the median level of abundance for the set of human interacting proteins (top 16 tissues shown). This median value was then compared with the distribution of abundance values for the full proteome in each tissue and summarized as a Z-score from which a p-value was calculated and adjusted for multiple tests. (d) Distribution of correlation of protein level changes during SARS-CoV-2 infection for pairs of viral-human proteins. (e) Significance of the overlap of human interacting proteins between SARS-CoV-2 and other pathogens.
Figure 4: The SARS-CoV-2 interactome reveals novel aspects of SARS-CoV-2 biology that can be targeted pharmacologically.
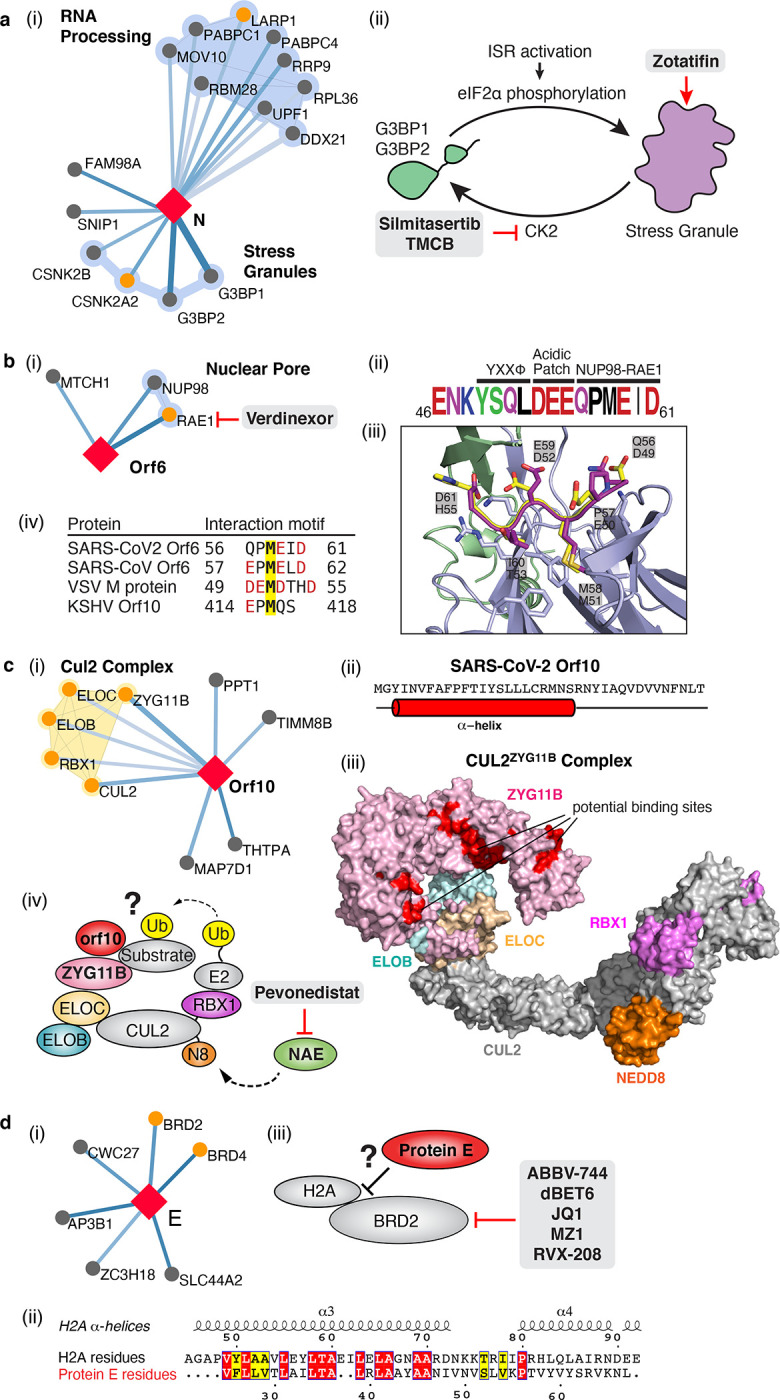
(a) Protein N targets stress granule proteins. (i) Protein N interactome. (ii) Model for therapeutic targeting of N interactions in the formation of stress granules (SGs). SGs are known to exhibit antiviral activity, with the integrative stress response (ISR) inducing eIF2α phosphorylation and SG formation, and Casein kinase II (CK2) disrupting and preventing the formation of SGs. By activating SG formation, or inhibiting CK2, the cellular environment could potentially shift to a more antiviral state. (b) Orf6 interacts with an interferon-inducible mRNA nuclear export complex. (i) Orf6 interactome including small molecule inhibitors for RAE. (ii) Annotated C-terminal sequence of SARS-CoV-2 Orf6, highlighting previously described trafficking motifs and the putative NUP98-RAE1 binding sequence. Colors indicate chemical properties of amino acids: polar (G,S,T,Y,C, green), neutral (Q,N, purple), basic (K, R, H, blue), acidic (D, E, red), and hydrophobic (A, V, L, I, P, W, F, M, black). (iii) SARS-CoV-2 Orf6 carboxy-terminal peptide modeled into the binding site of the VSV M protein-NUP98-RAE1 complex (PDB ID: 4OWR). Orf6 shown in dark purple, M protein in yellow, NUP98 in green, and RAE1 in light purple. Orf6 and M protein residues labeled. RAE1 hydrophobic residues contacting the key methionine and basic patch residues of RAE1 and NUP98 are shown. (iv) Putative NUP98-RAE1 interaction motifs present in proteins from several viral species. The consensus motif consists of negatively charged residues (red) surrounding a conserved methionine (yellow). (c) Orf10 interacts with the CUL2ZYG11B complex. (i) Orf10 interactome. (ii) The secondary structure of Orf10 contains an alpha helix motif. (iii) Surface representation of the homology model for CUL2ZYG11B complex, residues that are conserved amongst ZYG11B orthologues from various species are indicated in red are likely protein interaction surfaces for binding substrates and other proteins. (iv) A possible model of how Orf10 binds to the CUL2ZYG11B complex to hijack the complex for ubiquitination or viral restriction factors and how it can be targeted pharmacologically. (d) Envelope (E) interacts with bromodomain proteins. (i) E interactome. (ii) Sequence alignment of highlighted regions of E and Histone 2A (H2A). The positions with identical and similar amino acid residues are highlighted in red and yellow, respectively. Note the greater hydrophobicity of E may indicate a part of the alignment represents a transmembrane segment. (iii) Model of how E might mimic the BRD2 native interaction partner Histone 2A and how BRD2 can be targeted pharmacologically.
While the cells used for these AP-MS experiments, HEK-293T kidney cells, are permissive to SARS-CoV-2 infection31, they do not represent the primary physiological site of infection. Therefore, we asked whether the host proteins bound by SARS-CoV-2 might be specifically relevant to the virus’s typical environment, lung tissue. We tested if the interacting human proteins were preferentially highly expressed, at the protein level, in any of 29 human tissues32, which identified the lung as the tissue with the highest expression of the prey proteins relative to the average proteome (Fig. 2c). In accordance to this, when compared to overall RefSeq gene expression in the lung (median=3.198 TPM), the interacting proteins were more highly expressed (median=25.52 TPM, p=0.0007; t-test, Extended Data Fig. 4) and were also specifically enriched in lung expression relative to other tissues (Extended Data Fig. 5). These data support the hypothesis that SARS-CoV-2 preferentially hijacks proteins available in lung tissue and the degree of tissue specific expression may facilitate the drug targeting of host factors with fewer systemic side-effects; this observation is particularly compelling given its derivation from HEK293 cells, which are themselves not lung derived. To further study the relevance of our map, we analyzed the recently reported protein abundance changes during SARS-CoV-2 infection33. We calculated, when possible, the correlation between changes in abundance of viral proteins and their human interaction partners across the 4 timepoints reported. Interacting pairs tended to have stronger correlated changes than other pairs of viral-human proteins (Fig. 2d, KS test p-value=4.8e–05), arguing that the AP-MS derived interactions are of high relevance for the target tissue and the infection context. We compared our SARS-CoV-2 interaction map with those derived for 10 other pathogens (Fig. 2e), identifying West Nile Virus (WNV)23 and Mycobacterium tuberculosis (Mtb)27 as having the most similar host protein interaction partners. The association with Mtb is particularly interesting considering it also infects lung tissue.
Finally, we studied the evolutionary properties of the host proteins. Successful virus spread in a diverse population in theory could be facilitated by a reliance on conserved host molecular components, therefore we analyzed the conservation of the human proteins identified in the SARS-CoV-2 interactome. Relative to a control sample of genes, the 332 SARS-CoV-2 interacting human proteins had depleted missense and premature stop mutations in gnomAD34, indicating that they have reduced genetic variation in human populations (Extended Data Fig. 6).
The SARS-CoV-2 interactome reveals novel aspects of SARS-CoV-2 biology
Our study highlighted interactions between SARS-CoV-2 proteins and human proteins with a range of functions including DNA replication (Nsp1), epigenetic and gene expression regulators (Nsp5, Nsp8, Nsp13, E), vesicle trafficking (Nsp6, Nsp7, Nsp10, Nsp13, Nsp15, Orf3a, E, M, Orf8), lipid modification (Spike), RNA processing and regulation (Nsp8, N), ubiquitin ligases (Orf10), signaling (Nsp8, Nsp13, N, Orf9b), nuclear transport machinery (Nsp9, Nsp15, Orf6), cytoskeleton (Nsp1, Nsp13), mitochondria (Nsp4, Nsp8, Orf9c), and extracellular matrix (Nsp9) (Fig. 3).
A prominent number of interactions were related to lipid modifications and vesicle trafficking. Interestingly, the Spike protein (S) interacts with the GOLGA7-ZDHHC5 acyl-transferase complex, which likely mediates palmitoylation on its cytosolic tail (see also Appendix)35. Palmitoylation has been reported to facilitate membrane fusion by SARS-CoV Spike and suggests a potential target for therapeutic inhibition36. Interestingly, ZDHHC5 also has a published role in allowing anthrax toxin to enter cells, suggesting that inhibition of this enzyme could have broad utility37. Host interactions of Nsp8 (signal recognition particle), Orf8 (endoplasmic reticulum quality control), M (ER structural morphology proteins), Nsp13 (golgins) may facilitate the dramatic reconfiguration of ER/Golgi trafficking during coronavirus infection, and interactions in peripheral compartments by Nsp6 and M (vacuolar ATPase), Nsp7 (Rabs), Nsp10 (AP2), E (AP3), and Orf3a (HOPS) may also modify endomembrane compartments to favor coronavirus replication.
We identified protein-protein interactions with the main protease Nsp5, using both wild-type and catalytic dead (C145A) constructs. For wild-type Nsp5, we identified one high-confidence interaction, the epigenetic regulator histone deacetylase 2 (HDAC2), and predicted a cleavage site between the HDAC domain and the nuclear localization sequence, suggesting that Nsp5 may inhibit HDAC2 transport into the nucleus (Extended Data Fig. 7), potentially impacting the published functions of HDAC2 in mediating inflammation and interferon response38,39. We also identified an interaction of Nsp5 (C145A) with tRNA methyltransferase 1 (TRMT1), which is responsible for synthesis of the dimethylguanosine (m2,2G) base modification on both nuclear and mitochondrial tRNAs40. We predict TRMT1 is also cleaved by Nsp5, removing its zinc finger and nuclear localization signal and likely resulting in an exclusively mitochondrial localization (Extended Data Fig. 7).
SARS-CoV-2 interacts with multiple innate immune pathways
We identified a number of cellular proteins implicated in innate immune signaling that are targeted by several SARS-CoV-2 viral proteins. Interestingly, we found that Nsp13 interacts with two key players of IFN signaling pathway including TANK-binding kinase 1 (TBK1) and TANK-binding kinase 1-binding protein 1 (TBKBP1/SINTBAD). SINTBAD acts as a critical adaptor protein between TBK1 and IKKi and therefore mediates induction of IRF-dependent transcription41. Further, Nsp13 interacts with multiple proteins of the TLE family, which are known to modulate NF-κB inflammatory response42–44. RNF41/Nrdp1, an E3 ubiquitin ligase is targeted by Nsp15 protein which promotes activation of TBK1 and IRF3 and therefore increases type I interferon production45. Two other E3 ubiquitin ligases, TRIM59 and MIB1 regulate antiviral innate immune signaling and are usurped by Orf3a and Nsp9, respectively46,47. Orf9c protein was found to interact with multiple proteins that modulate IkB kinase and NF-kB signaling pathway including NLRX1, F2RL1, NDFIP248–50. We also found that Orf9b interacts with a mitochondrial import receptor, Tom70, which acts as an essential adaptor linking MAVS to TBK1/IRF3, resulting in the activation of IRF-351.
N targets stress granule protein G3BP1, an essential antiviral protein which is known to induce innate immune response through multiple mechanisms52–54. Common among coronaviridae is the manipulation of stress granules (SG) and related RNA biology, possibly leading to suppression of stress granules and host translation shutoff55. This functionality seems to benefit viral replication, as stress granules are inhibitory to replication of MERS-CoV56 and other viruses57. The SARS-CoV-2 nucleocapsid (N) interactome includes many host mRNA binding proteins, including the SG related factors G3BP1/2, the mTOR translational repressors LARP1, and the protein kinases CK2 (Fig. 4a). SGs are induced by protein kinase R (PKR)-mediated phosphorylation of eIF2α upon viral dsRNA recognition57. Promoting G3BP aggregation via the eIF4A inhibitor Zotatafin58,59 or reducing SG disassembly by Silmitasertib inhibition of CK260 warrant investigation for treatment of SARS-CoV-2. The mTOR inhibitor rapamycin disrupts the binding of LARP1 to mTORC161 and has been shown to reduce MERS infection by ~60% in vitro62, another drug that could be tested for repurposing.
Orf6 of SARS-CoV has been shown to play a role in antagonizing host interferon signaling63; we identified a novel, high-confidence interaction between SARS-CoV-2 Orf6 and NUP98-RAE1, an interferon-inducible mRNA nuclear export complex64 that is hijacked or degraded by multiple viruses, including VSV, Influenza-A, KSHV, and Polio, and is a restriction factor for Influenza-A infection58,60,62,65. The X-ray structure of VSV M protein complexed with NUP98-RAE166 reveals key binding interactions that include a buried methionine residue on the M-protein packing into a hydrophobic pocket in RAE1, as well as neighboring acidic residues interacting with a basic patch on the NUP98-RAE1 complex (Fig 4b). These binding features are also present in a conserved motif in the C-terminal region of SARS-CoV-2 Orf6 (Fig. 4b, Extended Data Fig. 8), providing a structural hypothesis for the observed SARS-CoV-2-NUP98-RAE1 interaction. Moreover, a peptide containing the binding region of the VSV M protein was previously shown to outcompete RNA binding to NUP98-RAE1, suggesting a role in interfering with mRNA export66. These observations suggest a viral strategy to target the RNA nuclear export activity of RAE1, potentially revealing a mode of interferon antagonism by SARS-CoV-2.
The novel Orf10 of SARS-CoV-2 interacts with a Cullin ubiquitin ligase complex
Viruses commonly hijack ubiquitination pathways for replication and pathogenesis67. The novel Orf10 of SARS-CoV-2 interacts with multiple members of a Cullin 2 (CUL2) RING E3 ligase complex (Fig. 4c), specifically the CUL2ZYG11B complex. ZYG11B, a substrate adapter of CUL2 that targets substrates with exposed N-terminal glycines for degradation65, is the highest scoring protein in the Orf10 interactome suggesting its direct interaction with Orf10. Orf10 may bind to the CUL2ZYG11B complex and hijack it for ubiquitination and degradation of restriction factors. The ubiquitin transfer to a substrate requires neddylation of CUL2 via NEDD8-activating enzyme (NAE), a druggable target that can be inhibited by the small molecule Pevonedistat68 (Fig. 4c).
SARS-CoV-2 envelope interacts with bromodomain proteins
Surprisingly, we find that the transmembrane protein E binds to the bromodomain-containing proteins BRD2 and BRD4 (Fig. 4d, Extended Data Fig. 9), potentially disrupting BRD-histone binding by mimicking histone structure. BRD2 is a member of the bromodomain and extra-terminal (BET) domain family whose members bind acetylated histones to regulate gene transcription69. The N-terminus of histone 2A shares local sequence similarity over an alpha-helix of approximately 15 residues, some of which are in a transmembrane segment, of Protein E (Fig. 4d). Moreover, this matching region of the histone is spanned by acetylated lysine residues shown to bind BRD270. This analysis may suggest that Protein E mimics the histone to disrupt its interaction with BRD2, thus inducing changes in host’s protein expression that are beneficial to the virus.
For a more comprehensive overview of the virus-host interactions we detected, see Supplemental Discussion.
Identification of existing drugs targeting SARS-CoV-2 human host factors.
To identify small molecules targeting human proteins in the SARS-CoV-2 interactome, we sought ligands known to interact with the human proteins, often directly but also by pathway and complexes, drawing on chemoinformatics databases and analyses (Methods). Molecules were prioritized by the statistical significance of the interaction between the human and viral proteins; by their status as approved drugs, investigational new drugs (INDs, “clinical” in Table 1a,b), or as preclinical candidates; by their apparent selectivity; and by their availability (for purchase availability notes, see Supplemental Tables 3 and 4). Chemoinformatics searches yielded 15 approved drugs, four investigational new drugs (clinical), and 18 pre-clinical candidates (Table 1a), while specialist knowledge revealed 12 approved drugs, 10 investigational new drugs (clinical), and 10 preclinical candidates (Table 1b). Of the 332 human targets that interact with the viral bait proteins with high significance, 63 have drugs/INDs/preclinical molecules that modulate them (Fig. 3). If we reduce our protein interaction score threshold slightly, we find an additional four human targets, revealing a total of 67 human targets (Supplementary Tables 3 and 4). The drug-human protein associations may be overlaid on top of our protein interaction network, highlighting potentially druggable host interactions (Fig. 5a).
Table 1a.
Literature-deriveda drugs and reagents that modulate SARS-Cov-2 interactors.
| Compound Name | Compound Structure | Human Gene | Viral Bait | Drug Status | Activity (nM) |
|---|---|---|---|---|---|
|
| |||||
| JQ1109 |
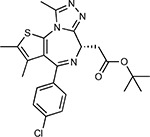
|
BRD2/4 | E | Pre-clinical | BRD inhibitor IC50 = 40–120 |
| RVX-208109 |
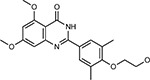
|
BRD2/4 | E | Clinical Trial | BRD inhibitor IC50 = 50–1800 |
| Silmitasertib110,111 |
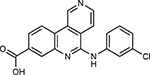
|
CSNK2A2 | N | Approved (Cancer) | CK2 inhibitor IC50 = 1 |
| TMCB112 |
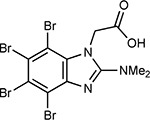
|
CSNK2A2 | N | Pre-clinical | Multi-targeted protein kinase inhibitor Ki = 21 |
| Apicidin113 |
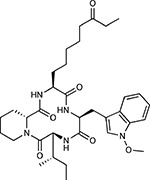
|
HDAC2 | Nsp5 | Pre-clinical | HDAC inhibitor IC50 = 120 |
| Valproic Acid114–116 |
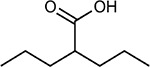
|
HDAC2 | Nsp5 | Approved (CNS diseases, Cancer) | HDAC2 inhibitor IC50 = 62,000 |
| Bafilomycin A1117 |
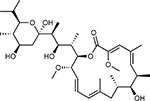
|
ATP6AP1 ATP6V1A | Nsp6 M | Pre-clinical | ATPase inhibitor IC50 = 100 |
| E-52862118 |
|
SIGMAR1 | Nsp6 | Clinical Trial | Sigma 1 antagonist IC50 = 17 |
| PD-144418119 |
|
SIGMAR1 | Nsp6 | Pre-clinical | Sigma 1 antagonist Ki = 0.8 |
| RS-PPCC120 |
|
SIGMAR1 | Nsp6 | Pre-clinical | Sigma 1 agonist Ki = 1.5 |
| PB28121 |
|
SIGMAR1 TMEM97 | Nsp6 Orf9c | Pre-clinical | Sigma 1/2 modulator IC50 = 15 |
| Haloperidol72 |
|
SIGMAR1 TMEM97 | Nsp6 Orf9c | Approved (CNS diseases) | Sigma 1/2 modulator Ki = 2–12 |
| Entacapone122,123 |
|
COMT | Nsp7 | Approved (Parkinson’s disease) | COMT inhibitor IC50 = 151 |
| Indomethacin124 |
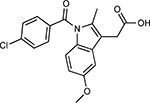
|
PTGES2 | Nsp7 | Approved (Inflammation, Pain) | Prostaglandin E2 synthase inhibitor IC50 = 750 |
| Metformin125 |
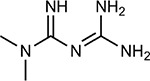
|
NDUFs | Nsp7 Orf9c | Approved (Diabetes) | MRC 1 inhibitor (indirect) |
| Ponatinib126 |
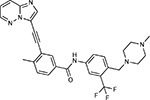
|
RIPK1 | Nsp12 | Approved (Cancer) | RIPK1 inhibitor IC50 = 12 |
| H-89127 |
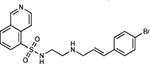
|
PRKACA | Nsp13 | Pre-clinical | Protein kinase A inhibitor KD = 48 |
| Merimepodib128 |
|
IMPDH2 | Nsp14 | Clinical Trial | IMPDH inhibitor Ki = 10 |
| Migalastat129 |
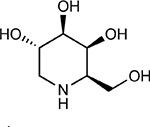
|
GLA | Nsp14 | Approved (Fabry disease) | α-Gal inhibitor IC50 = 40 |
| Mycophenolic acid130 |
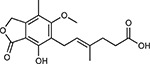
|
IMPDH2 | Nsp14 | Approved (Organ rejection) | IMPDH inhibitor IC50 = 20 |
| Ribavirin131 |
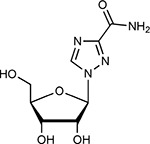
|
IMPDH2 | Nsp14 | Approved (Viral infection) | IMPDH inhibitor IC50 = 100–250 |
| XL413132 |
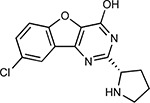
|
DNMT1 | Orf8 | Clinical Trial | CDC7 inhibitor IC50 = 3.4 |
| CCT 365623133 |
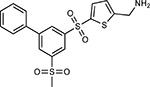
|
LOX | Orf8 | Pre-clinical | LOXL2 inhibitor IC50 = 1500 |
| Midostaurin134 |
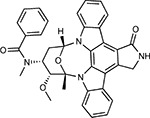
|
MARK2/3 | Orf9b | Approved (Cancer) | Protein kinase inhibitor MARK1 KD = 100 MARK3 KD = 23 |
| Ruxolitinib135 |
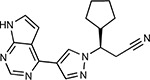
|
MARK2/3 | Orf9b | Approved (Myelofibrosis) | Protein kinase inhibitor MARK1 KD = 660 MARK3 KD > 10000 |
| ZINC1775962367136 |
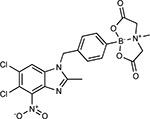
|
DCTPP1 | Orf9b | Pre-clinical | dCTPase inhibitor IC50 = 47 |
| ZINC4326719137 |
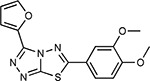
|
DCTPP1 | Orf9b | Pre-clinical | DCTPP1 inhibitor IC50 = 19 |
| ZINC4511851138 |
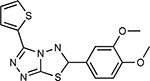
|
DCTPP1 | Orf9b | Pre-clinical | dCTPase inhibitor IC50 = 20 |
| ZINC95559591139 |
|
MARK3 TBK1 | Orf9b Nsp13 | Pre-clinical | Protein kinase inhibitor MARK3 IC50 = 12 TBK1 IC50 = 6 |
| AC-55541140 |
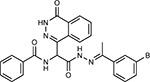
|
F2RL1 | Orf9c | Pre-clinical | PAR agonist pEC50 = 6.7 |
| AZ8838141 |
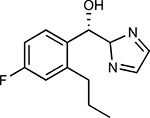
|
F2RL1 | Orf9c | Pre-clinical | PAR antagonist IC50 = 344 |
| Daunorubicin142 |
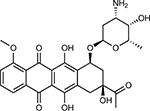
|
ABCC1 | Orf9c | Approved (Cancer) | Topoisomerase inhibitor Ki = 70 |
| GB110143 |
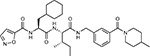
|
F2RL1 | Orf9c | Pre-clinical | PAR2 agonist EC50 = 280 |
| S-verapamil144 |
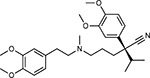
|
ABCC1 | Orf9c | Approved (Hypertension) | Ca2+ channel inhibitor and drug efflux transporter inhibitor Ki = 113 |
| AZ3451141 |
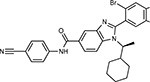
|
F2RL1 | Orf9c | Pre-clinical | PAR2 negative allosteric modulator pKD = 15 |
Table 1b.
Expert-identifieda drugs and reagents that modulate SARS-CoV-2 interactors.
| Compound Name | Compound Structure | Human Gene/Process | Viral Bait | Drug Status | Activity (nM) |
|---|---|---|---|---|---|
|
| |||||
| ABBV-74469 |
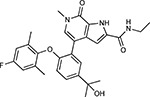
|
BRD2/4 | E | Clinical Trial | BRD inhibitor KD = 2.1 |
| dBET6146 |
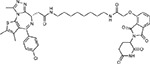
|
BRD2/4 | E | Pre-clinical | Degrades BRD proteins IC50 < 10000 |
| MZ1147 |
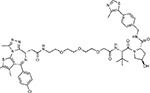
|
BRD2/4 | E | Pre-clinical | Degrades BRD proteins KD = 120–228 |
| CPI-0610148 |
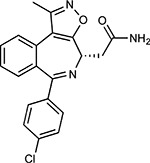
|
BRD2/4 | E | Clinical Trial | BRD2/4 inhibitor BRD2 IC50 = 25 BRD4 IC50 = 18 |
| Sapanisertib61,149 |
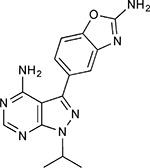
|
LARP1 | N | Clinical Trial | mTOR inhibitor IC50 = 1 |
| Rapamycin61,150 |
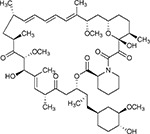
|
LARP1 FKBP15 FKBP7/10 | N Nsp2 Orf8 | Approved (Organ rejection) | mTOR inhibitor (with FKBP) IC50 = 2.0 |
| Zotatifin151 |
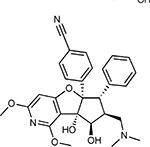
|
EIF4E2/H | Nsp2 | Clinical Trial | EIF4a inhibitor IC50 = 1.5 |
| Verdinexor152 |
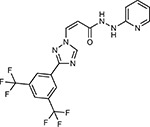
|
NUPs RAE1 | Nsp4 Nsp9 Orf6 | Clinical Trial | XPO1 nuclear export inhibitor IC50 = 960 |
| Chloroquine153 |
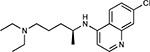
|
SIGMAR1 | Nsp6 | Approved (Malaria) | Sigma 1 binder Ki = 100 |
| Dabrafenib154 |
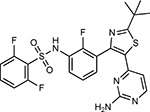
|
NEK9 | Nsp9 | Approved (Cancer) | NEK9 inhibitor IC50 = 1 |
| WDB002 |
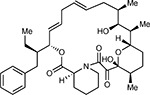
|
CEP250 | Nsp13 | Clinical Trial | CEP250 inhibitor (with FKBP) Kd = 0.29 |
| Sanglifehrin A73 |
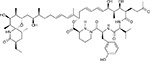
|
IMPDH2 | Nsp14 | Pre-clinical | PPIA-IMPDH2 modulator PPIA KD = 0.2 IDPDH2 Binding EC50 = 11.5 (with PPIA) |
| FK-506155 |
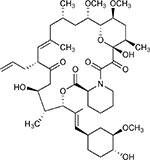
|
FKBP7 FKBP10 | Orf8 | Approved (Organ rejection) | FKBP binder |
| Pevonedistat68 |
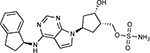
|
CUL2 | Orf10 | Clinical Trial | NEDD8-activating enzyme inhibitor IC50 = 4.7 |
| Ternatin 4156 |
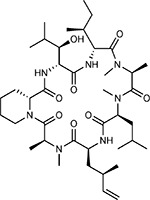
|
Translation | Pre-clinical | eEF1A inhibitor IC50 = 71 | |
| 4E2RCat157 |
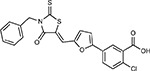
|
Translation | Pre-clinical | eIF4E/G PPI inhibitor IC50 = 13500 | |
| Tomivosertib158,159 |
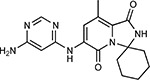
|
Translation | Clinical Trial | MNK1/2 inhibitor IC50 = 2.4 | |
| Compound 2160 |
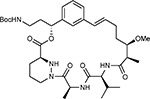
|
Viral Transcription | Pre-clinical | Cyclophilin inhibitor KD = 24 | |
| Compound 10161 |
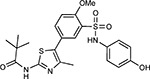
|
Viral Transcription | Pre-clinical | PI4K-IIIβ inhibitor IC50 = 3.4 | |
| PS306130 |
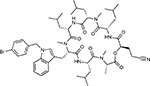
|
ER protein processing | Pre-clinical | Sec61 inhibitor IC50 = 20–500 | |
| IHVR-19029162,163 |
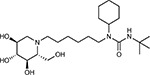
|
ER protein processing | Clinical Trial | Antiviral activity IC50 = 1200 | |
| Captopril164 |
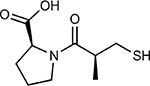
|
Cell Entry | Approved (Hypertension) | ACE inhibitor Ki = 3 | |
| Lisinopril165 |
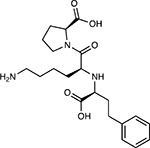
|
Cell Entry | Approved (Hypertension) | ACE inhibitor Ki = 0.27 | |
| Camostat166,167 |
|
Cell Entry | Approved (Pancreatitis) | Serine protease 1 inhibitor IC50 < 1000 | |
| Nafamostat166,168 |
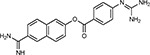
|
Cell Entry | Approved (Anticoagulant) | Serine protease 1 inhibitor IC50 = 100 | |
| Chloramphenicol169 |
|
Mitochondrial ribosome | Approved (Bacterial infection) | Mitochondrial ribosome inhibitor IC50 = 7400 | |
| Tigecycline170 |
|
Mitochondrial ribosome | Approved (Bacterial infection) | Mitochondrial ribosome inhibitor IC50 = 3300 | |
| Linezolid171 |
|
Mitochondrial ribosome | Approved (Bacterial infection) | Mitochondrial ribosome inhibitor IC50 = 16000 | |
These molecules derive from expert analysis of human protein interactors of SARS-Co-V2 and reagents and drugs that modulate them; not readily available from the chemoinformatically-searchable literature.
Figure 5: Drug-human target network.
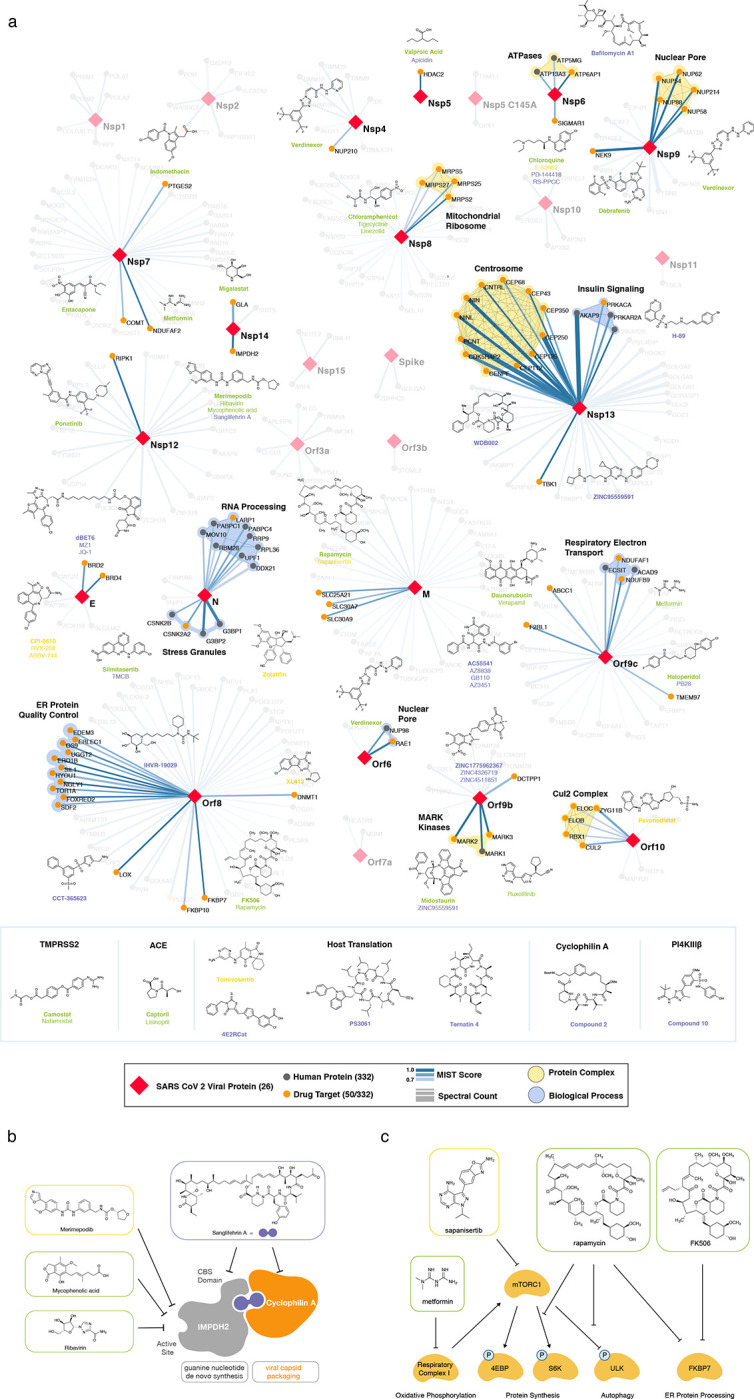
(a) Significant interactions identified by AP-MS between SARS-CoV-2 baits (red diamonds) and human prey proteins (orange circles) are shown as in Fig 3. Chemoinformatic and expert analysis identified FDA approved drugs (green), clinical candidates (yellow), and preclinical candidates (purple) with experimental activities against the host proteins and processes, with representative chemicals shown. (b) Inosine Monophosphate Dehydrogenase 2 (IMPDH2) regulates de novo nucleic acid biosynthesis. It is a target for proliferative diseases including cancer81 and autoimmune disorders, for instance by the approved drug mycophenolic acid82, and as a broad spectrum antiviral by Ribavirin83. While Ribavirin has activity against SARS in vitro84, it has low tolerability, something that might be addressed by the more selective Merimepodib, which is in phase II clinical trials85. (c) The mammalian target of Rapamycin (mTOR) pathway is a master regulator of cell proliferation and autophagy, which viruses including Influenza A are known to modulate86,87. Several proteins that interact with SARS-CoV-2 baits, including components of the Respiratory complex 1 by Nsp7, Nsp12, and Orf9c, the leucine importer B(0)AT2 (SLC6A15)88,89 by Nsp6 and LARP1 by N (not shown). In addition to Rapamycin, the mTOR pathway can be indirectly modulated by metformin, a widely prescribed diabetes drug, and by Sapanisertib, a drug in clinical trials for solid tumors61.
There are several mechanistically interesting, and potentially disease-relevant drug-target interactions revealed in the chemoinformatic network (Fig. 5a). Among them, the well-known chemical probe, Bafilomycin A1, is a potent inhibitor of the V1-ATPase, subunits of which interact with Nsp6 and M. Bafilomycin’s inhibition of this cotransporter acts to prevent the acidification of the lysosome, inhibiting autophagy and endosome trafficking pathways, which may impact the viral life-cycle. Similarly, drugs exist to target several well-known epigenetic regulators prominent among the human interactors, including HDAC2, BRD2 and BRD4, which interact with viral proteins nsp5 and E, respectively (Figs. 3 and 5a). The approved drug Valproic acid (an anticonvulsant) and the pre-clinical candidate Apicidin inhibit HDAC2 with affinities of 62 μM and 120 nM, respectively. Clinical compounds ABBV-744 and CPI-0610 act on BRD2/4, with an affinity of 2 nM or 39 nM, respectively -- several preclinical compounds also target bromodomain-containing proteins (Table 1a,b). As a final example, we were intrigued to observe that the SARS-CoV-2 Nsp6 protein interacts with the Sigma receptor, which is thought to regulate ER stress response71. Similarly, the Sigma2 receptor interacted with the vial protein orf9. Both Sigma1 and Sigma2 are promiscuous receptors that interact with many non-polar, cationic drugs. We prioritized several of these drugs based on potency or potential disease relevance, including the antipsychotic Haloperidol, which binds in the low nM range to both receptors72, and Chloroquine, which is currently in clinical trials for COVID-19 and has mid-nM activity vs the Sigma1 receptor, and low μM activity against the Sigma2 receptor. Because many patients are already treated with drugs that have off-target impact on Sigma receptors, associating clinical outcomes accompanying treatment with these drugs may merit investigation, a point to which we return. Finally, in addition to the druggable host factors, a few of which we have highlighted here, the SARS-CoV-2-human interactome reveals many traditionally “undruggable” targets. Among these, for instance, are components of the centriole such as CEP250, which interacts with the viral Nsp13. Intriguingly, a very recent patent disclosure revealed a natural product, WDB002, that directly and specifically targets CEP250. As a natural product, WDB002 would likely be harder to source than the molecules on which we have focused on here, but may well merit investigation. Similarly, other “undruggable” targets may be revealed to have compounds that could usefully perturb the viral-human interaction network, and act as leads to therapeutics.
Beyond direct interactions, several drug-pathway interactions seemed noteworthy. The human purine biosynthesis enzyme Inosine-5′-monophosphate dehydrogenase (IMPDH2) interacts with the viral protein nsp14. Several chemically diverse compounds inhibit IMPDH2, including the clinically approved mycophenolic acid (20 nM), the approved antiviral drug ribavirin (200 nM), and the investigational new drug Merimepodib (10 nM) (Table 1a). Intriguingly, the preclinical molecule Sanglifehrin A (Table 1b) is known to act as a molecular glue linking IMPDH with cyclophilin A (Fig. 5b)73, which itself is implicated in viral capsid packaging, even though it itself is not a human “prey” in the viral-human protein interactome. Similarly, direct viral-human interactions with proteins regulated by the mTORC1 pathway, such as LARP1, and FKBP7, which interact with the viral N and Orf8 proteins, led us to inhibitors of mTORC1, even though that kinase itself is not found to directly interact with a viral protein (Fig. 5c). Sapanisertib and rapamycin are low nM inhibitors of mTORC1, while metformin is an indirect modulator of this protein complex.
Discussion
We have used affinity purification-mass spectrometry to identify 332 high-confidence SARS-CoV-2-human PPIs. We find the viral proteins connected to a wide array of biological processes, including protein trafficking, translation, transcription and ubiquitination regulation. Using a combination of a systematic chemoinformatic drug search with a pathway centric analysis, we uncovered close to 70 different drugs and compounds, including FDA approved drugs, compounds in clinical trials as well as preclinical compounds, targeting parts of the resulting network. We are currently testing these compounds for antiviral activity and encourage others to do the same as well as extract insights from the map that could have therapeutic value.
More generally, this proteomic/chemoinformatic analysis is not only identifying drug and clinical molecules that might perturb the viral-human interactome, it gives these potentially therapeutic perturbations a mechanistic context. Among those that may be infection relevant are the inhibition of lysosomal acidification and trafficking with Bafilomycin A1, via inhibition of V-ATPase74, and modulation of the ER stress and the protein unfolding response pathway by targeting the Sigma1 and Sigma2 receptor by drugs like haloperidol (Fig. 5a, Tables 1a,b). Indeed, several of the human proteins in the interactome are targeted by drugs that have emerged phenotypically as candidate therapeutics for treating COVID-19, such as chloroquine75,76. While we do not pretend to have identified the molecular basis of chloroquine’s putative activity, we do note that this drug targets the Sigma1 and Sigma2 receptors at mid-nM and low μM concentrations, respectively. Similarly, antibiotics like azithromycin have also been mooted as treatments for COVID-19. While this too remains to be demonstrated, we note that Azithromycin has off-target activity against human mitochondrial ribosomes, components of which interact with the SARS-CoV-2 Nsp8 protein (MRPS5, MRPS27, MRPS2, and MRPS25). Other antibiotics that also have an off-target effect on mitochondrial ribosomes, such as chloramphenicol, tigecycline, and Linezolid77,78 may also merit study for efficacy. Indeed, this logic may be extended. Many COVID-19 patients will be on the drugs identified here, treating pre-existing conditions. It may be useful to correlate clinical outcomes with the taking of these drugs, cross-referencing with the networks described here. In some senses, this is already occurring phenomenologically, leading to concerns about ACE inhibitors such as captopril and enalapril, and for NSAIDs. What this study provides is a systematic schema for clinical/drug associations going forward, giving them a mechanistic context that allows investigators to seek them directly.
Systematic validation using genetic-based approaches79,80 will be key to determine the functional relevance of these interactions and if the human proteins are being used by the virus or are fighting off infection, information that would inform future pharmacological studies. It is important to note that pharmacological intervention with the agents we identified in this study could be either detrimental or beneficial for infection. For instance, the HDAC2 inhibitors may compound the potential action of the Nsp5 protease to hydrolyze this human protein. Future work will involve generation of protein-protein interaction maps in different human cell types, as well as bat cells, and the study of related coronaviruses including SARS-CoV, MERS-CoV and the less virulent OC435, data that will allow for valuable cross-species and viral evolution studies. Targeted biochemical and structural studies will also be crucial for a deeper understanding of the viral-host complexes, which will inform more targeted drug design.
Along with SARS-CoV-2, we have previously utilized global affinity purification-mass spectrometry (AP-MS) analysis to map the host-pathogen interfaces of a number of human pathogens including Ebola22, Dengue30, Zika30, Herpesvirus29, Hepatitis C28, Tuberculosis27, Chlamydia26 , Enteroviruses25, HIV19, HPV24, and West Nile Fever23. Excitingly, we have uncovered both shared and unique mechanisms in which these pathogens co-opt the host machinery during the course of infection. Although host-directed therapy is often not explored for combatting pathogenic infections, it would be interesting to use this information to identify host factors that could serve as targets that would harbor pan-pathogenic activity so that when the next virus undergoes zoonosis, we will have treatment options available.
MATERIALS AND METHODS
Genome annotation.
The genbank sequence for SARS-CoV-2 isolate 2019-nCoV/USA-WA1/2020, accession MN985325, was downloaded on January 24, 2020. In total, we identified 29 open reading frames and proteolytically mature proteins encoded by SARS-CoV-21,16. Proteolytic products resulting from Nsp3 and Nsp5-mediated cleavage of the Orf1a / Orf1ab polyprotein were predicted based on the protease specificity of SARS-CoV proteases90, and 16 predicted nonstructural proteins (Nsps) were subsequently cloned (Nsp1-Nsp16). For the proteases Nsp3 (papain-like / Plpro) and Nsp5 (3Clike / 3CLpro), we also designed catalytic dyad/triad mutants: Nsp3 C857A91 and Nsp5 C145A 92,93. Open reading frames at the 3’ end of the viral genome annotated in the original genbank file included 4 Structural proteins: S, E, M, N, and the additional open reading frames Orf3a, Orf6, Orf7a, Orf8, and Orf10. Based on analysis of open reading frames in the genome and comparisons with other annotated SARS-CoV open reading frames, we annotated a further four open reading frames: Orf3b, Orf7b, Orf9b, and Orf9c.
Cell culture.
HEK293T cells were cultured in Dulbecco’s Modified Eagle’s Medium (Corning) supplemented with 10% Fetal Bovine Serum (Gibco, Life Technologies) and 1% Penicillin-Streptomycin (Corning) and maintained at 37°C in a humidified atmosphere of 5% CO2.
Transfection.
For each affinity purification, ten million HEK293T cells were plated per 15-cm dish and transfected with up to 15 μg of individual Strep-tagged expression constructs after 20–24 hours. Total plasmid was normalized to 15 μg with empty vector and complexed with PolyJet Transfection Reagent (SignaGen Laboratories) at a 1:3 μg:μl ratio of plasmid to transfection reagent based on manufacturer’s recommendations. After more than 38 hours, cells were dissociated at room temperature using 10 ml Dulbecco’s Phosphate Buffered Saline without calcium and magnesium (D-PBS) supplemented with 10 mM EDTA for at least 5 minutes and subsequently washed with 10 ml D-PBS. Each step was followed by centrifugation at 200 ×g, 4°C for 5 minutes. Cell pellets were frozen on dry ice and stored at − 80°C. At least three biological replicates were independently prepared for affinity purification.
Affinity purification.
Frozen cell pellets were thawed on ice for 15–20 minutes and suspended in 1 ml Lysis Buffer [IP Buffer (50 mM Tris-HCl, pH 7.4 at 4°C, 150 mM NaCl, 1 mM EDTA) supplemented with 0.5% Nonidet P 40 Substitute (NP40; Fluka Analytical) and cOmplete mini EDTA-free protease and PhosSTOP phosphatase inhibitor cocktails (Roche)]. Samples were then frozen on dry ice for 10–20 minutes and partially thawed at 37°C before incubation on a tube rotator for 30 minutes at 4°C and centrifugation at 13,000 ×g, 4°C for 15 minutes to pellet debris. After reserving 50 μl lysate, up to 48 samples were arrayed into a 96-well Deepwell plate for affinity purification on the KingFisher Flex Purification System (Thermo Scientific) as follows: MagStrep “type3” beads (30 μl; IBA Lifesciences) were equilibrated twice with 1 ml Wash Buffer (IP Buffer supplemented with 0.05% NP40) and incubated with 0.95 ml lysate for 2 hours. Beads were washed three times with 1 ml Wash Buffer and then once with 1 ml IP Buffer. To directly digest bead-bound proteins as well as elute proteins with biotin, beads were manually suspended in IP Buffer and divided in half before transferring to 50 μl Denaturation-Reduction Buffer (2 M urea, 50 mM Tris-HCl pH 8.0, 1 mM DTT) and 50 μl 1x Buffer BXT (IBA Lifesciences) dispensed into a single 96-well KF microtiter plate, respectively. Purified proteins were first eluted at room temperature for 30 minutes with constant shaking at 1,100 rpm on a ThermoMixer C incubator. After removing eluates, on-bead digestion proceeded (below). Strep-tagged protein expression in lysates and enrichment in eluates were assessed by western blot and silver stain, respectively. The KingFisher Flex Purification System was placed in the cold room and allowed to equilibrate to 4°C overnight before use. All automated protocol steps were performed using the slow mix speed and the following mix times: 30 seconds for equilibration/wash steps, 2 hours for binding, and 1 minute for final bead release. Three 10 second bead collection times were used between all steps.
On-bead digestion.
Bead-bound proteins were denatured and reduced at 37°C for 30 minutes and after bringing to room temperature, alkylated in the dark with 3 mM iodoacetamide for 45 minutes and quenched with 3 mM DTT for 10 minutes. Proteins were then incubated at 37°C, initially for 4 hours with 1.5 μl trypsin (0.5 μg/μl; Promega) and then another 1–2 hours with 0.5 μl additional trypsin. To offset evaporation, 15 μl 50 mM Tris-HCl, pH 8.0 were added before trypsin digestion. All steps were performed with constant shaking at 1,100 rpm on a ThermoMixer C incubator. Resulting peptides were combined with 50 μl 50 mM Tris-HCl, pH 8.0 used to rinse beads and acidified with trifluoroacetic acid (0.5% final, pH < 2.0). Acidified peptides were desalted for MS analysis using a BioPureSPE Mini 96-Well Plate (20mg PROTO 300 C18; The Nest Group, Inc.) according to standard protocols.
Mass spectrometry data acquisition and analysis.
Samples were re-suspended in 4% formic acid, 2% acetonitrile solution, and separated by a reversed-phase gradient over a nanoflow C18 column (Dr. Maisch). Each sample was analyzed on two different mass spectrometers. First, a 75 min acquisition, in which peptides were directly injected via a Easy-nLC 1200 (Thermo) into a Q-Exactive Plus mass spectrometer (Thermo), with all MS1 and MS2 spectra collected in the orbitrap. For all acquisitions, QCloud was used to control instrument longitudinal performance during the project94. All proteomic data was searched against the human proteome (uniprot reviewed sequences downloaded February 28th, 2020), EGFP sequence, and the SARS-CoV-2 protein sequences using the default settings for MaxQuant95,96. Detected peptides and proteins were filtered to 1% false discovery rate in MaxQuant, and identified proteins were then subjected to protein-protein interaction scoring with both SAINTexpress20 and MiST19,97. We applied a two step filtering strategy to determine the final list of reported interactors which relied on two different scoring stringency cutoffs. In the first step, we chose all protein interactions that possess a MiST score ≥ 0.7, a SAINTexpress BFDR ≤ 0.05 and an average spectral count ≥ 2. For all proteins that fulfilled these criteria we extracted information about stable protein complexes they participate in from the CORUM98 database of known protein complexes. In the second step we then relaxed the stringency and recovered additional interactors that (1) form complexes with interactors determined in filtering step 1 and (2) fulfill the following criteria: MiST score ≥ 0.6, SAINTexpress BFDR ≤ 0.05 and average spectral counts ≥ 2. Proteins that fulfilled filtering criteria in either step 1 or step 2 were considered to be HC-PPIs and visualized with cytoscape99. Using this filtering criteria, nearly all of our baits recovered a number of HC-PPIs in close alignment with previous datasets reporting an average of ~6 PPIs per bait100. However, for a subset of baits (Orf8, Nsp8, Nsp13, and orf9c) we observed a much higher number of PPIs passing these filtering criteria. For these four baits, the MiST scoring was instead performed using an larger in-house database of 87 baits that were prepared and processed in an analogous manner to this SARS-CoV-2 dataset. This was done to provide a more comprehensive collection of baits for comparison, to minimize the classification of non-specifically binding background proteins as HC-PPIs. All mass spectrometry raw data and search results files have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD018117101,102. PPI networks have also been uploaded to NDEx.
Gene Ontology Over-representation Analysis.
The targets of each bait were tested for enrichment of Gene Ontology (GO Biological Process) terms. The over-representation analysis (ORA) was performed using the enricher function of clusterProfiler package in R with default parameters. The gene ontology terms were obtained from the c5 category of Molecular Signature Database (MSigDBv6.1). Significant GO terms (1% FDR) were identified and further refined to select non-redundant terms. In order to select non-redundant gene sets, we first constructed a GO term tree based on distances (1-Jaccard Similarity Coefficients of shared genes) between the significant terms. The GO term tree was cut at a specific level (h=0.99) to identify clusters of non-redundant gene sets. For a bait with multiple significant terms belonging to the same cluster, we selected the broadest term i.e. largest gene set size.
Virus Interactome Similarity Analysis.
Interactome similarity was assessed by comparing the number of shared human interacting proteins between pathogen pairs, using a hypergeometric test to calculate significance. The background gene set for the test consisted of all unique proteins detected by mass spectrometry across all pathogens (N=10,181 genes).
Orf6 peptide modeling.
The proposed interaction between Orf6 and the NUP98-RAE1 complex was modeled in PyRosetta 4103 (release v2020.02-dev61090) using the crystal structure of Vesicular stomatitis virus matrix (M) protein bound to NUP98-RAE1 as a template66 (PDB 4OWR downloaded from the PDB-REDO server104). The M protein chain (C) was truncated after residue 54 to restrict the model to the putative interaction motif in Orf6 (M protein residues 49–54, sequence DEMDTH). These residues were mutated to the Orf6 sequence, QPMEID, using the mutate_residue function in the module pyrosetta.toolbox, without repacking at this initial step. After all six residues were mutated, the full model was relaxed to a low energy conformation using the FastRelax protocol in the module pyrosetta.rosetta.protocols.relax. FastRelax was run with constraints to starting coordinates and scored with the ref2015 score function. The resulting model was inspected for any large energetic penalties associated with the modeled peptide residues or those NUP98 and RAE1 residues interacting with the peptide, and was found to have none. The model was visualized in PyMOL (The PyMOL Molecular Graphics System, Version 2.3.4 Schrödinger, LLC.).
CUL2ZYG11B homology model generation.
The CRL2ZYG11B homology model was built with Swissmodel105 and Modeller106 by using the homology template of each domain from PDB database (PDB codes: 4b8o, 5jh5,1g03, and 6r7n). The ZYG11B model has two structured domains: a leucine rich repeat (LRR) and Armadillo Repeat (ARM) at the N and C-terminus respectively. The linker between each domain was not modelled due to high flexibility between residues 32 to 49 and residues 304 to 322. Putative protein interaction surfaces on ZYG11B were modelled based on contiguous surface exposed residues that are conserved in ZYG11B orthologues from C. elegans to H. sapiens (ZY11B_HUMAN; ZY11B_MOUSE; F1M8P2_RAT; ZYG11_XENLA; ZYG11_DANRE; ZYG11_CAEEL) and located at typical substrate binding sites in the homologous structures of LRR and ARM domain co-complexes.
Alignment of Protein E and Histone H2A.
In order to align protein E and histone H2A, the structure of the protein E SARS-CoV homolog (PDB ID: 2MM4) was compared to the human nucleosome structure (PDB ID: 6K1K). Protein E was structurally aligned to the histone subunits using Pymol’s “align” function (https://pymolwiki.org/index.php/Align). Align performs a sequence alignment followed by a structural superposition, and then carries out zero or more cycles of refinement in order to reject structural outliers found during the fit. The best superposition was obtained for H2A residues 49–60 & 63–70 and Protein E residues 25–44 at an RMSD of 2.8Å, as reported in Figure 4d.
Chemoinformatic Analysis of SARS-CoV2 Interacting Partners.
To identify drugs and reagents that modulate the 332 host factors interacting with SARS-CoV-2-HEK293T (MiST >= 0.70), we used two approaches: 1) a chemoinformatic analysis of open-source chemical databases and 2) a target- and pathway-specific literature search, drawing on specialist knowledge within our group. Chemoinformatically, we retrieved 2,472 molecules from the IUPHAR/BPS Guide to Pharmacology (2020–3-12) that interacted with 30 human “prey” proteins (38 approved, 71 in clinical trials), and found 10,883 molecules (95 approved, 369 in clinical trials) from the ChEMBL25 database107 (Supplementary Tables 5, 6). For both approaches, molecules were prioritized on their FDA approval status, activity at the target of interest better than 1 μM, and commercial availability, drawing on the ZINC database108. FDA approved molecules were prioritized except when clinical candidates or preclinical research molecules had substantially better selectivity or potency on-target. In some cases, we considered molecules with indirect mechanisms of action on the general pathway of interest based solely on literature evidence (e.g., captopril modulates ACE2 indirectly via its direct interaction with Angiotensin Converting Enzyme, ACE). Finally, we predicted 6 additional molecules (2 approved, 1 in clinical trials) for proteins with MIST scores between 0.7–0.6 to viral baits (Supplemental Tables 3 and 4). Complete methods can be found here (www.github.com/momeara/BioChemPantry/vignette/COVID19).
ACKNOWLEDGEMENTS
We thank Joshua Sarlo for his artistic contributions to the manuscript.
This research was funded by grants from the National Institutes of Health (P50AI150476, U19AI135990, U19AI135972, R01AI143292, R01AI120694, P01A1063302, and R01AI122747 to N.J.K.; R35GM122481 to B.K.S.; 1R01CA221969 and 1R01CA244550 to K.M.S.; K08HL124068 to J.S.C.; 1F32CA236347-01 to J.E.M.); funding from F. Hoffmann-La Roche and Vir Biotechnology and gifts from The Ron Conway Family. K.M.S is an investigator of the Howard Hughes Medical Institute.
Abbreviations:
- HC-PPIs
High confidence protein-protein interactions
- PPIs
protein-protein interaction
- AP-MS
affinity purification-mass spectrometry
- COVID-19
Coronavirus Disease-2019
- ACE2
angiotensin converting enzyme 2
- Orf
open reading frame
- Nsp3
papain-like protease
- Nsp5
main protease
- Nsp
nonstructural protein
- TPM
transcripts per million
Footnotes
Conflicts:
The Krogan Laboratory has received research support from Vir Biotechnology and F. Hoffmann-La Roche. Kevan Shokat has consulting agreements for the following companies involving cash and/or stock compensation: Black Diamond Therapeutics, BridGene Biosciences, Denali Therapeutics, Dice Molecules, eFFECTOR Therapeutics, Erasca, Genentech/Roche, Janssen Pharmaceuticals, Kumquat Biosciences, Kura Oncology, Merck, Mitokinin, Petra Pharma, Qulab Inc. Revolution Medicines, Type6 Therapeutics, Venthera, Wellspring Biosciences (Araxes Pharma). Jack Taunton is a cofounder and shareholder of Global Blood Therapeutics, Principia Biopharma, Kezar Life Sciences, and Cedilla Therapeutics. Jack Taunton and Phillip P. Sharp are listed as inventors on a provisional patent application describing PS3061.
Supplementary Information is available for this paper.
Reprints and permissions information is available at www.nature.com/reprints
The authors have not filed for patent protection on the SARS-CoV-2 host interactions or the use of predicted drugs for treating COVID-19 to ensure all the information is freely available to accelerate the discovery of a treatment.
SUPPLEMENTARY INFORMATION
Supplementary table 1: Scoring results for all baits and all proteins
Supplementary table 2: SARS-CoV 2 high confidence interactors
Supplementary table 3: Literature-derived drugs and reagents that modulate SARS-Cov-2 interactors. Drug-target associations drawn from chemoinformatic searches of the literature, including information about purchasability
Supplementary table 4: Expert-identified drugs and reagents that modulate SARS-CoV-2 interactors. Drug-target associations drawn from expert knowledge of human protein interactors of SARS-Co-V2 and reagents and drugs that modulate them; not readily available from the chemoinformatically-searchable literature
Supplementary table 5: Raw chemical associations to prey proteins IUPHAR/BPS Guide to Pharmacology (2020–3-12)
Supplementary table 6: Raw chemical associations to prey proteins ChEMBL25
Supplementary Methods: Computational methods used to propagate tables and supplemental figures
Supplementary Discussion: In depth look at the SARS-CoV-2 individual bait subnetworks
REFERENCES
- 1.Wu F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Novel Coronavirus (2019-nCoV) situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports.
- 3.Wang C., Horby P. W., Hayden F. G. & Gao G. F. A novel coronavirus outbreak of global health concern. The Lancet vol. 395 470–473 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu N. et al. China Novel Coronavirus Investigating and Research Team. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su S. et al. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 24, 490–502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wit E. de, de Wit E., van Doremalen N., Falzarano D. & Munster V. J. SARS and MERS: recent insights into emerging coronaviruses. Nature Reviews Microbiology vol. 14 523–534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson R. M., Heesterbeek H., Klinkenberg D. & Déirdre Hollingsworth T. How will country-based mitigation measures influence the course of the COVID-19 epidemic? The Lancet (2020) doi: 10.1016/s0140-6736(20)30567-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gates B. Responding to Covid-19 — A Once-in-a-Century Pandemic? New England Journal of Medicine (2020) doi: 10.1056/nejmp2003762. [DOI] [PubMed] [Google Scholar]
- 9.Zarin D. A., Tse T., Williams R. J., Califf R. M. & Ide N. C. The ClinicalTrials. gov results database—update and key issues. N. Engl. J. Med. 364, 852–860 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sheahan T. P. et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 11, 222 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison C. Coronavirus puts drug repurposing on the fast track. Nature Biotechnology (2020) doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- 12.Sheahan T. P. et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 and multiple endemic, epidemic and bat coronavirus. bioRxiv 2020.03.19.997890 (2020) doi: 10.1101/2020.03.19.997890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paton J. Moderna’s Coronavirus Vaccine Trial Set to Begin This Month. Bloomberg News (2020). [Google Scholar]
- 14.Hoffmann M. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell (2020) doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prussia A., Thepchatri P., Snyder J. P. & Plemper R. K. Systematic approaches towards the development of host-directed antiviral therapeutics. Int. J. Mol. Sci. 12, 4027–4052 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan J. F.-W. et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 9, 221–236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fehr A. R. & Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol. Biol. 1282, 1–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chinese SARS Molecular Epidemiology Consortium. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303, 1666–1669 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Jäger S. et al. Global landscape of HIV-human protein complexes. Nature 481, 365–370 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teo G. et al. SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics 100, 37–43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.SARS-CoV-2 infected host cell proteomics reveal potential therapy targets. In Review (2020). [Google Scholar]
- 22.Batra J. et al. Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 175, 1917–1930.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M. et al. Identification of antiviral roles for the exon-junction complex and nonsense-mediated decay in flaviviral infection. Nat Microbiol (2019) doi: 10.1038/s41564-019-0375-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eckhardt M. et al. Multiple Routes to Oncogenesis Are Promoted by the Human Papillomavirus-Host Protein Network. Cancer Discov. 8, 1474–1489 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diep J. et al. Enterovirus pathogenesis requires the host methyltransferase SETD3. Nat Microbiol 4, 2523–2537 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirrashidi K. M. et al. Global Mapping of the Inc-Human Interactome Reveals that Retromer Restricts Chlamydia Infection. Cell Host Microbe 18, 109–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penn B. H. et al. An Mtb-Human Protein-Protein Interaction Map Identifies a Switch between Host Antiviral and Antibacterial Responses. Mol. Cell 71, 637–648.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramage H. R. et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 57, 329–340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Davis Z. H. et al. Global mapping of herpesvirus-host protein complexes reveals a transcription strategy for late genes. Mol. Cell 57, 349–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah P. S. et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 175, 1931–1945.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harcourt J. et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with 2019 Novel Coronavirus Disease, United States. Emerg. Infect. Dis. 26, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D. et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.SARS-CoV-2 infected host cell proteomics reveal potential therapy targets. In Review (2020) doi: 10.21203/rs.3.rs-17218/v1. [DOI] [Google Scholar]
- 34.Karczewski K. J. et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 531210 (2019) doi: 10.1101/531210. [DOI] [Google Scholar]
- 35.Chamberlain L. H. & Shipston M. J. The physiology of protein S-acylation. Physiol. Rev. 95, 341–376 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petit C. M. et al. Palmitoylation of the cysteine-rich endodomain of the SARS-coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology 360, 264–274 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sergeeva O. A. & van der Goot F. G. Anthrax toxin requires ZDHHC5-mediated palmitoylation of its surface-processing host enzymes. Proc. Natl. Acad. Sci. U. S. A. 116, 1279–1288 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes P. J. Role of HDAC2 in the pathophysiology of COPD. Annu. Rev. Physiol. 71, 451–464 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Xu P. et al. NOS1 inhibits the interferon response of cancer cells by S-nitrosylation of HDAC2. J. Exp. Clin. Cancer Res. 38, 483 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dewe J. M., Fuller B. L., Lentini J. M., Kellner S. M. & Fu D. TRMT1-Catalyzed tRNA Modifications Are Required for Redox Homeostasis To Ensure Proper Cellular Proliferation and Oxidative Stress Survival. Mol. Cell. Biol. 37, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryzhakov G. & Randow F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J. 26, 3180–3190 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramasamy S. et al. Tle1 tumor suppressor negatively regulates inflammation in vivo and modulates NF-κB inflammatory pathway. Proc. Natl. Acad. Sci. U. S. A. 113, 1871–1876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X., Li X., Ning F., Shang Y. & Hu X. TLE4 acts as a corepressor of Hes1 to inhibit inflammatory responses in macrophages. Protein Cell 10, 300–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tetsuka T. et al. Inhibition of nuclear factor-kappaB-mediated transcription by association with the amino-terminal enhancer of split, a Groucho-related protein lacking WD40 repeats. J. Biol. Chem. 275, 4383–4390 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Wang C. et al. The E3 ubiquitin ligase Nrdp1 ‘preferentially’ promotes TLR-mediated production of type I interferon. Nat. Immunol. 10, 744–752 (2009). [DOI] [PubMed] [Google Scholar]
- 46.Kondo T., Watanabe M. & Hatakeyama S. TRIM59 interacts with ECSIT and negatively regulates NF-κB and IRF-3/7-mediated signal pathways. Biochem. Biophys. Res. Commun. 422, 501–507 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Li S., Wang L., Berman M., Kong Y.-Y. & Dorf M. E. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35, 426–440 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia X. et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity 34, 843–853 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanke T. et al. Proteinase-activated receptor-2-mediated activation of stress-activated protein kinases and inhibitory kappa B kinases in NCTC 2544 keratinocytes. J. Biol. Chem. 276, 31657–31666 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Matsuda A. et al. Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene 22, 3307–3318 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Liu X.-Y., Wei B., Shi H.-X., Shan Y.-F. & Wang C. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. 20, 994–1011 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Reineke L. C. & Lloyd R. E. The stress granule protein G3BP1 recruits protein kinase R to promote multiple innate immune antiviral responses. J. Virol. 89, 2575–2589 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang W. et al. G3BP1 inhibits RNA virus replication by positively regulating RIG-I-mediated cellular antiviral response. Cell Death Dis. 10, 946 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim S. S.-Y., Sze L., Liu C. & Lam K.-P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-β response. J. Biol. Chem. 294, 6430–6438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raaben M., Groot Koerkamp M. J. A., Rottier P. J. M. & de Haan C. A. M. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell. Microbiol. 9, 2218–2229 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakagawa K., Narayanan K., Wada M. & Makino S. Inhibition of Stress Granule Formation by Middle East Respiratory Syndrome Coronavirus 4a Accessory Protein Facilitates Viral Translation, Leading to Efficient Virus Replication. J. Virol. 92, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ivanov P., Kedersha N. & Anderson P. Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb. Perspect. Biol. 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Slaine P. D., Kleer M., Smith N. K., Khaperskyy D. A. & McCormick C. Stress Granule-Inducing Eukaryotic Translation Initiation Factor 4A Inhibitors Block Influenza A Virus Replication. Viruses 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thompson P. A. et al. Abstract 2698: eFT226, a potent and selective inhibitor of eIF4A, is efficacious in preclinical models of lymphoma. Experimental and Molecular Therapeutics (2019) doi: 10.1158/1538-7445.am2019-2698. [DOI] [Google Scholar]
- 60.Reineke L. C. et al. Casein Kinase 2 Is Linked to Stress Granule Dynamics through Phosphorylation of the Stress Granule Nucleating Protein G3BP1. Mol. Cell. Biol. 37, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fonseca B. D., Jia J. J., Hollensen A. K. & Pointet R. LARP1 is a major phosphorylation substrate of mTORC1. bioRxiv (2018). [Google Scholar]
- 62.Kindrachuk J. et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 59, 1088–1099 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Frieman M. et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81, 9812–9824 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Faria P. A. et al. VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol. Cell 17, 93–102 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Timms R. T. et al. A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 365, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quan B., Seo H.-S., Blobel G. & Ren Y. Vesiculoviral matrix (M) protein occupies nucleic acid binding site at nucleoporin pair (Rae1 • Nup98). Proc. Natl. Acad. Sci. U. S. A. 111, 9127–9132 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mahon C., Krogan N. J., Craik C. S. & Pick E. Cullin E3 ligases and their rewiring by viral factors. Biomolecules 4, 897–930 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Soucy T. A. et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Faivre E. J. et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 578, 306–310 (2020). [DOI] [PubMed] [Google Scholar]
- 70.Filippakopoulos P. et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214–231 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mitsuda T. et al. Sigma-1Rs are upregulated via PERK/eIF2α/ATF4 pathway and execute protective function in ER stress. Biochem. Biophys. Res. Commun. 415, 519–525 (2011). [DOI] [PubMed] [Google Scholar]
- 72.Hellewell S. B. et al. Rat liver and kidney contain high densities of sigma 1 and sigma 2 receptors: characterization by ligand binding and photoaffinity labeling. Eur. J. Pharmacol. 268, 9–18 (1994). [DOI] [PubMed] [Google Scholar]
- 73.Pua K. H., Stiles D. T., Sowa M. E. & Verdine G. L. IMPDH2 Is an Intracellular Target of the Cyclophilin A and Sanglifehrin A Complex. Cell Rep. 18, 432–442 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Hanada H., Moriyama Y., Maeda M. & Futai M. Kinetic studies of chromaffin granule H+-ATPase and effects of bafilomycin A1. Biochem. Biophys. Res. Commun. 170, 873–878 (1990). [DOI] [PubMed] [Google Scholar]
- 75.Wang M. et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 30, 269–271 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao J., Tian Z. & Yang X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 14, 72–73 (2020). [DOI] [PubMed] [Google Scholar]
- 77.McKee E. E., Ferguson M., Bentley A. T. & Marks T. A. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob. Agents Chemother. 50, 2042–2049 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Skrtić M. et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 20, 674–688 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hultquist J. F. et al. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 17, 1438–1452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gordon D. E. et al. A Quantitative Genetic Interaction Map of HIV Infection. Mol. Cell (2020) doi: 10.1016/j.molcel.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Naffouje R. et al. Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story. Cancers 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Franklin T. J. & Cook J. M. The inhibition of nucleic acid synthesis by mycophenolic acid. Biochem. J 113, 515–524 (1969). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Organization, W. H. & Others. World Health Organization model list of essential medicines: 21st list 2019. https://apps.who.int/iris/handle/10665/325771 (2019).
- 84.Tan E. L. C. et al. Inhibition of SARS coronavirus infection in vitro with clinically approved antiviral drugs. Emerg. Infect. Dis. 10, 581–586 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Markland W., McQuaid T. J., Jain J. & Kwong A. D. Broad-spectrum antiviral activity of the IMP dehydrogenase inhibitor VX-497: a comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob. Agents Chemother. 44, 859–866 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Le Sage V., Cinti A., Amorim R. & Mouland A. J. Adapting the Stress Response: Viral Subversion of the mTOR Signaling Pathway. Viruses 8, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ranadheera C., Coombs K. M. & Kobasa D. Comprehending a Killer: The Akt/mTOR Signaling Pathways Are Temporally High-Jacked by the Highly Pathogenic 1918 Influenza Virus. EBioMedicine 32, 142–163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cota D. et al. Hypothalamic mTOR signaling regulates food intake. Science 312, 927–930 (2006). [DOI] [PubMed] [Google Scholar]
- 89.Hägglund M. G. A. et al. B(0)AT2 (SLC6A15) is localized to neurons and astrocytes, and is involved in mediating the effect of leucine in the brain. PLoS One 8, e58651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang D. & Leibowitz J. L. The structure and functions of coronavirus genomic 3’ and 5’ ends. Virus Res. 206, 120–133 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barretto N. et al. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79, 15189–15198 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yang H. et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 100, 13190–13195 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thiel V. et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 84, 2305–2315 (2003). [DOI] [PubMed] [Google Scholar]
- 94.Chiva C. et al. QCloud: A cloud-based quality control system for mass spectrometry-based proteomics laboratories. PLoS One 13, e0189209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cox J. & Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Cox J. et al. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Verschueren E. et al. Scoring Large-Scale Affinity Purification Mass Spectrometry Datasets with MiST. Curr. Protoc. Bioinformatics 49, 8.19.1–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Giurgiu M. et al. CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. 47, D559–D563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huttlin E. L. et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162, 425–440 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vizcaíno J. A. et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deutsch E. W. et al. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 45, D1100–D1106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chaudhury S., Lyskov S. & Gray J. J. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics 26, 689–691 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Joosten R. P., Long F., Murshudov G. N. & Perrakis A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Waterhouse A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Webb B. & Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gaulton A. et al. The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sterling T. & Irwin J. J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 55, 2324–2337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McLure K. G. et al. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS One 8, e83190 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pierre F. et al. Discovery and SAR of 5-(3-chlorophenylamino)benzo[c][2,6]naphthyridine-8-carboxylic acid (CX-4945), the first clinical stage inhibitor of protein kinase CK2 for the treatment of cancer. J. Med. Chem. 54, 635–654 (2011). [DOI] [PubMed] [Google Scholar]
- 111.Siddiqui-Jain A. et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 70, 10288–10298 (2010). [DOI] [PubMed] [Google Scholar]
- 112.Janeczko M., Orzeszko A., Kazimierczuk Z., Szyszka R. & Baier A. CK2α and CK2α’ subunits differ in their sensitivity to 4,5,6,7-tetrabromo- and 4,5,6,7-tetraiodo-1H-benzimidazole derivatives. Eur. J. Med. Chem. 47, 345–350 (2012). [DOI] [PubMed] [Google Scholar]
- 113.Khan N. et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J 409, 581–589 (2008). [DOI] [PubMed] [Google Scholar]
- 114.Göttlicher M. et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 20, 6969–6978 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Krämer O. H. et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 22, 3411–3420 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fass D. M. et al. Effect of Inhibiting Histone Deacetylase with Short-Chain Carboxylic Acids and Their Hydroxamic Acid Analogs on Vertebrate Development and Neuronal Chromatin. ACS Med. Chem. Lett. 2, 39–42 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gagliardi S. et al. 5-(5,6-Dichloro-2-indolyl)-2-methoxy-2,4-pentadienamides: novel and selective inhibitors of the vacuolar H+-ATPase of osteoclasts with bone antiresorptive activity. J. Med. Chem. 41, 1568–1573 (1998). [DOI] [PubMed] [Google Scholar]
- 118.Díaz J. L. et al. Synthesis and biological evaluation of the 1-arylpyrazole class of σ(1) receptor antagonists: identification of 4-{2-[5-methyl-1-(naphthalen-2-yl)-1H-pyrazol-3-yloxy]ethyl}morpholine (S1RA, E-52862). J. Med. Chem. 55, 8211–8224 (2012). [DOI] [PubMed] [Google Scholar]
- 119.Akunne H. C. et al. The pharmacology of the novel and selective sigma ligand, PD 144418. Neuropharmacology 36, 51–62 (1997). [DOI] [PubMed] [Google Scholar]
- 120.Prezzavento O. et al. Novel sigma receptor ligands: synthesis and biological profile. J. Med. Chem. 50, 951–961 (2007). [DOI] [PubMed] [Google Scholar]
- 121.Azzariti A. et al. Cyclohexylpiperazine derivative PB28, a sigma2 agonist and sigma1 antagonist receptor, inhibits cell growth, modulates P-glycoprotein, and synergizes with anthracyclines in breast cancer. Mol. Cancer Ther. 5, 1807–1816 (2006). [DOI] [PubMed] [Google Scholar]
- 122.Nissinen E., Lindén I. B., Schultz E. & Pohto P. Biochemical and pharmacological properties of a peripherally acting catechol-O-methyltransferase inhibitor entacapone. Naunyn. Schmiedebergs. Arch. Pharmacol. 346, 262–266 (1992). [DOI] [PubMed] [Google Scholar]
- 123.De Santi C., Giulianotti P. C., Pietrabissa A., Mosca F. & Pacifici G. M. Catechol-O-methyltransferase: variation in enzyme activity and inhibition by entacapone and tolcapone. Eur. J. Clin. Pharmacol. 54, 215–219 (1998). [DOI] [PubMed] [Google Scholar]
- 124.Vane J. R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 231, 232–235 (1971). [DOI] [PubMed] [Google Scholar]
- 125.Wheaton W. W. et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 3, e02242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Najjar M. et al. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 10, 1850–1860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chijiwa T. et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J. Biol. Chem. 265, 5267–5272 (1990). [PubMed] [Google Scholar]
- 128.Sintchak M. D. & Nimmesgern E. The structure of inosine 5’-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacology 47, 163–184 (2000). [DOI] [PubMed] [Google Scholar]
- 129.Asano N. et al. In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur. J. Biochem. 267, 4179–4186 (2000). [DOI] [PubMed] [Google Scholar]
- 130.Carter S. B. et al. Mycophenolic acid: an anti-cancer compound with unusual properties. Nature 223, 848–850 (1969). [DOI] [PubMed] [Google Scholar]
- 131.Wittine K. et al. Novel 1,2,4-triazole and imidazole derivatives of L-ascorbic and imino-ascorbic acid: synthesis, anti-HCV and antitumor activity evaluations. Bioorg. Med. Chem. 20, 3675–3685 (2012). [DOI] [PubMed] [Google Scholar]
- 132.Koltun E. S. et al. Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorg. Med. Chem. Lett. 22, 3727–3731 (2012). [DOI] [PubMed] [Google Scholar]
- 133.Leung L. et al. Anti-metastatic Inhibitors of Lysyl Oxidase (LOX): Design and Structure-Activity Relationships. J. Med. Chem. 62, 5863–5884 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Karaman M. W. et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 (2008). [DOI] [PubMed] [Google Scholar]
- 135.Davis M. I. et al. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 (2011). [DOI] [PubMed] [Google Scholar]
- 136.Llona-Minguez S. et al. Discovery of the First Potent and Selective Inhibitors of Human dCTP Pyrophosphatase 1. J. Med. Chem. 59, 1140–1148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Llona-Minguez S. et al. Identification of Triazolothiadiazoles as Potent Inhibitors of the dCTP Pyrophosphatase 1. J. Med. Chem. 60, 2148–2154 (2017). [DOI] [PubMed] [Google Scholar]
- 138.Llona-Minguez S. et al. Diverse heterocyclic scaffolds as dCTP pyrophosphatase 1 inhibitors. Part 2: Pyridone- and pyrimidinone-derived systems. Bioorg. Med. Chem. Lett. 27, 3219–3225 (2017). [DOI] [PubMed] [Google Scholar]
- 139.McIver E. G. et al. Synthesis and structure-activity relationships of a novel series of pyrimidines as potent inhibitors of TBK1/IKKε kinases. Bioorg. Med. Chem. Lett. 22, 7169–7173 (2012). [DOI] [PubMed] [Google Scholar]
- 140.Seitzberg J. G. et al. Discovery of potent and selective small-molecule PAR-2 agonists. J. Med. Chem. 51, 5490–5493 (2008). [DOI] [PubMed] [Google Scholar]
- 141.Cheng R. K. Y. et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 545, 112–115 (2017). [DOI] [PubMed] [Google Scholar]
- 142.Priebe W. et al. Doxorubicin- and Daunorubicin-Glutathione Conjugates, but Not Unconjugated Drugs, Competitively Inhibit Leukotriene C4Transport Mediated byMRP/GS-XPump. Biochemical and Biophysical Research Communications vol. 247 859–863 (1998). [DOI] [PubMed] [Google Scholar]
- 143.Barry G. D. et al. Novel agonists and antagonists for human protease activated receptor 2. J. Med. Chem. 53, 7428–7440 (2010). [DOI] [PubMed] [Google Scholar]
- 144.Perrotton T., Trompier D., Chang X.-B., Di Pietro A. & Baubichon-Cortay H. (R)- and (S)-verapamil differentially modulate the multidrug-resistant protein MRP1. J. Biol. Chem. 282, 31542–31548 (2007). [DOI] [PubMed] [Google Scholar]
- 145.Armstrong J. F. et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2020: extending immunopharmacology content and introducing the IUPHAR/MMV Guide to MALARIA PHARMACOLOGY. Nucleic Acids Res. 48, D1006–D1021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xu L. et al. Targetable BET proteins- and E2F1-dependent transcriptional program maintains the malignancy of glioblastoma. Proc. Natl. Acad. Sci. U. S. A. 115, E5086–E5095 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zengerle M., Chan K.-H. & Ciulli A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 10, 1770–1777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Albrecht B. K. et al. Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) Family as a Candidate for Human Clinical Trials. J. Med. Chem. 59, 1330–1339 (2016). [DOI] [PubMed] [Google Scholar]
- 149.Schenone S., Brullo C., Musumeci F., Radi M. & Botta M. ATP-competitive inhibitors of mTOR: an update. Curr. Med. Chem. 18, 2995–3014 (2011). [DOI] [PubMed] [Google Scholar]
- 150.Toral-Barza L. et al. Characterization of the cloned full-length and a truncated human target of rapamycin: activity, specificity, and enzyme inhibition as studied by a high capacity assay. Biochem. Biophys. Res. Commun. 332, 304–310 (2005). [DOI] [PubMed] [Google Scholar]
- 151.Thompson P. A. et al. Abstract 2698: eFT226, a potent and selective inhibitor of eIF4A, is efficacious in preclinical models of lymphoma. in Experimental and Molecular Therapeutics 2698–2698 (American Association for Cancer Research, 2019). [Google Scholar]
- 152.Jorquera P. A. et al. Verdinexor (KPT-335), a Selective Inhibitor of Nuclear Export, Reduces Respiratory Syncytial Virus Replication In Vitro. J. Virol. 93, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tesei A. et al. Sigma Receptors as Endoplasmic Reticulum Stress ‘Gatekeepers’ and their Modulators as Emerging New Weapons in the Fight Against Cancer. Front. Pharmacol. 9, 711 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Phadke M. et al. Dabrafenib inhibits the growth of BRAF-WT cancers through CDK16 and NEK9 inhibition. Mol. Oncol. 12, 74–88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nakamura T. et al. Molecular cloning, characterization, and chromosomal localization of FKBP23, a novel FK506-binding protein with Ca2+-binding ability. Genomics 54, 89–98 (1998). [DOI] [PubMed] [Google Scholar]
- 156.Carelli J. D. et al. Ternatin and improved synthetic variants kill cancer cells by targeting the elongation factor-1A ternary complex. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Cencic R. et al. Blocking eIF4E-eIF4G interaction as a strategy to impair coronavirus replication. J. Virol. 85, 6381–6389 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Reich S. H. et al. Structure-based Design of Pyridone-Aminal eFT508 Targeting Dysregulated Translation by Selective Mitogen-activated Protein Kinase Interacting Kinases 1 and 2 (MNK1/2) Inhibition. J. Med. Chem. 61, 3516–3540 (2018). [DOI] [PubMed] [Google Scholar]
- 159.Xu Y. et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat. Med. 25, 301–311 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mackman R. L. et al. Discovery of a Potent and Orally Bioavailable Cyclophilin Inhibitor Derived from the Sanglifehrin Macrocycle. J. Med. Chem. 61, 9473–9499 (2018). [DOI] [PubMed] [Google Scholar]
- 161.Rutaganira F. U. et al. Design and Structural Characterization of Potent and Selective Inhibitors of Phosphatidylinositol 4 Kinase IIIβ. J. Med. Chem. 59, 1830–1839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ma J. et al. Ester Prodrugs of IHVR-19029 with Enhanced Oral Exposure and Prevention of Gastrointestinal Glucosidase Interaction. ACS Med. Chem. Lett. 8, 157–162 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ma J. et al. Enhancing the antiviral potency of ER α-glucosidase inhibitor IHVR-19029 against hemorrhagic fever viruses in vitro and in vivo. Antiviral Res. 150, 112–122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Michaud A., Williams T. A., Chauvet M. T. & Corvol P. Substrate dependence of angiotensin I-converting enzyme inhibition: captopril displays a partial selectivity for inhibition of N-acetyl-seryl-aspartyl-lysyl-proline hydrolysis compared with that of angiotensin I. Mol. Pharmacol. 51, 1070–1076 (1997). [DOI] [PubMed] [Google Scholar]
- 165.Natesh R., Schwager S. L. U., Sturrock E. D. & Acharya K. R. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature 421, 551–554 (2003). [DOI] [PubMed] [Google Scholar]
- 166.Hoffmann M. et al. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. bioRxiv 2020.01.31.929042 (2020) doi: 10.1101/2020.01.31.929042. [DOI] [Google Scholar]
- 167.Kawase M., Shirato K., van der Hoek L., Taguchi F. & Matsuyama S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 86, 6537–6545 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yamamoto M. et al. Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus S Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother. 60, 6532–6539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kandasamy J. et al. Increased selectivity toward cytoplasmic versus mitochondrial ribosome confers improved efficiency of synthetic aminoglycosides in fixing damaged genes: a strategy for treatment of genetic diseases caused by nonsense mutations. J. Med. Chem. 55, 10630–10643 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Martin T. D. et al. A Role for Mitochondrial Translation in Promotion of Viability in K-Ras Mutant Cells. Cell Rep. 20, 427–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nagiec E. E. et al. Oxazolidinones inhibit cellular proliferation via inhibition of mitochondrial protein synthesis. Antimicrob. Agents Chemother. 49, 3896–3902 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]