

Abstract
SARS-CoV-2, a novel coronavirus (CoV), has recently emerged causing an ongoing outbreak of viral pneumonia around the world. While genetically distinct from the original SARS-CoV, both group 2B CoVs share similar genome organization and origins to coronaviruses harbored in bats. Importantly, initial guidance has used insights from SARS-CoV infection to inform treatment and public health strategies. In this report, we evaluate type-I Interferon (IFN-I) sensitivity of SARS-CoV-2 relative to the original SARS-CoV. Our results indicate that while SARS-CoV-2 maintains similar viral replication kinetics to SARS-CoV in Vero cell, the novel CoV is much more sensitive to IFN-I pretreatment. Examining transcriptional factor activation and interferon stimulated gene (ISG) induction, SARS-CoV-2 in the context of type I IFN induces phosphorylation of STAT1 and increased ISG proteins. In contrast, the original SARS-CoV has no evidence for STAT1 phosphorylation or ISG protein increases even in the presence of type I IFN pretreatment. Next, we examined IFN competent Calu3 2B4 cells finding SARS-CoV-2 had reduced viral replication relative to SARS-CoV and induced STAT1 phosphorylation late during infection. Finally, we examined homology between SARS-CoV and SARS-CoV-2 in viral proteins shown to be interferon antagonist. The absence of open reading frame (ORF) 3b and significant changes to ORF6 suggest the two key IFN antagonists may not maintain equivalent function in SARS-CoV-2. Together, the results identify key differences in susceptibility to the IFN-I response between SARS-CoV and SARS-CoV-2. that could help inform disease progression, treatment options, and animal model development.
Keywords: Coronavirus, 2019-nCoV, SARS-CoV-2, COVID-19, SARS-CoV, type I interferon, IFN
Article Summary:
SARS-CoV-2 has similar replication kinetics to SARS-CoV, but demonstrates significant sensitivity to type I interferon treatment.
Introduction
At the end of 2019, a cluster of patients in Hubei Province, China was diagnosed with a viral pneumonia of unknown origins. With community links to the Hunnan seafood market in Wuhan, the disease cluster had echoes of the severe acute respiratory syndrome coronavirus (SARS-CoV) outbreak that emerged at the beginning of the century (1). The 2019 etiologic agent was identified as a novel coronavirus, 2019-nCoV, and subsequently renamed SARSCoV-2 (2). The new virus has nearly 80% nucleotide identity to the original SARS-CoV and the corresponding CoV disease, COVID-19, has many of the hallmarks of SARS-CoV disease including fever, breathing difficulty, bilateral lung infiltration, and death in the most extreme cases (3, 4). In addition, the most severe SARS-CoV-2 disease corresponded to old age (>50 years old), health status, and health care workers, similar to both SARS and MERS-CoV (5). Together, the results indicate SARS-CoV-2 infection and disease have strong similarity to the original SARS-CoV epidemic occurring nearly two decades earlier.
In the wake of the outbreak, major research efforts have sought to rapidly characterize the novel CoV to aid in treatment and control. Initial modeling studies predicted (6) and subsequent cell culture studies confirmed that spike protein of SARS-CoV-2 utilizes human angiotensin converting enzyme 2 (ACE2) for entry, the same receptor as SARS-CoV (7, 8). Extensive case studies indicated a similar range of disease onset and severe symptoms seen with SARS-CoV (5). Notably, less severe SARS-CoV-2 cases have also been observed and were not captured in the original SARS-CoV outbreak. Importantly, screening and treatment guidance has relied on previous CoV data generated with SARS-CoV and MERS-CoV. Treatments with both protease inhibitors and type I interferon (IFN-I) have been employed (4); similarly, remdesivir, a drug targeting viral polymerases, has been reported to have efficacy against SARS-CoV-2 similar to findings with both SARS- and MERS-CoV (9–12). Importantly, several vaccine efforts have been initiated with a focus on the SARS-CoV-2 spike protein as the major antigenic determinate (13). Together, the similarities with SARS-CoV have been useful in responding to the newest CoV outbreak.
The host Innate immune response is initiated when viral products are recognized by host cell pattern recognition receptors, including Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs) (14, 15). This response ultimately results in production of IFN-I and other cytokines, which together are essential for an effective antiviral response (16). IFN-I then triggers its own signaling cascade via its receptor, in autocrine or paracrine manner, which induces phosphorylation of signal transducers and activators of transcription 1 (STAT1) and STAT2. Together, STAT1, STAT2, and a third transcription factor, IRF9, form the Interferon Stimulated Gene Factor 3 (ISGF3) complex, which is essential for induction of many IFN-stimulated genes (ISGs), and ultimately and effective antiviral response (17, 18). To establish productive replication, viruses have developed different mechanisms to escape this antiviral response targeting different parts of the IFN-I response machinery (19).
In this study, we further characterize SARS-CoV-2 and compare it to the original SARS-CoV. Using Vero E6 cells, we demonstrate that SARS-CoV-2 maintains similar viral replication kinetics as SARS-CoV following a low dose infection. In contrast, we find that SARS-CoV-2 is much more sensitive to IFN-I pretreatment as compared to SARS-CoV. Examining further, we determined that SARS-CoV-2 induces STAT1 phosphorylation and ISG expression, which is absent in SARS-CoV. Similarly, infection of IFN competent Calu3 2B4 cells resulted in reduced SARS-Cov-2 replication and STAT1 phosphorylation at late times. These results suggest distinct changes between the CoVs in terms of IFN antagonism and we subsequently examined sequence homology between the SARS-CoV and SARS-CoV-2 viral proteins that may be responsible for these differences. Together, the results suggest SARS-CoV-2 lacks the same capacity to control the IFN-I response as SARS-CoV.
Results
Our initial studies infected Vero E6 cells using a low multiplicity of infection (MOI) to explore the viral replication kinetics of SARS-CoV-2 relative to SARS-CoV. Following infection, we find that both SARS-CoV and SARS-CoV-2 replicate with similar kinetics, peaking 48 hours post infection (Fig. 1A). While SARS-CoV-2 titer had slightly lower viral titers at 24 hours post infection, the results were statistically different between the novel CoV and the original epidemic strain. By 48 hours, replication of both viruses had plateaued and significant cytopathic effect (CPE) was observed for both SARS-CoV and SARS-CoV-2 infections. Together, the results indicated that SARS-CoV and SARS-CoV-2 replicate with similar replication kinetics in Vero E6 cells.
Figure 1. SARS-CoV-2 sensitive to type I IFN pretreatment.
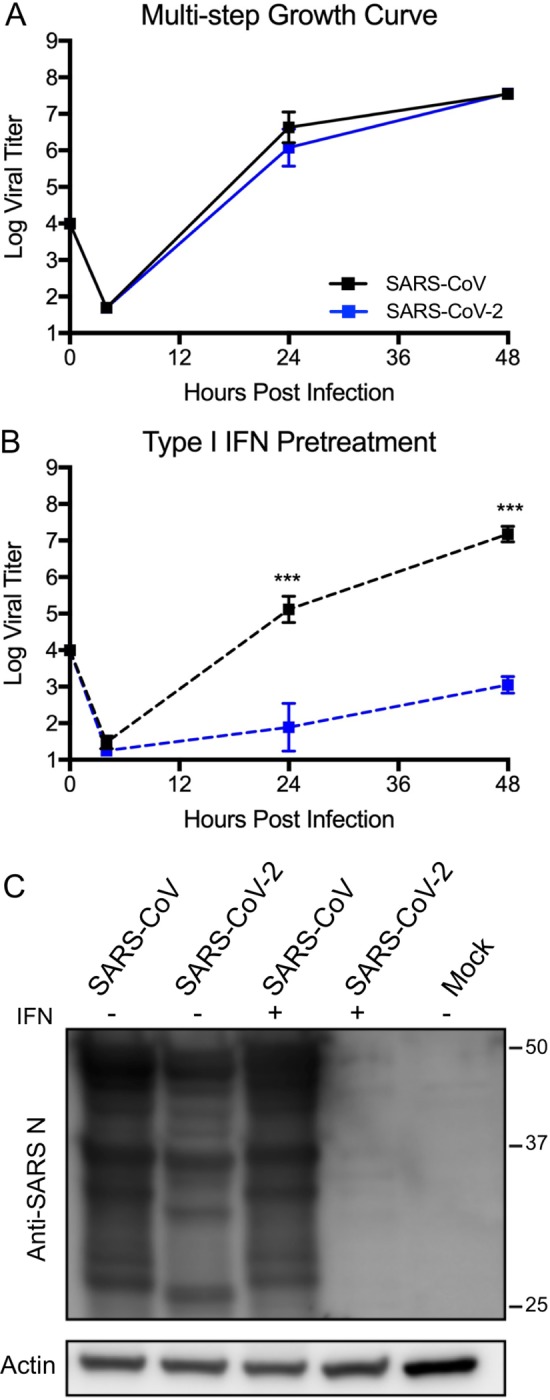
A) Vero E6 cells infected with either SARS-CoV WT (black) or SARS-CoV-2 (blue) at an MOI of 0.01. Media harvested at 4, 24, and 48 hours post infection. B) Vero E6 cells were treated with 1000 units recombinant type I IFN or mock for 18 hours prior to infection. Cells were subsequently infected at with either SARS-CoV WT (black) or SARS-CoV-2 (blue) at an MOI of 0.01 as described above. Each point on the line graph represents the group mean, N=6 for 24 and 48HPI, N=3 for 3HPI. All error bars represent SD. The two tailed students t-test was used to determine P-values: *** P < 0.001. C) Cell protein lysates from IFN treated and untreated cells were probed 48 hours post infection by using Western blotting with rabbit polyclonal anti-SARS N antibody or actin.
We next evaluated the susceptibility of SARS-CoV-2 to IFN-I pretreatment. Treatment with IFN-I (recombinant IFNα) has been attempted as an antiviral approach for a wide variety of pathogens including hepatitis B and C viruses as well as HIV (20). During both the SARS and MERS-CoV outbreaks, IFN-I has been employed with limited effect (21, 22). In this study, we pretreated Vero E6 cells with 1000 units of recombinant IFN-I (IFN-α) 18 hours prior to infection. Vero E6 lack the capacity to produce IFN-I, but are able to respond to exogenous treatment (23). Following pretreatment with IFN-I, SARS-CoV infection has a modest reduction in viral titer (1.5 log plaque forming units (PFU) as compared to untreated control 24 hours post infection (Fig. 1B). However, by 48 hours, SARS-CoV has nearly equivalent viral yields as the untreated conditions (7.2 log PFU versus 7.5 log PFU). In contrast, SARS-CoV-2 shows a significant reduction in viral replication following IFN-I treatment. At both 24 and 48 hours post infection, SARS-CoV-2 had massive 3-log (24 HPI) and 4-log (48 HPI) drops in viral titer as compared to control untreated cells. Finally, we examined viral protein production finding a major deficit in nucleocapsid protein production in IFN-I treated cells following SARS-CoV-2 infection (Fig. 1C). In contrast, viral proteins were robustly expressed for SARS-CoV-2 in untreated cells and for SARS-CoV in both conditions. Together, the results demonstrate a clear sensitivity to a primed IFN-I response in SARS-CoV-2, which is not observed with SARS-CoV.
SARS-CoV-2 fails to attenuate STAT1 phosphorylation and ISG production.
To explore differences in IFN-I antagonism between SARS-CoV and SARS-CoV, we examined both STAT1 activation and IFN stimulated gene (ISG) expression following IFN pretreatment and infection. Examining Vero cell protein lysates, we found that IFN-I treated cells infected with SARS-CoV-2 induced phosphorylated STAT-1 by 48 hours post infection (Fig. 2). STAT1 phosphorylation was absent in untreated cells infected with SARS-CoV-2 and suggest the novel CoV is unable to inhibit a IFN-I preprimed response. In contrast, SARS-CoV had no evidence for STAT1 phosphorylation in either IFN-I treated or untreated cells, illustrating robust control over IFN-I induction pathways. Examining further, STAT1, IFIT2, and TRIM25, known ISGs (17), had increased protein expression in the context of SARS-CoV-2 infection following IFN pretreatment (Fig. 2). Basal STAT1 and TRIM25 levels are reduced during SARS-CoV and SARS-CoV-2 infection relative to control likely due to the mRNA targeting activity of nonstructural protein 1 (NSP1) (24). However, IFN-I treatment results in augmented protein levels for both ISGs following SARS-CoV-2 infection as compared to untreated control. In contrast, IFN treated SARS-CoV had no significant increase in ISG protein relative to control infection. Together, the STAT1 phosphorylation, ISG production, and viral protein levels indicate that SARS-CoV-2 lacks the same capacity to modulate a primed type I IFN response as the original SARS-CoV.
Figure 2. SARS-CoV-2 infection induces STAT1 phosphorylation and ISG production.

Vero cell protein lysates from IFN-I treated and untreated cells were probed 48 hours post infection by Western blotting for phosphorylated STAT1 (Y701), STAT1, IFIT2, TRIM25, and Actin.
SARS-CoV-2 induces STAT1 phosphorylation in interferon competent cells.
While capable of responding to exogenous type I IFN, Vero cells lack the capacity to produce type I IFN following infection which likely plays a role in robust replication of a wide range of viruses []. To evaluate SARS-CoV-2 in a type I IFN responsive cell type, we infected Calu3 2B4 cells, a lung epithelial cell line sorted for ACE2 expression and previously used in coronavirus and influenza research (25). Using an MOI of 1, we examined the viral replication kinetics of SARS-CoV-2 relative to SARS-CoV in Calu3 cells. We found that both SARS-CoV and SARS-CoV-2 replicate with similar overall kinetics, peaking 24 hours post infection (Fig. 3A). However, SARS-CoV-2 replication is slightly attenuated relative to SARS-CoV at 24 hours post infection (0.82 log reduction). The attenuation in viral replication expands at 48 hours (1.4 log reduction) indicating a significant change in total viral titers between SARS-CoV and SARS-CoV-2. Notably, no similar attenuation was observed in untreated Vero cells (Fig. 1A) suggesting possible immune modulation of SARS-CoV-2 infection.
Figure 3. SARS-CoV-2 induces STAT1 phosphorylation in IFN competent cells.

A) Calu 2B4 cells were infected with either SARS-CoV WT (black) or SARS-CoV-2 (blue) at an MOI of 1. Media harvested at 4, 24, and 48 hours post infection. Each point on the line graph represents the group mean, N=3. All error bars represent SD. The two tailed students t-test was used to determine P-values: *** P < 0.001. B) Calu3 cell protein lysates were probed 48 hours post infection by Western blotting for phosphorylated STAT1 (Y701), STAT1, IFIT2, TRIM25, and Actin.
To further evaluate type I IFN induction, we examined both STAT1 phosphorylation and ISG expression following infection of Calu3 2B4 cells at 48 hours. Examining Calu3 cell protein lysates, we found cells infected with SARS-CoV-2 induced phosphorylated STAT-1 by 48 hours post infection (Fig. 3B). These results correspond to type I IFN treated Vero cell findings (Fig. 2) and suggest that the novel CoV is unable to completely inhibit the IFN-I response. In contrast, SARS-CoV had no evidence for STAT1 phosphorylation Calu3 cells, illustrating robust control over IFN-I induction pathways. Similar to the Vero IFN pretreatment, augmented levels of total STAT1 was observed in SARS-CoV-2 relative SARS-CoV, although with not as dramatic an increase. Similarly, TRIM25 was found to be reduced in both SARS-CoV and SARS-CoV-2 indicating that both viruses disrupt host protein production, likely due to mRNA antagonism by CoV NSP1 (24). Overall, the data from Calu3 cells confirm that SARS-CoV-2 is unable to maintain similar control over the IFN-I response as SARS-CoV.
Conservation of IFN antagonists across SARS-CoV and SARS-CoV-2
Considering the sensitivity to IFN-I, we next sought to evaluate changes between SARS-CoV and SARS-CoV-2 viral proteins. Previous work has established several key IFN antagonist in the SARS-CoV genome including NSP1, NSP3, ORF3b, ORF6, and others (26). Therefore, we compared the sequence homology across viral proteins from SARS-CoV, SARS-CoV-2, and several bat SARS-like viruses including WIV16-CoV (27), SHC014-CoV (28), and HKU3.1-CoV (29). Using sequence analysis, we found several changes to SARS-CoV-2 that potentially contribute to its type I IFN sensitivity (Fig. 4). For SARS-CoV structural proteins including the nucleocapsid (N) and matrix (M) protein, a high degree of sequence homology (>90%AA identity) suggests that their reported IFN antagonism is likely maintained in SARS-CoV-2 and other SARS-like viruses. Similarly, the ORF1ab poly-protein retains high sequence identity in SARS-CoV-2 and several known antagonists contained within the poly-protein (NSP1, NSP7, NSP14–16) are highly conserved relative to SARS-CoV. One notable exception is the large papain-like proteases, NSP3, which only 76% conserved between SARS-CoV and SARS-CoV-2. However, SARS-CoV-2 does maintain a deubiquitinating domain thought to confer IFN resistance (30). For SARS-CoV ORF3b, a 154 amino acid (AA) protein known to antagonize the type I IFN responses by blocking IRF3 phosphorylation (31), sequence alignments indicates that the SARS-CoV-2 equivalent ORF3b contains a premature stop codon resulting in a truncated 24 AA protein. Similarly, HKU3.1-CoV also has a premature termination resulting in a predicted 39 AA protein. Both WIV16-CoV and SHC014-CoV, the most closely related bat viruses to SARS-CoV, encode longer 114 AA truncated protein with >99% homology with SARS-CoV ORF3b suggesting that IFN antagonism might be maintained in these specific group 2B CoV strains. In addition, SARS-CoV ORF6 has been shown to be an IFN antagonist that disrupts karyopherin transportation of transcriptions factors like STAT1 (31, 32). In contrast to ORF3b, all five surveyed group 2B CoVs maintain ORF6; however, SARS-CoV-2 had only 69% homology with SARS-CoV while the other three group 2B bat CoVs had >90% conservation. Importantly, SARS-CoV-2 has a two amino acid truncation in its ORF6; previous work has found that alanine substitution in this C-terminal of SARS-CoV ORF6 resulted in ablated antagonism (32). Together, the sequence homology analysis suggests that differences in NSP3, ORF3b, and/or ORF6 may be key drivers of SARS-CoV-2 type I IFN susceptibility.
Figure 4. Conservation of SARS-CoV IFN antagonists.

Viral protein sequences of the indicated viruses were aligned according to the bounds of the SARS-CoV open reading frames for each viral protein. Sequence identities were extracted from the alignments for each viral protein, and a heat map of percent sequence identity was constructed using EvolView (www.evolgenius.info/evolview) with SARS-CoV as the reference sequence. TR = truncated protein.
Discussion
With the ongoing outbreak of COVID-19 caused by SARS-CoV-2, viral characterization remains a key factor in responding to the emergent novel virus. In this report, we describe differences in the IFN-I sensitivity between SARS-CoV-2 and the original SARS-CoV. While both viruses maintain similar replication in untreated Vero E6 cells, SARS-CoV-2 has a significant decrease in viral protein and replication following IFN-I pretreatment. The decreased SARS-CoV-2 replication correlates with phosphorylation of STAT1 and augmented ISG expression largely absent following SARS-CoV infection despite IFN-I pretreatment. Notably, infection of IFN competent Calu3 2B4 cells also resulted in reduced SARS-CoV-2 replication relative to SARS-CoV. This modest reduction in viral replication corresponded to STAT1 phosphorylation in SARS-CoV-2 infected Calu3 cells and indicated an inability to block type I IFN activation. the sensitivity to IFN-I is distinct from the original SARS-CoV and suggests that the novel CoV has distinct host interactions driving disease outcomes. Analysis of viral proteins finds SARS-CoV-2 has several changes that potentially impact its capacity to modulate the type I IFN response, including loss of ORF3b and a short truncation of ORF6, both known as IFN-I antagonists for SARS-CoV (31). Together, our results suggest SARS-CoV and SARS-CoV-2 have differences in their ability to antagonize the IFN-I response once initiated and that they may have major implication for COVID-19 disease and treatment.
With a similar genome organization and disease symptoms in humans, the SARS-CoV-2 outbreak has drawn insights from the closely related SARS-CoV. However, the differences in sensitivity to IFN-I pretreatment illustrate a clear distinction between the two CoVs. Coupled with a novel furin cleavage site (33), robust upper airway infection (8), and potential transmission prior to symptomatic disease (34), the differences between SARS-CoV and SARS-CoV-2 could prove important in disrupting the ongoing spread of COVID-19. For SARS-CoV, in vitro studies have consistently found that wild-type SARS-CoV is indifferent to IFN-I pretreatment (35, 36). Similarly, in vivo SARS-CoV studies have found that the loss of IFN-I signaling had no significant impact on disease (37), suggesting that this virus is not sensitive to the antiviral effects of IFN-I. However, more recent reports suggest that host genetic background may majorly influence this finding (38). For SARS-CoV-2, our results suggest that IFN-I pretreatment produces a 3 – 4 log drop in viral titer and correlates to STAT1 phosphorylation. This level of sensitivity is similar to MERS-CoV and suggests that the novel CoV lacks the same capacity to escape a primed IFN-I response as SARS-CoV (39, 40). Notably, the sensitivity to IFN-I does not completely ablate viral replication; unlike SARS-CoV 2’O methyl-transferase mutants (35), SARS-CoV-2 is able to replicate to low, detectable levels even in the presence of IFN-I. This finding could help explain positive test in patients with minimal symptoms and the range of disease observed. In addition, while SARS-CoV-2 is sensitive to IFN-I pretreatment, both SARS-CoV and MERS-CoV employ effective means to disrupt virus recognition and downstream signaling until late during infection (25). While SARS-CoV-2 may employ a similar mechanism early during infection, STAT1 phosphorylation and reduced viral replication are observed in IFN competent Calu3 indicating that the novel CoV does not as effectively block IFN-I signaling as the original SARS-CoV
For SARS-CoV-2, the sensitivity to IFN-I indicates a distinction from SARS-CoV and suggests differential host innate immune modulation between the viruses. The loss of ORF3b and truncation/changes in ORF6 could signal a reduced capacity of SARS-CoV-2 to interfere with type I IFN responses. For SARS-CoV ORF6, the N-terminal domain has been shown to have a clear role in its ability to disrupt karyopherin transport (32); in turn, the loss of ORF6 function for SARS-CoV-2 would likely render it much more susceptible to IFN-I pretreatment as activated STAT1 has the capacity to enter the nucleus and induce ISGs and the antiviral response. In these studies, we have found that following IFN-I pretreatment, STAT1 phosphorylation is induced following SARS-CoV-2 infection. The increase in ISG proteins (STAT1, IFIT2, TRIM25) suggests that SARS-CoV-2 ORF6 does not effectively block nuclear transport as well as SARS ORF6. For SARS-CoV ORF3b, the viral protein has been shown to disrupt phosphorylation of IRF3, a key transcriptional factor in the induction of IFN-I and the antiviral state (31). While its mechanism of action is not clear, the ORF3b absence in SARSCoV-2 infection likely impacts its ability to inhibit the IFN-I response and eventual STAT1 activation. Similarly, while NSP3 deubiquitinating domain remains intact, SARS-CoV-2 has a 24 AA insertion upstream of this deubiquitinating domain that could potentially alter that function (30). While other antagonists are maintained with high levels of conservation (>90%), single point mutations in key locations could modify function and contribute to increased IFN sensitivity. Overall, the sequence analysis suggests that differences between SARS-CoV and SARS-CoV-2 viral proteins may drive attenuation in the context of type I IFN pretreatment.
The increased sensitivity of SARS-CoV-2 suggests utility in treatment using type I IFN. While IFN-I has been used in response to chronic viral infection (41), previous examination of SARS-CoV cases found inconclusive effect for type I IFN treatment (42). However, the findings from the SARS-CoV outbreak were complicated by combination therapy of type I IFN with other treatments including ribavirin/steroids and lack of a regimented protocol. While type I IFN has been utilized to treat MERS-CoV infected patients, no conclusive data yet exists to determine efficacy (43). Yet, in vivo studies with MERS-CoV has found that early induction with type I IFN can be protective in mice (44); importantly, the same study found that late type I IFN induction can be detrimental for MERS-CoV disease (44). Similarly, early reports have described treatments using type I IFN in combination for SARS-CoV-2 infection; yet the efficacy of these treatments and the parameters of their use are not known (45). Overall, sensitivity data suggest that type I IFN treatment may have utility for treating SARS-CoV-2 if the appropriate parameters can be determined. In addition, use of type III IFN, which is predicted to have utility in the respiratory tract, could offer another means for effective treatment for SARS-CoV-2.
In addition to treatment, the sensitivity to type I IFN may also have implications for animal model development. For SARS-CoV, mouse models that recapitulate human disease were developed through virus passage in immune competent mice (46). Similarly, mouse models for MERS-CoV required adaptation in mice that had genetic modifications of their dipeptidyl-peptidase 4 (DPP4), the receptor for MERS-CoV (47, 48). However, each of these MERS-CoV mouse models still retained full immune capacity. In contrast, SARS-CoV-2 sensitivity to type I IFN may signal the need to use an immune deficient model to develop relevant disease. While initial work has suggested incompatibility to SARS-CoV-2 infection in mice based on receptor usage (8), the type I IFN response may be a second major barrier that needs to be overcome. Similar to the emergent Zika virus outbreak, the use of type I IFN receptor knockout mice or type I IFN receptor blocking antibody may be necessary to develop a useful SARS-CoV-2 animal models for therapeutic testing (49).
Overall, our results indicate that SARS-CoV-2 has a much higher sensitivity to type I IFN than the previously emergent SARS-CoV. This augmented type I IFN sensitivity is likely due to changes in viral proteins between the two epidemic CoV strains. Moving forward, these data could provide important insights for both the treatment of SARS-CoV-2 as well as developing novel animal models of disease. In this ongoing outbreak, the results also highlight a distinction between the highly related viruses and suggest insights from SARS-CoV must be verified for SARS-CoV-2 infection and disease.
Methods
Viruses and cells.
SARS-CoV-2 USA-WA1/2020, provided by the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA) and was originally obtained from the USA Centers of Disease Control as described(50). SARS-CoV-2 and mouse-adapted recombinant SARS-CoV (MA15) (46) were titrated and propagated on VeroE6 cells, grown in DMEM with 5% fetal bovine serum and 1% antibiotic/antimytotic (Gibco). Calu3 2B4 cells were grown in DMEM with 10% defined fetal bovine serum, 1% sodium pyruvate (Gibco), and 1% antibiotic/antimitotic (Gibco). Standard plaque assays were used for SARS-CoV and SARS-CoV-2 (51, 52). All experiments involving infectious virus were conducted at the University of Texas Medical Branch (Galveston, TX) in approved biosafety level 3 (BSL) laboratories with routine medical monitoring of staff.
Infection and type I IFN pretreatment.
Viral replication in Vero E6 and Calu3 2B4 cells were performed as previously described (35, 53).Briefly, cells were washed with two times with PBS and inoculated with SARS-CoV or SARS-CoV-2 at an multiplicity of infection (MOI) 0.01 for 60 minutes at 37 °C. Following inoculation, cells were washed 3 times, and fresh media was added to signify time 0. Three or more biological replicates were harvested at each described time. No blinding was used in any sample collections, nor were samples randomized. For type I IFN pretreatment, experiments were completed as previously described (35). Briefly, Vero E6 cells were incubated with 1000 units/mL of recombinant type I IFN alpha (PBL Assay Sciences) 18 hours prior to infection (35). Cells were infected as described above and type I IFN was not added back after infection.
Phylogenetic Tree and Sequence Identity Heat Map.
Heat maps were constructed from a set of representative group 2B coronaviruses by using alignment data paired with neighbor-joining phylogenetic trees built in Geneious (v.9.1.5). Sequence identity was visualized using EvolView (http://evolgenius.info/) and utilized SARS-CoV Urbani as the reference sequence. Tree shows the degree of genetic similarity of SARS-CoV-2 and SARS-CoV across a selected group 2B coronaviruses
Immunoblot Analysis and Antibodies:
Viral and host protein analysis were evaluated as previously described (50, 54). Briefly, cell lysates were resolved on 7.5% Mini-PROTEAN TGX SDS-PAGE gels and then transferred to polyvinylidene difluoride (PVDF) membranes using a Trans-Blot Turbo transfer system (BioRad). Membranes were blocked with 5% (w/v) non-fat dry milk in TBST (TBS with 0.1% (v/v) Tween-20) for 1 hr, and then probed with the indicated primary antibody in 3% (w/v) BSA in TBST at 4°C overnight. Following overnight incubation, membranes were probed with the following secondary antibodies in 5% (w/v) non-fat dry milk in TBST for 1 hr at room temperature: anti-rabbit or anti-mouse IgG-HRP conjugated antibody from sheep (both 1:10,000 GE Healthcare). Proteins were visualized using ECL or SuperSignal West Femto chemiluminescence reagents (Pierce) and detected by autoradiography. The following primary antibodies were used: anti-pSTAT1 (Y701) (1:1000 9171L Cell Signaling Technologies), antiSTAT1 D1K9Y (1:1000 14994P Cell Signaling Technologies), anti-IFIT2 (1:2000 PA3–845 Invitrogen), anti-TRIM25 (1:1000 610570 BD Biosciences), anti-SARS-CoV Nucleocapsid (1:1000), and anti-β-Actin (1:1000 ab8227 Abcam).
Statistical analysis.
All statistical comparisons in this manuscript involved the comparison between 2 groups, SARS-CoV or SARS-CoV-2 infected groups under equivalent conditions. Thus, significant differences in viral titer were determined by the unpaired two-tailed students T-Test.
Importance.
With the ongoing outbreak of COVID-19 disease, differences between the SARS-CoV-2 and the original SARS-CoV could be leveraged to inform disease progression and eventual treatment options. In addition, these findings could have key implications for animal model development as well as further research into how SARS-CoV-2 modulates the type I IFN response early during infection.
Acknowledgements.
Research was supported by grants from NIA and NIAID of the NIH (U19AI100625 and R00AG049092 to VDM; R24AI120942 to WRCEVA; R01AI134907 to RR; and T32 AI060549 to AH). Research was also supported by STARs Award provided by the University of Texas System to VDM and trainee funding provided by the McLaughlin Fellowship Fund at UTMB.
References
- 1.Gralinski LE, Menachery VD. 2020. Return of the Coronavirus: 2019-nCoV. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, Haagmans BL, Lauber C, Leontovich AM, Neuman BW, Penzar D, Perlman S, Poon LLM, Samborskiy DV, Sidorov IA, Sola I, Ziebuhr J, Coronaviridae Study Group of the International Committee on Taxonomy of V. 2020. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology doi: 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus I, Research T. 2020. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N Engl J Med 382:727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B. 2020. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Z, McGoogan JM. 2020. Characteristics of and Important Lessons From the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases From the Chinese Center for Disease Control and Prevention. JAMA doi: 10.1001/jama.2020.2648 [DOI] [PubMed] [Google Scholar]
- 6.Xu X, Chen P, Wang J, Feng J, Zhou H, Li X, Zhong W, Hao P. 2020. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci 63:457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Letko M, Marzi A, Munster V. 2020. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol doi: 10.1038/s41564-020-0688-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature doi: 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Wit E, Feldmann F, Cronin J, Jordan R, Okumura A, Thomas T, Scott D, Cihlar T, Feldmann H. 2020. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc Natl Acad Sci U S A doi: 10.1073/pnas.1922083117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G. 2020. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019nCoV) in vitro. Cell Res doi: 10.1038/s41422-020-0282-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheahan TP, Sims AC, Leist SR, Schafer A, Won J, Brown AJ, Montgomery SA, Hogg A, Babusis D, Clarke MO, Spahn JE, Bauer L, Sellers S, Porter D, Feng JY, Cihlar T, Jordan R, Denison MR, Baric RS. 2020. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat Commun 11:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sheahan TP, Sims AC, Graham RL, Menachery VD, Gralinski LE, Case JB, Leist SR, Pyrc K, Feng JY, Trantcheva I, Bannister R, Park Y, Babusis D, Clarke MO, Mackman RL, Spahn JE, Palmiotti CA, Siegel D, Ray AS, Cihlar T, Jordan R, Denison MR, Baric RS. 2017. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmed SF, Quadeer AA, McKay MR. 2020. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medzhitov R, Janeway CA Jr. 1997. Innate immunity: the virtues of a nonclonal system of recognition. Cell 91:295–298. [DOI] [PubMed] [Google Scholar]
- 15.Meylan E, Tschopp J. 2006. Toll-like receptors and RNA helicases: two parallel ways to trigger antiviral responses. Mol Cell 22:561–569. [DOI] [PubMed] [Google Scholar]
- 16.Akira S. 2006. TLR signaling. Curr Top Microbiol Immunol 311:1–16. [DOI] [PubMed] [Google Scholar]
- 17.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5:375–386. [DOI] [PubMed] [Google Scholar]
- 19.Rajsbaum R, Garcia-Sastre A. 2013. Viral evasion mechanisms of early antiviral responses involving regulaqtion of ubiquitin pathways. Trends Microbiol 21:421–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin FC, Young HA. 2014. Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev 25:369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zumla A, Hui DS, Perlman S. 2015. Middle East respiratory syndrome. Lancet 386:995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song Z, Xu Y, Bao L, Zhang L, Yu P, Qu Y, Zhu H, Zhao W, Han Y, Qin C. 2019. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaz MO, Ziemin S, Le Beau MM, Pitha P, Smith SD, Chilcote RR, Rowley JD. 1988. Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell lines. Proc Natl Acad Sci U S A 85:5259–5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayanan K, Ramirez SI, Lokugamage KG, Makino S. 2015. Coronavirus nonstructural protein 1: Common and distinct functions in the regulation of host and viral gene expression. Virus Res 202:89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menachery VD, Eisfeld AJ, Schafer A, Josset L, Sims AC, Proll S, Fan S, Li C, Neumann G, Tilton SC, Chang J, Gralinski LE, Long C, Green R, Williams CM, Weiss J, Matzke MM, Webb-Robertson BJ, Schepmoes AA, Shukla AK, Metz TO, Smith RD, Waters KM, Katze MG, Kawaoka Y, Baric RS. 2014. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 5:e01174–01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Totura AL, Baric RS. 2012. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol 2:264–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang XL, Hu B, Wang B, Wang MN, Zhang Q, Zhang W, Wu LJ, Ge XY, Zhang YZ, Daszak P, Wang LF, Shi ZL. 2015. Isolation and Characterization of a Novel Bat Coronavirus Closely Related to the Direct Progenitor of Severe Acute Respiratory Syndrome Coronavirus. J Virol 90:3253–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge XY, Li JL, Yang XL, Chmura AA, Zhu G, Epstein JH, Mazet JK, Hu B, Zhang W, Peng C, Zhang YJ, Luo CM, Tan B, Wang N, Zhu Y, Crameri G, Zhang SY, Wang LF, Daszak P, Shi ZL. 2013. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503:535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau SK, Woo PC, Li KS, Huang Y, Tsoi HW, Wong BH, Wong SS, Leung SY, Chan KH, Yuen KY. 2005. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc Natl Acad Sci U S A 102:14040–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez-Santos YM, Wang J, Takayama J, Ghosh AK, Li K, Mesecar AD, Baker SC. 2010. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J Virol 84:4619–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, Baric RA, Palese P. 2007. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol 81:548–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. 2007. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol 81:9812–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. 2020. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res 176:104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong ZD, Tang A, Li KF, Li P, Wang HL, Yi JP, Zhang YL, Yan JB. 2020. Potential Presymptomatic Transmission of SARS-CoV-2, Zhejiang Province, China, 2020. Emerg Infect Dis 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Menachery VD, Yount BL Jr., Josset L, Gralinski LE, Scobey T, Agnihothram S, Katze MG, Baric RS. 2014. Attenuation and restoration of severe acute respiratory syndrome coronavirus mutant lacking 2′-o-methyltransferase activity. J Virol 88:4251–4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thiel V, Weber F. 2008. Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev 19:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frieman MB, Chen J, Morrison TE, Whitmore A, Funkhouser W, Ward JM, Lamirande EW, Roberts A, Heise M, Subbarao K, Baric RS. 2010. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PLoS Pathog 6:e1000849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S. 2016. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 19:181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menachery VD, Gralinski LE, Mitchell HD, Dinnon KH 3rd, Leist, Yount Jr., Graham RL, McAnarney ET, Stratton KG, Cockrell AS, Debbink K, Sims AC, Waters KM, Baric RS. 2017. Middle East Respiratory Syndrome Coronavirus Nonstructural Protein 16 Is Necessary for Interferon Resistance and Viral Pathogenesis. mSphere 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Falzarano D, de Wit E, Martellaro C, Callison J, Munster VJ, Feldmann H. 2013. Inhibition of novel beta coronavirus replication by a combination of interferon-alpha2b and ribavirin. Sci Rep 3:1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finter NB, Chapman S, Dowd P, Johnston JM, Manna V, Sarantis N, Sheron N, Scott G, Phua S, Tatum PB. 1991. The use of interferon-alpha in virus infections. Drugs 42:749–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stockman LJ, Bellamy R, Garner P. 2006. SARS: systematic review of treatment effects. PLoS Med 3:e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Wit E, van Doremalen N, Falzarano D, Munster VJ. 2016. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M, Sompallae R, McCray PB Jr., Meyerholz DK, Perlman S. 2019. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J Clin Invest 130:3625–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pang J, Wang MX, Ang IYH, Tan SHX, Lewis RF, Chen JI, Gutierrez RA, Gwee SXW, Chua PEY, Yang Q, Ng XY, Yap RK, Tan HY, Teo YY, Tan CC, Cook AR, Yap JC, Hsu LY. 2020. Potential Rapid Diagnostics, Vaccine and Therapeutics for 2019 Novel Coronavirus (2019-nCoV): A Systematic Review. J Clin Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts A, Deming D, Paddock CD, Cheng A, Yount B, Vogel L, Herman BD, Sheahan T, Heise M, Genrich GL, Zaki SR, Baric R, Subbarao K. 2007. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog 3:e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cockrell A Y B, Scobey T, Jensen K, Douglas M, Beall A, Tang X-C, Marasco WA, Heise MT, Baric RS 2016. A Mouse Model for MERS Coronavirus Induced Acute Respiratory Distress Syndrome. . Nature Microbiology In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li K, Wohlford-Lenane CL, Channappanavar R, Park JE, Earnest JT, Bair TB, Bates AM, Brogden KA, Flaherty HA, Gallagher T, Meyerholz DK, Perlman S, McCray PB Jr. 2017. Mouse-adapted MERS coronavirus causes lethal lung disease in human DPP4 knockin mice. Proc Natl Acad Sci U S A 114:E3119–E3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS. 2016. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 19:720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harcourt J, Tamin A, Lu X, Kamili S, Sakthivel SK, Murray J, Queen K, Tao Y, Paden CR, Zhang J, Li Y, Uehara A, Wang H, Goldsmith C, Bullock HA, Wang L, Whitaker B, Lynch B, Gautam R, Schindewolf C, Lokugamage KG, Scharton D, Plante JA, Mirchandani D, Widen SG, Narayanan K, Makino S, Ksiazek TG, Plante KS, Weaver SC, Lindstrom S, Tong S, Menachery VD, Thornburg NJ. 2020. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with 2019 Novel Coronavirus Disease, United States. Emerg Infect Dis 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sims AC, Tilton SC, Menachery VD, Gralinski LE, Schäfer A, Matzke MM, WebbRobertson BJ, Chang J, Luna ML, Long CE, Shukla AK, Bankhead AR, Burkett SE, Zornetzer G, Tseng CT, Metz TO, Pickles R, McWeeney S, Smith RD, Katze MG, Waters KM, Baric RS. 2013. Release of severe acute respiratory syndrome coronavirus nuclear import block enhances host transcription in human lung cells. J Virol 87:3885–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Josset L, Menachery VD, Gralinski LE, Agnihothram S, Sova P, Carter VS, Yount BL, Graham RL, Baric RS, Katze MG. 2013. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. MBio 4:e00165–00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheahan T, Rockx B, Donaldson E, Corti D, Baric R. 2008. Pathways of cross-species transmission of synthetically reconstructed zoonotic severe acute respiratory syndrome coronavirus. J Virol 82:8721–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Tol S, Atkins C, Bharaj P, Johnson KN, Hage A, Freiberg AN, Rajsbaum R. 2020. VAMP8 Contributes to the TRIM6-Mediated Type I Interferon Antiviral Response during West Nile Virus Infection. J Virol 94. [DOI] [PMC free article] [PubMed] [Google Scholar]