

Abstract
Tools based on RNA interference (RNAi) and the recently developed clustered regularly short palindromic repeats (CRISPR) system enable the selective modification of gene expression, which also makes them attractive therapeutic reagents for combating HIV infection and other infectious diseases. Several parallels can be drawn between the RNAi and CRISPR-Cas9 platforms. An ideal RNAi or CRISPR-Cas9 therapeutic strategy for treating infectious or genetic diseases should exhibit potency, high specificity and safety. However, therapeutic applications of RNAi and CRISPR-Cas9 have been challenged by several major limitations, some of which can be overcome by optimal design of the therapy or the design of improved reagents. In this review, we will discuss some advantages and limitations of anti-HIV strategies based on RNAi and CRISPR-Cas9 with a focus on the efficiency, specificity, off-target effects and delivery methods.
Keywords: RNA interference, CRISPR-Cas, HIV, gene therapy, lentiviral vector, polymerase III promoter
HIV and AIDS
More than 30 years after the discovery of the human immunodeficiency virus type 1 (HIV-1) as the causative agent of the acquired immunodeficiency syndrome (AIDS), HIV remains one of the most serious infectious diseases in the world [1, 2]. Despite significant research efforts, there is still no vaccine available that protects against HIV infection. A combination of several drugs, known as combined antiretroviral therapy (cART), is currently used to keep HIV replication under control and to successfully treat HIV patients. Although cART can suppress viral replication, a cure is never achieved because the virus persists in some cells [3]. As a consequence, patients need to take the antiviral drugs for the rest of their life. Daily medication can create difficulties with adherence, and drug-associated side effects can occur over time [4]. Thus, the search for alternative strategies for combating HIV is warranted. An attractive approach concerns a gene therapy to deliver antiviral gene reagents that interfere with viral replication to cells that can be infected by HIV. Technologies to modify gene expression, either gene silencing at the RNA level by RNA interference (RNAi) or genome editing at the DNA level by the recently developed clustered regularly short palindromic repeats (CRISPR)-based tools offers new possibilities to inhibit HIV, a retrovirus that uses both RNA and DNA forms of genetic information [5–8]. We previously reviewed the antiviral RNAi approaches [9] and will now use this as background for a discussion of the more novel CRISPR-based anti-HIV strategies.
RNAi and CRISPR-Cas9 against HIV
RNAi provides a powerful tool to elucidate gene function by silencing the expression of a specific gene at the RNA level. The control of disease-associated genes makes RNAi an attractive choice for future therapeutics. The recently discovered CRISPR-Cas9 bacterial immune system can be repurposed to easily create gene mutations and replacements in the mammalian genome and has revolutionized the field of genome engineering and reinforced the field of gene therapy research. Both RNAi and CRISPR-Cas9 approaches enable the selective modification of gene expression and are therefore intriguing therapeutic reagents for combating HIV and other infectious diseases [5, 7, 8, 10]. Many parallels can be drawn between the RNAi and CRISPR-Cas9 platforms. A clinically ideal RNAi or CRISPR-Cas9 reagent for treating infectious or genetic diseases should exhibit potency, high specificity and safety. However, therapeutic applications of RNAi and CRISPR-Cas9 have been challenged by several major limitations, but some of these limitations can be overcome by the optimal design of the therapy or development of improved reagents. In this chapter, we will discuss advantages and limitations of RNAi and CRISPR-Cas9 with a focus on the efficiency, specificity, off-target effects and delivery methods.
The efficiency and sequence specificity of the RNAi and CRISPR-Cas9 systems make them attractive strategies to interrupt the expression of disease-associated genes or pathogenic viruses [6–8, 10]. As shown in Figure 1, RNAi and CRISPR-Cas9 can be used to target viral or host functions that play critical roles at during HIV replication. Many studies reported efficient suppression of HIV replication by these two approaches [3, 10–21]. A recent paper demonstrated that CRISPR treatment can eliminate infectious HIV in a subset of infected humanized mice [22]. Nevertheless, there are challenges that must be overcome before the RNAi and CRISPR-Cas therapeutics can fulfill their clinical potential. These issues include possible off-target effects, improvement of delivery strategies and approaches to reduce the risk of viral escape [8, 23].
Figure 1.
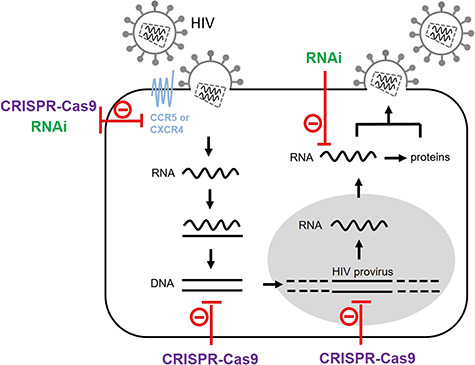
HIV targeting by RNAi or CRISPR-Cas9. HIV infects cells of the immune system, in particular the CD4-positive T cells. The HIV particle contains two genomic RNA copies. The virion attaches to the membrane of target T cells by binding to the CD4 and CCR5/CXCR4 receptors. Upon viral entry, the viral RNA genome is converted into double-stranded DNA (dsDNA) by the HIV reverse transcriptase. The resulting DNA is actively transported into the nucleus and integrated in the host cell genome. This integrated DNA or provirus uses the host cell transcription machinery to produce new viral RNAs, which serve as mRNA for protein production or as genomic RNAs that are packaged into new viral particles, which are released from the cell by budding. RNAi can target the RNA transcripts that encode the HIV receptors to block viral entry. In addition, RNAi can target the viral RNA produced during HIV replication. The receptor-encoding genes can be targeted by CRISPR-Cas9, which can also target the HIV dsDNA that is formed upon reverse transcription of the viral RNA and the integrated proviral DNA.
RNAi
Efficiency
Since the first application of RNAi in mammalian cells, the expression of shRNA molecules for targeted gene silencing has become a benchmark technology. The RNAi pathway can be triggered by artificial RNAi effectors that mimic the intermediates of miRNA processing, including natural small interfering RNAs (siRNAs) [24] and the man-made short hairpin RNAs (shRNAs) [25]. The latter—expressed from a transgene introduced in the cell—have the potential to achieve stable gene silencing. In Figure 2, we describe two possible processing routes for a shRNA substrate [26, 27]. A regular shRNA (left side) features a ~21-base pair (bp) stem with a 5–9-nucleotide (nt) loop and is processed by Dicer into a siRNA duplex of ~20 bp, of which one strand is preferentially loaded into the Argonaute 2 protein (Ago2) complex to form the RNA-induced silencing complex (RISC). The thermodynamic properties determine the selection of the guide strand, and the passenger strand is cleaved and degraded. Perfect complementarity between guide and the target results in cleavage of the messenger RNA (mRNA). We recently described the alternatively processed AgoshRNA molecule (right side) with a ~18 bp stem and 3–5 nt loop [26]. This molecule bypasses Dicer recognition and is processed by Ago2 in between bp 10 and 11 at the 3′ side of the stem to generate an extended ~30 nt miRNA that subsequently instructs RISC for gene silencing.
Figure 2.
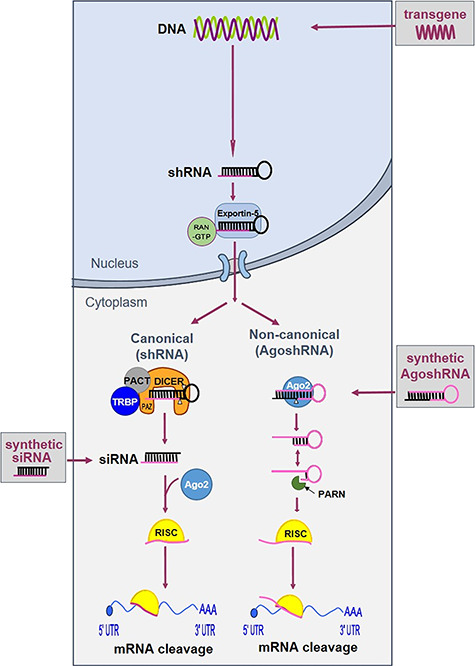
Schematic of two RNAi pathways available for shRNA processing. shRNA transcripts can be expressed from an episomal or stably integrated expression cassette. The shRNAs are then exported from the nucleus to the cytoplasm for processing: regular shRNAs by Dicer (left) and the shorter AgoshRNAs by Ago2 (right).
Intensive investigations indicated that usually only a small percentage of chosen targets allow efficient gene silencing [28, 29]. Rational target design algorithms were subsequently developed to increase the success rate [30–34]. Another important aspect concerns the development of highly active shRNA molecules. Many factors influence the shRNA activity, including the loop sequence and thermodynamic properties of the hairpin, and guidelines for optimal shRNA design were proposed [27, 35, 36].
Specificity and off-target effects
The efficiency and sequence specificity of RNAi make this strategy ideal to interrupt the expression of RNAs encoded by pathogenic viruses [9, 37]. RNAi can tolerate imperfections in the RNA–RNA duplex, and, as a consequence, off-target effects can be induced on non-related mRNAs. Efforts have been made to minimize this, such as careful target selection with the help of computational algorithms and optimization of the shRNA design [38–40]. Other adverse effects were reported for shRNAs. First, high-level shRNA expression can saturate the cellular RNAi machinery and cause toxicity [41, 42]. Second, either strand of the siRNA duplex can be loaded into RISC and adverse RNAi side effects can thus be induced by the passenger strand [39, 43]. In addition, shRNA expression was also reported to activate the dsRNA-induced protein kinase R (PKR)/interferon pathways [44, 45]. Strategies have been developed to minimize these problems. For instance, the use of a miRNA backbone or a reduction of the level of shRNA transcription by the use of a weak promoter can alleviate cellular toxicity issues [46, 47] and AgoshRNAs may prevent saturation of the cellular miRNA pathway as Dicer is bypassed. Importantly, the AgoshRNA design yields only a single guide strand, thus avoiding adverse effects caused by the passenger strand of a regular shRNA. Due to their smaller hairpin size, AgoshRNA may exhibit an improved safety profile concerning induction of the interferon response. Overall, AgoshRNAs exhibit some advantages by reducing off-target effects compared to regular shRNAs [26, 47], but their general value requires additional tests in different laboratories.
Delivery
In most cases, the Ago/shRNA therapeutic is synthesized in the target cells upon delivery of a DNA vector for transient or stable gene expression [36]. Transient Ago/shRNA expression can be realized by straightforward transfection or electroporation of DNA vectors. These methods are generally safe because of the transient nature of transgene expression, but an important drawback is the poor delivery efficiency in a broad range of cell types. Viral vectors like the lentiviral vector (LV) and adeno-associated virus (AAV) vector are popular tools to deliver shRNA constructs to both dividing and non-dividing cells, including quiescent and difficult-to-transduce cells. In particular, LV-mediated delivery of antiviral genes holds promise for a durable HIV therapy because this vector integrates into the host genome, thus ensuring long-term transgene expression. These integrating vectors remain promising for durable gene therapy applications, despite the small risk of insertional mutagenesis and eventually oncogenesis. But LV has been successfully applied in the clinic to produce cancer-specific chimeric antigen receptor T lymphocytes to treat leukemia and glioblastoma [48, 49]. AAV vectors hold potential as gene delivery vectors because of their non-pathogenicity, low immunogenicity and non-integrating nature. Several gene transfer clinical trials have recently been performed with AAV vectors [50].
CRISPR-Cas9
Efficiency
Based on differences in the components and mechanisms of action, CRISPR systems can be divided into two major systems: class 1 (type I, III, IV) requires a large complex of several effector proteins, while class 2 (type II, V, VI) only needs a single RNA-guided endonuclease (e.g. Cas9 in type II and Cas12a in type V) [51, 52]. Class 2 systems are therefore more attractive for genome editing applications. Adaptation of the bacterial CRISPR-Cas systems has facilitated application in mammalian cells, e.g. codon optimization of the Cas gene and generation of a chimeric guide RNA (gRNA) by fusion of the CRISPR RNA (crRNA) and the trans-activating crRNA (tracrRNA) components [53, 54]. The most widely used CRISPR-Cas9 system uses the Streptococcus pyogenes Cas9 (SpCas9) endonuclease and a gRNA (Figure 3). A 20 nt sequence on the gRNA 5′ end is designed to be complementary to the target DNA, which contains the protospacer adjacent motif (PAM) immediately downstream of the target site (e.g. NGG for SpCas9) (Table 1). Thus, by customizing a 20 nt region of the gRNA to pair with the DNA target of interest, Cas9 can essentially target any PAM-containing genomic locus. The efficiency depends on PAM-dependent DNA binding and gRNA complementarity to the target DNA [55]. Further studies unraveled that the genomic context of the target DNA (the local GC content) and the secondary structure of the gRNA also influence the cleavage efficiency [56–59]. Tools have been designed to predict the gRNA targeting efficiency [60]. The CRISPR-Cas9 efficiency can also be improved by modification of the gRNA module: e.g. by mutation, extension or truncation. For example, extension of the gRNA duplex or mutation of the T4 stretch in the gRNA backbone can improve the efficiency by changing the gRNA structure and/or transcription rate [59].
Figure 3.
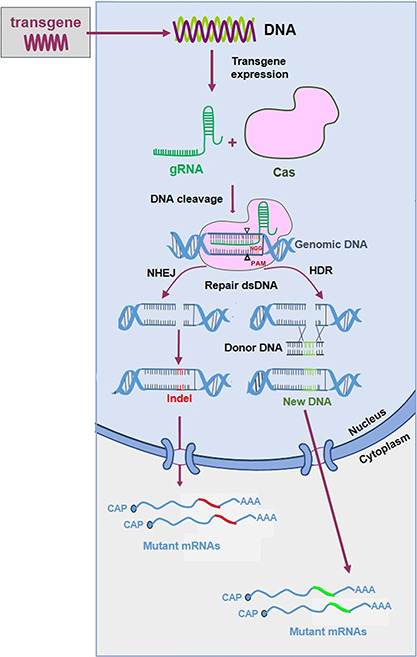
Schematic of the CRISPR-Cas9-mediated genome editing process. CRISPR-Cas9 requires expression of a gRNA (green line) and the Cas9 endonuclease (pink shape). The gRNA instructs Cas9 for cleavage of a complementary DNA target with an adjacent PAM sequence. Cas generates a dsDNA break that is repaired by the NHEJ or HDR pathways. See the text for further details.
Table 1.
In silico-guided prediction tools
| In silico prediction tools | Resources |
|---|---|
| CRISPR-Design Tools | http://crispr.mit.edu/ |
| CasFinder | http://arep.med.harvard.edu/CasFinder/ |
| Cas-OFFinder | http://www.rgenome.net/cas-offinder/ |
| E-CRISP | http://www.e-crisp.org/E-CRISP/ |
| CRISPOR | http://crispor.tefor.net |
| Benchling CRISPR Guide Design Software | https://www.benchling.com/crispr/ |
| CHOPCHOP | http://chopchop.cbu.uib.no |
| Breaking-Cas | http://bioinfogp.cnb.csic.es/tools/breakingcas/ |
The CRISPR-Cas9-induced double-stranded breaks (DSBs) in the cellular DNA can induce two repair pathways: non-homologous end joining (NHEJ) and homology-directed repair (HDR) (Figure 3) [61, 62]. NHEJ ligates the two DSB ends, but frequently inserts or deletes nucleotides. HR repairs DSB lesions with the requirement of a homologous piece of donor DNA. The CRISPR-Cas9 system thus allows not only gene disruption but also accurate gene editing with a ‘repair DNA donor’.
Specificity and off-target effects
CRISPR-Cas9 can tolerate imperfections in the RNA–DNA duplex, and consequently, cleavage can take place at unintended off-target sites, which can sometimes be identified through in silico-guided predictions (Table 1) and genome-wide assays [63–68].
Given the complexity and large size of the human genome (~3.2 billion bp), off-target DNA cleavage poses a non-negligible risk for knockout of essential or haplo-insufficient genes or mutation of tumor-suppressor genes. These shortcomings must be overcome before CRISPR-Cas9 can be applied for somatic gene therapy applications in humans. Germline editing is not considered safe for the moment [69]. A number of approaches have been developed to reduce off-target effects, including online tools for in silico gRNA design and prediction of off-target sites [60]. gRNA optimization and Cas9 nuclease engineering have led to significant improvement of the on/off-target ratio. Truncation of the base-pairing region (spacer) of 20 nt to 17 or 18 nt was reported to increase the targeting specificity [70]. Variation in the Cas9 nuclease include the use of a paired nickase [71], fusion of the catalytically dead Cas9 to the FokI restriction enzyme [72] and development of a high-fidelity Cas9 variant (HF-Cas9) [73]. Alternatively, more specific nucleases with reduced off-target effects may be identified in natural Cas9 variants. The recently described Cas12a nuclease (formerly called Cpf1) was reported to exhibit these properties [74, 75]. Because Cas9 and other CRISPR-based endonucleases are derived from bacteria, these systems will likely elicit a host immune response. This factor of immunological risk must be considered as CRISPR-Cas9 systems advance toward clinical trials, especially for applications that require long-term Cas9 expression. Several solutions to reduce the immunogenicity risks were recently proposed [76].
Delivery
A variety of delivery options can be considered for CRISPR-Cas9. The cargo for transient CRISPR-Cas9 delivery includes DNA vectors, Cas9 mRNA and gRNA or Cas9/gRNA ribonucleoprotein complexes [77–82]. These nucleic acids and proteins can be transfected into the cell by a variety of methods such as electroporation, cationic lipid and polymer-based transfection or lipid-based nanoparticles [83, 84]. Transient methods have a safety advantage, but will obviously not support long-term CRISPR-Cas activity that may be needed to achieve an HIV cure. Future developments in the field of nanotechnology may contribute to improving the delivery and minimizing the toxicity with highly specific cell-targeting capability. Viral vectors such as LV and AAV vectors have been widely used for the delivery of CRISPR-Cas9-encoding cassettes to a variety of cell types in vitro and in vivo [84–86]. One common problem with viral vectors is the limited packaging capacity that hinders the efficient delivery of large transgene cassettes like CRISPR-Cas9 [87–90]. For example, by delivery of the popular SpCas9/gRNA components, the LV transduction titer was significantly reduced [89]. The AAV packaging capacity of ~4.7 kb is also not sufficient for versatile CRISPR-SpCas9 applications [91, 92]. The search for alternative CRISPR-Cas systems of smaller size remains important to address these packaging problems. A smaller CRISPR-Cas9 homolog from S. aureus (SaCas9) could mitigate the AAV packaging problem, but this recently described system has limited efficiency and the complicated PAM sequence restricts the number of candidate target sites [57]. An alternative approach would be to minimize the modulatory elements of the CRISPR-Cas9 expression cassette, including the transcriptional promoter and the transcription termination signals.
Improvement of the new Cas12a system
The recently discovered Cas12a system was reported to have several distinct features compared to Cas9. An increased specificity could mitigate off-target problems, and a smaller size of the cassette encoding the nuclease and matching crRNA could facilitate more efficient delivery [74, 75]. However, the Cas12a system exhibits reduced gene editing efficiency compared to Cas9 [93–95]. Inspection of the crRNA sequence raised some uncertainty about the actual 5′ and 3′ ends, and we noticed that the currently used crRNA expression approach may have some flaws [96]. This triggered us to attempt to improve the Cas12a system by focusing on optimized crRNA expression. Our polymerase (Pol) III transcription study demonstrated that specific nucleotides (+1A/G) are required for efficient production of small RNAs with a precise 5′ end and that Pol III-generated small RNAs have a 3′ U-tail of variable length [97]. These results provided useful insights on how to design more precise therapeutic RNAs. We improved the CRISPR-Cas12a system by inclusion of a self-cleaving ribozyme to create a precise crRNA 3′ end [96]. This alteration enhanced the Cas12a-mediated gene editing efficiency and allowed us more recently to move towards prolonged HIV inhibition experiments (unpublished). The ribozyme addition also improved the Cas12a-based gene activation platform.
Comparing the CRISPR systems, there is a pertinent difference regarding the actual DNA cleavage event. Cas9-induced DNA cleavage occurs in the PAM-proximal sequence region that is critical for gRNA binding and target DNA cleavage, while Cas12a-triggered DNA cleavage occurs in the distal region of the PAM sequence that is less critical for target binding and cleavage. This feature of Cas12a is potentially beneficial for gene inactivation because the edited DNA sequence—cleaved and repaired with the frequent inclusion of insertions or deletions (indels)—can likely be retargeted. A subsequent round of cleavage and DNA repair is likely to create a larger indel type of mutation, which is extremely beneficial for maximal HIV provirus inactivation and may prevent the generation of escape virus variants with a minimal mutation, whereas HIV genomes with a more dramatic mutation are more likely to be replication-incompetent. This concept should be tested experimentally as it could spur the route towards HIV genome inactivation and possibly a cure. We therefore anticipate that Cas12a may become a superior anti-HIV tool over Cas9.
The design of a single promoter-driven Cas9 system
While improving Pol III-mediated AgoshRNA/crRNA expression strategies, we accidently discovered that the commonly used Pol III promoters (7SK, U6 and H1) are capable of transcribing extended mRNA transcripts that express Firefly luciferase. We subsequently demonstrated that these Pol III promoters for shRNA/gRNA expression also possess Pol II activity for mRNA transcription, but to a varying extent with H1 being the most active Pol II promoter [97, 98]. Thus, these promoters are dual-active for both polymerases II and III. We described that there is competition between these two polymerases for binding to overlapping DNA sequences of the promoter. For instance, a TATA box mutation abolished the Pol III activity and consequently enhanced the Pol II activity. Further studies are needed to map the regulatory promoter sequences that contribute to the Pol II and III activity. This should offer intriguing new possibilities to fine-tune the Pol II/III activity for the manipulation of transgene expression cassettes.
These dual-polymerase active promoter systems may be a valuable asset as they can simultaneously express a small non-coding RNA and a protein-coding mRNA. The H1 promoter seems most interesting as it exhibits abundant Pol II activity, even stronger than the standard SV40 early promoter [98]. We hypothesized that H1 can be used to express both the gRNA and Cas9 protein of the CRISPR-Cas9 system and established a single H1 promoter-driven CRISPR-Cas9 system [99]. Although this single promoter system produces less gRNA and Cas9 compared to the regular system, efficient gene editing was achieved. Most importantly, the novel cassette of reduced size should minimize the delivery problem of the viral vectors with limited packaging capacity. We indeed demonstrated that the new H1-gRNA-Cas9 system provides a significant LV titer advantage over the regular CRISPR-Cas9 system. This H1 advantage should benefit the scaled-up vector production required in preparation for future clinical trials. We expect that this novel H1-expression strategy will have similar benefits in combination with other viral expression systems such as AAV vectors. One may be able to further improve these vectors by identification of even shorter H1 promoter fragments that maintain the optimal Pol II and Pol III activity.
Key Points
DNA/RNA modification tools such as RNAi and CRISPR-Cas9 present new possibilities for treatment of a variety of genetic and infectious diseases, including HIV/AIDS. Despite rapid optimization of these two tools, many challenges remain.
RNAi and CRISPR-Cas9 can be used to either directly target viral components or indirectly host cell functions that fulfill a critical role during HIV-1 replication. Efficient suppression of HIV-1 replication has been described for both approaches.
Several challenges must be overcome before the RNAi and CRISPR-Cas therapeutics can fulfill their clinical potential. The issues involved include ways to prevent or minimize possible off-target effects and optimization of the delivery strategies in order to reduce the risk of viral escape.
Author’s perspective
Comparing the antiviral RNAi and CRISPR-Cas approaches, the latter method has the unique ability to directly target the integrated HIV provirus, which is the main cause of viral persistence under cART therapy and the viral rebound that inevitably occurs when therapy is stopped. Impressive results have been reported concerning the complete inactivation of infectious HIV in a simple, but powerful in vitro cell culture model [100] and in vivo in a small subset of CRISPR-treated humanized mice [22]. But at least two major challenges lie ahead of us. One, how can we target all or at least a significant fraction of the HIV-reservoir cells in an infected individual? This requires a more complete description of the tissues and cell types in which HIV can hide. The reservoir includes resting T cells and the recently described CD32a-positive T cells [101], but likely additional cell types. Second, it seems unsafe to deliver the functional CRISPR machinery by a vector system that persists and continues to produce the foreign endonuclease. Although CRISPR acts in a sequence-specific manner, it could over time start to modify non-HIV off-target sites in the human genome. Such genetic changes can eventually trigger oncogenesis. Thus, it would seem important to focus on transient CRISPR systems and to check if the same antiviral efficiency can be achieved. At the end, anti-HIV efficacy and safety can hopefully go hand in hand to deliver the first man-made HIV cure.
Dr Elena Herrera-Carrillo is a senior postdoc in HIV/AIDS research at Amsterdam UMC. Her primary research interest is the development of a gene therapy against chronic virus infections.
Dr Zongliang Gao worked as a PhD student on small therapeutic RNAs at Amsterdam UMC. His primary research interest concerns the use of CRISPR-Cas tools in a gene therapy setting.
Prof. Ben Berkhout is the head of the Laboratory of Experimental Virology at Amsterdam UMC. He has long-standing expertise in studying molecular mechanisms of HIV-1 replication and the design of novel antiviral strategies.
Funding
This research line was supported by the National Institutes of Health (NIH) under award number 1R01AI145045-01.
References
- 1. Gallo RC, Sarin PS, Gelmann E, et al. . Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 1983;220:865–867. [DOI] [PubMed] [Google Scholar]
- 2. Maartens G, Celum C, Lewin SR. HIV infection: epidemiology, pathogenesis, treatment, and prevention. Lancet 2014;384:258–271. [DOI] [PubMed] [Google Scholar]
- 3. Archin NM, Sung JM, Garrido C, et al. . Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat Rev Microbiol 2014;12:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carr A. Toxicity of antiretroviral therapy and implications for drug development. Nat Rev Drug Discov 2003;2:624. [DOI] [PubMed] [Google Scholar]
- 5. Herrera-Carrillo E, Berkhout B. Attacking HIV-1 RNA versus DNA by sequence-specific approaches: RNAi versus CRISPR-Cas. Biochem Soc Trans 2016;44:1355–1365. [DOI] [PubMed] [Google Scholar]
- 6. Badia R, Ballana E, Esté JA, et al. . Antiviral treatment strategies based on gene silencing and genome editing. Curr Opin Virol 2017;24:46–54. [DOI] [PubMed] [Google Scholar]
- 7. Swamy MN, Wu H, Shankar P. Recent advances in RNAi-based strategies for therapy and prevention of HIV-1/AIDS. Adv Drug Deliv Rev 2016;103:174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang G, Zhao N, Berkhout B, et al. . CRISPR-Cas based antiviral strategies against HIV-1. Virus Res 2017. [DOI] [PubMed] [Google Scholar]
- 9. Herrera-Carrillo E, Berkhout B. Gene therapy strategies to block HIV-1 replication by RNA interference. Adv Exp Med Biol 2015;848:71–95. [DOI] [PubMed] [Google Scholar]
- 10. Bobbin ML, Burnett JC, Rossi JJ. RNA interference approaches for treatment of HIV-1 infection. Genome Med 2015;7:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ter Brake O, Konstantinova P, Ceylan M, et al. . Silencing of HIV-1 with RNA interference: a multiple shRNA approach. Mol Ther 2006;14:883–892. [DOI] [PubMed] [Google Scholar]
- 12. McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat Rev Genet 2002;3:737. [DOI] [PubMed] [Google Scholar]
- 13. Kumar P, Ban H-S, Kim S-S, et al. . T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell 2008;134:577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. An DS, Donahue RE, Kamata M, et al. . Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci U S A 2007;104:13110–13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hultquist JF, Schumann K, Woo JM, et al. . A Cas9 ribonucleoprotein platform for functional genetic studies of HIV-host interactions in primary human T cells. Cell Rep 2016;17:1438–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xiao A, Cheng Z, Kong L, et al. . CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics 2014;30:1180–1182. [DOI] [PubMed] [Google Scholar]
- 17. Yin C, Zhang T, Qu X, et al. . In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol Ther 2017;25:1168–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi J-G, Bharaj P, Abraham S, et al. . Multiplexing seven miRNA-based shRNAs to suppress HIV replication. Mol Ther 2015;23:310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu Z, Chen S, Jin X, et al. . Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4+ T cells from HIV-1 infection. Cell Biosci 2017;7:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu L, Yang H, Gao Y, et al. . CRISPR/Cas9-mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Mol Ther 2017;25:1782–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Z, Pan Q, Gendron P, et al. . CRISPR/Cas9-derived mutations both inhibit HIV-1 replication and accelerate viral escape. Cell Rep 2016;15:481–489. [DOI] [PubMed] [Google Scholar]
- 22. Dash PK, Kaminski R, Bella R, et al. . Sequential LASER ART and CRISPR treatments eliminate HIV-1 in a subset of infected humanized mice. Nat Commun 2019;10:2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kang XJ, Caparas CIN, Soh BS, et al. . Addressing challenges in the clinical applications associated with CRISPR/Cas9 technology and ethical questions to prevent its misuse. Protein Cell 2017;8:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elbashir SM, Harborth J, Lendeckel W, et al. . Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494–498. [DOI] [PubMed] [Google Scholar]
- 25. Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002;296:550–553. [DOI] [PubMed] [Google Scholar]
- 26. Liu YP, Schopman NC, Berkhout B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res 2013;41:3723–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herrera-Carrillo E, Berkhout B. Dicer-independent processing of small RNA duplexes: mechanistic insights and applications. Nucleic Acids Res 2017;45:10369–10379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taxman DJ, Livingstone LR, Zhang J, et al. . Criteria for effective design, construction, and gene knockdown by shRNA vectors. BMC Biotechnol 2006;6:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Reynolds A, Leake D, Boese Q, et al. . Rational siRNA design for RNA interference. Nature Biotechnol 2004;22:326. [DOI] [PubMed] [Google Scholar]
- 30. Ding Y, Chan CY, Lawrence CE. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res 2004;32:W135–W141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ui-Tei K, Naito Y, Saigo K. Guidelines for the selection of effective short-interfering RNA sequences for functional genomics. Methods Mol Biol 2007;361:201–216. [DOI] [PubMed] [Google Scholar]
- 32. Shao Y, Chan CY, Maliyekkel A, et al. . Effect of target secondary structure on RNAi efficiency. RNA 2007;13:1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reynolds A, Leake D, Boese Q, et al. . Rational siRNA design for RNA interference. Nat Biotechnol 2004;22:326–330. [DOI] [PubMed] [Google Scholar]
- 34. Pei Y, Tuschl T. On the art of identifying effective and specific siRNAs. Nat Methods 2006;3:670–676. [DOI] [PubMed] [Google Scholar]
- 35. Bofill-De Ros X, Gu S. Guidelines for the optimal design of miRNA-based shRNAs. Methods 2016;103:157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taxman DJ, Moore CB, Guthrie EH, et al. . Short hairpin RNA (shRNA): design, delivery, and assessment of gene knockdown. RNA Therap. Springer 2010;139–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Qureshi A, Tantray VG, Kirmani AR, et al. . A review on current status of antiviral siRNA. Rev Med Virol 2018;28:e1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gu S, Jin L, Zhang Y, et al. . The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell 2012;151:900–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gu S, Jin L, Zhang F, et al. . Thermodynamic stability of small hairpin RNAs highly influences the loading process of different mammalian Argonautes. Proc Natl Acad Sci U S A 2011;108:9208–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matveeva OV, Kang Y, Spiridonov AN, et al. . Optimization of duplex stability and terminal asymmetry for shRNA design. PloS One 2010;5:e10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Snøve O, Rossi JJ. Toxicity in mice expressing short hairpin RNAs gives new insight into RNAi. Genome Biol 2006;7:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van Gestel M, Van Erp S, Sanders L, et al. . shRNA-induced saturation of the microRNA pathway in the rat brain. Gene Ther 2014;21:205. [DOI] [PubMed] [Google Scholar]
- 43. Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell 2003;115:209–216. [DOI] [PubMed] [Google Scholar]
- 44. Bridge AJ, Pebernard S, Ducraux A, et al. . Induction of an interferon response by RNAi vectors in mammalian cells. Nat Genet 2003;34:263. [DOI] [PubMed] [Google Scholar]
- 45. Judge AD, Sood V, Shaw JR, et al. . Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat Biotechnol 2005;23:457. [DOI] [PubMed] [Google Scholar]
- 46. McBride JL, Boudreau RL, Harper SQ, et al. . Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc Natl Acad Sci U S A 2008;105:5868–5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grimm D, Wang L, Lee JS, et al. . Argonaute proteins are key determinants of RNAi efficacy, toxicity, and persistence in the adult mouse liver. J Clin Invest 2010;120:3106–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maude SL, Frey N, Shaw PA, et al. . Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Johnson LA, Scholler J, Ohkuri T, et al. . Rational development and characterization of humanized anti–EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 2015;7:275ra222–275ra222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Naso MF, Tomkowicz B, Perry WL, et al. . Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 2017;31:317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Makarova KS, Wolf YI, Alkhnbashi OS, et al. . An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 2015;13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shmakov S, Abudayyeh OO, Makarova KS, et al. . Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell 2015;60:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cong L, Ran FA, Cox D, et al. . Multiplex genome engineering using CRISPR/Cas systems. Science 2013;339:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mali P, Yang L, Esvelt KM, et al. . RNA-guided human genome engineering via Cas9. Science 2013;339:823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cong L, Ran FA, Cox D, et al. . Multiplex genome engineering using CRISPR/Cas systems. Science 2013;1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liu X, Homma A, Sayadi J, et al. . Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci Rep 2016;6:19675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ran FA, Cong L, Yan WX, et al. . In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015;520:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moreno-Mateos MA, Vejnar CE, Beaudoin J-D, et al. . CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 2015;12:982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dang Y, Jia G, Choi J, et al. . Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol 2015;16:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. G-h C, Wang Q-L, Liu Q. In silico meets in vivo: towards computational CRISPR-based sgRNA design. Trends Biotechnol 2017;35:12–21. [DOI] [PubMed] [Google Scholar]
- 61. Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet 2011;45, 45:247–271. [DOI] [PubMed] [Google Scholar]
- 62. Ceccaldi R, Rondinelli B, D'Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol 2016;26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kim D, Bae S, Park J, et al. . Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 2015;12:237–243231 p following 243. [DOI] [PubMed] [Google Scholar]
- 64. Tsai SQ, Nguyen NT, Malagon-Lopez J, et al. . CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 2017;14:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cameron P, Fuller CK, Donohoue PD, et al. . Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods 2017;14:600–606. [DOI] [PubMed] [Google Scholar]
- 66. Tsai SQ, Zheng Z, Nguyen NT, et al. . GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 2015;33:187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Frock RL, Hu J, Meyers RM, et al. . Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 2015;33:179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yan WX, Mirzazadeh R, Garnerone S, et al. . BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat Commun 2017;8:15058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lander ES, Baylis F, Zhang F, et al. . Adopt a moratorium on heritable genome editing. Nature 2019;567:165–168. [DOI] [PubMed] [Google Scholar]
- 70. Fu Y, Sander JD, Reyon D, et al. . Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature Biotechnol 2014;32:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ran FA, Hsu PD, Lin C-Y, et al. . Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013;154:1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tsai SQ, Wyvekens N, Khayter C, et al. . Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 2014;32:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kleinstiver BP, Pattanayak V, Prew MS, et al. . High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016;529:490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zetsche B, Gootenberg JS, Abudayyeh OO, et al. . Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015;163:759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kim D, Kim J, Hur JK, et al. . Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 2016;34:863. [DOI] [PubMed] [Google Scholar]
- 76. Chew WL. Immunity to CRISPR Cas9 and Cas12a therapeutics. Wiley Interdiscip Rev Syst Biol Med 2018;10:e1408. [DOI] [PubMed] [Google Scholar]
- 77. Chuang CK, Chen CH, Huang CL, et al. . Generation of GGTA1 mutant pigs by direct pronuclear microinjection of CRISPR/Cas9 plasmid vectors. Anim Biotechnol 2017;28:174–181. [DOI] [PubMed] [Google Scholar]
- 78. Raveux A, Vandormael-Pournin S, Cohen-Tannoudji M. Optimization of the production of knock-in alleles by CRISPR/Cas9 microinjection into the mouse zygote. Sci Rep 2017;7:42661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kim S, Kim D, Cho SW, et al. . Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 2014;24:1012–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Qin W, Dion SL, Kutny PM, et al. . Efficient CRISPR/Cas9-mediated genome editing in mice by zygote electroporation of nuclease. Genetics 2015;200:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Yang H, Wang H, Shivalila CS, et al. . One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 2013;154:1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sun W, Ji W, Hall JM, et al. . Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew Chem Int Ed Engl 2015;54:12029–12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li L, He Z-Y, Wei X-W, et al. . Challenges in CRISPR/CAS9 delivery: potential roles of nonviral vectors. Hum Gene Ther 2015;26:452–462. [DOI] [PubMed] [Google Scholar]
- 84. Gori JL, Hsu PD, Maeder ML, et al. . Delivery and specificity of CRISPR/Cas9 genome editing technologies for human gene therapy. Hum Gene Ther 2015;26:443–451. [DOI] [PubMed] [Google Scholar]
- 85. Long C, Amoasii L, Mireault AA, et al. . Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016;351:400–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Carroll KJ, Makarewich CA, McAnally J, et al. . A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc Natl Acad Sci U S A 2016;113:338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Al Yacoub N, Romanowska M, Haritonova N, et al. . Optimized production and concentration of lentiviral vectors containing large inserts. J Gene Med 2007;9:579–584. [DOI] [PubMed] [Google Scholar]
- 88. Kumar M, Keller B, Makalou N, et al. . Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther 2001;12:1893–1905. [DOI] [PubMed] [Google Scholar]
- 89. Canté-Barrett K, Mendes RD, Smits WK, et al. . Lentiviral gene transfer into human and murine hematopoietic stem cells: size matters. BMC Res Notes 2016;9:312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Roehm PC, Shekarabi M, Wollebo HS, et al. . Inhibition of HSV-1 replication by gene editing strategy. Sci Rep 2016;6:23146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Senís E, Fatouros C, Große S, et al. . CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol J 2014;9:1402–1412. [DOI] [PubMed] [Google Scholar]
- 92. Swiech L, Heidenreich M, Banerjee A, et al. . In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 2015;33:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim D, Kim J, Hur JK, et al. . Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 2016;34:863–868. [DOI] [PubMed] [Google Scholar]
- 94. Kim D, Kim J, Hur JK, et al. . Erratum: genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 2016;34:888. [DOI] [PubMed] [Google Scholar]
- 95. Kleinstiver BP, Tsai SQ, Prew MS, et al. . Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol 2016;34:869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gao Z, Herrera-Carrillo E, Berkhout B. Improvement of the CRISPR-Cpf1 system with ribozyme-processed crRNA. RNA Biol 2018;15:1458–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Herrera-Carrillo E, Gao ZL, Harwig A, et al. . The influence of the 5-terminal nucleotide on AgoshRNA activity and biogenesis: importance of the polymerase III transcription initiation site. Nucleic Acids Res 2017;45:4036–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gao Z, Herrera-Carrillo E, Berkhout B. RNA Polymerase II Activity of type 3 pol III promoters, Mol Ther Nucleic Acids 2018;12:135–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gao Z, Herrera-Carrillo E, Berkhout B. A single H1 promoter can drive both guide RNA and endonuclease expression in the CRISPR-Cas9 system. Mol Ther Nucleic Acids 2019;14:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang G, Zhao N, Berkhout B, et al. . A combinatorial CRISPR-Cas9 attack on HIV-1 DNA extinguishes all infectious provirus in infected T cell cultures. Cell Rep 2016;17:2819–2826. [DOI] [PubMed] [Google Scholar]
- 101. Abdel-Mohsen M, Kuri-Cervantes L, Grau-Exposito J, et al. . CD32 is expressed on cells with transcriptionally active HIV but does not enrich for HIV DNA in resting T cells. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]