

Abstract
Invading cancer cells extend cell protrusions, which guide cancer-cell migration and invasion, eventually leading to metastasis. The formation and activity of cell protrusions involve the localization of molecules and organelles at the cell front; however, it is challenging to precisely isolate these subcellular structures at the single-cell level for molecular analysis. Here, we describe a newly developed microfluidic platform capable of high-throughput isolation of cell protrusions at single-cell precision for profiling subcellular gene expression. Using this microfluidic platform, we demonstrate the efficient generation of uniform cell-protrusion arrays (more than 5000 cells with protrusions) for a series of cell types. We show precise isolation of cell protrusions with high purity at single-cell precision for subsequent RNA-Seq analysis, which was further validated by RT-qPCR and RNA FISH. Our highly controlled protrusion isolation method opens a new avenue for the study of subcellular functional mechanisms and signaling pathways in metastasis.
Keywords: analytical methods, cell protrusion, microfluidics, gene expression, RNA sequencing
Graphical Abstract
Subcellular-localized biomolecules, such as messenger RNA (mRNA) and proteins, are significant drivers of biological processes that regulate cellular functions, including embryonic development, synaptic interactions, wound healing, and metastasis.[1–3] Cell protrusions are dynamic structures that are critical for multiple cellular processes, in particular, cancer metastasis.[4, 5] Localized biomolecules at the leading edge of protrusions are thought to contribute to the generation and maintenance of cell asymmetry during migration and invasion.[5, 6] The study of subcellular gene expression in cell protrusions may help further characterize the underlying molecular mechanisms of cancer metastasis and may reveal a unique therapeutic approach to prevent tumor progression by controlling the cell-protrusion process.
Several approaches can be utilized to interrogate gene expression in the protrusions of migrating cancer cells. Imaging-based methods that directly label target mRNAs using complementary oligonucleotide probes conjugated with fluorophores, including fluorescent in situ hybridization (FISH),[7] the MS2 system,[8] and padlock probes,[9] allow for the visualization and analysis of only a limited number of localized mRNAs. Although sequential FISH,[10] multiplexed error-robust FISH,[11] and fluorescent in situ RNA sequencing[12] were developed to improve throughput, they are still limited by low efficiency and sophisticated image analysis. Global gene-expression analysis of isolated cell protrusions has been attempted to identify protrusion-enriched mRNAs using transwell-based protrusion fractionation,[13, 14] aspiration,[15] or microdissection,[16] combined with microarray or RNA sequencing (RNA-Seq). However, due to the technical limitations, the above methods often encounter challenges in operation controllability, sample purity, isolation precision, and so on. Furthermore, existing single-cell RNA-Seq techniques require the quantity of input mRNAs from at least one cell.[17] However, high-throughput isolation of a large number of cell protrusions while retaining high purity is still a challenge.
Recent developments in microfluidics allow for high-throughput and precise manipulation at the single-cell level, leading to more accurate subcellular isolation.[18–22] Here, we present a high-throughput microfluidic platform, called protrusion-generating chip (PG-Chip), an in vitro assay to mimic mesenchymal cell migration and isolate highly pure cell protrusions for comprehensive subcellular biomolecule profiling. Large-area uniform cell-protrusion arrays (more than 5000 cells with protrusions) were generated within the PG-Chips for various cell types, including a series of cancer cells, fibroblasts, endothelial cells, and neuronal cells. These cell-protrusion arrays allowed for the high-throughput isolation of highly pure cell protrusions, ranging from one to thousands, for downstream molecular analysis using laser capture microdissection (LCM). As a demonstration, we isolated mRNA from cell protrusions and performed RNA-Seq to identify protrusion-localized gene expression patterns, which were validated by quantitative real-time polymerase chain reaction (RT-qPCR) and RNA FISH.
Cytology patterns of cells at the tumor front invading into surrounding tissues often show active cell protrusions, which assist the typical mesenchymal mode migration (Figure 1 A).[5] To design an in vitro assay that provides the mechanical confinements that mimic cancer cell migration and thereby allows for the isolation of cell protrusions in high purity, we generated the PG-Chip using the cell-printing method (Figure 1 B–D).[23] The PG-Chip consists of a polydimethylsiloxane (PDMS) mold placed on a polyethylene naphthalate (PEN) membrane that enables infrared (IR) laser capturing and ultraviolet (UV) laser cutting (Figure 2 A).[24] Within the PG-Chip, arrayed micro-hooks were designed along one side of the flow channels with 3-mm gaps to enable the trapping of single cells (Supporting Information, Figure S1 A). We used MDA-MB-231 cells, which are mesenchymal-like breast cancer cells, in this study. Accordingly, we designed the width and height of the micro-hooks to be 14 μm and 20 μm, respectively, and the width of the flow channels to be 43 μm. These parameters are designed following the established principle[23, 25] and according to the diameter of cells to achieve high loading efficiency. By applying a negative pressure (approximately 1 psi) from the outlet, about 10 μL of cell suspensions with a concentration of about 5 × 106 cells mL−1 at the inlet flowed into the channels, and single cells were trapped by the micro-hooks, forming single-cell arrays (Figure 2 B and Supporting Information, Movie S1). A large-area single-cell array could be routinely prepared with approximately 97 % loading efficiency (Figure 2 C and Supporting Information, Figure S1 B).
Figure 1.

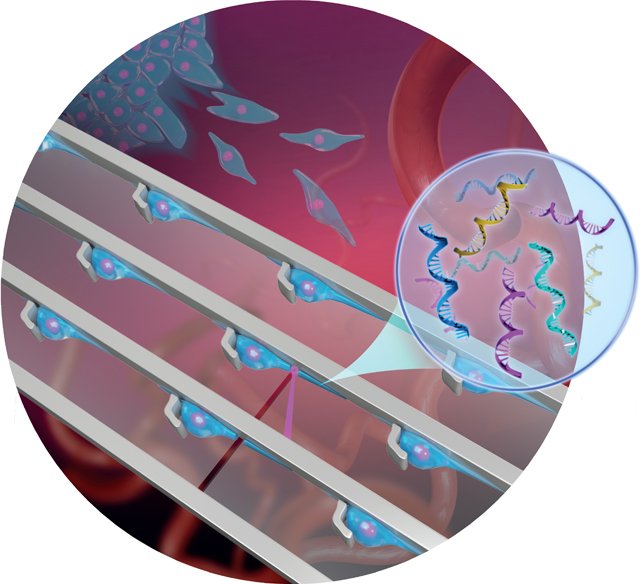
Overview of the microfluidic platform for molecular analysis of cell protrusions. A) Schematic illustration of invasive cells in a migrating tumor front. B) The generation of uniform cell-protrusion arrays in microfluidic chips, which provide an in vitro model for cancer cell migration. C,D) After removing the microfluidic mold and fixing, selected cell protrusions are cut using LCM at single-cell precision, collected on the surface of LCM caps, and lysed for biomolecular analysis.
Figure 2.
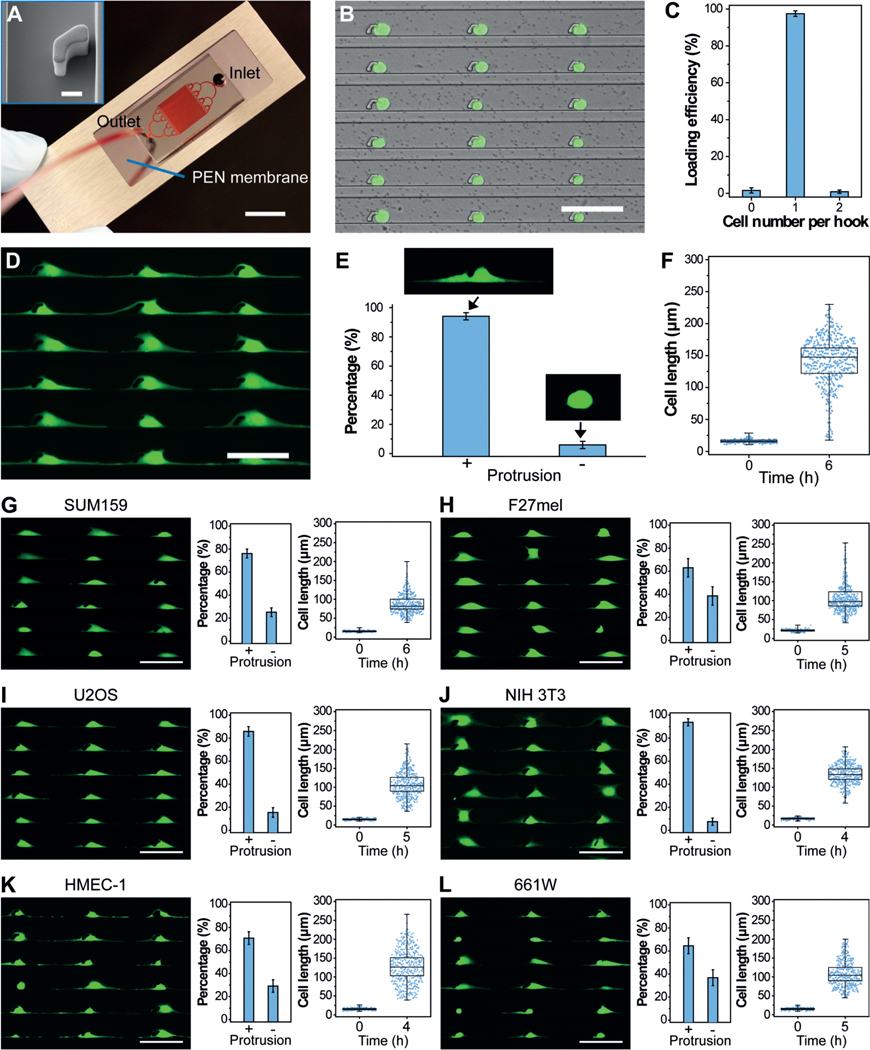
PG-Chip for the generation of uniform cell-protrusion arrays. A) A typical PG-Chip consisted of a PDMS microfluidic mold and a PEN membrane slide. Scale bar, 1 cm. Red dye was injected to visualize the channel networks. Insert image shows the morphology of the micro-hook (Scale bar, 10 mm). B) A representative image and C) loading efficiency of the generated array of single MDA-MB-231/GFP cells using the PG-Chip. Scale bar, 100 μm. D) Uniform cell-protrusion arrays after incubation of the single-cell arrays within the PG-Chip for 6 h. Scale bar, 100 μm. E) Bar graph illustrating the percentage of cells with and without protrusions. Data are expressed as mean standard deviation (SD) F) Analysis of the cell lengths before and after incubation. Whiskers illustrate the range. G–L) Images of uniform cell-protrusion arrays, quantification of the percentage of cells with and without protrusions, and lengths of G) SUM159 breast cancer cells, H) F27 melanoma cells, I) U2OS osteosarcoma cells, J) NIH 3T3 fibroblast cells, K) HEMC-1 endothelial cells, and L) 661W photoreceptor cells after incubation. Data is expressed as mean SD. Whiskers indicate the range. Scale bars, 100 μm.
One critical aspect of mesenchymal-like tumor-cell migration is the asymmetrical extension of cell protrusions toward the leading front. Similar to this process, the trapped single cells in the PG-Chip migrated by extending cell protrusions to both the left and right sides of the micro-hooks in the confined spaces (Supporting Information, Movie S2), leading to the formation of a uniform cell-protrusion array after incubation for 6 hours (Figure 2 D). Importantly, we found that the cytophilic coating of PEN membranes and the distances between adjacent micro-hooks significantly affected the generation of uniform cell-protrusion arrays. As shown in Figure S2 A, cells on the PEN membranes coated with extracellular matrix proteins, including basement membrane extract (BME), poly-L-lysine (PLL), and fibronectin (FN), extended distinct protrusions. Among them, FN-coated PEN membranes had the best performance. The distances between adjacent micro-hooks also influenced the generation of cell protrusions. Short distances, such as 50 μm, did not allow enough space for the cells to extend distinct protrusions. After the distances were increased to 100–200 μm, most of the trapped cells (approximately 94 %) extended distinct cell protrusions and formed uniform cell-protrusion arrays (Figure 2 E,F and Supporting Information, Figure S2 B). About 5000 cells with uniform cell protrusions accumulated in one chip (Supporting Information, Figure S3). Consistent with published literature,[26] the cell protrusions were composed of cytoskeletal components, including F-actin and tubulin (Supporting Information, Figure S4).
Further to MDA-MB-231 breast cancer cells, the protrusions of other cancer and non-cancer cell types are essential in numerous biological processes. Therefore, the generation of uniform protrusion arrays for use with many cell types is crucial for the global study of protrusion-localized functional molecules. Using the PG-Chip, we generated single-cell arrays (Supporting Information, Figure S5) and cell-protrusion arrays (Figure 2 G–L) with high efficiency for a variety of cell types, including SUM159 breast cancer cells (Figure 2 G), F27 melanoma (F27mel) cells (Figure 2 H), U2OS osteosarcoma cells (Figure 2 I), NIH 3T3 fibroblast cells (Figure 2 J), HEMC-1 endothelial cells (Figure 2 K), and 661W photoreceptor cells (Figure 2 L). Owing to the genotype and phenotype of each cell type, they show the various capability to generate cell protrusions. For example, SUM159 cells, which are less invasive than MDA-MB-231 cells, show shorter cell lengths, indicating the ability of cell protrusion generation relevant to cell migration and invasion. Therefore, such uniform cell-protrusion arrays provide an opportunity for the in vitro study of protrusion-based molecular mechanisms in metastasis, angiogenesis, wound healing, and other biological processes, demonstrating the versatility and broad utility of the PG-Chip.
High-throughput isolation of cell protrusions with high purity presents a great challenge, using current methods, for the subsequent molecular analysis of protrusion-localized biomolecules. This challenge could be overcome by the combined utilization of our PG-Chip and the LCM technique, which has been widely used to isolate single cells with high purity via direct microscopic visualization.[27] This strategy allowed us to specifically select and isolate cell protrusions with single-cell precision (Figure 3 A). To prevent cross-contamination of the cytosol during the LCM process, we fixed the cell-protrusion arrays before isolation. As shown in Figure S6A and Movie S3 in the Supporting Information, the selected cell protrusions were automatically captured by an IR laser, cut with high efficiency and accuracy by a UV laser, and collected on the surface of the LCM cap; meanwhile, cell bodies were left on the PEN membranes.
Figure 3.

Precise isolation of cell protrusions at single-cell precision using LCM. A) Representative images of isolated cell protrusions (upper panel) and corresponding cell bodies remaining on the PEN membranes (lower panel), indicating the high purity of the collected cell protrusions. Actin was stained with Alexa Fluor 488-Phalloidin, and nuclei were stained with Hoechst. B) High-throughput isolation of cell protrusions, ranging from a single protrusion to hundreds, in one run. Scale bars, 100 μm.
To verify the purity of the isolated cell protrusions, we labeled F-actin with phalloidin and nuclei with Hoechst and the nuclear marker histone deacetylase 1 (HDAC1). We observed that only cell protrusions were isolated, and all nuclei remained on the PEN membranes, demonstrating the high purity of the isolated cell protrusions (Figure 3 B and Supporting Information, Figure S6B). Importantly, LCM isolation of cell protrusions was controllable, and any number of cell protrusions, ranging from one to hundreds, at single-cell precision were successfully isolated in a high-throughput manner (Figure 3 C). This is currently impossible to achieve using other existing methods. Therefore, our system provides a robust and highly selective way to isolate highly pure cell protrusions from uniform cell-protrusion arrays in a high-throughput manner, superior to existing sampling-based methods for protrusion isolation.
Cell protrusions isolated using our novel method could be used to globally profile functional molecules (for example, microRNAs, mRNA, mitochondrial DNA (mtDNA), proteins, and even organelles) in the protrusions using advanced analytical techniques including RNA-Seq or mass spectrometry.[28] As a demonstration, we assessed the gene-expression patterns in the protrusions of MDA-MB-231 cells. Although we could isolate a single protrusion, existing commercial single-cell RNA-Seq techniques require the amount of mRNA from at least one cell and to achieve this amount of input RNA, hundreds of cell protrusions may be required.
To investigate how many cell protrusions would be needed to fulfil the requirement for a standard single-cell RNA-Seq protocol, we isolated several samples, containing approximately 20, 100, 200, or 600 cell protrusions, for mRNA extraction. Of note, isolation of intact mRNAs from fixed single cells was still challenging due to the high rate of rapid mRNA degradation during the experimental protocol. To obtain high-quality protrusion-localized mRNAs (Supporting Information, Figure S7), we employed a recently reported method, which was developed for mRNA purification and transcriptomic profiling of fixed single cells.[29] The extracted mRNAs were then amplified using the Smart-Seq2 protocol,[30] and the subsequently generated complementary DNAs (cDNAs) were evaluated using a Bioanalyzer. Representative traces (Figure 4 A) illustrated the successful isolation of high-quality mRNA from the isolated cell protrusions. For samples containing only 20 cell protrusions, we detected signals; however, they could not satisfy the criteria for a standard single-cell RNA-Seq protocol.
Figure 4.

Profiling gene-expression patterns in cell protrusions by RNA-Seq. A) Bioanalyzer traces of amplified cDNA from mRNAs that are enriched in cell protrusions. Samples with various numbers of cell protrusions were analyzed. B) Quantification of the number of genes detected in various cell-protrusion samples. C,D) Gene-expression correlation C) between cell-protrusion samples and D) between protrusion samples and cell-body samples with Pearson correlation coefficients (r). E) Validation of protrusion-localized genes by RT-qPCR analyses. The RNA localization ratios were calculated as the relative expression level in the protrusions fraction compared with the cell-body fraction and were normalized to ACTB. Data are expressed as mean SD. F) Validation of protrusion-localized genes by FISH. White arrows indicate the RAB13 mRNAs in cell protrusions. Scale bar, 50 μm.
We then performed RNA-Seq with equal amounts of mRNA from cell protrusions and cell bodies to identify enriched mRNA species in cell protrusions. We used a different number of cell protrusions as a starting material to perform RNA-Seq. Our results show that the use of 600 protrusions consistently allowed for the detection of more than 8000 genes (Figure 4 B, for reliably detected genes with transcripts per million (TPM) > 1). Less than 4000 genes were detected from samples containing 20 cell protrusions. We speculate that such a difference can be attributed to the bias and limited sensitivity of PCR, which leads to the loss of transcripts with low copy numbers in these samples. The correlations between different protrusion samples containing 600 cell protrusions were quite high using Pearson correlation coefficients (ranging from 0.84–0.89) (Figure 4 C), indicating the high reproducibility of our method. Furthermore, we found many genes enriched in the cell body (Figure 4 D). This is consistent with the fact that the cell bodies contain nuclei and other organelles that are enriched in mRNA molecules.
To determine whether spatial control of gene expression occurs in cell protrusions, we analyzed protrusion-enriched mRNAs by combining results from two R packages commonly used for differential gene-expression analysis (limma and samr) and identified 439 genes with at least two-fold increased expression in protrusions (Data file S1 in the Supporting Information). To validate the RNA-Seq results, we carried out RT-qPCR to quantify several protrusion-enriched mRNAs identified by RNA-Seq, including CENPB, RAB13, ZEB1, ANP32B, CORO1A, and PINK1, using ACTB (β-Actin) as a reference gene and ARPC3, a non-enriched gene, as a control. Consistent with the RNA-Seq results, we observed protrusion-localized enrichment of CENPB (2.3-fold), RAB13 (8.1-fold), ZEB1 (1.6-fold), ANP32B (2.0-fold), CORO1A (4.6-fold), and PINK1 (1.7-fold, Figure 4 E). We then performed FISH to image the subcellular localization of specific mRNAs. As shown in Figure 4 F and Figure S8 in the Supporting Information, RAB13 transcripts are prominently localized at the leading edges of cell protrusions, and PINK1 transcripts are also present in the cell protrusions, consistent with our RNA-Seq and RT-qPCR results.
In summary, we developed a microfluidic platform capable of the high-throughput generation and precise isolation of hundreds of highly pure cell protrusions for profiling the gene expression in protrusions of migrating cancer cells. Compared with the existing approaches, our microfluidics-based approach has several advantages. First, the PG-Chip is high-throughput, with the ability to array thousands of cells in one run. It precisely aligns the cell bodies and protrusions of various cell types (for example, cancer cells, fibroblasts, endothelial cells, and neuronal cells). This level of uniformity cannot be achieved by any other current method. Second, up to thousands of cell protrusions can be precisely and rapidly isolated with high purity, which is essential for subsequent functional analysis of microRNAs, mRNAs, proteins, and even organelles, amongst others. Finally, we isolated protrusion-localized mRNAs and performed RNA-Seq to profile subcellular gene expression. The analysis and validation of the RNA-Seq data reveal consistency among independent samples, indicating that our method is an accurate and reproducible way to profile subcellular gene expression. Our results indicate that the comprehensive analysis of other protrusion-localized molecules, even from single protrusions, is highly possible with the application of appropriate advanced analytical techniques. We believe that this microfluidic platform has promising potential for the comprehensive understanding of cell-protrusion-related signaling during various physiological processes.
Supplementary Material
Acknowledgements
We thank Dr. Muayyad Al-Ubaidi from the University of Houston for kindly providing 661W cells. We thank Dr. Rongfu Wang from Houston Methodist Research Institute for generously providing F27mel cells. We are grateful for funding support from the original R21 CA191179 and its supplement and R01 DA035868.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201903694.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Pengchao Zhang, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA) and Department of Cell and Developmental Biology, Weill Medical College of Cornell University New York, NY 10065 (USA).
Xin Han, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA) and Department of Cell and Developmental Biology, Weill Medical College of Cornell University New York, NY 10065 (USA); School of Medicine and Life Sciences, Nanjing University of Chinese Medicine, Nanjing, 210023 (P. R. China).
Jun Yao, Department of Genetics, The University of Texas MD Anderson Cancer Center Houston, TX 77030 (USA).
Ning Shao, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA) and Department of Cell and Developmental Biology, Weill Medical College of Cornell University New York, NY 10065 (USA).
Kai Zhang, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA) and Department of Cell and Developmental Biology, Weill Medical College of Cornell University New York, NY 10065 (USA).
Yufu Zhou, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA) and Department of Cell and Developmental Biology, Weill Medical College of Cornell University New York, NY 10065 (USA).
Youli Zu, Department of Pathology and Genomic Medicine, Houston Methodist Research Institute, Houston, TX 77030 (USA).
Bin Wang, Department of Molecular and Cellular Oncology, The University of Texas MD Anderson Cancer Center Houston, TX 77030 (USA).
Lidong Qin, Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX 77030 (USA) and Department of Cell and Developmental Biology, Weill Medical College of Cornell University New York, NY 10065 (USA).
References
- [1].Holt CE, Bullock SL, Science 2009, 326, 1212–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Martin KC, Ephrussi A, Cell 2009, 136, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lundberg E, Borner GHH, Nat. Rev. Mol. Cell Biol 2019, 20, 285–302. [DOI] [PubMed] [Google Scholar]
- [4].Ridley AJ, Cell 2011, 145, 1012–1022. [DOI] [PubMed] [Google Scholar]
- [5].Friedl P, Wolf K, Nat. Rev. Cancer 2003, 3, 362–374. [DOI] [PubMed] [Google Scholar]
- [6].Wang T, Hamilla S, Cam M, Aranda-Espinoza H, Mili S, Nat. Commun 2017, 8, 896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Femino AM, Fay FS, Fogarty K, Singer RH, Science 1998, 280, 585–590. [DOI] [PubMed] [Google Scholar]
- [8].Tyagi S, Nat. Methods 2009, 6, 331–338. [DOI] [PubMed] [Google Scholar]
- [9].Larsson C, Grundberg I, Soderberg O, Nilsson M, Nat. Methods 2010, 7, 395–397. [DOI] [PubMed] [Google Scholar]
- [10].Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, Cai L, Nat. Methods 2014, 11, 360–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chen KH, Boettiger AN, Moffitt JR, Wang S, Zhuang X, Science 2015, 348, aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, Terry R, Jeanty SSF, Li C, Amamoto R, Peters DT, Turczyk BM, Marblestone AH, Inverso SA, Bernard A, Mali P, Rios X, Aach J, Church GM, Science 2014, 343, 1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mili S, Moissoglu K, Macara IG, Nature 2008, 453, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jakobsen KR, Sørensen E, Brøndum KK, Daugaard TF, Thomsen R, Nielsen AL, Mol J.. Signaling 2013, 8, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moccia R, Chen D, Lyles V, Kapuya E, Kalachikov YES, Spahn CMT, Frank J, Kandel ER, Barad M, Martin KC, J. Neurosci 2003, 23, 9409–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zivraj KH, Tung YCL, Piper M, Gumy L, Fawcett JW, Yeo GSH, Holt CE, Neurosci J.. 2010, 30, 15464 – 15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Grün D, van Oudenaarden A, Cell 2015, 163, 799–810. [DOI] [PubMed] [Google Scholar]
- [18].Sackmann EK, Fulton AL, Beebe DJ, Nature 2014, 507, 181–189. [DOI] [PubMed] [Google Scholar]
- [19].Duncombe TA, Tentori AM, Herr AE, Nat. Rev. Mol. Cell Biol 2015, 16, 554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ma Y, Han X, de Castro RB, Zhang P, Zhang K, Hu Z, Qin L, Sci. Adv 2018, 4, eaas9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hou S, Zhao L, Shen Q, Yu J, Ng C, Kong X, Wu D, Song M, Shi X, Xu X, OuYang W-H, He R, Zhao X-Z, Lee T, Brunicardi FC, Garcia MA, Ribas A, Lo RS, Tseng H-R, Angew. Chem. Int. Ed 2013, 52, 3379–3383; Angew. Chem. 2013, 125, 3463 – 3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rakszewska A, Stolper RJ, Kolasa AB, Piruska A, Huck WTS, Angew. Chem. Int. Ed 2016, 55, 6698–6701; Angew. Chem. 2016, 128, 6810 – 6813. [DOI] [PubMed] [Google Scholar]
- [23].Zhang K, Chou C-K, Xia X, Hung M-C, Qin L, Proc. Natl. Acad. Sci. USA 2014, 111, 2948–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Espina V, Wulfkuhle JD, Calvert VS, VanMeter A, Zhou W, Coukos G, Geho DH, Petricoin EF, Liotta LA, Nat. Protoc 2006, 1, 586–603. [DOI] [PubMed] [Google Scholar]
- [25].Zhang K, Han X, Li Y, Li SY, Zu Y, Wang Z, Qin L, J. Am. Chem. Soc 2014, 136, 10858 – 10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shankar J, Messenberg A, Chan J, Underhill TM, Foster LJ, Nabi IR, Cancer Res. 2010, 70, 3780–3790. [DOI] [PubMed] [Google Scholar]
- [27].Simone NL, Bonner RF, Gillespie JW, Emmert-Buck MR, Liotta LA, Trends Genet. 1998, 14, 272–276. [DOI] [PubMed] [Google Scholar]
- [28].Zhang L, Vertes A, Angew. Chem. Int. Ed 2018, 57, 4466–4477; Angew. Chem. 2018, 130, 4554 – 4566. [DOI] [PubMed] [Google Scholar]
- [29].Thomsen ER, Mich JK, Yao Z, Hodge RD, Doyle AM, Jang S, Shehata SI, Nelson AM, Shapovalova NV, Levi BP, Ramanathan S, Nat. Methods 2016, 13, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, Sandberg R, Nat. Methods 2013, 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.