

Abstract
The first synthesis of the tetrasaccharide fragment of the anthracycline natural product Arugomycin is described. A reagent controlled dehydrative glycosylation method involving cyclopropenium activation was utilized to synthesize the α-linkages with complete anomeric selectivity. The synthesis was completed in 20 total steps, and in 2.5% overall yield with a longest linear sequence of 15 steps.
Graphical Abstract

2,6-Dideoxy sugars are often found in natural products of therapeutic significance,1,2 and alterations of the sugar composition of these natural products have been shown to significantly impact their biological activity.3–7 While this approach holds promise for new avenues for therapeutic development, it is impeded by the inherent difficulties in deoxy-sugar oligosaccharide synthesis.2,8 Conducting α-selective glycosylations of 2,6-dideoxy sugars is a synthetic challenge, primarily due to the lack of chemical handles at both the C-6 position to promote conformational bias9,10 and at the C-2 position for the use of a chiral auxiliary.11–13 Deoxy sugar donor intermediates are also more sensitive to hydrolysis than their fully substituted sugar counterparts,14 and species such as halides and trichloroacetimidates have to be either freshly prepared and used immediately or generated in situ to function as effective donors in direct glycosylations.15−20 A variety of novel direct,21−30 indirect,31−39 and de novo40−46 glycosylation methods have emerged in recent years to address these drawbacks, but the extension of these methods to complex oligosaccharide synthesis remains at the frontier of carbohydrate synthesis.
Our group has had an enduring interest in developing methods for reagent-controlled direct dehydrative glycosylation using shelf-stable 2-deoxy hemiacetal donors. In these approaches, the stereochemical outcome of the reaction is controlled by the promoter system, with sulfonyl chloride promoters affording β-linked structures47,48 and a combination of a cyclopropene-1-thione and oxalyl bromide affording α- linked compounds.49−51 Both our lab and other groups have applied the former approach to complex β-linked 2-deoxy oligosaccharide synthesis,52−56 but the utility of the latter method in α-linked oligosaccharide synthesis has yet to be established. In order to determine if this chemistry would be useful in oligosaccharide synthesis, we decided to apply it to the synthesis of the tetrasaccharide fragment of Arugomycin (Scheme 1).
Scheme 1.

Retrosynthesis of Arugomycin Tetrasaccharide

Arugomycin, first isolated from the bacteria Streptomyces violaceochromogenes in 1983, was shown to have moderate activity against Gram-Positive bacteria (S. aureus, B. subtilis, M. luteus, MIC = 12.5 μg/mL),57−59 and it also has potential as an antitumor agent from its ability to intercalate DNA similar to other anthracyclines.60 Notably, this compound possesses tetrasaccharide and trisaccharide deoxy-sugar chains, which represent challenging synthetic targets for reasons outlined above. We particularly viewed the tetrasaccharide chain, which consists of α-linked L-oliose configured residues with a fumaric acid moiety at the nonreducing end, as an ideal model system to test our method. Here we report the first synthesis of this tetrasaccharide, complete with fumarate attachment, starting from L-fucose.
From our retrosynthetic analysis of target compound 1, we considered an approach where the fumarate moiety could be appended to tetrasaccharide 3 at the later stage. Tetrasaccharide 3 could then arise from a [2 + 2] coupling of disaccharide donor 5 and acceptor 4. Both disaccharides could originate from monosaccharide coupling partners 6, 7, and 8, which would in turn be derived from L-fucal.
We began our synthesis with the construction of L-fucal derivative 11 as a precursor to the monosaccharide coupling partners (Scheme 2A). Commercially available L-fucose was peracetylated with acetic anhydride and DMAP to afford compound 9 in 96% yield. Compound 9 was then halogenated at the anomeric position with PBr3 and subsequently subjected to a modified Fischer-Zach protocol to afford fucal 10 in 70% yield.61 Compound 10 was then deacetylated with catalytic sodium methoxide, silylated at the C3 position with tert butyldimethylsilyl chloride (TBSCl), and then C-4 was protected as a 2-naphthylmethyl (Nap) ether using 2- naphthylmethyl bromide (NapBr) and sodium hydride to afford common core fucal 11 in 68% yield over three steps.62
Scheme 2.

Synthesis of the Starting Monosaccharides: Common Core Fucal 9 (A), Oliose Donors 7 and 8 (B), and Oliose Acceptor 6 (C)
With the requisite fucal in hand, we turned our attention to the synthesis of the monosaccharide coupling partners. Hemiacetal donor 7 was produced by hydration of fucal 11 with catalytic triphenylphosphine hydrobromide (Scheme 2B).44 To obtain 8, fucal 11 was desilylated with tetrabutylammonium fluoride (TBAF) to afford fucal 12 in 84% yield, followed by methylation at the C3 position with methyl iodide in 91% yield.63 Finally, hydration of the glycal with catalytic triphenyl phosphine hydrobromide provided 8 in 73% yield.64
Synthesis of monosaccharide acceptor 6 commenced with dehydrative glycosylation of donor 8 using a coactivation based variant of our published methodology for secondary alcohol acceptors.62 To this end, thione 14 was activated with oxalyl bromide in (5.5:1) TCE/CH2Cl2, followed by the sequential addition of para-methoxyphenol (PMP) at −10 °C, and donor 8. This order of activation minimized the formation of trehalose byproducts and afforded 13 in 93% yield exclusively as the α-anomer (Scheme 2C). Subsequent deprotection of the Nap group at C4 with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) produced monosaccharide acceptor 6 in 89% yield. With the necessary monosaccharides in hand, we turned our attention to disaccharide synthesis.
We initially investigated coupling of 8 and 6 using our previously optimized conditions for secondary alcohol acceptors at rt.51 These conditions afforded α-linked disaccharide 15 in a low yield of 14% (Table 1, entry 1). The major byproducts of the reaction were an α,α-trehalose derivative of 8, and a α- PMP glycoside derivative of 8, which we posit arose from aglycone elimination/transfer from acceptor 6.65 Lowering the temperature to 0 °C led to a slight increase in yield (Table 1, entry 2). Switching to a more basic proton scavenger 2,6-di-tert-butyl-4-methylpyridine (DTBMP) negatively impacted the yield and led to increased formation of PMP transfer byproduct (Table 1, entry 3). The use of THF as a cosolvent, a beneficial practice in our prior study with thione promoter reagent 14,51 also led to aglycone transfer and decreased our yield relative to the TCE/CH2Cl2 solvent condition (Table 1, entry 4). Unable to limit the formation of byproducts by solvent or scavenger adjustments, we elected to switch the stoichiometry of the glycosylation to make the donor the limiting reagent, which led to a slight drop in byproduct formation (Table 1, entry 5). Further increasing the stoichiometry of the acceptor resulted in a marked increase in yield to 64% (Table 1, entry 6). Reducing the temperature to −10 °C led to a further increase in yield to 74% (Table 1, entry 7). These latter conditions were used to scale up the reaction (Table 1, entry 8).
Table 1.
Optimization of Disaccharide 15
 | ||||
|---|---|---|---|---|
| entry | donor/acceptor | scavenger | temp | yield (α-only) |
| 1a | 2:1 | TTBP | rt | 14%b |
| 2a | 2:1 | TTBP | 0 °C | 44%b |
| 3a | 2:1 | DTBMP | 0 °C | 11%b |
| 4c | 2:1 | TTBP | 0 °C | 18%b |
| 5d | 1:2 | TTBP | 0 °C | 47%b |
| 6d | 1:3 | TTBP | 0 °C | 64%b |
| 7d | 1:3 | TTBP | −10 °C | 74%e |
| 8d,f | 1:3 | TTBP | −10 °C | 69%e |
TCE/CH2Cl2 (2.7:1).
NMR yield, product identified in a complex with α,α-trehalose.
TCE/CH2Cl2/THF (6.5:1:3.2).
TCE/CH2Cl2 (1.6:1).
Isolated yield.
300 mg scale of 8.
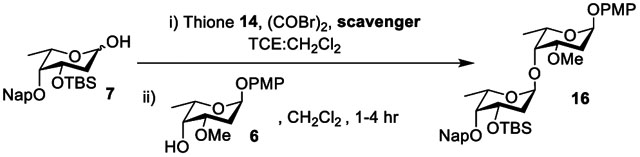
We applied a similar optimization strategy to the coupling of donor 7 and acceptor 6. Applying a 1:3 donor/acceptor ratio at 0 °C afforded α-linked disaccharide 16 in 55% yield by NMR integration (Table 2, entry 1). Again α,α-trehalose and derivatives of the donor 7 arising from intramolecular aglycone transfer were the major byproducts of the reaction. Furthermore, free p-methoxyphenol coeluted with 16 and could not be separated. Running the reaction at room temperature increased byproduct formation, and the use of THF as a cosolvent did not improve the yield (Table 2, entries 2 and 3). Rationalizing that byproduct formation was the result of inefficient proton scavenging, we again tried DTBMP, but this was again detrimental to the yield (Table 2, entry 4). Seeing the detrimental effect of increasingly Lewis basic scavengers on our substrates, we then investigated the alkene 4-allyl-1,2-dimethoxybenzene (ADB) as an alternate type of scavenger,66 but the use of this scavenger led to a complex mixture (Table 2, entry 5). Inspired by our last optimization, we then lowered the temperature of the system to −10 °C and we were pleased to observe an increase in yield of 16 to 87% with no decrease in α-selectivity (Table 2, entry 6). These latter conditions were used for scale-up with minimal impact on the yield (Table 2, entries 7 and 8).
Table 2.
Optimization of Disaccharide 16
 | ||||
|---|---|---|---|---|
| entry | donor/acceptor | scavenger | temp | yield (α-only)a |
| 1b | 1:3 | TTBP | 0 °C | 55% |
| 2b | 1:3 | TTBP | rt | 11% |
| 3c | 1:3 | TTBP | 0 °C | 53% |
| 4b | 1:3 | DTBMP | 0 °C | 38% |
| 5b | 1:3 | ADB | 0 °C | 2% |
| 6b | 1:3 | TTBP | −10 °C | 87% |
| 7b,d | 1:3 | TTBP | −10 °C | 74% |
| 8b,e | 1:3 | TTBP | −10 °C | 65% |
NMR yield, product identified in a complex with p-methoxyphenol.
TCE/CH2Cl2 (1.6:1).
TCE/CH2Cl2/THF (9.5:1:4.5).
400 mg scale 7.
1 g scale 7.
With disaccharides 15 and 16 in hand, we turned our attention to converting them to the requisite disaccharide coupling partners 5 and 4 (Scheme 3). To this end, DDQ deprotection of the Nap group of 16 gave disaccharide acceptor 4 in 89% yield, and we gratifyingly did not observe anomerization of the PMP group in the absence of a proton scavenger for this reaction.64 Disaccharide donor 5 was then generated with ceric ammonium nitrate in 64% yield. In this latter reaction we found that addition of sodium bicarbonate to this reaction was essential to limit the acid-catalyzed hydrolysis of the disaccharide.67
Scheme 3.

Disaccharide Deprotections
With the stage set for tetrasaccharide formation, we initially examined the [2 + 2] glycosylation using the optimal conditions for disaccharide 16 formation. Using our preactivation protocol, we reacted donor 5 and acceptor 4 in a 1:3 ratio at −10 °C and obtained tetrasaccharide 3 as a single α-anomer in 28% yield (Table 3, entry 1). Under these conditions donor 5 was not fully consumed, and we identified the major byproduct of this reaction as the α,α-trehalose derivative of donor 5. We additionally could quantitatively recover and reuse excess acceptor 4 through flash chromatography, reducing the impact of these low yields. In an attempt to improve the reaction outcome, we switched to the coactivation protocol described above, which had limited trehalose formation in the PMP glycosylation of monosaccharide donor 8 (Scheme 2C). This method resulted in a complex mixture of products and a significant amount of α-PMP glycoside of donor 5 was produced, signifying aglycone transfer (Table 3, entry 2). Reasoning that the majority of trehalose would be formed during the activation of the donor in the absence of the acceptor, we then cooled the reaction to −30 °C and then allowed the system to warm to −10 °C upon addition of the acceptor, but this failed to increase the yield (Table 3, entry 3), and the donor consumption was sluggish at this lower temperature. Increasing the reaction time to 23 h did not improve the yield (Table 3, entry 4), and we again explored nonbasic proton scavengers as a means of controlling byproduct formation without causing elimination of the PMP substituent of 4. Isobutylene oxide (IBO), which was reported to be effective in preventing halo acid formation in direct glycosylation methods involving halide ion release,27,68 did not benefit our system, instead affording a complex mixture of products (Table 3, entry 5). We next examined the strained alkene β-Pinene as an acid scavenger;69−71 however, this too afforded a complex mixture (Table 3, entry 6). Having established a fully selective, albeit low-yielding [2 + 2] glycosylation method, we decided to move forward with the synthesis.
Table 3.
Tetrasaccharide Optimization
 | ||||
|---|---|---|---|---|
| entryd | activation method | scavenger | temp | yield (α-only)a |
| 1 | preactivation | TTBP | −10 °C | 28% |
| 2 | coactivation | TTBP | −10 °C | trace |
| 3b | preactivation | TTBP | −30 °C → −10 °C | 31% |
| 4c | preactivation | TTBP | −10 °C | 30% |
| 5 | preactivation | IBO | −10 °C | trace |
| 6 | preactivation | β-Pinene | −10 °C | trace |
Isolated yield.
4 added at −30 °C.
rxn time 23 h,
1:3 donor/acceptor ratio for all reactions.
To complete the synthesis of tetrasaccharide 1, compound 3 was first quantitatively deprotected at the nonreducing end with DDQ in the presence of β-Pinene, which was necessary to prevent PMP anomerization (Scheme 4).64 The alcohol derivative of 3 was then coupled to fumarate monoester 2 using 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDCI·HCl) and N,N-diisopropylethylamine (DIPEA) to give tetrasaccharide ester 17 in 74% yield.72 The sensitivity of the glycosidic linkages of 17 made removal of the TBS masking group challenging, and several conventional approaches failed (e.g., acid-buffered TBAF, HF·pyridine complex). Fortunately, we were able to remove the TBS with an excess of triethylamine trihydrofluoride to afford alcohol-ester 1 in 64% yield.73
Scheme 4.

Synthesis of Tetrasaccharide Ester 1
In conclusion, we have achieved the first synthesis of the Arugomycin tetrasaccharide fragment by a convergent [2 + 2] route, relying solely on our third-generation promoter system of aryl cyclopropene-1-thione and oxalyl bromide to conduct fully α-selective glycosylations with secondary alcohol acceptors. In our application of this method to more complex glycosylations of 2,6-dideoxy mono- and disaccharides, we addressed the formation of yield-limiting byproducts through adjustments to temperature, stoichiometry, and addition method to ensure good to excellent yields in our disaccharide couplings. Our challenges in extending this method further to tetrasaccharide glycosylation are illustrative of the sensitivity of 2-deoxy sugar linkages and the need for both effective and innocuous proton scavengers. Taken together, the strategies and results reported herein may inform the synthesis of other 2,6-dideoxy-sugar containing oligosaccharides.
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Science Foundation (NSF CHE-1566233) and National Institutes of Health (R01-GM115779 and U01-GM120414) for generous financial support
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.0c01153.
Experimental details and characterization data (PDF)
FAIR data, including the primary NMR FID files, for compounds 1−10 (ZIP)
FAIR data, including the primary NMR FID files, for compounds 11−17, S3, S5, and S8 (ZIP)
The authors declare no competing financial interest.
REFERENCES
- (1).Elshahawi SI; Shaaban KA; Kharel MK; Thorson JS A Comprehensive Review of Glycosylated Bacterial Natural Products. Chem. Soc. Rev 2015, 44, 7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bennett CS; Galan MC Methods for 2-Deoxyglycoside Synthesis. Chem. Rev 2018, 118, 7931–7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nicolaou KC; Li Y; Sugita K; Monenschein H; Guntupalli P; Mitchell HJ; Fylaktakidou KC; Vourloumis D; Giannakakou P; O’Brate A Total Synthesis of Apoptolidin: Completion of the Synthesis and Analogue Synthesis and Evaluation. J. Am. Chem. Soc 2003, 125, 15443–15454 [DOI] [PubMed] [Google Scholar]
- (4).Wehlan H; Dauber M; Fernaud MTM; Schuppan J; Keiper S; Mahrwald R; Garcia MEJ; Koert U Apoptolidin A: Total Synthesis and Partially Glycosylated Analogues. Chem. - Eur. J 2006, 12, 7378–7397. [DOI] [PubMed] [Google Scholar]
- (5).Langenhan JM; Peters NR; Guzei IA; Hoffmann FM; Thorson JS Enhancing the Anticancer Properties of Cardiac Glycosides by Neoglycorandomization. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 12305–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Iyer AKV; Zhou M; Azad N; Elbaz H; Wang L; Rogalsky DK; Rojanasakul Y; O’Doherty GA; Langenhan JM A Direct Comparison of the Anticancer Activities of Digitoxin MeON-Neoglycosides and O-Glycosides. ACS Med. Chem. Lett 2010, 1, 326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ostash B; Rix U; Rix LLR; Liu T; Lombo F; Luzhetskyy A; Gromyko O; Wang C; Braña AF; Méndez C; et al. Generation of New Landomycins by Combinatorial Biosynthetic Manipulation of the LndGT4 Gene of the Landomycin E Cluster in S. Globisporus. Chem. Biol 2004, 11, 547–555 [DOI] [PubMed] [Google Scholar]
- (8).Hou D; Lowary TL Recent Advances in the Synthesis of 2- Deoxy-Glycosides. Carbohydr. Res 2009, 344, 1911–1940. [DOI] [PubMed] [Google Scholar]
- (9).Imamura A; Ando H; Korogi S; Tanabe G; Muraoka O; Ishida H; Kiso M Di-Tert-Butylsilylene (DTBS) Group-Directed α- Selective Galactosylation Unaffected by C-2 Participating Functionalities. Tetrahedron Lett. 2003, 44, 6725–6728. [Google Scholar]
- (10).Imamura A; Matsuzawa N; Sakai S; Udagawa T; Nakashima S; Ando H; Ishida H; Kiso M The Origin of High Stereoselectivity in Di-Tert-Butylsilylene-Directed α-Galactosylation. J. Org. Chem 2016, 81, 9086–9104. [DOI] [PubMed] [Google Scholar]
- (11).Boltje TJ; Kim JH; Park J; Boons GJ Chiral-Auxiliary- Mediated 1,2-Cis-Glycosylations for the Solid-Supported Synthesis of a Biologically Important Branched α-Glucan. Nat. Chem 2010, 2, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Boltje TJ; Kim JH; Park J; Boons GJ Stereoelectronic Effects Determine Oxacarbenium vs β-Sulfonium Ion Mediated Glycosylations. Org. Lett 2011, 13, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fang T; Mo KF; Boons GJ Stereoselective Assembly of Complex Oligosaccharides Using Anomeric Sulfonium Ions as Glycosyl Donors. J. Am. Chem. Soc 2012, 134, 7545–7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Overend WG; Rees CW; Sequeira JS Reactions at Position 1 of Carbohydrates. Part III. The Acid-Catalyzed Hydrolysis of Glycosides. J. Chem. Soc 1962, 1, 3429–3439. [Google Scholar]
- (15).Noecker L; Duarte F; Bolton SA; Mcmahon WG; Diaz MT; Giuliano RM Glycosylation of Branched Amino and Nitro Sugars. 2. Synthesis of the Cororubicin Trisaccharide. J. Org. Chem 1999, 64, 6275–6282. [Google Scholar]
- (16).Lam SN; Gervay-Hague J Efficient Route to 2-Deoxy β-O-Aryl-D-Glycosides via Direct Displacement of Glycosyl Iodides. Org. Lett 2003, 5, 4219–4222. [DOI] [PubMed] [Google Scholar]
- (17).Tanaka H; Yoshizawa A; Takahashi T Direct and Stereoselective Synthesis of β-Linked 2,6- Deoxyoligosaccharides. Angew. Chem., Int. Ed 2007, 46, 2505–2507. [DOI] [PubMed] [Google Scholar]
- (18).Kaneko M; Herzon SB Scope and Limitations of 2-Deoxy- and 2,6-Dideoxyglycosyl Bromides as Donors for the Synthesis of β−2- Deoxy- and β−2,6-Dideoxyglycosides. Org. Lett 2014, 16, 2776–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tanaka H; Yoshizawa A; Chijiwa S; Ueda JY; Takagi M; Shin-ya K; Takahashi T Efficient Synthesis of the Deoxysugar Part of Versipelostatin by Direct and Stereoselective Glycosylation and Revision of the Structure of the Trisaccharide Unit. Chem. - Asian J 2009, 4, 1114–1125. [DOI] [PubMed] [Google Scholar]
- (20).Crimmins MT; Long A Enantioselective Synthesis of Apoptolidin Sugars. Org. Lett 2005, 7, 4157–4160. [DOI] [PubMed] [Google Scholar]
- (21).Han Z; Zheng Z; Cai L; Zhou D; Li C; Sui Q; Liu S; Gao Q Synthesis of Flavonoid 2-Deoxyglucosides via the Mitsunobu Reaction. Tetrahedron Lett. 2018, 59, 3773–3776. [Google Scholar]
- (22).Beale TM; Moon PJ; Taylor MS Organoboron- Catalyzed Regio- and Stereoselective Formation of β−2-Deoxyglycosidic Linkages. Org. Lett 2014, 16, 3604–3607. [DOI] [PubMed] [Google Scholar]
- (23).Balmond EI; Benito-Alifonso D; Coe DM; Alder RW; McGarrigle EM; Galan MCA 3,4-Trans-Fused Cyclic Protecting Group Facilitates α-Selective Catalytic Synthesis of 2-Deoxyglycosides. Angew. Chem., Int. Ed 2014, 53, 8190–8194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ruei JH; Venukumar P; Ingle AB; Mong KKT C6 Picoloyl Protection: A Remote Stereodirecting Group for 2-Deoxy-b- Glycoside Formation. Chem. Commun 2015, 51, 5394–5397. [DOI] [PubMed] [Google Scholar]
- (25).Palo-Nieto C; Sau A; Galan MC Gold(I)-Catalyzed Direct Stereoselective Synthesis of Deoxyglycosides from Glycals. J. Am. Chem. Soc 2017, 139, 14041–14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Palo-Nieto C; Sau A; Williams R; Galan MC Cooperative Brønsted Acid-Type Organocatalysis for the Stereoselective Synthesis of Deoxyglycosides. J. Org. Chem 2017, 82, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Park Y; Harper KC; Kuhl N; Kwan EE; Liu RY; Jacobsen EN Macrocyclic Bis-Thioureas Catalyze Stereospecific Glycosylation Reactions. Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chen JH; Ruei JH; Mong KKT Iterative α- Glycosylation Strategy for 2-Deoxy- and 2,6-Dideoxysugars: Application to the One-Pot Synthesis of Deoxysugar-Containing Oligosaccharides. Eur. J. Org. Chem 2014, 2014, 1827–1831. [Google Scholar]
- (29).Hoang KM; Lees NR; Herzon SB Programmable Synthesis of 2-Deoxyglycosides. J. Am. Chem. Soc 2019, 141, 8098–8103. [DOI] [PubMed] [Google Scholar]
- (30).Zhu D; Baryal KN; Adhikari S; Zhu J Direct Synthesis of 2-Deoxy-β-Glycosides via Anomeric O-Alkylation with Secondary Electrophiles. J. Am. Chem. Soc 2014, 136, 3172–3175. [DOI] [PubMed] [Google Scholar]
- (31).Roush WR; Gung BW; Bennett CE 2-Deoxy-2-Iodo- and 2-Deoxy-2-Bromo-α-Glucopyranosyl Trichloroacetimidates: Highly Reactive and Stereoselective Donors for the Synthesis of 2-Deoxy-β-Glycosides. Org. Lett 1999, 1, 891–893. [DOI] [PubMed] [Google Scholar]
- (32).Handa M; Smith WJ; Roush WR Studies on the Synthesis of Apoptolidin A. 2. Synthesis of the Disaccharide Unit. J. Org. Chem 2008, 73, 1036–1039. [DOI] [PubMed] [Google Scholar]
- (33).Bucher C; Gilmour R Fluorine-Directed Glycosylation. Angew. Chem., Int. Ed 2010, 49, 8724–8728. [DOI] [PubMed] [Google Scholar]
- (34).Sirion U; Purintawarrakun S; Sahakitpichan P; Saeeng R An Efficient Method for the Selective Synthesis of 2-Deoxy-2-Iodo- Glycosides by O-Glycosidation of d-Glucal Using I2−Cu(OAc)2. Carbohydr. Res 2010, 345 (16), 2401–2407. [DOI] [PubMed] [Google Scholar]
- (35).Yang X; Fu B; Yu B Total Synthesis of Landomycin A, a Potent Antitumor Angucycline Antibiotic. J. Am. Chem. Soc 2011, 133, 12433–12435. [DOI] [PubMed] [Google Scholar]
- (36).Wang H; Tao J; Cai X; Chen W; Zhao Y; Xu Y; Yao W; Zeng J; Wan Q Stereoselective Synthesis of α-Linked 2-Deoxy Glycosides Enabled by Visible-Light-Mediated Reductive Deiodination. Chem. - Eur. J 2014, 20, 17319–17323. [DOI] [PubMed] [Google Scholar]
- (37).Thiem J; Schöttmer B Formation of Aureolic Acid Oligodeoxy Mono-, Di-, and Trisaccharides Employing the Dibromomethyl Methyl Ether Reaction. J. Carbohydr. Chem 2017, 36, 279–293. [Google Scholar]
- (38).Mestre J; Collado D; Benito-Alifonso D; Rodríguez MA; Matheu MI; Díaz Y; Castillόn S; Boutureira O Highly Reactive 2-Deoxy-2-Iodo-d-: Allo and d- Gulo Pyranosyl Sulfoxide Donors Ensure β-Stereoselective Glycosylations with Steroidal Aglycones. RSC Adv. 2018, 8, 30076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ge J-T; Zhou L; Luo T; Lv J; Dong H A One-Pot Method for Removal of Thioacetyl Group via Desulfurization under Ultraviolet Light To Synthesize Deoxyglycosides. Org. Lett 2019, 21, 5903–5906. [DOI] [PubMed] [Google Scholar]
- (40).Babu RS; Zhou M; O’Doherty GA Novo Synthesis of Oligosaccharades Using a Palladium-Catalyzed Glycosylation Reaction. J. Am. Chem. Soc 2004, 126, 3428–3429. [DOI] [PubMed] [Google Scholar]
- (41).Zhou M; O’Doherty GA Novo Approach to 2-Deoxy-β- Glycosides: Asymmetric Syntheses of Digoxose and Digitoxin. J. Org. Chem 2007, 72, 2485–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Zhou M; O’Doherty GA Novo Synthesis of the Trisaccharide Subunit of Landomycins A and E. Org. Lett 2008, 10, 2283–2286. [DOI] [PubMed] [Google Scholar]
- (43).Babu RS; Chen Q; Kang SW; Zhou M; O’Doherty GA Novo Asymmetric Synthesis of All-d-, All-l-, and d-/l-Oligosaccharides Using Atom-Less Protecting Groups. J. Am. Chem. Soc 2012, 134, 11952–11955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).McDonald FE; Reddy KS Convergent Synthesis of Digitoxin: Stereoselective Synthesis and Glycosylation of the Digoxin Trisaccharide Glycal. Angew. Chem., Int. Ed 2001, 40, 3653–3655. [DOI] [PubMed] [Google Scholar]
- (45).Cutchins WW; McDonald FE Stereoselective Synthesis of Vancosamine and Saccharosamine Glycals via Tungsten-Catalyzed Alkynol Cycloisomerization. Org. Lett 2002, 4, 749–752. [DOI] [PubMed] [Google Scholar]
- (46).Balthaser BR; McDonald FE Brønsted Acid-Promoted Glycosylations of Disaccharide Glycal Substructures of the Saccharomicins. Org. Lett 2009, 11, 4850–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Issa JP; Lloyd D; Steliotes E; Bennett CS Reagent Controlled Beta-Specific Dehydrative Glycosylation Reactions with 2-Deoxy-Sugars. Org. Lett 2013, 15, 4170–4173. [DOI] [PubMed] [Google Scholar]
- (48).Issa JP; Bennett CS A Reagent-Controlled SN2- Glycosylation for the Direct Synthesis of Beta-Linked 2-Deoxy- Sugars. J. Am. Chem. Soc 2014, 136, 5740–5744. [DOI] [PubMed] [Google Scholar]
- (49).Nogueira JM; Nguyen SH; Bennett CS Cyclopropenium Cation Promoted Dehydrative Glycosylations Using 2-Deoxy- and 2,6-Dideoxy-Sugar Donors. Org. Lett 2011, 13, 2814–2817. [DOI] [PubMed] [Google Scholar]
- (50).Nogueira JM; Issa JP; Chu AHA; Sisel JA; Schum RS; Bennett CS Halide Effects on Cyclopropenium Cation Promoted Glycosylation with Deoxy Sugars: Highly α-Selective Glycosylations Using a 3,3-Dibromo-1,2- Diphenylcyclopropene Promoter. Eur. J. Org. Chem 2012, 2012, 4927–4930. [Google Scholar]
- (51).Nogueira JM; Bylsma M; Bright DK; Bennett CS Reagent-Controlled α-Selective Dehydrative Glycosylation of 2,6- Dideoxy- and 2,3,6-Trideoxy Sugars. Angew. Chem., Int. Ed 2016, 55, 10088–10092. [DOI] [PubMed] [Google Scholar]
- (52).Lloyd D; Bennett CS An Improved Approach to the Direct Construction of 2-Deoxy-β-Linked Sugars: Applications to Oligosaccharide Synthesis. Chem. - Eur. J 2018, 24, 7610–7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Mizia JC; Bennett CS Reagent Controlled Direct Dehydrative Glycosylation with 2-Deoxy Sugars: Construction of the Saquayamycin Z Pentasaccharide. Org. Lett 2019, 21, 5922–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Yalamanchili S; Lloyd D; Bennett CS Synthesis of the Hexasaccharide Fragment of Landomycin A Using a Mild, Reagent-Controlled Approach. Org. Lett 2019, 21, 3674–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Lee J; Kang J; Lee S; Rhee YH Flexible Total Synthesis of 11-Deoxylandomycins and Their Non-Natural Analogues by Way of Asymmetric Metal Catalysis. Angew. Chem., Int. Ed 2020, 59, 2349–2353. [DOI] [PubMed] [Google Scholar]
- (56).Xie T; Zheng C; Chen K; He H; Gao S Asymmetric Total Synthesis of the Complex Polycyclic Xanthone FD-594. Angew. Chem., Int. Ed 2020, 59, 4360–4364. [DOI] [PubMed] [Google Scholar]
- (57).Kawai H; Hayakawa Y; Nakagawa M; Imamura K; Tanabe K; Shimazu A; Seto H; Ōtake N Arugomycin, a New Anthracycline Antibiotic. J. Antibiot 1983, 36, 1569–1571. [DOI] [PubMed] [Google Scholar]
- (58).Kawai H; Hayakawa Y; Nakagawa M; Furihata K; Seto H; Otake N Studies on Arugomycin, a New Anthracycline Antibiotic Part II. Structural Elucidation of Arugomycin. Tetrahedron Lett. 1984, 25, 1941–1944. [Google Scholar]
- (59).Kawai H; Hayakawa Y; Nakagawa M; Furihata K; Furihata K; Shimazu A; Seto H; Otake N Arugomycin, a New Anthracycline Antibiotic. I. Taxonomy, Fermentation, Isolation and Physico-Chemical Properties. J. Antibiot 1987, 40, 1266–1272. [DOI] [PubMed] [Google Scholar]
- (60).Searle MS; Bicknell W; Wakelin LPG; Denny WA Anthracycline Antibiotic Arugomycin Binds in Both Grooves of the DNA Helix Simultaneously: An NMR and Molecular Modelling Study. Nucleic Acids Res. 1991, 19, 2897–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Fischer E; Zach K Reduktion Der Acetobromglucose Und Ähnlicher Stoffe. K. Prouss. Akad. Wiss 1913, 16, 311. [Google Scholar]
- (62).Soliman SE; Bennett CS Reagent-Controlled Synthesis of the Branched Trisaccharide Fragment of the Antibiotic Saccharomicin B. Org. Lett 2018, 20, 3413–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Henderson AS; Bower JF; Galan MC Carbohydrate- Based N-Heterocyclic Carbenes for Enantioselective Catalysis. Org. Biomol. Chem 2014, 12, 9180–9183. [DOI] [PubMed] [Google Scholar]
- (64).Lloyd D; Bylsma M; Bright DK; Chen X; Bennett CS Mild Method for 2-Naphthylmethyl Ether Protecting Group Removal Using a Combination of 2,3-Dichloro-5,6-Dicyano-1,4-Benzoquinone (DDQ) and β-Pinene. J. Org. Chem 2017, 82, 3926–3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Li Z; Gildersleeve JC Mechanistic Studies and Methods to Prevent Aglycon Transfer of Thioglycosides. J. Am. Chem. Soc 2006, 128, 11612–11619. [DOI] [PubMed] [Google Scholar]
- (66).Gildersleeve JC; Smith A; Sakurai K; Raghavan S; Kahne D Scavenging Byproducts in the Sulfoxide Glycosylation Reaction: Application to the Synthesis of Ciclamycin 0. J. Am. Chem. Soc 1999, 121, 6176–6182. [Google Scholar]
- (67).Pachamuthu K; Vankar YD Ceric Ammonium Nitrate- Catalyzed Tetrahydropyranylation of Alcohols and Synthesis of 2- Deoxy-O-Glycosides. J. Org. Chem 2001, 66, 7511–7513. [DOI] [PubMed] [Google Scholar]
- (68).Yu F; Li J; DeMent PM; Tu YJ; Schlegel HB; Nguyen HM Phenanthroline-Catalyzed Stereoretentive Glycosylations. Angew. Chem., Int. Ed 2019, 58, 6957–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Saliba RC; Wooke ZJ; Nieves GA; Chu AHA; Bennett CS; Pohl NLB Challenges in the Conversion of Manual Processes to Machine-Assisted Syntheses: Activation of Thioglycoside Donors with Aryl(Trifluoroethyl)Iodonium Triflimide. Org. Lett 2018, 20, 800–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Gu X; Chen L; Wang X; Liu X; You Q; Xi W; Gao L; Chen G; Chen YL; Xiong B; et al. Direct Glycosylation of Bioactive Small Molecules with Glycosyl Iodide and Strained Olefin as Acid Scavenger. J. Org. Chem 2014, 79, 1100–1110. [DOI] [PubMed] [Google Scholar]
- (71).Chen G; Yin Q; Yin J; Gu X; Liu X; You Q; Chen YL; Xiong B; Shen J Strained Olefin Enables Triflic Anhydride Mediated Direct Dehydrative Glycosylation. Org. Biomol. Chem 2014, 12, 9781–9785. [DOI] [PubMed] [Google Scholar]
- (72).Fu J; Laval S; Yu B Total Synthesis of Nucleoside Antibiotics Plicacetin and Streptcytosine A. J. Org. Chem 2018, 83, 7076–7084. [DOI] [PubMed] [Google Scholar]
- (73).Matsuo I; Wada M; Ito Y Desilylation under High Pressure. Tetrahedron Lett. 2002, 43, 3273–3275. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.