

Abstract
Biologic therapies have shown high efficacy in psoriasis, but individual response varies and is poorly understood. To inform biomarker discovery in the Psoriasis Stratification to Optimise Relevant Therapy (PSORT) study, we evaluated a comprehensive array of omics platforms across three time-points and multiple tissues in a pilot investigation of ten severe psoriasis patient, treated with the tumor necrosis factor (TNF) inhibitor, etanercept. We used RNA-sequencing to analyse mRNA and small-RNA transcriptome in blood, lesional and non-lesional skin and the Somascan platform to investigate the serum proteome. Using an integrative systems biology approach, we identified signals of treatment response in genes and pathways associated with TNF signalling, psoriasis pathology and the MHC region. Notably, we found association between clinical response and TNF-regulated genes in blood and skin. Using a combination of differential expression testing, upstream regulator analysis, clustering techniques, and predictive modelling, we demonstrate that baseline samples are indicative of patient response to biologic therapies, including signals in blood, which have traditionally been considered unreliable for inference in dermatology. In conclusion, our pilot study provides both an analytical framework and empirical basis to estimate power for larger studies, specifically the ongoing PSORT study, which we demonstrate as powered for biomarker discovery and patient stratification.
Introduction
The introduction of biologic therapies into clinical practice has led to major improvements for patients with severe psoriasis. However, optimal, cost-effective provision of these therapies in a resource-limited healthcare system will necessitate a stratified approach (Griffiths, 2017, Lebwohl, 2016).
Translational research has been revolutionized by the availability of technologies to measure features of the genome, transcriptome, and proteome (so called “omics”), primarily facilitated by high-throughput sequencing (HTS, formerly next generation sequencing). It is likely that data generated by these new technologies will inaugurate an era of stratified care founded on comprehensive cellular profiles, rather than individual biomarker molecules (Johnston et al., 2017). Such laboratory methods have already been employed in dermatological research to identify biomarkers of treatment response in inflammatory skin disease (Correa da Rosa et al., 2017, Ungar et al., 2017), but validation of those markers remains elusive and unlike our colleagues in oncology, clinical dermatologists have yet to see the integration of omics into daily practice.
Methodological problems have in part hampered the translation of pharmacogenomic results into clinical success in dermatology (Jorgensen and Williamson, 2008). Well-designed and adequately powered prospective studies are required to identify clinically robust biomarkers. Psoriasis Stratification to Optimise Relevant Therapy (PSORT) is an academic-industrial UK stratified medicine consortium funded by the Medical Research Council and devoted to developing a stratifier of response prediction to biologics, scalable for clinical use for those with moderate to severe disease (Griffiths et al., 2015).
In order to inform the analytical strategy of PSORT, we conducted a pilot study to evaluate response to a biologic in psoriasis patients using lesional (PP) and non-lesional (PN) skin and blood, and a range of omic platforms and different analysis pipelines. In addition to comparing the performance of each platform and tissue, we used our preliminary data to obtain an empirical estimate of the required sample size to adequately power the full PSORT study. Here we report the results of a comprehensive multi-omic pilot study (Figure 1), including RNA sequencing (RNA-Seq) of mRNA PP and PN skin, RNA-Seq from mRNA and from miRNA from blood as well as Somascan proteomic data from blood.
Figure 1.
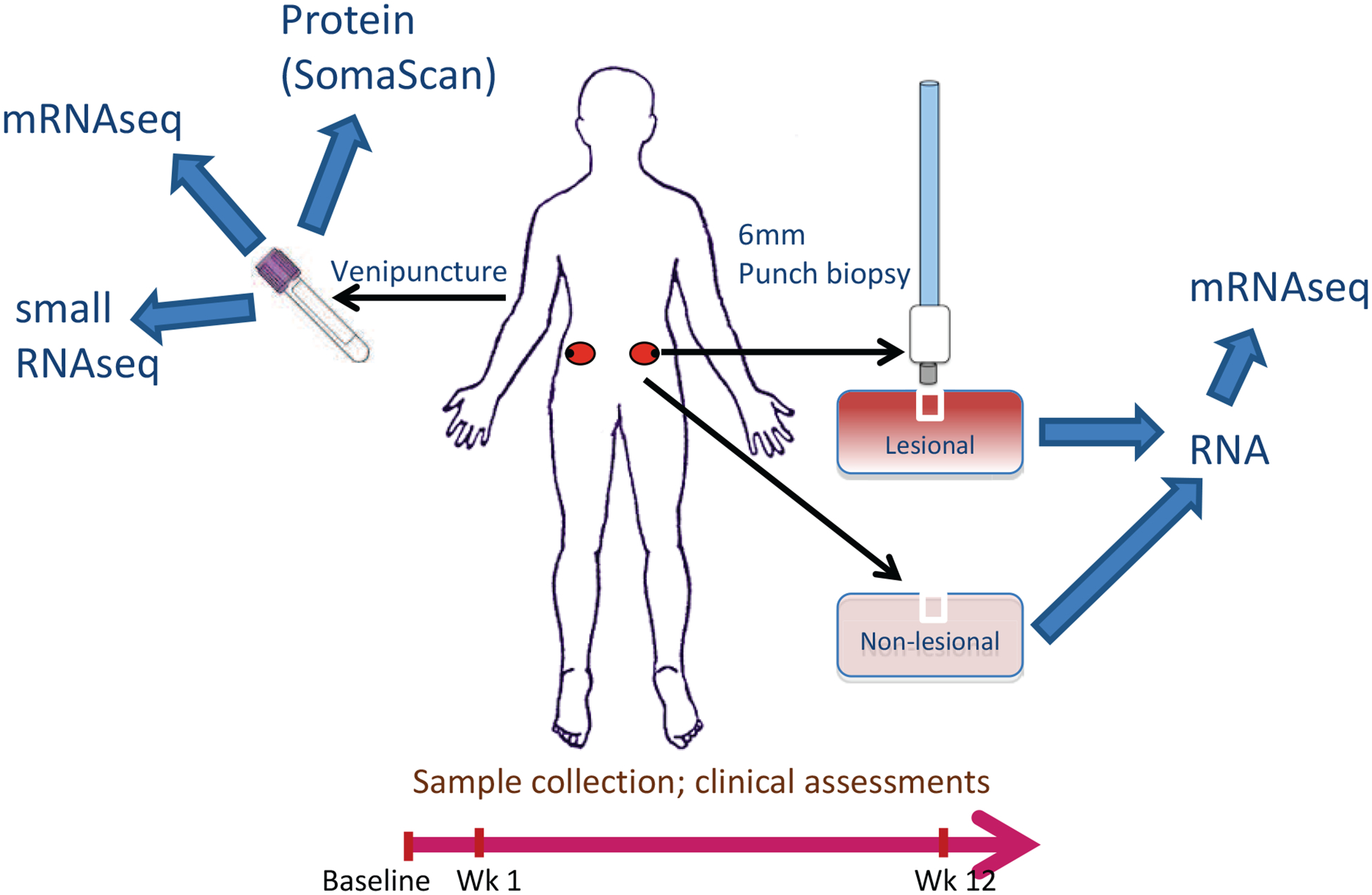
Study Overview
Participants were assessed at baseline, week one and week 12 of therapy. Participant sampling comprised blood testing, urine collection, lesional and non-lesional skin biopsies (from photoprotected sites on the lower back/buttock, from the edge of plaques and at a minimum distance from previous biopsy sites). RNA-Seq was conducted on mRNA from blood, lesional and non-lesional skin and miRNA from blood. Proteomic assessment was conducted on serum.
Our tightly phenotyped, rigorously controlled cohort of patients with severe chronic plaque psoriasis commenced biologic therapy with the tumor necrosis factor inhibitor (TNFi), etanercept. We evaluated the relative merits of each platform and demonstrate a workflow for scaled use on large datasets. We provide not only open data but open access to our complete analysis scripts and a fully executable R Markdown document for colleagues to evaluate and exactly reproduce the workflow themselves (Foulkes et al., 2017). Multi-omic analysis is a highly resource intensive process, particularly with the breadth of approaches described here, which are beyond the resources of most projects. We use this pilot study to comment on the relative merits of multi-omic approaches and highlight platforms that show particular promise in predicting response to therapy.
Results
Patient Characteristics and Analysis of Clinical Response
Ten patients commencing etanercept therapy were recruited from a prospective clinical observational study entitled pharmacogenomic signatures of treatment response in psoriasis. Figure 1 provides an overview of the study and patient characteristics for included participants are shown in Table 1. Participants were assessed at baseline, week one and week 12 of therapy and response to therapy was determined using the Psoriasis Area and Severity Index (PASI), with a response defined as a reduction of PASI by at least 75% from baseline (PASI75) and non-response defined as failure to achieve a reduction of at least 50% from baseline (PASI50). Supplementary Figure 2 demonstrates a scatterplot of PASI at baseline vs. PASI at week 12.
Table 1.
Summary of clinical characteristics of included participants
| Variable | Patients (n = 10) |
|---|---|
| Age, mean (years) | 43 |
| Sex | F 2, M 8 |
| Weight, kg (mean ± SD) | 94.3 ± 17.7 |
| BMI (mean ± SD) | 30.6 ± 5.5 |
| Age at onset of psoriasis (years) mean ± SD | 17 ± 11 |
| Baseline PASI; mean ± SD | 20.3 ± 8.8 |
| PASI at week 12; mean ± SD | 6.8 ± 3.9 |
| Baseline DLQI; mean ± SD | 20.1 ± 9.3 |
| DLQI at week 12; mean ± SD | 4.5 ± 3.4 |
Multi-omic analysis
Using samples from each participant at each time-point (one biopsy sample per library), we performed RNA-Seq on mRNA from PP skin (60m paired reads/sample), PN skin (60m paired reads/sample), and blood (30m paired reads/sample). We additionally performed RNA-Seq on miRNA from blood (10m single reads/sample) and Somalogic proteomic assessment on serum samples. As exploratory data analysis is a key first step in multi-omics, we first constructed a sample similarity matrix to compare mRNA transcriptome across tissues, by calculating the pairwise Euclidean distance between all mRNA samples (Supplementary Figure 2). Samples were clearly separated by tissue, although less distinction was seen between PP and PN skin samples, in part reflecting strong intra-subject effects and treatment effects between baseline and 12 weeks. Next we examined transcriptome structure on a tissue-by-tissue basis using two different projection methods. Principal component analysis (PCA) demonstrated clear separation between skin and blood along the first principal component, as expected (Supplementary Figure 3a). The second principal component separated PP from PN samples, albeit with one data point corresponding to a participant’s week 12 observation. This patient showed good response to therapy, suggesting putative detection of remission at mRNA transcriptome level. Similar, although less distinct tissue separation was seen by another projection method, t-distributed stochastic neighbour embedding (t-SNE) (Supplementary Figure 3b). Tissue-wise projection plots across all platforms were dominated by intra-subject signatures (Supplementary Figures 4 and 5). These unsupervised methods do not appear to separate patients by treatment response, indicating that supervised techniques may be required to detect a response signal in these data.
Response differential expression analysis by platform
Differential expression analyses (DEA) to investigate the effects of etanercept treatment over time were performed for each platform (mRNA-Seq, miRNA-Seq and Somascan proteomic assessment), and tissue type (PP skin, PN skin and blood) using a common limma analytical framework. Access to our complete analysis script and fully executable R Markdown document allows reproduction of this workflow with evaluation of these results (see Materials and Methods and Supplementary File). We imposed a 10% false discovery rate (FDR) threshold. We selected this cut-off because power calculations suggest the modest sample size of the study will impede our sensitivity to detect differential expression. A 10% FDR threshold is therefore likely to underestimate the true number of differentially expressed genes or proteins in our dataset.
A summary of differential expression of mRNA, miRNA and protein across time and across tissue types may be found in Figure 2, whilst all differentially expressed molecules are summarised in supplementary table 1.
Figure 2.
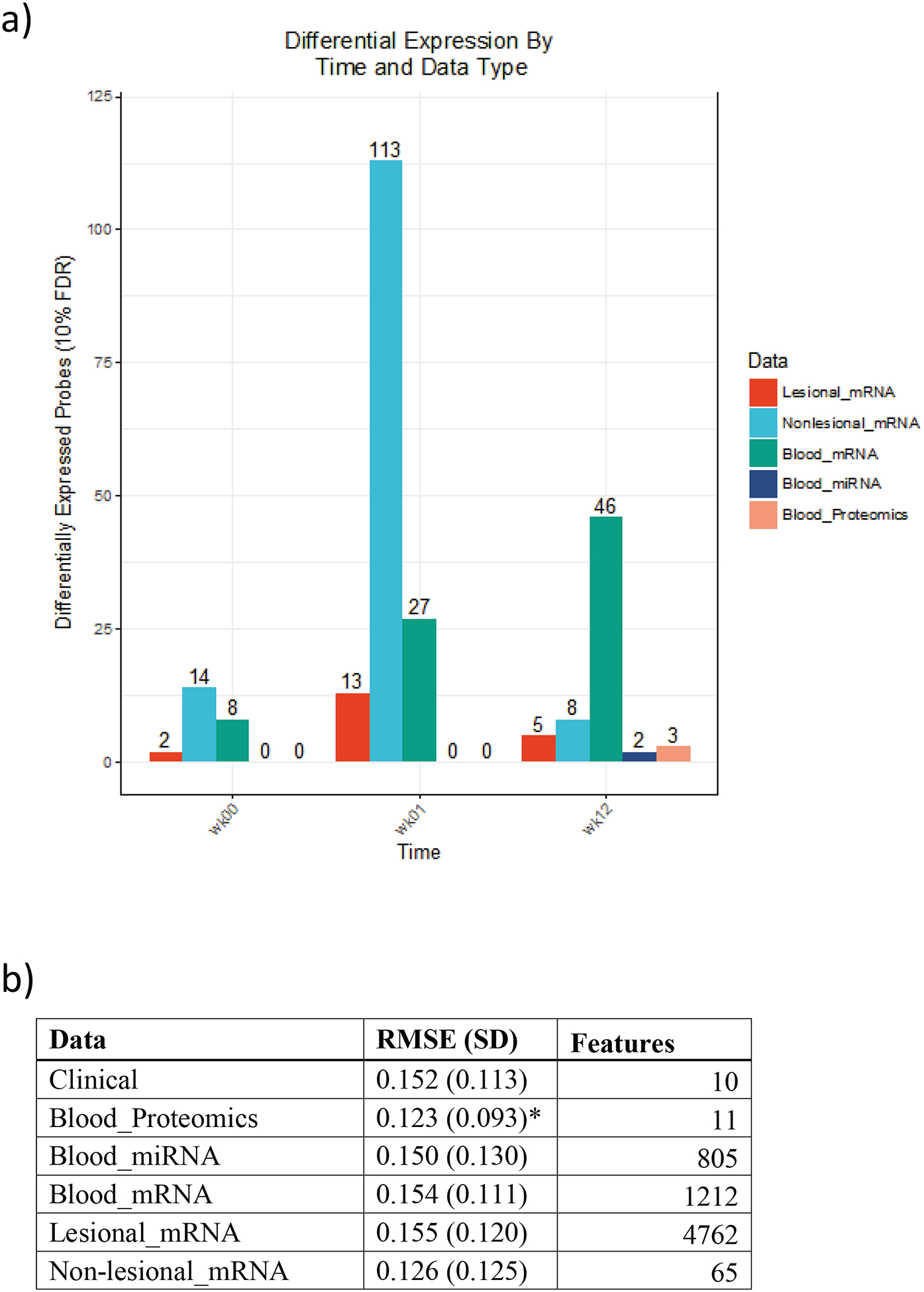
Differential expression of mRNA, miRNA and protein across time and across tissue. a) The number of biomolecules declared differentially expressed between responders and non-responders at 10% FDR for each tissue, time point, and platform. The number of tests vary between platforms, mRNA (19304), miRNA (3632), protein (1129) b) Model metrics for random forests; we report mean (SD) predictive error and number of features retained after recursive feature elimination for each data platform and response type. Continuous response models were evaluated using root mean square error (RMSE), while categorical models were tuned with cross entropy loss. Asterisks denote the top performing data platform for each class of random forests.
Heatmaps of the top 1% gene expression changes from PP skin, PN skin, and blood are shown in Supplementary Figure 6. The top 1% of genes cluster by response to treatment across PP, PN skin and blood. Similar results were seen with supervised and unsupervised cluster assignments.
Upstream regulator analysis
Acknowledging that our study is not powered for discovery, we used the Upstream Regulator Analysis function in Ingenuity Pathway Analysis (IPA) to evaluate upstream regulator signals at a systems-level that may be responsible for the observed gene expression changes. Upstream regulators are defined as any molecule that can affect the expression of another molecule, including transcription factors, cytokines, miRNAs and drugs. The activation state for each regulator was predicted based on global direction of changes in the DEA for previously published targets of this regulator. The predicted top 30 regulators across all tissues and time points are shown in a hierarchically clustered heatmap in Figure 3. Results demonstrate a range of pro-inflammatory signalling and drug pathways, including a highly conserved, pan-tissue TNF signature, strongest at baseline in blood and at week-1 in PP and PN skin, and substantially diminished at week-12 across all tissues. A similar pattern is also seen in Figure 3 hierarchically clustered with TNF in Interferon α−2 and γ signalling, in addition to NFΚβ signalling. This is an interesting proof of concept of the ability to detect a biologic drug response at a systems level, which we discuss further below.
Figure 3.

Top upstream regulators across genes differentially expressed in relation to etanercept differential expression (p<0.05) response in psoriasis. Top 30 upstream regulators demonstrated. The prediction of activation state is based on the global direction of changes of genes with differential expression p<0.05. The nominal limit of significance (z-score < −2 or > 2) is indicated by the Activation z-score colour scale.
Platform comparison
Baseline Omic Platform Concordance
We performed supervised and unsupervised PAM (Partitioning Around Medoids) clustering on baseline samples for each tissue across all platforms, relating the differentially expressed genes from each platform to response and informing where drivers of prediction to response have commonalities. Cross-platform concordance was evaluated using the mutual information between cluster assignments, indicating a wide range of concordance values among supervised clusters (Figure 4a). Lesional mRNA and blood mRNA concordance was highest at 0.88 bits.
Figure 4.
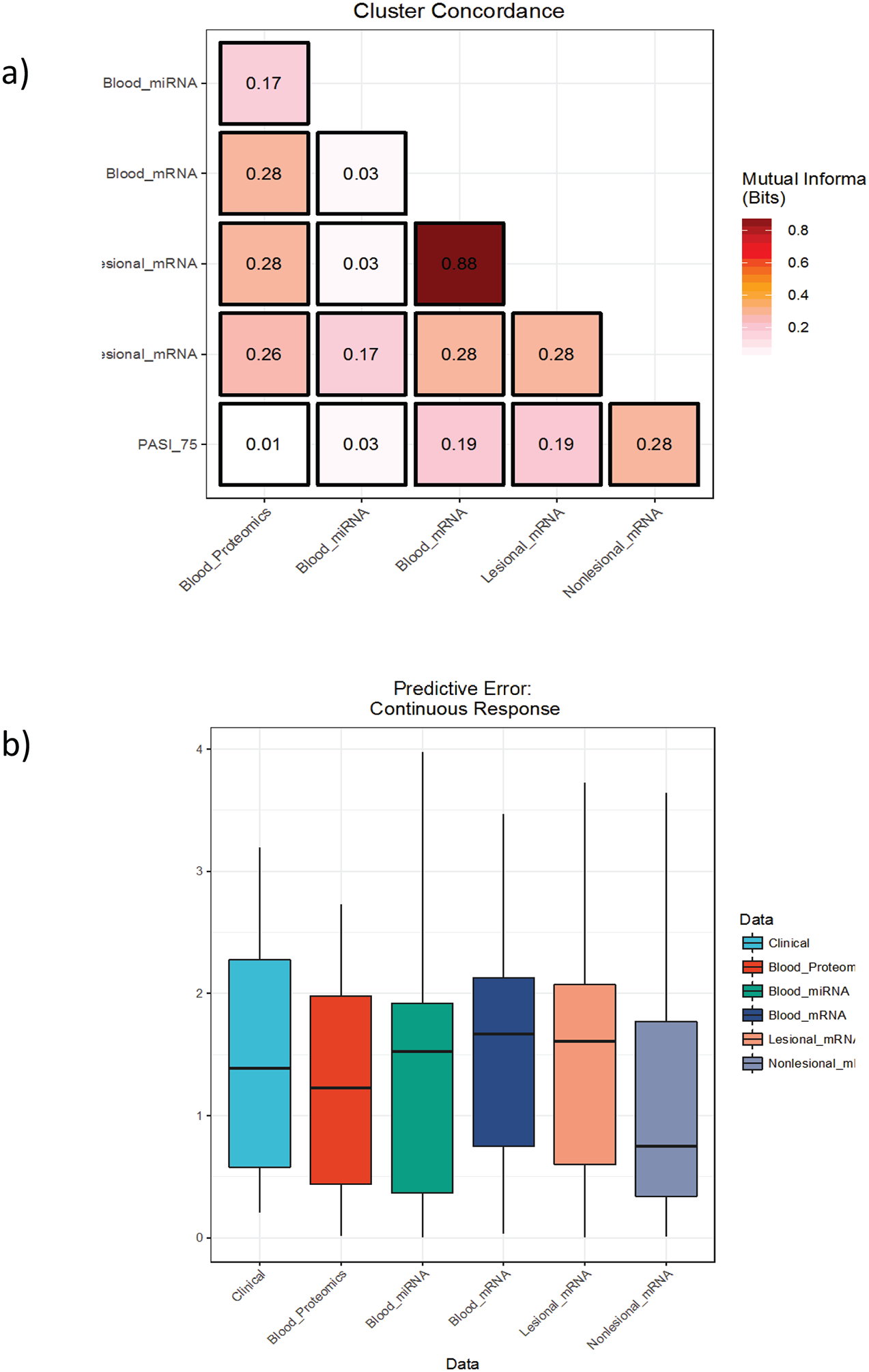
Concordance of platforms at prediction of PASI 75. a) Heatmap depicting the concordance of cluster assignments across platforms as determined by supervised methods. b) Box plots demonstrate the distribution of cross-validated root mean square error (RMSE) over ten folds for a series of random forests models with recursive feature elimination trained to predict the change in PASI using only baseline samples. Lower RMSE values indicate more predictive models
Machine Learning Models
We built a series of random forest models to predict continuous response using baseline data from each tissue-type and platform (Figure 4b). Predictive power was detected across platforms using this methodology, demonstrating additional signal to the differential expression analyses. The proteomics assay, in which we found no significant differentially expressed proteins at baseline using traditional marginal techniques (i.e., looking at each feature separately), proved the most predictive platform for response when modelled using random forests; however differences between data types were generally insignificant. The recursive feature elimination algorithm we used for these models (see Methods) may provide an alternative approach to biomarker discovery, offering insight into omic signatures of response. Our top-performing model achieved a root-mean-square error (RMSE) of 0.123, which is just less than 75% of the standard deviation of our (winsorised) delta PASI distribution.
Power calculations for a prospective observational study
Using the method of Guo et al. (Guo et al., 2014) and parameters derived from this pilot study, we calculated the requisite sample size to achieve 90% power to detect differential expression associated with response. Using the pilot data presented here as a guide, we project that 17,000 genes are likely to pass a reasonable expression filter, and that some 1% of these genes will prove prognostic in a sufficiently large cohort. The top 1% of genes in our baseline measures had an average read count of ~100 prior to normalisation; a minimum log fold change of approximately 0.72 after modelling; and a global dispersion estimate of 0.137, as estimated by the empirical Bayes procedure of McCarthy et al. (2012). Imposing a 5% FDR threshold and a target log fold change of 1.5, we find that a study would require 41 subjects to achieve 90% power to identify transcriptomic markers of biologic response for patients with chronic plaque psoriasis. Relaxing the number of differentially expressed genes to 5%, we can maintain 90% power with 34 subjects. We present power curves projected across an expected range of fold changes at 1% and 5% DE in supplementary figure 7.
Discussion
These results were presented at the 66th annual Montagna Symposium on the Biology of Skin. In this study we present a framework for multi-omic analysis of biologic response. Our results are transparent and fully reproducible via companion markdown documents. This makes our analysis framework suitable for larger studies of similar nature, such as the PSORT program. We emphasise that this proof of concept study is not powered for discovery; however, our results do suggest that signals of response to therapy in patients with severe psoriasis treated with the TNFi etanercept may be systemically detectable in lesional skin, non-lesional skin and blood at baseline, prior to commencement of therapy. Evidence of differential expression correlated with treatment response was observed across all tissue types and time points, but differed across omic platforms.
The choice of the TNFi etanercept related to the timing of study design and the observed rates of etanercept response were within the range observed in studies of larger cohorts (Leonardi et al., 2003). Prior pharmacogenomic evaluations of patient cohorts have centred on the use of genetic or genomic techniques, predominantly using skin biopsies, although several studies have used skin and blood (Chow et al., 2016, Suárez-Fariñas et al., 2012), with consideration of detection of response early in treatment. Whilst no prospective biomarkers have yet been validated in adequately powered cohorts, there has been substantial progress, with the creation of predictors or classifiers of response (Correa da Rosa et al., 2017).
Our focus was RNA-Seq technology as the gold standard for gene expression profiling. RNA sequencing provides counts of all the genes expressed in a sample including microRNAs (miRNAs) and other potentially important noncoding RNA species. Use of high quality RNA inputs (RIN>8) ensured high quality libraries, which passed relatively stringent QC thresholds. We selected an RNA-Seq platform to enable direct comparison with other open access research data and to data from a future larger validation cohort. RNA-Seq is now becoming the platform of choice for transcriptome analysis; especially as costs of HTS techniques reduce over time. Our use of RNA-Seq allowed for the same technique to be applied across evaluation of tissue types, directly comparing samples from lesional skin, non-lesional skin and blood, in addition to proteomics assessment. We have evaluated a range of exploratory visualisations of our pilot data, which showed differing performance, for example a comparison of PCA and t-SNE visualisation of lesional vs. non-lesional skin highlighted the former method’s greater sensitivity to local effects.
The individual genes identified in differential expression analyses were not further evaluated, since our study is not powered for discovery and this approach has been comprehensively reported elsewhere (Li et al., 2014). However, at a systems level, upstream regulator analysis (IPA) of DEGs associated with clinical response across tissues and time points indicated that changes in genes controlled by the target of the drug, TNF, were the most predictive of response. Although this might seem intuitive, previous reports have linked etanercept response to interleukin (IL)-17 signalling rather than TNF early response genes (Zaba et al., 2009). In blood, in addition to TNF regulation, we also saw a strong interferon signature associated with response to etanercept, which has previously been reported in association with etanercept response in skin (Johnston et al., 2014) and also with TNF activation in inflammatory diseases (Mavragani et al., 2007, Zou et al., 2003). Comparison of TNF and interferon signatures across time points in association with response also shows an interesting pattern, with strong signals in blood at baseline, and in skin at 1 week, potentially indicating the genomic response to TNFi therapy (Figure 3). Concordance of baseline omic platforms in prediction of response demonstrated the strongest association between lesional skin mRNA and blood mRNA. Few response associated genes were seen in common across tissues and time points, notably all genes associated in more than one tissue were located in the major histocompatibility complex (MHC) (Supplementary Table 1). This correlates with previous genetic findings (Talamonti et al., 2013) supporting an immunologic basis to both treatment response and psoriasis pathology (Krueger, 2002).
Whilst it is difficult to either identify or validate stable subgroups within small cohorts, we are confident this approach will be more informative in a larger study and preliminary evidence here, suggests that blood biomarkers may be an informative and less invasive predictor of response. We used our dataset to empirically inform a power calculation for the prospective study PSORT; where 80 participants are being recruited for assessment of each of adalimumab and ustekinumab. This demonstrates that the PSORT study is adequately powered to detect moderate to large treatment effects in most scenarios.
We encourage researchers to access our data in ArrayExpress (accession numbers E-MTAB-6428, E-MTAB-6555 and E-MTAB-6556) and review our supplementary R Markdown documents on GitHub to learn more about our pipeline and to fully reproduce our results. Data sharing and open source analytics are the obvious solution to the reproducibility crisis that plagues clinical and omic research today, and is becoming more commonplace in fields which are advancing stratified medicine (Omberg et al., 2013).We believe that open access to data and code should be the norm in life science research, not the exception (Foulkes et al., 2017).
Our study went beyond analysis of a single technology appraisal of treatment prediction in one cohort to provide a scalable framework for predictive and inferential analysis of multi-omic data for clinical dermatology. Despite our small sample size, we were able to detect consistent signals of differential expression and build machine-learning models that in adequately powered studies may offer complementary information to clinical factors in the prediction of outcome. We suggest this ability to detect signals is in part due to the use of a single clinician for cohort ascertainment and sample processing thereby minimising clinical confounders and batch issues and allowing bioinformatics expertise to synergise with clinical research strategy from conception through analysis. These results have implications for ongoing studies. Our exploration has provided the framework for the generation of a large-scale omics dataset from PSORT. The signals we have detected will be examined for validity using the same robust analytical pipeline in PSORT, which we demonstrate is substantially powered to detect true biomarkers of response to therapy. Likewise as omics techniques are applied to other dermatological diseases such as atopic eczema (Suarez-Farinas et al., 2015) at the same time as an expansion in biologic therapies is occurring (Blauvelt et al., 2017), genomic approaches to personalisation and stratification of therapies may have broad applicability.
Materials and Methods
Prospective observational study
Ten participants commencing etanercept therapy (50mg by subcutaneous injection administered once weekly) were recruited (providing written, informed consent) to take part in a prospective clinical observational study entitled ‘Pharmacogenomic signatures of treatment response in psoriasis’ (UK Research Ethics Committee reference 11/NW/0500; protocol available in supplementary materials). Patients had a diagnosis of chronic plaque psoriasis of early onset (≤ 40 years) disease, were White of European ancestry (to third generation) and had not received prior systemic or biologic treatments in at least two weeks (or four × t½ of last treatment, whichever was longer). Of the 10 participants, nine were naïve to biologic therapy. Patients completed detailed demographic questioning, including reporting information on comorbidities and concomitant medication. Disease severity and response to therapy were assessed using the PASI, Physician Global Assessment (PGA) and DLQI. Clinical samples including blood and skin biopsies were collected at baseline, one week (following the second injection of etanercept) and 12 weeks of treatment. Adherence to therapy was assessed, including witnessed/administered injections at the initial visit, self-reporting of timings of injections between visits and monitored drug levels at the final visit. The same physician and research nurse conducted all research visits (ACF and JH).
Laboratory methodology
For skin and blood sampling techniques, RNA sequencing details and Somalogic proteomic analysis methodology, please refer to Supplementary Materials and Methods.
Genomic data analysis workflow
The RNA-Seq data is available Array Express (accession number). The analysis code is available in our public GitHub repository (https://github.com/C4TB/PSORT_ETN_pilot). Executable R scripts and R Markdown documents are available in order to allow complete reproduction of our analysis workflow. All analyses were conducted in R version 3.4.0.
Upstream regulator analysis
Functional analysis of systems-level upstream regulators responsible for observed differential gene expression related to response was performed using the Upstream Regulator function in Ingenuity Pathways Analysis (IPA; Ingenuity Systems), using all genes with nominal response p ≤ 0.05 as input. For all gene set enrichment analyses, a right-tailed Fisher’s exact test was used to calculate a pathway p-value determining the probability that each biological function assigned to that data set was due to chance alone. All enrichment scores were calculated in IPA using all transcripts that passed QC as the background data set. Upstream regulator analysis is based on prior knowledge of expected effects between regulators and their known target genes according to the IPA database. The prediction of activation state is based on the global direction of changes of differentially expressed genes, a z-score is calculated and determines whether gene expression changes for known targets of each regulator are correlated with what is expected from the literature for an activation of this pathway. In this exploratory analysis we emphasized power over type 1 error, using a nominal z score threshold of z > 2 to indicate activation or z < −2 to indicate inhibition. For definition of response and differential expression analyses, please see Supplementary Materials and Methods.
Clustering
Supervised and unsupervised clusters differ with respect to how genes were filtered across the two groupings. For our supervised analysis, we filtered out the bottom half of probes by association with biologic response, as determined by moderated t-tests. With unsupervised clusters, we filtered by the leading fold change between each sample pair, as implemented in limma (Ritchie et al., 2015). Next, we projected the data in two dimensions using t-SNE (Van Der Maaten et al., 2008). Finally, we clustered the samples using k-medoids, also known as the PAM algorithm (Kaufman and Rousseeuw, 1990). Ideally, optimal cluster number k would be established via a resampling procedure such as consensus clustering (Monti et al., 2003). However, given our limited sample size, we chose to fix k = 2, separating samples into two groups that would ideally correspond to responders and non-responders. Cross-platform concordance was evaluated using the mutual information between cluster assignments, a dependency metric that ranges from 0 to 1 bit when k = 2.
Predictive Models
We built and evaluated a series of random forest models using continuous response measures to compare the predictive power associated with different platforms. To do so, we created a pipeline using tools from the caret package for classification and regression training (Kuhn and Johnson, 2013).
Continuous models, designed to predict a patient’s percent change in PASI, were tuned using the root mean square error (RMSE) loss function, which is standard for linear regressions. Response was defined by a winsorised the delta PASI distribution, as explained above. We selected variables using the two-loop RFE algorithm outlined in (Kuhn and Johnson, 2013). For each platform, we tested 20 different subsets of probes, with dimensionality determined by an exponential function so that relatively low-dimensional subsets of the feature space were explored more closely than high-dimensional subsets. Performance was evaluated using 10-fold cross-validation. Lower RMSE values indicate more predictive models.
Supplementary Material
Supplementary Table 1. Summary of differential expression of mRNA, miRNA and protein (q<0.1) across time and across tissue types (Serum, Blood, Lesional Skin, Non-Lesional Skin)
Supplementary Figure 1. Clinical response observed over 12 weeks of TNFi therapy; Baseline PASI vs. PASI at week 12 of therapy. This figure plots baseline vs. 12 week PASI scores for each patient. The black line has an intercept of 0 and a slope of 1, representing zero improvement over the course of treatment. The blue line has an intercept of 0 and a slope corresponding to the line of best fit through the data points. To obtain a least squares estimate of our study’s average delta PASI, we calculate the difference in slope between the black and blue lines: 69%. (The mean of our delta PASI distribution is 64%A).
Supplementary Figure 2. Exploratory data analysis of mRNA transcriptome data. Sample similarity matrix depicting samples clustered by pairwise Euclidean distance.
Supplementary Figure 3. Exploratory visualisation of the skin and blood transcriptomes. a) Principal component analysis of all skin and blood mRNA samples across all time points, b) t-stochastic neighbour embedding (t-SNE) clustering of all skin and blood samples across all time points.
Supplementary Figure 4. Exploratory visualisation of the psoriasis skin transcriptome. a) Principal component analysis of lesional and non-lesional skin mRNA samples; b) t-stochastic neighbour embedding (t-SNE) of mRNA from lesional and non-lesional skin samples
Supplementary figure 5. Exploratory visualisation of blood mRNA and miRNA transcriptome and proteome. a) Principal component analysis (PCA) of mRNA from blood samples; b) PCA of miRNA from blood samples; c) PCA of proteome from blood samples; d) t-stochastic neighbour embedding (t-SNE) of mRNA from blood samples; e) t-SNE of miRNA from blood samples; f) t-SNE of proteome from blood samples.
Supplementary Figure 6. Heatmaps depicting the top 1% of gene expression changes measured by mRNA-Seq in association with the change in PASI in lesional skin (4a), non-lesional skin (4b) and blood (4c). Cells are colored by scaled Pearson distance. Annotation tracks atop the figures show continuous and categorical response, as well as supervised and unsupervised cluster assignments.
Supplementary figure 7. Power curves projected across an expected range of fold changes based on the assumption that a) 1% or b) 5% of genes are likely to prove prognostic. Power calculations were performed using the method of Guo et al. (2014) and parameters derived from this pilot study; we calculated the requisite sample size to achieve 90% power (grey dotted line on plot) to detect differential expression associated with response.
Acknowledgements
We would like to thank Professors M. Kulesz-Martin and J. Tolar, Symposium Director and Program Chair of the Montagna Symposium on the Biology of Skin for their assistance. We acknowledge the support of the National Institute of Arthritis and Musculoskeletal and Skin Diseases and all co-funding support provided the National Institute on Aging in grant 5R13AR009431-52. We would like to thank the medical and nursing staff at Salford Royal NHS Foundation Trust, in particular Sister J. Howe and at Broadgreen Hospital Liverpool, in particular Dr A. Al-Sharqi and Sister B. Dever for their assistance in recruitment. We would like to thank Carole Todd, ICM, Newcastle University for her help in optimising RNA extraction from skin, Dr K. Maratou at GSK Stevenage for performance of RNA-Seq (skin samples), and Dr M. Hughes and Dr A. Lucaci of the Centre for Genomic Research Liverpool for performance of RNA-Seq (blood samples). We thank Dr I. Strickland at MedImmune for his assistance. We thank Dr M. Jani for performing week 12 serum etanercept levels. CEMG and MP are National Institute for Health Research (NIHR) Senior Investigators. NJR’s research/laboratory is funded in part by the Newcastle NIHR Biomedical Research Centre (BRC) and the Newcastle MRC/EPSRC Molecular Pathology Node. CEMG and RBW are funded in part by the Manchester NIHR BRC. MRB and DW were funded by the NIHR as part of the portfolio of translational research of the NIHR BRC at Barts and The London School of Medicine and Dentistry. This project was enabled through access to the MRC eMedLab Medical Bioinformatics infrastructure supported by the Medical Research Council [grant number MR/L016311/1]. The investigators are partially funded by the PSORT Medical Research Council, grant MR/1011808/1.
Disclosure
ACF has received educational support to attend conferences from or acted as a consultant or speaker for Abbvie, Almirall, Eli Lilly, Leo Pharma, Novartis, Pfizer, Janssen and UCB. NJR has received honoraria, travel support, and/or research grants (Newcastle University) from Abbvie, Almirall, Amgen, AstraZeneca, Bristol-Myers Squibb, Celgene, Genentech, Janssen, Leo-Pharma Research Foundation, Novartis, Pfizer, and Stiefel GSK. CEMG has acted as a consultant and/or speaker for Abbvie, Almirall, Janssen, Novartis, Sandoz, Rock Creek Pharma, Pfizer, Eli Lilly, Sun Pharmaceuticals, UCB, Leo Pharma, Galderma and Celgene. RBW has acted as a consultant and/or speaker for Abbvie, Amgen, Almirall, Boehringer, Medac, Eli Lilly, Janssen, Leo Pharma, Pfizer, Novartis, Sun Pharma, Valeant, Schering-Plough (now MSD) and Xenoport.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Blauvelt A, de Bruin-Weller M, Gooderham M, Cather JC, Weisman J, Pariser D, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet 2017;389(10086):2287–303. [DOI] [PubMed] [Google Scholar]
- Chow M, Lai K, Ahn R, Gupta R, Arron S, Liao W. Effect of Adalimumab on Gene Expression Profiles of Psoriatic Skin and Blood. Journal of Drugs in Dermatology 2016;15(8):988–94. [PMC free article] [PubMed] [Google Scholar]
- Correa da Rosa J, Kim J, Tian S, Tomalin LE, Krueger JG, Suarez-Farinas M. Shrinking the Psoriasis Assessment Gap: Early Gene-Expression Profiling Accurately Predicts Response to Long-Term Treatment. The Journal of investigative dermatology 2017;137(2):305–12. [DOI] [PubMed] [Google Scholar]
- Foulkes AC, Watson DS, Griffiths CEM, Warren RB, Huber W, Barnes MR. Research Techniques Made Simple: Bioinformatics for Genome-Scale Biology. The Journal of investigative dermatology 2017;137(9):e163–e8. [DOI] [PubMed] [Google Scholar]
- Griffiths CE. Systems medicine and psoriasis. Br J Dermatol 2017;176(3):560–2. [DOI] [PubMed] [Google Scholar]
- Griffiths CE, Barnes MR, Burden AD, Nestle FO, Reynolds NJ, Smith CH, et al. Establishing an Academic-Industrial Stratified Medicine Consortium: Psoriasis Stratification to Optimize Relevant Therapy. The Journal of investigative dermatology 2015;135(12):2903–7. [DOI] [PubMed] [Google Scholar]
- Guo Y, Zhao S, Li CI, Sheng Q, Shyr Y. RNAseqPS: A web tool for estimating sample size and power for RNAseq experiment. Cancer informatics 2014;13:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston A, Guzman AM, Swindell WR, Wang F, Kang S, Gudjonsson JE. Early tissue responses in psoriasis to the antitumour necrosis factor-alpha biologic etanercept suggest reduced interleukin-17 receptor expression and signalling. Br J Dermatol 2014;171(1):97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston A, Sarkar MK, Vrana A, Tsoi LC, Gudjonsson JE. The Molecular Revolution in Cutaneous Biology: The Era of Global Transcriptional Analysis. The Journal of investigative dermatology 2017;137(5):e87–e91. [DOI] [PubMed] [Google Scholar]
- Jorgensen A, Williamson P. Methodological quality of pharmacogenetic studies: Issues of concern. Statistics in Medicine 2008;27(30):6547–69. [DOI] [PubMed] [Google Scholar]
- Kaufman L, Rousseeuw PJ. Partitioning Around Medoids (Program PAM) Finding Groups in Data: John Wiley & Sons, Inc.; 1990. p. 68–125. [Google Scholar]
- Krueger JG. The immunologic basis for the treatment of psoriasis with new biologic agents. J Am Acad Dermatol 2002;46(1):1–23; quiz −6. [DOI] [PubMed] [Google Scholar]
- Kuhn M, Johnson K. Applied Predictive Modeling. 2013. [Google Scholar]
- Lebwohl M Psoriasis Therapy: Breakthroughs in Pharmacogenomics or in Pharmacology? The Journal of investigative dermatology 2016;136(12):2339–40. [DOI] [PubMed] [Google Scholar]
- Leonardi CL, Powers JL, Matheson RT, Goffe BS, Zitnik R, Wang A, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003;349(21):2014–22. [DOI] [PubMed] [Google Scholar]
- Li B, Tsoi LC, Swindell WR, Gudjonsson JE, Tejasvi T, Johnston A, et al. Transcriptome analysis of psoriasis in a large case-control sample: RNA-seq provides insights into disease mechanisms. The Journal of investigative dermatology 2014;134(7):1828–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavragani CP, Niewold TB, Moutsopoulos NM, Pillemer SR, Wahl SM, Crow MK. Augmented interferon-alpha pathway activation in patients with Sjogren’s syndrome treated with etanercept. Arthritis Rheum 2007;56(12):3995–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti S, Tamayo P, Mesirov J, Golub T. Consensus clustering: A resampling-based method for class discovery and visualization of gene expression microarray data. Machine Learning 2003;52:91–118. [Google Scholar]
- Omberg L, Ellrott K, Yuan Y, Kandoth C, Wong C, Kellen MR, et al. Enabling transparent and collaborative computational analysis of 12 tumor types within The Cancer Genome Atlas. Nat Genet 2013;45(10):1121–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez-Fariñas M, Li K, Fuentes-Duculan J, Hayden K, Brodmerkel C, Krueger JG. Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate-to-severe psoriasis. Journal of Investigative Dermatology 2012;132(11):2552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Farinas M, Ungar B, Correa da Rosa J, Ewald DA, Rozenblit M, Gonzalez J, et al. RNA sequencing atopic dermatitis transcriptome profiling provides insights into novel disease mechanisms with potential therapeutic implications. J Allergy Clin Immunol 2015;135(5):1218–27. [DOI] [PubMed] [Google Scholar]
- Talamonti M, Botti E, Galluzzo M, Teoli M, Spallone G, Bavetta M, et al. Pharmacogenetics of psoriasis: HLA-Cw6 but not LCE3B/3C deletion nor TNFAIP3 polymorphism predisposes to clinical response to interleukin 12/23 blocker ustekinumab. British Journal of Dermatology 2013;169(2):458–63. [DOI] [PubMed] [Google Scholar]
- Ungar B, Garcet S, Gonzalez J, Dhingra N, Correa da Rosa J, Shemer A, et al. An Integrated Model of Atopic Dermatitis Biomarkers Highlights the Systemic Nature of the Disease. The Journal of investigative dermatology 2017;137(3):603–13. [DOI] [PubMed] [Google Scholar]
- Van Der Maaten L, Hinton G, van der Maaten GH. Visualizing Data using t-SNE. Journal of Machine Learning Research 2008;9:2579–605. [Google Scholar]
- Zaba LC, Suárez-Fariñas M, Fuentes-Duculan J, Nograles KE, Guttman-Yassky E, Cardinale I, et al. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. Journal of Allergy and Clinical Immunology 2009;124(5):1022–10.e1– 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Rudwaleit M, Brandt J, Thiel A, Braun J, Sieper J. Up regulation of the production of tumour necrosis factor alpha and interferon gamma by T cells in ankylosing spondylitis during treatment with etanercept. Ann Rheum Dis 2003;62(6):561–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Summary of differential expression of mRNA, miRNA and protein (q<0.1) across time and across tissue types (Serum, Blood, Lesional Skin, Non-Lesional Skin)
Supplementary Figure 1. Clinical response observed over 12 weeks of TNFi therapy; Baseline PASI vs. PASI at week 12 of therapy. This figure plots baseline vs. 12 week PASI scores for each patient. The black line has an intercept of 0 and a slope of 1, representing zero improvement over the course of treatment. The blue line has an intercept of 0 and a slope corresponding to the line of best fit through the data points. To obtain a least squares estimate of our study’s average delta PASI, we calculate the difference in slope between the black and blue lines: 69%. (The mean of our delta PASI distribution is 64%A).
Supplementary Figure 2. Exploratory data analysis of mRNA transcriptome data. Sample similarity matrix depicting samples clustered by pairwise Euclidean distance.
Supplementary Figure 3. Exploratory visualisation of the skin and blood transcriptomes. a) Principal component analysis of all skin and blood mRNA samples across all time points, b) t-stochastic neighbour embedding (t-SNE) clustering of all skin and blood samples across all time points.
Supplementary Figure 4. Exploratory visualisation of the psoriasis skin transcriptome. a) Principal component analysis of lesional and non-lesional skin mRNA samples; b) t-stochastic neighbour embedding (t-SNE) of mRNA from lesional and non-lesional skin samples
Supplementary figure 5. Exploratory visualisation of blood mRNA and miRNA transcriptome and proteome. a) Principal component analysis (PCA) of mRNA from blood samples; b) PCA of miRNA from blood samples; c) PCA of proteome from blood samples; d) t-stochastic neighbour embedding (t-SNE) of mRNA from blood samples; e) t-SNE of miRNA from blood samples; f) t-SNE of proteome from blood samples.
Supplementary Figure 6. Heatmaps depicting the top 1% of gene expression changes measured by mRNA-Seq in association with the change in PASI in lesional skin (4a), non-lesional skin (4b) and blood (4c). Cells are colored by scaled Pearson distance. Annotation tracks atop the figures show continuous and categorical response, as well as supervised and unsupervised cluster assignments.
Supplementary figure 7. Power curves projected across an expected range of fold changes based on the assumption that a) 1% or b) 5% of genes are likely to prove prognostic. Power calculations were performed using the method of Guo et al. (2014) and parameters derived from this pilot study; we calculated the requisite sample size to achieve 90% power (grey dotted line on plot) to detect differential expression associated with response.