

Abstract
Amphipathic agents are widely used in various fields including biomedical sciences. Micelle-forming detergents are particularly useful for in vitro membrane protein characterization. As many conventional detergents are limited in their ability to stabilize membrane proteins, it is necessary to develop novel detergents to facilitate membrane protein research. In the current study, we developed novel trimaltoside detergents with an alkyl pendant-bearing terphenyl unit as a hydrophobic group, designated terphenyl-cored maltosides (TPMs). We found that the geometry of the detergent hydrophobic group substantially impacts detergent self-assembly behaviour, as well as detergent efficacy for membrane protein stabilization. TPM-Vs with a bent terphenyl group were superior to the linear counterparts (TPM-Ls) at stabilizing multiple membrane proteins. The favourable protein stabilization efficacy of these bent TPMs is likely associated with a binding mode with membrane proteins distinct from conventional detergents and facial amphiphiles. When compared to n-dodecyl-β-D-maltoside (DDM), most TPMs were superior or comparable to this gold standard detergent at stabilizing membrane proteins. Notably, TPM-L3 was particularly effective at stabilizing the human β2 adrenergic receptor (β2AR), a G-protein coupled receptor, and its complex with Gs protein. Thus, the current study not only provides novel detergent tools useful for membrane protein study, but also suggests a critical role for detergent hydrophobic group geometry in governing detergent efficacy.
Keywords: detergent design, detergent geometry, self-assembly, aromatic-aromatic interactions, protein stabilization
Graphical Abstract
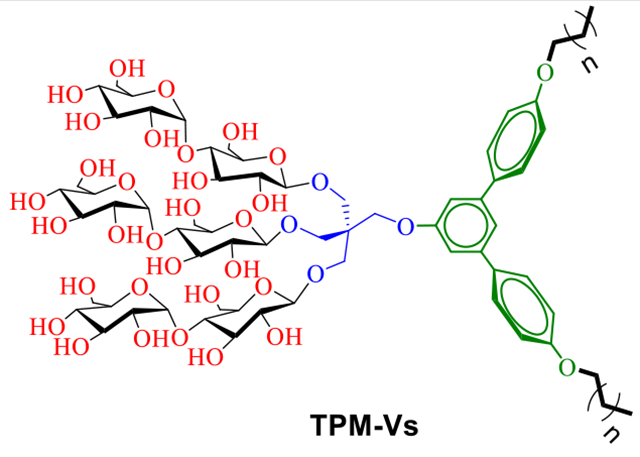
A comparative study of two sets of new amphiphiles revealed a large difference in detergent efficacy for membrane protein stabilization between TPM-Vs and TPM-Ls. This result indicates the importance of detergent hydrophobic group geometry in membrane protein stability.
Introduction
The biomimetic materials capable of reproducing the architecture and properties of native biological membranes are attracting much interest in the field of biomedical sciences.[1,2] Some natural or synthetic amphiphiles can simulate the behaviours of cell membrane components by self-assembling into organized structures in aqueous environments.[3,4] Self-assembled structures formed by amphiphiles vary from simple micelles to highly organized large aggregates such as fibres, tubes, and helices and are growingly used in material chemistry, nanotechnology, and medicinal chemistry.[5–8] Among these amphipathic agents, carbohydrate-bearing agents such as n-octyl-β-D-glucoside (OG), n-decyl-β-D-maltoside (DM) and n-dodecyl-β-D-maltoside (DDM) are particularly useful for membrane protein characterization. However, membrane proteins encapsulated even in these popular detergents are often prone to denaturation and aggregation.[9] Thus, it is difficult to conduct downstream protein characterization such as functional studies, spectroscopic analysis, or crystallization trials. As membrane proteins play critical roles in a variety of cellular processes and are major targets of pharmaceuticals, understanding of their structures and functions is of great importance. Thus, we need to develop new amphiphilic molecules or membrane-mimetic systems with enhanced protein stabilization efficacy to advance membrane protein research.[10]
Recent years have witnessed increasing efforts to develop new amphiphilic systems with the ability to maintain the native structures of membrane proteins. Several innovative approaches such as lipid nanodiscs (NDs),[11] styrene-maleic acid copolymers (SMAs),[12] amphiphilic polymers (APols),[13] lipopeptide detergents (LPDs)[14] and β-peptides (BPs) has been developed[15] and some of these (e.g., NDs and APols) have found broad applications in membrane protein biochemistry. However, the repertoire of amphiphiles that have been successfully used for protein crystallization is still quite limited. In addition, they are generally inefficient at extracting proteins from the membranes and are often difficult to synthesize on a bulk scale. Small amphiphilic agents with chemically well-defined structures have been invented for protein extraction and purification as well as protein crystallization. Representatives include tripod amphiphiles (TPAs),[16] glucose or maltose neopentyl glycols (GNGs/MNGs),[17,18] steroid-based amphiphiles (e.g., chobimalt[19] and GDN[20]), hemifluorinated surfactants (HFSs).[21] Of these amphiphiles, GNG-3 and MNG-3 have facilitated the crystal structure determinations of ~40 new membrane proteins including several classes of G-protein coupled receptors (GPCRs).[22] We recently reported several synthetic amphiphiles such as mannitol-based amphiphiles (MNAs),[23] xylene or mesitylene-based amphiphiles (XMAs/MGAs),[24] neopentyl glycol triglucosides (NDTs),[25] penta-saccharide-bearing amphiphiles (PSEs),[26] norbornane-based maltosides (NBMs)[27] and dendronic trimaltosides (DTMs).[28] As a part of our long-term efforts, we describe here a class of small amphiphilic agents with a non-conventional hydrophobic group. This class features a trimaltoside head group and a central terphenyl group with short alkyl appendages, designated terphenyl-cored maltosides (TPMs). Depending on the geometry of the central terphenyl group (bent vs linear), these agents exhibited markedly different self-assembly behaviour and architecture. In an evaluation with four model membrane proteins including a G-protein coupled receptor (GPCR), the TPMs conferred enhanced stability to those proteins compared to DDM. In addition, the geometry of the hydrophobic group significantly affected detergent efficacy for membrane protein stabilization. Of the TPMs, TPM-L3 was particularly useful at stabilizing a GPCR-GS complex, allowing clear visualization of the complex via negative stain EM.
Results and Discussion
Detergent structures and physical characterizations
The newly designed amphiphiles commonly have a trimaltoside head group conjugated to a lipophilic group via a pentaerythritol linker (Scheme 1). All the detergents also share a benzene trimer (i.e., terphenyl group) with alkyl pendants as the lipophilic group but vary in the geometry of the group. One set has a linear benzene trimer (para-terphenyl group) as a central hydrophobic scaffold (TPM-Ls), while the other set contains a bent benzene trimer (meta-terphenyl group) (TPM-Vs) in the same region. Thus, these two sets of the TPMs have distinct geometries in their lipophilic groups (linear vs bent (V-shaped)). We hypothesized that this geometrical difference in the lipophilic group leads to a significant variation in amphiphile efficacy for membrane protein stabilization, despite their identical molecular formula. Detergent hydrophobicity varied through the attachment of either a propyl (C3) or butyl (C4) chain at both terminals of the terphenyl groups via ether linkages, as indicated in the detergent designation. This alkyl chain length variation is essential for optimizing the hydrophile-lipophile balance (HLB),[29] known to be crucial in detergent efficacy.[16a] Because of the coexistence of the rigid terphenyl group and the flexible alkyl chains in the lipophilic region, the new agents not only have modulated flexibility, but may also customize their interactions with individual membrane proteins, as observed previously with the lithocholic acid-based facial amphiphiles (LFA).[30] It is important to note that the new agents structurally differ from recently developed novel agents. First, there is no report of terphenyl group-bearing detergents for membrane protein study. In considering the facial segregation of the hydrophilic group from the hydrophobic group, the TPM-Ls can be classified into facial amphiphiles like FAs and TFAs.[31] However, these linear TPMs bear three consecutive aromatic rings flanked by two short alkyl chains, different from previous facial amphiphiles. In addition, the hydrophilic group is rather localized at the centre of the TPMs while the corresponding hydrophilic groups in FAs and TFAs are dispersedly distributed along the hydrophobic dimensions of the molecules. Compared to the TPM-Ls, the TPM-Vs would lack faciality due to the bent architecture of the hydrophobic group. However, the TPM-Vs are the first examples of detergents with a curved hydrophobic surface, which is potentially suited for stabilizing membrane proteins with curved hydrophobic surfaces.
Scheme 1.
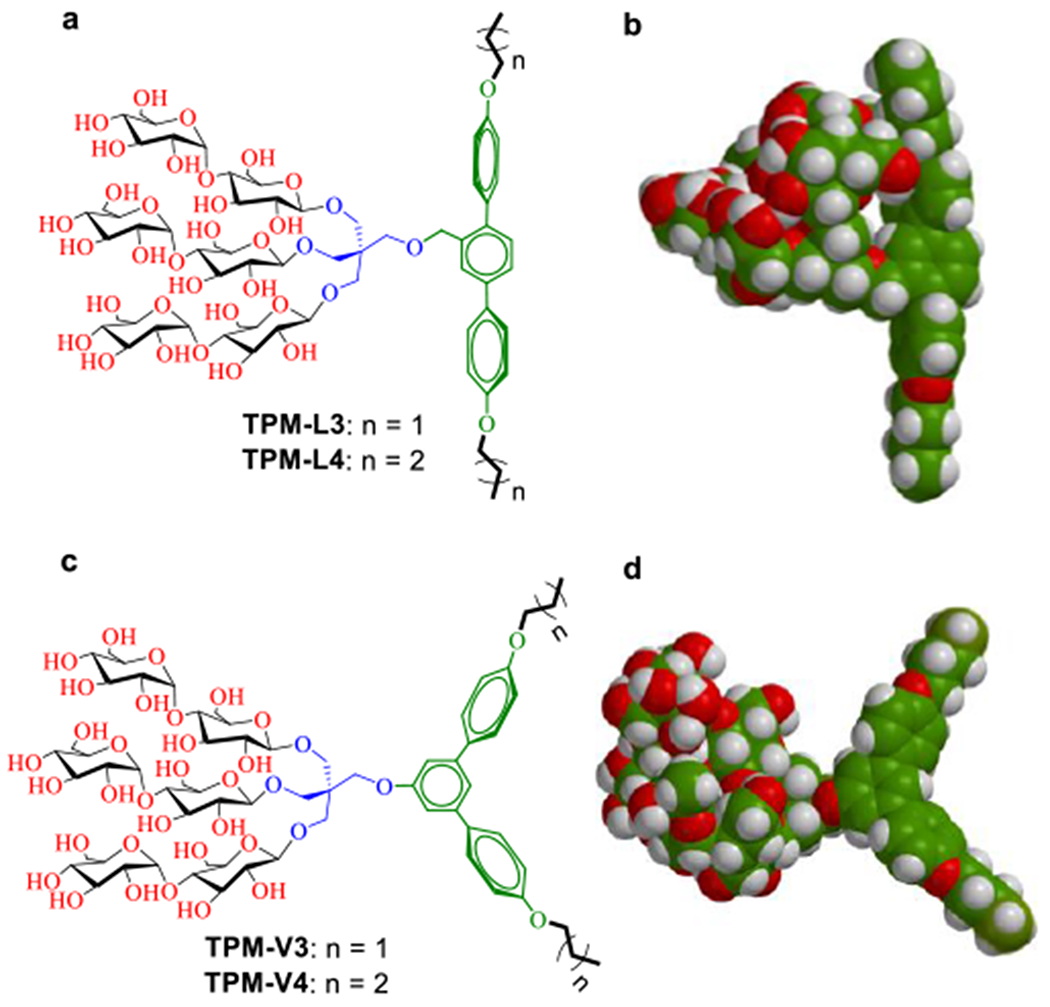
(a,c) Chemical structures of terphenyl-cored maltosides (TPMs) and (b,d) space-filing models for the energy-minimized structures of TPM-L4 and TPM-V4. These amphiphiles commonly contain a trimaltoside head group connected to the lipophilic group using a neopentyl glycol linker. The lipophilic group features with three consecutive phenyl rings (i.e., terphenyl group) with alkyl chain appendages at both terminals. The three phenyl rings are organized in a linear or in a bent arrangement (V-shaped), and are thus designated TPM-Ls (a,b) and TPM-Vs (c,d), respectively. Two short alkyl chains (propyl (C3) and butyl (C4)) were introduced as the terminal units, as indicated in the detergent designation. The energy-minimized structures of TPM-L4 and TPM-V4 as obtained by DFT calculations (B3LYP/6–31G* level) in water (model, space filling). Carbon, hydrogen and oxygen are indicated in green, white and red, respectively.
We compared these two sets in terms of their hydrophobic group dimensions as the hydrophobic dimensions of a detergent molecule should be compatible with those of membrane proteins for protein stability. The width of the total hydrophobic group of TPM-L4 was calculated to be 23.3 Å while TPM-V4 has a hydrophobic group width of 19.4 Å (Figure S1), both substantially shorter than a typical range of the hydrophobic thickness of membrane proteins (28~32 Å). Thus, two molecules of these agents would need to assemble side by side to form a dimeric pair that can span the hydrophobic region of membrane proteins. The dimeric pairs of these two agents (TPM-L4 and TPM-V4) appear to have substantially large hydrophobic lengths, compared to the hydrophobic thickness of membrane proteins. Because of the presence of the flexible alkyl chain at both sides of the rigid terphenyl groups, however, it is possible that the dimeric pair of TPM-L4/V4 has a range of effective hydrophobic thickness via alkyl chain overlap and/or adoption of non-anti-staggered (i.e., gauche) chain conformation.[30] Alternatively, to maximize detergent-protein interactions, these agents could assemble around membrane protein surfaces in an arrangement different from facial detergents. We also measured the dimensions of the central terphenyl group of TPM-L4/V4. The linear terphenyl group of TPM-L4 has a length of 11.3 Å, longer than that of the bent aromatic group in TPM-V4 (9.8 Å) (Figure S1). The thicknesses of these rigid hydrophobic groups were estimated to be 4.3 Å for TPM-L4 and 5.0/3.9 Å for TPM-V4 (Figure S2)
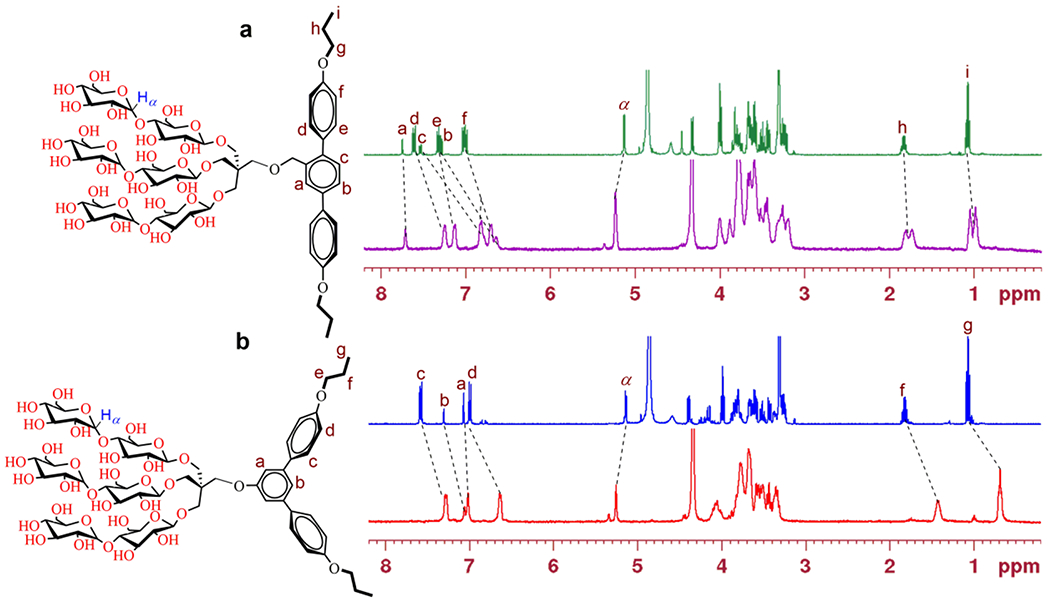
These novel agents were prepared through a synthetic protocol comprising four/five synthetic steps using commercially available boronic acid derivatives (see Supplementary schemes 1 & 2). The synthetic route for the TPMs contains three key steps: (i) coupling of the boronic acid derivatives with an aromatic bromide via palladium-catalyzed Suzuki coupling; (ii) synthesis of the tri-ol derivatives with a pentaerythritol linker; and (iii) stereo-selective glycosylation (Scheme 2). For preparation of TPM-L4, cross-coupling of 4-butoxyphenylboronic acid was carried out with methyl 2,5-dibromobenzoate in the presence of Pd[PPh3]4 in a water-THF solvent system. The resulting terphenyl group-bearing alcohol (A) was reacted with a pentaerythritol derivative to produce a triol compound (C). For TPM-V4 synthesis, a similar protocol was used, but in this case 3,5-dibromophenol was used as a starting material to obtain the V-shaped mono-ol (B) and triol compound (D), respectively. The tri-ol derivative (C or D) was then used as a substrate for glycosylation where silver triflate (AgOTf) and perbenzyolated maltosylbromide were used as promotor and glycosyl donor, respectively. Such Lewis acid-based glycosylation is known to afford a stereo-selective β-anomer via neighbouring group participation. The β-stereochemistry for the newly formed glycosidic bonds was confirmed by the individual 1H or 13C NMR spectra of the TPMs in CD3OD (Figures S3 & S4). For instance, the 1H NMR spectrum of TPM-L3 showed a sharp peak at 4.33 ppm as a doublet, with a vicinal coupling constant (3Jaa) of 8.0 Hz (Figure 1). These peak features are typical of a β-anomeric axial hydrogen (Ha), demonstrating exclusive β-glycosidic bond formation in the glycosylation. Note that α-anomeric proton produces a peak downfield shifted to 5.13 ppm with a relatively small coupling constant (3Jae = 4.0 Hz), as also observed in the NMR spectrum of TPM-L3 due to the pre-existence of an α-glycosidic bond in the maltose unit. A consistent stereochemistry was observed in the 13C NMR spectrum of this amphiphile where two peaks corresponding to the anomeric carbons appeared at 105.1 and 103.0 ppm (Figure 1, bottom). Because of the high efficiency of each synthetic step, the final amphipathic compounds could be prepared in high overall yields, making them feasible for preparation in multigram quantities.
Scheme 2.
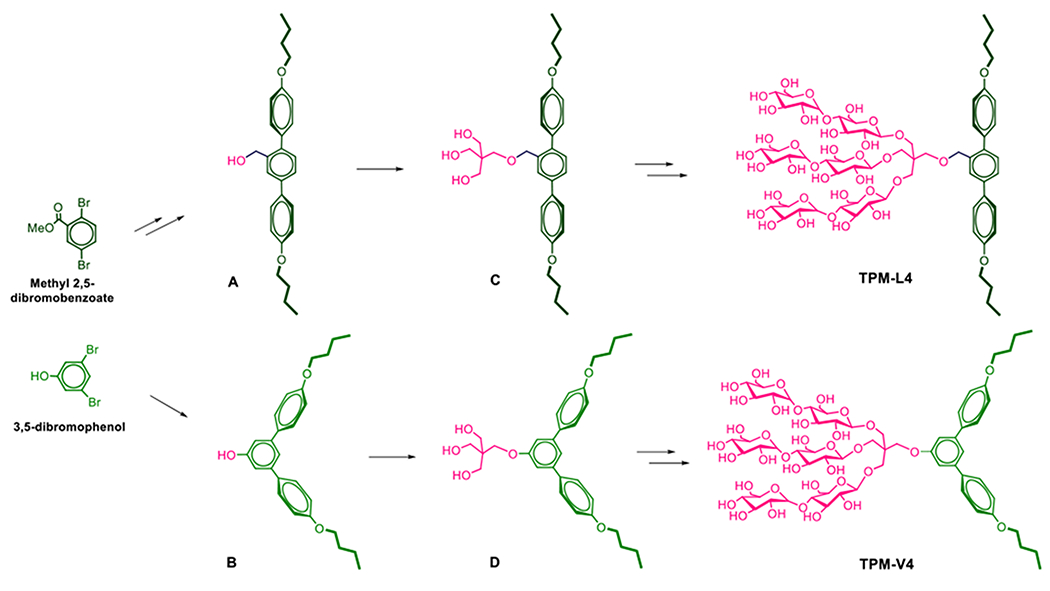
Synthetic scheme for the preparation of two TPMs (TPM-L4 (top) and TPM-V4 (bottom)). Two different starting materials, methyl 2,5-dibromobenzoate and 3,5-dibromophenol (left), were used to synthesize TPM-L4 and TPM-V4, respectively. Rigid aromatic segments (i.e., terphenyl group) were built from these starting materials via cross-coupling reactions with commercially available boronic acid derivatives with an alkyl pendant (Suzuki-coupling reactions). The resulting mono-ol derivatives (A and B) were coupled with a neopentyl glycol linker to give the tri-ol derivatives (C and D, respectively) used to introduce three maltose groups (glycosylation).
Figure 1.
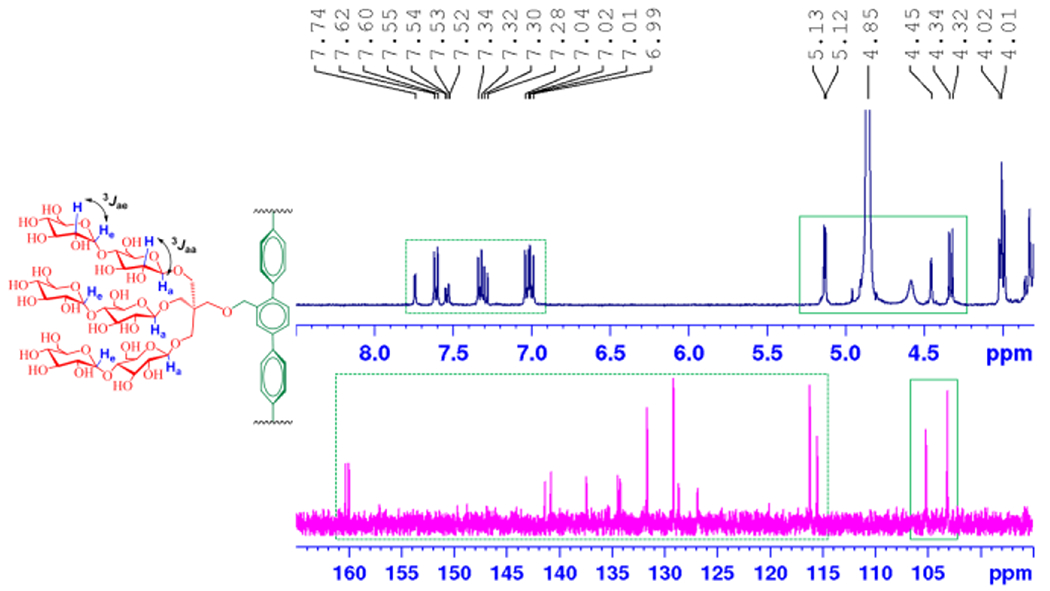
Partial 1H and 13C NMR spectra of TPM-L3 focusing on the anomeric and aromatic regions (see Fig. S2 for the full range of the spectra), (top) 1H NMR spectrum of TPM-L3 gave a doublet at 4.33 ppm, along with a coupling constant (3Jaa) of 8.0 Hz, a typical peak characteristic for β-anomeric proton (Ha). This agent also contains α-anomeric protons (He), giving a doublet at 5.13 ppm with a reduced coupling constant (3Jae = 4.0 Hz). Ha and He indicate anomeric protons in the axial and equatorial positions, respectively. Aromatic protons of the terphenyl group were well resolved in the region from 6.9 to 7.8 ppm in this spectrum. (bottom) The anomeric protons (Ha and He) of TPM-L3 gave peaks at 103.0 and 105.1 ppm in the 13C NMR spectrum while peaks corresponding to the aromatic protons dispersedly appeared in a range of 126 to 161 ppm. The chemical structure of TPM-L3 including the anomeric and aromatic regions is shown to illustrate the anomeric protons of interest. Dotted and solid boxes on the spectra represent the aromatic and anomeric regions, respectively.
All the novel agents were highly soluble in water (>10 % w/v), a which is prerequisite for biophysical studies with membrane proteins. The aggregates formed by these agents were stable enough to give clear solutions for one month at room temperature. Aggregation behaviours of the TPMs were investigated by measuring critical micelle concentrations (CMCs) and hydrodynamic diameters (Dh) of the micelles in aqueous solution. CMCs were estimated by monitoring solubilization of a hydrophobic dye (i.e., diphenylhexatriene (DPH)),[32] while Dh values of detergent micelles were determined by dynamic light scattering (DLS) measurements. The results for the TPMs along with DDM are summarized in Table 1. Like DDM, these new detergents have defined CMCs in a sub-millimolar range that varied depending on the hydrophobicity/geometry of the hydrophobic groups. All the TPMs gave smaller CMCs than DDM (0.17 mM). An increase in alkyl chain length from propyl (C3) to butyl (C4) resulted in a decrease in the CMC values due to increased hydrophobicity. While the CMC of TPM-L4 was only slightly lower than that of TPM-L3 (~0.020 vs 0.025 mM), a large drop in the CMC was found for the TPM-Vs; the CMC was reduced three-fold with a chain length increase from C3 to C4 (~0.040 vs ~0.012 mM). Micelle sizes formed by the new agents were measured at 1.0 wt% detergent concentration. All the agents self-organized into well-defined small assemblies in water, with a range of hydrodynamic diameters (Dh) of 5.0 to 7.4 nm, suggesting that the aggregates are likely to be micelles rather than liposomes or other aggregates (Table 1).
Table 1.
Molecular weights (MWs), critical micelle concentrations (CMCs), and aggregation numbers (ANs) of TPMs (TPM-L3/L4 and TPM-V3/V4) along with the conventional detergent DDM and their micelle sizes in terms of hydrodynamic diameters (Dh) (mean ± S.D., n = 5) in water.
| Detergent | MWa | CMC (mM) | Dh (nm)b | AN |
|---|---|---|---|---|
| TPM-L3 | 1467.5 | ~0.025 | 5.0±0.6 | ~5 |
| TPM-L4 | 1495.5 | ~0.020 | 7.4±1.4 | ~8 |
| TPM-V3 | 1453.5 | ~0.040 | 5.6±0.1 | ~14 |
| TPM-V4 | 1481.5 | ~0.012 | 6.4±0.2 | ~30 |
| DDM | 510.6 | ~0.17 | 6.8±0.0 | ND |
Molecular weight of detergents.
Hydrodynamic diameters of detergents measured at 1.0 wt% by dynamic light scattering.
ND = not determined.
The micelle sizes of the TPMs were of a similar size or smaller than those of DDM (~6.8 nm), with the exception of TPM-L4 which formed larger micelles (~7.4 nm). The micelle sizes tend to increase with increasing alkyl chain length of the new agents, as similar trends observed for other facial agents.[14,31b] With a change of the alkyl pendant from C3 to C4, the micelle sizes formed by the TPM-Ls increased from 5.0 to 7.4 nm. This increase in micelle size is likely caused by association of a greater number of detergent molecules upon micelle formation (i.e., higher aggregation numbers (ANs)). The micelles formed by the TPM-Vs exhibited only a small increase in size from ~5.6 to ~6.4 nm, indicating that the AN of TPM-V4 should be similar to that of TPM-V3. Due to the presence of a longer arm, the intermolecular distance of the V4 compound would be larger than that of the V3 agent. When we switched the solvent from water to Tris buffer, detergent micelle sizes were in the same range, pariticularly in the case of the TPM-Ls (Table S1). The detergent micelle sizes remained constant over a 19-day incubation at room temperature or temperature variation from 15 to 65 °C (Figure 2a,b), indicating a high thermal stability of the TPM micelles. Further analysis revealed that, like DDM, there is little variation in micelle size for the TPM-Vs with increasing detergent concentration from 0.1 to 2.0 wt% (Figure 2c). In contrast, micelles formed by TPM-L3/L4 became enlarged with increasing detergent concentration. Thus, detergent micelle behaviours such as CMC and concentration-dependent micelle size were substantially different between the TPM-Ls and TPM-Vs, indicating a clear effect of the geometry of the hydrophobic group (linear vs V-shaped) on detergent self-assembly. Detergent micelles were further analyzed in terms of size distribution of micellar populations. The number- or volume-weighted DLS profiles of the TPM-L/Vs showed a singlet set of micellar populations, supporting the small and highly homogeneous micelle formation (Figure S5). Due to the extreme sensitivity of scattered light intensity to a large particle, large aggregates were detected in the intensity-weighted DLS profiles of these TPMs.[33] The ANs of micelles formed by the TPMs in Tris buffer (pH 7.4) were determined via size exclusion chromatography (SEC) equipped with a triple-detector system (UV, light scattering, and refractive index) (Figure S6). The ANs of the TPM-L3 and TPM-L4 were estimated to be ~5 and ~8, respectively, consistent with the DLS data under similar conditions (Table 1). The larger AN of TPM-L4 is in agreement with the formation of larger micelles than that of TPM-L3 (5.0 vs 7.4 nm). As for the TPM-Vs, a similar range of ANs was expected as these bent TPMs form micelles with sizes similar to those of the TPM-Ls. However, the ANs of these TPMs were unexpectedly large (~14 for TPM-V3 and ~30 for TPM-V4), inconsistent with DLS data. This discrepancy might be the result of a higher tendency of the TPM-Vs to aggregate within the SEC column containing beads with a large solid surface area.
Figure 2.
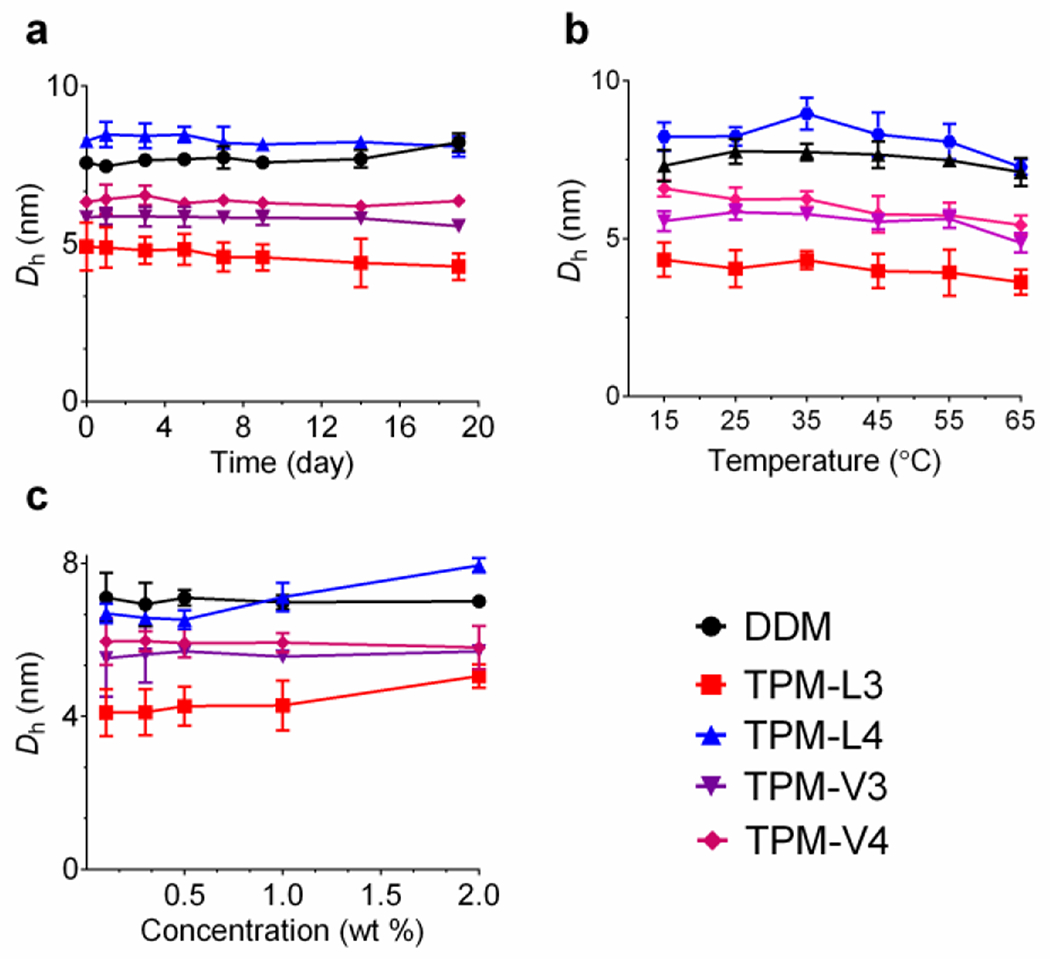
Variations in micelle hydrodynamic diameters (Dh) of TPMs (TPM-L3/L4 and TPM-V3A/4) depending on incubation time (a), temperature (b), and detergent concentration (c) in water. Micelle sizes were measured at a detergent concentration of 1.0 wt% over the course of a 19-day incubation at room temperature (a) or with increasing temperature from 15 to 65 °C (b). (c) Detergent micelle sizes were measured with increasing detergent concentrations from 0.1 to 2.0 wt% at room temperature. Error bars are standard deviations (SD), n = 4–5.
Detailed intermolecular interactions driving these self-assembly formations were addressed via chemical shift analysis using 1H NMR spectroscopy.[34,35] When dissolved in CD3OD at room temperature, TPM-L3 showed six well-resolved signals in a range of 7.74 to 6.99 ppm, corresponding to the aromatic protons (Figure 3a, top). When D2O instead of CD3OD was used as a solvent at room temperature, all NMR peaks were largely broadened and aggregated, consistent with self-assembly formation in aqueous solution (Figure S7a). Recording the NMR spectra in an increased temperature of 60 °C resulted in a significant improvement in peak resolution allowing correct assignment of each aromatic peak. With the solvent exchange and temperature variation from CD3OD (room temperature) to D2O (60 °C), all aromatic signals of TPM-L3 underwent significant upfield shifts (Figure 3a, bottom). Hb and Hc on the central phenyl ring showed prominent upfield shifts of their signals (Δδ = −0.66 and −0.77 ppm, respectively), indicating that these aromatic protons locating at the hydrophobic core are efficiently shielded by the aromatic terphenyl group (Table S2). Intermediate upfield shifts were observed for other aromatic protons (Hd, He, and Hf) attached to the peripheral phenyl rings (Δδ = −0.20 ~ −0.34 ppm). In contrast, an aromatic proton (Ha) directed to the hydrophilic surface gave only a minor peak shift (Δδ = −0.04 ppm), implying little shielding of this proton by the aromatic system. Thus, this result indicates that TPM-L3 forms self-assemblies where the terphenyl groups strongly interact with each other to form an aromatic shell in the self-assembled interior (aromatic-aromatic interaction). A similar result was observed for TPM-V3 (Figure 3b). Under the same conditions, the aromatic signals of this agent were broadened and shifted upfield probably due to the formation of self-assemblies with aromatic group packing (Figure S7b). However, the signal shift values observed for this agent were generally a little smaller than those of TPM-L3 (Table S2), which could be ascribed to relatively weak aromatic-aromatic interactions or a small shielding effect of the bent aromatic group relative to the linear group. Again, the aromatic signal (Ha) close to the hydrophilic surface gave only a minor upfield shift (Δδ = −0.04 ppm). Interestingly, these two TPMs (TPM-L3 and TPM-V3) in D2O showed different patterns in the signals of the alkyl pendants. The alkyl chain protons (Hh and Hi) of TPM-L3 gave only small upfield shifts of their NMR peaks (Δδ = −0.04 ppm), contrast to the large upfield shifts observed for the alkyl proton peaks of TPM-V3 (Hf and Hg; Δδ = −0.4 ppm). This spectral difference indicates that both alkyl chains of TPM-L3 were positioned outside the packing of the terphenyl groups (i.e., aromatic shell) in the micelle interior, whereas those of TPM-V3 were effectively encapsulated in the aromatic shell of the self-assemblies. Additionally, the two alkyl chains of TPM-L3 gave different chemical shifts (two signals) when D2O was used as the NMR solvent, while those of TPM-V3 appeared to give a single peak under the same conditions. Thus, the two alkyl chains of TPM-L3 were, otherwise identical, differentiated by assembly formation. In other words, the two alkyl chains of this linear TPM were in a different environment (asymmetric) within assembly architecture. This was different from the alkyl chains of TPM-V3 in an identical environment (symmetric). These different behaviours of the alkyl chains between TPM-L3 and TPM-V3 in the self-assembly formation would originate from a geometrical variation in their hydrophobic groups (linear vs V-shaped). Apart from the aromatic or alkyl proton signals, α-anomeric protons (Hα) yielded a small downfield peak shift (Δδ = +0.10 ppm) in the case of both TPM-L3 and TPM-V3. This small downfield shift may be an indication of interaction of these anomeric protons with water molecules present in the surfaces of the self-assemblies.[36] Self-assembly behaviors of TPM-L4 and TPM-V4 were investigated under the same conditions and the results very similar to those of TPM-L3 and TPM-V3, respectively (Figure S8). This finding indicates that the change in alkyl chain length from C3 to C4 has little effect on the structures of their self-assemblies. Based on the DLS, NMR and SEC results, along with molecular geometry of the detergent hydrophobic groups, the TPM-Ls would facially interact with each other to form nanocylinder-like micelles, while the bent TPM-Vs are likely to be associated to form nanocapsules, which appears reasonably consistent with the assembly architectures of structurally related amphiphiles.[37] A more detailed study is necessary to clarify unambiguously the structures of the self-assemblies formed by these new TPMs.
Figure 3.
(a) 1H NMR spectrum (400 MHz) of (a) TPM-L3 and (b) TPM-V3 at 1.0 mM in CD3OD (top) or in D2O (bottom). The spectra in CD3OD were measured at room temperature while those in D2O were measured at 60 °C. The chemical structures of the amphiphiles (TPM-L3 and TPM-V3) were given to show proton assignment of interest. Peak shifts induced by the solvent change from CD3OD to D2O were indicated by dotted lines in the spectra. Tetramethylsilane (TMS) was used as an internal standard.
Detergent evaluation with membrane proteins
The new agents were evaluated with a set of membrane proteins to investigate their abilities to maintain a membrane protein in a soluble and functional form. The TPMs were first evaluated using the Rhodobacter (R.) capsulatus super-assembly, comprising light harvesting complex I and the reaction centre complex (LHI-RC).[38] To start with, the complexes were extracted by 1.0 wt% DDM from the membranes and isolated in the same detergent using Ni2+-NTA affinity column. The DDM-purified LHI-RC was diluted into buffer solutions containing individual detergents to give final detergent concentrations of CMCs+0.05 wt%. The thermal stability of the protein samples was investigated by incubation at 25 °C for the first 10 days and then at an elevated temperature of 35 °C for the next 10 days. Absorbance at 875 nm (A875) was used as a criterion to assess the complex integrity in the individual detergents as the native complex contains the collection of cofactors (e.g., chlorophylls and carotenoids) embedded in the protein interior.[39] As can be shown in Figure 4a, DDM-solubilized complexes gradually lost their integrity over time and reached ~55% intact structure after the 10-day incubation at 25 °C. Use of the TPM-Ls led to enhanced protein stability, attaining ~90% retention under the same conditions.
Figure 4.
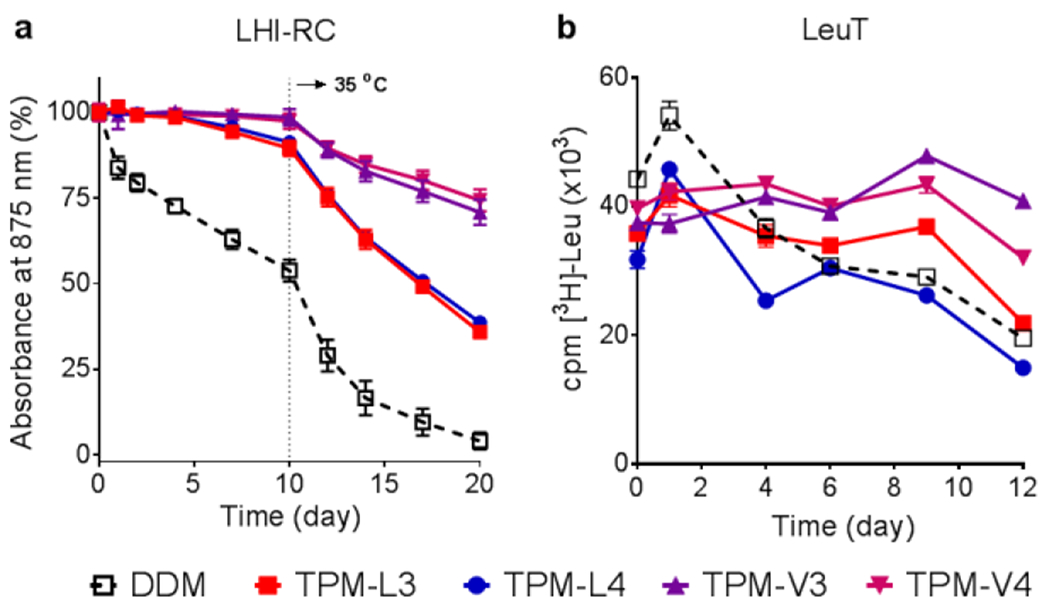
Time course stability of (a) LHI-RC complex (b) LeuT solubilized in novel agents (TPM-L3, TPM-L4, TPM-V3 and TPM-V4). A conventional detergent, DDM, was used as control. LHI-RC and LeuT stability assays were carried out at detergent concentrations of CMC+0.05 wt% and CMCs+0.04 wt%, respectively. LHI-RC stability was assessed by monitoring the absorbance of the complexes at 875 nm (A875) at regular intervals during a 20-day incubation. The samples were stored at room temperature for the first 10-day incubation and the incubation temperature was increased and maintained at 35 °C for the next 10 days. Error bars, SEM, n = 2. LeuT stability was assessed by measuring the ability of the transporter to bind the radio-labelled substrate (3[H]-leucine (Leu)) via scintillation proximity assay (SPA) and was monitored at regular intervals over the course of a 12-day incubation at room temperature. Error bars, SEM, n = 2–3.
When the TPM-Vs were used, the LHI-RC fully retained protein integrity over the course of the initial 10-day incubation at 25 °C. Increasing the incubation temperature to 35 °C resulted in accelerated degradation of the LHI-RC solubilized in the individual detergents. At the end of the test period (day 20), the DDM-solubilized LHI-RC had little intact structure. In contrast, The TPMs showed the enhanced ability to maintain complex integrity, with a better performance observed for the TPM-Vs than TPM-Ls. The TPM-Ls and TPM-Vs gave ~40% and ~70% retention at day 20, respectively. There was no appreciable difference within each set (TPM-L3/L4 or TPM-V3/V4). The enhanced efficacy of the TPMs could be further verified by the change in the colour of the complex over time. The DDM-solubilized LHI-RC was colourless after the 20-day incubation whereas TPM-V4 almost fully retained a pink colour under the same conditions. TPM-L4 gave an orangish-pink colour, indicative of substantial complex degradation (Figure S9). Overall, the TPM-Vs/Ls were more effective than DDM at maintaining the integrity of the LHI-RC complex.
We next investigated these agents with the leucine transporter (LeuT) from bacteria Aquifex aeolicus.[40] After extraction from the membranes using DDM, the transporter was isolated in 0.05% same detergent, which was used for sample dilution in the next step. Final concentrations of the individual TPMs and DDM were CMCs+0.04 wt%. LeuT stability was assessed by monitoring the ability of the transporter to bind the radiolabelled substrate ([3H]-leucine (Leu)) via scintillation proximity assay (SPA).[41] The substrate binding ability of the transporter was measured at regular intervals during a 12-day incubation at room temperature. Upon detergent dilution, all the new agents yielded initial transporter activity a little lower than DDM, but this initial activity was well maintained in the presence of the individual TPMs during the first 9-day incubation, particularly for the TPM-Vs (TPM-V3 and TPM-V4) (Figure 4b). However, all transporters solubilized in DDM or the individual TPMs gradually lost activity during the last three-day of incubation (day 9 to 12). Overall, most of the TPMs were more effective than DDM at preserving the substrate binding ability of the transporter over time.
Encouraged by the results with the LHI-RC complex and LeuT, we were further evaluated the TPMs against a G-protein-coupled receptor (GPCR),[42] the human β2 adrenergic receptor (β2AR). For this experiment, the receptor was extracted from the membranes using DDM and purified in 0.1% of the same detergent. Detergent exchange from DDM to each TPM was carried out by diluting the DDM-purified receptor into each detergent-containing buffer solution. At final detergent concentrations of CMCs+0.2 wt%, protein stability was assessed by measuring the ability of the receptor to bind the radio-active antagonist ([3H]-dihydroalprenolol (DHA)).[43] A preliminary result was obtained by measuring receptor activity after a 30-min detergent exchange. As can be seen in Figure 5a, β2AR solubilized in the TPM-Vs (TPM-V3/V4) gave little ligand binding activity compared to the receptor solubilized in DDM whereas the TPM-Ls (TPM-L3/L4) were at least comparable to DDM in this regard. Based on this result, we selected the linear TPMs (TPM-LS and TPM-L4) for further evaluation with regards to long-term receptor stability. The receptor solubilized in each linear agent was incubated for three days at room temperature and the ability to bind the ligand was measured at regular intervals over the incubation (Figure 5b). The DDM-solubilized receptor rapidly lost activity over time. In contrast, the receptor was markedly more stable in TPM-L3/L4. Use of TPM-L3 led to ~70% retention in receptor activity at the end of incubation (t = 3 day). TPM-L3 was further evaluated for its potential utility in cryo-electron microscopy (CryoEM)-based structural analysis of membrane protein complex.[44] The β2AR-GS complex in DDM micelle prepared from agonist-bound β2AR and Gs protein was subjected to detergent exchange from DDM to TPM-L3. The β2AR-GS complex isolated in TPM-L3 micelles produced mostly monodisperse particles, with little aggregation observed by negative stain EM (Figure 5c),[44] in contrast to a substantial particle aggregation previously observed for the DDM-purified complex.[45] Moreover, the negative stained β2AR-GS complex in TPM-L3 was suitable for the generation of 2D class averages showing well-defined densities for the individual subunits of the complex (Figure 5d). This result indicates that TPM-L3 could be a promising agent for visualizing membrane protein complexes by EM.
Figure 5.
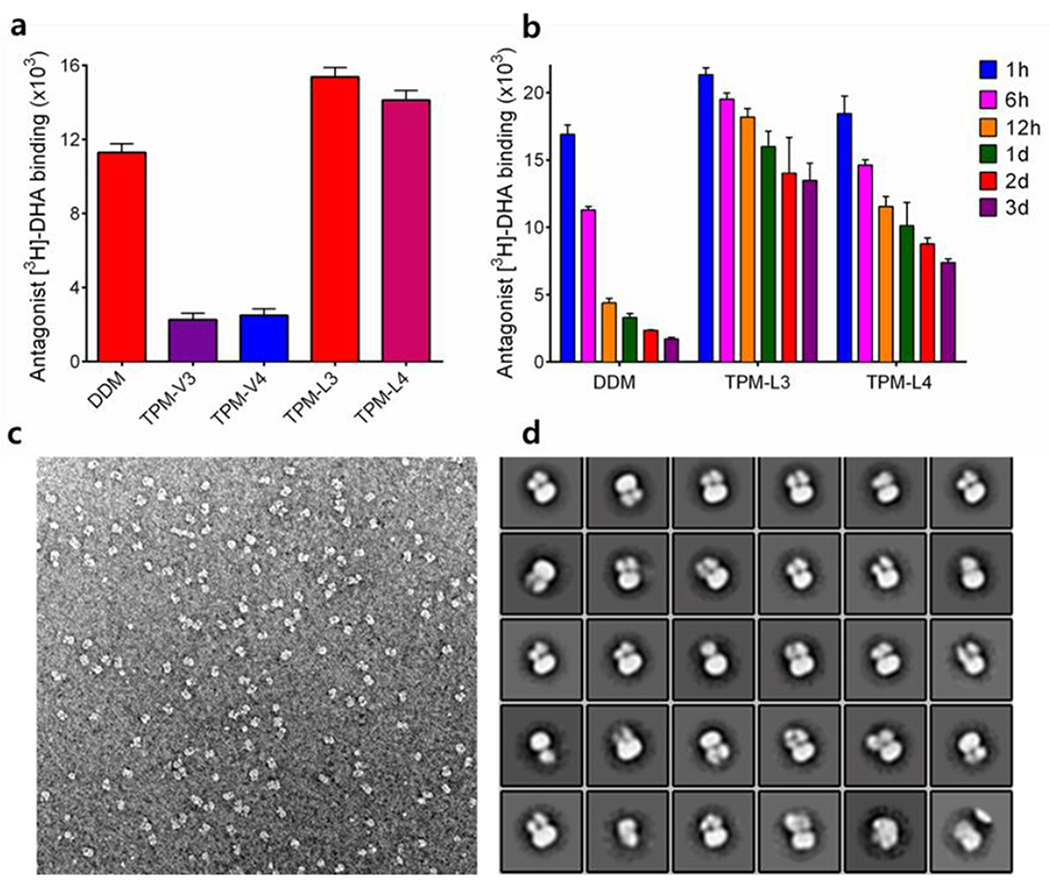
(a) initial and (b) long-term β2AR stability in indicated detergents, (c) A representative EM raw image and (d) 2D classification of β2AR-GS complex purified in TPM-L3. For the stability assay, DDM-purified β2AR was subjected to detergent exchange by diluting the samples into the buffer solutions containing individual detergents. The final detergent concentrations were CMCs+0.2 wt%. Protein stability was assessed by measuring the receptor ability to bind the radio-labelled antagonist ([3H]-dihydroalprenolol (DHA)) during a 3-day incubation at room temperature. Error bars, SEM, n = 3. For EM study, the complex solubilized in TPM-L3 was stained using 0.75% uranyl formate.
To investigate the TPM efficiency for protein extraction from the membranes, we turned to the melibiose permease of Salmonella typhimurium (MelBst).[46] Escherichia coli membranes expressing MelBst at 10 mg/mL were treated with DDM or individual TPMs and incubated for 90 min at 25 °C. As can be seen in Figure S10, all TPMs were less efficient at extracting MelBst from the membranes. Of the TPMs, TPM-L4 was most efficient, giving ~80% soluble MelBst. A similar result was obtained with an elevated incubation temperature of 45 °C. When we continued to increase incubation temperature to 55 °C, most of the TPMs (TPM-L4 and TPM-V3/V4) yielded amounts of soluble MelBst at least comparable to DDM. Combined together, the TPM-L/Vs were relatively poor at efficiently extracting MelBst from the membranes but appeared to be a little better than DDM at maintaining the transporter in a soluble state.
Here we describe the design and synthesis of a novel class of maltoside amphiphiles (TPMs) with a short alkyl chain-attached terphenyl aromatic scaffold as the lipophilic group. The feature of the TPMs that is distinct from previously developed detergents, is the presence of the rigid aromatic group with an ability to form intermolecular aromatic-aromatic interactions. Rigid aromatic groups have been extensively studied for their mesophase behaviours,[47,48] and monomolecular film formation at the air/water interface,[49] but haven’t yet been incorporated into detergent structures for membrane protein study. Due to the presence of the rigid aromatic group at the central part of the molecules, the hydrophobic groups of the TPMs have limitations in their conformational flexibility. Thus, these agents might be poor at adopting a conformation suitable for effective interactions with membrane proteins. This contrasts with a typical conventional detergent that contains a long and flexible alkyl chain and thus can readily adopt an optimal chain conformation for protein stability. As a result, for membrane protein stability, the new agents with rather rigid conformation should possess the hydrophobic dimensions more rigorously matching those of the protein hydrophobic surfaces than conventional detergents. The TPMs also have synthetic modularity utilized for structural modification. Instead of propyl (C3) and butyl (C4) chains, for example, versatile alkyl/aromatic groups can be attached to the central aromatic skeleton as pendants using boronic acid derivatives with the alkyl/aromatic pendant. This structural variation is important as it allows us to optimize detergent properties toward protein stability. Furthermore, from a synthetic point of view, these agents are more accessible than most other novel amphiphiles, an important feature for a widespread use in membrane protein study.
The TPMs formed self-assemblies with intermolecular aromatic-aromatic interaction, a feature likely to be associated with their enhanced effects on protein stability observed here. The new agents were better than DDM, a gold standard conventional detergent, at stabilizing the multiple membrane proteins tested here. Despite the presence of the very similar chemical groups, the two amphiphile sets (TPM-Vs and TPM-Ls) were very different in detergent self-assembly characteristics and protein stabilization efficacy. The only molecular difference between the two groups of detergents is the geometry of their hydrophobic groups: linear (TPM-Ls) vs bent (TPM-Vs). The TPM-Ls tended to increase their micelle sizes with increasing detergent concentration while the micelle size formed by the TPM-Vs was invariant under the same conditions. The two sets of detergents gave a large variation in detergent efficacy for protein stabilization in most cases, likely due to the difference in self-assembly. The TPM-Vs were superior to the TPM-Ls in stabilizing LHI-RC and LeuT, while an opposite trend was observed for β2AR stability. At this point, we don’t know a precise reason why some proteins (e.g., LHI-RC, LeuT and β2AR) have a preference for one geometrical arrangement of the detergent. However, it is notable that the curved TPM-Vs were superior to the linear TPM-Ls for LHI-RC and LeuT stability. Since it has been reported that facial amphiphiles such as the TPM-Ls can effectively interact with cylindrical membrane protein surfaces, we expected that this linear set with full faciality would be better than the TPM-Vs with limited faciality. This unexpected result in the current study suggests that detergent faciality may not be the optimal detergent property for membrane protein study and TPM-Vs could interact with membrane proteins in a distinct and effectivey way.
It would be difficult to precisely know how these TPM-Ls/Vs arrange around membrane protein surfaces as detergent-protein interactions remain elusive. However, plausible binding modes of these detergents with membrane proteins can be conceived by noting the respective structural features of the TPM-Ls and TPM-Vs. Due to the facial property, the TPM-Ls are likely to associate with protein surfaces like other facial amphiphiles where the axis of detergent hydrophobic group aligns parallel with the long axis of a cylindrical membrane protein with a range of hydrophobic widths from 28 to 32 υ (facial binding, Figure 6).[31] However, this facial binding mode would be suboptimal for the TPM-Vs because it produces large empty spaces at the interfaces between protein and detergent micelles, leading to a decrease in the detergent-protein interactions. Consequently, the TPM-Vs likely prefer to interact with protein surfaces in another way. Here we suggest a circular mode where detergent hydrophobic groups surround the cylindrical hydrophobic surfaces of membrane proteins in a circular arrangement (circular binding, Figure 6). As both proteins and the TPM-Vs have curved hydrophobic surfaces, these bent TPMs could fit into the curved protein surfaces by adopting this interaction mode. So far there is no report of a detergent utilizing circular binding with membrane proteins for protein stability. Conclusively, we propose that the detergent binding mode could vary depending on the geometry of detergent hydrophobic groups: conventional detergents with a linear alkyl chain (prolate),[50] facial amphiphiles with linear hydrophobic surface (facial), a detergent with bent hydrophobic surface (e.g., TPM-Vs) (circular). Although more evidences are necessary to further support this proposition, the current study can be the first showcase of introduction for a circular binding mode between a detergent and membrane proteins.
Figure 6.
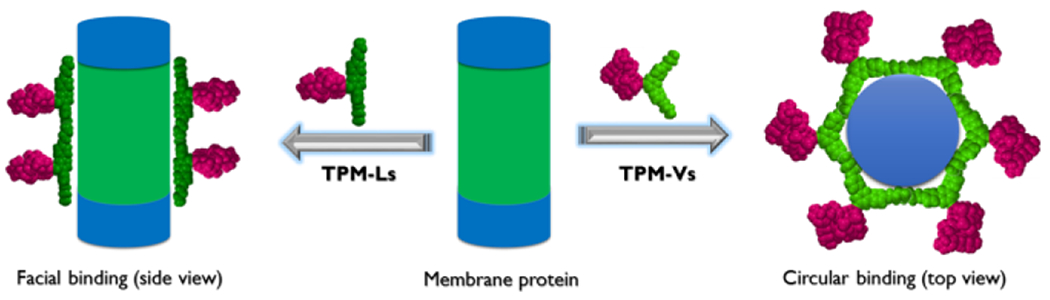
Plausible binding modes of TPM-Ls (left) and TPM-Vs (right) with a membrane protein. Due to the full facial nature, the TPM-Ls would be facially associated with a membrane protein (facial binding) while the TPM-Vs with a curved hydrophobic group likely surround protein surface in a circular way (circular binding). These two binding modes give effective interactions between the TPM-Ls/Vs and membrane protein surface, essential for protein stability.
Conclusions
Here we designed and prepared a novel class of terphenyl-based amphiphiles (TPMs) for membrane protein study. These aromatic group-cored amphiphiles tended to form small and stable self-assemblies (i.e., micelles) with different architecture depending on the geometry of their hydrophobic groups. The linear TPM-Ls likely formed nanocylinder-like assemblies while the bent TPM-Vs appeared to form a nanocapsule-like assemblies. Such molecular geometry-based effect was also found in detergent evaluation with multiple membrane proteins. The TPM-Vs were overall more effective than the TPM-Ls at stabilizing LHI-RC and LeuT, while the TPM-Ls were superior to the bent analogs for β2AR stability. An unprecedented binding mode of the TPM-Vs with protein surfaces (i.e., circular binding) proposed here could be associated with the favourable behaviours of these agents for LHI-RC and LeuT stability. Notably we found a couple of the TPMs that conferred enhanced stability to all the membrane proteins targeted here, including β2AR-GS complex, compared to DDM. Thus, this study not only introduces new biochemical tools with unique architecture for membrane protein study, but also first provides the geometrical effect of detergent hydrophobic group on protein stability. The design principles and new protein-binding mode introduced here should enrich the future development of novel amphiphiles with diverse structures.
Experimental Section
Experimental Details can be found in the Supporting information, including the synthesis and characterization of the new detergents, and membrane protein stability assay.
Supplementary Material
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) (2016R1A2B2011257 and 2018R1A6A1A03024231 to P.S.C.), and NIH Awards (R01GM122759 and R21NS105863 to L.G.).
Footnotes
Supporting information for this article is given via a link at the end of the document. See DOI: 10.1002/chem.201902468.
References
- [1].Zhu B, Li J and Xu D, Phys. Chem. Chem. Phys 2011, 13, 10584. [DOI] [PubMed] [Google Scholar]
- [2].Reimhult E, Baumann M, Kaufmann S, Kumar K, Spycher P, Biotechnol. Genet. Eng. Rev 2010, 27, 185. [DOI] [PubMed] [Google Scholar]
- [3].Mehta SK, Sharma S, Mehta N, Cameotra SS, Adv. Exp. Med. Biol 2010, 672, 102. [DOI] [PubMed] [Google Scholar]
- [4].Kwak M, Herrmann A, Chem. Soc. Rev 2011, 40, 5745. [DOI] [PubMed] [Google Scholar]
- [5].Shimizu T, Masuda M, Minamikawa H, Chem. Rev 2005, 105, 1401. [DOI] [PubMed] [Google Scholar]
- [6].Fuhrhop J-H, Koning J, Membranes and Molecular Assemblies: The Synkinetic Approach; Monographs in Supramolecular Chemistry; Royal Society of Chemistry: Cambridge, 1994. [Google Scholar]
- [7].Mezei A, Pérez L, Pinazo A, Comelles F, Infante MR, Pons R, Langmuir 2012, 28, 16761. [DOI] [PubMed] [Google Scholar]
- [8].a) Cui H, Webber MJ, Stupp SI, Biopolymers 2010, 94, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) van Bommel KJC, Friggeri A, Shinkai S, Angew. Chem., Int. Ed 2003, 42, 980; [DOI] [PubMed] [Google Scholar]; c) Luk Y-Y, Abbott NL, Curr. Opin. Colloid Interface Sci 2002, 7, 267; [Google Scholar]; d) Soussan E, Cassel S, Blanzat M, Rico-Lattes, Angew. Chem., Int. Ed 2009, 48, 274; [DOI] [PubMed] [Google Scholar]; e) Berti D, Curr. Opin. Colloid Interface Sci 2006, 11, 74. [Google Scholar]
- [9].a) Prive GG, Methods 2007, 41, 388; [DOI] [PubMed] [Google Scholar]; b) Carpenter EP, Beis K, Cameron AD, Iwata S, Curr. Opin. Struct. Biol 2008, 18, 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Loll JP, J. Struct. Biol 2003, 142, 144; [DOI] [PubMed] [Google Scholar]; b) White SH. Protein Sci 2004, 13, 1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nath A, Atkins WM, Sligar SG, Biochemistry 2007, 46, 2059. [DOI] [PubMed] [Google Scholar]
- [12].Swainsbury DJK, Scheidelaar S, van Grondelle R, Killian JA, Jones MR, Angew. Chem. Int. Ed 2014, 53, 11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Popot JL, Althoff T, Bagnard D, Ban’eres JL, Bazzacco P, Billon-Denis E, Catoire LJ, Champeil P, Charvolin D, Cocco MJ, Cremel G, Dahmane T, de la Maza LM, Ebel C, Gabel F, Giusti F, Gohon Y, Goormaghtigh E, Guittet E, Kleinschmidt JH, Kuhlbrandt W, Le Bon C, Martinez KL, Picard M, Pucci B, Sachs JN, Tribet C, van Heijenoort C, Wien F, Zito F, Zoonens M, Annu. Rev. Biophys 2011, 40, 379. [DOI] [PubMed] [Google Scholar]
- [14].McGregor C-L, Chen L, Pomroy NC, Hwang P, Go S, Chakrabartty A, Prive GG, Nat. Biotechnol 2003, 21, 171. [DOI] [PubMed] [Google Scholar]
- [15].Tao H, Lee SC, Moeller A, Roy RS, Siu FY, Zimmermann J, Stevens RC, Potter CS, Carragher B, Zhang Q, Nat. Methods 2013, 10, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Chae PS, Kruse AC, Gotfryd K, Rana RR, Cho KH, Rasmussen SGF, Bae HE, Chandra R, Gether U, Guan L, Kobilka BK, Loland CJ, Byrne B, Gellman SH, Chem.-Eur. J, 2013, 19, 15645; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cho KH, Hariharan P, Mortensen JS, Du Y, Nielsen AK, Byrne B, Kobilka BK, Loland CJ, Guan L, Chae PS, ChemBioChem 2016, 17, 2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chae PS, Rana RR, Gotfryd K, Rasmussen SGF, Kruse AC, Cho KH, Capaldi S, Carlsson E, Kobilka BK, Loland CJ, Gether U, Banerjee S, Byrne B, Lee JK, Gellman SH, Chem. Commun, 2013, 49, 2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot J-L, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka BK, Gellman SH, Nat. Methods 2010, 7, 1003; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cho KH, Byrne B, Chae PS, ChemBioChem 2013, 14, 452; [DOI] [PubMed] [Google Scholar]; c) Cho KH, Husri M, Amin A, Gotfryd K, Lee HJ, Go J, Kim JW, Loland CJ, Guan L, Byrne B, Chae PS, Analyst 2015, 140, 3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Howell SC, Mittal R, Huang L, Travis B, Breyer RM, Sanders CR, Biochemistry 2010, 49, 9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Kruse AC, Manglik A, Cho KH, Nurva S, Gether U, Guan L, Loland CJ, Byrne B, Kobilka BK, Gellman SH, Chem.-Eur. J, 2012, 18, 9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Abla M, Unger S, Keller S, Bonnete F, Ebel C, Pucci B, Breyton C, Durand G, Colloid Interface Sci J. 2015, 445, 127. [DOI] [PubMed] [Google Scholar]
- [22].a) Rosenbaum DM, Zhang C, Lyons J, Holl R, Aragao D, Arlow DH, Rasmussen SGF, Choi H-J, DeVree BT, Sunahara RK, Chae PS, Gellman SH, Dror RO, Shaw DE, Weis WI, Caffrey M, Gmeiner P, Kobilka BK, Nature 2011, 469, 236; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T Nature 2012, 482, 547; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic-Jeremic J, Shah P, Shiloach J, Tate CG, Grisshammer R, Nature 2012, 490, 508; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK, Nature 2013, 504, 101; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Miller PS, Aricescu AR, Nature 2014, 512, 270; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Karakas E, Furukawa H, Science 2014, 344, 992; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Kane Dickson V, Pedi L, Long SB, Nature 2014, 516, 213; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Shukla AK, Westfield GH, Xiao K, Reis RI, Huang L-Y, Tripathi-Shukla, Qian J, Li S, Blanc A, Oleskie AN, et al. Nature 2014, 512, 218; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Taniguchi R, Kato HE, Font J, Deshpande CN, Wada M, Ito K, Ishitani R, Jormakka M, Nureki O, Nat Commun 2015, 6, 8545; [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Dong YY, et al. , Science 2015, 347, 1256; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Paulsen CE, Jean-Paul A, Gao Y, Cheng Y, Julius D, Nature 2015, 520, 511; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Schmidt HR, Zheng S, Gurpinar E, Koehl A, Manglik A, Kruse AC, Nature 2016, 532, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hussain H, Du Y, Scull NJ, Mortensen JS, Tarrasch J, Bae ΗE, Loland CJ, Byrne B, Kobilka BK, Chae PS, Chem.-Eur. J, 2016, 22, 7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Cho KH, Du Y, Scull NJ, Hariharan P, Gotfryd K, Loland CJ, Guan L, Byrne B, Kobilka BK, Chae PS, Chem.-Eur. J, 2015, 21, 10008; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cho KH, Ribeiro O, Du Y, Tikhonova E, Mortensen JS, Markham K, HarihSaran P, Loland CJ, Guan L, Kobilka BK, Byrne B, Chae PS, Chem.-Eur. J, 2016, 22, 18833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sadat A, Mortensen JS, Capaldi S, Tikhonova E, Hariharan P, Ribeiro O, Loland CJ, Guan L, Byrne B, Chae PS, Chem. Sci, 2016, 7, 1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ehsan M, Du Y, Scull NJ, Tikhonova E, Tarrasch J, Mortensen JS, Loland CJ, Skiniotis G, Guan L, Byrne B, Kobilka BK, Chae PS, J. Am. Chem. Soc, 2016, 138, 3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Das M, Du Y, Ribeiro O, Hariharan P, Mortensen JS, Patra D, Skiniotis G, Loland CJ, Guan L, Kobilka BK, Byrne B, Chae PS, J. Am. Chem. Soc, 2017, 139, 3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sadat A, Du Y, Santillan C, Mortensen JS, Molist I, Seven AB, Hariharan P, Skiniotis G, Loland CJ, Kobilka BK, Guan L, Byrne B, Chae PS, Chem. Sci, 2017, 8, 8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Israelachvili JN, Intermolecular and surface forces, Academic Press, London, 2nd Ed., 1992. [Google Scholar]
- [30].Das M, Du Y, Mortensen JS, Bae ΗE, Byrne B, Loland CJ, Kobilka BK, Chae PS, Chem.-Eur. J, 2018, 24, 9860. [DOI] [PubMed] [Google Scholar]
- [31].a) Lee SC, Bennett BC, Hong WX, Fu Y, Baker KA, Marcoux J, Robinson CV, Ward AB, Halpert JR, Stevens RC, Stout CD, YeagSer MJ, Zhang Q, Proc. Natl. Acad. Sci. U. S. A, 2013, 110, E1203; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chae PS, Gotfryd K, Pacyna J, MSiercke LJW, Rasmussen SGF, Robbins RA, Rana RR, LSoland CJ, Kobilka BK, Stroud R, Byrne B, Gether U, Gellman SH, J. Am. Chem. Soc, 2010, 132, 16750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chattopadhyay A, London E, Anal. Biochem 1984, 139, 408. [DOI] [PubMed] [Google Scholar]
- [33].Plum MA, Steffen W, Fytas G, Knoll W and Menges B, Opt. Express 2009, 17, 10364. [DOI] [PubMed] [Google Scholar]
- [34].a) Funasaki N, Ishikawa S, Neya S, J. Phys. Chem. B 2004, 108, 9593; [Google Scholar]; b) Nordstierna L, Furo I, Stilbs P, J. Am. Chem. Soc, 2006, 128, 6704; [DOI] [PubMed] [Google Scholar]; c) Cui X, Mao S, Liu M, Yuan H, Du Y, Langmuir 2008, 24, 10771; [DOI] [PubMed] [Google Scholar]; d) Cui XH, Jiang Y, Yang CS, Lu XY, Chen H, Mao SZ, Liu ML, Yuan ΗZ, Luo PY, DSu YR, J. Phys. Chem. B 2010, 114, 7808. [DOI] [PubMed] [Google Scholar]
- [35].a) Schnell I, Spiess HW, J. Magn. Reson 2001, 151, 153; [DOI] [PubMed] [Google Scholar]; b) Mafra L, Santos MS, Siegel R, Alves I, Almeida Paz FA, Dudenko D, Spiess HW, J. Am. Chem. Soc, 2011, 134, 71. [DOI] [PubMed] [Google Scholar]
- [36].Wu S, Shi F, Zhang Q, Bubeck C, Macromolecules 2009, 42, 4110. [Google Scholar]
- [37].a) Okazawa Y, Kondo K, Akita M, Yoshizawa M, J. Am. Chem. Soc 2015, 137, 98; [DOI] [PubMed] [Google Scholar]; b) Okazawa Y Kondo K, Akita M, Yoshizawa M, Chem. Sci, 2015, 6, 5059; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huang Z, Lee H, Lee E, Kang S-K, Nam J-M, Lee M, Nat. Comm 2011, 2: 459. [DOI] [PubMed] [Google Scholar]
- [38].Laible PD, Kirmaier C, Udawatte SMC, Hofman SJ, Holten D, Hanson DK, Biochemistry 2003, 42, 1718. [DOI] [PubMed] [Google Scholar]
- [39].Cho KH, Husri M, Amin A, Gotfryd K, Lee HJ, Go J, Kim JW, Loland CJ, Guan L, Byrne B, Chae PS, Analyst 2015, 140, 3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Decked G, Warren PV, Gaasterland T, Young WG, Lenox AL, Graham DE, Overbeek R, Snead MA, Keller M, Aujay M, Huber R, Feldman RA, Shod JM, Olsen GJ, Swanson RV, Nature 1998, 392, 353. [DOI] [PubMed] [Google Scholar]
- [41].(a) Had ΗE, Greenwald EB, Mol. Immunol, 1979, 16, 265; [DOI] [PubMed] [Google Scholar]; b) Quick M, Javitch JA, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Bobilka BK, Science 2007, 318, 1266. [DOI] [PubMed] [Google Scholar]
- [43].a) Yao X, Parnot C, Deupi X, Ratnala VRP, Swaminath G, Farrens D, Kobilka B, Nat. Chem. Biol 2006, 2, 417; [DOI] [PubMed] [Google Scholar]; b) Swaminath G, Steenhuis J, Kobilka B, Lee TW, Mol. Pharmacol 2002, 61, 65. [DOI] [PubMed] [Google Scholar]
- [44].Peisley A, Skiniotis G, Filizola M (ed.), Methods in Molecular Biology 2015, 1335, 29. [DOI] [PubMed] [Google Scholar]
- [45].a) Rasmussen SGF, Choi HJ, Fung JJ, ParSdon E, Casarosa P, Chae PS, DeVrSee BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaeri J, Weis WI, Kobilka BK, Nature 2011, 469, 175; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Murray CW, Blundell TL, Curr. Opin. Struct. Biol 2010, 20, 497. [DOI] [PubMed] [Google Scholar]
- [46].a) Guan L, Nurva S, Ankeshwarapu SP, J. Biol. Chem 2011, 286, 6367; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ethayathulla AS, Yousef MS, Amin A, Leblanc G, Kaback HR, Guan L, Nat. Commun 2014, 5, 3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen B, Baumeister U, Pelzl G, Das MK, Zeng X, Ungar G, Tschierske C, J. Am. Chem. Soc, 2005, 127, 16578. [DOI] [PubMed] [Google Scholar]
- [48].a) Tschierske C, J. Mater. Chem 2001, 11, 2647; [Google Scholar]; b) Tschierske C, Chem. Soc. Rev 2007, 36, 1930; [DOI] [PubMed] [Google Scholar]; c) Chen B, Zeng XB, Baumeister U, Diele S, Ungar G, Tschierske C, Angew. Chem., Int. Ed 2004, 43, 4621. [DOI] [PubMed] [Google Scholar]
- [49].a) Schröter JA, Plehneri R, Tschierske C, Katholy S, Janietz D, Penacorada F, Brehmer L, Langmuir 1997, 13, 796; [Google Scholar]; b) Plehneri R, Schroter JA, Tschierske C, Langmuir 1998, 14, 5245; [Google Scholar]; c) Plehneri R, Schroter JA, Tschierske C, Langmuir 1999, 15, 3773. [Google Scholar]
- [50].le Maire M, Champeil P, Möller JV, Biochim. Biophysica. Acta 2000, 1508, 86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.