

Abstract
This chapter reviews the history of glycogen-related research and discusses in detail the structure, regulation, chemical properties and subcellular distribution of glycogen and its associated proteins, with particular focus on these aspects in brain tissue.
2.1. Introduction
In the 1840s, the French physiologist Claude Bernard made an astonishing observation: that sugar could be synthesized de novo by the liver of a dog that was exclusively fed meat (Bernard 1850). In 1857, he described the isolation of this sugar-forming substance, which he aptly called la matière glycogène (Bernard 1857). Soon afterward, glycogen in muscle was reported, shown to diminish with exercise, and discovered to “ferment” into lactic acid. Over the century that followed, the structure and regulation of glycogen were extensively characterized in the context of liver and muscle, leading to multiple Nobel prizes throughout the 20th century. Although the mechanisms governing glycogen metabolism have been deeply studied, the work is not yet complete. Meticulous investigations of Nobel laureates Otto Meyerhof, Carl and Gerty Cori, Luis Leloir, Edwin Krebs and Edmond Fischer, and countless others have paved the way for an even more profound understanding of glycogen metabolism, even in organs where glycogen constitutes a smaller fraction of total weight, such as the brain. Despite glycogen metabolism being a mainstay in textbooks, new aspects of glycogen structure, metabolism and tissue-specific roles are still being defined, aided by modern technologies and the mechanistic investigations of glycogen storage diseases.
This chapter discusses the historical progression of glycogen research and its current condition with particular emphasis on how it has benefitted the overarching field of biochemistry, why the study of brain glycogen has lagged, and what gaps remain in our understanding of glycogen metabolism. There are aspects of glycogen structure and metabolism that are applicable to all types of glycogen, but some aspects are specific to organisms, tissues, and/or nutritional state, and insights can be gleaned through study of the other types of carbohydrates, particularly plant starch. Given the similarities and connections, it is useful to review the literature on glycogen (and starch) to understand what can be extrapolated to brain glycogen. This chapter provides a detailed overview of our current understanding of glycogen structure, architecture, associated proteins, tissue-specific properties and subcellular distribution. Later chapters of this book are devoted to the structure and regulation of glycogen synthase and phosphorylase (Chapters 3 and 4), so they will not be discussed in depth here. Glycogen storage diseases inform our studies of glycogen in ways that would not otherwise be possible; insights into glycogen metabolism gained from these diseases are discussed in this chapter. The entire disease class has been recently reviewed elsewhere (Adeva-Andany et al. 2016).
2.2. Discovery of glycogen and its associated proteins
2.2.1. Claude Bernard and early reports of brain glycogen
Glycogen constitutes up to 8% of liver volume and up to 2% of muscle volume, and its content is higher in these tissues than in any other tissue of the adult mammal (Brown 2004). It is not surprising that the fundamental study of its structure and metabolism began in liver and muscle. Claude Bernard, through a series of fundamental experiments, discovered the primary role of the liver in supplying glucose to other organs through the bloodstream (reviewed by Young 1937, 1957; Grmek 1968). In 1848, Bernard noticed that after death, a large amount of glucose was released from the hepatic veins of a dog, even when the dog had not been fed any carbohydrates (Grmek 1968). He concluded that the liver could produce glucose, and by 1857, reported that he could extract glycogen (a term that literally means “sugar-former”) by plunging the tissue in boiling water shortly after death and precipitating the material with alcohol (Bernard 1857) (Fig. 2.1). Through fermentation with yeast and basic chemical tests, he confirmed that its main constituent was glucose (Young 1957). He also noticed that the glycogen yielded a red wine color in the presence of iodine, which facilitated its histological observation in other tissues (Foster 1899; Larner 1967). Very shortly afterward, Sanson reported the isolation of “a glycogen-like substance analogous to dextrin” from the spleen, muscle and kidneys of a horse (translated, Sanson 1857) (Fig. 2.1). By 1875, Bernard and others reported glycogen in mammalian muscle, embryos, and trace amounts in other tissues, but it was difficult to know for certain that those trace amounts were indeed glycogen (Carpenter et al. 1876).
Figure 2.1. Timeline of landmark discoveries in the history of glycogen-related research.
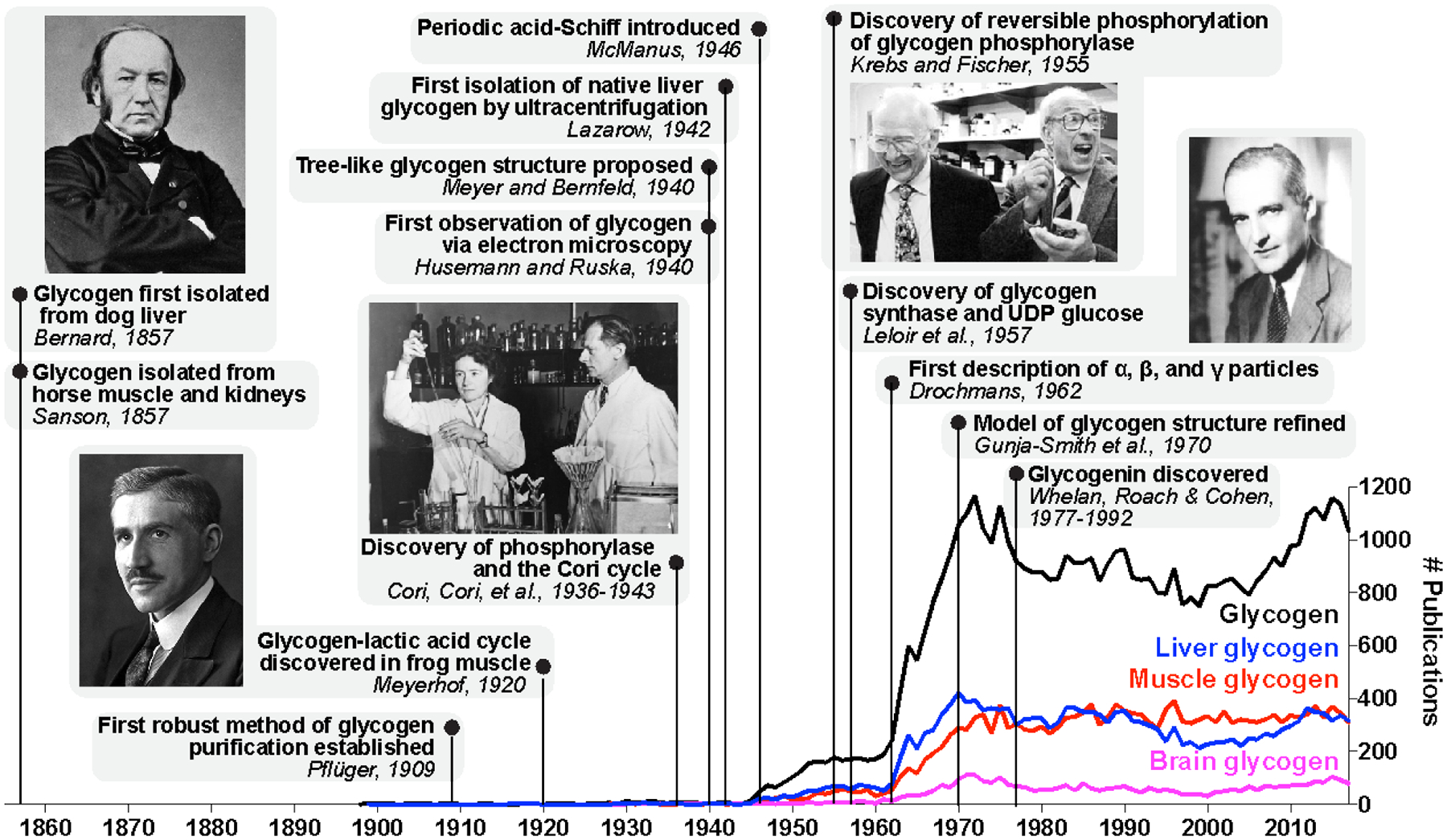
Notable discoveries and papers are shown. Claude Bernard, the physiologist who discovered glycogen and its function in liver, and Nobel laureates are pictured. PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) searches were performed using the keywords shown (glycogen, liver glycogen, muscle glycogen, or brain glycogen), and the number of publications per year for each set of keywords are shown. Key terms related to glycogen synthase kinase (GSK, GSK3, GSK3 beta) were excluded from search results since much of the GSK-related literature is not directly relevant to glycogen metabolism.
Soon after Bernard’s discovery of glycogen, it was noted that glycogen in muscle diminished upon contraction, and Bernard reported that “muscle glycogen always undergoes a lactic acid fermentation, and this is the only change that muscle glycogen ever undergoes, either in the living animal or after death” (Carpenter et al. 1876; Young 1957). In the early twentieth century, a landmark paper from Fletcher and Hopkins demonstrated that lactic acid was formed in muscle under anaerobic conditions (Fletcher and Hopkins 1907). Then, a German physician named Otto Meyerhof published a series of experiments demonstrating that the lactic acid produced by frog muscle was derived from glycogen, and that in the presence of oxygen, it could be either oxidized, or reconverted to glycogen (Meyerhof 1920a; Meyerhof 1920b, c) (Fig. 2.1). “Meyerhof’s brilliant analysis of the glycogen-lactic acid cycle and its relation to respiration explained the course of the heat production and, for the first time, established the cyclic character of energy transformations in the living cell” (Nachmanson et al. 1960). For this discovery and his seminal work on glycolysis, Meyerhof and his colleague A.V. Hill were awarded the 1922 Nobel Prize in Physiology or Medicine.
The German physiologist Eduard Pflüger is credited as the first to establish a method for obtaining pure fractions of glycogen (Fig. 2.1), and modifications of his method are still used today (Pflüger 1909; Young 1937; Passonneau et al. 1967). While Pflüger, Bernard and most others were unable to detect glycogen in the nervous system, traces of brain glycogen in the normal and diabetic human brain were reported by Cramer in 1880 and Pavy in 1881 (Gage 1917). Similar findings were published in an 1890 edition of The Lancet: “Dr. Fütterer has examined various organs of a diabetic person, finding glycogen in the medulla oblongata, spinal cord, and kidney in large quantities, and a little in the liver. A careful examination of the cerebral cortex showed that the vessels were full of glycogen. He concluded that extensive disturbances of nutrition were bound to result from this” (The Lancet, 1890).
Although nearly all studies of glycogen in the early 1900s were in the liver and muscle of various organisms, as well as in yeast, a few groups reported glycogen in the central nervous system (CNS) of animals by staining tissues with an iodine solution. Glycogen was observed in the CNS, often in the retina, of the lancelet, lamprey, frog, pigeon, rabbit, and dog (Gage 1917; Holmes and Holmes 1926). Gage asserted its presence in the CNS of lower organisms, in the dorsal root ganglia of the pig, and in the choroid plexus of human embryos (Fig. 2.2). He validated the identity of the “mahogany-red substance” by incubating the sections with saliva, which contained amylases that digested the glycogen so that it no longer stained. “With the higher vertebrates, glycogen in demonstrable amount is not found in the nervous system after the embryonic period, the liver and muscles then assuming the main glycogenic function.” Gage was hopeful that with the proper techniques and material, glycogen would also be found in the adult human CNS (Gage 1917).
Figure 2.2. Glycogen in the CNS of invertebrates and vertebrates.
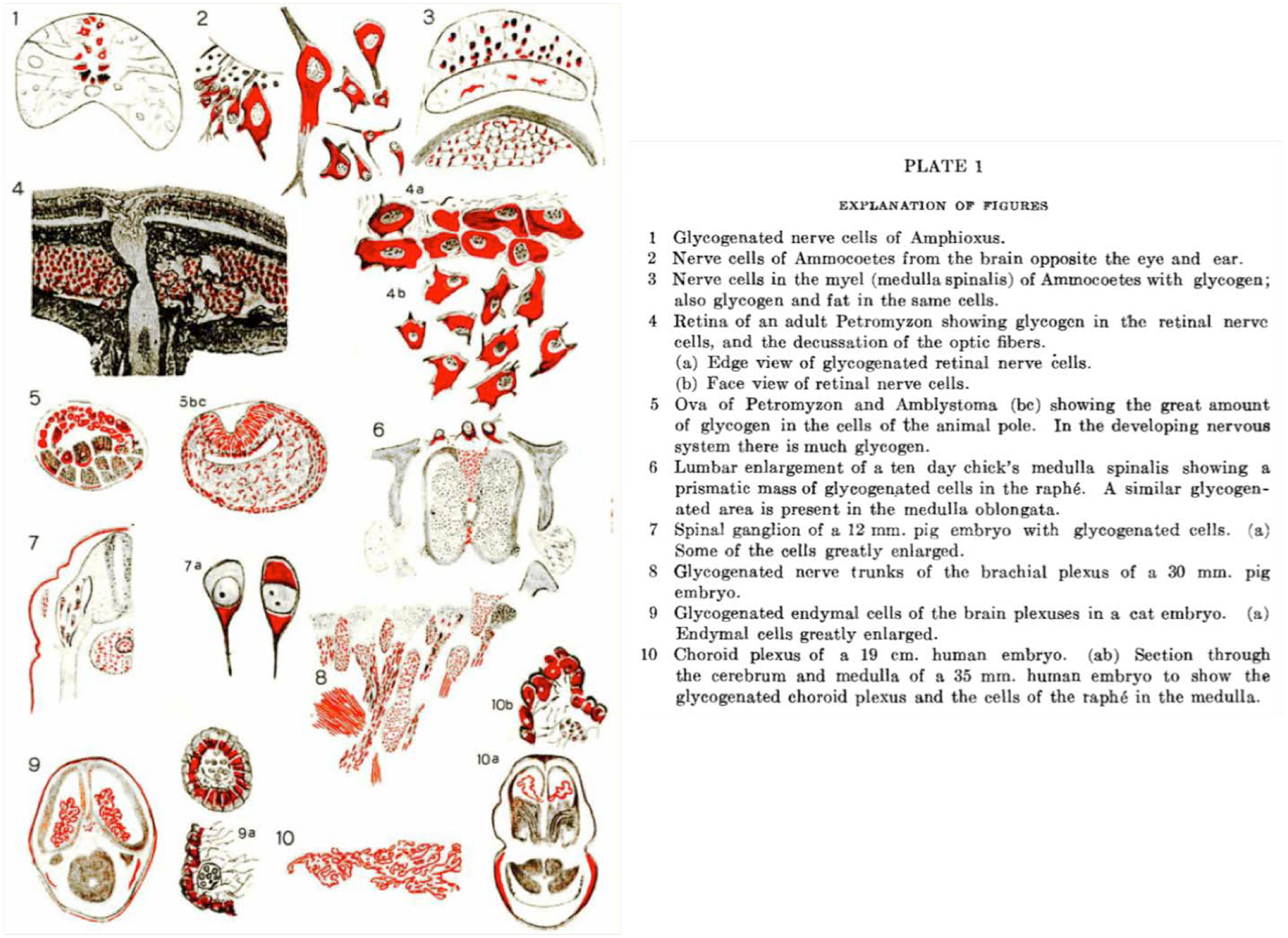
Drawings illustrating glycogen in the CNS of multiple organisms detected via iodine staining (Gage 1917). Copyright © 2005 John Wiley and Sons, Inc. Used with permission.
With the relationship between glycogen and lactic acid demonstrated in muscle by Meyerhof, a few groups began investigating this relationship in brain. However, brain glycogen and lactic acid remained difficult to isolate and detect, and results were variable. Some groups reported a reduction in glycogen with insulin-stimulated convulsions or anesthetics, while others reported a rise in glycogen after convulsions produced by methylguanidine; Holmes and Holmes in reviewing these studies argued that the low values reported for brain glycogen were within the normal range of variation, and that brain glycogen is unlikely to produce lactic acid under normal conditions (Holmes and Holmes 1926). They astutely commented that “it is possible that the low values which we, in common with other workers, find for the glycogen content of the brain, may be due, not to a slow or inadequate breakdown mechanism, but to an extremely rapid one.” This was indeed the case, and methods for capturing brain glycogen improved over the years; however, it was not until the end of the 20th century that the most sophisticated methods for measuring brain glycogen emerged. “It seemed to us, therefore, that it is quite possible that glycogen plays a comparatively unimportant (or at least quite obscure) part in the carbohydrate metabolism of mammalian brain” (Holmes and Holmes 1926). This sentiment prevailed for many years, and it did not become clear until the following century that the role of glycogen in brain was more obscure than it was unimportant. Meanwhile, studies on glycogen enzymology had just begun, which led to a series of fundamental discoveries with major impacts on the wider field of biochemistry.
2.2.2. The Cori years: advances in glycogen enzymology and glycogen structure
Soon after Meyerhof’s Nobel prize was awarded, Carl and Gerty Cori, Czech-born medical doctors who had emigrated to the United States, started working on the effects of insulin and epinephrine on liver glycogen. They reported that the hyperglycemia observed in the blood after epinephrine injections could not be accounted for solely by liver glycogen, and that lactic acid, which would be derived from muscle, could give rise to newly formed liver glycogen (Cori and Cori 1929). They theorized that there was a “cycle of carbohydrates” linking liver glycogen and muscle glycogen, with the intermediates between the two being glucose and lactic acid (Fig. 2.3a). Their subsequent work demonstrated this theory to be true, and it became known as the Cori cycle.
Figure 2.3. The Cori cycle and the tree-like structure of glycogen.
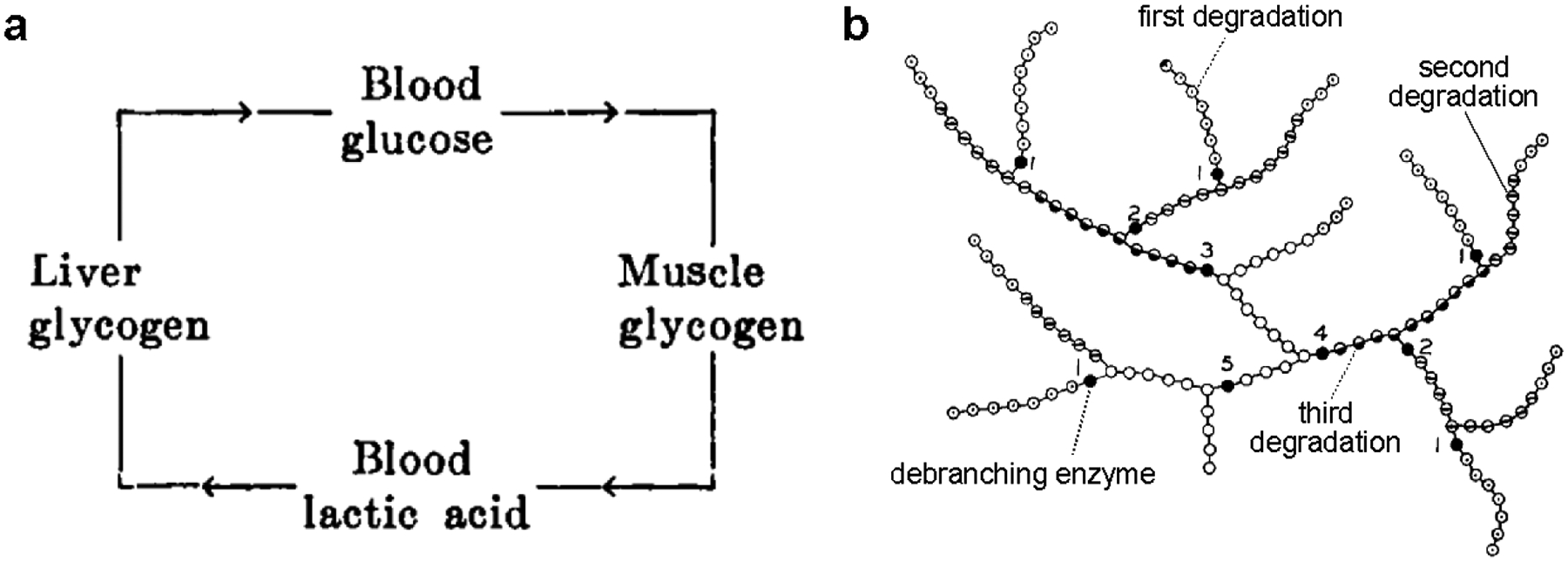
(a) The cycle of carbohydrates between liver and muscle proposed by Cori and Cori (1929) when they observed that ingestion or subcutaneous injection of sodium d-lactate led to glycogen deposition in the liver. Their subsequent work validated and elaborated this model, which became known as the Cori cycle. (b) A segment of the tree-like structure of glycogen based on Meyer’s model and the results of the Cori group. Glucose residues are represented by circles; dotted, bisected, and half-filled circles correspond to glucose residues released by the first, second, and third rounds of degradation with phosphorylase, as indicated. Filled circles represent glucose residues released by α−1,6 glucosidase (i.e. debranching enzyme). Tiers are numbered. Modified from Larner et al. 1952. Copyright © American Society for Biochemistry and Molecular Biology. Used with permission.
The Coris made a series of major discoveries in the 1930s (Fig. 2.1). In 1936 they isolated a novel ester, glucose-1-phosphate, from a mixture of glycogen, inorganic phosphate, and aqueous muscle extract (Cori and Cori 1936). So significant was this discovery that the compound became known as the Cori ester (Larner 1967). In subsequent years, the Coris showed that a phosphorylating enzyme found in muscle, heart, liver, brain, and yeast extracts catalyzed the release of glucosyl moieties from glycogen by esterifying them with inorganic phosphate. The resulting glucose-1-phosphate was converted to glucose-6-phosphate by an enzyme they named phosphoglucomutase (Cori et al. 1937; Cori et al. 1938). In a 1939 article in Science, they reported that the enzyme producing the Cori ester (which they aptly named phosphorylase) could also catalyze the reverse reaction, in order to synthesize polysaccharide (Cori et al. 1939). Almost simultaneously, phosphorolytic enzymes catalyzing polysaccharide synthesis were being described in the extracts of peas and potatoes and in yeast (Kiessling 1939; Hanes 1940). In 1943, Cori, Cori and Green crystallized glycogen phosphorylase and published a series of seminal papers on its properties (Cori et al. 1943; Cori and Cori 1943; Cori and Green 1943; Green and Cori 1943). In reward for their pioneering work, the Coris shared the 1946 Nobel Prize in Physiology or Medicine with Bernardo Houssay, who also made important contributions to the field of carbohydrate metabolism through his study of the role of the pituitary gland in hormonal regulation.
The discovery of phosphorylase in mammals, plants and yeast was a major breakthrough in the overarching field of biochemistry (Larner 1967). Phosphorylase was the first enzyme demonstrated to synthesize polysaccharide, and glycogen was the first macromolecule to be synthesized in vitro (Manners 1963; Whelan 2007). However, while mammals and yeast utilize glycogen as an energy source, plants synthesize starch, a glucose polymer with very different properties. Investigations of plant starch had been underway years before Bernard’s discovery of glycogen and were progressing in parallel with those of glycogen. By the 1940s it had become clear that starch was composed of two distinct polymeric fractions: amylose, which was composed of long linear chains of glucose and gave an intense blue-black color with iodine, and amylopectin, which was a branched macromolecule and gave a purplish to reddish color (Bates et al. 1943). While the Coris were working out glycogen enzymology, others were studying the polymeric nature of both glycogen and starch. Haworth and Percival established a method for identifying the chemical linkages in glucose polymers through methylation of the free hydroxyls of the glucose polymer prior to hydrolysis with acid (Haworth and Percival 1932). They found that since most of the product had unmethylated hydroxyls at the 1- and 4- positions, the majority of the glucose units in glycogen must be linked by α−1,4 glycosidic bonds. A small proportion of glucose was methylated at the 4-hydroxyl position, which would represent what is called the non-reducing end of a glucose chain (i.e. the end lacking an aldehyde group). Based on the proportion of the methylated products, the average linear chain in glycogen was proposed to contain 12 glucose units. Similar work was done on several varieties of plant starch, and the average chain length in starch was estimated to be 24–30 units (Bawn et al. 1940). In 1947 and 1949, the α−1,6-linked disaccharide isomaltose was isolated from hydrolyzed starch and glycogen, providing definitive evidence for the chemical identity of the branch points (Montgomery et al. 1949; Wolfrom et al. 1951).
For many years it was believed that phosphorylase catalyzed both the synthesis and degradation of glycogen and starch, although synthesis by phosphorylase always required a primer, i.e. the addition of a small amount of carbohydrate upon which phosphorylase could act (Larner et al. 1952; Larner 1967). Curiously, phosphorylase from brain, heart and liver extracts produced branched polysaccharides that yielded a similar color to glycogen when stained with iodine, while the enzyme from potato and muscle extracts synthesized a linear, unbranched polysaccharide resembling amylose (Cori and Cori 1943). It was hypothesized that the former preparations contained a contaminating enzyme that was capable of introducing the α−1,6-linked branch points. A branching enzyme had been identified in potato, and in 1953, Larner characterized the branching enzyme from rat liver and muscle extracts. Using isotopic labeling, he showed that potato, liver and muscle branching enzymes transferred a 1,4-linked chain of 6–11 glucose moieties to create the 1,6-linked branch. Thus, branching enzyme is considered a transglucosidase (Larner 1953). His approach was novel in that he utilized radioactive isotopes rather than measuring branching by a shift in iodine color, susceptibility to phosphorylase, or change in end-group. With the combination of phosphorylase and branching enzyme, glycogen could be synthesized in vitro, and for the most part, it resembled the glycogen purified from tissues (Parodi et al. 1969).
Similarly, it was observed that while muscle phosphorylase could completely digest glycogen, recrystallized preparations of muscle phosphorylase and potato phosphorylase degraded glycogen only partially, stopping at the branch points, and leaving what was called a limit dextrin (Cori and Larner 1951). A second enzyme must be present in the crude extracts that could remove the branch points. In 1951, Cori and Larner discovered the contaminating enzyme, showing that it cleaved α−1,6 linkages and released free glucose (Cori and Larner 1951). Walker and Whelan later found that the phosphorylase limit dextrin contained not one, but four glucose units attached to branch points (Walker and Whelan 1960). It was then discovered that the enzyme they had identified, which they called amylo-1,6-glucosidase, also had transglucosidase activity, favoring the transfer of three glucose units from one chain to another (Brown and Illingworth 1962). It is now well established that when phosphorolysis terminates four units away from a branch point, the glycogen debranching enzyme, which possesses both transferase and glucosidase activities, moves three glucose units to another chain and then releases the single branched glucosyl moiety (Huijing 1975).
It appeared at that time that all of the enzymes required for glycogen synthesis and degradation had been identified: glycogen phosphorylase, branching enzyme, and debranching enzyme. Enzymes that were either analogous or distinct to these had also been identified in plants. It became possible to utilize these enzymes to more precisely understand the structure of the glycogen and amylopectin molecules. With such useful tools and insights in common, the fields of starch and glycogen metabolism continued to progress together and overlapped considerably. In 1940 a tree-like structure for glycogen and amylopectin was proposed by Meyer and Bernfeld based on serial degradation with various enzymes (Meyer and Bernfeld 1940) (Fig. 2.1). This model varied from the prior structures proposed by Staudinger and Haworth, which were based solely on data obtained by chemical methods. The Cori group used stepwise enzymatic degradation of various glycogens and amylopectins with phosphorylase and debranching enzyme to test the various models. It was evident that glycogen was composed of “tiers,” and each successive tier could only be accessed by phosphorylase after removal of the branch points by the debranching enzyme; degradation of multiple tiers required alternating treatments of the two enzymes. “The Staudinger and the Haworth models would yield a constant percentage of the total branch points in each tier during successive enzymatic degradation, while the Meyer model would yield a diminishing percentage as one progresses from the outer to the inner tiers, such as is actually indicated by the data in Table II. Only one kind of model could be made to fit this arrangement of branch points; namely, one which represents the polysaccharides as multibranched, tree-like structures” (Larner et al. 1952) (Fig. 2.3b). It was also noted in this paper that the average chain length of liver and muscle glycogen (15 glucose units) were shorter than those of wheat and corn amylopectin (18 and 24 glucose units, respectively). We now know that chain length is one of the major features that distinguish glycogen from amylopectin. Meyer’s model was the accepted model for glycogen and amylopectin until experiments from Whelan’s group in 1970 showed the absence of very long chains and refined the model (Fig. 2.1) (Gunja-Smith et al. 1970; Gunja-Smith et al. 1971). The currently accepted model for glycogen is based on Whelan’s work, but recent data indicates this model requires further refinement (see Section 2.3.1).
2.2.3. Emerging technologies with staying power: Periodic acid-Schiff, the ultracentrifuge, and the electron microscope
When the Coris began their work, the most common procedure for purifying glycogen was based on the 1909 Pflüger method. The protocol is similar to what Bernard used and involves boiling tissue in hot concentrated alkali and then precipitating the glycogen with alcohol (Pflüger 1909). Modifications of this procedure, especially those of Somogyi (Somogyi 1934), were used by the Coris and are still the most commonly utilized protocols for glycogen extraction. Another frequently used method, introduced in 1934 (Willstätter and Rohdewald 1934), is extraction with cold trichloroacetic acid (TCA); however, it has been observed that not all glycogen can be extracted by this method (see Section 2.6.2), and a slightly different molecular weight is observed for the purified glycogen (Lazarow 1942; Calder 1991). In 1936, the first reliable procedure for extracting brain glycogen was established utilizing Somogyi’s method (Kerr 1938). Kerr found that he obtained the highest yields when the brains of dogs were rapidly excised under amytal anesthesia and crushed directly into 60% potassium hydroxide. He also obtained high yields when the brains were frozen in situ in liquid air. Importantly, he showed that the purified brain glycogen “is indistinguishable from liver glycogen prepared by Somogyi’s method. It dissolves readily in water to form a transparent solution, which is opalescent in reflected light. This when treated with iodine solution gives a Burgundy red color identical with that obtained with a solution of liver glycogen.” The similar chemical properties of brain and liver glycogen were important to establish, and made it reasonable to extend the principles of glycogen structure and regulation that were being so thoroughly defined in liver and muscle to brain glycogen.
Quantifications of brain glycogen were now reproducible. Kerr and other groups showed that brain glycogen levels dropped during hypoglycemia in dogs, cats and rabbits (Carter and Stone 1961). Once a robust method for quantifying low levels of brain lactate was also established (Carr 1947), informative experiments on the effects of various stimuli on brain glycogen and lactate could begin. Using a circular saw to rapidly excise the brains of mice, Chance and Yaxley showed that lactate was elevated with both subconvulsive and convulsive levels of various seizure-inducing stimulants, while glycogen was only elevated when a convulsion was reached (Chance and Yaxley 1950). They also carefully defined the rapid decrease of glycogen and lactate levels after death in both normal and convulsed mice, demonstrating that most of the glycogen was lost after five minutes (Fig. 2.4a). Based on their own measurements and those of their contemporaries, they estimated that “a large number of vertebrates normally possess between 10 and 30 mg of glycogen per 100 g of brain tissue” and that glycogen distribution within the brain was heterogeneous. These values are in agreement with current estimates (Brown 2004). A few groups also began studying glycogen metabolism in isolated cerebral tissues and the effects of sleep on brain glycogen (LeBaron 1955; McIlwain and Tresize 1956; Svorad 1958).
Fig. 2.4. Early studies of brain glycogen.

(a) Rapid glycogen loss following extraction from brain of normal and convulsed mice; glycogen was measured using a modification of the method by Kerr (1938) (from Chance and Yaxley 1950). Copyright © Company of Biologists, Ltd. Used with permission. (b) Staining of glycogen (dark stain) in the perfused rabbit brain using the lead-tetra-acetate-Schiff method, a modification of PAS (Shimizu and Kumamoto 1952). The identity of stained regions as glycogen was confirmed by salivary digestion of comparable sections. Left: glycogen is abundant in the granular and molecular layers of the cerebellum, but Purkinje neurons lack glycogen. Upper right: ependymal cells of the hypothalamus lining the third ventricle show intense staining for glycogen, and granules are abundant in the neuropil, but nerve cells lack glycogen in this region. Lower right: some small nerve cells in the lateral hypothalamic nucleus appear to contain glycogen. Scale bars have been approximated based on magnification. Copyright © 2004 John Wiley and Sons, Inc. Used with permission.
As glycogen quantification methods improved, so did microscopic techniques. Glycogen had long been visualized with iodine using the light microscope, but the stain was not sensitive enough to detect the small amounts found in the normal adult brain. Additionally, alcoholic fixation was also critical, as glycogen would dissolve in aqueous solutions (Gage 1917). Some attempts were made to visualize glycogen with Best’s carnitine or the Bauer reaction, but these methods were also criticized for their lack of sensitivity (Kerr 1938; Lillie 1950; Mowry and Bangle 1951). In 1946, McManus introduced the periodic acid-Schiff (PAS) technique as a delicate and convenient way to stain carbohydrates, including glycogen, in tissue sections: the periodic acid reacts with the 1,2 glycol linkage of carbohydrates, producing an aldehyde that can be colored with Schiff reagent (McManus 1946, 1948) (Fig. 2.1). Although PAS stained most polysaccharides, including glycoproteins and mucins, its specificity for glycogen could be tested by incubation with diastase (a general term for amylase) or saliva (which contains amylase) to digest the glycogen and distinguish it from other types of polysaccharides, which were left intact after digestion (Mowry and Bangle 1951). Using PAS and a modification of this stain (lead-tetra-acetate-Schiff), Shimizu and Kumamoto published a series of papers in the 1950s on glycogen deposition in the normal brain and with pathological insults (Shimizu and Kumamoto 1952; Shimizu 1955; Shimizu and Kubo 1957; Shimizu and Hamuro 1958). Glycogen was detected in the area postrema (an area populated primarily by glial cells) in mammalian brains, particularly around blood vessels, and in ependymal cells of the supraoptic crest in rodents; it could also be detected in nerve cells of the hypothalamic nucleus and other regions when the brain was perfused with appropriate fixatives (Shimizu and Kumamoto 1952) (Fig. 2.4b). The PAS technique, usually in combination with diastase (PASD), is now the most widely used histochemical stain for glycogen detection in tissues (Bancroft and Gamble 2008).
Another extremely useful technology in visualizing glycogen structure and distribution was born with the invention of the electron microscope in 1931 by Ernst Ruska (Ruska and Knoll 1931). The electron microscope achieved superior resolution to that of the light microscope. In 1934, Marton developed a histological technique that allowed biological specimens to be visualized by the electron microscope without destruction (Marton 1934). With this technique, the fine structure of subcellular organelles could be visualized. The first report of glycogen using electron microscopy (EM) was from Husemann and Helmut Ruska, Ernst Ruska’s brother, in 1940 (Husemann and Ruska 1940) (Fig. 2.1). The spherical particles, which were derivatized to produce better scattering of the x-ray beam, were about 15–30 nm in diameter. But visualization of glycogen was difficult for a number of reasons. Firstly, polysaccharides contain primarily light atoms (carbon, oxygen and hydrogen) and are intrinsically not very electron dense. Secondly, after the identification of ribonucleoprotein particles by EM, particulate components with a diameter of ~150 Å free in the cytoplasm or associated with the endoplasmic reticulum were typically interpreted as ribonucleoprotein (Revel et al. 1960; Revel 1964). Unknown tissue components were sometimes identified by comparing the thin EM sections with thicker, stained sections viewed under the light microscope, but this too had its limits.
The ultracentrifuge, invented by Theodor Svedburg in 1925, became a very powerful tool in the field of biochemistry and allowed glycogen to be isolated its native form. Lazarow showed in 1942 a species which he called “particulate glycogen” could be isolated from liver via ultracentrifugation without the use of harsh chemicals (Lazarow 1942) (Fig. 2.1). Its sedimentation constant suggested a molecular weight much larger than previous reports for Pflüger-purified glycogen, and the particle, which he showed was made of pure glycogen, could be dispersed by heating, trichloroacetic acid (TCA), or potassium hydroxide treatment. Lazarow concluded that “clearly, then, particulate glycogen is an aggregate of smaller glycogen units.” In 1962, Drochmans, combining these two state-of-the art technologies and utilizing a negative staining technique with phosphotungstate, elegantly described the ultrastructure of glycogen particles purified by differential ultracentrifugation (Drochmans 1962) (Fig. 2.1). He defined three levels of glycogen structure in the rat liver visible by EM: the largest structure was the α particle, which was 60–200 nm in diameter and had a rosette like appearance; the β particle, 20–40 nm in diameter, which associated to form α particles; and the γ particle, which referred to the 3 nm fine filaments comprising the β particle (Fig. 2.5a,b). The α particles could be dispersed into β particles in low pH. It became clear that Drochmans’s α particle was equivalent to Lazarow’s particulate glycogen, and that these particles represent the primary native form of glycogen in the liver. This helped to explain the discrepancies in molecular weights that others had observed for glycogen purified by different methods. In 1968, Wanson and Drochmans also visualized differentially centrifuged glycogen from rabbit skeletal muscle, showing the presence of solely β particles (Wanson and Drochmans 1968) (Fig. 2.5c). Dozens of EM studies on glycogen in various tissues and organisms followed.
Fig. 2.5. Glycogen ultrastructure as demonstrated by electron microscopy.

(a, b) Negative staining of natively purified rat liver glycogen from reveals the presence of α, β and γ particles (Drochmans 1962). Copyright © 1962 Elsevier. Used with permission. (c) Typically, purified muscle glycogen exists only as β particles (Wanson and Drochmans 1968). Scale bars have been approximated based on magnification. Copyright © Rockerfeller University Press. Used with permission. (d,e) Glycogen in the rat cerebral cortex visualized by EM (Maxwell and Kruger 1965). (d) In sections of normal cortex, glycogen β particles (g) can be found in astrocyte processes (AP) and astrocytic end feet applied to the basement membrane (b). An astrocytic bundle of fibrils (f) is labeled. (e) One day after irradiation with ionizing particles, end feet (ef) are enlarged and contain numerous glycogen granules and mitochondria (m). An endothelial cell lining the capillary is also labeled (E). Copyright © Rockerfeller University Press. Used with permission.
The use of EM on sections of the mammalian brain in the 1960s and later years made it clear that the vast majority of brain glycogen was present in the form of β particles in astrocytic processes (Maxwell and Kruger 1965; Cataldo and Broadwell 1986) (Fig. 2.5d). Multiple groups showed striking accumulations of glycogen in reactive astrocytes in response to various traumas (Shimizu and Kubo 1957; Shimizu and Hamuro 1958; Maxwell and Kruger 1965) (Fig. 2.5e). Most groups reported that in the adult mammalian brain, neurons and microglia contained virtually no glycogen except under certain pathological conditions; however, in a few studies of normal tissue, glycogen was found in nerve cells (reviewed by Koizumi 1974). During this time, numerous groups began using PAS and EM to visualize the microscopic characteristics of polyglucosan bodies, abnormal carbohydrate structures found in a variety of tissues. Polyglucosan bodies were identified in both astrocytes and neurons, in the context of glycogen storage diseases, epilepsy, neurodegenerative disorders, and aging (Cavanagh 1999; Duran and Guinovart 2015). Although glycogen still received little attention in the brain compared to other tissues, its significance was becoming increasingly evident. In 1961, A.W. Merrick stated, “A substantial amount of Russian work within the past decade has been directed toward brain glycogen changes during various function and biochemical states of the animal. The opinion is offered by many of these investigators (several of whom are cited in this paper) that glycogen takes an active part in brain metabolism” (Merrick 1961).
2.2.4. Sugar nucleotides, reversible phosphorylation, and a primer for glycogen synthesis
In 1950, the Argentinian chemist Luis Leloir discovered the first sugar nucleotide: uridine-diphosphate (UDP) glucose (Caputto et al. 1950). It soon became apparent that the sugar nucleotides were found throughout nature and were essential for the interconversion of carbohydrates, occupying “a central position in carbohydrate metabolism” (Hassid et al. 1959). In 1957, Leloir made the shocking discovery that the true catalyst of glycogen synthesis was not glycogen phosphorylase, but a glucosyl transferase utilizing UDP-glucose as a glucosyl donor, which became known as glycogen synthase (Leloir and Cardini 1957) (Fig. 2.1). In the words of Larner, “Nature seems to have found a new more powerful glucose donor by making a pyrophosphate derivative of the Cori ester” (Larner 1967). Leloir’s group went on to show that glycogen synthesized in vitro using glycogen synthase in combination with branching enzyme was identical to natively purified glycogen β particles from muscle; in contrast, glycogen synthesized by phosphorylase and branching enzyme was not exactly equivalent to native glycogen: it differed in its stability and response to various types of degradative treatments (Mordoh et al. 1966; Parodi et al. 1967). These results were confirmed and expanded upon by numerous groups, and Leloir was awarded the 1970 Nobel Prize in Chemistry for his discovery of the sugar nucleotides.
It became apparent that both glycogen phosphorylase and synthase are peculiar enzymes. The Coris observed that glycogen phosphorylase existed in two forms, an active a form, and inactive b form, which were differentially activated by adenylic acid (Cori and Cori 1946). The Coris had identified another enzyme capable of converting the a form to the b form, calling this the “prosthetic-group-removing” enzyme (Cori and Green 1943). Kinase activity, the covalent addition of phosphate to one protein by another, was first described in 1954 (Burnett and Kennedy 1954), and in 1955, Fisher and Krebs showed that phosphorylase a could be converted to phosphorylase b in the presence of adenosine triphosphate (ATP) by an enzyme from muscle that became known as phosphorylase kinase (PhK) (Fischer and Krebs 1955). Concurrently, Wosilait and Sutherland also demonstrated the presence of an equivalent converting enzyme in liver (Sutherland and Wosilait 1955). These were the first ever demonstrations of reversible phosphorylation, a momentous discovery that has reverberated throughout the life sciences. Fischer and Krebs published a series of papers on the details of this novel mechanism that earned them the 1970 Nobel Prize in Physiology or Medicine, and Sutherland received the 1971 Nobel Prize for his discovery of cyclic adenosine monophosphate (cAMP) and its role in hormonal regulation (Cohen 2002). A few years later, Larner and others showed that glycogen synthase is also subject to allosteric control, that it exists in two forms, and that the interconversion of these forms requires phosphorylation by another kinase (Friedman and Larner 1963). In fact, unlike phosphorylase, which has one phospho-site and one kinase, synthase is hierarchically phosphorylated at multiple sites by multiple kinases, inextricably linking glycogen metabolism to hormonal regulation, energy status, and a milieu of intracellular signals (see Chapter 3) (Roach 1990). One of the kinases discovered to phosphorylate synthase, glycogen synthase kinase 3 (GSK3), was later found to regulate a much greater array of cellular processes and play an important role in many pathologies including cancer, Alzheimer’s disease, Parkinson’s disease and diabetes (Cohen and Frame 2001; Jope and Johnson 2004). Many of the roles of GSK3 are quite unrelated to glycogen metabolism, leading to an unfortunately misleading keyword in literature searches.
While more pieces in the puzzle of glycogen regulation had fallen into place, other pieces were not even known to be missing. One enzyme must be mentioned at this point that has received insufficient attention despite its discovery in 1963. It was discovered as the deficient enzyme in Pompe disease, one of the glycogen storage diseases (GSDs). GSDs, also known as glycogenoses, are a group of diverse pathologies characterized by glycogen accumulation in various tissues. GSDs were first described in the early 1900s, and over the decades, the enzyme deficiencies causing many of these disorders were identified through biochemical analyses of patient tissues. Typically, patients were lacking one of the basic enzymes involved in glycogen metabolism (reviewed by Huijing 1975). However, in one of the most severe GSDs, Pompe disease, all of the known glycogen-related enzymes had normal activity. Lysosomes had been recently discovered by de Duve with the use of the ultracentrifuge (De Duve et al. 1955), and in 1963, Hers demonstrated that Pompe patients were lacking an enzyme called acid α-glucosidase or maltase, which was found in lysosomes and converts glycogen or maltose to glucose (Hers 1963). He identified Pompe disease as the first of the lysosomal storage diseases, now known to have a combined incidence of 1 in 5,000–10,000 (Fuller et al. 2006; Raben et al. 2012). Brown, Brown and Jeffrey demonstrated that the lysosomal enzyme discovered by Hers could cleave both 1,4 and 1,6 glycosidic linkages (Brown et al. 1970; Jeffrey et al. 1970a, b). The role of this enzyme in the degradation of glycogen was unclear, but presumably it was important since its absence resulted in a fatal condition. It is now well established, but not broadly known, that glycogen can be degraded within lysosomes, but the details of this pathway are still being defined (see Section 2.5).
The discoveries of Leloir, Krebs and Fischer spurred a major wave of glycogen-related research in the decade that followed (Fig. 2.1). By 1970, these fervent investigations were waning, as it seemed to most that the work on glycogen was virtually complete. In 1971, Ryman and Whelan synthesized an exhaustive review of glycogen metabolism, which spanned 158 pages and cited nearly 900 publications. They stated in their introduction:
“The field of glycogen metabolism is one that to the outside observer has long seemed in a settled condition, with new discoveries being only likely to add gloss to existing facets. This is because it has been possible since the early 1940’s to draw metabolic maps that seem to explain the process fully and satisfactorily, these maps being based on highly satisfactory in vitro experiments carried out with purified or semi-purified enzymes. This has been the polymer par excellence as far as in vitro work is concerned. This apparently settled condition is, in fact, illusory, and this has probably worked to the detriment of progress, since potentially interested investigators have almost certainly turned to other pursuits, feeling that no more major advances would be forthcoming. The true situation is the exact opposite, and the ferment of activity now going on testifies to the growing realization that studies of glycogen metabolism have thrown up key discoveries that have the widest implications throughout biochemistry”
.
One aspect that remained unclear was how glycogen synthesis was initiated in vivo. Both the Coris and Leloir observed that in vitro glucose polymerization by glycogen phosphorylase or glycogen synthase required the addition of a pre-formed carbohydrate primer (Swanson and Cori 1948; Leloir and Cardini 1957). Some investigators believed that glycogen synthesis could be initiated in vivo by glycogen synthase (Salsas and Larner 1975); others reported that a protein acted as the priming factor (Krisman and Barengo 1975). In 1977, Whelan’s group discovered a protein covalently bound to glycogen in liver, and later showed that the protein, which became known as glycogenin, was linked to glycogen via a novel tyrosine-glucose linkage (Butler et al. 1977; Rodriguez and Whelan 1985) (Fig. 2.1). At first, Whelan’s discovery was doubted, but his subsequent work and work from the laboratories of Cohen and Roach provided very strong support for this protein acting as the true primer of glycogen biogenesis. These groups established that glycogenin was a glucosyltransferase using UDP-glucose, and that after glycosylating itself, it could also extend the glucose chain to ~8 glucose residues, which would then be acted upon by glycogen synthase and branching enzyme (Lomako et al. 1988; Pitcher et al. 1988; Lomako et al. 1990; Viskupic et al. 1992). The initiation of glycogen synthesis by glycogenin is now widely accepted and has been extensively characterized, and similar protein primers for starch synthesis have also been described (reviewed by Roach 2002; D’Hulst and Mérida 2010; Roach et al. 2012).
2.2.5. Reviving interest in brain glycogen metabolism
In recent years there has been a growing interest in studying brain metabolism, in part due to some important technological advancements. Firstly, in the 1970s, neurochemists began using focused microwave irradiation to rapidly and irreversibly inactivate enzymes in rodent brains in order to more accurately measure labile metabolites (reviewed by Schneider et al. 1981; Marani 1998). This technique was elegantly applied to determine the regional distribution of glycogen in the rat brain in the 1980s (Sagar et al. 1987; Swanson et al. 1989) and more recently in the mouse brain (Oe et al. 2016). Secondly, after it was suggested that elevated glycogen in a GSD patient could be observed by 1H NMR, Choi et al. introduced the use of NMR spectroscopy to noninvansively measure the turnover of 13C-labelled glycogen in vivo in rodents (Salvan et al. 1997; Choi et al. 1999). Öz and colleagues applied this technique to humans, publishing a series of studies on the role of glycogen in normal human brain metabolism and during hypoglycemia (Öz et al. 2003; Öz et al. 2007; Öz et al. 2009).
Another reason for the growing interest in brain glycogen is the emerging theme of metabolic coupling of astrocytes and neurons, particularly involving the transfer of lactate. These interactions are fascinating, complex, and highly debated. They have been recently discussed by multiple excellent reviews (Barros 2013; Dienel and Cruz 2016; Alberini et al. 2018; Bak et al. 2018; Magistretti and Allaman 2018) and in the subsequent chapters of this book. The coupling between astrocytes and neurons is reminiscent of the Cori cycle and the shuttling of lactate between liver and muscle; however, these interactions are more complicated and difficult to study. Glycogen metabolism is intimately linked to granule size, architecture, and its various associated proteins, which are still being investigated. Such topics are not frequently discussed in the context of brain glycogen metabolism. These nuanced aspects of glycogen regulation, in addition to its surprisingly dynamic nature, subcellular distribution, and alternative degradation pathways, are introduced in the next sections.
2.3. Glycogen Structure
2.3.1. Basic structure of glycogen and other glucose polymers
Glycogen is one of many types of polysaccharides that exist in biology. A polysaccharide refers to any large polymer comprised of many covalently linked monosaccharides; the bonds between them are known as glycosidic linkages. Glycogen and the two primary constituents of plant starch, amylopectin and amylose, are all homopolymers of glucose designed for energy storage in various organisms. We and others refer to these glucose homopolymers as polyglucans. Glycogen and amylopectin contain branches, while amylose is almost exclusively composed of linear chains. The linear chains of glucose are connected by α−1,4 glycosidic linkages, and the branch points are comprised of α−1,6 glycosidic linkages (Fig. 2.6a). Because of the tree-like arrangement of branching, glycogen and amylopectin contain fewer reducing ends (i.e. ends with a free aldehyde group) than nonreducing ends, which are oriented outward. The α configuration of the glycosidic linkage causes a linear α−1,4 linked chain to twist, and when the chains are long enough, and uninhibited by branch points, they can form single or double helices (Gessler et al. 1999) (Fig. 2.6b). An alternative β-linked configuration of glucose polymers favors very straight chains that can associate into tight fibrils; this is characteristic of structural polyglucans such as cellulose. Mammals do not synthesize or hydrolyze β-glycosidic linkages, so they cannot digest cellulose, but they are well equipped to degrade glycogen and starch (Berg et al. 2002).
Fig. 2.6. Structure and synthesis of polyglucans.
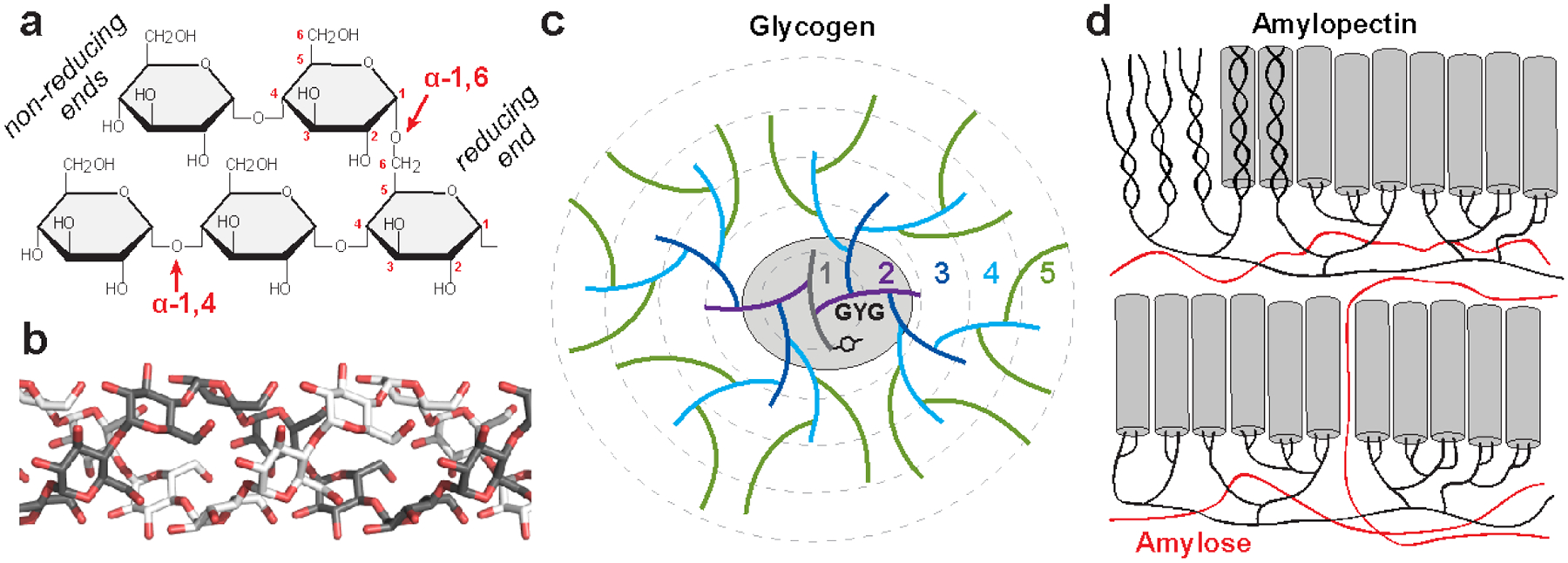
(a) Starch and glycogen contain linear chains of glucose joined by α−1,4 glycosidic linkages, and α−1,6 glycosidic linkages constitute the branch points. The reducing ends (containing an aldehyde group) are oriented near the interior of the polysaccharide molecule, while metabolic enzymes work on the nonreducing ends, which are oriented outward. (b) The α conformation of α−1,4 linked polyglucans gives them a propensity to twist, and long unbranched chains can form single or double helices; a model of a double helix is shown. (c) A model of the first five tiers of the glycogen particle according to the Whelan model. A tyrosine residue of glycogenin (GYG) is shown, which is covalently linked to the glucan chain making up the first tier. (d) A recent model for starch structure according to (Bertoft 2017). In amylopectin, the branched component of starch, branching is clustered, and the long linear chains form double helices (grey cylinders) that make up the crystalline regions of starch. Amylose, the unbranched component of starch, is believed to occupy areas around the amylopectin molecules.
Although the models of glycogen and amylopectin structure are both based on the tree-like arrangement originally proposed by Meyer and Bernfeld, the models were individually refined as it became increasingly evident that the arrangement and length of the branches of these glucose polymers were quite different. Glycogen contains a high degree of branching and short linear chains. By enzymatic analyses, there are, on average, 13 glucose units per α−1,4-linked linear chain, and 8% of the total glycosidic linkages are α−1,6 branch points (Cori 1952; Illingworth et al. 1952; Melendez-Hevia et al. 1993; Melendez et al. 1997; Roach et al. 2012) (Fig. 2.6c). Branching in glycogen is believed to be continuous, meaning the branch points are evenly distributed within the molecule. The presently accepted model for glycogen is based on the revisions of Whelan’s group (Calder 1991), but high resolution studies of chain length distribution (CLD) suggests this model may still not be completely accurate (discussed below). Typically, a single glycogen is depicted as originating from a single chain covalently attached to a glycogenin molecule, which gives rise to all subsequent chains (Roach et al. 2012; Prats et al. 2018) (Fig. 2.6c). Enzymatic experiments indicate 3 to 4 glucose units between each branch point, so each linear chain would yield two branches (Illingworth et al. 1952; Calder 1991). Each new set of branches is referred to as a tier (Fig. 2.6c), and since the number of linear chains doubles with each successive tier, the outermost tier would theoretically contain about one-third of the total glucose in the molecule, which is consistent with empirical observations (Larner et al. 1952; Melendez et al. 1997). Mathematical modeling has demonstrated that due to physical constraints, glycogen molecules can theoretically contain up to 12 tiers, corresponding to ~55,000 glucose molecules; beyond this size, the outer chains would become so crowded they would be inaccessible to enzymes (Melendez-Hevia et al. 1993). Indeed, the empirically observed upper limit of glycogen β-particles (44 nm) is consistent with the theoretical maximal diameter of a 12-tiered glycogen molecule (42 nm) (Shearer and Graham 2004; Prats et al. 2018). The continuous branching within glycogen prevents, or dramatically limits, the formation of double helices, which would lead to insolubility of glycogen and inaccessibility of the glucan chains to enzymatic degradation (Emanuelle et al. 2016). Mathematical modeling studies have also demonstrated that the empirically observed average chain length and branching degree are optimal for maintaining solubility and structural homogeneity (Melendez et al. 1998).
Amylopectin is also a branched polyglucan, but it contains longer chains of 20–25 glucose units per chain with infrequent and clustered branching (Manners 1989; Buleon et al. 1998) (Fig. 2.6d). The clustering of branch points means there are regions of long, unbranched chains that associate to form crystalline, water-excluding double helices (Fig. 2.6b). How these crystalline regions are arranged is still debated, but the most recent data suggests they are arranged along a polyglucan “backbone” (Bertoft 2017). The alternating crystalline and amorphous layers within starch are advantageous as it allows for very dense glucose packing within starch granules, which can be as large as 100 μm in diameter (Emanuelle et al. 2016). Amylose is believed to be interspersed among amylopectin molecules (Fig. 2.6d). Glycogen, amylopectin, and amylose have diverse structural properties due to differences in chain length and degree of branching. Glycogen has the shortest average chain length and the highest degree of branching, usually 8%. The degree of branching in amylopectin is nearly half that of glycogen, only 4–6% (Manners 1991; Buleon et al. 1998). Amylose is generally considered to be entirely composed of very long, linear chains, although infrequent branching has been reported (Manners 1989).
2.3.2. Glycogen architecture
It is well established that amylopectin and glycogen differ in chain length and degree of branching. But a diverse array of polyglucans exist in nature with structures that vary beyond these two simple parameters. It is becoming increasingly apparent that the distribution of chain lengths and the arrangement of branch points within a polyglucan are variable, producing distinct biological and physiochemical properties. Furthermore, not only do amylopectin and glycogen differ significantly from each other, but each polyglucan comes in a variety of shapes and properties based on species, tissue of origin, and even the nutritional state of the tissue. Polyglucan diversity in the context of starch has been exhaustively studied and thoroughly parameterized (Pérez and Bertoft 2010; Nakamura 2015). We will use the term “architecture” to refer to the unique chain length distribution, branching frequency and arrangement, and quantity and distribution of non-glucose moieties (notably phosphate) characteristic of a certain type of glycogen or amylopectin.
Since the days of Bernard, investigators have utilized iodine to study polyglucan architecture. Bernard observed in 1877 that newly synthesized glycogen in rabbit muscle following exhaustive exercise gave a bluish coloration with iodine, in contrast to the reddish color of glycogen from rested muscle or well-fed liver (Young 1937). This colorimetric test can be made quantitative by an absorbance spectral scan: the wavelength of maximal absorption (λmax) increases with longer chains and less branching, reflected by the staining color (Swanson 1948; Krisman 1962; Banks et al. 1971). Glycogen from different sources yields a range of colors from yellow to reddish brown with iodine and produces a λmax of 420–490 nm; amylopectin gives a more intense color ranging from red to lavender with λmax of 490–570 nm; and amylose yields a blue to green color with a λmax of 580–640 nm (Archibald et al. 1961; Bailey and Whelan 1961; Krisman and Alfredo 1991). The iodine method is not perfectly quantitative, due to the various effects of the iodine and salt concentrations, temperature, and polysaccharide structure and source (Morris 1946; Archibald et al. 1961). Additionally, although chain length and degree of branching describe different aspects of glycogen architecture, both parameters are related to λmax (Archibald et al. 1961; Bailey and Whelan 1961; Hirai et al. 1994). It is difficult to deconvolve the various architectural parameters of polyglucans using just iodine, but it remains a convenient technique that yields useful information.
High-performance anion exchange chromatography (HPAEC) was introduced in the 1990s to determine the chain length distribution (CLD) of enzymatically debranched polysaccharides with very high resolution (Rani et al. 1992). The CLD for various glycogens is asymmetrically unimodal: in bovine and rabbit liver glycogens, chains ranged from 3 to 35 degrees of polymerization (DP), peaking at DP 6–10 or 7–15, respectively (Rani et al. 1992; Matsui et al. 1993) (Fig. 2.7a). This range of chain lengths suggests the architecture of glycogen may be intermediary between the Meyer and Whelan models. Amylopectin CLD profiles display a strikingly different bimodal CLD, with distinct groupings of short and long chains, further evidence for amylopectin chains being organized very differently than glycogen (Fig. 2.7b) (Hanashiro et al. 1996). CLD profiles for skeletal muscle and brain glycogen from mice have also been described recently. The Minassian group showed that the CLD profiles of mouse skeletal muscle and brain glycogen are nearly identical, ranging from DP 2–35 or more, with most chains being DP 4–15 (Nitschke et al. 2013; Nitschke et al. 2017). Additionally, gradual isoamylolysis of muscle glycogen suggests that the internal chains are longer than the external chains (Nitschke et al. 2013). The Roach group showed a CLD for mouse skeletal muscle glycogen ranging from DP 3–45, with most chains being 7–15 units long (Irimia et al. 2015). After exercise and a 1 hour recovery, the CLD shifted toward long chains (Fig. 2.7c). Similarly, muscle glycogen had an increased λmax after exercise and recovery, which was gradually restored over the course of 6 days. These data suggest that newly synthesized glycogen is less branched with longer chains that are remodeled over time (Fig. 2.7d).
Fig. 2.7. Chain length distribution (CLD) of glycogen and amylopectin demonstrated by HPAEC.
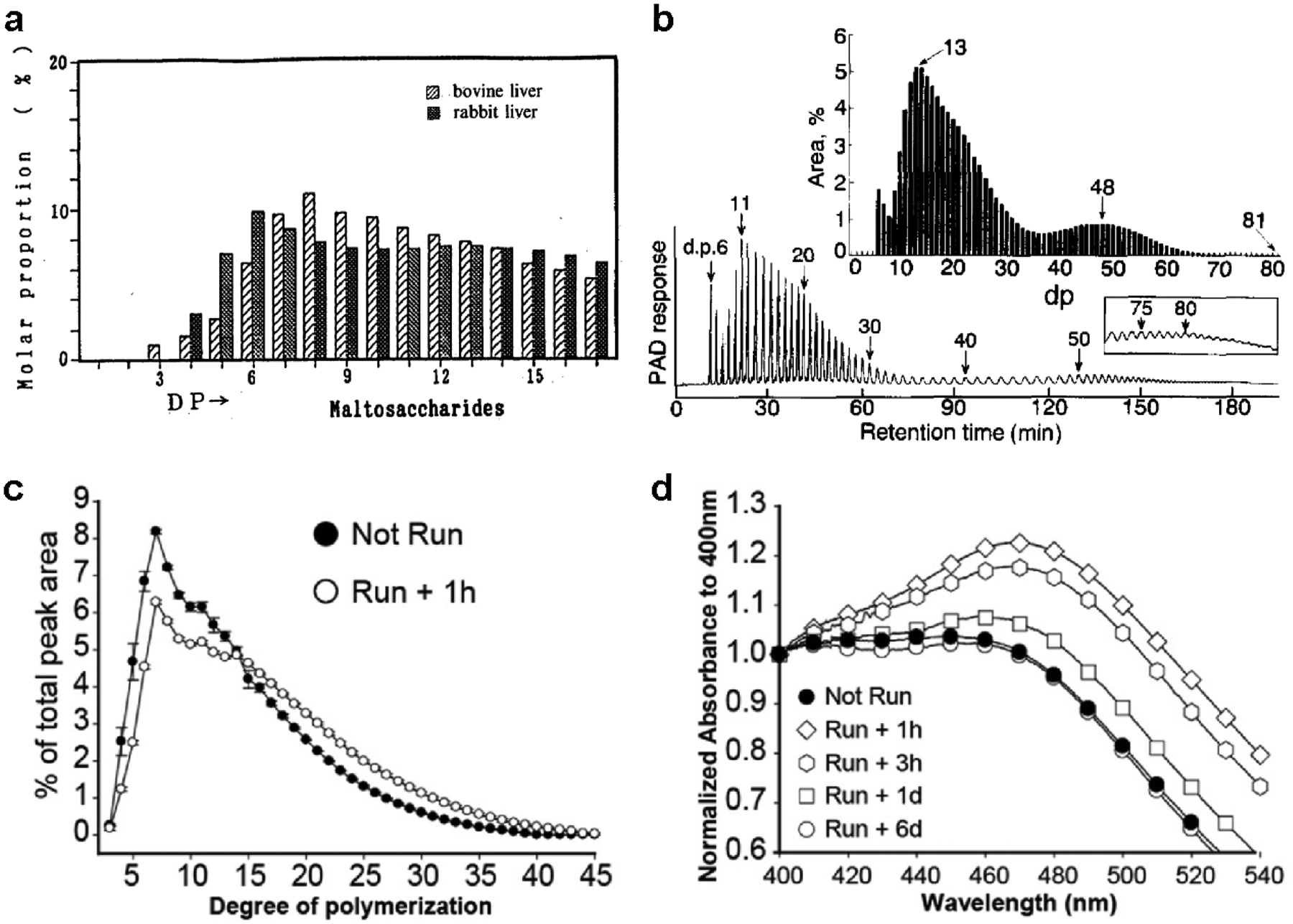
(a) CLD of bovine liver and rabbit liver glycogen determined by HPAEC (Matsui et al. 1993). Copyright © 1993 Japan Society for Bioscience and Agrochemistry, reprinted by permission of Taylor & Francis, Ltd. (b) HPAEC profile and CLD of potato amylopectin (Hanashiro et al. 1996). Copyright © 1996 Elsevier. Used with permission. (c) CLD of skeletal muscle glycogen from rested mice (not run) and 1 hour following exhaustive exercise (Irimia et al. 2015). (d) Iodine spectra of skeletal muscle glycogen from rested mice and 1 hour, 3 hour, 1 day, and 6 days post-exercise (Irimia et al. 2015). Copyright © 2015 American Society for Biochemistry and Molecular Biology. Used with permission.
It appears that glycogen molecule is not as homogenously structured as the Whelan model suggests, and chain length and branching appear to fluctuate based on nutritional state. Gerty Cori stated in a 1952 lecture that “the relationship of structure to nutritional state has not yet been fully explored, but it seems that glycogen freshly deposited after a fasting period is least branched and has long outer chains whereas the opposite is true of ‘old’ glycogens” (Cori 1952). Additionally, mammalian and non-mammalian glycogens with similar average chain lengths produce differences in iodine spectra, suggesting that mammalian glycogens contain long chains in the interior of the granule that are not present in non-mammalian glycogens (Manners et al. 1983). Whole glycogen particles also display a continuum of sizes, and their size and ultrastructure vary with tissue type (see Section 2.6.1).
2.3.3. Glycogen structure and phosphate: insights from Lafora disease
Fontana first demonstrated that [32P]-labeled glycogen could be purified from the livers of rats after the administration of radioactive phosphoric acid (Fontana 1980). Whelan’s group subsequently reported that mammalian muscle glycogen contains about 0.064% phosphorus by weight (Lomako et al. 1993b; Lomako et al. 1994). However, much like Whelan’s reports of a proteinaceous component of glycogen that were later substantiated with the discovery of glycogenin, phosphate was initially treated as a contaminant. The physiological relevance of glycogen phosphate became evident through basic scientific investigations of Lafora disease (LD), a fatal, inherited childhood epilepsy and non-classical glycogen storage disease. LD is characterized by accumulations of abnormal polysaccharides known as Lafora bodies that cause neurodegeneration (recently reviewed by Gentry et al. 2018) (Fig. 2.8a). In the 1960s and 1970s, it was demonstrated that LBs contain high levels of phosphorus and histologically resemble plant amylopectin (Yokoi et al. 1968; Sakai et al. 1970). In 1998 and 2003 the genes associated with the disease were identified, and it is now well established that virtually all LD patients carry recessive mutations in either the EPM2A or EPM2B gene (Minassian et al. 1998; Serratosa et al. 1999; Chan et al. 2003). EPM2A encodes laforin, a glycogen phosphatase (Worby et al. 2006; Tagliabracci et al. 2007), and EPM2B encodes malin, an E3 ubiquitin ligase (Gentry et al. 2005; Lohi et al. 2005). Epm2a−/− and Epm2b−/− mouse models recapitulate the disease with respect to LB formation and neurodegeneration (Ganesh et al. 2002; DePaoli-Roach et al. 2010; Valles-Ortega et al. 2011; Criado et al. 2012; Tiberia et al. 2012; Duran et al. 2014) (Fig. 2.8b). Purified polysaccharides from LD mice have a shifted λmax reminiscent of amylopectin (Valles-Ortega et al. 2011), and their CLD profile shows a higher proportion of long chains than wild-type mice, most prominently in brain tissue (Nitschke et al. 2017) (Fig. 2.8c). Unlike glycogen, purified LBs are quite large (>1 μm in diameter) and insoluble; they are presumed to be aggregates of a polysaccharide with abnormal architecture resulting from aberrant glycogen metabolism (Gentry et al. 2018).
Fig. 2.8. Insights from Lafora disease, a nonclassical glycogen storage disease.
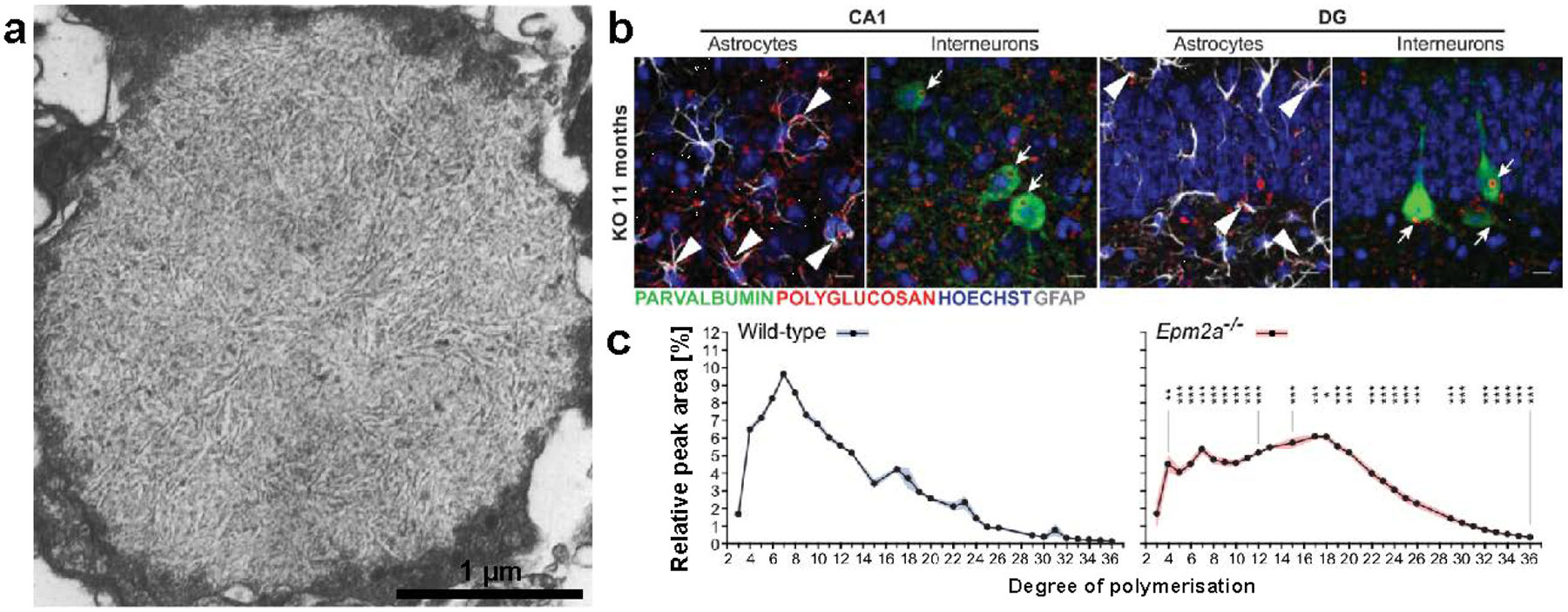
(a) A typical LB visualized by electron microscopy in the human retina (Berard-Badier et al. 1980). Scale bar has been approximated based on magnification. Copyright © 1980 Springer Nature. Used with permission. (b) Neuronal (arrows) and astrocytic (arrowheads) polyglucosan accumulations visualized by immunostaining in the dentate gyrus (DG) and CA1 of the hippocampus in malin knockout (KO) mice (Valles-Ortega et al. 2011). Scale bars = 10 μm. (c) Normal and abnormal CLD of purified polysaccharides from WT and LD (Epm2a−/−) mice determined by HPAEC (Nitschke et al. 2017).
Laforin and malin regulate two aspects of glycogen architecture that may be intertwined: chain length and phosphate level. Through studies of its role in LD, laforin was discovered to be the founding member of a class of enzymes known as glucan phosphatases that directly dephosphorylate carbohydrate substrates (Worby et al. 2006; Gentry et al. 2007; Gentry and Pace 2009; Vander Kooi et al. 2010; Meekins et al. 2013; Meekins et al. 2015). Laforin is the only known glucan phosphatase in mammals, and decreased laforin activity results in glycogen hyperphosphorylation. Malin ubiquitinates enzymes involved in glycogen metabolism, but the effects of ubiquitination are not clear (see Section 2.5.2). Surprisingly, the absence of malin also leads to hyperphosphorylation, and the absence of either enzyme leads to the accumulation of glycogen with abnormally long chains, which is puzzling (Sullivan et al. 2017). Recent results from mouse models overexpressing a catalytically inactive laforin demonstrate that the inactive laforin rescues the LD phenotype in the Epm2a-deficient mouse model (Gayarre et al. 2014; Nitschke et al. 2017). As a result, the relevance of laforin’s catalytic activity has been questioned, despite the very high conservation of its catalytic residues and the presence of glucan phosphatases across multiple kingdoms (Gentry et al. 2007; Raththagala et al. 2015). It has also been suggested that laforin and malin form a ubiquitination-targeting complex involved in the disposal of glycogen molecules with aberrantly long chains that could crystallize and cause the molecule to precipitate (Sullivan et al. 2017). A growing body of evidence suggest that laforin and malin are an integral part of an alternative route for glycogen degradation known as glycophagy, involving members of the autophagic pathway and lysosomal α-glucosidase (see Section 2.5.2). However, glycophagy is probably not reserved only for aberrantly structured glycogen molecules, since the accumulated lysosomal glycogen in acid α-glucosidase deficiency (i.e. Pompe disease) is of normal structure based on iodine staining (Levin et al. 1968; Mahler 1969). Additional evidence for a physiological role of glycogen phosphate comes from the Roach lab and a substantial body of work from the field of starch metabolism.
The Roach lab analyzed glycogen CLD and phosphate levels of laforin-deficient mice pre- and post-exercise. When muscle glycogen phosphate was depleted, phosphate levels remained suppressed even after total glycogen and CLD returned to normal (Irimia et al. 2015). Laforin-deficient mice displayed exercise-induced glycogen depletion identical to wild-type mice, but phosphate levels remained elevated and iodine spectra suggested a delay in glycogen remodeling post-recovery. This study demonstrated that laforin dephosphorylates glycogen during exercise-induced cytosolic glycogeolysis, providing strong evidence for its physiological role as a glycogen phosphatase. Whelan suggested in 1994 that phosphate was a marker for the age of a glycogen molecule; the suppression of glycogen phosphate after exhaustive exercise in wild-type mice does indeed suggest that phosphate accumulates with age, possibly from multiple cycles of glycogen degradation and re-synthesis (Lomako et al. 1994; Irimia et al. 2015). The role of phosphate in glycogen metabolism is not clear, but studies from the starch field demonstrate that phosphate plays an important physiochemical and biological role in starch architecture and metabolism (Hejazi et al. 2008; Blennow and Engelsen 2010; Kotting et al. 2010).
In plants, phosphorylation is necessary for the proper synthesis of starch, and reversible phosphorylation is an integral part of starch breakdown. Only amylopectin contains significant amounts of phosphate, which is enriched in the amorphous regions (Takeda and Hizukuri 1982; Blennow et al. 2000). Root and tuber starches, which exhibit less densely packed crystalline helices than other types of starches, have high phosphate levels (Lim et al. 1994; Blennow et al. 1998). Starch phosphate is covalently linked to the C3 and C6 hydroxyls of glucose moieties; about 70–80% of the phosphate is esterified to C6 (Ritte et al. 2006). Phosphate, particularly at the C3 position, disrupts the crystalline helices in amylopectin by introducing steric hindrance, promoting their solubilization (Hansen et al. 2009). Starch degradation is believed to be a cyclic process involving reversible phosphorylation and the concerted action of multiple enzymes: glucan dikinases, amylases, and glucan phosphatases (Emanuelle et al. 2016). Two dikinases that respectively phosphorylate the C6 and C3 position in that order are glucan, water dikinase and phosphoglucan, water dikinase. Phosphorylation promotes solubilization of the glucan chains and facilitates their access by plant amylases. However, the amylases cannot proceed past a phosphate, so phosphate removal is achieved by the glucan phosphatases Starch Excess 4 (SEX4) and Like Sex Four 2 (LSF2), named after the plant phenotype that results from their deficiency and part of the same family as the glycogen phosphatase laforin (Edner et al. 2007; Kotting et al. 2009; Santelia et al. 2011; Gentry et al. 2016; Meekins et al. 2016). Crystal structures and structure-function studies of SEX4 and LSF2 preceded those of laforin (Vander Kooi et al. 2010; Meekins et al. 2013; Meekins et al. 2014; Raththagala et al. 2015) and the cooperative study of both systems has led to a wealth of insight about polyglucan architecture and phosphorylation (Gentry et al. 2009; Emanuelle et al. 2016; Gentry et al. 2016).
Through studies of LD, the Roach and Minassian groups determined that normal glycogen contains about 1 phosphate moiety per 600–2500 glucose units, depending on species and tissue type (Tagliabracci et al. 2007; Tagliabracci et al. 2008; Turnbull et al. 2010; Tiberia et al. 2012; DePaoli-Roach et al. 2014). Phosphates are present as monoesters linked to the C2, C3 and C6 hydroxyls of glucose moieties, in approximately equal quantities (Tagliabracci et al. 2011; Nitschke et al. 2013; DePaoli-Roach et al. 2014). Data from multiple groups using various approaches strongly suggest the phosphate is concentrated at the interior of glycogen molecules (Tagliabracci et al. 2007; Nitschke et al. 2013; Irimia et al. 2015). Liver glycogen contains less phosphate than muscle glycogen, while C6 phosphate has been detected in brain glycogen at similar levels to C6 phosphate in muscle glycogen (Lomako et al. 1994; Tagliabracci et al. 2007; Turnbull et al. 2010; Nitschke et al. 2017). The source of glycogen phosphate remains a mystery: although it has been reported that glycogen synthase can incorporate the β-phosphate of UDP-glucose into glycogen in a rare side reaction, this mechanism could only account for C2 and C3 phosphate, and the rate of incorporation is very low (1 in 10,000 catalytic cycles) compared to the actual levels observed in glycogen (Tagliabracci et al. 2011; Contreras et al. 2016). Although no glucan dikinase has been identified in mammals, the physiological role of glycogen phosphate will eventually be defined, likely through the continued investigations of starch metabolism and the molecular mechanisms of LD.
2.3.4. Glucosamine
Glycogen also contains small amounts of covalent glucosamine. In the 1960s and 70s, Maley et al. reported the incorporation of glycosidically linked [14C]glucosamine into glycogen from [14C]galactosamine, and showed that glycogen synthase could catalyze glucosamine incorporation in vitro by using the substrate UDP-glucosamine instead of UDP-glucose (Maley et al. 1966; Tarentino and Maley 1976). Whelan also reported that intraperitoneal injection of [14C]galactosamine led to the incorporation of [14C]glucosamine into glycogen, replacing as much as 10% of the glucose residues (Romero et al. 1980). Covalently linked glucosamine in glycogen apparently would not block phosphorolysis, since it could be released as glucosamine-1-phosphate by phosphorylase. Whelan reported that normal pig and rabbit liver contained 86–266 nmol glucosamine per g glycogen, which was randomly distributed throughout the molecule (Kirkman and Whelan 1986). It is worth noting that an earlier study also demonstrating the incorporation of [14C] into glycogen from injected [14C]glucosamine showed that glucosyl residues were labeled, indicating [14C]glucosamine was converted to [14C]glucose through a deamination reaction prior to glycogen incorporation (Khac et al. 1972).
Additional links between glycogen and glucosamine have been reported. Glycogen synthase can be modified by N-acetyl-glucosamine (GlcNAc), and it has been suggested that glycogenin could also be modified by GlcNAc (Parker et al. 2003; Tavridou and Agius 2003). Furthermore, studies have suggested that glycogen could be a carbohydrate source for protein glycosylation, for which glucosamine is a major precursor, and hypoglycosylation has been reported in GSDs (McMahon and Frost 1996; Hayee et al. 2011; Tegtmeyer et al. 2014; Ondruskova et al. 2018). A physiological role for glycogen in protein glycosylation and the relevance of covalently linked glucosamine await further investigation.
2.4. Cytosolic glycogen synthesis and degradation
2.4.1. Glycogenin
Glycogen synthesis begins with glycogenin [UDP-α-glucose:glycogenin α-glucosyltransferase, EC 2.4.1.186], a member of glucosyltransferase family 8 (Campbell et al. 1997)(www.cazy.org). The enzyme possesses two distinct enzymatic activities: self-glucosylation and chain elongation, both requiring UDP-glucose as a glucosyl donor and both occurring at the same active site, though the chemistries of the reactions are different (Alonso et al. 1995; Lomako et al. 2004). For this reason the two activities were initially believed to belong to separate enzymes, until Whelan and Cohen discovered that both were accomplished by glycogenin (Lomako et al. 1988; Pitcher et al. 1988). Crystal structures and biochemical studies show that glycogenin functions as an obligate dimer and requires Mn2+ for activity (Gibbons et al. 2002; Chaikuad et al. 2011). Autoglucosylation on tyrosine creates a glucose-1-O-tyrosyl linkage, a chemical linkage rarely found in nature, then glycogenin synthesizes a short oligosaccharide primer of at least 7–8 glucose units (Smythe and Cohen 1991; Alonso et al. 1995). Even if this tyrosine is mutated, glycogenin is still capable of glucoslyation, just not of itself (Cao et al. 1993a; Alonso et al. 1994). Whether glucosyl additions occur via intra- or intersubunit reactions has been debated, but recent studies suggest that the first four glucosyl units may be added via an intrasubunit reaction, and longer chains require intersubunit interaction (Chaikuad et al. 2011). It has been suggested that although glycogenin functions as a dimer, at low enough concentrations the two subunits would dissociate after chain initiation, giving rise to two separate glycogen molecules (Lin et al. 1999). The presence of a single chain giving rise to an entire β-particle is supported by structural studies of glycogen (Calder 1991).
In most mammals, there is only one isoform of glycogenin that is widely expressed, but humans have two isoforms: glycogenin-1 (GYG1), expressed in all tissues, and glycogenin-2 (GYG2) which is primarily expressed only in liver, with minor expression in heart, pancreas, and adipose tissue (Mu et al. 1997; Roach 2002; www.proteinatlas.org). There is some evidence that glycogenin is phosphorylated, but it has not been corroborated (Lomako et al. 2004). Glycogenin can interact directly with glycogen synthase, and a crystal structure of the interaction provides evidence for their cooperation in the initiation of the glycogen granule (Skurat et al. 2006; Zeqiraj et al. 2014). Additionally, insufficiently glucosylated glycogenin does not serve as an efficient primer for glycogen synthase, and phosphorylase can reduce the glucosylation state of glycogenin, making it a less effective substrate for synthase (Cao et al. 1993b; Skurat et al. 1993). Phosphorylase induces the dissociation of glycogen synthase from a proteoglycan fraction in hepatocytes, presumably containing glycogenin (Tavridou and Agius 2003). Thus, priming through glycogenin may be regulated indirectly by the signals that govern glycogen synthase and phosphorylase activity (see Chapters 3 and 4), rather than by direct regulation of glycogenin. Glycogenin interacting proteins (GNIPs) have been suggested to stimulate the activity of glycogenin (Graham et al. 2010). Paradoxically, glycogenin mutations in humans and mice lead to glycogen accumulation in some tissues and muscle weakness, indicating that without glycogenin, glycogen synthesis can still occur, but in a dysregulated and pathogenic manner (Malfatti et al. 2014; Testoni et al. 2017).
2.4.2. Glycogen synthase and phosphorylase
Glycogen synthase [UDPglucose:glycogen α−4-glucosyltransferase, EC 2.4.1.11] is a member of the glycosyltransferase family 3 that catalyzes the addition of alpha-1,4-linked glucosyl units to a glycogen chain, using UDP-glucose as the donor and releasing UDP as product (www.cazy.com). Two isoforms of synthase exist in mammals: GYS1, encoding muscle glycogen synthase, highly expressed in all tissues including brain, and GYS2, restricted to liver (Roach et al. 2012)(www.proteinatlas.org). Muscle glycogen synthase has nine phosphorylation sites and was one of the first examples of a hierarchically phosphorylated protein (Roach 1990). Although liver glycogen synthase is also multiply phosphorylated, its activity appears to be regulated by only one phospho-site (Ros et al. 2009). Phosphorylation inhibits synthase activity, although activity can be fully restored in the presence of the potent allosteric activator glucose-6-phosphate. Numerous kinases are responsible for phosphorylating synthase, including protein kinase A, protein kinase C, AMP-activated protein kinase (AMPK), caseine kinase 2, and glycogen synthase kinase 3 (GSK3). Thus, control of glycogen metabolism is inextricably linked to intracellular energy status, glucose homeostasis, insulin signaling, and other external signals through diverse signaling cascades (Roach 1990; Lawrence and Roach 1997; Roach et al. 2012). The structure and regulation of glycogen synthase warrant much attention and are discussed in Chapter 3.
Glycogen phosphorylase [1,4-α-glucan:orthophosphate α-glycosyltransferase, EC 2.4.1.1] is a member of the glycosyltransferase family 35, and catalyzes the transfer of glucose moieties from the glycogen molecule to inorganic phosphate, releasing the product glucose-1-phospate (www.cazy.com). Glucose-1-phosphate is converted to glucose-6-phosphate by phosphoglucomutase, facilitating its entry into glycolysis (see Chapter 6). In the presence of excess glucose-1-phosphate, phosphorylase can catalyze the reverse reaction (Cori and Cori 1943). There are three isoforms of phosphorylase corresponding to the tissues in which they are enriched, but not restricted: muscle (PYGM), liver (PYGL) and brain (PYGB) (www.proteinatlas.org). Only the brain isoform is expressed in fetal tissues, and all are expressed to some extent in adult brain (Pfeiffer-Guglielmi et al. 2000). A single site on phosphorylase is phosphorylated by only one kinase: phosphorylase kinase (PhK), a multisubunit enzyme with muscle, liver and brain isoforms. Both phosphorylase and PhK are activated by phosphorylation and allosteric modulators, and an excellent review on their regulation in brain has been published recently (Nadeau et al. 2018). Crystal structures of all phosphorylase isoforms have been determined (Rath et al. 2000; Lukacs et al. 2006; Mathieu et al. 2016). The structure and regulation of brain phosphorylase are also given a comprehensive review in Chapter 4.
2.4.3. Glycogen branching and debranching enzymes
Like glycogenin, and unlike synthase and phosphorylase, glycogen branching enzyme (GBE) and debranching enzyme (GDE) do not appear to be regulated by phosphorylation or allosteric modulation. There is only one isoform of each, and both are highly expressed ubiquitously (www.proteinatlas.org). Due to their abundance, these enzymes are not considered to be rate-limiting under normal circumstances (Geddes 1986; Melendez et al. 1997; Roach et al. 2012). However, they are clearly essential for maintaining proper glycogen structure, as deficiencies in either enzyme result in GSDs characterized by distinctly abnormal glycogen deposits with pathological consequences (Adeva-Andany et al. 2016). Crystal structures of human GBE and yeast GDE have been determined and the effects of GSD-causing mutations defined, providing molecular level insights into how they affect glycogen structure (Froese et al. 2015; Zhai et al. 2016).
GBE (α−1,4-glucan:α−1,4-glucan 6-glycosyltransferase, EC 2.4.1.18) belongs to the subfamily 8 of the GH13 family of glucosyl hydrolases. Like other members of this subfamily, it contains a carbohydrate binding module, CBM48 (Janecek et al. 2011; www.cazy.com). Two reaction steps occur successively in its central catalytic core: hydrolysis and transglucosylation (Froese et al. 2015). In the first step, the enzyme cleaves an α−1,4 linkage on a glucan chain, forming a covalent enzyme-glycosyl intermediate; in the second step, an α−1,6 linkage is formed from the same chain or one nearby. The minimum length transferred is 6 or 7 glucosyl units (Brown and Brown 1966; Verhue and Hers 1966), and the average distance between chains is 3 or 4 glucose units (Gibson et al. 1971; Calder 1991). This chain length requirement is supported by the crystal structure of GBE in complex with a heptasaccharide (Froese et al. 2015).
Mammalian GBE can convert amylose or amylopectin to a glycogen-like polysaccharide. Krisman showed that incubation of amylopectin or amylose with GBE purified from rat liver led to a displacement of λmax to a value that coincided with that of glycogen (Krisman 1962). However, GBE did not change the λmax of rabbit liver glycogen, suggesting the molecule already had been maximally branched. Indeed, comparative studies on mammalian and plant branching enzymes indicate that the branching degree in polysaccharides is determined by intrinsic properties of the branching enzyme used to synthesize them (Tolmasky and Krisman 1987; Kuriki et al. 1997). In other words, the optimal structure of the mammalian glycogen molecule may be primarily attributed to the inherent “equilibrium” of the mammalian branching enzyme. Not surprisingly, synthetic glycogens produced in vitro by branching enzymes from different organisms have similar, but not identical, structural properties to natural glycogens (Kajiura et al. 2008; Takata et al. 2009; Kajiura et al. 2010). Perhaps these observations explain the delay in glycogen remodeling reported by the Roach lab after exercise (Irimia et al. 2015): glycogen super-compensation, characterized by high glycogen synthase activity, results in longer chains because branching enzyme has not yet introduced the appropriate branch points. Only over time does branching enzyme act so that glycogen structure returns to an equilibrium, with CLD and λmax at basal levels. Indeed, it has been suggested that a chronic imbalance of glycogen synthase and GBE activities leads to abnormal polysaccharide formation in pathological conditions (Raben et al. 2001; Tagliabracci et al. 2008; Kakhlon et al. 2013). Also, the ubiquitous expression of a single GBE isoform may help to explain why brain glycogen has a similar CLD to muscle glycogen (Nitschke et al. 2017). Presumably all healthy tissues in a single species would reach the same CLD at “equilibrium.”
GDE is required for cytosolic glycogen degradation since phosphorylase terminates four glucosyl residues away from a branch point. Mammalian GDE has two catalytic activities: 4-α-glucanotransferase activity (EC 2.4.1.25) involves transfer of a chain of three glucosyl units from the branch to a nearby nonreducing end, leaving a single α−1,6 glucosyl unit. Its amylo-α−1,6-glucosidase activity (EC 3.2.1.33) follows, and the α−1,6 linkage is hydrolyzed to release free glucose (Ryman and Whelan 1971). The release of free glucose instead of glucose-1-phosphate makes it very convenient to compare the levels of α−1,6 and α−1,4 linkages in polysaccharides. GDE is considered an indirect debranching enzyme; direct debranching enzymes such as isoamylase exclusively cleave α−1,6-linkages and release intact oligosaccharide chains. Direct debranching enzymes occur in plants, bacteria and yeast but not in mammals (Ryman and Whelan 1971). These enzymes are very useful in polysaccharide analysis, especially in determining CLD.
GDE catalytic mutants display a loss of either glucosidase or transferase activity, but not both, strongly suggesting disparate catalytic sites (Nakayama et al. 2001). This was confirmed when the crystal structure of yeast GDE was determined, revealing an elongated structure with catalytic domains at either end of the molecule (Zhai et al. 2016). Structures of both the free wild-type enzyme and a transferase-deficient mutant in complex with maltopentaose were reported. Maltopentaose molecules were bound at both catalytic domains and other sites, illustrating the specificity of GDE transferase activity for a glucan of five units or less and the necessity of non-catalytic binding sites for glycogen association. The data also suggested that the product of the transferase reaction, a chain containing a single α−1,6-linked glucosyl unit, completely dissociates from GDE prior to recruitment to the glucosidase catalytic site. Multiple binding sites are a recurring theme among glycogen- and starch-associated proteins (Baskaran et al. 2010; Meekins et al. 2013; Froese et al. 2015; Raththagala et al. 2015; Zhai et al. 2016). In observing that oligosaccharides were bound both to catalytic and non-catalytic sites in the crystal structure of GBE, Froese et al. stated: “[Non-catalytic binding sites] may provide GBEs the capability to anchor a complex glycogen granule and, as proposed previously, determine the chain length specificity for the branching reaction as a ‘molecular ruler’. This agrees with the emerging concept of glycogen serving not only as the substrate and product of its metabolism but also as a scaffold for all acting enzymes” (2015) (See Section 2.7).
2.5. Glycophagy
2.5.1. Lessons from Pompe disease
Phosphorolysis is not the sole means of glycogen degradation in cells. This became evident through studies of Pompe disease, a lysosomal storage disorder characterized by the buildup of vacuole-bound glycogen in virtually all tissues (Hers 1963; Raben et al. 2012). The deficient enzyme in Pompe disease is lysosomal acid α-glucosidase (called GAA or acid maltase) [EC 3.2.1.3] which hydrolyzes both α−1,4 and α−1,6 linkages, optimally at low pH (Hermans et al. 1991). It is a widely expressed enzyme, with a promoter characteristic of a housekeeping gene (Hoefsloot et al. 1990). GAA is synthesized as a precursor protein in the endoplasmic reticulum and undergoes extensive processing before it achieves optimal catalytic activity and reaches its lysosomal destination (Hermans et al. 1991; Wisselaar et al. 1993). It can fully hydrolyze glycogen as well as short oligosaccharides, maltose and other polyglucans to glucose (Hers 1963; Palmer 1971; Matsui et al. 1984). GAA is exo-acting, cleaving single glucosyl units successively from the non-reducing ends of glycogen. However, rather than releasing glucose-6-phosphate like phosphorylase, GAA liberates free glucose, which can translocate into the cytosol via glucose transporters in the lysosomal membrane (Mancini et al. 1990).
How glycogen gets into the lysosome is not well defined, but recent work demonstrates that it involves the autophagic machinery. Autophagy, or ‘self-eating,’ refers to multiple cellular pathways converging on the lysosomal breakdown of cellular components such as long-lived proteins and organelles (reviewed by Cuervo et al. 2005; Mizushima and Komatsu 2011; Schneider and Cuervo 2014). Autophagy is critical both for general cellular maintenance and for nutrient release in times of energy stress. It is especially critical for turning over worn or damaged cellular components in neurons and other post-mitotic cells (Cuervo et al. 2005; Komatsu et al. 2006; Mizushima and Komatsu 2011). The best characterized autophagic pathway is macroautophagy, activated in response to starvation; ‘autophagy’ and ‘macroautophagy’ are frequently used interchangeably. Macroautophagy is a highly conserved pathway in eukaryotes involving a family of autophagy-related (Atg) proteins and the formation of a structure with a characteristic double membrane known as the autophagosome, which engulfs substrates and fuses with the lysosome. Microautophagy and chaperone-mediated autophagy are alternative pathways in which cellular components are targeted directly to lysosomes in the absence of autophagosome formation. In recent years, numerous terms for the selective degradation of specific substrates (via any of the autophagic pathways) have been introduced; e.g. mitophagy (of mitochondria), pexophagy (of perioxisomes), nucleophagy (of nuclear components), reticulophagy (of ER), and xenophagy (of pathogens) (Mizushima and Komatsu 2011; Zhao et al. 2018). In 2011, the Roach group introduced the term ‘glycophagy’ to refer to the selective autophagic degradation of glycogen (Jiang et al. 2011). Ironically, the term ‘glycogenosome’ had already been used by some investigators to refer to the glycogen-laden lysosomes found in newborn rat liver, aged neural tissue, and glycogen storage diseases (Iwamasa et al. 1980; Iwamasa et al. 1983; Cavanagh and Jones 2000), but the term did not gain traction.
In addition to glycogenosomes, which have a single membrane characteristic of a lysosome, Pompe (GAA−/−) tissues also display accumulated autophagosomes filled with ubiquitinated proteins and cellular debris. In order to study the effect of autophagy on lysosomal glycogen, Raben and colleagues generated a muscle-specific knockout of a critical autophagosome gene, Atg5, on the GAA−/− background. These mice had just as many glycogenosomes as the GAA−/− mice, but they had a reduced number of autophagosomes and no improvement in pathology. Ubiquitinated proteins were increased, but they were not surrounded by the double membrane as in the GAA−/− mice (Raben et al. 2008; Lim et al. 2017). The study suggested that the autophagy defect was caused by inefficient fusion of the autophagosomes with the glycogen-laden lysosomes, which formed by an independent pathway. In a later study, the group selectively inactivated a closely related gene, Atg7, in fast-twitch GAA−/− muscle (Raben et al. 2010). No autophagosomes were formed, glycogenosomes were still present, and there was no change in pathology; however, the glycogen level was reduced. These two studies demonstrate that macroautophagy contributes to some, but not all, of the delivery of glycogen to the lysosome (Raben et al. 2010). The group also illustrated that the muscle pathology of Pompe disease resulted largely from defective autophagic flux. The observation that Atg7, but not Atg5, abrogated glycogen accumulation may be explained by the fact that there are two primary conjugation systems in autophagy, and while Atg5 is only involved in one, Atg7 is required for both (Nakatogawa et al. 2009).
In 2002, Janeček identified a putatitive CBM20 in a protein of unknown function called starch binding domain-containing protein 1 (Stbd1, also called genethonin 1) (Janeček 2002). The only other proteins in mammals with a CBM20 are laforin and a glycosyltransferase (glycerophosphocholine phosphodiesterase 1, GPCPD1). It was known that the laforin CBM facilitates laforin binding to glycogen (Wang et al. 2002). The Roach group later showed that Stbd1 is highly expressed in glycogen-rich tissues such as muscle, liver, and heart, with trace amounts in brain, kidney and pancreas, and that Stbd1 levels mirrored glycogen levels in fed, fasted and transgenic mice (Jiang et al. 2010). Stbd1 binds glycogen in vitro and co-localizes with the lysosomal-associated membrane protein LAMP1 and the Atg8 homolog GABARAPL1 in cell culture, but not with the microtubule-associated light chain 3 (LC3), a marker for auotophagosomes (Jiang et al. 2010; Jiang et al. 2011). Demetriadou et al. later showed that when overexpressed in HeLa cells, Stbd1 was N-myristoylated, and this modification facilitates its recruitment with glycogen to subcellular domains associated with autophagy (Demetriadou et al. 2017). The Sun group showed that Stbd1 is elevated in muscle tissue of GAA−/− mice, but not in liver or heart, but shRNA-mediated knockdown of Stbd1 did not alter glycogen levels in any tissues (Yi et al. 2013). A full knockout of Stbd1 in the Pompe mice (Stbd1−/− GAA−/−) did not change lysosomal accumulation of glycogen in muscle or heart tissue, but reduced liver glycogen by 73% (Sun et al. 2016). Glycogen accumulation could be restored by exogenous expression of human Stbd1, but not a mutant lacking the CBM20. Their study demonstrated that Stbd1 may significantly participate in glycophagy in the liver but not in the heart or skeletal muscle, and that the CBM20 is essential for its function.
2.5.2. Lessons from Lafora disease
Another link between glycogen and autophagy emerged through studies of LD. Laforin- and malin-deficient mouse models were generated and analyzed by six different groups. Most observed autophagy defects in the brain of these mice (Criado et al. 2012; Puri et al. 2012; Duran et al. 2014; Gayarre et al. 2014). Some groups observed reduced conversion of LC3 from its cyotosolic form (LC3-I) to lipidated form (LC-II), which is associated with its recruitment to autophagosomal membranes (Criado et al. 2012; Gayarre et al. 2014). Both mouse models also displayed an increase in p62 (Criado et al. 2012; Puri et al. 2012; Duran et al. 2014), a protein that normally binds to both ubiquitinated cargo and LC3, is incorporated into the autophagosome, and degraded (Mizushima and Komatsu 2011). Reduced LC3-II and increased p62 levels strongly suggested defects in autophagic flux. Similar defects were also observed in liver of laforin-deficient mice, but not in muscle (Aguado et al. 2010; Irimia et al. 2015). In contrast, the Minassian group did not observe autophagy defects in muscle or brain in either mouse model (Wang et al. 2016; Nitschke et al. 2017).
The Roach, Minassian and Guinovart groups generated LD mouse models with genetically reduced glycogen synthesis and found that LB formation was reduced and neuropathology was abrogated (Turnbull et al. 2011a; Pederson et al. 2013; Duran et al. 2014; Turnbull et al. 2014). The Guinovart group elegantly demonstrated that the autophagy impairment was secondary to polyglucosan accumulation by crossing malin-deficient mice with mice specifically lacking muscle glycogen synthase in the brain, which eliminated cerebral glycogen synthesis, LB formation, autophagy defects, gliosis, and susceptibility to epilepsy (Duran et al. 2014). These studies indicate that polyglucosan accumulation drives the autophagy impairment and neurological phenotype in LD. Although some groups have argued that laforin and malin regulate autophagy independent of glycogen, this is unlikely to be the case in vivo (Gentry et al. 2018). However, as was demonstrated with the Pompe mice, insufficient clearance of glycogen via glycophagy may indeed provoke pathological damage by obstructing overall autophagic flux. This is a very prominent theme among lysosome storage disorders, which frequently have severe neurological phenotypes; this disease class is reviewed in depth elsewhere (Lieberman et al. 2012; Seranova et al. 2017). Corroborating this, recent studies from the Sanz group showed that in LD, mitophagy is impaired due to a global autophagic defect, and endocytic recycling of the astrocytic glutamate transporter GLT1 is altered (Munoz-Ballester et al. 2016; Garcia-Gimeno et al. 2018), likely also resulting from impaired autophagy.
The possibility that laforin and malin are involved in glycophagy builds on work from many labs showing that malin, typically in concert with laforin, ubiquitinates glycogen-associated substrates. Malin is an E3 ubiquitin ligase that was initially demonstrated to bind and ubiquitinate laforin in cell culture (Gentry et al. 2005; Lohi et al. 2005). Malin also promotes the ubiquitination of multiple proteins that bind to glycogen molecules: glycogen synthase, protein targeting to glycogen (PTG), R6, AMPK and GDE (see Section 2.7). In most of these studies, ubiquitination was shown to require laforin, which is believed to act as a scaffold for malin interaction with its substrate (Cheng et al. 2007; Vilchez et al. 2007; Worby et al. 2008; Moreno et al. 2010; Rubio-Villena et al. 2013). Although ubiquitination led to the degradation of these substrates in cell culture, PTG, R6 and AMPK levels were unchanged in LD mouse models; in contrast, glycogen synthase accumulated in the insoluble glycogen fraction, as did laforin in malin-deficient mice (Wang et al. 2007; DePaoli-Roach et al. 2010; Valles-Ortega et al. 2011; Tiberia et al. 2012; Gentry, Sun, Dukhande, unpublished results). Importantly, whenever the ubiquitin linkages incorporated by malin are defined, they are K63-linked (Moreno et al. 2010; Roma-Mateo et al. 2011a; Rubio-Villena et al. 2013; Sanchez-Martin et al. 2015). K63-linked ubiquitination is a substrate for p62 binding and specifically linked to selective autophagy (Tan et al. 2008; Shaid et al. 2013). The presence of a laforin-malin ubiquitination complex has been highly debated (Gentry et al. 2013; Sullivan et al. 2017; Gentry et al. 2018).
Interestingly, the Roach group reported that there was no change in the levels of GABARAPL1 (a member of the Atg8 family and a homolog of LC3; see Lee and Lee 2016) and LAMP1 in laforin-deficient fibroblasts, but these proteins were decreased in malin-deficient fibroblasts and in fibroblasts with both laforin and malin knocked out (Garyali et al. 2014). These results suggest laforin is upstream of malin in the path from glycogen to the lysosome. The Sanz group showed that laforin, malin and p62 form a complex, and that malin mediates the interaction between laforin and p62, likely through ubiquitination (Sanchez-Martin et al. 2015). Additionally, malin and p62 do not co-localize in cells without the expression of laforin. It is well known that laforin binds to and co-localizes with glycogen in vitro, in cell culture, and in vivo.
A model to explain these studies that is compatible with available data posits: laforin binds to glycogen and recruits malin; the laforin-malin complex promotes K63-linked ubiquitination of various glycogen-bound proteins; p62 binds to K63-linked ubiquitin moieties, recruiting LC3 (or its homolog GABARAPL1) and the autophagosome machinery, which engulfs the glycogen particle and fuses with the lysosome. Since glycogen synthase and laforin expression parallel glycogen levels and these proteins accumulate with glycogen in LD mouse models, they may also be turned over inside the lysosome. The fact that a catalytically inactive form of laforin rescues laforin-deficient mouse models supports this scaffolding role for laforin (Gayarre et al. 2014; Nitschke et al. 2017), but does not exclude the possibility that glycogen dephosphorylation by laforin is still relevant, as during cytosolic glycogenolysis (Irimia et al. 2015). Also, phosphorylation of laforin by AMPK has been shown to enhance the interaction of laforin with malin (Roma-Mateo et al. 2011b); this regulation event could be one way to stimulate glycophagy in times of energy stress, when AMPK becomes activated. Finally, it is well established that laforin preferentially binds to polysaccharides with long chains and localizes to LBs in malin-deficient mice (Chan et al. 2004; Criado et al. 2012; Raththagala et al. 2015). Thus, laforin would selectively recruit the glycophagy machinery to precipitation-prone glycogen molecules with precariously long chains, as has been proposed (Sullivan et al. 2017).
It is worth noting that delivery of glycogen to lysosomes may involve multiple pathways, possibly with cell-specific relevance. Perhaps Stbd1 and laforin, each containing a CBM20, function as distinct receptors for glycophagy. It is interesting that although LBs occupy most tissues in LD, they are enriched in certain cell types within these tissues. For example, in LD mice, 97% of LBs in skeletal muscle are found in fast-twitch type IIb muscle fibers and 2% in slow-twitch type I fibers (Turnbull et al. 2011b). In human skin biopsy, LBs are only found in duct cells, but not secretory cells (Andrade et al. 2003). In human liver, LBs are enriched in the periportal regions, but not the perivenous regions (Carpenter et al. 1974). All of these cell types appear to make glycogen (Montagna et al. 1951; Vøllestad et al. 1984; Quistorff 1985), but the differences in whether or not they make LBs suggest the glycogen is handled differently. Cell-specific glycogen metabolism and polyglucosan body formation are of particular interest in the brain. LBs were first reported in neurons (Lafora 1911), and historically were believed to occupy this cell type almost exclusively (Minassian 2001). However, this observation has been perplexing since the vast majority of brain glycogen is stored in astrocytes. Recent work has demonstrated that neurons possess an active glycogen metabolism (Vilchez et al. 2007; Saez et al. 2014), and LD mouse models accumulate both LBs in neurons and a distinct polyglucosan body known as corpora amylacea in astrocytes (Augé et al. 2018; Rubio-Villena et al. 2018). Early literature on human patients corroborate these findings. Corpora amylacea in addition to LBs have been reported in multiple studies (Van Heycop Ten Ham 1975). Small inclusions, which may also correspond to corpora amylacea, have been identified in astrocytes and oligodendrocytes (Schwarz and Yanoff 1965). Recently, it has been asserted that the striking appearance of neuronal LBs left glial LBs largely overlooked over the years; upon careful review of the literature, small LBs that were assumed to be in neuronal processes are likely to actually be astrocytic (Minassian, personal communication). This topic is further discussed in Chapter 10.
2.5.3. The benefits of glycophagy
The lysosomal glycogen pool plays an important role in circumstances when a burst of glucose is needed, such as during the neonatal starvation period (Kotoulas et al. 2004; Kotoulas et al. 2006; Schiaffino et al. 2008). Glycogen-rich autophagosomes have been observed in the neonatal liver, muscle and heart, but they are depleted within hours after birth. A unique role for glycophagy in the heart is also emerging (Reichelt et al. 2013; Delbridge et al. 2015). While liver and muscle normally deplete their glycogen stores with fasting, cardiac tissues accumulate glycogen and upregulate glycogen autophagy under nutrient stress (Reichelt et al. 2013). An advantage of lysosomal glycogen is that it can be mobilized independently or in parallel with cytosolic glycogen by a distinct set of cellular stimuli. For example, glycophagy could be specifically regulated by lysosomal uptake of Ca2+, which enhances GAA activity (Kotoulas et al. 2006; Delbridge et al. 2015). Very little is known about the role of glycophagy in brain tissue, besides the report of glycogenosomes in the aging brain (Cavanagh and Jones 2000). It has been suggested that glycogen of large molecular weight (i.e. α particles) is specifically degraded in lysosomes, and proceeds by a random, rather than an ordered degradation mechanism (Geddes and Stratton 1977a; Devos and Hers 1980). Based on glycogen degradation patterns it has been speculated that brain glycogen is degraded primarily by phosphorolysis and not by lysosomal hydrolysis (Chee et al. 1983).
2.6. Glycogen ultrastructure and subcellular distribution
In over a century of glycogen-related research many researchers have debated the existence of multiple forms of glycogen that differ in size, structure, associated proteins, and metabolic activity. The distinction between glycogen α and β particles came into focus with studies using the electron microscope and ultracentrifugation, and some progress has been made on defining the nature of these particles. Acid-soluble and acid-insoluble fractions of glycogen have also been observed, but the physiological relevance of these distinct pools has been highly debated. These topics, as well as a possible role for glycogen metabolic enzymes in the nucleus, are discussed in the next sections.
2.6.1. Glycogen α, β, and γ particles
Drochmans and colleagues were the first to describe the various levels of glycogen structure in native glycogen isolated from liver and muscle, and their observations were corroborated by others (Drochmans 1962; Revel 1964; Wanson and Drochmans 1968; Ryu et al. 2009). The largest glycogen particle, named the α particle, is a rosette-shaped association of several smaller units, known as β particles, that can reach up to 300 nm in diameter and over 108 Daltons in molecular weight (Gilbert and Sullivan 2014; Prats et al. 2018). Glycogen β particles typically range in size from 10–44 nm and have a molecular weight of 106-107 Daltons (Shearer and Graham 2002; Sullivan et al. 2012). It is generally accepted that a β particle corresponds to a single molecular unit of glycogen that can reach up to 12 tiers as modeled by Whelan and others (See Section 2.3.1) (Fig. 2.6c). Drochmans designated the 3 nm filaments making up the β particles as γ particles (Drochmans 1962). However, ‘γ particle’ has also been used to refer to protein-rich subunits on the glycogen β particles (see Section 2.7) (Prats et al. 2018). Generally speaking, glycogen is primarily stored as α particles (or a mixture of α and β particles) in the liver and as β particles in muscle, brain, and other tissues (reviewed below). Recent work has demonstrated a mixture of α and β particles in cardiac tissue (Besford et al. 2012). It is apparent that these norms become disrupted in pathological conditions.
It is estimated that liver α particles contain, on average, 20 to 40 β particle subunits (Devos et al. 1983). Liver glycogen is typically cytoplasmic and often reported to closely associate with smooth endoplasmic reticulum (Bruni and Porter 1965; Cardell and Cardell 1990). In muscle, β particles are associated with sarcoplasmic reticulum, concentrated either in subsarcolemmal regions, between myofibrils, or within the myofibrils; these pools respond differently to metabolic cues (Graham et al. 2010). Aggregates up to 60 nm in diameter, apparently composed of 4–6 β particles, were also observed via EM by Wanson and Drochmans in muscle, and others have reported α particles in rat muscle and insect flight muscle (Wanson and Drochmans 1968; Calder 1991). In skin and adipose tissue, β particles appear as cytoplasmic granules interspersed between ribonucleoprotein particles (Napolitano and Fawcett 1958; Hashimoto and DiBella 1967). In adipose tissue, they are only prominent after refeeding following a fast (Eichner 1984). In leukocytes and thymus, β particles are scattered throughout the cytoplasm and are fairly homogenous in size (Scott and Still 1968; Salmoral et al. 1990). In the retina, cytoplasmic β particles and occasional small α particles (60 nm) have been observed in the cone, but not rod, photoreceptor cells, and in Müller cells (Okubo et al. 1998).
In the brain, glycogen exists primary as cytoplasmic β particles in the cell bodies and processes of astrocytes (Maxwell and Kruger 1965; Sotelo and Palay 1968; Phelps 1975; Cataldo and Broadwell 1986). Normally, they are sparse to nonexistent in neurons, microglia, or oligodendrocytes, but β particles have appeared in neuronal axons and dendrites following fasting or trauma (Phelps 1975; Cataldo and Broadwell 1986). Astroglial β particles become apparent in early embryos and increase in both abundance and diameter with development (Gadisseux and Evrard 1985). Small α particles (80–100 nm in diameter, composed of 4–8 β particle subunits by EM) have also been reported in rat astrocytes (Sotelo and Palay 1968). Biochemical studies of glycogen isolated via a mild cold-water extraction method show significant heterogeneity in molecular weight: a fraction of particles had molecular weights suggestive of small α-particles composed of 3–6 β particle subunits (Chee et al. 1983). In several cases of human glycogenoses, small (60–120 nm in diameter) and large (150–350 nm in diameter) α particles have been reported in astrocytes or neurons (Résibois-Grégoire and Dourov 1966; Kornfeld and LeBaron 1984; Towfighi et al. 1989). Aggregates of α or β particles have also been reported in Alzheimer’s disease and the aging brain (Gertz et al. 1985; Mann et al. 1987), which are distinct from polyglucosan bodies (Cavanagh 1999). Reactive astrocytes are known to accumulate glycogen β particles in response to various pathological stimuli, including: brain infarction (Kajihara et al. 2001), methionine sulfoxide (Phelps 1975; Delorme and Hevor 1985), X-ray irradiation (Maxwell and Kruger 1965), and barbituates (Phelps 1975). Occasionally, intramitochondrial glycogen has also been observed in rat retinal cells (Ishikawa and Pei 1965), in the human and canine myocardium (Buja et al. 1972; Maron and Ferrans 1975), and in astrocytes (Sotelo and Palay 1968; Alaraj et al. 2004).
β particle size exists around a normal distribution, with most granules being 20–30 nm (Calder 1991; Marchand et al. 2002) (Fig. 2.9a). Since each additional tier is estimated to contribute 3.8 nm to the diameter (Melendez-Hevia et al. 1993; Marchand et al. 2002), the observed size distribution implies that most β particles have only about 7–9 tiers (Fig. 2.9a,b,c). It is still not clear why most β particles do not reach the maximum diameter, since such large granules can store a significantly greater amount of glucose (Graham et al. 2010). A growing body of evidence suggests that glycogenin regulates β particle size. In cell culture and in tissue, glycogenin does not appear to exist in a free form; it is always attached to glycogen (Alonso et al. 1995; Skurat et al. 1997; Testoni et al. 2017). Overexpression of glycogenin in cell culture did not change total glycogen levels, but it increased the total number of glycogen molecules, which were of a smaller size (Skurat et al. 1997). The Guinovart group showed that mice lacking glycogenin display reduced exercise endurance and an accumulation of glycogen particles in muscle that were larger than those in wild-type mice (Testoni et al. 2017). Also, crystal structures and data from the Sicheri lab suggest glycogen particle size is regulated by the length of the linker between the glycogenin catalytic domain and the region that binds to glycogen synthase; this region is alternatively spliced and varies in length across species (Zeqiraj et al. 2014; Zeqiraj and Sicheri 2015).
Fig. 2.9. Size and structure of glycogen α and β particles, proglycogen and macroglycogen.
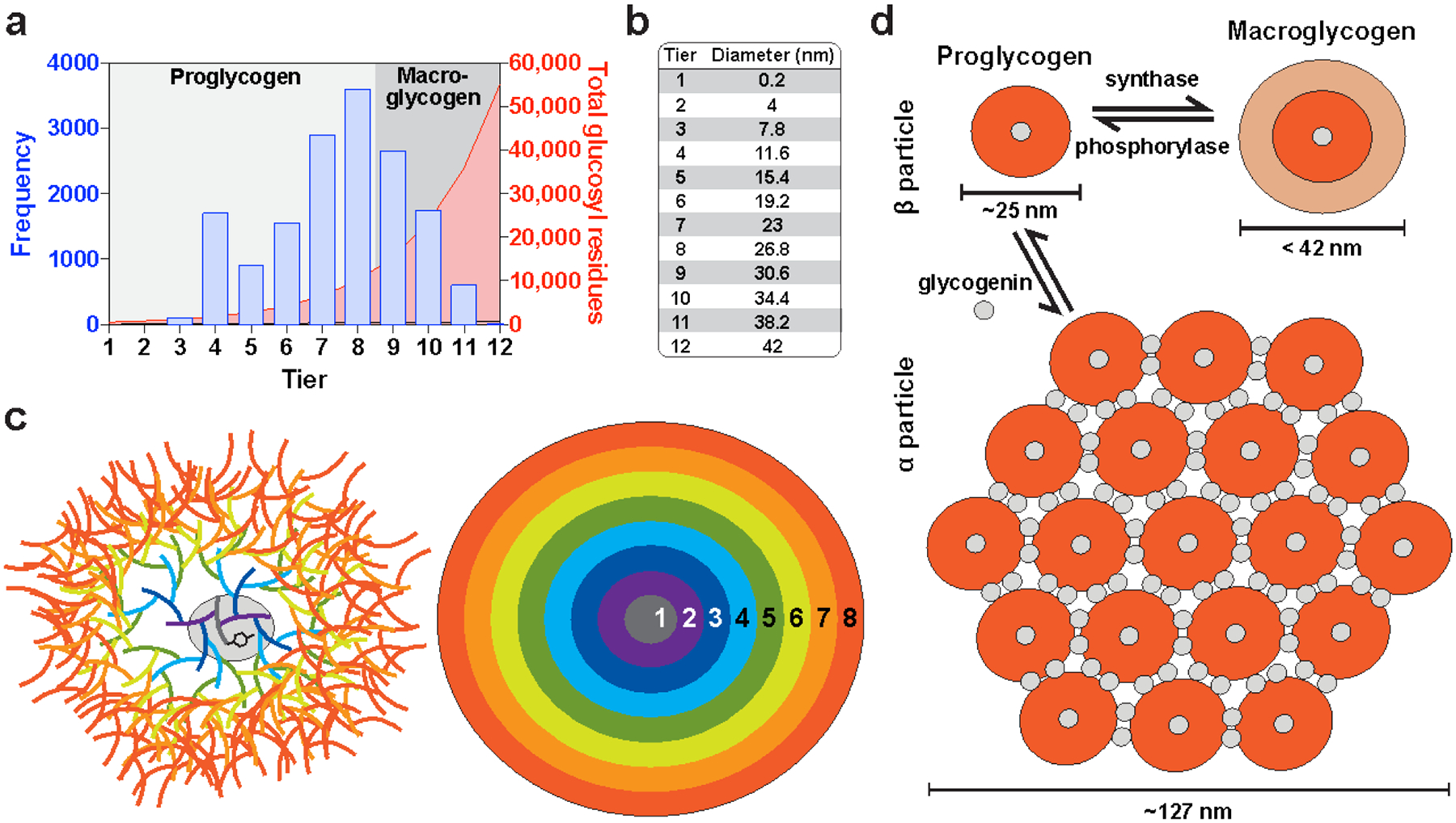
(a) Distribution of β glycogen particle size in muscle (Marchand et al. 2002) compared to the estimated number of glucosyl residues per glycogen molecule (Shearer and Graham 2002). The theoretical threshold between proglycogen and macroglycogen is after tier 8. (b) Estimated diameter of the tiers of glycogen particles. (c) Schematic diagrams of an 8-tiered glycogen molecule based on the studies of Whelan and mathematical modeling of (Melendez-Hevia et al. 1993). (d) A possible relationship between proglycogen and macroglycogen and α and β particles and approximated sizes. Grey circles represent glycogenin molecules. Diagram of α particle was adapted from (Tan et al. 2018).
In healthy organisms, α particles seem to be a unique feature of the liver. It is well established that total liver glycogen depletes with fasting and exceeds basal levels upon refeeding, a phenomenon known as glycogen super-compensation that is also observed in muscle and brain (Nilsson and Hultman 1973; Terjung et al. 1974; Matsui et al. 2012). Additionally, hepatic glycogen levels and the activities of glycogen metabolizing enzymes undergo a diurnal rhythm (Halberg et al. 1960; Roesler and Khandelwal 1985). The Gilbert lab analyzed glycogen particles by size exclusion chromatography and EM and proposed a model for diurnal glycogen cycling in liver based on their data and earlier work (Sullivan et al. 2014). Starved livers maintain low glycogen in the form of small β particles. Upon refeeding, glycogen is quickly synthesized on these preexisting β particles, which provide optimal surface-to-volume ratio for rapid resynthesis. Once glycogen levels peak, the β particles are assembled into α particles. The α particles persist as glycogen levels begin to gradually fall, but they are gradually disassembled into β particles, which are preferentially degraded. Thus it appears that the primarily role of α particles is to facilitate a slower release of glucose than β particles (Chandramouli et al. 2015). This indeed seems optimal for the organ whose primary role is to maintain blood glucose homeostasis. Additionally, α particles in diabetic livers are more fragile than those of healthy mice, suggesting they could more easily be broken into β particles and contribute to hyperglycemia (Deng et al. 2015; Hu et al. 2018).
The nature of α particle assembly has remained elusive for decades. Drochmans observed that purified α particles could be dissociated into β particles under acidic conditions (Drochmans 1962). Lazarow, Orrell and Bueding previously showed using ultracentrifugation that acid, alkali and heat drastically changed the sedimentation of liver glycogen, suggesting these treatments disrupt α particle structure (Lazarow 1942; Orrell and Bueding 1964). The Gilbert lab has devoted a significant body of work to elucidating the nature of the α particle bond due to its relevance to diabetes (Sullivan et al. 2010; Sullivan et al. 2012; Deng et al. 2015; Deng et al. 2016; Tan et al. 2018). The lack of significant dissociation of normal α particles in dimethyl sulfoxide, 2-mercaptoethanol, or sodium dodecyl sulfate suggested the association is not through hydrogen or disulfide bonds or weak protein-protein interactions (Sullivan et al. 2010; Sullivan et al. 2012). It was reported that unexpectedly high levels of glycogenin were found in a proteomic study of liver glycogen, suggesting that glycogenin was present on the granule surface in addition to the granule core (Stapleton et al. 2010). The Gilbert lab then used proteomics to identify glycogenin as the molecular “glue” holding α particles together (Tan et al. 2018) (Fig. 2.9d).
2.6.2. Proglycogen and macroglycogen
There is a lot of controversy surrounding proglycogen and macroglycogen, which have gone by different terms over the years and have been discussed in great detail by others (Lomako et al. 1993a; Alonso et al. 1995; Rybicka 1996; Shearer and Graham 2002). Although the precise nature of these pools is debated, there does appear to be sufficient evidence for their distinct physiological roles in glycogen turnover (Shearer and Graham 2002; Graham et al. 2010). The relationship between proglycogen/macroglycogen and α/β particles is not completely clear as these distictions are discussed seperately. However, since they are relevant to tissues generally lacking α particles, proglycogen and macroglycogen likely represent two states of β particle synthesis (Alonso et al. 1995).
When the TCA extraction method was first introduced for the purification of glycogen, it was observed that a portion of the glycogen was resistant to extraction by cold TCA (Willstätter and Rohdewald 1934). It could not be released unless the tissue was treated with heat, alkali, or protease, so it was concluded that this resistant fraction was attached to protein, and the two fractions were called lyo- (“free”) and desmo- (“fixed”) glycogen (Willstätter and Rohdewald 1934; Van Heijningen and Kemp 1955). Such observations also gave rise to the terms acid-soluble, i.e. extractable, glycogen, and acid-insoluble, i.e. residual, glycogen (Russell and Bloom 1955; Stetten and Stetten 1960). In a 1960 literature review, Stetten and Stetten noted that while the quantity of free glycogen was widely variable with nutritional state, the quantity of fixed glycogen remained constant. However, experiments suggested the latter was more metabolically active, since injected [14C]glucose was more readily incorporated into fixed glycogen (Stetten and Stetten 1960). For decades, researchers continued to debate whether such observations were artefactual, merely reflecting the association of known metabolic enzymes with glycogen (Whelan 1976). Whelan revisited the subject in the late 20th century while investigating the origin of glycogen synthesis. Using cultured primary astrocytes, his group showed that fixed glycogen, which was re-named proglycogen, contained 10% protein by weight and was rapidly resynthesized upon refeeding after glucose starvation, reaching a size of 400,000 Da. The proglycogen was later converted to macroglycogen, as demonstrated by pulse-chase labeling (Lomako et al. 1993a). The group proposed that proglycogen was an intermediate between glycogenin and the mature glycogen granule (Lomako et al. 1993a; Alonso et al. 1995). Resynthesis of proglycogen prior to macroglycogen following exhaustive exercise was also confirmed in vivo in humans (Adamo 1998).
It has been suggested that the upper limit of proglycogen size corresponds to a diameter of ~30 nm, a molecule of approximately 8 tiers; anything larger would be considered macroglycogen (Melendez et al. 1997; Marchand et al. 2002; Shearer and Graham 2002) (Fig. 2.9a,d). However, Whelan’s group estimated that the upper limit of proglycogen was 400 kDa (Lomako et al. 1993a; Alonso et al. 1995), which corresponds to a glycogen molecule with ~2500 glucose residues, i.e. approximately 5 tiers. Whelan’s group also suggested that proglycogen and macroglycogen are synthesized by different enzymes, however it may be that the different activities associated with the pro- and macroglycogen fractions both correspond to glycogen synthase. Two independent groups have demonstrated that glycogen synthase in the proglycogen fraction has a higher affinity for UDP-glucose than synthase in the macroglycogen fraction (Curtino and Lacoste 2000, Tavridu 2003). Curtino and Lacoste proposed that the insolubility of proglycogen in TCA was due to its heavy association with glycogen synthase, not with glycogenin, as Whelan had proposed (Curtino and Lacoste 2000). Tavridu and Agnius showed that the hormone insulin increased the association of glycogen synthase with the proglycogen fraction, and glucagon had the opposite effect (Tavridou and Agius 2003). Insulin is known to stimulate dephosphorylation (and activation) of glycogen synthase, and the effects of both hormones are well known to regulate glycogen synthesis through signaling cascades, so the difference in synthase activity may be due to its phosphorylation state. Indeed, phosphorylation-dependent stimulation of glycogen synthase and phosphorylase has been shown to induce translocation of these proteins to actin-rich structures proximal to sarcoplasmic reticulum in skeletal muscle (Prats et al. 2005; Prats et al. 2009). In liver, newly synthesized glycogen has also been reported to be associated with a protein backbone (Geddes and Stratton 1977b) and smooth endoplasmic reticulum (Cardell and Cardell 1990). Additionally, glycogenin has been shown to bind to actin (Baqué et al. 1997). In vitro studies suggest that glycogen synthase is more active when it is bound to glycogenin (Pitcher et al. 1987; Pitcher et al. 1988). When glycogen content decreases, glycogen synthase has been shown to translocate from a glycogen-enriched membrane fraction to the cytoskeleton, concurrent with a decrease in activity (Nielsen et al. 2001). In isolated hepatocytes, glucose stimulated the cytoskeleton-dependent translocation of synthase from the cytosol to the plasma membrane, and newly synthesized glycogen molecules always appeared first at the cell membrane, but moved inward as they were replaced by newer molecules (García-Rocha et al. 2001; Fernández-Novell et al. 2002; Ferrer et al. 2003).
Proglycogen may represent a membrane-associated and/or actin-bound form of glycogen that is primed and ready for active anabolism/catabolism. When it grows to a certain size, it becomes macroglycogen, synthesis levels off, and it is released into the cytosol. A similar model has been proposed recently (Prats et al. 2018) and prompts further discussion of glycogen and its associated proteins as a dynamic subcellular entity rather than a static molecule (See Section 2.7) (Rybicka 1996). It is still not clear whether synthesis and degradation can occur simultaneously on the same granule, but proximity of these activities is suggested (Prats et al. 2005). The spatiotemporal regulation of glycogen metabolism is an interesting area of research requiring further elucidation.
2.6.3. Nuclear glycogen
Dozens of microscopy studies have reported intranuclear glycogen deposits, most often in pathological conditions of the liver (Chipps and Duff 1942; Sheldon et al. 1962; Tanikawa 1965). In a 1975 case report, Ferrans et al. beautifully reviewed 24 prior studies on intranuclear glycogen either in the context of diabetes, glycogen storage diseases, or as an incidental feature in healthy humans and animals (Ferrans et al. 1975). In these reports, nuclear glycogen was usually in the form of β particles, sometimes dispersed, other times clustered; occasionally a few α particles were reported. The findings of Ferrans et al. were unusual in that nuclear glycogen was observed in the diseased myocardium. Intranuclear glycogen was only found in 6 out of 90 patients with cardiac diseases, and only in a few nuclei per patient. Most of the glycogen was present as β particles, often aggregated and filling the entire nucleoplasm. The presence of nuclear glycogen was not associated with any cellular damage. The authors proposed three mechanisms for the formation of nuclear glycogen, but shrewdly concluded that only one was possible: (1) there was no ultrastructural evidence supporting phagocytosis or endocytosis of glycogen particles by the nuclear membrane; (2) glycogen particles were too large to pass through the nuclear pore complex; (3) the glycogen must be synthesized in situ, which had been previously demonstrated in normal Müller cells of the rat retina (Amemiya 1970) and in Novikoff hepatoma cells (Karasaki 1971). Ferrans et al. proposed that conditions favoring increased permeability of the nuclear membrane facilitated the entry of the metabolic enzymes into the nucleus, but that they became trapped when permeability returned to normal. This implies that atypical trapping of glycogenic enzymes into the nucleus may result in excessive levels of glycogen. Nuclear α and β particles have also been reported in the context of various cancers: human gastric adenocarcinoma (Ohyumi and Takano 1977), sublines of the Ehrlich-Lettré mouse ascites tumor (Kopun et al. 1989), chicken sarcoma (Binggeli 1959) and human and mouse hepatoma (Leduc and Wilson 1959; Ghandially and Parry 1966). Nuclear glycogen synthase activity was also detected in the Ehrlich-Lettré subline (Granzow et al. 1981; Kopun et al. 1982). In the brain, intranuclear glycogen deposits have been observed in the abnormal astrocytes of Alzheimer’s disease (Horita et al. 1981; Miyakawa et al. 1982) and in the pituicytes of aged rats (Lafarga et al. 1991). Nuclear polyglucosan was also reported in a neuronal cell culture model of GBE deficiency (Kakhlon et al. 2013).
In more recent years, Guinovart, Gentry and others have shown that nearly all the central glycogen metabolic enzymes can be found in the nucleus: glycogenin (Miozzo et al. 1996; Baqué et al. 1997), glycogen synthase (Ferrer et al. 1997; Cid et al. 2005; Vilchez et al. 2007) glycogen phosphorylase (Sun, Dukhande, and Gentry, unpublished results), and debranching enzyme (Cheng et al. 2007). Guinovart’s group published a series of studies demonstrating the translocation of the muscle isoform of glycogen synthase from the cytosol to the nucleus in conditions of glucose deprivation, which required glycogen binding (Ferrer et al. 1997; Cid et al. 2005; Díaz et al. 2011). They also showed that neurons display primarily nuclear localization of glycogen synthase, characteristic of cells lacking glycogen, in contrast to glycogen-rich astrocytes with cytosolic synthase (Vilchez et al. 2007). Additionally, GDE and glucokinase, a key glucose sensing enzyme, also shuttle to the nucleus upon glucose deprivation (Fernández-Novell et al. 1999; Cheng et al. 2007). In contrast, glucose starvation has an opposite effect on the gluconeogenic enzyme fructose bisphosphatase, inducing its translocation to the cytosol (Yáñez et al. 2004). It has been suggested that the nuclear sequestration of these enzymes is yet another way to regulate their activity, preventing them from acting inappropriately in times of energy stress (Jurczak et al. 2008). Laforin and AMPK, which both contain CBMs and bind to glycogen in vitro and in cell culture, also translocate to the nucleus with glucose deprivation and glycogen depletion (Cheng et al. 2007; Singh et al. 2012) (Fig. 2.10a,b).
Fig. 2.10. Nuclear-cytoplasmic shuttling and colocalization of glycogen associated enzymes.
(a) In media containing glucose and metformin, laforin colocalizes with glycogen in Neuro2A cells (Singh et al. 2012). (b) When expressed in COS-7 cells, laforin and AMPK colocalize (with glycogen) in the cytosol in the presence of glucose, and translocate to the nucleus upon glucose starvation (Singh et al. 2012). For (a,b), scale bars = 10 μm. Copyright © 2012 American Society for Microbiology. Used with permission. (c) Immunolabeling of glycogen synthase, phosphorylase, and glycogen itself in tilapia gill sections (Chang et al. 2007). Scale bars = 20 μm. Copyright © Company of Biologists. Used with permission.
Nuclear-cytoplasmic shuttling has also been observed for yeast glycogen synthase. In a review discussing these data, it was postulated that “the freeing of glycogen synthase from its cytoplasmic tether to the glycogen particle as these stores reduced would therefore be a signal that carbon and energy reserves were low. The uptake of glycogen synthase into the nucleus might therefore represent a form of molecular ‘fuel gauge.’ It is possible that glycogen synthase could regulate transcription in response to energy availability by some as yet undetermined means” (Wilson et al. 2010). ‘Moonlighting’ of metabolic enzymes in the nucleus is not a new theme. Moonlighting proteins perform multiple unrelated functions, expanding the functional repertoire of the cell without expanding the number of genes, and the first to be described was the lens structural protein ε-crystallin, also known as lactate dehydrogenase (Hendriks et al. 1988). All essential glycolytic enzymes have been observed in the nucleus, as well as some mitochondrial proteins and enzymes involved in methylation and acetylation (Kim and Dang 2005; Boukouris et al. 2016). These energy-sensing proteins have been shown to regulate transcription by diverse means in a “metabolism-epigenetic axis” that is critical for cellular homeostasis (Boukouris et al. 2016). Although the epigenetic role of glycolytic proteins is well described, it is still not clear whether glycolysis occurs in the nucleus, although its products ATP and NADH could certainly be useful. Glycogen-associated enzymes may also have both an epigenetic and energy producing role, and perhaps excessive glycogen deposits result from misregulated nuclear sequestration. AMP-activated protein kinase (AMPK), a master regulator of cellular energy known to bind to and sense glycogen levels (McBride et al. 2009) (see Section 2.7), also translocates to the nucleus and regulates transcription and the circadian clock through phosphorylation (Leff 2003; Lamia et al. 2009).
2.7. The glycogen granule and its associated proteins
In 1968, Scott and Still recognized the dynamic nature of glycogen and introduced the term “glycosome” to refer to the molecule and its associated proteins, stating that the nature of glycogen as a cellular organelle had not been appreciated (Scott and Still 1968). Rybicka reintroduced the term in a detailed historical review of the evidence for such a classification (Rybicka 1996). Unfortunately, the term “glycosome” has been used far more frequently to refer to the glycolytic enzyme-containing peroxisomes of trypanosomatids, a family of unicellular parasites (Michels et al. 2006), which creates confusion. Also, in the strict definition, organelles are membrane-bound entities (Klausner et al. 1992; Mellman and Warren 2000), and glycogen is primarily cytosolic. Perhaps it is safest to simply recognize that a host of proteins are associated with the glycogen granule, its structure is complex and highly regulated, and it is a dynamic participant in cellular metabolism.
The γ particle named by Drochmans was a description of the ramifying fine fibers (3 nm in diameter) comprising β particle structure, likely corresponding to glucan chains (Drochmans 1962). Later authors came to identify protein-rich subdomains on the glycogen molecule as γ particles, which may be a misinterpretation (Prats et al. 2018). Protein is more electron dense than carbohydrate, so with lead and uranyl acetate staining, protein-rich regions appear as darkly stained clusters about 2–3 nm in diameter; alternatively, staining with the histochemical method of Thiéry using silver proteinate results in electron-dense clusters of the same size corresponding to vicinal glycols (Ferrans et al. 1973; Rybicka 1996; Prats et al. 2018). Rybicka refers to the electrodense vicinal glycols as γ particles, which is likely consistent with the meaning originally intended by Drochmans. Due to their very small size, both γ particles and protein-rich subdomains are only visible by EM. It is not yet clear why they exist as discrete, regular entities and which proteins correspond to the protein subdomains. However, it is well established that all of the primary glycogen metabolic enzymes (glycogenin, synthase, phosphorylase, GDE and GBE) bind to and colocalize with glycogen granules in cell culture and in vivo (Chang et al. 2007; Cheng et al. 2007; Vilchez et al. 2007; Stapleton et al. 2013; Zhai et al. 2016)(Fig. 2.10c, Fig. 2.11). In fact, due to its very tight association, glycogen synthase is frequently used as a marker for glycogen and polyglucosan bodies (Criado et al. 2012; Augé et al. 2018). There are a few antibodies that directly bind to the polysaccharide chains of glycogen, but they are not yet commercially available and are still being characterized (Nakamura-Tsuruta et al. 2012; Oe et al. 2016). Other proteins known to closely associate with the glycogen molecule all contain a glycogen-binding CBM: laforin and Stbd1 (Section 2.5), the β-subunit of AMPK, and the glycogen-targeting subunits of protein phosphatase 1 (discussed below) (Fig. 2.10b,c). Phosphorylase kinase also binds to glycogen (Chebotareva et al. 2009; Nadeau et al. 2018).
Fig. 2.11. Crystal structures and relative sizes of glycogen-associated proteins.
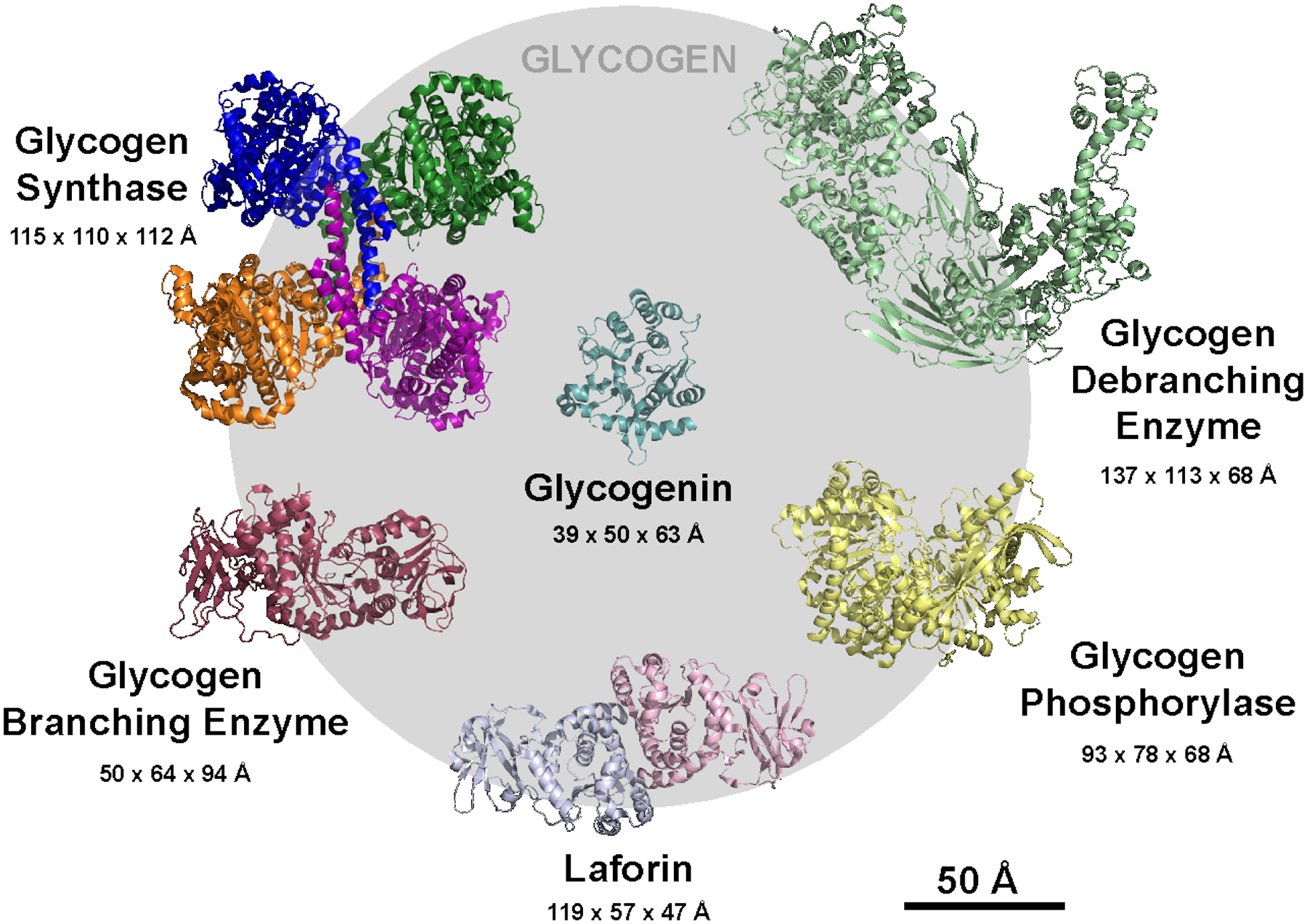
Crystal structures of yeast glycogen synthase (PDB: 3NAZ), yeast glycogen debranching enzyme (PDB:5D06), human branching enzyme (PDB: 4BZY), human brain glycogen phosphorylase (PDB: 5IKO), rabbit muscle glycogenin (PDB: 1LL2), and human laforin (PDB: 4RKK). Structures were superimposed manually in PyMol and are shown at equivalent zoom. The size of an average glycogen β particle (diameter = 250 Å) is shown for scale to illustrate the relative sizes of the proteins and glycogen particle. Crystal structure dimensions were calculated based on an inertia axis aligned bounding box in PyMol. In multimeric structures, individual subunits are colored separately.
Protein phosphatase 1 (PP1) catalyzes a vast array of dephosphorylation events in cells, regulation of which is achieved by diverse targeting subunits that bring the catalytic subunit (PP1c) into the vicinity of its target (Bollen et al. 2010; Heroes et al. 2013). Seven glycogen-targeting subunits of PP1 have been identified (PPP1R3A-G), but four have been most extensively characterized as primary regulators of glycogen synthesis (Newgard et al. 2000; Roach et al. 2012). GM (also known as RGL, encoded by PPP1R3A) expression is restricted to heart and skeletal muscle; GL (encoded by PPP1R3B) is primarily expressed in liver; and Protein Targeting to Glycogen (PTG, also known as R5, encoded by PPP1R3C) and R6 (encoded by PPP1R3D) are more ubiquitously expressed, with enrichment in glycogen-rich tissues (Roach et al. 2012; www.proteinatlase.org). All these subunits possess both a PP1-binding domain and a putative CBM21 and recruit PP1 to glycogen granules (Newgard et al. 2000; Christiansen et al. 2009; www.cazy.com). Dephosphorylation by PP1 has opposing effects on synthase (activating) and phosphorylase (inhibitory), leading to net glycogen synthesis (Newgard et al. 2000; Greenberg et al. 2006; Vilchez et al. 2007). Overexpression of PTG, GM, GL, and R6 leads to glycogen accumulation in cell culture and mouse models (Printen et al. 1997; Gasa et al. 2000; Greenberg et al. 2006; Worby et al. 2008; Montori-Grau et al. 2011; Duran et al. 2014).
AMPK, a cellular energy sensor with multiple intracellular targets, is also considered an important glycogen sensor (Hardie 2014). AMPK is activated by elevated levels of AMP relative to ATP reflecting energy deficits. It is a heterotrimeric complex composed of a catalytic α subunit and regulatory β and γ subunits; the β subunit contains a CBM48 and targets AMPK to glycogen granules (Polekhina et al. 2003; McBride et al. 2009; Janecek et al. 2011). AMPK phosphorylates both isoforms of glycogen synthase, inhibiting its activity (Bultot et al., 2012; Jørgensen et al., 2004). It has been speculated that recruitment of activated AMPK to glycogen granules would lead to decreased glycogen synthesis. There is also significant evidence for regulation of AMPK activity by glycogen via the β subunit (Hardie and Sakamoto 2006). AMPK is particularly inhibited by the presence of a single α−1,6 linked glucose unit or by a glycogen molecule that has been partially degraded by phosphorylase (McBride et al. 2009; Koay et al. 2010). A single glucose unit at a branch point could be exposed during debranching, since GDE is likely to dissociate from glycogen between its transferase and glucosidase activities (Zhai et al. 2016). Thus, glycogen, especially when being actively degraded, is a potent inhibitor of AMPK, and there is likely to be reciprocal regulation between glycogen and AMPK.
Proteomic studies have been performed on glycogen isolated from mouse and rat liver and the mouse adipocyte cell line 3T3-L1 (Stapleton et al. 2010; Stapleton et al. 2013). Synthase, phosphorylase, GBE, GDE, and glycogenin were all abundant, as well as PP1 regulatory subunits. Stbd1, laforin, and proteins associated with endoplasmic reticulum were only found in the hepatic proteome; lysosomal α amylase was identified only in the 3T3-L1 proteome, indicative of cell-specific differences in glycogen metabolism. The lack of AMPK or PhK binding was consistent with earlier studies and attributed to the stringency of the isolation procedure and the nutritional state of the liver (Parker et al. 2007). A number of proteins associated with mitochondria, ribosomes and nuclei were also identified in lower abundance, suggestive of both a heterogeneous subcellular distribution and the link between glycogen and a variety of cellular processes, as has been discussed in previous sections.
It should be noted that many structures of glycogen-associated enzymes have been determined, and they are quite large relative to the size of the glycogen granule (Fig. 2.11). The dimeric structure of rabbit muscle glycogenin is ~80 kDa in solution and the crystal structure of the monomer is 63 Å at its widest point (Gibbons et al. 2002). This corresponds to one fourth of the diameter of the average glycogen β particle (25 nm, i.e. 250 Å). Yeast glycogen synthase is tetrameric, measuring 115 Å at its widest point, approximately half the diameter of a glycogen granule; each subunit is about 90 Å long (Baskaran et al. 2010). Monomeric human brain phosphorylase is 78 Å long, but it also is known to function as a dimer (Mathieu et al. 2016). Yeast GDE and human GBE monomers are 137 Å and 94 Å long, respectively (Froese et al. 2015; Zhai et al. 2016). The elongated human laforin dimer measures 119 Å in length both in crystals and in solution (Raththagala et al. 2015). PhK and AMPK are both very large multisubunit complexes (Hardie 2014; Nadeau et al. 2018). Although a complete structure of AMPK is not yet available, structures of the PhK holoenzyme and PhK in complex with phosphorylase determined by cryo-EM reveal that these complexes are 270 Å by 225 Å and 310 Å by 250 Å, respectively, roughly the same dimensions as glycogen β particles (Vénien-Bryan et al. 2009). With this scale in mind, one can begin to envision that the surface of glycogen molecules is likely to be very crowded, and enzymes may compete for access to glucan chains. Molecular crowding has been shown to influence the interaction of PhK with phosphorylase and with the glycogen molecule (Chebotareva et al. 2009). Glycogen and its associated proteins may function as molecular scaffolds, binding to one another and recruiting a variety of enzymes that respond to glycogen levels and regulate energy homeostasis.
2.8. The future of brain glycogen research
The groundbreaking discoveries of the 20th century greatly expanded our understanding of glycogen metabolism and monumentally impacted the broader field of biochemistry. As a result, there is a widespread belief that glycogen metabolism is an antiquated area of research that requires no further investigation. However, a milieu of interesting questions remain unresolved regarding glycogen, particularly on its complex yet elusive role in the mammalian brain. Defects in glycogen metabolism are a hallmark of many diseases, including neurodegenerative diseases. The advent of new technologies make it possible to better study brain glycogen and will most certainly lead to a better understanding of its architecture and metabolism in a variety of contexts.
References
- Adeva-Andany MM, González-Lucán M, Donapetry-García C, Fernández-Fernández C, Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin. 2016;5:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado C, Sarkar S, Korolchuk VI, Criado O, Vernia S, Boya P et al. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet. 2010;19(14):2867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaraj M, Gajkowska B, Cholewinski M, Lazarewicz JW. Hyperglycaemia and intramitochondrial glycogen granules in the brain of mice. Ultrastructural study. Folia neuropathologica. 2004;42(2):113–8. [PubMed] [Google Scholar]
- Alberini CM, Cruz E, Descalzi G, Bessières B, Gao V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. Glia. 2018;66(6):1244–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso MD, Lomako J, Lomako WM, Whelan WJ. Tyrosine-194 of glycogenin undergoes autocatalytic glucosylation but is not essential for catalytic function and activity. FEBS letters. 1994;342(1):38–42. [DOI] [PubMed] [Google Scholar]
- Alonso MD, Lomako J, Lomako WM, Whelan WJ. A new look at the biogenesis of glycogen. The FASEB journal. 1995;9(12):1126–37. [DOI] [PubMed] [Google Scholar]
- Amemiya T. INTRANUCLEAR POLYSACCHARIDE SYNTHESIS IN MÜLLER’S CELLS OF THE NORMAL RAT RETINA. Acta histochemica et cytochemica. 1970;3(2):41–51. [Google Scholar]
- Andrade DM, Ackerley CA, Minett TSC, Teive HAG, Bohlega S, Scherer SW et al. Skin biopsy in Lafora disease: genotype–phenotype correlations and diagnostic pitfalls. Neurology. 2003;61(11):1611–4. [DOI] [PubMed] [Google Scholar]
- Archibald AR, Fleming ID, Liddle AM, Manners DJ, Mercer GA, Wright A. 232. α−1, 4-Glucosans. Part XI. The absorption spectra of glycogen–and amylopectin–iodine complexes. Journal of the Chemical Society (Resumed). 1961:1183–90. [Google Scholar]
- Augé E, Pelegrí C, Manich G, Cabezón I, Guinovart JJ, Duran J et al. Astrocytes and neurons produce distinct types of polyglucosan bodies in Lafora Disease. Glia. 2018;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JM, Whelan WJ. Physical properties of starch I. Relationship between iodine stain and chain length. Journal of Biological Chemistry. 1961;236(4):969–73. [PubMed] [Google Scholar]
- Bak LK, Walls AB, Schousboe A, Waagepetersen HS. Astrocytic glycogen metabolism in the healthy and diseased brain. J Biol Chem. 2018;293(19):7108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft JD, Gamble M. Theory and Practice of Histological Techniques. Churchill Livingstone; 2008. [Google Scholar]
- Banks W, Greenwood CT, Khan KM. The interaction of linear, amylose oligomers with iodine. Carbohydrate Research. 1971;17(1):25–33. [Google Scholar]
- Baqué S, Guinovart JJ, Ferrer JC. Glycogenin, the primer of glycogen synthesis, binds to actin. FEBS letters. 1997;417(3):355–9. [DOI] [PubMed] [Google Scholar]
- Barros LF. Metabolic signaling by lactate in the brain. Trends in neurosciences. 2013;36(7):396–404. [DOI] [PubMed] [Google Scholar]
- Baskaran S, Roach PJ, DePaoli-Roach AA, Hurley TD. Structural basis for glucose-6-phosphate activation of glycogen synthase. Proceedings of the National Academy of Sciences. 2010;107(41):17563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates FL, French D, Rundle RE. Amylose and amylopectin content of starches determined by their iodine complex formation. Journal of the American Chemical Society. 1943;65(2):142–8. [Google Scholar]
- Bawn CEH, Hirst EL, Young GT. The nature of the bonds in starch. Transactions of the Faraday Society. 1940;36:880–5. [Google Scholar]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. W.H. Freeman; 2002.
- Bernard C. Sur une nouvelle fonction du foie chez l’homme et les animaux. Institut de France; 1850. [Google Scholar]
- Bernard C. Sur le mécanisme physiologique de la formation du sucre dans le foie. C R Acad Sci (Paris). 1857;44:578–86. [Google Scholar]
- Bertoft E. Understanding Starch Structure: Recent Progress. Agronomy. 2017;7(3). [Google Scholar]
- Besford QA, Sullivan MA, Zheng L, Gilbert RG, Stapleton D, Gray-Weale A. The structure of cardiac glycogen in healthy mice. International journal of biological macromolecules. 2012;51(5):887–91. [DOI] [PubMed] [Google Scholar]
- Binggeli MF. Abnormal intranuclear and cytoplasmic formations associated with a chemically induced, transplantable chicken sarcoma. The Journal of biophysical and biochemical cytology. 1959;5(1):143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow A, Bay-Smidt AM, Olsen CE, Møller BL. Analysis of starch-bound glucose 3-phosphate and glucose 6-phosphate using controlled acid treatment combined with high-performance anion-exchange chromatography. Journal of Chromatography A. 1998;829(1–2):385–91. [Google Scholar]
- Blennow A, Bay-Smidt AM, Olsen CE, Møller BL. The distribution of covalently bound phosphate in the starch granule in relation to starch crystallinity. International journal of biological macromolecules. 2000;27(3):211–8. [DOI] [PubMed] [Google Scholar]
- Blennow A, Engelsen SB. Helix-breaking news: fighting crystalline starch energy deposits in the cell. Trends Plant Sci. 2010;15(4):236–40. [DOI] [PubMed] [Google Scholar]
- Bollen M, Peti W, Ragusa MJ, Beullens M. The extended PP1 toolkit: designed to create specificity. Trends in biochemical sciences. 2010;35(8):450–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boukouris AE, Zervopoulos SD, Michelakis ED. Metabolic enzymes moonlighting in the nucleus: metabolic regulation of gene transcription. Trends in biochemical sciences. 2016;41(8):712–30. [DOI] [PubMed] [Google Scholar]
- Brown AM. Brain glycogen re-awakened. J Neurochem. 2004;89(3):537–52. [DOI] [PubMed] [Google Scholar]
- Brown BI, Brown DH, Jeffrey PL. Simultaneous absence of α−1, 4-glucosidase and α−1, 6-glucosidase activities (pH 4) in tissues of children with type II glycogen storage disease. Biochemistry. 1970;9(6):1423–8. [DOI] [PubMed] [Google Scholar]
- Brown DH, Brown BI. Action of a muscle branching enzyme on polysaccharides enlarged from UDP [14C] glucose. Biochimica et Biophysica Acta (BBA)-General Subjects. 1966;130(1):263–6. [Google Scholar]
- Brown DH, Illingworth B. The properties of an oligo-1, 4→ 1, 4-glucantransferase from animal tissues. Proceedings of the National Academy of Sciences. 1962;48(10):1783–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruni C, Porter KR. The fine structure of the parenchymal cell of the normal rat liver: I. General observations. The American journal of pathology. 1965;46(5):691. [PMC free article] [PubMed] [Google Scholar]
- Buja LM, Ferrans VJ, Levitsky S. Occurrence of intramitochondrial glycogen in canine myocardium after prolonged anoxic cardiac arrest. Journal of molecular and cellular cardiology. 1972;4(3):237–42. [DOI] [PubMed] [Google Scholar]
- Buleon A, Colonna P, Planchot V, Ball S. Starch granules: structure and biosynthesis. Int J Biol Macromol. 1998;23(2):85–112. [DOI] [PubMed] [Google Scholar]
- Burnett G, Kennedy EP. The enzymatic phosphorylation of proteins. Journal of Biological Chemistry. 1954;211(2):969–80. [PubMed] [Google Scholar]
- Butler NA, Lee EYC, Whelan WJ. A protein-bound glycogen component of rat liver. Carbohydrate research. 1977;55(1):73–82. [DOI] [PubMed] [Google Scholar]
- Calder PC. Glycogen structure and biogenesis. International journal of biochemistry. 1991;23(12):1335–52. [DOI] [PubMed] [Google Scholar]
- Campbell JA, Davies GJ, Bulone V, Henrissat B. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochemical Journal. 1997;326(Pt 3):929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Mahrenholz AM, DePaoli-Roach AA, Roach PJ. Characterization of rabbit skeletal muscle glycogenin. Tyrosine 194 is essential for function. Journal of Biological Chemistry. 1993a;268(20):14687–93. [PubMed] [Google Scholar]
- Cao Y, Skurat AV, DePaoli-Roach AA, Roach PJ. Initiation of glycogen synthesis. Control of glycogenin by glycogen phosphorylase. Journal of Biological Chemistry. 1993b;268(29):21717–21. [PubMed] [Google Scholar]
- Caputto R, Leloir LF, Cardini CE, Paladini AC. Isolation of the coenzyme of the galactose phosphate-glucose phosphate transformation. Journal of Biological Chemistry. 1950;184(1):333–50. [PubMed] [Google Scholar]
- Cardell RR, Cardell EL. Heterogeneity of glycogen distribution in hepatocytes. Journal of electron microscopy technique. 1990;14(2):126–39. [DOI] [PubMed] [Google Scholar]
- Carpenter S, Karpati G, Andermann F, Jacob JC, Andermann E. Lafora’s disease: peroxisomal storage in skeletal muscle. Neurology. 1974;24(6):531–8. [DOI] [PubMed] [Google Scholar]
- Carpenter WB, Smith FG, Meneses HP. Principles of Human Physiology. Henry C. Lea; 1876. [Google Scholar]
- Carr C. Practical Physiological Chemistry. By Philip B. Hawk, Bernard L. Oser, and William H. Summerson. The Journal of Physical and Colloid Chemistry. 1947;51(5):1214–5. [Google Scholar]
- Carter SH, Stone WE. Effect of convulsants on brain glycogen in the mouse. Journal of neurochemistry. 1961;7(1):16–9. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions: I. Neurons and glia. Journal of Electron Microscopy Technique. 1986;3(4):413–37. [DOI] [PubMed] [Google Scholar]
- Cavanagh JB. Corpora-amylacea and the family of polyglucosan diseases. Brain Res Brain Res Rev. 1999;29(2–3):265–95. [DOI] [PubMed] [Google Scholar]
- Cavanagh JB, Jones HB. Glycogenosomes in the aging rat brain: their occurrence in the visual pathways. Acta neuropathologica. 2000;99(5):496–502. [DOI] [PubMed] [Google Scholar]
- Chaikuad A, Froese DS, Berridge G, von Delft F, Oppermann U, Yue WW. Conformational plasticity of glycogenin and its maltosaccharide substrate during glycogen biogenesis. Proceedings of the National Academy of Sciences. 2011;108(52):21028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Ackerley CA, Lohi H, Ianzano L, Cortez MA, Shannon P et al. Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum Mol Genet. 2004;13(11):1117–29. [DOI] [PubMed] [Google Scholar]
- Chan EM, Young EJ, Ianzano L, Munteanu I, Zhao X, Christopoulos CC et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35(2):125–7. [DOI] [PubMed] [Google Scholar]
- Chance MRA, Yaxley DC. Central Nervous Function and Changes in Brain Metabolite Concentration: I. Glycogen and Lactate in Convulsing Mice. Journal of Experimental Biology. 1950;27(3):311–23. [DOI] [PubMed] [Google Scholar]
- Chandramouli C, Varma U, Stevens EM, Xiao RP, Stapleton DI, Mellor KM et al. Myocardial glycogen dynamics: new perspectives on disease mechanisms. Clin Exp Pharmacol Physiol. 2015;42(4):415–25. [DOI] [PubMed] [Google Scholar]
- Chang JC-H, Wu S-M, Tseng Y-C, Lee Y-C, Baba O, Hwang P-P. Regulation of glycogen metabolism in gills and liver of the euryhaline tilapia (Oreochromis mossambicus) during acclimation to seawater. Journal of Experimental Biology. 2007;210(19):3494–504. [DOI] [PubMed] [Google Scholar]
- Chebotareva NA, Meremyanin AV, Makeeva VF, Eronina TB, Kurganov BI. Glycogen phosphorylase b and phosphorylase kinase binding to glycogen under molecular crowding conditions. Inhibitory effect of FAD. Biochemistry (moscow). 2009;74(5):562–8. [DOI] [PubMed] [Google Scholar]
- Chee NP, Geddes R, Wills PR. Metabolic heterogeneity in rabbit brain glycogen. Biochimica et Biophysica Acta (BBA)-General Subjects. 1983;756(1):9–12. [DOI] [PubMed] [Google Scholar]
- Cheng A, Zhang M, Gentry MS, Worby CA, Dixon JE, Saltiel AR. A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori’s disease. Genes Dev. 2007;21(19):2399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipps HD, Duff GL. Glycogen infiltration of the liver cell nuclei. The American journal of pathology. 1942;18(4):645. [PMC free article] [PubMed] [Google Scholar]
- Choi IY, Tkáčc I, Ugurbil K, Gruetter R. Noninvasive measurements of [1-13C] glycogen concentrations and metabolism in rat brain in vivo. Journal of neurochemistry. 1999;73(3):1300–8. [DOI] [PubMed] [Google Scholar]
- Christiansen C, Abou Hachem M, Janecek S, Vikso-Nielsen A, Blennow A, Svensson B. The carbohydrate-binding module family 20--diversity, structure, and function. Febs j. 2009;276(18):5006–29. [DOI] [PubMed] [Google Scholar]
- Cid E, Cifuentes D, Baqué S, Ferrer JC, Guinovart JJ. Determinants of the nucleocytoplasmic shuttling of muscle glycogen synthase. The FEBS journal. 2005;272(12):3197–213. [DOI] [PubMed] [Google Scholar]
- Cohen P. The origins of protein phosphorylation. Nature cell biology. 2002;4(5):E127. [DOI] [PubMed] [Google Scholar]
- Cohen P, Frame S. The renaissance of GSK3. Nature reviews Molecular cell biology. 2001;2(10):769. [DOI] [PubMed] [Google Scholar]
- Contreras CJ, Segvich DM, Mahalingan K, Chikwana VM, Kirley TL, Hurley TD et al. Incorporation of phosphate into glycogen by glycogen synthase. Arch Biochem Biophys. 2016;597:21–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cori CF, Colowick SP, Cori GT. The isolation and synthesis of glucose-1-phosphoric acid. J biol Chem. 1937;121:465–77. [Google Scholar]
- Cori CF, Cori GT. Glycogen formation in the liver from d-and l-lactic acid. Journal of Biological Chemistry. 1929;81(2):389–403. [Google Scholar]
- Cori CF, Cori GT. Mechanism of formation of hexosemonophosphate in muscle and isolation of a new phosphate ester. Proceedings of the Society for Experimental Biology and Medicine. 1936;34(5):702–5. [Google Scholar]
- Cori CF, Cori GT. Carbohydrate metabolism. Annual review of biochemistry. 1946;15(1):193–218. [DOI] [PubMed] [Google Scholar]
- Cori CF, Cori GT, Green AA. Crystalline muscle phosphorylase III. Kinetics. Journal of Biological Chemistry. 1943;151(1):39–55. [Google Scholar]
- Cori CF, Schmidt G, Cori GT. The synthesis of a polysaccharide from glucose-1-phosphate in muscle extract. Science. 1939;89(2316):464–5. [DOI] [PubMed] [Google Scholar]
- Cori GT. Glycogen structure and enzyme deficiencies in glycogen storage disease. Harvey lectures. 1952;48:145. [PubMed] [Google Scholar]
- Cori GT, Colowick SP, Cori CF. The enzymatic conversion of glucose-1-phosphoric ester to 6-ester in tissue extracts. Journal of Biological Chemistry. 1938;124(2):543–55. [Google Scholar]
- Cori GT, Cori CF. Crystalline muscle phosphorylase IV. Formation of glycogen. Journal of Biological Chemistry. 1943;151(1):57–63. [Google Scholar]
- Cori GT, Green AA. Crystalline muscle phosphorylase II. Prosthetic group. Journal of Biological Chemistry. 1943;151(1):31–8. [Google Scholar]
- Cori GT, Larner J. Action of amylo-1, 6-glucosidase and phosphorylase on glycogen and amylopectin. Journal of Biological Chemistry. 1951;188(1):17–29. [PubMed] [Google Scholar]
- Criado O, Aguado C, Gayarre J, Duran-Trio L, Garcia-Cabrero AM, Vernia S et al. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol Genet. 2012;21(7):1521–33. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Bergamini E, Brunk UT, Dröge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining” clean” cells. Autophagy. 2005;1(3):131–40. [DOI] [PubMed] [Google Scholar]
- Curtino JA, Lacoste ER. Two glycogen synthase activities associated with proteoglycogen in retina. Neurochemical research. 2000;25(1):129–32. [DOI] [PubMed] [Google Scholar]
- D’Hulst C, Mérida Á. The priming of storage glucan synthesis from bacteria to plants: current knowledge and new developments. New phytologist. 2010;188(1):13–21. [DOI] [PubMed] [Google Scholar]
- De Duve C, Pressman B, Gianetto R, Wattiaux R, Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochemical Journal. 1955;60(4):604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbridge LMD, Mellor KM, Taylor DJR, Gottlieb RA. Myocardial autophagic energy stress responses—macroautophagy, mitophagy, and glycophagy. American Journal of Physiology-Heart and Circulatory Physiology. 2015;308(10):H1194–H204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme P, Hevor TK. Glycogen particles in methionine sulfoximine epileptogenic rodent brain and liver after the administration of methionine and actinomycin D. Neuropathology and applied neurobiology. 1985;11(2):117–28. [DOI] [PubMed] [Google Scholar]
- Demetriadou A, Morales-Sanfrutos J, Nearchou M, Baba O, Kyriacou K, Tate EW et al. Mouse Stbd1 is N-myristoylated and affects ER-mitochondria association and mitochondrial morphology. J Cell Sci. 2017:jcs. 195263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng B, Sullivan MA, Chen C, Li J, Powell PO, Hu Z et al. Molecular structure of human-liver glycogen. PloS one. 2016;11(3):e0150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng B, Sullivan MA, Li J, Tan X, Zhu C, Schulz BL et al. Molecular structure of glycogen in diabetic liver. Glycoconjugate journal. 2015;32(3–4):113–8. [DOI] [PubMed] [Google Scholar]
- DePaoli-Roach AA, Contreras CJ, Segvich DM, Heiss C, Ishihara M, Azadi P et al. Glycogen phosphomonoester distribution in mouse models of the progressive myoclonic epilepsy, Lafora disease. J Biol Chem. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, Roach PJ. Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J Biol Chem. 2010;285(33):25372–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos P, Baudhuin P, Van Hoof F, Hers H-G. The alpha particulate liver glycogen. A morphometric approach to the kinetics of its synthesis and degradation. Biochemical Journal. 1983;209(1):159–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos P, Hers H-G. Glycogen in rat adipose tissue: sequential synthesis and random degradation. Biochemical and biophysical research communications. 1980;95(3):1031–6. [DOI] [PubMed] [Google Scholar]
- Díaz A, Martínez-Pons C, Fita I, Ferrer JC, Guinovart JJ. Processivity and subcellular localization of glycogen synthase depend on a non-catalytic high-affinity glycogen-binding site. Journal of Biological Chemistry. 2011:jbc. M111. 236109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel GA, Cruz NF. Aerobic glycolysis during brain activation: adrenergic regulation and influence of norepinephrine on astrocytic metabolism. Journal of neurochemistry. 2016;138(1):14–52. [DOI] [PubMed] [Google Scholar]
- Drochmans P. Morphologie du glycogène: Etude au microscope électronique de colorations négatives du glycogène particulaire. Journal of ultrastructure research. 1962;6(2):141–63. [DOI] [PubMed] [Google Scholar]
- Duran J, Gruart A, Garcia-Rocha M, Delgado-Garcia JM, Guinovart JJ. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet. 2014;23(12):3147–56. [DOI] [PubMed] [Google Scholar]
- Duran J, Guinovart JJ. Brain glycogen in health and disease. Mol Aspects Med. 2015;46:70–7. [DOI] [PubMed] [Google Scholar]
- Edner C, Li J, Albrecht T, Mahlow S, Hejazi M, Hussain H et al. Glucan, water dikinase activity stimulates breakdown of starch granules by plastidial beta-amylases. Plant Physiol. 2007;145(1):17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner RD. Adipose tissue glycogen synthesis. International Journal of Biochemistry. 1984;16(3):257–61. [DOI] [PubMed] [Google Scholar]
- Emanuelle S, Brewer MK, Meekins DA, Gentry MS. Unique carbohydrate binding platforms employed by the glucan phosphatases. Cell Mol Life Sci. 2016;73(14):2765–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Novell JM, Castel S, Bellido D, Ferrer JC, Vilaró S, Guinovart JJ. Intracellular distribution of hepatic glucokinase and glucokinase regulatory protein during the fasted to refed transition in rats. FEBS letters. 1999;459(2):211–4. [DOI] [PubMed] [Google Scholar]
- Fernández-Novell JM, López-Iglesias C, Ferrer JC, Guinovart JJ. Zonal distribution of glycogen synthesis in isolated rat hepatocytes. FEBS letters. 2002;531(2):222–8. [DOI] [PubMed] [Google Scholar]
- Ferrans VJ, Buja LM, Jones M. Ultrastructure and cytochemistry of glycogen in cardiac diseases. Recent advances in studies on cardiac structure and metabolism. 1973;3:97. [PubMed] [Google Scholar]
- Ferrans VJ, Maron BJ, Buja LM, Ali N, Roberts WC. Intranuclear glycogen deposits in human cardiac muscle cells: ultrastructure and cytochemistry. Journal of molecular and cellular cardiology. 1975;7(6):381–6. [DOI] [PubMed] [Google Scholar]
- Ferrer JC, Baqué S, Guinovart JJ. Muscle glycogen synthase translocates from the cell nucleus to the cytosol in response to glucose. FEBS letters. 1997;415(3):249–52. [DOI] [PubMed] [Google Scholar]
- Ferrer JC, Favre C, Gomis RR, Fernández-Novell JM, García-Rocha M, de la Iglesia N et al. Control of glycogen deposition. FEBS letters. 2003;546(1):127–32. [DOI] [PubMed] [Google Scholar]
- Fischer EH, Krebs EG. Conversion of phosphorylase b to phosphorylase a in muscle extracts. Journal of Biological Chemistry. 1955;216(1):121–32. [PubMed] [Google Scholar]
- Fletcher WM, Hopkins FG. Lactic acid in amphibian muscle 1. The Journal of physiology. 1907;35(4):247–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana JD. The presence of phosphate in glycogen. FEBS Lett. 1980;109(1):85–92. [DOI] [PubMed] [Google Scholar]
- Foster M. Claude Bernard. Unwin; 1899. [Google Scholar]
- Friedman DL, Larner J. Studies on UDPG-α-glucan transglucosylase. III. Interconversion of two forms of muscle UDPG-α-glucan transglucosylase by a phosphorylation-dephosphorylation reaction sequence. Biochemistry. 1963;2(4):669–75. [DOI] [PubMed] [Google Scholar]
- Froese DS, Michaeli A, McCorvie TJ, Krojer T, Sasi M, Melaev E et al. Structural basis of glycogen branching enzyme deficiency and pharmacologic rescue by rational peptide design. Human molecular genetics. 2015;24(20):5667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller M, Meikle PJ, Hopwood JJ. Epidemiology of lysosomal storage diseases: an overview. 2006. [PubMed]
- Gadisseux J-F, Evrard P. Glial-neuronal relationship in the developing central nervous system. Developmental neuroscience. 1985;7(1):12–32. [DOI] [PubMed] [Google Scholar]
- Gage SH. Glycogen in the nervous system of vertebrates. Journal of Comparative Neurology. 1917;27(3):451–65. [Google Scholar]
- Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y et al. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 2002;11(11):1251–62. [DOI] [PubMed] [Google Scholar]
- Garcia-Gimeno MA, Knecht E, Sanz P. Lafora Disease: A Ubiquitination-Related Pathology. 2018;7(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Rocha M, Angela R, de la Iglesia N, Otto B, Fernández-Novell JM, Ferrer JC et al. Intracellular distribution of glycogen synthase and glycogen in primary cultured rat hepatocytes. Biochemical Journal. 2001;357(1):17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garyali P, Segvich DM, DePaoli-Roach AA, Roach PJ. Protein degradation and quality control in cells from laforin and malin knockout mice. J Biol Chem. 2014;289(30):20606–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasa R, Jensen PB, Berman HK, Brady MJ, DePaoli-Roach AA, Newgard CB. Distinctive regulatory and metabolic properties of glycogen targeting subunits of protein phosphatase-1 (PTG, GL, GM/RGl) expressed in hepatocytes. Journal of Biological Chemistry. 2000. [DOI] [PubMed] [Google Scholar]
- Gayarre J, Duran-Trio L, Criado Garcia O, Aguado C, Juana-Lopez L, Crespo I et al. The phosphatase activity of laforin is dispensable to rescue Epm2a−/− mice from Lafora disease. Brain. 2014;137(Pt 3):806–18. [DOI] [PubMed] [Google Scholar]
- Geddes R. Glycogen: a metabolic viewpoint. Bioscience reports. 1986;6(5):415–28. [DOI] [PubMed] [Google Scholar]
- Geddes R, Stratton GC. The influence of lysosomes on glycogen metabolism. Biochemical Journal. 1977a;163(2):193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes R, Stratton GC. Molecular and metabolic heterogeneity of liver glycogen. Carbohydrate research. 1977b;57:291–9. [DOI] [PubMed] [Google Scholar]
- Gentry MS, Brewer MK, Vander Kooi CW. Structural biology of glucan phosphatases from humans to plants. Curr Opin Struct Biol. 2016;40:62–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Dixon JE, Worby CA. Lafora disease: insights into neurodegeneration from plant metabolism. Trends Biochem Sci. 2009;34(12):628–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Dowen RH 3rd, Worby CA, Mattoo S, Ecker JR, Dixon JE. The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J Cell Biol. 2007;178(3):477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Guinovart JJ, Minassian BA, Roach PJ, Serratosa JM. Lafora disease offers a unique window into neuronal glycogen metabolism. J Biol Chem. 2018;293(19):7117–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Pace RM. Conservation of the glucan phosphatase laforin is linked to rates of molecular evolution and the glycogen metabolism of the organism. BMC Evol Biol. 2009;9(1):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Roma-Mateo C, Sanz P. Laforin, a protein with many faces: glucan phosphatase, adapter protein, et alii. FEBS J. 2013;280(2):525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Worby CA, Dixon JE. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A. 2005;102(24):8501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz HJ, Cervos-Navarro J, Frydl V, Schultz F. Glycogen accumulation of the aging human brain. Mechanisms of ageing and development. 1985;31(1):25–35. [DOI] [PubMed] [Google Scholar]
- Gessler K, Uson I, Takaha T, Krauss N, Smith SM, Okada S et al. V-Amylose at atomic resolution: X-ray structure of a cycloamylose with 26 glucose residues (cyclomaltohexaicosaose). Proc Natl Acad Sci U S A. 1999;96(8):4246–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandially FN, Parry EW. Ultrastructure of a human hepatocellular carcinoma and surrounding non-neoplastic liver. Cancer. 1966;19(12):1989–2004. [DOI] [PubMed] [Google Scholar]
- Gibbons BJ, Roach PJ, Hurley TD. Crystal structure of the autocatalytic initiator of glycogen biosynthesis, glycogenin. Journal of molecular biology. 2002;319(2):463–77. [DOI] [PubMed] [Google Scholar]
- Gibson WB, Brown BI, Brown DH. Glycogen branching enzyme. Preparation and properties of α−1, 4-glucan-α−1, 4-glucan 6-glycosyltransferase and its action on the characteristic polysaccharide of the liver of children with type IV glycogen storage disease. Biochemistry. 1971;10(23):4253–62. [DOI] [PubMed] [Google Scholar]
- Gilbert RG, Sullivan MA. The molecular size distribution of glycogen and its relevance to diabetes. Australian Journal of Chemistry. 2014;67(4):538–43. [Google Scholar]
- Graham TE, Yuan Z, Hill AK, Wilson RJ. The regulation of muscle glycogen: the granule and its proteins. Acta Physiol (Oxf). 2010;199(4):489–98. [DOI] [PubMed] [Google Scholar]
- Granzow C, Kopun M, Zimmermann H-P. Role of nuclear glycogen synthase and cytoplasmic UDP glucose pyrophosphorylase in the biosynthesis of nuclear glycogen in HD33 Ehrlich-Lettré ascites tumor cells. The Journal of cell biology. 1981;89(3):475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AA, Cori GT. Crystalline muscle phosphorylase I. Preparation, properties, and molecular weight. Journal of Biological Chemistry. 1943;151(1):21–9. [Google Scholar]
- Greenberg CC, Danos AM, Brady MJ. Central role for protein targeting to glycogen in the maintenance of cellular glycogen stores in 3T3-L1 adipocytes. Mol Cell Biol. 2006;26(1):334–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grmek MD. First steps in Claude Bernard’s discovery of the glycogenic function of the liver. Journal of the History of Biology. 1968;1(1):141–54. [Google Scholar]
- Gunja-Smith Z, Marshall JJ, Mercier C, Smith EE, Whelan WJ. A revision of the Meyer-Bernfeld model of glycogen and amylopectin. FEBS Lett. 1970;12(2):101–4. [DOI] [PubMed] [Google Scholar]
- Gunja-Smith Z, Marshall JJ, Smith EE. Enzymatic determination of the unit chain length of glycogen and related polysaccharides. FEBS Lett. 1971;13(5):309–11. [DOI] [PubMed] [Google Scholar]
- Halberg F, Albrecht PG, Barnum CP Jr. Phase shifting of liver-glycogen rhythm in intact mice. American Journal of Physiology-Legacy Content. 1960;199(3):400–2. [DOI] [PubMed] [Google Scholar]
- Hanashiro I, Abe J-i, Hizukuri S. A periodic distribution of the chain length of amylopectin as revealed by high-performance anion-exchange chromatography. Carbohydrate Research. 1996;283:151–9. [Google Scholar]
- Hanes CS. The reversible formation of starch from glucose-1-phosphate catalysed by potato phosphorylase. Proc R Soc Lond B. 1940;129(855):174–208. [Google Scholar]
- Hansen PI, Spraul M, Dvortsak P, Larsen FH, Blennow A, Motawia MS et al. Starch phosphorylation—Maltosidic restrains upon 3′-and 6′-phosphorylation investigated by chemical synthesis, molecular dynamics and NMR spectroscopy. Biopolymers: Original Research on Biomolecules. 2009;91(3):179–93. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMPK—sensing energy while talking to other signaling pathways. Cell metabolism. 2014;20(6):939–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, DiBella RJ. Electron microscopic studies of normal and abnormal elastic fibers of the skin. Journal of Investigative Dermatology. 1967;48(5):405–23. [DOI] [PubMed] [Google Scholar]
- Hassid WZ, Neufeld EF, Feingold DS. Sugar nucleotides in the interconversion of carbohydrates in higher plants. Proceedings of the National Academy of Sciences. 1959;45(7):905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haworth WN, Percival EGV. 324. Polysaccharides. Part XI. Molecular structure of glycogen. Journal of the Chemical Society (Resumed). 1932:2277–82. [Google Scholar]
- Hayee BH, Antonopoulos A, Murphy EJ, Rahman FZ, Sewell G, Smith BN et al. G6PC3 mutations are associated with a major defect of glycosylation: a novel mechanism for neutrophil dysfunction. Glycobiology. 2011;21(7):914–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejazi M, Fettke J, Haebel S, Edner C, Paris O, Frohberg C et al. Glucan, water dikinase phosphorylates crystalline maltodextrins and thereby initiates solubilization. Plant J. 2008;55(2):323–34. [DOI] [PubMed] [Google Scholar]
- Hendriks W, Mulders JW, Bibby MA, Slingsby C, Bloemendal H, De Jong WW. Duck lens epsilon-crystallin and lactate dehydrogenase B4 are identical: a single-copy gene product with two distinct functions. Proceedings of the National Academy of Sciences. 1988;85(19):7114–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans MM, Kroos MA, Van Beeumen J, Oostra BA, Reuser AJ. Human lysosomal alpha-glucosidase. Characterization of the catalytic site. Journal of Biological Chemistry. 1991;266(21):13507–12. [PubMed] [Google Scholar]
- Heroes E, Lesage B, Görnemann J, Beullens M, Van Meervelt L, Bollen M. The PP1 binding code: a molecular-lego strategy that governs specificity. The FEBS journal. 2013;280(2):584–95. [DOI] [PubMed] [Google Scholar]
- Hers HG. α-Glucosidase deficiency in generalized glycogen-storage disease (Pompe’s disease). Biochemical Journal. 1963;86(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai M, Hirai T, Ueki T. Effect of branching of amylopectin on complexation with iodine as steric hindrance. Polymer. 1994;35(10):2222–5. [Google Scholar]
- Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJJ, Oostra BA. Characterization of the human lysosomal α-glucosidase gene. Biochemical journal. 1990;272(2):493–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EG, Holmes BE. Contributions to the study of brain metabolism: Carbohydrate metabolism relationship of glycogen and lactic acid. Biochemical Journal. 1926;20(6):1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horita N, Matsushita M, Ishii T, Oyanagi S, Sakamoto K. Ultrastructure of Alzheimer type II glia in hepatocerebral disease. Neuropathology and applied neurobiology. 1981;7(2):97–102. [DOI] [PubMed] [Google Scholar]
- Hu Z, Deng B, Tan X, Gan H, Li C, Nada SS et al. Diurnal changes of glycogen molecular structure in healthy and diabetic mice. Carbohydrate polymers. 2018;185:145–52. [DOI] [PubMed] [Google Scholar]
- Huijing F. Glycogen metabolism and glycogen-storage diseases. Physiological reviews. 1975;55(4):609–58. [DOI] [PubMed] [Google Scholar]
- Husemann E, Ruska uH. Versuche zur Sichtbarmachung von Glykogenmolekülen. Journal für Praktische Chemie. 1940;156(1–3):1–10. [Google Scholar]
- Illingworth B, Larner J, Cori GT. Structure of glycogens and amylopectins. I. Enzymatic determination of chain length. J Biol Chem. 1952;199(2):631–40. [PubMed] [Google Scholar]
- Irimia JM, Tagliabracci VS, Meyer CM, Segvich DM, DePaoli-Roach AA, Roach PJ. Muscle glycogen remodeling and glycogen phosphate metabolism following exhaustive exercise of wild type and laforin knockout mice. J Biol Chem. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Pei YF. Intramitochondrial glycogen particles in rat retinal receptor cells. The Journal of cell biology. 1965;25(2):402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamasa T, Fukuda S, Tokumitsu S, Ninomiya N, Matsuda I, Osame M. Myopathy due to glycogen storage disease: pathological and biochemical studies in relation to glycogenosome formation. Experimental and molecular pathology. 1983;38(3):405–20. [DOI] [PubMed] [Google Scholar]
- Iwamasa T, Tsuru T, Hamada T, Takeuchi T. Physicochemical and ultrastructural studies on glycogenosomes in newborn rat hepatocytes. Pathology-Research and Practice. 1980;167(2–4):363–73. [DOI] [PubMed] [Google Scholar]
- Janeček Š. A motif of a microbial starch-binding domain found in human genethonin. Bioinformatics. 2002;18(11):1534–7. [DOI] [PubMed] [Google Scholar]
- Janecek S, Svensson B, MacGregor EA. Structural and evolutionary aspects of two families of non-catalytic domains present in starch and glycogen binding proteins from microbes, plants and animals. Enzyme Microb Technol. 2011;49(5):429–40. [DOI] [PubMed] [Google Scholar]
- Jeffrey PL, Brown DH, Brown BI. Lysosomal α-glucosidase. I. Purification and properties of the rat liver enzyme. Biochemistry. 1970a;9(6):1403–15. [DOI] [PubMed] [Google Scholar]
- Jeffrey PL, Brown DH, Brown BI. Lysosomal α-glucosidase. II. Kinetics of action of the rat liver enzyme. Biochemistry. 1970b;9(6):1416–22. [DOI] [PubMed] [Google Scholar]
- Jiang S, Heller B, Tagliabracci VS, Zhai L, Irimia JM, DePaoli-Roach AA et al. Starch binding domain containing protein 1/genethonin 1 is a novel participant in glycogen metabolism. Journal of Biological Chemistry. 2010:jbc. M110. 150839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Wells CD, Roach PJ. Starch-binding domain-containing protein 1 (Stbd1) and glycogen metabolism: Identification of the Atg8 family interacting motif (AIM) in Stbd1 required for interaction with GABARAPL1. Biochemical and biophysical research communications. 2011;413(3):420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3. Trends in biochemical sciences. 2004;29(2):95–102. [DOI] [PubMed] [Google Scholar]
- Jurczak MJ, Danos AM, Rehrmann VR, Brady MJ. The role of protein translocation in the regulation of glycogen metabolism. Journal of cellular biochemistry. 2008;104(2):435–43. [DOI] [PubMed] [Google Scholar]
- Kajihara H, Tsutsumi E, Kinoshita A, Nakano J, Takagi K, Takeo S. Activated astrocytes with glycogen accumulation in ischemic penumbra during the early stage of brain infarction: immunohistochemical and electron microscopic studies. Brain research. 2001;909(1–2):92–101. [DOI] [PubMed] [Google Scholar]
- Kajiura H, Kakutani R, Akiyama T, Takata H, Kuriki T. A novel enzymatic process for glycogen production. Biocatalysis and Biotransformation. 2008;26(1–2):133–40. [Google Scholar]
- Kajiura H, Takata H, Kuriki T, Kitamura S. Structure and solution properties of enzymatically synthesized glycogen. Carbohydrate research. 2010;345(6):817–24. [DOI] [PubMed] [Google Scholar]
- Kakhlon O, Glickstein H, Feinstein N, Liu Y, Baba O, Terashima T et al. Polyglucosan neurotoxicity caused by glycogen branching enzyme deficiency can be reversed by inhibition of glycogen synthase. J Neurochem. 2013;127(1):101–13. [DOI] [PubMed] [Google Scholar]
- Karasaki S. Cytoplasmic and nuclear glycogen synthesis in Novikoff ascites hepatoma cells. Journal of ultrastructure research. 1971;35(1–2):181–96. [DOI] [PubMed] [Google Scholar]
- Kerr SE. The carbohydrate metabolism of brain VI. Isolation of glycogen. Journal of Biological Chemistry. 1938;123(2):443–9. [Google Scholar]
- Khac LD, Eboué-bonis D, Chambaut AM, Clauser H. The Use of Glucosamine as a Metabolic Probe in the Rat Diaphragm: The Effect of Insulin on Its Metabolic Fate. European journal of biochemistry. 1972;31(1):86–94. [DOI] [PubMed] [Google Scholar]
- Kiessling W. Über ein neues Fermentprotein der Hefe und eine reversible enzymatische Synthese des Glykogens. Naturwissenschaften. 1939;27(8):129–30. [Google Scholar]
- Kim J-w, Dang CV. Multifaceted roles of glycolytic enzymes. Trends in biochemical sciences. 2005;30(3):142–50. [DOI] [PubMed] [Google Scholar]
- Kirkman BR, Whelan WJ. Glucosamine is a normal component of liver glycogen. FEBS Lett. 1986;194(1):6–11. [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. The Journal of cell biology. 1992;116(5):1071–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koay A, Woodcroft B, Petrie EJ, Yue H, Emanuelle S, Bieri M et al. AMPK beta subunits display isoform specific affinities for carbohydrates. FEBS Lett. 2010;584(15):3499–503. [DOI] [PubMed] [Google Scholar]
- Koizumi J. Glycogen in the central nervous system. Progress in histochemistry and cytochemistry. 1974;6(4):III–35. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J-i, Tanida I et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880. [DOI] [PubMed] [Google Scholar]
- Kopun M, Granzow C, Krisman CR. Comparative study of nuclear and cytoplasmic glycogen isolated from mutant HD33 ascites cells. Journal of cellular biochemistry. 1989;39(2):185–95. [DOI] [PubMed] [Google Scholar]
- Kopun M, Spring H, Granzow C. Nuclear glycogen synthase—fact or artifact? FEBS letters. 1982;147(2):207–10. [DOI] [PubMed] [Google Scholar]
- Kornfeld M, LeBaron M. Glycogenosis type VIII. Journal of Neuropathology & Experimental Neurology. 1984;43(6):568–79. [DOI] [PubMed] [Google Scholar]
- Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy. Microscopy research and technique. 2004;64(1):10–20. [DOI] [PubMed] [Google Scholar]
- Kotoulas OB, Kalamidas SA, Kondomerkos DJ. Glycogen autophagy in glucose homeostasis. Pathology-Research and Practice. 2006;202(9):631–8. [DOI] [PubMed] [Google Scholar]
- Kotting O, Kossmann J, Zeeman SC, Lloyd JR. Regulation of starch metabolism: the age of enlightenment? Curr Opin Plant Biol. 2010;13(3):321–9. [DOI] [PubMed] [Google Scholar]
- Kotting O, Santelia D, Edner C, Eicke S, Marthaler T, Gentry MS et al. STARCH-EXCESS4 Is a Laforin-Like Phosphoglucan Phosphatase Required for Starch Degradation in Arabidopsis thaliana. Plant Cell. 2009;21(1):334–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisman CR. A method for the colorimetric estimation of glycogen with iodine. Anal Biochem. 1962;4:17–23. [DOI] [PubMed] [Google Scholar]
- Krisman CR, Alfredo CJ. Corn Starch (α1, 4-α1, 6) Glucopolysaccharides-Correlation Between Amylose: Amylopectin Ratios and Physical Properties of the Grains. Starch-Stärke. 1991;43(8):291–4. [Google Scholar]
- Krisman CR, Barengo R. A precursor of glycogen biosynthesis: α-1, 4-glucan-protein. European journal of Biochemistry. 1975;52(1):117–23. [DOI] [PubMed] [Google Scholar]
- Kuriki T, Stewart DC, Preiss J. Construction of chimeric enzymes out of maize endosperm branching enzymes I and II: activity and properties. Journal of Biological Chemistry. 1997;272(46):28999–9004. [DOI] [PubMed] [Google Scholar]
- Lafarga M, Berciano MT, Pérez-Fígares JM, Andrés MA, Maquiera E. Influence of age on nuclear bodies and nuclear volume in pituicytes of the rat neurohypophysis. The Anatomical Record. 1991;230(3):319–24. [DOI] [PubMed] [Google Scholar]
- Lafora GR. Uber des Vorkommen amyloider KJrperchen im innern der Ganglienzellen. Virchows Arch f Path Anat. 1911;205:295. [Google Scholar]
- Lamia KA, Sachdeva UM, DiTacchio L, Williams EC, Alvarez JG, Egan DF et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326(5951):437–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Lancet. J. Onwhyn; 1890.
- Larner J. The action of branching enzymes on outer chains of glycogen. Journal of Biological Chemistry. 1953;202(2):491–503. [PubMed] [Google Scholar]
- Larner J. The discovery of glycogen and glycogen today. Claude Bernard and Experimental Medicine, Schenkman Publ Cambridge, Massachusetts. 1967:p135. [Google Scholar]
- Larner J, Illingworth B, Cori GT, Cori CF. Structure of glycogens and amylopectins II. Analysis by stepwise enzymatic degradation. Journal of Biological Chemistry. 1952;199(2):641–51. [PubMed] [Google Scholar]
- Lawrence JC, Roach PJ. New insights into the role and mechanism of glycogen synthase activation by insulin. Diabetes. 1997;46(4):541–7. [DOI] [PubMed] [Google Scholar]
- Lazarow A. Particulate glycogen: A submicroscopic component of the guinea pig liver cell; its significance in glycogen storage and the regulation of blood sugar. The Anatomical Record. 1942;84(1):31–50. [Google Scholar]
- LeBaron FN. The resynthesis of glycogen by guinea pig cerebral-cortex slices. Biochemical Journal. 1955;61(1):80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc EH, Wilson JW. An electron microscope study of intranuclear inclusions in mouse liver and hepatoma. The Journal of Cell Biology. 1959;6(3):427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-K, Lee J-A. Role of the mammalian ATG8/LC3 family in autophagy: differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB reports. 2016;49(8):424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leff T. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Portland Press Limited; 2003. [DOI] [PubMed] [Google Scholar]
- Leloir LF, Cardini CE. BIOSYNTHESIS OF GLYCOGEN FROM URIDINE DIPHOSPHATE GLUCOSE1. Journal of the American Chemical Society. 1957;79(23):6340–1. [Google Scholar]
- Levin B, Burgess EA, Mortimer PE. Glycogen storage disease type IV, amylopectinosis. Archives of disease in childhood. 1968;43(231):548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8(5):719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie RD. Further exploration of the HIO4-Schiff reaction with remarks on its significance. The Anatomical Record. 1950;108(2):239–53. [DOI] [PubMed] [Google Scholar]
- Lim J-A, Zare H, Puertollano R, Raben N. Atg5flox-derived autophagy-deficient model of Pompe disease: does it tell the whole story? Molecular Therapy-Methods & Clinical Development. 2017;7:11–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ST, Kasemsuwan T, Jane J-L. Characterization of phosphorus in starch by 31P-nuclear magnetic resonance spectroscopy. Cereal Chem. 1994;71:488–93. [Google Scholar]
- Lin A, Mu J, Yang J, Roach PJ. Self-glucosylation of glycogenin, the initiator of glycogen biosynthesis, involves an inter-subunit reaction. Archives of biochemistry and biophysics. 1999;363(1):163–70. [DOI] [PubMed] [Google Scholar]
- Lohi H, Ianzano L, Zhao X-C, Chan EM, Turnbull J, Scherer SW et al. Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum Mol Genet. 2005;14(18):2727–36. [DOI] [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Kirkman BR, Whelan WJ. The role of phosphate in muscle glycogen. Biofactors. 1994;4(3–4):167–71. [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Whelan WJ. A self-glucosylating protein is the primer for rabbit muscle glycogen biosynthesis. The FASEB journal. 1988;2(15):3097–103. [DOI] [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Whelan WJ. The biogenesis of glycogen: nature of the carbohydrate in the protein primer. Biochemistry international. 1990;21(2):251–60. [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Whelan WJ. Glycogenin: the primer for mammalian and yeast glycogen synthesis. Biochimica et Biophysica Acta (BBA)-General Subjects. 2004;1673(1–2):45–55. [DOI] [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Whelan WJ, Dombro RS, Neary JT, Norenberg MD. Glycogen synthesis in the astrocyte: from glycogenin to proglycogen to glycogen. The FASEB journal. 1993a;7(14):1386–93. [DOI] [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Whelan WJ, Marchase RB. Glycogen contains phosphodiester groups that can be introduced by UDPglucose: glycogen glucose 1-phosphotransferase. FEBS Lett. 1993b;329(3):263–7. [DOI] [PubMed] [Google Scholar]
- Lukacs CM, Oikonomakos NG, Crowther RL, Hong LN, Kammlott RU, Levin W et al. The crystal structure of human muscle glycogen phosphorylase a with bound glucose and AMP: an intermediate conformation with T-state and R-state features. Proteins. 2006;63(4):1123–6. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. 2018;19(4):235–49. [DOI] [PubMed] [Google Scholar]
- Mahler R. Glycogen storage diseases. Journal of Clinical Pathology Supplement (Ass Clin Path). 1969;2:32. [Google Scholar]
- Maley F, McGarrahan JF, DelGiacco R. Galactosamine: a precursor of glycogen glucosamine. Biochemical and biophysical research communications. 1966;23(1):85–91. [DOI] [PubMed] [Google Scholar]
- Malfatti E, Nilsson J, Hedberg-Oldfors C, Hernandez-Lain A, Michel F, Dominguez-Gonzalez C et al. A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Annals of neurology. 2014;76(6):891–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini GM, Beerens CE, Verheijen FW. Glucose transport in lysosomal membrane vesicles. Kinetic demonstration of a carrier for neutral hexoses. Journal of Biological Chemistry. 1990;265(21):12380–7. [PubMed] [Google Scholar]
- Mann DMA, Sumpter PQ, Davies CA, Yates PO. Glycogen accumulations in the cerebral cortex in Alzheimer’s disease. Acta neuropathologica. 1987;73(2):181–4. [DOI] [PubMed] [Google Scholar]
- Manners DJ. Enzymic synthesis and degradation of starch and glycogen. Advances in carbohydrate chemistry. Elsevier; 1963. p. 371–430. [Google Scholar]
- Manners DJ. Recent developments in our understanding of amylopectin structure. Carbohydrate Polymers. 1989;11(2):87–112. [Google Scholar]
- Manners DJ. Recent developments in our understanding of glycogen structure. Carbohydrate polymers. 1991;16(1):37–82. [Google Scholar]
- Manners DJ, Stark JR, Thambyrajah V. Iodine Staining Properties of Branched α-(14)-D-Glucanst. Journal of the Japanese Society of Starch Science. 1983;30(1):13–8. [Google Scholar]
- Marani E. Microwave applications in neuromorphology and neurochemistry: safety precautions and techniques. Methods. 1998;15(2):87–99. [DOI] [PubMed] [Google Scholar]
- Marchand I, Chorneyko K, Tarnopolsky M, Hamilton S, Shearer J, Potvin J et al. Quantification of subcellular glycogen in resting human muscle: granule size, number, and location. Journal of Applied Physiology. 2002;93(5):1598–607. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Ferrans VJ. Intramitochondrial glycogen deposits in hypertrophied human myocardium. Journal of molecular and cellular cardiology. 1975;7(9):697–8. [DOI] [PubMed] [Google Scholar]
- Marton L. Electron microscopy of biological objects. Physical Review. 1934;46(6):527. [Google Scholar]
- Mathieu C, De La Sierra-Gallay IL, Duval R, Xu X, Cocaign A, Léger T et al. Insights into brain glycogen metabolism: the structure of human brain glycogen phosphorylase. Journal of Biological Chemistry. 2016:jbc. M116. 738898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Sasaki M, Takemasa E, Kaneta T, Chiba S. Kinetic studies on the substrate specificity and active site of rabbit muscle acid α-glucosidase. The Journal of Biochemistry. 1984;96(4):993–1004. [DOI] [PubMed] [Google Scholar]
- Matsui M, Kakuta M, Misaki A. Comparison of the unit-chain distributions of glycogens from different biological sources, revealed by anion exchange chromatography. Bioscience, biotechnology, and biochemistry. 1993;57(4):623–7. [Google Scholar]
- Matsui T, Ishikawa T, Ito H, Okamoto M, Inoue K, Lee Mc et al. Brain glycogen supercompensation following exhaustive exercise. The Journal of physiology. 2012;590(3):607–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell DS, Kruger L. The fine structure of astrocytes in the cerebral cortex and their response to focal injury produced by heavy ionizing particles. The Journal of cell biology. 1965;25(2):141–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride A, Ghilagaber S, Nikolaev A, Hardie DG. The glycogen-binding domain on the AMPK β subunit allows the kinase to act as a glycogen sensor. Cell metabolism. 2009;9(1):23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlwain H, Tresize MA. The glucose, glycogen and aerobic glycolysis of isolated cerebral tissues. Biochemical Journal. 1956;63(2):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon RJ, Frost SC. Glycogen: a carbohydrate source for GLUT-1 glycosylation during glucose deprivation of 3T3-L1 adipocytes. American Journal of Physiology-Endocrinology and Metabolism. 1996;270(4):E640–E5. [DOI] [PubMed] [Google Scholar]
- McManus JFA. Histological demonstration of mucin after periodic acid. Nature. 1946;158(4006):202. [DOI] [PubMed] [Google Scholar]
- McManus JFA. Histological and histochemical uses of periodic acid. Stain technology. 1948;23(3):99–108. [DOI] [PubMed] [Google Scholar]
- Meekins DA, Guo HF, Husodo S, Paasch BC, Bridges TM, Santelia D et al. Structure of the Arabidopsis glucan phosphatase like sex four2 reveals a unique mechanism for starch dephosphorylation. Plant Cell. 2013;25(6):2302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meekins DA, Raththagala M, Auger KD, Turner BD, Santelia D, Kotting O et al. Mechanistic Insights into Glucan Phosphatase Activity Against Polyglucan Substrates. J Biol Chem. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meekins DA, Raththagala M, Husodo S, White CJ, Guo HF, Kotting O. Phosphoglucan-bound structure of starch phosphatase Starch Excess4 reveals the mechanism for C6 specificity. 2014;111(20):7272–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meekins DA, Vander Kooi CW, Gentry MS. Structural mechanisms of plant glucan phosphatases in starch metabolism. Febs j. 2016;283(13):2427–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez R, Melendez-Hevia E, Cascante M. How did glycogen structure evolve to satisfy the requirement for rapid mobilization of glucose? A problem of physical constraints in structure building. J Mol Evol. 1997;45(4):446–55. [DOI] [PubMed] [Google Scholar]
- Melendez R, Melendez-Hevia E, Mas F, Mach J, Cascante M. Physical constraints in the synthesis of glycogen that influence its structural homogeneity: a two-dimensional approach. Biophys J. 1998;75(1):106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez-Hevia E, Waddell TG, Shelton ED. Optimization of molecular design in the evolution of metabolism: the glycogen molecule. Biochem J. 1993;295 (Pt 2):477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellman I, Warren G. The road taken: past and future foundations of membrane traffic. Cell. 2000;100(1):99–112. [DOI] [PubMed] [Google Scholar]
- Merrick AW. Encephalic glycogen differences in young and adult rats. The Journal of physiology. 1961;158(3):476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KH, Bernfeld P. Recherches sur l’amidon V. L’arnylopectine. Helv chim Acta. 1940;23:875. [Google Scholar]
- Meyerhof O. Die Energieumwandlungen im Muskel. Pflüger’s Archiv für die gesamte Physiologie des Menschen und der Tiere. 1920a;182(1):232–83. [Google Scholar]
- Meyerhof O II. Das Schicksal der Milchsäure in der Erholungsperiode des Muskels. Pflügers Arch. 1920b;182:284. [Google Scholar]
- Meyerhof O. Kohlenhydrat und Milchsäureumsatz im Froschmuskel. Pflüg Arch ges Physiol. 1920c;185:11–20. [Google Scholar]
- Michels PAM, Bringaud F, Herman M, Hannaert V. Metabolic functions of glycosomes in trypanosomatids. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2006;1763(12):1463–77. [DOI] [PubMed] [Google Scholar]
- Minassian BA. Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol. 2001;25(1):21–9. [DOI] [PubMed] [Google Scholar]
- Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ et al. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 1998;20(2):171–4. [DOI] [PubMed] [Google Scholar]
- Miozzo MC, Maldonado C, Curtino JA. Cellular and subcellular localization of glycogenin in chicken retina. IUBMB Life. 1996;40(1):173–80. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Kuramoto R, Shimoji A, Higuchi Y. Fine structure of inclusion body in the nucleus of Alzheimer glia type II in the brain of hepatocerebral degeneration. Acta neuropathologica. 1982;56(4):315–9. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41. [DOI] [PubMed] [Google Scholar]
- Montagna W, Chase HB, Hamilton JB. The distribution of glycogen and lipids in human skin. Journal of Investigative Dermatology. 1951;17(3):147–57. [DOI] [PubMed] [Google Scholar]
- Montgomery EM, Weakley FB, Hilbert GE. Isolation of 6-[α-D-Glucopyranosyl]-D-glucose (Isomaltose) from Enzymic Hydrolyzates of Starch2. Journal of the American Chemical Society. 1949;71(5):1682–7. [Google Scholar]
- Montori-Grau M, Guitart M, García-Martínez C, Orozco A, Gómez-Foix AM. Differential pattern of glycogen accumulation after protein phosphatase 1 glycogen-targeting subunit PPP1R6 overexpression, compared to PPP1R3C and PPP1R3A, in skeletal muscle cells. BMC biochemistry. 2011;12(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordoh J, Krisman CR, Leloir LF. Further studies on high molecular weight liver glycogen. Archives of biochemistry and biophysics. 1966;113(2):265–72. [DOI] [PubMed] [Google Scholar]
- Moreno D, Towler MC, Hardie DG, Knecht E, Sanz P. The laforin-malin complex, involved in Lafora disease, promotes the incorporation of K63-linked ubiquitin chains into AMP-activated protein kinase beta subunits. Mol Biol Cell. 2010;21(15):2578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DL. Colorimetric determination of glycogen. Disadvantages of the iodine method. Journal of Biological Chemistry. 1946;166:199–203. [PubMed] [Google Scholar]
- Mowry RW, Bangle J Raymond. Histochemically Demonstrable Glycogen in the Human Heart: With Special Reference to Glycogen Storage Disease and Diabetes Mellitus. The American journal of pathology. 1951;27(4):611. [PMC free article] [PubMed] [Google Scholar]
- Mu J, Skurat AV, Roach PJ. Glycogenin-2, a novel self-glucosylating protein involved in liver glycogen biosynthesis. Journal of Biological Chemistry. 1997;272(44):27589–97. [DOI] [PubMed] [Google Scholar]
- Munoz-Ballester C, Berthier A, Viana R, Sanz P. Homeostasis of the astrocytic glutamate transporter GLT-1 is altered in mouse models of Lafora disease. Biochim Biophys Acta. 2016;1862(6):1074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachmanson D, Ochoa S, Lipmann FA. Otto Meyerhof-1884–1951: A biographical memoir. Washington DC: National Academy of Sciences; 1960. [Google Scholar]
- Nadeau OW, Fontes JD, Carlson GM. The regulation of glycogenolysis in the brain. J Biol Chem. 2018;293(19):7099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, editor. Starch: Metabolism and Structure. Akita, Japan: Springer; 2015. [Google Scholar]
- Nakamura-Tsuruta S, Yasuda M, Nakamura T, Shinoda E, Furuyashiki T, Kakutani R et al. Comparative analysis of carbohydrate-binding specificities of two anti-glycogen monoclonal antibodies using ELISA and surface plasmon resonance. Carbohydr Res. 2012;350:49–54. [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nature reviews Molecular cell biology. 2009;10(7):458. [DOI] [PubMed] [Google Scholar]
- Nakayama A, Yamamoto K, Tabata S. Identification of the catalytic residues of bifunctional glycogen debranching enzyme. Journal of Biological Chemistry. 2001;276(31):28824–8. [DOI] [PubMed] [Google Scholar]
- Napolitano L, Fawcett D. The fine structure of brown adipose tissue in the newborn mouse and rat. The Journal of Cell Biology. 1958;4(6):685–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard CB, Brady MJ, O’Doherty RM, Saltiel AR. Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes. 2000;49(12):1967–77. [DOI] [PubMed] [Google Scholar]
- Nielsen JN, Derave W, Kristiansen S, Ralston E, Ploug T, Richter EA. Glycogen synthase localization and activity in rat skeletal muscle is strongly dependent on glycogen content. The Journal of Physiology. 2001;531(3):757–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson LH, Hultman E. Liver Glycogen in Man–-the Effect of Total Starvation or a Carbohydrate-Poor Diet Followed by Carbohydrate Refeeding. Scandinavian journal of clinical and laboratory investigation. 1973;32(4):325–30. [DOI] [PubMed] [Google Scholar]
- Nitschke F, Sullivan MA, Wang P, Zhao X, Chown EE, Perri AM et al. Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease. 2017;9(7):906–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke F, Wang P, Schmieder P, Girard JM, Awrey DE, Wang T et al. Hyperphosphorylation of glucosyl c6 carbons and altered structure of glycogen in the neurodegenerative epilepsy lafora disease. Cell Metab. 2013;17(5):756–67. [DOI] [PubMed] [Google Scholar]
- Oe Y, Baba O, Ashida H, Nakamura KC, Hirase H. Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns. Glia. 2016;64(9):1532–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyumi M, Takano S. Intranuclear synthesized and native glycogen particles in human gastric cancer: ultrastructure and histochemistry. Histochemistry. 1977;50(3):239–50. [DOI] [PubMed] [Google Scholar]
- Okubo A, Sameshima M, Unoki K, Uehara F, Ohba N. Ultracytochemical demonstration of glycogen in cone, but not in rod, photoreceptor cells in the rat retina. Annals of Anatomy-Anatomischer Anzeiger. 1998;180(4):307–14. [DOI] [PubMed] [Google Scholar]
- Ondruskova N, Honzik T, Kolarova H, Pakanova Z, Mucha J, Zeman J et al. Aberrant apolipoprotein C-III glycosylation in glycogen storage disease type III and IX. Metabolism. 2018;82:135–41. [DOI] [PubMed] [Google Scholar]
- Orrell SA, Bueding E. A comparison of products obtained by various procedures used for the extraction of glycogen. Journal of Biological Chemistry. 1964;239(12):4021–6. [PubMed] [Google Scholar]
- Öz G, Henry P-G, Seaquist ER, Gruetter R. Direct, noninvasive measurement of brain glycogen metabolism in humans. Neurochemistry international. 2003;43(4–5):323–9. [DOI] [PubMed] [Google Scholar]
- Öz G, Kumar A, Rao JP, Kodl CT, Chow L, Eberly LE et al. Human brain glycogen metabolism during and following hypoglycemia. Diabetes. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öz G, Seaquist ER, Kumar A, Criego AB, Benedict LE, Rao JP et al. Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. American Journal of Physiology-Endocrinology and Metabolism. 2007;292(3):E946–E51. [DOI] [PubMed] [Google Scholar]
- Palmer TN. The substrate specificity of acid α-glucosidase from rabbit muscle. Biochemical Journal. 1971;124(4):701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker GJ, Koay A, Gilbert-Wilson R, Waddington LJ, Stapleton D. AMP-activated protein kinase does not associate with glycogen α-particles from rat liver. Biochemical and biophysical research communications. 2007;362(4):811–5. [DOI] [PubMed] [Google Scholar]
- Parker GJ, Lund KC, Taylor RP, McClain DA. Insulin resistance of glycogen synthase mediated by O-linked N-acetylglucosamine. Journal of Biological Chemistry. 2003;278(12):10022–7. [DOI] [PubMed] [Google Scholar]
- Parodi AJ, Krisman CR, Leloir LF, Mordoh J. Properties of synthetic and native liver glycogen. Archives of biochemistry and biophysics. 1967;121(3):769–78. [DOI] [PubMed] [Google Scholar]
- Parodi AJ, Mordoh J, Krisman CR, Leloir LF. In vitro synthesis of particulate glycogen from uridine diphosphate glucose. Archives of biochemistry and biophysics. 1969;132(1):111–7. [DOI] [PubMed] [Google Scholar]
- Passonneau JV, Gatfield PD, Schulz DW, Lowry OH. An enzymic method for measurement of glycogen. Analytical biochemistry. 1967;19(2):315–26. [DOI] [PubMed] [Google Scholar]
- Pederson BA, Turnbull J, Epp JR, Weaver SA, Zhao X, Pencea N et al. Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann Neurol. 2013;74(2):297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez S, Bertoft E. The molecular structures of starch components and their contribution to the architecture of starch granules: A comprehensive review. Starch-Stärke. 2010;62(8):389–420. [Google Scholar]
- Pfeiffer-Guglielmi B, Bröer S, Bröer A, Hamprecht B. Isozyme pattern of glycogen phosphorylase in the rat nervous system and rat astroglia-rich primary cultures: electrophoretic and polymerase chain reaction studies. Neurochemical research. 2000;25(11):1485–91. [DOI] [PubMed] [Google Scholar]
- Pflüger E. Meine Methode der quantitativen Analyse des Glykogenes und die Arteigenthümlichkeit der Substanzen des Thierleibes. Archiv für die gesamte Physiologie des Menschen und der Tiere. 1909;129(6–7):362–78. [Google Scholar]
- Phelps CH. An ultrastructural study of methionine sulphoximine-induced glycogen accumulation in astrocytes of the mouse cerebral cortex. Journal of neurocytology. 1975;4(4):479–90. [DOI] [PubMed] [Google Scholar]
- Pitcher J, Smythe C, Campbell DG, Cohen P. Identification of the 38-kDa subunit of rabbit skeletal muscle glycogen synthase as glycogenin. European journal of biochemistry. 1987;169(3):497–502. [DOI] [PubMed] [Google Scholar]
- Pitcher J, Smythe C, Cohen P. Glycogenin is the priming glucosyltransferase required for the initiation of glycogen biogenesis in rabbit skeletal muscle. European journal of biochemistry. 1988;176(2):391–5. [DOI] [PubMed] [Google Scholar]
- Polekhina G, Gupta A, Michell BJ, van Denderen B, Murthy S, Feil SC et al. AMPK β subunit targets metabolic stress sensing to glycogen. Current biology. 2003;13(10):867–71. [DOI] [PubMed] [Google Scholar]
- Prats C, Cadefau JA, Cussó R, Qvortrup K, Nielsen JN, Wojtaszewki JFP et al. Phosphorylation-dependent translocation of glycogen synthase to a novel structure during glycogen re-synthesis. Journal of Biological Chemistry. 2005. [DOI] [PubMed] [Google Scholar]
- Prats C, Graham TE, Shearer J. The dynamic life of the glycogen granule. Journal of Biological Chemistry. 2018;293(19):7089–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prats C, Helge JW, Nordby P, Qvortrup K, Ploug T, Dela F et al. Dual regulation of muscle glycogen synthase during exercise by activation and compartmentalization. Journal of Biological Chemistry. 2009;284(23):15692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printen JA, Brady MJ, Saltiel AR. PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science. 1997;275(5305):1475–8. [DOI] [PubMed] [Google Scholar]
- Puri R, Suzuki T, Yamakawa K, Ganesh S. Dysfunctions in endosomal-lysosomal and autophagy pathways underlie neuropathology in a mouse model for Lafora disease. Hum Mol Genet. 2012;21(1):175–84. [DOI] [PubMed] [Google Scholar]
- Quistorff B. Gluconeogenesis in periportal and perivenous hepatocytes of rat liver, isolated by a new high-yield digitonin/collagenase perfusion technique. Biochemical Journal. 1985;229(1):221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Danon M, Lu N, Lee E, Shliselfeld L, Skurat AV et al. Surprises of genetic engineering: A possible model of polyglucosan body disease. Neurology. 2001;56(12):1739–45. [DOI] [PubMed] [Google Scholar]
- Raben N, Hill V, Shea L, Takikita S, Baum R, Mizushima N et al. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Human molecular genetics. 2008;17(24):3897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Schreiner C, Baum R, Takikita S, Xu S, Xie T et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder—murine Pompe disease. Autophagy. 2010;6(8):1078–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, Wong A, Ralston E, Myerowitz R, editors. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten American Journal of Medical Genetics Part C: Seminars in Medical Genetics; 2012: Wiley Online Library. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani MRS, Shibanuma K, Hizukuri S. The fine structure of oyster glucogen. Carbohydrate research. 1992;227:183–94. [Google Scholar]
- Rath VL, Ammirati M, LeMotte PK, Fennell KF, Mansour MN, Danley DE et al. Activation of human liver glycogen phosphorylase by alteration of the secondary structure and packing of the catalytic core. Molecular cell. 2000;6(1):139–48. [PubMed] [Google Scholar]
- Raththagala M, Brewer MK, Parker MW, Sherwood AR, Wong BK, Hsu S et al. Structural mechanism of laforin function in glycogen dephosphorylation and lafora disease. Mol Cell. 2015;57(2):261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt ME, Mellor KM, Curl CL, Stapleton D, Delbridge LMD. Myocardial glycophagy—a specific glycogen handling response to metabolic stress is accentuated in the female heart. Journal of molecular and cellular cardiology. 2013;65:67–75. [DOI] [PubMed] [Google Scholar]
- Résibois-Grégoire A, Dourov N. Electron microscopic study of a case of cerebral glycogenosis. Acta neuropathologica. 1966;6(1):70–9. [DOI] [PubMed] [Google Scholar]
- Revel JP. Electron microscopy of glycogen. Journal of Histochemistry & Cytochemistry. 1964;12(2):104–14. [DOI] [PubMed] [Google Scholar]
- Revel JP, Napolitano L, Fawcett DW. Identification of glycogen in electron micrographs of thin tissue sections. The Journal of Cell Biology. 1960;8(3):575–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritte G, Heydenreich M, Mahlow S, Haebel S, Kotting O, Steup M. Phosphorylation of C6- and C3-positions of glucosyl residues in starch is catalysed by distinct dikinases. FEBS Lett. 2006;580(20):4872–6. [DOI] [PubMed] [Google Scholar]
- Roach PJ. Control of glycogen synthase by hierarchal protein phosphorylation. The FASEB Journal. 1990;4(12):2961–8. [PubMed] [Google Scholar]
- Roach PJ. Glycogen and its Metabolism. Current Molecular Medicine. 2002;2:101–20. [DOI] [PubMed] [Google Scholar]
- Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem J. 2012;441(3):763–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez IR, Whelan WJ. A novel glycosyl-amino acid linkage: rabbit-muscle glycogen is covalently linked to a protein via tyrosine. Biochemical and biophysical research communications. 1985;132(2):829–36. [DOI] [PubMed] [Google Scholar]
- Roesler WJ, Khandelwal RL. Diurnal variations in the activities of the glycogen metabolizing enzymes in mouse liver. The International journal of biochemistry. 1985;17(1):81–5. [DOI] [PubMed] [Google Scholar]
- Roma-Mateo C, Moreno D, Vernia S, Rubio T, Bridges TM, Gentry MS et al. Lafora disease E3-ubiquitin ligase malin is related to TRIM32 at both the phylogenetic and functional level. BMC Evol Biol. 2011a;11:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roma-Mateo C, Solaz-Fuster Mdel C, Gimeno-Alcaniz JV, Dukhande VV, Donderis J, Worby CA et al. Laforin, a dual-specificity phosphatase involved in Lafora disease, is phosphorylated at Ser25 by AMP-activated protein kinase. Biochem J. 2011b;439(2):265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero PA, Smith EE, Whelan WJ. Glucosamine as a substitute for glucose in glycogen metabolism. Biochemistry International. 1980;1(1):1–9. [Google Scholar]
- Ros S, García-Rocha M, Domínguez J, Ferrer JC, Guinovart JJ. Control of liver glycogen synthase activity and intracellular distribution by phosphorylation. Journal of Biological Chemistry. 2009;284(10):6370–8. [DOI] [PubMed] [Google Scholar]
- Rubio-Villena C, Garcia-Gimeno MA, Sanz P. Glycogenic activity of R6, a protein phosphatase 1 regulatory subunit, is modulated by the laforin-malin complex. Int J Biochem Cell Biol. 2013;45(7):1479–88. [DOI] [PubMed] [Google Scholar]
- Rubio-Villena C, Viana R, Bonet J, Garcia-Gimeno MA, Casado M, Heredia M et al. Astrocytes: new players in progressive myoclonus epilepsy of Lafora type. Hum Mol Genet. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruska E, Knoll M. Die magnetische Sammelspule für schnelle Elektronenstrahlen. The magnetic concentrating coil for fast electron beams) Z techn Physik. 1931;12:389–400. [Google Scholar]
- Russell JA, Bloom WL. Extractable and residual glycogen in tissues of the rat. American Journal of Physiology-Legacy Content. 1955;183(3):345–55. [DOI] [PubMed] [Google Scholar]
- Rybicka KK. Glycosomes--the organelles of glycogen metabolism. Tissue Cell. 1996;28(3):253–65. [DOI] [PubMed] [Google Scholar]
- Ryman BE, Whelan WJ. New aspects of glycogen metabolism. Adv Enzymol Relat Areas Mol Biol. 1971;34:285–443. [DOI] [PubMed] [Google Scholar]
- Ryu JH, Drain J, Kim JH, McGee S, Gray-Weale A, Waddington L et al. Comparative structural analyses of purified glycogen particles from rat liver, human skeletal muscle and commercial preparations. Int J Biol Macromol. 2009;45(5):478–82. [DOI] [PubMed] [Google Scholar]
- Saez I, Duran J, Sinadinos C, Beltran A, Yanes O, Tevy MF et al. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J Cereb Blood Flow Metab. 2014;34(6):945–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar SM, Sharp FR, Swanson RA. The regional distribution of glycogen in rat brain fixed by microwave irradiation. Brain research. 1987;417(1):172–4. [DOI] [PubMed] [Google Scholar]
- Sakai M, Austin J, Witmer F, Trueb L. Studies in myoclonus epilepsy (Lafora body form). II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology. 1970;20(2):160–76. [DOI] [PubMed] [Google Scholar]
- Salmoral EM, Tolmasky DS, Krisman CR. Evidence for the presence of glycogen in rat thymus. Cellular and molecular biology. 1990;36(2):163–74. [PubMed] [Google Scholar]
- Salsas E, Larner J. Glycogen synthase can use glucose as an acceptor. Journal of Biological Chemistry. 1975;250(5):1833–7. [PubMed] [Google Scholar]
- Salvan AM, Vion-Dury J, Confort-Gouny S, Dano P, Cozzone PJ. Increased cerebral glycogen detected by localized 1H-magnetic resonance spectroscopy in a patient with suspected McArdle’s disease. European neurology. 1997;37(4):251–3. [DOI] [PubMed] [Google Scholar]
- Sanchez-Martin P, Roma-Mateo C, Viana R, Sanz P. Ubiquitin conjugating enzyme E2-N and sequestosome-1 (p62) are components of the ubiquitination process mediated by the malin-laforin E3-ubiquitin ligase complex. Int J Biochem Cell Biol. 2015;69:204–14. [DOI] [PubMed] [Google Scholar]
- Sanson MA. Note sur la formation physiologique du sucre dans l’economie animale. Comptes rendus des seances de l’Academie des Sciences. 1857;44:1323–5. [Google Scholar]
- Santelia D, Kotting O, Seung D, Schubert M, Thalmann M, Bischof S et al. The phosphoglucan phosphatase like sex Four2 dephosphorylates starch at the C3-position in Arabidopsis. Plant Cell. 2011;23(11):4096–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiaffino S, Mammucari C, Sandri M. The role of autophagy in neonatal tissues: just a response to amino acid starvation? Autophagy. 2008;4(5):727–30. [DOI] [PubMed] [Google Scholar]
- Schneider DR, Felt BT, Goldman H. Microwave radiation energy: a probe for the neurobiologist. Life sciences. 1981;29(7):643–53. [DOI] [PubMed] [Google Scholar]
- Schneider JL, Cuervo AM. Autophagy and human disease: emerging themes. Current opinion in genetics & development. 2014;26:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz GA, Yanoff M. Lafora’s Disease. Distinct Clinico-Pathologic Form of Unverricht’s Syndrome. Archives of neurology. 1965;12:172–88. [DOI] [PubMed] [Google Scholar]
- Scott RB, Still WJS. Glycogen in human peripheral blood leukocytes: II. The macromolecular state of leukocyte glycogen. The Journal of clinical investigation. 1968;47(2):353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seranova E, Connolly KJ, Zatyka M, Rosenstock TR, Barrett T, Tuxworth RI et al. Dysregulation of autophagy as a common mechanism in lysosomal storage diseases. Essays in biochemistry. 2017;61(6):733–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serratosa JM, Gomez-Garre P, Gallardo ME, Anta B, de Bernabe DB, Lindhout D et al. A novel protein tyrosine phosphatase gene is mutated in progressive myoclonus epilepsy of the Lafora type (EPM2). Hum Mol Genet. 1999;8(2):345–52. [DOI] [PubMed] [Google Scholar]
- Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell death and differentiation. 2013;20(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer J, Graham TE. New perspectives on the storage and organization of muscle glycogen. Canadian journal of applied physiology. 2002;27(2):179–203. [DOI] [PubMed] [Google Scholar]
- Shearer J, Graham TE. Novel aspects of skeletal muscle glycogen and its regulation during rest and exercise. Exerc Sport Sci Rev. 2004;32(3):120–6. [DOI] [PubMed] [Google Scholar]
- Sheldon H, Silverberg M, Kerner I. On the differing appearance of intranuclear and cytoplasmic glycogen in liver cells in glycogen storage disease. The Journal of cell biology. 1962;13(3):468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N. Histochemical studies of glycogen of the area postrema and the allied structures of the mammalian brain. Journal of Comparative Neurology. 1955;102(2):323–39. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Hamuro Y. Deposition of glycogen and changes in some enzymes in brain wounds. Nature. 1958;181(4611):781. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Kubo Z. Histochemical studies on brain glycogen of the guinea pig and its alteration following electric shock. Journal of neuropathology and experimental neurology. 1957;16(1):40–7. [DOI] [PubMed] [Google Scholar]
- Shimizu N, Kumamoto T. Histochemical studies on the glycogen of the mammalian brain. The Anatomical Record. 1952;114(3):479–97. [DOI] [PubMed] [Google Scholar]
- Singh PK, Singh S, Ganesh S. The laforin-malin complex negatively regulates glycogen synthesis by modulating cellular glucose uptake via glucose transporters. Mol Cell Biol. 2012;32(3):652–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurat AV, Cao Y, Roach PJ. Glucose control of rabbit skeletal muscle glycogenin expressed in COS cells. Journal of Biological Chemistry. 1993;268(20):14701–7. [PubMed] [Google Scholar]
- Skurat AV, Dietrich AD, Roach PJ. Interaction between glycogenin and glycogen synthase. Archives of biochemistry and biophysics. 2006;456(1):93–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurat AV, Lim SS, Roach PJ. Glycogen biogenesis in rat 1 fibroblasts expressing rabbit muscle glycogenin. European journal of biochemistry. 1997;245(1):147–55. [DOI] [PubMed] [Google Scholar]
- Smythe C, Cohen P. The discovery of glycogenin and the priming mechanism for glycogen biogenesis EJB Reviews 1991. Springer; 1991. p. 149–55. [DOI] [PubMed] [Google Scholar]
- Somogyi M. The solubility and preparation of phosphorus-and nitrogen-free glycogen. Journal of Biological Chemistry. 1934;104(2):245–53. [Google Scholar]
- Sotelo C, Palay SL. The fine structure of the lateral vestibular nucleus in the rat: I. Neurons and neuroglial cells. The Journal of cell biology. 1968;36(1):151–79. [PubMed] [Google Scholar]
- Stapleton D, Nelson C, Parsawar K, Flores-Opazo M, McClain D, Parker G. The 3T3-L1 adipocyte glycogen proteome. Proteome Sci. 2013;11(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stapleton D, Nelson C, Parsawar K, McClain D, Gilbert-Wilson R, Barker E et al. Analysis of hepatic glycogen-associated proteins. Proteomics. 2010;10(12):2320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetten D Jr, Stetten MR. Glycogen metabolism. Physiological reviews. 1960;40(3):505–37. [DOI] [PubMed] [Google Scholar]
- Sullivan MA, Aroney ST, Li S, Warren FJ, Joo JS, Mak KS et al. Changes in glycogen structure over feeding cycle sheds new light on blood-glucose control. Biomacromolecules. 2014;15(2):660–5. [DOI] [PubMed] [Google Scholar]
- Sullivan MA, Nitschke S, Steup M, Minassian BA, Nitschke F. Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan. Int J Mol Sci. 2017;18(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MA, O’Connor MJ, Umana F, Roura E, Jack K, Stapleton DI et al. Molecular insights into glycogen α-particle formation. Biomacromolecules. 2012;13(11):3805–13. [DOI] [PubMed] [Google Scholar]
- Sullivan MA, Vilaplana F, Cave RA, Stapleton D, Gray-Weale AA, Gilbert RG. Nature of α and β particles in glycogen using molecular size distributions. Biomacromolecules. 2010;11(4):1094–100. [DOI] [PubMed] [Google Scholar]
- Sun T, Yi H, Yang C, Kishnani PS, Sun B. Starch binding domain-containing protein 1 plays a dominant role in glycogen transport to lysosomes in liver. Journal of Biological Chemistry. 2016;291(32):16479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland EW, Wosilait WD. Inactivation and activation of liver phosphorylase. Nature. 1955;175(4447):169. [DOI] [PubMed] [Google Scholar]
- Svorad D. Diurnal changes in the brain glycogen. Experientia. 1958;14(12):452-. [DOI] [PubMed] [Google Scholar]
- Swanson MA. Studies on the structure of polysaccharides; relation of the iodine color to the structure. J Biol Chem. 1948;172(2):825–37. [PubMed] [Google Scholar]
- Swanson MA, Cori CF. Studies on the structure of polysaccharides III. Relation of structure to activation of phosphorylases. Journal of Biological Chemistry. 1948;172(2):815–24. [PubMed] [Google Scholar]
- Swanson RA, Sagar SM, Sharp FR. Regional brain glycogen stores and metabolism during complete global ischaemia. Neurological research. 1989;11(1):24–8. [DOI] [PubMed] [Google Scholar]
- Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X et al. Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem. 2008;283(49):33816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Heiss C, Karthik C, Contreras CJ, Glushka J, Ishihara M et al. Phosphate incorporation during glycogen synthesis and Lafora disease. Cell Metab. 2011;13(3):274–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV et al. Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo. Proc Natl Acad Sci U S A. 2007;104(49):19262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata H, Kajiura H, Furuyashiki T, Kakutani R, Kuriki T. Fine structural properties of natural and synthetic glycogens. Carbohydrate research. 2009;344(5):654–9. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Hizukuri S. Location of phosphate groups in potato amylopectin. Carbohydrate Research. 1982;102(1):321–7. [Google Scholar]
- Tan JMM, Wong ESP, Dawson VL, Dawson T, Lim K-L. Lysine 63-linked polyubiquitin potentially partners with p62 to promote the clearance of protein inclusions by autophagy. Autophagy. 2008;4(2):251–3. [Google Scholar]
- Tan X, Sullivan MA, Nada SS, Deng B, Schulz BL, Gilbert RG. Proteomic Investigation of the Binding Agent between Liver Glycogen β Particles. Acs Omega. 2018;3(4):3640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanikawa K. Electron microscopic observation of the intranuclear glycogen of the liver in diabetes mellitus. The Kurume medical journal. 1965;11(4):177–81. [DOI] [PubMed] [Google Scholar]
- Tarentino AL, Maley F. Direct evidence that D-galactosamine incorporation into glycogen occurs via UDP-glucosamine. FEBS letters. 1976;69(1–2):175–8. [DOI] [PubMed] [Google Scholar]
- Tavridou A, Agius L. Phosphorylase regulates the association of glycogen synthase with a proteoglycogen substrate in hepatocytes. FEBS letters. 2003;551(1–3):87–91. [DOI] [PubMed] [Google Scholar]
- Tegtmeyer LC, Rust S, van Scherpenzeel M, Ng BG, Losfeld M-E, Timal S et al. Multiple phenotypes in phosphoglucomutase 1 deficiency. New England Journal of Medicine. 2014;370(6):533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terjung RL, Baldwin KM, Winder WW, Holloszy JO. Glycogen repletion in different types of muscle and in liver after exhausting exercise. American Journal of Physiology-Legacy Content. 1974;226(6):1387–91. [DOI] [PubMed] [Google Scholar]
- Testoni G, Duran J, García-Rocha M, Vilaplana F, Serrano AL, Sebastián D et al. Lack of glycogenin causes glycogen accumulation and muscle function impairment. Cell metabolism. 2017;26(1):256–66. e4. [DOI] [PubMed] [Google Scholar]
- Tiberia E, Turnbull J, Wang T, Ruggieri A, Zhao XC, Pencea N et al. Increased laforin and laforin binding to glycogen underlie Lafora body formation in malin-deficient Lafora disease. J Biol Chem. 2012;287(30):25650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolmasky DS, Krisman CR. The degree of branching in (α1, 4)-(α1, 6)-linked glucopolysaccharides is dependent on intrinsic properties of the branching enzymes. European journal of biochemistry. 1987;168(2):393–7. [DOI] [PubMed] [Google Scholar]
- Towfighi J, Yoss BS, Wasiewski WW, Vannucci RC, Bentz MS, Mamourian A. Cerebral glycogenosis, alpha particle type: morphologic and biochemical observations in an infant. Human pathology. 1989;20(12):1210–5. [DOI] [PubMed] [Google Scholar]
- Turnbull J, Depaoli-Roach AA, Zhao X, Cortez MA, Pencea N, Tiberia E et al. PTG Depletion Removes Lafora Bodies and Rescues the Fatal Epilepsy of Lafora Disease. PLoS Genet. 2011a;7(4):e1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Epp JR, Goldsmith D, Zhao X, Pencea N, Wang P et al. PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann Neurol. 2014;75(3):442–6. [DOI] [PubMed] [Google Scholar]
- Turnbull J, Girard JM, Pencea N, Zhao X, Graham TE, Wang P et al. Lafora bodies in skeletal muscle are fiber type specific. Neurology. 2011b;76(19):1674–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG et al. Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol. 2010;68(6):925–33. [DOI] [PubMed] [Google Scholar]
- Valles-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L et al. Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO Mol Med. 2011;3(11):667–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Heijningen AJMK, Kemp A. Free and fixed glycogen in rat muscle. Biochemical Journal. 1955;59(3):487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Heycop Ten Ham MW. Lafora disease, a form of progressive myoclonus epilepsy In: Vinken PJ, Bruyn GW, editors. Handbook of Clinical neurology. Holland, Amsterdam: North Holland Publishing Company; 1975. p. 382–422. [Google Scholar]
- Vander Kooi CW, Taylor AO, Pace RM, Meekins DA, Guo HF, Kim Y et al. Structural basis for the glucan phosphatase activity of Starch Excess4. Proc Natl Acad Sci U S A. 2010;107(35):15379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vénien-Bryan C, Jonic S, Skamnaki V, Brown N, Bischler N, Oikonomakos NG et al. The structure of phosphorylase kinase holoenzyme at 9.9 Å resolution and location of the catalytic subunit and the substrate glycogen phosphorylase. Structure. 2009;17(1):117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhue W, Hers HG. A study of the reaction catalysed by the liver branching enzyme. Biochemical Journal. 1966;99(1):222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia-Fojeda B et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10(11):1407–13. [DOI] [PubMed] [Google Scholar]
- Viskupic E, Cao Y, Zhang W, Cheng C, DePaoli-Roach AA, Roach PJ. Rabbit skeletal muscle glycogenin. Molecular cloning and production of fully functional protein in Escherichia coli. Journal of Biological Chemistry. 1992;267(36):25759–63. [PubMed] [Google Scholar]
- Vøllestad MK, Vaage ODD, Hermansen L. Muscle glycogen depletion patterns in type I and subgroups of type II fibres during prolonged severe exercise in man: Glycogen depletion in muscle fibres during exercise. Acta Physiologica Scandinavica. 1984;122(4):433–41. [DOI] [PubMed] [Google Scholar]
- Walker GJ, Whelan WJ. The mechanism of carbohydrase action. 8. Structures of the muscle-phosphorylase limit dextrins of glycogen and amylopectin. Biochemical Journal. 1960;76(2):264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Stuckey JA, Wishart MJ, Dixon JE. A unique carbohydrate binding domain targets the lafora disease phosphatase to glycogen. J Biol Chem. 2002;277(4):2377–80. [DOI] [PubMed] [Google Scholar]
- Wang P, Israelian L, Xue Y, Song S, Attisano L, Minassian BA. SGK1 (glucose transport), dishevelled2 (wnt signaling), LC3/p62 (autophagy) and p53 (apoptosis) proteins are unaltered in Lafora disease. The all results journals Biol. 2016;7(3):28. [PMC free article] [PubMed] [Google Scholar]
- Wang W, Lohi H, Skurat AV, Depaoli-Roach AA, Minassian BA, Roach PJ. Glycogen metabolism in tissues from a mouse model of Lafora disease. Arch Biochem Biophys. 2007;457(2):264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanson J-C, Drochmans P. RABBIT SKELETAL MUSCLE GLYCOGEN: A Morphological and Biochemical Study of Glycogen ß-Particles Isolated by the Precipitation-Centrifugation Method. The Journal of cell biology. 1968;38(1):130–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan WJ. On the origin of primer for glycogen synthesis. Trends in Biochemical Sciences. 1976;1(1):13–5. [Google Scholar]
- Whelan WJ. Why the linkage of glycogen to glycogenin was so difficult to determine. Biochemistry and Molecular Biology Education. 2007;35(5):313–5. [DOI] [PubMed] [Google Scholar]
- Willstätter R, Rohdewald M. Über den Zustand des Glykogens in der Leber, im Muskel und in Leukocyten.(Zur Kenntnis der Proteinbindung physiologisch wichtiger Stoffe.). Hoppe-Seyleŕ s Zeitschrift für physiologische Chemie. 1934;225(2–3):103–24. [Google Scholar]
- Wilson WA, Roach PJ, Montero M, Baroja-Fernández E, Muñoz FJ, Eydallin G et al. Regulation of glycogen metabolism in yeast and bacteria. FEMS microbiology reviews. 2010;34(6):952–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisselaar HA, Kroos MA, Hermans MM, Van Beeumen J, Reuser AJ. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. Journal of Biological Chemistry. 1993;268(3):2223–31. [PubMed] [Google Scholar]
- Wolfrom ML, Lassettre EN, O’Neill AN. Degradation of Glycogen to Isomaltose1. Journal of the American Chemical Society. 1951;73(2):595–9. [Google Scholar]
- Worby CA, Gentry MS, Dixon JE. Laforin: A dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem. 2006;281(41):30412–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Gentry MS, Dixon JE. Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J Biol Chem. 2008;283(7):4069–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez AJ, Garcia-Rocha M, Bertinat R, Droppelmann C, Concha II, Guinovart JJ et al. Subcellular localization of liver FBPase is modulated by metabolic conditions. FEBS letters. 2004;577(1–2):154–8. [DOI] [PubMed] [Google Scholar]
- Yi H, Fredrickson KB, Das S, Kishnani PS, Sun B. Stbd1 is highly elevated in skeletal muscle of Pompe disease mice but suppression of its expression does not affect lysosomal glycogen accumulation. Molecular genetics and metabolism. 2013;109(3):312–4. [DOI] [PubMed] [Google Scholar]
- Yokoi S, Austin J, Witmer F, Sakai M. Studies in myoclonus epilepsy (Lafora body form). I. Isolation and preliminary characterization of Lafora bodies in two cases. Arch Neurol. 1968;19(1):15–33. [DOI] [PubMed] [Google Scholar]
- Young FG. Claude Bernard and the theory of the glycogenic function of the liver. Annals of Science. 1937;2(1):47–83. [Google Scholar]
- Young FG. Claude Bernard and the discovery of glycogen. British Medical Journal. 1957;1(5033):1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeqiraj E, Sicheri F. Getting a handle on glycogen synthase–Its interaction with glycogenin. Molecular aspects of medicine. 2015;46:63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeqiraj E, Tang X, Hunter RW, García-Rocha M, Judd A, Deak M et al. Structural basis for the recruitment of glycogen synthase by glycogenin. Proceedings of the National Academy of Sciences. 2014:201402926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai L, Feng L, Xia L, Yin H, Xiang S. Crystal structure of glycogen debranching enzyme and insights into its catalysis and disease-causing mutations. Nature communications. 2016;7:11229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Tang M, Liu M, Chen L. Glycophagy: An emerging target in pathology. Clinica Chimica Acta. 2018. [DOI] [PubMed] [Google Scholar]