

Abstract
NahE and PhdJ are bifunctional hydratase-aldolases found in bacterial catabolic pathways for naphthalene and phenanthrene, respectively. Bacterial species with these pathways can use polycyclic aromatic hydrocarbons (PAHs) as sole sources of carbon and energy. Due to the many harmful properties of PAHs and their widespread distribution and persistence in the environment, there is great interest in understanding these degradative pathways including the mechanisms and specificities of the enzymes found in the pathways. This knowledge can be used to develop and optimize bioremediation techniques. Hydratase-aldolases catalyze a major step in the PAH degradative pathways, but their mechanisms are poorly understood. Sequence analysis identified NahE and PhdJ as members of the N-acetylneuraminate lyase (NAL) subgroup in the aldolase superfamily. Accordingly, both have a conserved lysine and tyrosine (for Schiff base formation) as well as a GXXGE motif (to bind the pyruvoyl carboxylate group). Herein, we report the structures of NahE, PhdJ, and PhdJ covalently bound to substrate via a Schiff base. Analysis shows the structural features that determine the individual specificities as well as the features that differentiate these enzymes from the other NAL subgroup members. In addition, the PhdJ complex structure suggests a potential mechanism for hydration of substrate and subsequent retro-aldol fission. The combined findings fill a gap in our understanding of the mechanisms of these enzymes as well as their place in the N-acetylneuraminate lyase (NAL) subgroup.
Graphical Abstract

INTRODUCTION
Naphthalene (1, Scheme 1), phenanthrene (2), and other polycyclic aromatic hydrocarbons [e.g., fluoranthene (3), and pyrene (4)] are persistent environmental contaminants that are responsible for many human health problems (1). Their effects can be direct or indirect (via reactive metabolites) (2). Polycyclic aromatic hydrocarbons (PAHs) are also toxic to marine and other aquatic organisms (3,4). Due to all of these adverse effects, there is much interest in the development of technologies for the removal of PAHs from the environment (5).
Scheme 1.

Representative Polycyclic Aromatic Hydrocarbons.
PAHs can be converted to useful cellular intermediates by bacterial catabolic pathways. The most extensively characterized of these pathways is the one for the degradation of naphthalene in Pseudomonas putida G7 (6). The phenanthrene catabolic pathway is not as well characterized and many proposed enzymatic activities (including the identification of the substrates and products) have not been experimentally verified at the biochemical level. Other reactions have only been partially characterized (7,8). The pathways for the higher molecular weight species (i.e., 3 and 4) are poorly characterized at best (9–13). As part of an effort to use these pathways in bioremediation efforts, we are carrying out a systematic characterization of the individual enzymatic steps.
One key reaction in bacterial catabolic pathways for PAHs is catalyzed by a bifunctional hydratase-aldolase. In the naphthalene catabolic pathway, trans-o-hydroxybenzylidenepyruvate hydratase-aldolase (designated NahE) converts trans-o-hydroxybenzylidenepyruvate (5, Scheme 2) to salicyaldehyde (9) and pyruvate (11) (6). In the phenanthrene catabolic pathway, trans-o-carboxybenzylidenepyruvate hydratase-aldolase (designated PhdJ), converts o-carboxybenzylidenepyruvate (6, Scheme 2) to 2-carboxybenzaldehyde (10) and pyruvate (8) (8,14). These enzymes process their respective substrates through the putative intermediates 7 or 8 (or the Schiff bases of 7 or 11) (8,14). Sequence analysis identified NahE and PhdJ as N-acetylneuraminate lyase (NAL) subgroup members in the Class I aldolase superfamily. Members of this subfamily show a conserved (β/α)8-barrel structure, two strictly conserved active site residues (tyrosine and lysine) that are involved in Schiff base formation, and a GXXGE motif that is associated with the binding of the α-keto acid moiety (15–20). Much of the remaining portion of the active site is tailored to accommodate the individual reactions catalyzed by the NAL subfamily members. For the NahE- and PhdJ-catalyzed reactions, this involves binding of the o-hydroxy- or o-carboxybenzylidene moiety and the addition of water at C4 (of 5 and 6, Scheme 2), to set up a retro-aldol fission.
Scheme 2.

The Hydratase-Aldolase Reactions in PAH Catabolic Pathways.
Herein, we report the structural characterization of NahE and PhdJ. The structures were determined by de novo phasing and molecular replacement to resolutions at 1.9 and 2.0 Å, respectively. Examination of a complex structure of PhdJ covalently linked to its substrate (i.e., 6) provides a structural basis for its substrate specificity when compared to that of NahE. Mutagenesis supports the explanation. Comparison of the structures identifies elements that differentiate NahE and PhdJ from the other NAL family members. Finally, a conserved water molecule near C4 of the substrate (see 6 in Scheme 2) suggests a reaction mechanism for the addition of water at C4.
EXPERIMENTAL PROCEDURES
Materials.
Chemicals, biochemicals, buffers, and solvents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO), Fisher Scientific Inc. (Pittsburgh, PA), Fluka Chemical Corp. (Milwaukee, WI), or EMD Millipore, Inc. (Billerica, MA). 2-Carboxybenzaldehyde (10) was obtained from Sigma-Aldrich Chemical Co. Phenyl-Sepharose 6 Fast Flow resin was obtained from GE Healthcare Bio-sciences (Pittsburgh, PA). The Econo-Column® chromatography columns were obtained from BioRad (Hercules, CA). The Amicon stirred cell concentrators and the ultrafiltration membranes (10,000 Da, MW cutoff) were purchased from EMD Millipore Inc. Oligonucleotide primers were synthesized by Sigma-Aldrich.
Bacterial Strains and Plasmids.
The plasmid designated pRE701 (carrying the nahE gene) was a gift from Dr. Richard Eaton. The Mycobacterium vanbaalenii PYR-1 genomic DNA was a gift from Dr. Carl Cerniglia (National Center for Toxicological Research, US Food and Drug Administration, Jefferson, AR). Escherichia coli ArcticExpress cells were obtained from Agilent Technologies (Santa Clara, CA).
General Methods.
The PCR amplification of DNA sequences was conducted in a GeneAmp 2700 thermocycler (Applied Biosystems, Carlsbad, CA). Techniques for restriction enzyme digestion, ligation, transformation, and other standard molecular biology manipulations were based on methods described elsewhere (21). DNA sequencing was performed in the DNA Sequencing Facility in the Institute for Cellular and Molecular Biology (ICMB) at the University of Texas at Austin. Electrospray ionization mass spectrometry (ESI-MS) was carried out on an LCQ electrospray ion-trap mass spectrometer (Thermo, San Jose, CA) in the Proteomics Facility in the ICMB. Steady-state kinetic assays were performed on an Agilent 8453 diode-array spectrophotometer at 22 °C. Non-linear regression data analysis was performed using the program Mathematica (Wolfram Research, Inc., Version 8.0, Champaign, Il.) Protein concentrations were determined by the Waddell method (22). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out on denaturing gels containing 12% polyacrylamide (23). Nuclear magnetic resonance (NMR) spectra were recorded on a Varian DirectDrive 600 MHz spectrometer (Palo Alto, CA). NMR signals were analyzed using the software program SpinWorks 3.1.6 (Copyright © 2009 Kirk Marat, University of Manitoba). The sequence alignments and secondary information were visualized using ESPript version 3.0. The dynamic light scattering experiments were carried out as described elsewhere (24).
Synthesis of trans-o-Hydroxybenzylidenepyruvate (5).
A mixture of salicylaldehyde (9, 1 g, 13mmol), ethyl triphenylphosphoranylidenepyruvate (13) (5 g, 13 mmol), and a catalytic amount of benzoic acid (~50 mg) was heated in anhydrous DMF (2.5 mL) under argon at 80°C, as described previously (25), to yield 14 (Scheme 3). After 15 h, water (25 mL) was added to the reaction mixture and it was extracted with hexanes/ethyl acetate (2:1). The organic layers were combined, dried over anhydrous Na2SO4, passed through a small silica plug column, and evaporated to dryness in vacuo. The residue was purified by flash column chromatography (4:1 hexanes/ethyl acetate). Fractions containing 14 were combined, dried over anhydrous Na2SO4, and evaporated to dryness under reduced pressure. The compound was suspended in H2O (5 mL) and the ester was hydrolyzed by the dropwise addition of 1M NaOH (and subsequent ethyl acetate extraction) without exceeding pH 10. The aqueous phase was then rapidly acidified to pH 2 (with 8.5% phosphoric acid) and the product was isolated by extraction with ethyl acetate. The organic layers were collected, dried over anhydrous Na2SO4, and evaporated to dryness to yield ~1 g of product. 5: 1H NMR (100 mM NaH2PO4, pH 7, 30 μL DMSO, 600 MHz) δ 6.77 (1H, d, J = 16.5 Hz), 6.80 (2H, m), 7.20 (1H, m), 7.47 (1H, dd, J = 7.8 Hz, 1.5 Hz), 7.79 (1H, d, J = 16.5 Hz).
Scheme 3.

Syntheses of Substrates 5 and 6.
Synthesis of trans-o-Carboxybenzylidenepyruvate (6).
The methyl ester of o-carboxybenzaldehyde (12, Scheme 3) was synthesized by mixing o-carboxybenzaldehyde (o-CBA, 10, 1.5 g, 10 mM) and dimethyl sulfate (1.9 g, 15 mM) with 2 eq of K2CO3. The reaction mixture was stirred at a gentle reflux in acetone (50 mL) overnight. After most of the acetone was removed under reduced pressure, the residue was diluted with ethyl acetate (~100 mL) and filtered to removed salts. The ethyl acetate was washed with water, the organic layer was collected, dried over anhydrous Na2SO4, and evaporated to dryness. For additional purity, the residue was purified by flash column chromatography (4:1 hexanes/ethyl acetate). Fractions containing 12 were combined, dried over anhydrous Na2SO4, and evaporated to dryness.
Subsequently, 12 (1g, 13 mmol) was treated with 13 (25), and processed as described above, to form 15. After the flash chromatography step and evaporation of the ethyl acetate, the compound was suspended in H2O (5 mL) and the esters hydrolyzed by the dropwise addition of 1 M NaOH without exceeding pH 10. If necessary, the pH was adjusted to ~8, concentrated to ~2 mL in vacuo, and passed through a G-25 gel filtration column (to desalt). Product (6) was eluted using H2O and identified by UV. Fractions containing 6 were combined and concentrated under reduced pressure to yield ~1 g of product (as the disodium salt). 6: 1H NMR (100 mM NaH2PO4, pH 7, 30 μL DMSO, 600 MHz) δ 6.72 (1H, d, J = 16.3 Hz), 7.31(3H, m), 7.62 (1H, d, 7.4 Hz), 7.86 (1H, d, J = 16.3).
Synthesis of trans-Benzylidenepyruvate (16).
The compound was synthesized following a literature procedure (26). 16: 1H NMR (100 mM NaH2PO4, pH 7, 30 μL DMSO, 600 MHz) δ 6.72 (1H, d, J = 16.5 Hz), 7.32 (3H, m), 7.52 (3H, m).
Construction of the NahE Expression Vector.
NahE was amplified from the pRE701 plasmid using the PCR with 5’-TAGTAGTAGCATATGTTGAATAAAG-3’ and 5’-GATGATGATCTCGAGTCATTATTATTTACTGTATTTAGCGTG-3’, as forward and reverse primers, respectively. The primers contained the XhoI and NdeI restriction sites (underlined). The PCR product and the pET24 vector were treated with the appropriate restriction enzymes, ligated, and processed to construct an expression vector for NahE. The PCR introduced three mutations, which were corrected (D265G, S275T, and P299S) using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) following the manufacturer’s instructions.
Expression and Purification of NahE.
The pET vector encoding nahE was transformed into E. coli ArcticExpress cells and used to inoculate LB media (450 mL) containing kanamycin (50 μg/mL), and gentimicin (20 μg/mL). After the cells had been shaken overnight at 37°C, 25 mL of the culture was used to inoculate each of 9 2-L Erlenmeyer flasks containing 500 mL of M9 minimal media with 0.4% glucose, kanamycin (50 μg/mL), and gentimicin (20 μg/mL) (21). The cells were allowed to grow until the OD600 reached ~0.5 (~3 h). In one preparation, the flasks containing the cells were put on ice for 30 min, followed by the addition of isopropyl β-D-thiogalactoside (IPTG) to make each one 1 mM in IPTG. After growing for ~65 h at 15 °C, cells were harvested by centrifugation (11,300 rpm for 20 min) and stored at −20 °C.
In a typical procedure, cells were thawed and suspended in 50 mM NaH2PO4 buffer (pH 7.0, 150 mL) that contained 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 5 mM EDTA, and 1.5 mg/mL lysozyme. Cells were lysed by sonication using a W-385 ultrasonicator made by Heat Systems (15 min at 30% duty, 5 s intervals). Ground (NH4)2SO4 was added to the lysate (30% saturation) before centrifugation (39000 g, 30 min). The supernatant was loaded onto a Phenyl Sepharose 6 (~55 mL) column equilibrated in 50 mM NaH2PO4 buffer (pH 7.5) made 15% in (NH4)2SO4. After the column was washed (>1 column volume), protein was eluted by using a linear gradient [15–0% in (NH4)2SO4 in the NaH2PO4 buffer, 200 mL]. The protein did not completely elute from the column, as indicated by a protein determination assay, so a 100 mL linear gradient was used (50 mM NaH2PO4 buffer to de-ionized water), which resulted in complete elution of protein. Fractions (150 drops) were collected and analyzed by SDS-PAGE for the appearance of a band (~35 kDa). Fractions were pooled based on the purity (assessed by SDS-PAGE). The protein was dialyzed into 50 mM NaH2PO4 buffer (pH 7.0) overnight. The dialyzed protein was loaded on a DEAE anion exchange column (~30 mL) equilibrated in 50 mM NaH2PO4 buffer (pH 7.0). Protein was eluted using a linear gradient (0–0.5 M NaCl in the NaH2PO4 buffer, ~100 mL). Fractions (~2 mL) were collected and pooled based on purity (as assessed by SDS-PAGE) and dialyzed into 50 mM NaH2PO4 buffer (pH 7.0).
Protein used for crystallography was concentrated and further purified by gel filtration chromatography using a HiLoad 16/60 Superdex (120 mL) column connected to a fast protein liquid chromatography (FPLC) system. The protein was eluted isocratically (1 mL/min) in 25 mM HEPES buffer (pH 7.5) made 0.1 M in NaCl. Fractions (1.0 mL) were collected, analyzed by SDS-PAGE, and pooled based on purity. The pooled fractions were concentrated and divided into small aliquots. These stocks were flash-frozen with liquid nitrogen, then stored at −80 °C. Typically the yield for this procedure is ~30 mg per L of culture or 3 mg per g of cells. The purity of NahE was determined to be >95% as assessed by SDS-PAGE and electrospray ionization mass spectroscopy (ESI-MS).
Preparation of SeMet-NahE.
The L-selenomethionine (SeMet) derivative of NahE was expressed and purified similar to wild type enzyme with the following modifications. Cells were grown in M9 minimal media with 0.2% glucose until the OD600 reached 0.5–1.0. To each flask were added lysine, phenylalanine, threonine (100 mg each); isoleucine, leucine, and valine (50 mg each); and selenomethionine (60 mg). Additionally, IPTG (1 mM) was added prior to incubation (16 h) at room temperature. The remainder of the purification protocol for the derivative was identical to that of wild-type except dithiothreitol (5 mM) was added to all buffers to prevent oxidation of selenium. The size-exclusion chromatography step was included in the purification.
Construction of the Expression Vectors for Native and Mutant PhdJ.
The gene, phdJ (Mvan_0469), was amplified from the M. vanbaalennii PYR-1 genomic DNA using the PCR with 5’-CGAGAGAGCATATGGTGCACGT-3’ and 5’-TCCTCAGGATCCGTGGTTCGAGAC-3’ as forward and reverse primers, respectively. The primers contained NdeI and BamHI restriction sites (underlined). The PCR product and the pET24 vector were treated with the appropriate restriction enzymes, ligated, and processed to construct the PhdJ expression vector. The PCR introduced three mutations, which were corrected (K44T, Q325E, and G335Stop) using the QuikChange Site-Directed Mutagenesis Kit following the manufacturer’s instructions. The S278N and D282E mutants of PhdJ were constructed using the same kit and the native PhdJ expression vector as the template. The plasmids were transformed into E. coli ArcticExpress cells (Agilent Technologies) following the manufacturer’s protocol. In all three plasmids, the ATG start codon is immediately before the GUG start codon found in the phdJ gene in M. vanbaalennii PYR-1. As a result, the expressed proteins had an additional valine on the N-terminus.
Expression and Purification of Native and Mutant PhdJ.
The E. coli ArcticExpress cells (containing the expression vector) were used to inoculate a starter culture containing 50 mL LB media with kanamycin (Kn, 50 μg/mL) and gentimicin (20 μg/mL). The cultures were grown at 37°C overnight. Subsequently, 4–2L Erlenmeyer flasks containing 400 mL ZYM-5052 auto-induction media (27) and Kn (100 μg/mL) were inoculated with 25 mL starter culture (each) and grown until the OD600 reading reached about 0.3 (~3 h). The cultures were then cooled to 12° C and shaken at 250 rpm for 65 h. Cells were harvested by centrifugation at 11,300 rpm for 20 min and stored at −20 °C. The cell pellet mass for 1.6 L of culture was 25.5 g.
Cells were lysed in 50 mM NaH2PO4 buffer (pH 7.0, 80 mL), as described above. The lysate was centrifuged (12,000 rpm for 30 min), the pellet discarded, and 30 mL of 50 mM NaH2PO4 buffer, pH 7.0, was added to the supernatant. The resulting solution was then centrifuged again (18,000 rpm for 30 min). The supernatant was loaded onto a DEAE column (17 mL) equilibrated with 50 mM NaH2PO4 buffer, pH 7.0. The column was washed (>1 column volume of the same buffer) and the protein was eluted with a linear gradient (0–0.5 M NaCl in the NaH2PO4 buffer, 85 mL) Fractions (1.4 mL) were pooled based on the presence of an SDS-PAGE band at ~35 kD and activity (with 6). Ammonium sulfate (to 30% saturation) was added slowly to the pooled fractions and stirred for 20 min on ice before centrifugation at 18,000 for 20 min. Ammonium sulfate was then added again to the supernatant to reach 40% saturation. After stirring for 20 min on ice, the solution was centrifuged (18,000 rpm, 20 min). The pellet was dissolved into 50 mM NaH2PO4 buffer, pH 7.0. A typical preparation yields ~4.9 mg of purified protein/L culture. An additional Phenyl Sepharose column was performed when purity was <95% (as assessed by SDS-PAGE). Fractions were divided into small aliquots (50–200 μL), flash-frozen with liquid nitrogen, and stored at −80°C.
PhdJ Activity with trans-o-Hydroxybenzylidenepyruvate (5), trans-o-Carboxybenzylidenepyruvate (6), and trans-Benzylidenepyruvate (16).
The enzymatic activities of PhdJ with 5, 6, and 16 were determined as follows. For 5, the substrate (2.7 mg of free acid) was dissolved in ethanol (1 mL). Diluted solutions (1.8, 1.4, 0.90, 0.54, and 0.36 mg/mL) were made in ethanol using the stock solution. Aliquots (6 μL) of these solutions were added to 50 mM NaH2PO4 buffer, pH 7.0 (942 μL) so that the final concentrations of 5 were 85, 56, 42, 28, 17, and 11 μM. The reactions were initiated by the addition of PhdJ (50 μL of a 0.51 mg/mL solution to give a final enzyme concentration of 0.71 μM). Reactions were monitored by following the decrease in absorbance at 296 nm (9,400 M−1cm−1). Spectra were recorded for 60 s at 5-s intervals. The extinction coefficient for 5 was determined by measuring the absorbance of 5 (68 μM) in ethanol (2.5%) in triplicate. The solution was made up by dissolving 5 (5.2 mg) in ethanol (10 mL) and diluting 25 μL of the stock solution into 50 mM NaH2PO4 buffer, pH 7.0 (970 μL). Initial rates were calculated using the differences in the extinction coefficients at 296 nm for 5 (10,700 M−1 cm−1) and salicylaldehyde (9, 1260 M−1cm−1). The extinction coefficient for 9 was determined by dissolving 6 mg in 50 mM NaH2PO4 buffer, pH 7.0 (3 mL). An aliquot (5 μL) was diluted into buffer (995 μL) to make a final concentration of 82.6 μM. The absorbance at 296 nm was measured in triplicate.
For 6, the substrate (2.0 mg) was added to 50 mM NaH2PO4 buffer, pH 7.0 (500 μL) to make a 16 mM stock solution (pH ~8). The stock solution was diluted in the same buffer to make various concentrations (8.1 mM, 4.1 mM, 2.4 mM, 1.6 mM, 1.1 mM, and 0.81 mM). Subsequently, aliquots (10 μL) of these diluted stock solutions were further diluted into 985 μL of buffer to give final concentrations of 81, 41, 24, 16, 11, and 8.1 μM. The reactions were initiated by the addition of PhdJ (5 μL of a 0.51 mg/mL solution to make a final enzyme concentration of 71 nM). Reactions were monitored by following the decrease in absorbance at 300 nm (ε = 9,050 M−1cm−1). Spectra were recorded for 60 s at 3-s intervals. The extinction coefficient for 6 was determined by measuring the absorbance of a solution of 6 (81 μM) in 50 mM NaH2PO4 buffer, pH 7.0 (in triplicate). This concentration is the initial absorbance of the highest concentration of 6 before the addition of enzyme. The initial rates were calculated using the difference between extinction coefficients at 300 nm (10,500 M−1cm−1) for 6 and ortho-carboxybenzaldehyde (10) (1450 M−1 cm−1). The extinction coefficient for 10 was measured by dissolving 3.6 mg in buffer (1 mL). An aliquot (5 μL) was further diluted in buffer (995 μL) and the absorbance was measured in triplicate.
For 16, the substrate (5.4 mg of free acid) was dissolved in ethanol (1 mL). This solution was diluted with ethanol to make stock solutions of 1.1, 0.72, 0.54, 0.36, 0.27, 0.18, and 0.12 mg/mL. Aliquots (7 μL) of the diluted stock solution were added to 50 mM NaH2PO4 buffer, pH 7.0 (943 μL) so that the final concentrations of 16 were 48, 32, 24, 16, 12, 7.9, and 5.3 μM. The reactions were initiated by the addition of PhdJ (10 μL of a 0.51 mg/mL solution to give a final concentration of 1.4 μM). Reactions were monitored by following the decrease in absorbance at 300 nm (20,000 M−1 cm−1). Spectra were recorded for 10 min at 20-s intervals. The extinction coefficient for 16 was determined by measuring the absorbance of a solution made up as follows. A solution of 16 (9.7 mg) in ethanol (1 mL) was diluted 10-fold with ethanol. An aliquot (5 μL) was added to 50 mM NaH2PO4 buffer, pH 7.0 (1 mL) and the absorbance was measured five times. Initial rates were calculated using the differences in the extinction coefficients at 300 nm for 16 (21,000 M−1 cm−1) and benzaldehyde (540 M−1 cm−1). The extinction coefficient for benzaldehyde was determined by diluting a solution of benzaldehyde (4.4 mg in 1 mL ethanol) 200-fold in 50 mM NaH2PO4 buffer, pH 7.0 (1 mL). The absorbance of the resulting solution was measured in triplicate.
S278N and D282E PhdJ Activities with 5 and 6.
For 5, aliquots (7 μL of 0.30–2.20 mg/mL stock solutions made up in ethanol) were diluted into the 50 mM NaH2PO4 buffer, pH 7.0 (986 μL) to make final concentrations (11 μM, 16 μM, 23 μM, 36 μM, 54 μM, and 80 μM) of 5. The reactions were initiated by the addition of S278N PhdJ (240 nM) or D282E PhdJ (310 nM). For 6, aliquots (7 μL of 0.52–2.75 mg/mL stock solutions) were diluted in 50 mM NaH2PO4 buffer, pH 7.0 (986 μL) to make final concentrations (19 μM, 28 μM, 42 μM, 70 μM, and 100 μM) of 6. The reactions were initiated by the addition of S278N PhdJ (340 nM) or D282E PhdJ (160 nM). Saturation could not be achieved for 6 so the kcat/Km values were obtained from a linear fit of the plot of vo vs [S] (28).
1H NMR Identification of the Products in the PhdJ-catalyzed Reaction.
Two PhdJ-catalyzed reactions were followed by 1H NMR spectroscopy (using 5 or 6). To an NMR tube containing 100 mM NaH2PO4 buffer (0.6 mL, pH 7.5) is added a quantity of 5 or 6 [4.5 mg in 30 μL dimethyl sulfoxide (DMSO)-d6)] to reach a final concentration of 30 mM. The final pH of the solution is 7.47. DMSO-d6 was used as the lock signal and for the standardization of the chemical shifts (at 2.49 ppm). The reaction was initiated by adding PhdJ (100 μL of a 0.51 mg/mL solution) for a final concentration of 1.9 μM. Spectra were recorded every 3 min over a 33 min period. 9: 1H NMR (H2O, 600 MHz) δ 9.56 (1H, s), 7.5 (1H, dd, J = 1.3 Hz, 7.7 Hz), 7.42 (1H, m), 6.89 (1H, m), 6.81 (1H, d, J = 8.4 Hz); 10: 1H NMR (H2O, 600 MHz) δ 9.80 (1H, s), 7.68 (1H, d, J = 7.7 Hz), 7.52 (1H, m), 7.39 (2H, m); 11: 1H NMR (H2O, 600 MHz) δ 2.17 (3H, s).
Covalent Modification of PhdJ by 6 in the Presence of NaCNBH3.
A reaction mixture was made up in 50 mM NaH2PO4 buffer (pH 7.0) containing PhdJ (0.5 mg/mL solution) and NaCNBH3 (15 mg/mL in 100 mM Na2HPO4). The final volume was 120 μL (before addition of aliquots of 6). The NaCNBH3 solution was made to 0.75 M in 100 Na2HPO4 buffer (pH ~9). Aliquots from a stock solution of 6 (260 μM) were added to the reaction mixture (1–15 μL increments) until enzyme activity had decreased more than 50% (2–3 h). All reactions were carried out at room temperature. Samples were prepared as described elsewhere and subjected to ESI-MS analysis (29).
Crystallization of the NahE SeMet variant.
Initial crystallization conditions for NahE were identified using sparse-matrix screening with a Phoenix crystallization robotic system (Art Robbins Instruments) and a protein concentration of 6.5 mg/mL in 50 mM sodium phosphate buffer at pH 7.0. The conditions that identified initial hits were then optimized by systematic optimization. The diffraction-quality crystals for the selenomethionine (SeMet) NahE variant were grown in a solution containing 0.1 M 2-morpholin-4-ylethanesulfonic acid (MES) buffer (pH 6.5–8.0) and 21–26% PEG 2000 monomethyl ether (MME) at room temperature.
Crystallization of PhdJ and Soaking with 6.
PhdJ at a concentration of 3.4 mg/mL was used for sparse-matrix crystal screening in the Phoenix crystallization robot (Art Robbins Instruments). Initial hits were identified and then optimized by systematic optimization. Diffracting quality crystals were obtained in the sitting drop using vapor-diffusion method at room temperature with condition of 0.1 M MES at pH 7–7.5 and 22–25% in PEG 3350.
The complex crystal of PhdJ with compound 6 was obtained by soaking PhdJ crystals in a solution of 6 at a concentration of 10 mM for 4 min at room temperature. The solution of 6 was made up by diluting a 10× stock solution (1.2 mg of 6 dissolved in 45μL of 100 mM Na2HPO4 buffer, pH 8–9) into mother liquor at pH 7.0. The crystals of NahE, PhdJ and PhdJ-ligand complex were cryo-protected by mother liquors containing 25–30% glycerol before vitrified in liquid nitrogen in preparation for data collection.
Data Collection, Processing, Structure Determination, and Refinement.
X-ray diffraction data for the SeMet-NahE were collected at Advanced Light Source (ALS) beamline 8.2.1 (Berkeley, CA) where diffractions in multiple wavelengths were captured. X-ray diffraction data of PhdJ and the PhdJ-substrate complex were obtained at Advanced Light Source beamline 5.0.3 (ALS, Berkeley, CA). Diffraction data were indexed, integrated, and scaled using HKL2000 (30). Data collection statistics are summarized in Table 1.
Table 1.
Crystallographic data and refinement statistics for NahE, PhdJ and PhdJ-6adduct.
| Data Collection | PhdJ | PhdJ-6 | SeMet-NahE |
|---|---|---|---|
| space group | P21 | P21 | P3221 |
| cell dimensions | |||
| a, b, c (Å) | 52.4, 137.0, 88.3 | 52.5, 137.2, 88.3 | 85.3, 85.3, 133.3 |
| α, β, γ (deg) | 90.0, 99.1, 90.0 | 90.0, 98.9, 90.0 | 90.0, 90.0, 120.0 |
| resolution (Å) | 40.00–2.05 (2.12–2.05)1 | 50.00–2.00 (2.07–2.00)1 | 40.0–1.94 (2.01–1.94)1 |
| Rsym | 0.104 (0.496) 1 | 0.087 (0.326) 1 | 0.150 (0.506)1 |
| I/σI | 11 (2.1) 1 | 14 (3.2)1 | 15 (4.5)1 |
| completeness (%) | 94.3 (83.6) 1 | 93.5 (88.7)1 | 100.0 (100.0)1 |
| Refinement | |||
| resolution (Å) | 39.90–2.05 | 45.29–2.00 | 38.09–1.94 |
| no. of unique reflections | 72626 | 77204 | 42138 |
| Rwork/Rfree (%) | 17.95/22.39 | 16.34/19.20 | 13.55/18.63 |
| no. of atoms | |||
| protein | 9893 | 9960 | 5053 |
| water | 894 | 1116 | 650 |
| Average B factor (Å2) | |||
| protein | 32.9 | 23.3 | 14.1 |
| water | 39.1 | 33.7 | 26.6 |
| rmsd | |||
| bond lengths (Å) | 0.002 | 0.003 | 0.010 |
| bond angles (deg) | 0.644 | 0.772 | 1.140 |
| Ramachandran plot (%) | |||
| most favored regions | 98.2 | 98.3 | 98.3 |
| allowed regions | 1.85 | 1.70 | 1.7 |
| disallowed regions | 0.00 | 0.00 | 0.0 |
Data for the last resolution shell are given in parentheses.
Rfree is calculated with 5% of data randomly omitted from refinement.
Phases for the SeMet-NahE were calculated using Autosol from the PHENIX suite of programs (31) using multiwavelength anomalous dispersion (MAD). The native PhdJ structure was determined by molecular replacement (MR) using Phaser (32) and Autobuild (33) from the PHENIX suite of programs. The monomeric NahE was used as a search model for the initial estimates of the PhdJ structure factors. Subsequently, the structure of the native PhdJ was used as a search model for PhdJ-substrate structure determinations. Structure refinement was carried out using PHENIX Refine (34), in which TLS parameter refinement was included in the refinement. The structure models were evaluated using Molprobity (35) and Procheck (36). The refinement statistics for all structures are summarized in Table 1. All figures were prepared with PyMol (37).
RESULTS AND DISCUSSION
Dynamic Light Scattering.
Two peaks were observed in the LC chromatograph (19.9 min and 22.4 min) for NahE. The first peak (97.5% mass fraction) had a molar mass moment of 145,700 Da. This species is likely the tetrameric form of NahE (calculated mass, 146,548 Da). The second peak (2.5% mass fraction) had a molar mass of 36,660 Da and this species is presumed to be monomeric NahE, as the calculated monomeric mass is 36,637 Da.
The calculated monomeric mass of PhdJ is 35,966.8 Da. Of the 4 absorbance peaks observed in the LC chromatogram for a sample, the largest peak (accounting for 88.0% of the sample mass) corresponded to a PhdJ tetramer (molar mass moment of 138,300). The fourth peak contained 4.8% of the total mass and is consistent with a trimer of tetramers (molar mass moment of 421,500). The other two peaks (with 7.2% of the mass of the sample) are likely impurities from the protein preparation. Thus, the predominant PhdJ molecule in solution is a tetramer.
Sequence Analysis.
An alignment of representative sequences in the N-acetylneuraminate lyase (NAL) subgroup was carried out using the program ESPript (Figure 1) (38). The alignment includes three hydratase-aldolase-catalyzed reactions in the bacterial catabolic pathways for phenanthrene (PhdJ and PhdG) and the naphthalene (NahE) (Scheme 2). (PhdG is proposed to catalyze an earlier step in phenanthrene catabolism.) The alignment shows the strict conservation of Lys-180 (using the PhdJ numbering system), Tyr-152, and the GXXGE motif (Gly-61, Thr-62, Phe-63, Gly-64, Glu-65) at the active site. These residues are the so-called primary residues involved in the common reaction of the NAL subgroup, which is Schiff base formation and binding of the α-keto acid moiety (of substrate). Three other residues are also conserved (Glu-74, Arg-298, Pro-300), but these are found outside the active site and might play a structural role.
Figure 1.

Sequence alignment of representative NAL subgroup members. The secondary structural elements for PhdJ are shown above the alignment. The α-helices and 310 (η) helices are indicated by large and small squiggles, respectively. β-strands are shown as arrows and β-turns are indicated by TT letters. Red letters indicate similar residues and white lettering with red fill indicates conserved residues. Similar and conserved residues are shown in blue boxes. The dots beneath residues indicate primary residues (blue) in the NAL subgroup, secondary residues (red) common in hydratase-aldolases, and secondary residues (green) that are specific to individual hydratase-aldolases, as discussed in the text. The red line indicates an insertion in the hydratase-aldolases that codes for α-helix 10, as discussed in the text. The sequences shown are M. vanbaalenii PYR-1 PhdJ (GI: 49072891), M. vanbaalenii PYR-1 PhdG (GI: 49072894), Pseudomonas putida NahE (GI: 483790), Escherichia coli NAL (GI: 216589), E. coli DHDPS (GI: 758861597), Sulfolobus solfataricus KDGA (GI: 27429444), and Agrobacterium fabrum KDGDH (GI: 1134942). The alignment was carried out and the Figure was generated using ESPript version 3.0 (38).
Three other residues, Asn-154, Trp-225, and Phe-274, are conserved in the three hydratase-aldolases (indicated by a red dot). Eight additional residues are specific to the individual hydratase-aldolase (indicated by a green dot). In PhdJ, these residues are Thr-62, Trp-125, Leu-182, Phe-271, Leu-275, Ser-278, Asp-282, and Gln-285. In NahE, the first four residues are the same, but the last four residues (Leu-275, Ser-278, Asp-282, Gln-285) are replaced by Ser, Asn, Glu, and Arg (respectively). All 11 residues are likely the so-called secondary residues that are responsible for the unique chemistry of the hydratase-aldolases and their specificities.
Covalent Modification of PhdJ in the Presence of Substrate and NaCNBH3.
PhdJ was incubated with substrate (6) in the presence of NaCNBH3 (for 2–3 h). Activity decreased significantly when compared to a control sample without compound or NaCNBH3. The ESI mass spectrum of the control sample showed a major signal at 35,967 Da whereas the mass spectrum of the treated sample showed a major signal at 36,173 Da (calculated, 36,169.8 Da). The mass difference, 206 Da, is consistent with the reduced imine of 6 attached to the enzyme. The loss of PhdJ activity (in the presence of NaCNBH3 and 6) coupled with a single covalent modification is consistent with the formation of a Schiff base between substrate and enzyme. A similar result for NahE has been previously reported (39).
1H NMR Characterization of the PhdJ-catalyzed Reactions.
The PhdJ-catalyzed reaction using 6 was monitored by 1H NMR spectroscopy in order to confirm the identities of the products. After 33 min, the spectra show signals consistent with the formation of 10 and 11 from 6 (Scheme 2). In a similar NMR experiment, the spectra show signals that indicate the formation of 9 and 11 from 5. The NMR experiments show that both substrates are processed by PhdJ and afford products that are consistent with the proposed reactions.
Kinetic Analysis.
The activity of PhdJ was measured with three substrates (5, 6, and 16) (Table 2). The substrates vary by the ortho-substituent on the ring (R = OH, CO2−, H). The best substrate is 6, as assessed by the highest kcat/Km value (~8.2 × 104 M−1 s−1). The value is consistent with those in the literature and the proposed role of PhdJ in the catabolic pathway (14). Replacing the ortho-carboxylate group with a hydroxyl group (i.e., 5, the substrate for NahE) results in 26-fold drop in the kcat/Km value (due mostly to the 23-fold drop in kcat). Finally, removing the ortho-substituent entirely (i.e., 16) causes a 1025-fold drop in the kcat/Km value. It is also not possible to saturate the enzyme with 16.
table 2.
Kinetic Parameter for PhdJ and Mutants Using Various Substratesa
| Substrate | Enzyme |
kcat (S−1) |
Km (μM) |
kcat/Km (M−1 s−1) |
|---|---|---|---|---|
 |
PhdJ | 0.28 ± 0.04 | 90 ±20 | (3.1 ± 0.8) x 103 |
| S278N PhdJ | <0.01 | 40 ± 6 | 130 ±40 | |
| D282E PhdJ | <0.01 | 40 ±10 | 200 ±60 | |
 |
PhdJ | 6.5 ± 0.4 | 80 ±7 | (8.2 ± 0.9) x 104 |
| S278N PhdJ | - | - | (3.6 ± 0.3) x 103 | |
| D282E PhdJ | - | - | 53 ±7 | |
 |
PhdJ | - | - | 80 ± 1 |
The steady-state kinetic parameters were determined under the conditions described in the text.
Crystal structure of NahE.
In order to gain structural insights into catalysis and specificity of the hydratase-aldolases, the structures of NahE, PhdJ, and a complex of PhdJ with 6 were determined. Due to the low sequence similarity between NahE and other NAL family members, the structure of NahE was solved by de novo phasing using the SeMet derivative of NahE (19). The final model is refined to 1.9 Å resolution with two monomers per asymmetric unit (Table 1). As evident from light scattering experiments, NahE functions as a tetramer in solution. In the crystal structure, NahE shows a configuration of dimer of dimers with two monomers forming a tight dimer and then the pair of dimers forming the physiologically functioning unit of a tetramer (Figure 2A). This results in two different dimer interfaces: a tight interface for the two monomers to form a dimer with 2009.3 Å2 accounting for 17.4% of the surface and a loose interface between the two dimers with only 746.1 Å2 for 5.4% of surface, as calculated by the PISA program (40). The overall fold of NahE is typical of the NAL subgroup members (15,19), which are composed of (α/β)8-barrels at the N-terminal catalytic moiety of the enzyme (residue 8–250), followed by three C-terminal helices tilted on the side of the barrel (residue 251–331) (Figure 2B). The active site of NahE is located at the heart of the barrel with the highly conserved features of the NAL subgroup (Lys-183, Tyr-155, and the GXXGE motif) (Figure 1 and Figure 2C) (15,19). In addition, hydrophobic residues (Ile-22, Phe-66, Met-122, Tyr-155, Trp-224) dominate the active site pocket. Trp-128, from the neighboring monomer, also participates the formation of the active site (Figure 2C).
Figure 2.
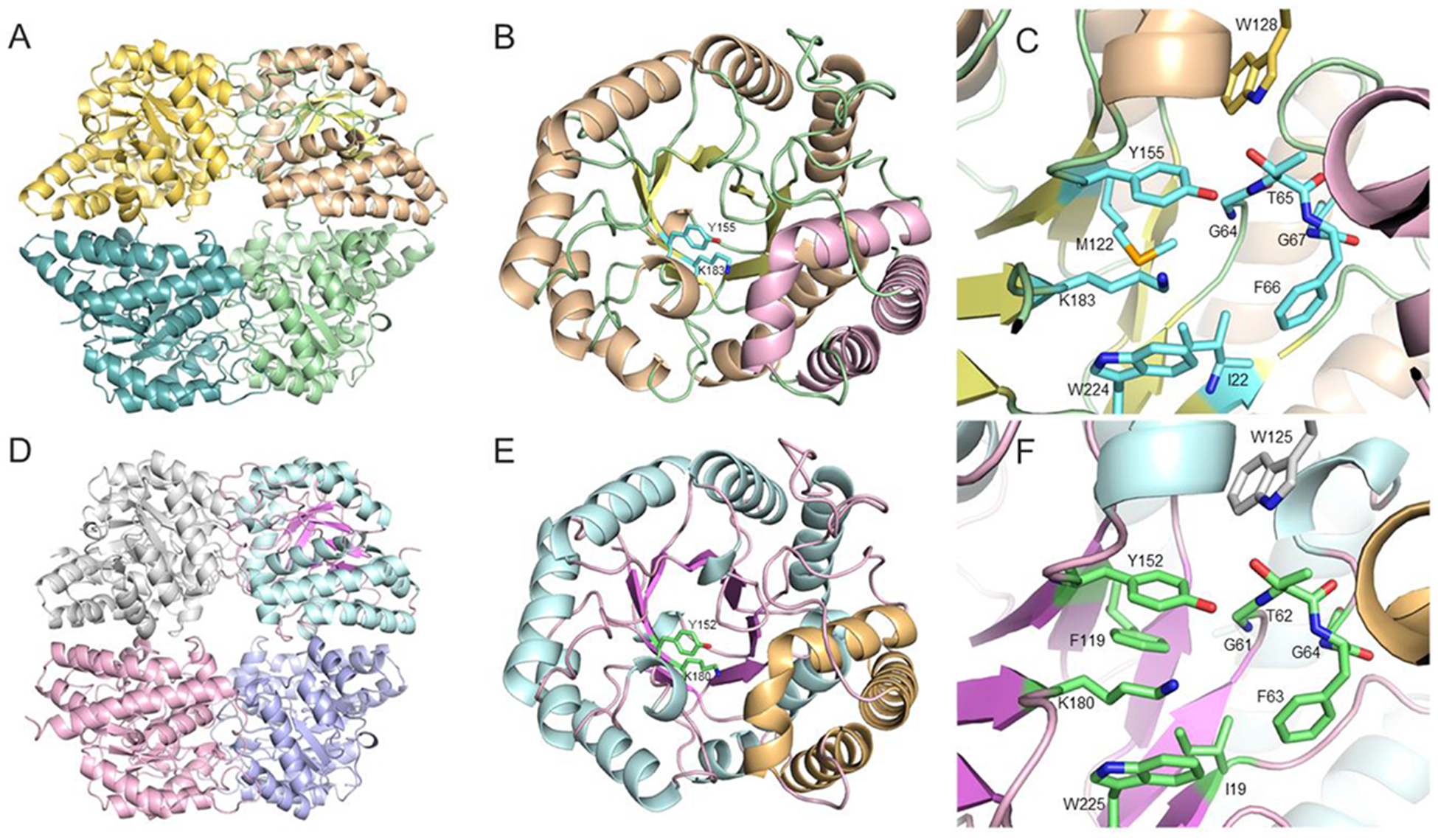
Overview of the NahE and PhdJ structures. A) NahE tetramer shown with a different color for each monomer. B) Overall structure of the NahE monomer shown as a ribbon diagram. The N-terminal TIM barrel domain is shown with the α-helices in wheat, β-strands in yellow, and coil in green. The C-terminal helical bundle is shown in pink. The highly conserved active site residues Tyr-155 and Lys-183 are highlighted in carbon colored in cyan and shown as sticks. C) The active site of NahE. Important residues are shown as sticks with carbon atoms colored cyan. The residue from the adjacent monomer is colored in gold. D) PhdJ tetramer shown with a different color for each monomer. E) Overall structure of PhdJ monomer shown as a ribbon diagram. The N-terminal TIM barrel domain is shown with the α-helices in cyan, β-strands in violet, and coil in pink. The C-terminal helical bundle is colored yellow. The highly conserved active site residues Tyr-152 and Lys-180 are shown as sticks with carbon atoms colored green. F) The active site of PhdJ. Important active site residues are shown as sticks with the carbon atoms colored green. The residue from the adjacent monomer is colored silver.
Crystal Structure of Native PhdJ.
The crystal structure of the native PhdJ was determined by molecular replacement using the NahE structure as the search model. The final structure was refined to 2.1 Å resolution (Table 1). Similar to NahE, PhdJ maintains the same architecture of tetramer formation as a dimer of dimers (Figure 2D), consistent with dynamic light scattering profile in solution. The dimer in between the monomers is 1983.1 Å2 (accounting for 15.2% of the surface), substantially larger than the interface between the dimers (635.5 Å2, 4.7% of the surface). For each monomer, a core (α/β)8-barrel (TIM barrel) forms the catalytic portion of the enzyme (residue 1–248) followed by a C-terminal helical bundle (residue 249–329) leaning against the barrel (Figure 2E). The conserved Lys-180, implicated in Schiff base formation and hydrolysis, is found at the center of the barrel on β-strand 6, and indicates the location of the active site. The conserved tyrosine (Tyr-154) is found nearby along with the GXXGE motif. Like NahE, the active site pocket is dominated by hydrophobic residues (e.g., Ile-19, Phe-63, Phe-119, Tyr-152, Trp-225), suggesting a preference for a hydrophobic substrate (Figure 2F). Similar to NahE, a conserved tryptophan residue (i.e., Trp-125) from neighboring monomer forms part of the active site.
Formation of the PhdJ complex with substrate 6.
To gain insight into the catalytic reaction carried out by PhdJ, we soaked the substrate 6 (10 mM) into pre-formed PhdJ crystals in mother liquor conditions for 4 min. The crystal data were collected and scaled to 2.0 Å resolution. The native PhdJ structure was used as the search model to find a solution for the structure (Table 1). The folding of the substrate-soaked protein shows an identical configuration to that of native PhdJ. However, strong positive density is observed in all four active sites (Figure 3A). The density extends from the side chain of Lys-180, suggesting the formation of a Schiff base between the side chain of Lys-180 and the 2-keto group of the substrate. Iterative cycles of ligand building were used to establish the identity of the ligand at the active site (Figure 3B). Strong and flat positive density prompted us to model the phenyl ring of 6 in the density. Further calculations reveal strong positive density connected between the Schiff base (at C-2) and the phenyl ring (Figures 3B and 3C). In three of the four active sites, the strong positive density at the active site is consistent with the formation of a Schiff base between Lys-180 and 6 (Figure 3D). Formation of the Schiff base between PhdJ and substrate is surprising because the wild type PhdJ is likely still active in the crystalline form. The thermal factors of the adduct moiety of Lys-180 (~30 Å2) are slightly higher than the main chain atoms of 20 Å2, but comparable to the overall average B factors of the whole molecule. We speculate that the short soaking time, high precipitant concentration, and crystal packing stabilize protein-substrate adduct formation and slow down the product turnover to allow the capture of the complex.
Figure 3.

The electron density maps at the active site of PhdJ after being soaked in substrate 6. A) The Fo–Fc map contoured to 2.5σ shown in green mesh extending from Lys-183. The structure of a Schiff base between the side chain of Lys-183 and the pyruvoyl moiety of substrate is modeled in the density. B) Continuous 2Fo–Fc electron density, contoured at 1.0 σ, shown for the Lys-183-pyruvoyl adduct in blue. The Fo–Fc map is contoured at 2.5 σ showing additional positive electron density. C. A phenyl ring is built into the positive density observed in 3B. The 2Fo–Fc map contoured to 1.0 σ shows complete coverage of the phenyl ring. The Fo–Fc map contoured at 3 σ indicates positive density consistent with the o-carboxylate group of 6 and linker atoms between the phenyl ring and the pyruvoyl adduct. D) 2 Fo–Fc map contoured at 1.0 σ superimposed with the Lys-183 forming an adduct with 6.
In the native enzyme, the ε-amino group of Lys-180 is sandwiched between Tyr-152 and Trp-225 with a distance of 3.5 Å from the nitrogen to each π system (Figure 4A). The same arrangement is observed in the NahE structure. This orientation could be responsible (in part) for lowering the pKa of the ε-amino group to allow deprotonation and thereby facilitate the nucleophilic attack at the C-2 carbonyl of substrate. Tyr-152 is found in all known NAL subgroup members and Trp-225 is conserved in the three hydratase-aldolases (Figure 1). The hydroxyl group of Tyr-152 is within hydrogen bond distance to the C-1 carboxylate group of the adduct and could stabilize adduct formation (Figure 4B) as well as assist in the formation and hydrolysis of the Schiff base (15,19,20,40). The substrate adduct is further stabilized by the highly conserved GXXGE motif with the carboxylate group forming hydrogen bonds with the backbone amides of Thr-62 and Phe-63 (Figure 4B). The phenyl ring of the substrate (i.e., 6) extends back to a hydrophobic portion of the active site composed of residues including Leu-265, Tyr-266, Phe-271, Phe-274 and Leu-275 (Figure 4C). These hydrophobic residues form an α-helix that provides a lid to cover the active site and sequester it from the bulk solvent.
Figure 4.

Interactions of active site residues in PhdJ with 6. A) The ε-amino group of Lys-180 is sandwiched between conserved residues Tyr-152 and Trp-225. B) Covalent adduct between Lys-180 and 6 shown in dark green. The hydrogen bond interaction between 6 and Tyr-152 and the GXXGE motif of PhdJ are shown in orange dotted lines. C) The hydrophobic helix in the active site of PhdJ is shown in yellow with residues Leu-275, Phe-274, Phe-271, Leu-265 and Tyr-266 depicted as sticks.
Interestingly, when PhdJ is compared with other proteins in this subgroup, the overall folding of PhdJ superimposes well with NAL (PDB entry 1NAL) and dihydrodipicolinate synthetase (DHDPS) (PDB entry 1DHP) (Figure 5A). The only major difference is the insertion of this hydrophobic helix in PhdJ (Figure 5A) (19). This structural insert in PhdJ versus NAL and DHDPS might account (in part) for the substrate specificities where PhdJ mediates the reaction of substrates with a hydrophobic portion and NAL and DHDPS process hydrophilic substrates. A comparison of the PhdJ and NahE structures shows that this helix is conserved (Figure 5B).
Figure 5.

The specificity and mechanism of PhdJ. A. Superimposition of PhdJ with NAL and DHDPS. PhdJ is shown with the α-helix in cyan and β-strand in pink for the N-terminal TIM barrel domain. The hydrophobic helix, found only in PhdJ, is shown in yellow. DHDPS is colored in dark grey and NAL is shown in white. B) Superimposition of PhdJ and NahE. The additional hydrophobic helix found in both proteins is colored yellow and cyan, respectively. C) Water molecules in the active site are conserved in their location in different molecules in the asymmetric unit. Hydrogen bonds are shown in dash lines with the distances indicated. The side chains of residues and the Schiff base between Lys-180 and 6 are represented by sticks. D) Superimposition of the active sites of PhdJ (cyan) and NahE (wheat). The hydrogen bonding between the o-carboxylate group of 6 and Ser278/Asp282 of PhdJ is shown with yellow dash lines.
This is consistent with the fact that the substrates for PhdJ and NahE have a phenyl ring (Scheme 4A), and those for the other sub-group members do not (Scheme 4B). Instead, the four substrates have hydrophilic groups.
Scheme 4.

Representative substrates for the NAL subgroup members.
Even though this helix confers hydrophobic character to the active site pocket, there are several water molecules present. Particularly, there is a water molecule (W1) anchored by Asn-154 through a hydrogen bond (distance ~2.8–3.0 Å) and another water molecule (W2) that forms hydrogen bonds with the backbone carbonyl group of Leu-275 (~2.7–3.1Å) (Figure 5C). The positions of these two water molecules are strictly conserved within the three active sites where the PhdJ-substrate adduct is observed (Figure 5C). This hydrogen bond network places W1 about 3.4 Å away from C-4 of substrate, in a position suitable for addition across the C3,C4 bond (Scheme 5).
Scheme 5.

One possible mechanism for PhdJ.
In contrast to the interactions between the hydrophobic helical moiety and the substrate, the o-carboxylate group on the phenyl ring of substrate forms polar interactions with Asp-282 and Ser-278 of PhdJ. The carboxylate group is tilted at an angle of about 60° from the planar phenyl ring (Figure 3D) in order to minimize steric clash with the adjacent substituent. This carboxylate group interacts with Asp-282 via a hydrogen bond through a distance of 2.8 Å, with the hydroxyl group of Ser-278 nearby (3.6–3.7 Å) (Figure 5D). In NahE, these two residues are replaced with Glu-285 (in place of Asp-282) and Asn-281 (in place of Ser-278). If the o-carboxylate group binds in the same configuration in the presence of these two groups, the longer side chain of Glu-285 and the proximity of Asn-281 (1.5 Å away) would introduce steric clashes and reduce activity.
In order to examine the consequences of such a scenario, Asp-282 and Ser-278 were replaced (in separate mutants) with their NahE counterparts, Glu-285 and Asn-281, respectively, and the resulting enzymes were examined for activity with 6. Neither mutant could be saturated with substrate, suggesting less optimal binding. However, based on the kcat/Km values, the D282E mutation is much more detrimental than the S278N mutation: the D282E mutant shows a 1547-decrease in kcat/Km, whereas the S278N mutant only shows a 22-fold decrease. Evidently, the longer side chain of Glu-285 is more damaging than is the replacement of the serine with an asparagine. Interestingly, the D282E mutant has better activity with 5 than it does with 6, as assessed by the 3.8-fold increase in kcat/Km values.
Mechanistic Implications.
The common chemistry in the NAL subgroup members (as well as in the Class I aldolase superfamily) is the utilization of a Schiff base intermediate that forms between the strictly conserved lysine and the α-keto acid moiety of substrate (15,19). Sequence and crystallographic analysis along with Na(CN)BH3 trapping experiments, reported here and elsewhere (39), support the Schiff base intermediate through Lys-183 (NahE) and Lys-180 (PhdJ). The carboxylate group of substrate 6 (in the PhdJ•6 structure) interacts with the backbone amides of Thr-62 and Phe-63 in the conserved GTFGE motif (Step 1, Scheme 5). The crystal structures of NahE and PhdJ also show that the ε-amino group of the conserved lysine is sandwiched between the non-polar aromatic portions of Trp-225 and Tyr-152 (in PhdJ). This positioning favors the neutral form, suggesting a mechanism to lower (in part) the pKa of the lysine such that it can function as a nucleophile. The proximity of the hydroxyl group of Tyr-180 suggests that it could also play a role in Schiff base formation and hydrolysis, as has been suggested (41).
What distinguishes NahE and PhdJ from the other NAL subgroup members is that they catalyze a hydration reaction that sets up the retro-aldol fission. The crystal structure of the PhdJ•6 complex suggests a possible mechanism. There are two strictly conserved water molecules (Figure 5C) in the vicinity of C-4 of 6, with one being ~3.4 Å away (and anchored by Asn-154). The nearby hydroxyl group of Tyr-152 might assist in the addition of one of these water molecules across the C3,C4 double bond, which is in conjugation with the imine of the Schiff base (Step 2, Scheme 5). Once added, Tyr-152 could initiate bond cleavage to release 10 (in the case of PhdJ) and 9 (in the case of NahE). Free enzyme and pyruvate (i.e., 11) are released by hydrolysis of the Schiff base (Step 3, Scheme 5). This proposed mechanism is under investigation.
Basis for Specificity.
In addition to the mechanistic distinction, the substrates for the hydratase-aldolases are different than those for the other NAL subgroup members. The substrates for NahE and PhdJ have a hydrophilic end (the α-keto acid group) and a hydrophobic end (the phenyl and olefin moiety) whereas the substrates for all the other members are hydrophilic all along the molecule (Scheme 4). The hydrophobic end appears to be accommodated by the “greasy” side chains from the α-helix insert (Figure 4C), observed only in NahE and PhdJ. The active site residues that bind the o-substituent of 5 and 6 determine the individual specificities of NahE and PhdJ. For NahE, these residues are Asn-281 and Glu-285, and for PhdJ, they are Ser-278 and Asp-282.
ACKNOWLEDGEMENTS
The protein mass spectrometry analysis was conducted in the Institute for Cellular and Molecular Biology Protein and Metabolite Analysis Facility at the University of Texas at Austin. We thank Steve D. Sorey (Department of Chemistry, University of Texas at Austin) for his expert assistance in the acquisition of the 1H NMR spectra. Instrumentation and technical assistance for this work were provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, and the Institute for Cellular and Molecular Biology at the University of Texas at Austin.The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Funding
This research was supported by the National Institutes of Health Grant (GM-41239 to CPW and GM-104896 to YJZ) and the Robert A. Welch Foundation (F-1334 to CPW and F-1778 to YJZ).
ABBREVIATIONS
- DHDPS
dihydrodipicolinate synthetase
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediamine tetraacetic acid
- ESI-MS
electrospray ionization mass spectrometry
- HEPES
4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid
- IPTG
isopropyl β-D-thiogalactoside
- Kn
kanamycin
- KDGA
2-keto-3-deoxygluconate aldolase
- KDGDH
D-5-keto-4-deoxyglucarate dehydratase
- LB
Luria-Bertani
- PAH
polycyclic aromatic hydrocarbon
- MAD
multi-wavelength anomalous dispersion
- MALDI-MS
matrix-assisted laser desorption/ionization-mass spectrometry
- MR
molecular replacement
- MME
monomethyl ether
- MES
2-morpholin-4-ylethane sulfonic acid
- NMR
nuclear magnetic resonance
- NAL
N-acetylneuraminate lyase
- PEG
polyethylene glycol
- PCR
polymerase chain reaction
- PMSF
phenylmethylsulfonyl fluoride
- SeMet
L-selenomethionine
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Footnotes
Notes
The authors declare no competing financial interest.
Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank: PDB entry 6DAO for SeMet-NahE variant, PDB entry 6DAN for native PhdJ, and PDB entry 6DAQ for the PhdJ•6 adduct.
REFERENCES
- 1.PAHs: An Ecotoxicological Perspective (Douben PET, Ed.) (2003) John Wiley & Sons, Ltd. [Google Scholar]
- 2.Penning TM, Burczynski ME, Hung C-F, McCoull KD, Palackal NT, and Tsuruda LS (1998) Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: generation of reactive and redox active o-quinones. Chem. Res. Toxicol 12, 1–18. [DOI] [PubMed] [Google Scholar]
- 3.Tuvikene A (1995) Responses of fish to polycyclic aromatic hydrocarbons (PAHs). Ann. Zool. Fennici 32, 295–309. [Google Scholar]
- 4.Ramesh A, Walker SA, Hood DB, Guillen MD, Schneider K, and Weyand EH (2004) Bioavailability and risk assessment of orally ingested polycyclic aromatic hydrocarbons. Int. J. Toxicol 23, 301–333. [DOI] [PubMed] [Google Scholar]
- 5.Atlas RM, and Hazen TC (2011) Oil biodegradation and bioremediation: A tale of the two worst spills in U.S. history. Environ. Sci. Technol 45, 6709–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eaton RW, and Chapman PJ (1992) Bacterial metabolism of naphthalene: construction and use of recombinant bacteria to study ring cleavage of 1,2-dihydroxynaphthalene and subsequent reactions. J. Bacteriol 174, 7542–7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kweon O, Kim S-J, Holland RD, Chen H, Kim D-W, Gao Y, Yu L-R, Baek S, Baek D-H, Ahn H, and Cerniglia CE (2011) Polycyclic aromatic hydrocarbon metabolic network in Mycobacterium vanbaalenii PYR-1. J. Bacteriol 193, 4326–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stingley RL, Khan AA, and Cerniglia CE (2004) Molecular characterization of a phenanthrene degradation pathway in Mycobacterium vanbaalenii PYR-1. Biochem. Biophys. Res. Commun 322, 133–146. [DOI] [PubMed] [Google Scholar]
- 9.Kelley I, and Cerniglia CE (1991) The metabolism of fluoranthene by a species of Mycobacterium. J. Indust. Microbiol 7, 19–26. [Google Scholar]
- 10.Kelley I, Freeman JP, Evans FE, and Cerniglia CE (1993) Identification of metabolites from the degradation of fluoranthene by Mycobacterium sp. strain PYR-1. Appl. Environ. Microbiol 59, 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kweon O, Kim SJ, Jones RC, Freeman JP, Adjei MD, Edmondson RD, and Cerniglia CE (2007) A polyomic approach to elucidate the fluoranthene-degradative pathway in Mycobacterium vanbaalenii PYR-1. J. Bacteriol 189, 4635–4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SJ, Kweon O, Jones RC, Freeman JP, Edmondson RD, and Cerniglia CE (2007) Complete and integrated pyrene degradation pathway in Mycobacterium vanbaalenii PYR-1 based on systems biology. J. Bacteriol 189, 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YH, Freeman JP, Moody JD, Engesser KH, and Cerniglia CE (2005) Effects of pH on the degradation of phenanthrene and pyrene by Mycobacterium vanbaalenii PYR-1. Appl. Microbiol. Biotechnol 67, 275–285. [DOI] [PubMed] [Google Scholar]
- 14.Iwabuchi T, and Harayama S (1998) Biochemical and genetic characterization of trans-2’-carboxybenzalpyruvate hydratase-aldolase from a phenanthrene-degrading Nocardioides strain. J. Bacteriol 180, 945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrence MC, Barbosa JA, Smith BJ, Hall NE, Pilling PA, Ooi HC, and Marcuccio SM (1997) Structure and mechanism of a sub-family of enzymes related to N-acetylneuraminate lyase. J. Mol. Biol 266, 381–399. [DOI] [PubMed] [Google Scholar]
- 16.Blickling S, Renner C, Laber B, Pohlenz HD, Holak TA, and Huber R (1997) Reaction mechanism of Escherichia coli dihydrodipicolinate synthase investigated by X-ray crystallography and NMR spectroscopy. Biochemistry 36, 24–33. [DOI] [PubMed] [Google Scholar]
- 17.Theodossis A, Walden H, Westwick EJ, Connaris H, Lamble HJ, Hough DW, Danson MJ, and Taylor GL (2004) The structural basis for substrate promiscuity in 2-keto-3-deoxygluconate aldolase from the Entner-Doudoroff pathway in Sulfolobus solfataricus. J. Biol. Chem 279, 43886–43892 [DOI] [PubMed] [Google Scholar]
- 18.Taberman H, Andberg M, Parkkinen T, Janis J, Penttila M, Hakulinen N, Koivula A, and Rouvinen J (2014) Structure and function of a decarboxylating Agrobacterium tumefaciens keto-deoxy-D-galactarate dehydratase. Biochemistry 53, 8052–8060. [DOI] [PubMed] [Google Scholar]
- 19.Barbosa JA, Smith BJ, DeGori R, Ooi HC, Marcuccio SM, Campi EM, Jackson WR, Brossmer R, Sommer M, and Lawrence MC (2000) Active site modulation in the N-acetylneuraminate lyase sub-family as revealed by the structure of the inhibitor-complexed Haemophilus influenzae enzyme. J. Mol. Biol 303, 405–421. [DOI] [PubMed] [Google Scholar]
- 20.Daniels AD, Campeotto I, van der Kamp MW, Bolt AH, Trinh CH, Phillips SE, Pearson AR, Nelson A, Mulholland AJ, and Berry A (2014) Reaction mechanism of N-acetylneuraminic acid lyase revealed by a combination of crystallography, QM/MM simulation, and mutagenesis. ACS Chem. Biol 9, 1025–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sambrook J, Fritsch EF, and Maniatis T (1989) Molecular Cloning: A Laboratory Manual, 2nd ed, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 22.Waddell WJ (1956) A simple ultraviolet spectrophotometric method for the determination of protein. J. Lab. Clin. Med 48, 311–314. [PubMed] [Google Scholar]
- 23.Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 24.LeVieux JA, Baas BJ, Kaoud TS, Davidson R, Babbitt PC, Zhang YJ, and Whitman CP (2017) Kinetic and structural characterization of a cis-3-chloroacrylic acid dehalogenase homologue in Pseudomonas sp. UW4: A potential step between subgroups in the tautomerase superfamily. Arch. Biochem. Biophys 636, 50–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hajipour G, Johnson WH, Dauben PD, Stolowich NJ, and Whitman CP (1993) Chemical and enzymic ketonization of 5-(carboxymethyl)-2-hydroxymuconate. J. Am. Chem. Soc 115, 3533–3542. [Google Scholar]
- 26.Reimer M (1931) Benzalpyruvic acid dibromide. J. Am. Chem. Soc 53, 3147–3149. [Google Scholar]
- 27.Studier FW (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- 28.Wang SC, Johnson WH Jr., and Whitman CP. (2003) The 4-oxalocrotonate tautomerase- and YwhB-catalyzed hydration of 3E-haloacrylates: implications for the evolution of new enzymatic activities. J. Am. Chem. Soc 125, 14282–14283. [DOI] [PubMed] [Google Scholar]
- 29.Wang SC, Person MD, Johnson WH Jr., and Whitman CP (2003) Reactions of trans-3-chloroacrylic acid dehalogenase with acetylene substrates: consequences of and evidence for a hydration reaction. Biochemistry 42, 8762–8773. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bunkoczi G, Echols N, McCoy AJ, Oeffner R, Adams PD, and Read RJ (2013) Phase.MRage: automated molecular replacement. Acta Crystallogr. D. Biol. Crystallogr 69, 2276–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Echols N, Moriarty NW, Klei HE, Afonine PV, Bunkoczi G, Headd JJ, McCoy AJ, Oeffner R, Read RJ, Terwilliger TC, and Adams PD (2014) Automating crystallographic structure solution and refinement of protein-ligand complexes. Acta Crystallogr. D. Biol. Crystallogr 70, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D. Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laskowski RA, MacArthur MW, Moss DS, and Thornton JM (1993) PROCHECK - a program to check the stereochemical quality of protein structures. J. Appl. Cryst 26, 283–291. [Google Scholar]
- 37.De Lano WL (2002) The PyMol molecular graphics system. DeLano Scientific, San Carlos, CA. [Google Scholar]
- 38.Robert X, and Gouet P (2014) Deciphering key features in protein structures with the new ENDscript server. Nucl. Acids Res 42, W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LeVieux JA, Johnson WH Jr., Zhang Y, and Whitman CP. (2016) The bacterial catabolism of polycyclic aromatic hydrocarbons: characterization of three hydratase/aldolase-catalyzed reactions. Perspect. Sci 9, 33–41. [Google Scholar]
- 40.Krissinel E, and Henrick K (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- 41.Choi KH, Lau V, Foster CE, Morris AJ, Tolan DR, and Allen KN (2006) New superfamily members identified for Schiff-base enzymes based on verification of catalytically essential residues. Biochemistry 45, 8546–8555. [DOI] [PubMed] [Google Scholar]