

Abstract
Oxidative stress is implicated in the etiology of many ethanol-induced pathologies. Oxidative stress has been shown to contribute to the development of endothelial dysfunction and cardiovascular disease, such as hypertension. This review details mechanisms of vascular function, the role of oxidative stress in vascular biology and how ethanol consumption may alter endothelial and smooth muscle cell function as well as microvascular function. Also reviewed are data from human investigations that have examined the association between alcohol consumption and changes in blood pressure and increased risk for hypertension.
Introduction
There is a growing body of evidence, that ethanol exposure is associated with the development of oxidative stress either directly (via stimulating the generation of free radicals) or indirectly (via increasing the susceptibility of the cell to other stressors) (Table 1) (1, 2). Consequently, oxidative stress is implicated in the etiology of many ethanol-induced pathologies (1, 3). The presence of reactive oxygen species (ROS) and the generation of an oxidative environment can accelerate vascular dysfunction, thereby contributing to the development of endothelial dysfunction leading to hypertension (HTN)(4). This review will focus on mechanisms of vascular function, the role of oxidative stress in vascular biology, and review how ethanol consumption may alter endothelial and vascular smooth muscle cell function as well as microvascular function. Also, briefly reviewed, are the translational effects of ethanol-induced vascular effects, specifically the association between alcohol consumption and changes in blood pressure (BP) and increased risk for HTN.
Table 1.
Potential ethanol-induced sources of reactive oxygen species.
| Ethanol Metabolism |
|
| Ethanol Effects on Antioxidant Proteins and Antioxidant Enzymes |
|
| Activation/Alteration in Neurohormonal Systems |
|
Abbreviations NADH/NAD+ - Nicotinamide adenine dinucleotide reduced/ nicotinamide adenine dinucleotide, ROS- reactive oxygen species.
Mechanisms of peripheral vascular function
The peripheral vascular system refers to all blood vessels external to the myocardium and consists of large and medium arteries (25 to 4 mm in diameter), small arteries (<400 um) and the microvasculature (arterioles < 150 μm in diameter). The function and structure of these vessels vary: large arteries distribute blood flow whereas the microvasculature is critical for maintaining vascular tone and peripheral resistance. Importantly, microvascular dysfunction may be critical in the development of many cardiovascular diseases including HTN and diabetes (5, 6). The inner layer of arteries consists of endothelial cells, which play a critical role in protecting and maintaining vessel wall integrity, regulating vasodilation and vasoconstriction, preventing vascular permeability and adhesion of circulating lipoproteins as well as platelets and leukocytes (7). Endothelial cells achieve vascular homeostasis through the release of several vasodilators including nitric oxide (NO) and prostacyclin (PGI2), endothelium-derived hyperpolarizing factor (EDHF) as well as vasoconstrictors including endothelin-1 (ET-1), angiotensin II (ANGII), and other prostaglandins. Imbalances in the release of these substances and the disruption of the physical barrier can lead to endothelial dysfunction, which is a key contributor to the development and progression of cardiovascular disease.
Endothelial cell-derived substances
Among endothelial cell-derived substances, NO is a potent vasodilator, has a short physiological half-life, and freely diffuses across cell membrane. Figure 1 outlines the sources of NO in the endothelium. NO is synthesized from L-arginine by endothelial NO synthases (eNOS) in response to various physiological (i.e., shear stress) or pharmacological (i.e., acetylcholine or norepinephrine) stimuli. Several co-factors, such as tetrahydrobiopterin (BH4) are important for NO synthesis. As endothelial cell intra-cellular calcium concentrations increase, thereby increasing calcium–calmodulin complex formation and displacement of caveolin from calmodulin, eNOS is translocated from caveolae to the cytosol and activated, leading to the production of NO (8, 9) (Figure 1). eNOS is also activated through a calcium-independent pathway, such as insulin-mediated Akt pathway (10–12). NO diffuses into vascular smoonth muscle cells (VSMC) and activates soluble guanylate cyclase, which increases cyclic guanosine monophosphate concentration and activates the protein kinase-G pathway including phosphorylation of big conductance calcium-activated potassium channels (BKCa), thus increasing intracellular potassium concentration and decreasing calcium concentration in VSMCs and hence relaxation (8, 13).
Figure 1.
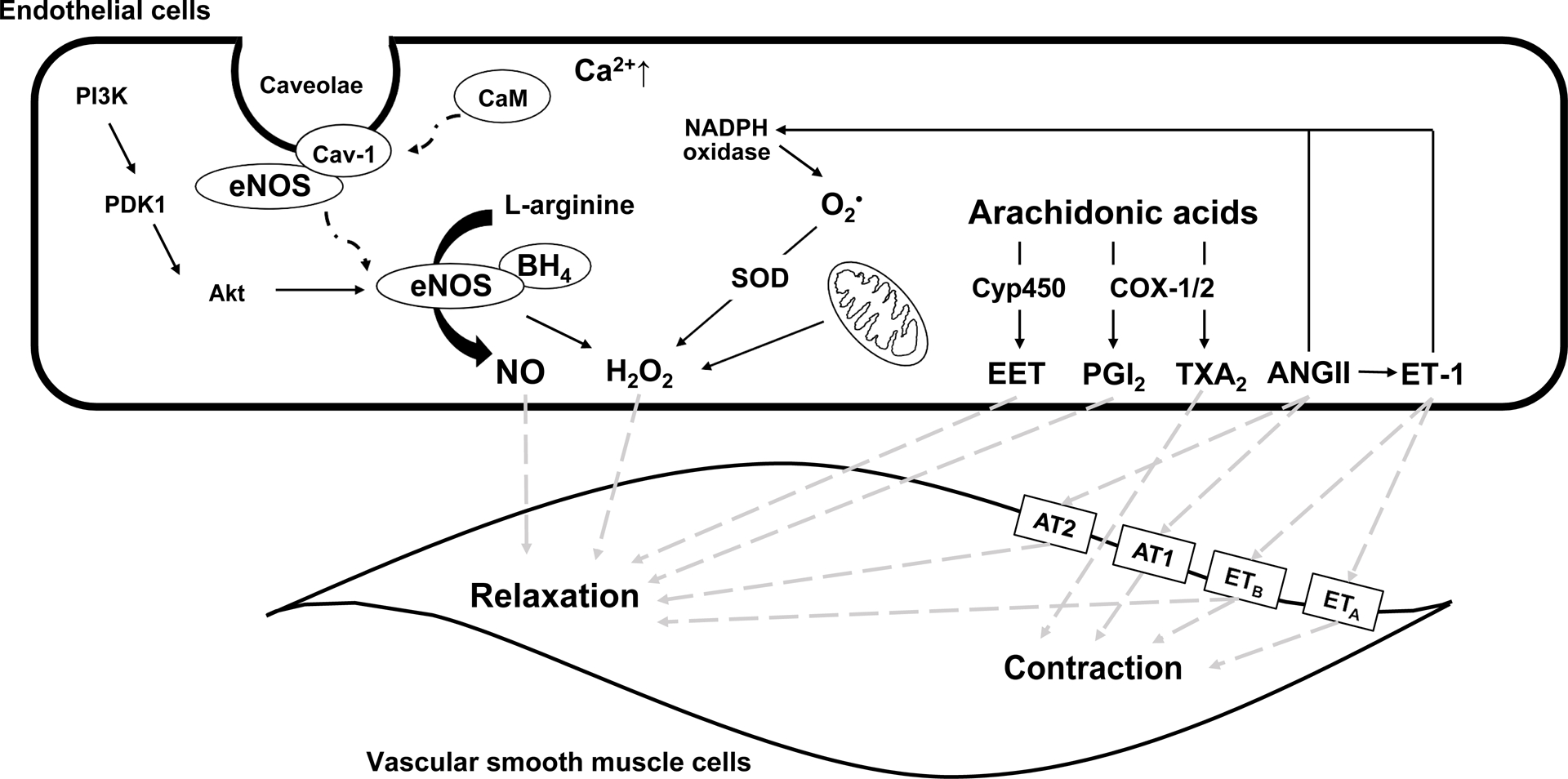
Endothelial cell-derived vasodilators and constrictors. ANGII, angiotensin II; AT1, ANGII receptor type 1; AT2, ANGII receptor type 2; BH4, tetrahydrobiopterin; Ca2+, calcium; Cav-1, caveolin-1; CaM, calmodulin; COX, cyclooxygenases; Cyp450, cytochrome p450; EET, epoxyeicosatrienoic acids; eNOS, endothelial nitric oxide synthase; ETA, endothelin receptors type A; ETB, endothelin receptors type B; ET-1, endothelin-1; H2O2, hydrogen peroxide; NADPH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; O2•, superoxide; PGI2, prostacyclin; PI3K, phosphotidylinositol-3-kinase; PDK1, phosphoinositide-dependent kinase-1; SOD, superoxide dismutase; TXA2, thromboxane A2.
PGI2 is produced by prostacyclin synthase and the cyclooxygenases (COX-1 and −2) from arachidonic acids in response to shear stress, hypoxia and several other stimuli (Figure 1). PGI2 has a physiological half-life of 2–3 minutes. PGI2 binds inositol phosphate receptors on VSMC membranes (14), which increases cyclic adenosine monophosphate concentration via adenylyl cyclase activity and activates protein kinase-A, leading to VSMC relaxation (15). PGI2 also has antiplatelet and antithrombotic effects (16). In contrast, COX-1/2 enzymes also produce thromboxane A2 and other prostaglandins, which cause VSMC contraction (17) (Figure 1).
EDHF is a generic term describing substances, other than NO and PGI2, such as H2O2 and epoxyeicosatrienoic acids (EETs). The contribution of EDHF-mediated relaxation depends on the size of vessel and is more significant in small resistance arteries than large elastic arteries (18) (Figure 1). H2O2 is relatively stable compared to other oxidants and serves as an important second messenger under physiological conditions (19). H2O2 can be produced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (20), during mitochondrial respiration, generated as a by-product with xanthine oxidase, or synthesized from superoxide (O2•) via superoxide dismutase. Under certain conditions, eNOS is a source of H2O2 (21). H2O2 activates various K+ channels such as BKCa channel directly (22) or indirectly through cAMP/protein kinase A pathway to stimulate VSMC relaxation (19). H2O2-mediated vasodilation can also be NO-dependent via p38 mitogen-activated protein kinase-activation of eNOS (20). EETs are generated by cytochrome p450 (Cyp450) epoxygenases from arachidonic acids in response to various stimuli such as acetylcholine and bradykinin (23). EETs can activate the cyclic adenosine monophospahte/protein kinase A pathway (24) or induce increases in intracellular Ca2+ (25), thus activating BKCa and/or adenosine triphosphate-sensitive potassium channels and causing hyperpolarization of VSMCs and thus vasodilation (26, 27).
ET-1 is a potent vasoconstrictor (28) and binds endothelin receptors type A (ETA) and type B (ETB) on VSMCs(29). Activation of VSMC ETA and ETB receptors by ET-1 leads to an increase in intracellular Ca2+ (30) and VSMC contraction and vasoconstriction. Activation of VSMC ETA receptors can also lead to the release of cytokines and formation of ROS. However, ET-1 activation of ETB receptors, leads to NO and PGI2 production and thus VSMC relaxation (31). ANGII is another vasoconstrictor produced via the renin-angiotensin system and plays a critical role in BP regulation. ANGII affects vascular function (32) via activation of ANGII type 1 (AT1) or type 2 (AT2) receptors located on VSMCs or the within the arterial adventitia (33). The activation of AT1 receptors induces vasoconstriction, VSMC proliferation, collagen formation, and inflammation, which are counterbalanced by the effect of AT2 which induces vasodilation (34). ANGII plays a role in the generation of ROS by activating specific enzymes such as NADPH oxidase in the endothelial and VSMCs and via release of the vasoconstrictor ET-1 (35).
Oxidative stress and the role of oxidative stress in vascular function
Oxidative stress
Oxidative stress and the subsequent generation of high levels of ROS has been identified as the key mechanism involved in vascular dysfunction (36). ROS are molecules with an unpaired electron such as O2•, H2O2, hydroxyl radical (•OH), and peroxynitrite (ONOO•). Under normal and stressful/pathological conditions (e.g., inflammation or injury), ROS is generated via several different types of cellular reactions. A significant amount of ROS is generated in the mitochondria (37) as well as produced by NADPH oxidase, an enzyme that regulates vascular tone in endothelial cells, VSMC, and adventitia (36, 38). There are other enzymatic pathways which may contribute to ROS production including xanthine oxidase, uncoupled eNOS, COX, lipoxygenase Cyp450 oxidases (36). Antioxidants neutralize ROS after their formation and serve to maintain appropriate ROS formation and levels. Most cells, including vascular cells have an elaborate system of antioxidant defense mechanisms to combat ROS. Important antioxidants include enzymes such as superoxide dismutase, glutathione peroxidase and catalase, as well as non-enzyme molecules such as albumin, bilirubin, glutathione, vitamin E and C (ascorbic acid) (39).
At appropriate concentrations, ROS act as important cellular signals, that serve to regulate cell growth and differentiation, vascular tone, and immune and inflammatory responses (36). ROS can also react with deoxyribonucleic acid (DNA), proteins, carbohydrates, and lipids within the cell in a destructive manner (37). Malondialdehyde and isoprostanes are the two most widely studied markers of lipid peroxidation in humans (36). Malondialdehyde is formed by peroxidation of polyunstaturated fatty acids and can be detected with thiobarbituric acid reactive substances (ie. TBAR assay). Elevated levels of TBAR are found with HTN, atherosclerosis, diabetes, heart failure, stroke and aging (36). The end product of lipid peroxidation is F2-isoprostanes, which can be measured in all human tissues and biological fluids, including urine, plasma, and cerebrospinal fluid (36). The elevations of circulating measures of malondialdehyde and isoprostanes are associated with reduced endothelial function and elevations in oxidative stress (40, 41).
The role of oxidative stress in vascular function
Low levels of free radicals, ROS, and reactive nitrogen species can participate in signaling pathways and maintain vascular homeostasis. For example, O2• generated during mitochondrial respiratory chains can react with superoxide dismutase, producing H2O2 (42), one of the EDHFs, leading to vasodilation (noted above). However, excessive ROS and the generation of an “oxidative environment” is associated with vascular dysfunction. Oxidative stress is known to lead to the development of atherosclerosis in medium and large arteries via different mechanisms. Oxidative stress stimulates the oxidation and accumulation of low density lipoprotein (LDL), which induces inflammation, macrophage migration, foam cell formation, and a series of vascular remodeling including arterial stiffness (43–45). Oxidative stress also increases arterial stiffness by activating p38 mitogen-activated protein kinase (46). Oxidized LDL activates NADPH oxidase leading to further ROS generation in endothelial cells (44). ROS, particularly O2•, decreases the amount of NO by rapidly oxidizing it to ONOO•, which is a strong oxidant. ONOO• contributes to the accumulation of oxidized LDL in the intima and reacts with proteins, thus changing their function and cellular signaling (47). Oxidative stress also accelerates microvasculature dysfunction and reduce tissue perfusion (48), which impact the oxygen demand and supply within the tissue and initiate a viscous inflammatory cycle, thereby facilitating the progression to atherosclerosis.
Oxidative stress is the major contributor to endothelial dysfunction. Endothelial dysfunction is characterized by reduced NO bioavailability and occurs prior to the development of atherosclerosis. Several of the signal-transduction steps in eNOS/NO-induced relaxation of VSMCs can be a target of ROS, leading to reduced vasodilation. In addition, ROS stimulates BH4 oxidation and induces S-glutathionylation of eNOS, both resulting in eNOS uncoupling (49, 50). Instead of producing NO, uncoupled eNOS becomes a source of O2− (51). ROS also inhibits dimethylarginine dimethylaminohydrolase, causing the loss of enzyme function to metabolize asymmetric dimethylarginine, an endogenous competitive inhibitor of eNOS (52, 53). Interestingly, when eNOS/NO-dependent vasodilation is impaired, the EDHF pathway becomes more predominant in mediating vasodilation (54, 55). With the onset of endothelial dysfunction and the pro-inflammatory responses associated with the adhesion of platelets and monocytes, numerous growth factors are secreted from these cells (e.g., basic fibroblast growth factor and platelet-derived growth factor) which promote VSMC proliferation and migration. These changes ultimately lead to an increased total peripheral resistance and elevation in BP.
Effects of alcohol consumption on endothelial function
Using endothelium-dependent flow-mediated dilation (FMD) and endothelium-independent (nitroglycerin) FMD techniques, investigators have examined the effects of different types of alcoholic beverages and level (moderate and high) on endothelial function. In order to make comparisons among studies, in this section we limit our review to those studies conducted in healthy human beings using noninvasive FMD techniques such as high resolution ultrasound. . Using brachial ultrasound, Vlachopoulos et al. examined endothelium-dependent FMD and endothelium-independent nitroglycerin (NTG) FMD in 12 healthy men (n=7) and women (n=5) following the consumption of 1 ounce of pure alcohol (mixed in grapefruit juice) (56). Thirty minutes after alcohol consumption, there was no change in endothelium-dependent FMD and endothelium-independent NTG FMD (56). In young healthy adults (n=83, mean age range 23 – 25, sex distribution not indicated), Tousoulis D et al. examined the ‘acute’ one-time effect of different alcoholic beverages containing the same amount of alcohol (30 grams) on endothelial function using gauge-strain plethysmography (57). Reactive hyperemia was significantly increased one hour after red wine and beer consumption, whereas no changes were found after water or whisky consumption (57). Among all alcohol groups, there were no differences in reactive hyperemia following nitroglycerin administration (only measured at 4 hours after alcohol consumption). Zilkens and colleagues in 2 different prospective, cross over studies enrolling healthy men (average age 51–53 years) examined the effects of 4 weeks of daily alcohol consumption (wine and beer, either moderate to low levels of consumption) and found no effects of either beverage on endothelium-dependent FMD and endothelium-independent NTG FMD (58, 59). Agewall et al. examined FMD in healthy adults (males n =8, females n= 4) consuming a onetime 250 ml of red wine or de-alcoholized red wine.(60) Flow-mediated FMD (an average of measurements at 30 and 60 mins after consumption) was increased only after de-alcoholized red wine (60).
In terms of the pattern of drinking, 2 studies have examined the effects of binge drinking. Hijmering et al. examined the effects of laboratory ‘simulated binge drinking (61). Healthy men (n=7) and women (n=3) (mean age 34–36 years) were assigned to drink 3 and 6 glasses of either Bacardi Breeze (11 grams of alcohol/drink) or red wine (Rioja, 11.4 grams of alcohol/glass) to test the effect of a low vs. high polyphenolic alcohol containing beverages, respectively as well as high level of alcohol consumption (~ 6 drinks)(61). After 3 and 6 glasses of both beverages, there were significant decrease in endothelium-mediated FMD (investigators did not obtain measurement of endothelium-independent NTG) FMD) (61). We also reported that in young adults (mean age 24 years), a history of repeated binge drinking associated with impaired endothelium-dependent FMD and endothelium-independent NTG FMD (62). To our knowledge, only one study examined FMD in individuals with a history of chronic alcohol use (mean age 31 years, 75 gm alcohol/day for at least 8 years), but who were abstinent for at least 3 months (63). Compared to age-matched controls, FMD was significantly lower (6.3 ± 3.67%) in abstinent subjects with history of heavy alcohol consumption compared to controls (13.7 ± 4.65%). There was no significant difference between groups in endothelium-independent NTG FMD (63).
Based upon the majority of findings noted above, an acute or 4 week period of low-to moderate alcohol consumption appears to have no effects on FMD (56, 58, 59). The one study which examined the ‘acute’ effects of alcohol consumption, reported improved FMD with red wine or beer, but no effects related to whisky consumption(57). Findings from one study, suggested other components in red wine, such as the polyphenols or flavonoids might affect FMD, a finding supported by others who found enhanced FMD responses in patients with coronary artery disease after the consumption of grape juice (64). Finally, based on available evidence, both binge drinking and long-term heavy alcohol consumption adversely affect vascular function (61, 63, 65).
Effects of alcohol consumption on increased blood pressure and incident hypertension
In healthy persons, a onetime low-to-moderate amount of alcohol consumption (~1–2 standard drinks [12–13 grams of ethanol]) has no appreciable effects on BP. However, consuming > 5 standard drinks in one sitting is associated with transient increases in BP that range between 4–7 mm Hg for systolic BP and 4–6 mm Hg for diastolic BP (66–68). In a randomized cross-over trial, others have shown that 4 weeks of high daily alcohol consumption (~ 2–3 standard [13 grams of ethanol] drinks/day) is associated with increases in systolic BP (SBP) and diastolic BP (DBP) (69). Among healthy pre-menopausal women (20 to 45 years of age), Mori et al. examined different levels of red wine consumption (eg., 42–73 g alcohol/week, ~0.5 to 1 standard drinks/day vs. 146–218 g alcohol/week, ~2–3 standard drinks/day) consumed over a 4 week period on 24-hour BP levels (69). Awake SBP and DBP was 2.3/1.3 mm Hg higher in women who consumed higher amounts of alcohol (~2–3 standard drinks/day) than lower levels (~0.5–1 standard drink/day) or no alcohol. Consuming the lower concentrations of red wine had no effect on BP. Using a similar cross-over randomized design, this same group of investigators examined the effects of 4 weeks of wine or beer consumption on ambulatory blood pressure in healthy men (N=24, mean age 53 years)(58). Men were regular drinkers (43±10 grams alcohol/day) for average duration of 21 years. Compared to the control-abstinence period, both red wine and beer (~ 4 standard drinks per day for 4 weeks) increased awake ambulatory SBP (2.9 mm Hg and 1.9 mm Hg, respectively), with no changes in DBP. This study included measurements of FMD and flow-independent NTG and found no changes among all the groups. In healthy males (mean age 51 years), Zilkens et al. have also reported that switching to low-alcohol beer (0.9%) for a period of 4 weeks, compared to consuming a moderate daily level of alcohol (drinking between 40 and 110 gm ethanol/day) was associated with significant reductions in SBP and DBP (moderate drinking: SBP 122 ± 3.5 mmHg vs. light drinking: 117 ± 3.5 and DBP 77 ±2.3 vs 73±2.3 mm Hg) (59).
The above data support the findings from meta-analyses which indicate that regular alcohol consumption is an important risk factor for the development of HTN (70, 71). In a systematic review and meta-analysis including prospective studies (n=16 studies) on the effects of alcohol consumption on the relative risk (RR) of HTN (SBP>140 mmHg/DBP >90 mmHg), Briasoulis et al. (2012) found that >20 g ethanol/day (~1–2 standard drinks/day) significantly increased RR of HTN in women, whereas higher amounts (31–40 g/day) increased RR of HTN in men (70). In women there was a J-shaped relationship: < 10 g/day was associated with a reduced RR for HTN, whereas in men the alcohol risk relationship was more linear. In recent updated meta-analysis, including cohort and randomized studies (n 20 studies), Roerecke et al. found that an average consumption of 1 to 2 drinks/day in men compared to abstainers was associated with an increased risk of HTN (SBP>140 mmHg), whereas women showed no different risk compared to abstainers (71). However, alcohol consumption exceeding 2 drinks/day was associated with an increased incidence of HTN in men and women. Similarly, results from another meta-analysis study, report a linear relationship for males and a J-shaped relationship for females (72). These data highlight how sex may be an important modifier of the “alcohol threshold level” and shape of the alcohol-risk relationship for BP.
Other modifiers of the alcohol-risk relationship include the pattern of drinking (i.e., binge and heavy episodic) (73). There are several reports that indicate, binge drinking in both young adults and middle-aged adults is associated with increases in BP and increased risk of HTN. Using data from the 2011–2014 National Health and Nutrition Examination Survey and after controlling for diet and physical activity, we reported that infrequent (1–12 times in the past year) and frequent binge drinking (≥ 12 times in the past year) in young adult men (18–45 years), were associated with significantly higher SBP (121.8 and 119.0 mmHg, respectively) compared to non-binge drinkers (117.5 mmHg)(74). Binge drinking was defined as consuming 4/5 drinks on one occasion. There were no effects of binge drinking on SBP in young adult women (74). Others have also reported that binge drinking is associated with higher SBP in young adults. Wellman et al. examined the relationship between BP and current and past binge drinking among young adults (mean age 24) participating in the longitudinal Nicotine Dependence in Teens study (75). Binge drinking was defined as consuming 5 or more drinks on one occasion. Subjects were recruited in 1999 (mean age 12), and follow-up assessments were measured in 2007–2008 (mean age 20) and 2011–2014 (mean age 24). Among 24-year-old subjects, both monthly and weekly binge drinkers had SBP values 2.61 mm Hg and 4.03 mm Hg greater, respectively, than non-binge drinkers (similar SBP increases were found in the 20-year-old subjects) (75). Using data from the 1999–2004 National Health and Nutrition Examination Survey, Fan et al. examined the relationship between BP and binge drinking (≥ 5 drinks on one occasion) and frequency of binge drinking (those who reported ≥ once per week binge drinking). In men and women (mean age 38 years), there was increased prevalence ratio (PR) for pre-HTN (SBP 120–140 and DBP 70–90 mmHg) in those reporting ≥ 1 episode of binge drinking per week (men PR: 1.26, 95% CI 1.08–1.53; women PR: 1.49, 95% CI, 0.87–2.56); binge drinking less than once a week was not associated with an increased PR for pre-HTN (76).
There are a few investigations of the relationship between binge drinking and risk for incident HTN in middle age to older adults. Using data from the baseline survey for the Health, Alcohol, and Psychosocial Factors in Eastern Europe study and data from men and women
(age range 55–57 years), Pajak et al. found a greater odds ratio (OR) of HTN (SBP ≥ 140/DBP ≥ 90 mm Hg) for binge drinking (100 g [men] and 60 g [women] of ethanol in one session at least once a month) in men (OR = 1.62, 95% confidence interval [CI] 1.45–1.82) and women (OR = 1.3, 95% CI 1.05–1.63) (77). However, after controlling for drinking frequency and volume, the association between HTN and binge drinking was not significant. These authors concluded that this finding may be unique to this study population, in which alcohol drinking amount, frequency, and binge pattern are high relative to other countries (77).
Collectively, these results suggest that binge drinking in both young adulthood and middle age is associated with increases in BP and the risk of pre-HTN and HTN. Results from a systematic review and meta-analysis support a reduction in alcohol consumption as a means to reduce BP. Roerecke et al. found that in individuals who drank more than 2 drinks per day, a reduction in alcohol consumption to less than 2 drinks per day was associated with a decrease in SBP (mean difference −5.50 mm Hg [CI: −6.70 to −4.30) and DBP (mean difference −3.97 mm Hg, CI: −4.70 to −3.25) (78). Considering that elevated BP is a strong predictor of future cardiovascular risk and outcomes (79), alcohol consumption-associated increases in BP may have important long-term clinical consequences.
Alcohol: antioxidant or oxidant
Oxidative stress and associated changes in nitric oxide signaling are suggested mechanisms underlying alcohol-induced negative effects on the balance between reduced vasodilation and increased vasoconstriction, of which may include elevations in BP and HTN (see summary of the prooxidant effects in Figure 2). As reviewed by Fernández-Checa and colleagues alcohol consumption can lead to oxidative stress and the generation of reactive oxygen species, through a number of indirect (via alcohol metabolism) and direct (effects on antioxidant proteins and enzymes) pathways or mechanisms in which alcohol may lead to an oxidative environment (Table 1)(80). However, there is also evidence from clinical studies in human beings that that alcohol consumption is associated with a decrease in markers of of oxidative stress.
Figure 2.
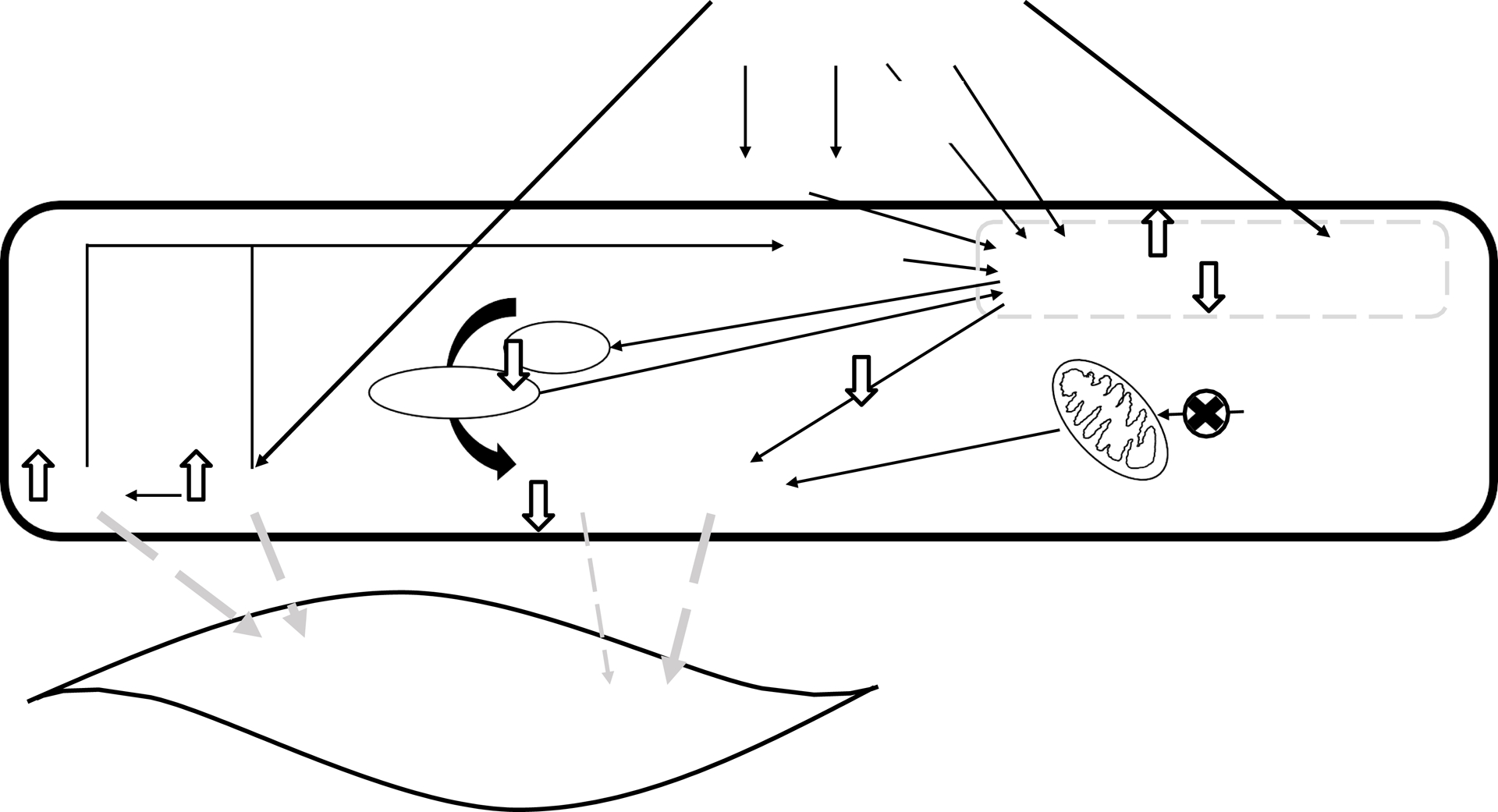
Summary of the pro oxidant effects of alcohol on the mechanisms of vascular relaxation and constriction. ADH, alcohol dehydrogenase; ANGII, angiotensin II; BH4, tetrahydrobiopterin; CYP2E1, cytochrome P450 2E1; eNOS, endothelial nitric oxide synthase; ET-1, endothelin-1; FAEE, fatty acid ethyl ester; H2O2, hydrogen peroxide; NADH/NAD+, nicotinamide adenine dinucleotide reduced/ nicotinamide adenine dinucleotide; NADPH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; O2•, superoxide; PLD, phospholipase D; ROS, reactive oxygen species; SOD, superoxide dismutase.
To date there have been several prospective clinical studies using different study designs (eg. randomized or non-randomized, cross-over designs) to examine the effects of short term (ie. generally 28–30 days) moderate alcohol consumption on changes in plasma markers of oxidative stress and corresponding changes in BP or coronary blood flow. Plasma markers have included malondialdehyde (MDA), super oxide dismutase (SOD), oxidized low density lipoproteins (oxLDL), reduced glutathione (GSH), glutathione peroxidase (GPx), reduced glutathione (GSH), thiobartitruic acid (TBARS), and total antioxidant capacity of plasma (AOC).
In men (N=40, mean age 38 years), Estruch et al. found 28 days of moderate red wine consumption (30 grams, ~ 2–3 standard drinks) was associated with a significant decrease in SOD, MDA, LDL peroxides and an increase in vitamin E levels, whereas no changes were found in GSH levels.(81) In this same study, no effects of gin (30 grams/day) consumption was found on the aforementioned plasma oxidative markers. Systolic BP decreased significantly after gin consumption (pre 121 ± 16 vs. post 118 ± 15 mm Hg), whereas no significant changes were found after red wine consumption (pre 119 ± 16 vs. post 120 ± 16 mmHg) (81). Using a cross-over design, Kiviniemi et al. examined the ‘acute’ one time effects of a moderate dose of red wine (0.5g/kg) and high dose of red wine (1.0 g/kg) in men (N=23) (mean age 23 years) on total plasma antioxidant levels (82). Measurements were made 30 mins after wine consumption (and only after the high dose red wine), significant increases were found in total plasma antioxidant levels (pre 265 ±35 μmol/L vs. post 333 ±29.3 μmol/L). Coronary fractional reserve, as a measure of coronary blood flow response was also measured and was unaltered after high wine consumption (82). Rajdl et al. conducted a field study in the Czech Republic, among men (N=42, mean age 41 years) with a history of moderate alcohol consumption. Men were assigned in a non-random fashion to consume white wine (375 ml/daily with dinner, ~ 1 month)(83). One month following daily white wine (49 g/day, ~ 3 drinks) consumption there were significant decreases in SOD, total antioxidant capacity of the plasma, advanced oxidation protein products, and TBARS and increases in GPx and GSH. However, no changes were found in SBP or DBP (83). In contrast to the three aforementioned studies, Addolorato et al. found 30 days of daily moderate (40 grams, ~ 2–3 drinks) on red wine, beer or spirit consumption in men (N=40, age range 28–31 years) was associated with significant increases in plasma MDA, and decreases in GSH and Vitamin E. Authors noted no changes were found BP levels (actual BP values were not reported) (84). Findings from all the latter studies suggest that BP and coronary flow reserve responses are not associated with plasma oxidative stress measures (either increases or decreases) after either a one-time or short-term period of low-to moderate alcohol consumption. Considering the findings from large scale epidemiologic and case control studies (summarized above), more research is needed to examine the effects of long-term heavy drinking and binge drinking on oxidative stress markers and corresponding changes in BP or other functional vascular measures. In addition, no women were included in any of the reviewed studies, limiting our understanding of the effects of sex on these parameters (81–84).
Animal models have also been used to investigate the associations of oxidative stress with acute and repeated alcohol administration. Tirapelli et al. have performed a series of studies to test the effect of a single dose of ethanol in rats on oxidative stress markers (85–87). In these studies, increased plasma levels of TBARS and increased aortic endothelial and VSMC O2• levels were found, along with increased membrane-to-cytosol fraction expression of the p47phox subunit of NADPH oxidase (87). Those same investigators found increases in plasma renin and angiotensin-converting enzyme activity and decreases in plasma and aortic (endothelial-intact) nitrate levels (87). Pre-treatment of animals with Losartan, an angiotensin receptor blocker, prevented the increase in oxidative stress markers. In addition, the alcohol-induced translocation of p47phox and NADPH oxidase activation was reduced by Losartan, suggesting a role for ANGII in mediating binge-induced oxidative stress in aortic tissue (87). It is known that high pressure itself may accelerate the activation of the renin angiotensin system and that ANGII can stimulate endothelial ROS (88) such that the activation of the renin-angiotensin system may be a critical component of ethanol consumption induced oxidative stress and HTN.
The effects of alcohol on oxidative stress may be related to cellular changes in antioxidant capacity. For example, a study by Das and Vasudevan showed that ethanol consumption in rats (0.8 to 2.0 g ethanol/kg/day; 4 weeks) increased SOD activity and reduced catalase activity in a time- and dose-dependent manner, an effect that would promote increases of H2O2 (89). Similarly, one study by Husain et al showed that superoxide dismutase activity was increased in the liver of rats fed ethanol (90). Ethanol may increase catalase activity and glutathione peroxidase an effect that would reduce H2O2 bioavailability in renal tissue (91). However, other studies have found that catalase and glutathione were decreased in the aorta of ethanol fed rats, an effect that would increase H2O2 generation (92). These effects of chronic alcohol consumption on antioxidant expression and activity appear to be vascular bed specific effects, since Simplico et al. found that 6 weeks of ethanol consumption (20% v/v) in rats had no effect on H2O2, catalase activity, or glutathione activity in the mesenteric circulation (93). Importantly, O2• generation in mesenteric homogenates was elevated in these studies. Although not measured directly, these results would promote the generation of ONOO-by reaction with available NO leading to further and more profound cell stress (50).
In another investigation, using a similar animal model (rats treated with 20% ethanol for 6 weeks), those investigators found that some, but not all, of the aforementioned oxidative stress markers were changed in mesenteric resistance vessels (85, 86). For example, increases were found in TBARS and O2• and NADPH oxidase expression (i.e., increase in the membrane/cytosol fraction ratio of p47phox). In contrast there were no changes found in H2 O2 , glutathione, superoxide dismutase or catalase activity after 48 hours following ethanol consumption (86).
Collectively, these data from animal models suggest that repeated and one-time binge drinking induces oxidative stress in both large (aortic) and resistance (mesenteric) vessels. However, the oxidative stress and BP responses seem to be modulated by the vascular bed, duration and pattern of drinking (ie., binge drinking). Repeated, chronic binge drinking is associated with a robust oxidative response, which correlates to both increases in BP and ANGII levels (94). Findings from other studies indicate that some of the oxidative stress changes can be prevented by pre-treatment with agents that block the effects of ANGII (95) or antioxidant/scavenging agents, such as vitamin C (86).
Our group has recently found microvascular dysfunction in young adults with a history of repeated binge drinking. In these studies we found that in addition to reductions in brachial artery FMD, microvascular flow-induced dilation (FID) is significantly reduced (62). Interestingly, arterioles isolated from subcutaneous adipose biopsies demonstrated FIDs that were reduced by about 40% (96). The remaining dilation was inhibited by the H2O2 scavenger catalase, but there was no effect of NOS inhibition with L-NAME (96). In contrast, vessels from abstainers showed a significant reduction in FIDs in the presence of L-NAME (96). These data suggest that the mechanism of the reduced dilation is dependent on the generation of the ROS, H2O2, and is particularly intriguing given that arterioles obtained from patients with coronary artery disease also rely on ROS for FID (97). These data further suggest enhancement of ROS in adults consuming alcohol chronically. This effect of ROS on maintained FID may be unique to the human microcirculation, however in our recent studies we have found that uncoupling of eNOS may be a contributing factor in the reduced NO dependent dilation in this population. For example, we found that the addition of BH4 (a cofactor for NO synthesis) restored NO generation in binge drinkers to similar levels found in arterioles from abstainers (unpublished results). Although more confirmatory data are necessary, these data suggest that binge drinking may induce microvascular dysfunction through oxidative stress mechanisms involving impairments in the endothelial-NO-generating system, such as eNOS uncoupling.
Conclusion
Oxidative stress may be an important pathophysiologic mechanism underlying vascular dysfunction associated with alcohol consumption. However, as reviewed above the amount, duration and pattern may be important modifiers of this relationship. In particular alcohol-induced microvascular dysfunction following binge or heavy alcohol consumption, may arise from changes in vascular wall biology which may include imbalances in the generation of NO and enzymes such as eNOS and other oxidase enzyme systems, such as NAPDH oxidase that generate ROS. The presence of ROS and the generation of an “oxidative environment” can accelerate microvasculature dysfunction and impaired microvascular dilator capacity and flow regulation. These effects can impact the oxygen demand and supply within the tissue and initiate a viscous inflammatory cycle, facilitating the development of inflammation and atherosclerosis in medium and larger arteries and the eventual development of cardiovascular diseases such as HTN. As reviewed above there is evidence that oxidative stress can induce many disturbances in vascular homeostasis and NO-signaling by reducing bioavailability of L-arginine and/or co-factors such BH4. There is a need for research examining upstream mechanisms contributing to reduce NO and microvascular dysfunction.
Data from several clinical studies reviewed herein, suggest low-to-moderate alcohol consumption in human beings is associated with a decrease in markers of oxidative stress. Yet these changes in oxidative markers were either associated with no change in BP or increases in BP. More research is needed examining the effects of heavy and binge drinking on circulating markers of oxidative stress and corresponding changes in BP and vascular function. In addition, future studies should address differences in the BP responses to alcohol between men and women. Data from several different types of clinical and epidemiologic studies indicate different levels of alcohol consumption is associated with BP elevations and risk for the development of HTN. Continuing to elucidate mechanisms and design studies to demonstrate cause and effect will lead to a better understanding of the relationship between alcohol consumption and cardiovascular health and disease.
Acknowledgements:
Due to page limitations, it is impossible to recognize that not all the excellent scientific work completed in the area of alcohol and the vascular system has been included. This review was supported in part by NIH grant T32-HL-139439 (AS) and R21AA024535 (SAP and MRP).
Footnotes
Disclosure(s): None.
References
- 1.Piano MR, Phillips SA. Alcoholic cardiomyopathy: pathophysiologic insights. Cardiovasc Toxicol. 2014;14(4):291–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandez-Checa JC, Kaplowitz N, Garcia-Ruiz C, Colell A. Mitochondrial glutathione: importance and transport. Seminars in liver disease. 1998;18(4):389–401. [DOI] [PubMed] [Google Scholar]
- 3.Zhu H, Jia Z, Misra H, Li YR. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: updated experimental and clinical evidence. J Dig Dis. 2012;13(3):133–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landmesser U, Harrison DG. Oxidative stress and vascular damage in hypertension. CoronArtery Dis. 2001;12(6):455–61. [DOI] [PubMed] [Google Scholar]
- 5.Jonk AM, Houben AJ, de Jongh RT, Serne EH, Schaper NC, Stehouwer CD. Microvascular dysfunction in obesity: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Physiology(Bethesda). 2007;22:252–60. [DOI] [PubMed] [Google Scholar]
- 6.Phillips SA, Mahmoud AM, Brown MD, Haus JM. Exercise interventions and peripheral arterial function: implications for cardio-metabolic disease. Progress in cardiovascular diseases. 2015;57(5):521–34. [DOI] [PubMed] [Google Scholar]
- 7.Yurdagul A Jr., Finney AC, Woolard MD, Orr AW. The arterial microenvironment: the where and why of atherosclerosis. The Biochemical journal. 2016;473(10):1281–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lloyd-Jones MDDM, Bloch MDKD. The Vascular Biology of Nitric Oxide and Its Role in Atherogenesis. Annual Review of Medicine. 1996;47(1):365–75. [DOI] [PubMed] [Google Scholar]
- 9.Vanhoutte PM, Zhao Y, Xu A, Leung SW. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ Res. 2016;119(2):375–96. [DOI] [PubMed] [Google Scholar]
- 10.Muniyappa R, Quon MJ. Insulin action and insulin resistance in vascular endothelium. Curr Opin Clin Nutr Metab Care. 2007;10(4):523–30. [DOI] [PubMed] [Google Scholar]
- 11.Mather KJ, Steinberg HO, Baron AD. Insulin resistance in the vasculature. J Clin Invest. 2013;123(3):1003–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuboki K, Jiang ZY, Takahara N, Ha SW, Igarashi M, Yamauchi T, et al. Regulation of endothelial constitutive nitric oxide synthase gene expression in endothelial cells and in vivo : a specific vascular action of insulin. Circulation. 2000;101(6):676–81. [DOI] [PubMed] [Google Scholar]
- 13.Furchgott RF. Role of endothelium in responses of vascular smooth muscle. Circ Res. 1983;53(5):557–73. [DOI] [PubMed] [Google Scholar]
- 14.Coleman RA, Smith WL, Narumiya S. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46(2):205–29. [PubMed] [Google Scholar]
- 15.Moncada S, Vane JR. Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2, and prostacyclin. Pharmacol Rev. 1978;30(3):293–331. [PubMed] [Google Scholar]
- 16.Moncada S, Korbut R, Bunting S, Vane JR. Prostacyclin is a circulating hormone. Nature. 1978;273(5665):767–8. [DOI] [PubMed] [Google Scholar]
- 17.Davidge ST. Prostaglandin H Synthase and Vascular Function. Circulation Research. 2001;89(8):650–60. [DOI] [PubMed] [Google Scholar]
- 18.Tomioka H, Hattori Y, Fukao M, Sato A, Liu M, Sakuma I, et al. Relaxation in different-sized rat blood vessels mediated by endothelium-derived hyperpolarizing factor: importance of processes mediating precontractions. J Vasc Res. 1999;36(4):311–20. [DOI] [PubMed] [Google Scholar]
- 19.Shimokawa H, Morikawa K. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Mol Cell Cardiol. 2005;39(5):725–32. [DOI] [PubMed] [Google Scholar]
- 20.Breton-Romero R, Gonzalez de Orduna C, Romero N, Sanchez-Gomez FJ, de Alvaro C, Porras A, et al. Critical role of hydrogen peroxide signaling in the sequential activation of p38 MAPK and eNOS in laminar shear stress. Free Radic Biol Med. 2012;52(6):1093–100. [DOI] [PubMed] [Google Scholar]
- 21.Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K, et al. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J Clin Invest. 2000;106(12):1521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, et al. H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation. Circ Res. 2012;110(3):471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nithipatikom K, Pratt PF, Campbell WB. Determination of EETs using microbore liquid chromatography with fluorescence detection. Am J Physiol Heart Circ Physiol. 2000;279(2):H857–62. [DOI] [PubMed] [Google Scholar]
- 24.Imig JD, Inscho EW, Deichmann PC, Reddy KM, Falck JR. Afferent arteriolar vasodilation to the sulfonimide analog of 11, 12-epoxyeicosatrienoic acid involves protein kinase A. Hypertension. 1999;33(1 Pt 2):408–13. [DOI] [PubMed] [Google Scholar]
- 25.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels. Circ Res. 2005;97(12):1270–9. [DOI] [PubMed] [Google Scholar]
- 26.Edwards G, Félétou M, Weston AH. Endothelium-derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch - Eur J Physiol. 2010;459(6):863–79. [DOI] [PubMed] [Google Scholar]
- 27.Coats P, Johnston F, MacDonald J, McMurray JJ, Hillier C. Endothelium-derived hyperpolarizing factor : identification and mechanisms of action in human subcutaneous resistance arteries. Circulation. 2001;103(12):1702–8. [DOI] [PubMed] [Google Scholar]
- 28.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–5. [DOI] [PubMed] [Google Scholar]
- 29.Ihara M, Noguchi K, Saeki T, Fukuroda T, Tsuchida S, Kimura S, et al. Biological profiles of highly potent novel endothelin antagonists selective for the ETA receptor. Life Sci. 1992;50(4):247–55. [DOI] [PubMed] [Google Scholar]
- 30.Marsden PA, Danthuluri NR, Brenner BM, Ballermann BJ, Brock TA. Endothelin action on vascular smooth muscle involves inositol trisphosphate and calcium mobilization. Biochem Biophys Res Commun. 1989;158(1):86–93. [DOI] [PubMed] [Google Scholar]
- 31.Schneider MP, Boesen EI, Pollock DM. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annual review of pharmacology and toxicology. 2007;47:731–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Touyz RM, Schiffrin EL. Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev. 2000;52(4):639–72. [PubMed] [Google Scholar]
- 33.Allen AM, Zhuo J, Mendelsohn FA. Localization and function of angiotensin AT1 receptors. Am J Hypertens. 2000;13(1 Pt 2):31S–8S. [DOI] [PubMed] [Google Scholar]
- 34.Katada J, Majima M. AT(2) receptor-dependent vasodilation is mediated by activation of vascular kinin generation under flow conditions. Br J Pharmacol. 2002;136(4):484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112(17):2668–76. [DOI] [PubMed] [Google Scholar]
- 36.Montezano AC, Dulak-Lis M, Tsiropoulou S, Harvey A, Briones AM, Touyz RM. Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. The Canadian journal of cardiology. 2015;31(5):631–41. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi G, Cortopassi G. Oxidative stress in inherited mitochondrial diseases. Free radical biology & medicine. 2015;88(Pt A):10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magenta A, Greco S, Gaetano C, Martelli F. Oxidative stress and microRNAs in vascular diseases. International journal of molecular sciences. 2013;14(9):17319–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siti HN, Kamisah Y, Kamsiah J. The role of oxidative stress, antioxidants and vascular inflammation in cardiovascular disease (a review). Vascular pharmacology. 2015;71:40–56. [DOI] [PubMed] [Google Scholar]
- 40.Carroll MF, Schade DS. Timing of antioxidant vitamin ingestion alters postprandial proatherogenic serum markers. Circulation. 2003;108(1):24–31. [DOI] [PubMed] [Google Scholar]
- 41.Kinlay S, Behrendt D, Fang JC, deLaGrange D, Morrow J, Witztum JL, et al. Long-term effect of combined vitamins E and C on coronary and peripheral endothelial function. JAmCollCardiol. 2004;43(4):629–34. [DOI] [PubMed] [Google Scholar]
- 42.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552(Pt 2):335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steinberg D Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem. 1997;272(34):20963–6. [DOI] [PubMed] [Google Scholar]
- 44.Rueckschloss U, Galle J, Holtz J, Zerkowski HR, Morawietz H. Induction of NAD(P)H oxidase by oxidized low-density lipoprotein in human endothelial cells: antioxidative potential of hydroxymethylglutaryl coenzyme A reductase inhibitor therapy. Circulation. 2001;104(15):1767–72. [DOI] [PubMed] [Google Scholar]
- 45.Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fonarow GC, et al. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. JLipid Res. 2004;45(6):993–1007. [DOI] [PubMed] [Google Scholar]
- 46.Martinez-Revelles S, Garcia-Redondo AB, Avendano MS, Varona S, Palao T, Orriols M, et al. Lysyl Oxidase Induces Vascular Oxidative Stress and Contributes to Arterial Stiffness and Abnormal Elastin Structure in Hypertension: Role of p38MAPK. Antioxid Redox Signal. 2017;27(7):379–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological reviews. 2007;87(1):315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gutterman DD, Chabowski DS, Kadlec AO, Durand MJ, Freed JK, Ait-Aissa K, et al. The Human Microcirculation: Regulation of Flow and Beyond. Circulation research. 2016;118(1):157–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468(7327):1115–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen W, Druhan LJ, Chen CA, Hemann C, Chen YR, Berka V, et al. Peroxynitrite induces destruction of the tetrahydrobiopterin and heme in endothelial nitric oxide synthase: transition from reversible to irreversible enzyme inhibition. Biochemistry. 2010;49(14):3129–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cosentino F, Patton S, d’Uscio LV, Werner ER, Werner-Felmayer G, Moreau P, et al. Tetrahydrobiopterin alters superoxide and nitric oxide release in prehypertensive rats. J Clin Invest. 1998;101(7):1530–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pope AJ, Karrupiah K, Kearns PN, Xia Y, Cardounel AJ. Role of dimethylarginine dimethylaminohydrolases in the regulation of endothelial nitric oxide production. J Biol Chem. 2009;284(51):35338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sydow K, Schwedhelm E, Arakawa N, Bode-Boger SM, Tsikas D, Hornig B, et al. ADMA and oxidative stress are responsible for endothelial dysfunction in hyperhomocyst(e)inemia: effects of L-arginine and B vitamins. Cardiovasc Res. 2003;57(1):244–52. [DOI] [PubMed] [Google Scholar]
- 54.Huang A, Sun D, Smith CJ, Connetta JA, Shesely EG, Koller A, et al. In eNOS knockout mice skeletal muscle arteriolar dilation to acetylcholine is mediated by EDHF. Am J Physiol Heart Circ Physiol. 2000;278(3):H762–8. [DOI] [PubMed] [Google Scholar]
- 55.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. American journal of physiology Heart and circulatory physiology. 2007;292(1):H93–100. [DOI] [PubMed] [Google Scholar]
- 56.Vlachopoulos C, Tsekoura D, Tsiamis E, Panagiotakos D, Stefanadis C. Effect of alcohol on endothelial function in healthy subjects. Vascular medicine (London, England). 2003;8(4):263–5. [DOI] [PubMed] [Google Scholar]
- 57.Tousoulis D, Ntarladimas I, Antoniades C, Vasiliadou C, Tentolouris C, Papageorgiou N, et al. Acute effects of different alcoholic beverages on vascular endothelium, inflammatory markers and thrombosis fibrinolysis system. Clinical nutrition (Edinburgh, Scotland). 2008;27(4):594–600. [DOI] [PubMed] [Google Scholar]
- 58.Zilkens RR, Burke V, Hodgson JM, Barden A, Beilin LJ, Puddey IB. Red wine and beer elevate blood pressure in normotensive men. Hypertension. 2005;45(5):874–9. [DOI] [PubMed] [Google Scholar]
- 59.Zilkens RR, Rich L, Burke V, Beilin LJ, Watts GF, Puddey IB. Effects of alcohol intake on endothelial function in men: a randomized controlled trial. Journal of hypertension. 2003;21(1):97–103. [DOI] [PubMed] [Google Scholar]
- 60.Agewall S, Wright S, Doughty RN, Whalley GA, Duxbury M, Sharpe N. Does a glass of red wine improve endothelial function? European heart journal. 2000;21(1):74–8. [DOI] [PubMed] [Google Scholar]
- 61.Hijmering ML, de Lange DW, Lorsheyd A, Kraaijenhagen RJ, van de Wiel A. Binge drinking causes endothelial dysfunction, which is not prevented by wine polyphenols: a small trial in healthy volunteers. The Netherlands journal of medicine. 2007;65(1):29–35. [PubMed] [Google Scholar]
- 62.Goslawski M, Piano MR, Bian JT, Church E, Szczurek M, Phillips SA. Binge Drinking Impairs Vascular Function in Young Adults. JAmCollCardiol. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maiorano G, Bartolomucci F, Contursi V, Minenna FS, Di Mise R, Palasciano A, et al. Noninvasive detection of vascular dysfunction in alcoholic patients. American journal of hypertension. 1999;12(2 Pt 1):137–44. [DOI] [PubMed] [Google Scholar]
- 64.Stein JH, Keevil JG, Wiebe DA, Aeschlimann S, Folts JD. Purple grape juice improves endothelial function and reduces the susceptibility of LDL cholesterol to oxidation in patients with coronary artery disease. Circulation. 1999;100(10):1050–5. [DOI] [PubMed] [Google Scholar]
- 65.Goslawski M, Piano MR, Bian JT, Church EC, Szczurek M, Phillips SA. Binge drinking impairs vascular function in young adults. Journal of the American College of Cardiology. 2013;62(3):201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Potter JF, Watson RD, Skan W, Beevers DG. The pressor and metabolic effects of alcohol in normotensive subjects. Hypertension. 1986;8(7):625–31. [DOI] [PubMed] [Google Scholar]
- 67.Seppa K, Sillanaukee P. Binge drinking and ambulatory blood pressure. Hypertension. 1999;33(1):79–82. [DOI] [PubMed] [Google Scholar]
- 68.Rosito GA, Fuchs FD, Duncan BB. Dose-dependent biphasic effect of ethanol on 24-h blood pressure in normotensive subjects. American journal of hypertension. 1999;12(2 Pt 1):236–40. [DOI] [PubMed] [Google Scholar]
- 69.Mori TA, Burke V, Beilin LJ, Puddey IB. Randomized Controlled Intervention of the Effects of Alcohol on Blood Pressure in Premenopausal Women. Hypertension. 2015;66(3):517–23. [DOI] [PubMed] [Google Scholar]
- 70.Briasoulis A, Agarwal V, Messerli FH. Alcohol consumption and the risk of hypertension in men and women: a systematic review and meta-analysis. J Clin Hypertens (Greenwich). 2012;14(11):792–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roerecke M, Tobe SW, Kaczorowski J, Bacon SL, Vafaei A, Hasan OSM, et al. Sex-Specific Associations Between Alcohol Consumption and Incidence of Hypertension: A Systematic Review and Meta-Analysis of Cohort Studies. J Am Heart Assoc. 2018;7(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taylor B, Irving HM, Baliunas D, Roerecke M, Patra J, Mohapatra S, et al. Alcohol and hypertension: gender differences in dose-response relationships determined through systematic review and meta-analysis. Addiction. 2009;104(12):1981–90. [DOI] [PubMed] [Google Scholar]
- 73.Piano MR, Mazzuco A, Kang M, Phillips SA. Cardiovascular Consequences of Binge Drinking: An Integrative Review with Implications for Advocacy, Policy, and Research. Alcoholism, clinical and experimental research. 2017;41(3):487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Piano MR, Burke L, Kang M, Phillips SA. Effects of Repeated Binge Drinking on Blood Pressure Levels and Other Cardiovascular Health Metrics in Young Adults: National Health and Nutrition Examination Survey, 2011–2014. J Am Heart Assoc. 2018;7(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wellman RJ, Vaughn JA, Sylvestre MP, O’Loughlin EK, Dugas EN, O’Loughlin JL. Relationships Between Current and Past Binge Drinking and Systolic Blood Pressure in Young Adults. The Journal of adolescent health : official publication of the Society for Adolescent Medicine. 2016;58(3):352–7. [DOI] [PubMed] [Google Scholar]
- 76.Fan AZ, Li Y, Elam-Evans LD, Balluz L. Drinking pattern and blood pressure among non-hypertensive current drinkers: findings from 1999–2004 National Health and Nutrition Examination Survey. Clinical epidemiology. 2013;5:21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pajak A, Szafraniec K, Kubinova R, Malyutina S, Peasey A, Pikhart H, et al. Binge drinking and blood pressure: cross-sectional results of the HAPIEE study. PloS one. 2013;8(6):e65856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Roerecke M, Kaczorowski J, Tobe SW, Gmel G, Hasan OSM, Rehm J. The effect of a reduction in alcohol consumption on blood pressure: a systematic review and meta-analysis. The Lancet Public health. 2017;2(2):e108–e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mourad JJ. The evolution of systolic blood pressure as a strong predictor of cardiovascular risk and the effectiveness of fixed-dose ARB/CCB combinations in lowering levels of this preferential target. Vascular health and risk management. 2008;4(6):1315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fernandez-Checa JC, Kaplowitz N, Colell A, Garcia-Ruiz C. Oxidative stress and alcoholic liver disease. Alcohol health and research world. 1997;21(4):321–4. [PMC free article] [PubMed] [Google Scholar]
- 81.Estruch R, Sacanella E, Mota F, Chiva-Blanch G, Antunez E, Casals E, et al. Moderate consumption of red wine, but not gin, decreases erythrocyte superoxide dismutase activity: a randomised cross-over trial. Nutrition, metabolism, and cardiovascular diseases : NMCD. 2011;21(1):46–53. [DOI] [PubMed] [Google Scholar]
- 82.Kiviniemi TO, Saraste A, Toikka JO, Saraste M, Raitakari OT, Parkka JP, et al. A moderate dose of red wine, but not de-alcoholized red wine increases coronary flow reserve. Atherosclerosis. 2007;195(2):e176–81. [DOI] [PubMed] [Google Scholar]
- 83.Rajdl D, Racek J, Trefil L, Siala K. Effect of white wine consumption on oxidative stress markers and homocysteine levels. Physiological research. 2007;56(2):203–12. [DOI] [PubMed] [Google Scholar]
- 84.Addolorato G, Leggio L, Ojetti V, Capristo E, Gasbarrini G, Gasbarrini A. Effects of short-term moderate alcohol administration on oxidative stress and nutritional status in healthy males. Appetite. 2008;50(1):50–6. [DOI] [PubMed] [Google Scholar]
- 85.Gonzaga NA, Callera GE, Yogi A, Mecawi AS, Antunes-Rodrigues J, Queiroz RH, et al. Acute ethanol intake induces mitogen-activated protein kinase activation, platelet-derived growth factor receptor phosphorylation, and oxidative stress in resistance arteries. Journal of physiology and biochemistry. 2014;70(2):509–23. [DOI] [PubMed] [Google Scholar]
- 86.Simplicio JA, Hipolito UV, Vale GT, Callera GE, Pereira CA, Touyz RM, et al. Acute Ethanol Intake Induces NAD(P)H Oxidase Activation and Rhoa Translocation in Resistance Arteries. Arquivos brasileiros de cardiologia. 2016:0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yogi A, Callera GE, Mecawi AS, Batalhao ME, Carnio EC, Antunes-Rodrigues J, et al. Acute ethanol intake induces superoxide anion generation and mitogen-activated protein kinase phosphorylation in rat aorta: a role for angiotensin type 1 receptor. Toxicol Appl Pharmacol. 2012;264(3):470–8. [DOI] [PubMed] [Google Scholar]
- 88.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. CircRes. 2002;91(5):406–13. [DOI] [PubMed] [Google Scholar]
- 89.Das SK, Vasudevan DM. Effect of ethanol on liver antioxidant defense systems: Adose dependent study. Indian J Clin Biochem. 2005;20(1):80–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Husain K, Scott BR, Reddy SK, Somani SM. Chronic ethanol and nicotine interaction on rat tissue antioxidant defense system. Alcohol. 2001;25(2):89–97. [DOI] [PubMed] [Google Scholar]
- 91.Dinu D, Nechifor MT, Movileanu L. Ethanol-induced alterations of the antioxidant defense system in rat kidney. Journal of biochemical and molecular toxicology. 2005;19(6):386–95. [DOI] [PubMed] [Google Scholar]
- 92.Husain K, Vazquez-Ortiz M, Lalla J. Down regulation of aortic nitric oxide and antioxidant systems in chronic alcohol-induced hypertension in rats. Hum Exp Toxicol. 2007;26(5):427–34. [DOI] [PubMed] [Google Scholar]
- 93.Simplicio JA, do Vale GT, Gonzaga NA, Leite LN, Hipolito UV, Pereira CA, et al. Reactive oxygen species derived from NAD(P)H oxidase play a role on ethanol-induced hypertension and endothelial dysfunction in rat resistance arteries. J Physiol Biochem. 2017;73(1):5–16. [DOI] [PubMed] [Google Scholar]
- 94.Husain K, Vazquez M, Ansari RA, Malafa MP, Lalla J. Chronic alcohol-induced oxidative endothelial injury relates to angiotensin II levels in the rat. Mol Cell Biochem. 2008;307(1–2):51–8. [DOI] [PubMed] [Google Scholar]
- 95.Yogi A, Callera GE, Hipolito UV, Silva CR, Touyz RM, Tirapelli CR. Ethanol-induced vasoconstriction is mediated via redox-sensitive cyclo-oxygenase-dependent mechanisms. Clinical science. 2010;118(11):657–68. [DOI] [PubMed] [Google Scholar]
- 96.Bian JT, Piano MR, Kotlo KU, Mahmoud AM, Phillips SA. MicroRNA-21 Contributes to Reduced Microvascular Function in Binge Drinking Young Adults. Alcoholism, clinical and experimental research. 2018;42(2):278–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Phillips SA, Hatoum OA, Gutterman DD. The mechanism of flow-induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. AmJPhysiol Heart CircPhysiol. 2007;292(1):H93–100. [DOI] [PubMed] [Google Scholar]
- 98.Vickers KC, Rye KA, Tabet F. MicroRNAs in the onset and development of cardiovascular disease. Clinical science. 2014;126(3):183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Miranda RC, Pietrzykowski AZ, Tang Y, Sathyan P, Mayfield D, Keshavarzian A, et al. MicroRNAs: master regulators of ethanol abuse and toxicity? Alcoholism, clinical and experimental research. 2010;34(4):575–87. [DOI] [PMC free article] [PubMed] [Google Scholar]