

Substrate-free and -bound states of Lon reveal distinct operational modes used by the enzyme to process protein substrates.
Abstract
Substrate-bound structures of AAA+ protein translocases reveal a conserved asymmetric spiral staircase architecture wherein a sequential ATP hydrolysis cycle drives hand-over-hand substrate translocation. However, this configuration is unlikely to represent the full conformational landscape of these enzymes, as biochemical studies suggest distinct conformational states depending on the presence or absence of substrate. Here, we used cryo–electron microscopy to determine structures of the Yersinia pestis Lon AAA+ protease in the absence and presence of substrate, uncovering the mechanistic basis for two distinct operational modes. In the absence of substrate, Lon adopts a left-handed, “open” spiral organization with autoinhibited proteolytic active sites. Upon the addition of substrate, Lon undergoes a reorganization to assemble an enzymatically active, right-handed “closed” conformer with active protease sites. These findings define the mechanistic principles underlying the operational plasticity required for processing diverse protein substrates.
INTRODUCTION
Adenosine triphosphatases (ATPases) associated with a variety of cellular activities (AAA+) enzymes constitute a broad superfamily of proteins defined by a structurally conserved domain that contains elements involved in nucleotide binding, sensing, and hydrolysis (1–4). Despite structural similarities, AAA+ proteins play distinct roles in regulating diverse cellular activities, including protein degradation, cytoskeleton remodeling, and DNA replication (1–6). AAA+ proteins have evolutionarily diverged into clades that are characterized by the incorporation of unique secondary structure elements into the canonical AAA+ domain (1, 3). Protein quality control serves as an excellent example of convergent functionality across distantly related clades of AAA+ proteins, as AAA+ proteases of both classical (i.e., FtsH and 26S proteasome) and HCLR (HslUV, ClpX, and Lon) families are required for protein degradation across all kingdoms of life (4).
AAA+ protein translocases assemble as hexamers, threading substrates through a narrow central pore, thereby forcing them to unravel. Recent cryo–electron microscopy (cryo-EM) structures of substrate-bound AAA+ proteins from both the classical and HCLR clades support a unified mechanism for adenosine 5′-triphosphate (ATP)–dependent substrate translocation (7–24). According to this mechanism, a spiral staircase configuration of the ATPase domains gives rise to a sequential, around-the-ring ATP hydrolysis cycle that leads to a constant pull on substrate. However, biochemical and biophysical studies of ClpXP, an AAA+ protein translocase belonging to the HCLR clade, indicate that a single active subunit within the hexamer is sufficient to drive degradation of certain substrates (25–30). These observations are incompatible with a strictly sequential mechanism. A plausible unifying explanation is that AAA+ proteins are able to alternate between different operational modes. However, the structural basis for this conformational switching has not yet been demonstrated nor has a mechanism for transitioning between operational modes been established.
The highly conserved AAA+ protease Lon is a representative member of the HCLR clade that is responsible for maintaining proteostasis in diverse subcellular environments, including the bacterial cytosol and the eukaryotic mitochondrial matrix (31–35). Lon’s role as an essential bacterial protease is of particular relevance in the development of antimicrobials (36), and mechanistic insights into its proteolytic activity could aid in the design of chemical inhibitors. The Lon polypeptide consists of an N-terminal domain (NTD) involved in substrate recognition, an ATPase domain that powers translocation, and a C-terminal serine protease domain. Numerous biophysical approaches have been used to gain insight into the specific nucleotide sensing and substrate translocation mechanism for this model protein degradation machine (31–40). Prior crystallographic studies of adenosine 5′-diphosphate (ADP)–bound Meiothermus taiwanensis Lon revealed a trimer-of-dimers configuration of the AAA+ domains, wherein subunits alternate between apo- and ADP-bound conformations (41). Alternatively, a crystal structure of Escherichia coli Lon with ADP in all six subunits suggests that its subunits assemble into a left-handed open lock washer configuration (42). A cryo-EM structure of a proteolytically inactive S679A mutant Lon solved in the absence of nucleotide also shows a similar left-handed conformation (43). Curiously, none of these structures are consistent with the right-handed helical arrangements that have recently been observed for several other AAA+ proteins (7–24, 44, 45).
We determined cryo-EM structures of Yersinia pestis Lon in the presence or absence of substrate and showed that, in the absence of substrate, the enzyme is trapped in a left-handed spiral organization that is both ADP-bound and proteolytically inactive. When bound to substrate, the Lon AAA+ domains adopt a “closed” right-handed spiral around the translocating polypeptide. Switching between the actively translocating, right-handed “on” state and the left-handed “off” state provides the structural basis for distinct, coexisting operational modes, reconciling a sequential substrate processing model with previous biochemical observations supporting a multimodal processing mechanism.
RESULTS
Structure of substrate-bound Lon is consistent with hand-over-hand translocation
Full-length Y. pestis Lon was incubated with an excess of Y2853, an 18-kDa putative sensory transduction regulator protein that is a robust Lon substrate (46). Substrate-bound complexes were separated from unbound substrate using size-exclusion chromatography (fig. S1A). Substrate engagement was increased via the introduction of a Walker B mutation (E424Q) into the Lon ATPase, slowing ATP hydrolysis by 90% compared to wild type (fig. S1E). Isolated complexes were incubated with saturating amounts of ATP (1 mM) and vitrified for single-particle cryo-EM analyses. This resulted in a reconstruction with an overall resolution of ~3.0 Å (fig. S2). Most of the Lon N-domain (1 to 252) was not visualized in the reconstruction, likely due to conformational flexibility of this region. However, an ~50-amino acid N-terminal domain three helix bundle (NTD3H) attached to the ATPase domains (253 to 305) was well resolved (Fig. 1, A and B, and fig S3). The cryo-EM density of this bundle, as well as the ATPase motor and protease domains, was of sufficient quality for atomic model building (figs. S2, E and F, and S3). An additional density corresponding to an extended seven-residue polypeptide was identified in the central pore of the ATPase hexamer, which we ascribe to an undefined segment of the Y2853 substrate (Figs. 1A and 2).
Fig. 1. Architectures of the substrate-bound LonENZ and substrate-free, LonOFF configurations.
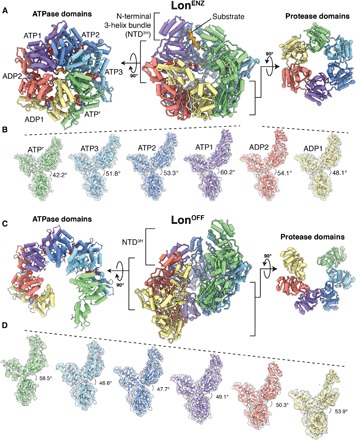
(A) Cutaway view of the substrate-bound Y. pestis LonENZ atomic model (center) flanked by orthogonal exterior views of the ATPase (left) and protease (right) domain rings. Each subunit of the homohexamer is assigned a distinct color depending on its position in the spiral staircase, and the cryo-EM density of the substrate is shown as a solid isosurface colored orange. Nucleotides are depicted using a sphere representation. (B) The different orientations of individual protomers relative to the protease domain, produced by orienting all the protease domains to a common view. Subunits are colored as in (A). The descending and ascending movements of the NTD3H and ATPase domains relative to their proteases are accentuated by dashed lines shown above the NTD3H. Dihedral angle measurements between ATPase and protease domains demonstrate a gradual expansion of descending subunits in the spiral staircase and compression in the two seam subunits. (C) Cutaway view of the substrate-free Y. pestis LonOFF atomic model (center) flanked by orthogonal exterior views of the ATPase (left) and protease (right) domain rings. Subunits are colored to correspond to their position in the LonENZ staircase architecture after transitioning. Nucleotides are depicted using a sphere representation. (D) The similar orientations of the individual protomers of the substrate-free structure are produced by orienting all the protease domains to a common view without changing axial position. The descending movement of the subunits is accentuated by dashed lines shown above the NTD3H. Dihedral angle measurements between ATPase and protease domains demonstrate subtle changes in the expansion and compression of the six descending subunits.
Fig. 2. Substrate translocation is mediated by residues in pore loop 1 and the NTD3H.
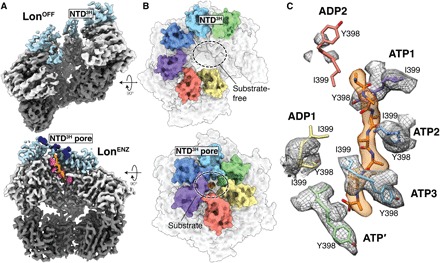
(A) Top: Cutaway view of the LonOFF cryo-EM density with the NTD3H colored light blue, ATPase domains colored light gray, and protease domains colored dark gray. Bottom: Cutaway view of the substrate-bound Lon cryo-EM density with NTD3H colored light blue, ATPase domains colored light gray, protease domains colored dark gray, and substrate colored orange. Sphere representations of spiraling methionine (M284) side chains in the NTD3H and tyrosine residues of pore loop 1 are colored dark blue and hot pink, respectively. (B) The NTD3H pores of substrate-free (top) and substrate-bound Lon (bottom) shown as a molecular surface representation and colored by subunit as in Fig. 1A. During substrate translocation, NTD3H forms a pore above the central channel. (C) A seven-residue polyalanine chain is modeled into the substrate density found in the closed Lon structure shown in a transparent orange surface representation. Y398 and I399 from pore loop 1 are shown using stick representations with associated cryo-EM density zoned around these residues in gray. While Y398 and I399 show intercalating, zipper-like interactions with substrate in ATP1 to ATP3, Y398, and I399 of pore loop 1 in ATP′ are positioned further away from the central channel. Pore loop 1 residues in the seam subunits (ADP1 and ADP2) are detached from the substrate.
As expected, the secondary structural elements of the Lon protomer in our cryo-EM reconstruction resembles previously determined crystal structures of substrate-free, ADP-bound M. taiwanensis Lon [Protein Data Bank (PDB): 4YPL; fig. S4]. Individually, the NTD3H, ATPase, and protease domains of our subunits aligned well with a nucleotide-free subunit of M. taiwanensis Lon [0.78, 1.26, and 1.01 Å Cα root mean square deviation (RMSD), respectively], demonstrating conservation of secondary structural elements within these three domains between bacterial Lon homologs. While the observed C6-symmetric organization of the serine protease domains in our substrate-bound cryo-EM structure is consistent with previously determined crystal structures, the quaternary organization of the ATPase domains is notably distinct. Whereas the crystal structure of M. taiwanensis Lon shows the NTD3H and ATPase domains arranged in a symmetric “trimer-of-dimers” configuration, our reconstruction shows these domains assembled into an asymmetric closed spiral staircase. Four subunits are in a continuous right-handed helical arrangement, while two “seam” subunits are found between the lowest and highest subunits of the staircase, thereby “closing” the spiral (Fig. 1, A and B, and movie S1). This asymmetric configuration of the Lon subdomains is largely consistent with recently determined cryo-EM structures of other substrate-bound AAA+ protein translocases of both the classical and HCLR clades (7–24). These structural similarities indicate that the staircase architecture is a conserved characteristic of the ATPase cassette in protein translocases across different clades and supports a generally conserved substrate processing mechanism.
Recent cryo-EM structures of substrate-bound AAA+ proteins have shown that pore loop aromatic residues protrude from the central channel of the ATPase staircase to interact with substrates (7–24, 44, 45). In agreement, the ATP-bound subunits that form the spiral within our substrate-bound Lon structure interact with the threaded substrate via the conserved pore loop 1 aromatic residue Y398, with one pore loop intercalating against the substrate backbone every two amino acids (Fig. 2, A to C, and movie S1). A semi-conserved hydrophobic pore loop 1 residue I399, which neighbors the aromatic Y398, contributes an additional intercalating interaction with substrate. Together, these two pore loop 1 residues in Lon flank both sides of the incoming substrate, allowing the pore loop 1 of each subunit to engage the polypeptide backbone with a pincer-like grasp to facilitate translocation into the peptidase chamber (Fig. 2C). Additional substrate-interacting residues have been observed in other AAA+ proteins (7, 9, 10, 12–16, 18, 22–24, 45), and consistent with a key role in substrate translocation, the I399A mutant of Lon exhibits defective degradation of Lon substrates HspQ and Y2853 while only minimally affecting ATP hydrolysis (fig. S1, G and H). Together, our results indicate that our structure of Lon trapped in the act of processing substrate is consistent with the hand-over-hand model for translocation driven by a sequential ATP hydrolysis cycle proposed for many other AAA+ proteins (7–24, 47, 48).
Substrate-free Lon forms an open, left-handed spiral
Most substrate-bound AAA+ translocase structures published to date have been solved using either nucleotide analogs or Walker B mutants to slow hydrolysis and effectively generate “substrate traps,” thus providing a snapshot of the enzyme stalled midtranslocation. However, these snapshots are unlikely to represent the complete conformational landscape of active AAA+ motors. For instance, the motors must transition through different stages of activity, including substrate engagement and release before and after translocation, respectively. Prior biochemical studies have suggested that Lon AAA+ protease accesses distinct conformational states depending on the presence or absence of substrate and is conformationally sensitive to substrate concentration and type (38, 49). As a result, the Lon AAA+ protease represents a unique opportunity to examine the structural basis for distinct conformational states that correlate to the different operational modes predicted to be required for AAA+ activity.
To access different potential operational modes of Lon, we imaged wild-type Lon protease in the absence of substrate but in the presence of physiological levels of ATP (1 mM). This structure revealed a distinct configuration of Lon that substantially deviates from our substrate-engaged structure. The resolution of our reconstruction (~3.8 Å) enabled atomic modeling based on a rigid-body fitting of the individual domains from our closed, substrate-bound structure (Fig. 1C and fig. S5). Our substrate-free Lon structure reveals a left-handed spiraling organization, which is opposite to that observed in the substrate-bound state but consistent with other Lon structures solved in the presence of ADP or a proteolytically inactive mutant in the absence of nucleotide (Fig. 1, C and D) (42, 43). The left-handed spiral is considerably steeper than the right-handed spiral, and there is a notable absence of any seam subunits, as all six subunits contribute to the helical arrangement. The protease domains—which assume a tightly interacting, sixfold symmetric organization in the substrate-bound configuration—are instead detached from one another and follow the left-handed spiraling trajectory of the ATPase domains, forming a double-ring open lock washer (Fig. 1, C and D). This extended complex presents a large aperture at the seam that is ~14 Å wide, exposing the central channel that was conspicuously devoid of substrate.
The open and closed Lon configurations contain distinct nucleotide states
ATP hydrolysis and exchange within the nucleotide pockets of AAA+ proteins are primarily responsible for inducing the conformational rearrangements required for substrate processing, as nucleotide state has been linked to rigid-body motions between the large and small subdomains of the AAA+ domain (8, 13, 15, 17–24). Similar to other AAA+ proteins (1–4), the nucleotide-binding pockets in the substrate-bound Lon complex are formed in the cleft between the large and small subdomains of the ATPase at the interface with a neighboring protomer, enabling nucleotide-dependent changes to allosterically affect the subdomains of both the nucleotide-bound subunit and its neighbor (fig. S6, A and B). These pockets contain conserved structural motifs such as the Walker A motif (356-GPPGVGKTS-364; important for nucleotide binding) and the Walker B motif (D423 and E424; important for nucleotide hydrolysis, mutated from E to Q in our substrate-bound structure; fig. S6, A and B) (1–4). The Lon nucleotide-binding pocket also contains a sensor-1 residue (N473) and a cis-acting sensor-2 residue (R542) (3). These residues are involved in positioning and stabilizing the ATP γ phosphate for hydrolysis (fig. S6, A and B). In addition, a trans-acting arginine finger (R484) from the clockwise adjacent subunit interacts with the γ phosphate and is critically important for ATP hydrolysis (fig. S6B) (50).
A mixture of nucleotide states is proposed to establish the asymmetric organization of the ATPase ring used by AAA+ protein translocases to process substrates (8, 13, 15, 17–24, 47, 48). The cryo-EM density in the nucleotide-binding pockets of our substrate-bound Lon reconstruction was of sufficient quality to identify the nucleotide state in each of the six subunits (fig. S6B). The four subunits that form the continuous right-handed staircase contain density that is consistent with an ATP-like molecule, with both the γ phosphate and magnesium ion present. These subunits are hereafter named ATP1, ATP2, ATP3, and ATP′, in order from the uppermost subunit of the ATPase staircase to the lowest (Fig. 1, A and B, and fig. S6B). The nucleotide density for the two seam subunits did not contain a γ phosphate (fig. S6B) and are hereafter named ADP1 and ADP2. These two subunits are displaced from the hexamer, with ADP1 at an intermediate position between the lowermost and uppermost staircase subunits and ADP2 at approximately the same height as ATP1 (Fig. 1B and fig. S6B). Intriguingly, the ADP1 nucleotide pocket contains density for a nucleotide-associated magnesium ion, while the ADP2 nucleotide does not (fig. S6B). This organization is consistent with a sequential ATP hydrolysis model, where the ADP1 and ADP2 subunits are in successive stages of phosphate release and nucleotide exchange as they ascend toward the top of the staircase to rebind ATP for another translocation cycle (Fig. 1B).
Prior cryo-EM studies of substrate-bound structures of AAA+ proteins demonstrated a direct correlation between nucleotide state and pore loop–mediated substrate interactions. In these structures, the conformations of the ATP-containing subunits were consistent with one another, while ATP hydrolysis induced large rigid-body domain rotations resulting in a loss of interactions between pore loops and substrate (8, 13, 15, 17, 18, 22–24). Similarly, in Lon, the dihedrals between the large and small ATPase subdomains in the ATP-bound subunits ATP1, ATP2, and ATP3 are indistinguishable in our substrate-bound structure (fig. S7A), and the pore loops of these subunits tightly interact with the substrate (Fig. 2C). However, the lowermost subunit, ATP′, displays a conspicuous 9° closure of the subdomains (fig. S7B), and its pore loop aromatic does not intercalate against the substrate backbone (Fig. 2C). In addition, its clockwise neighbor, ADP1, retains the compressed subdomain configuration and is displaced from the hexamer (Fig. 1, A and B, and fig. S7C). As a result, all inter-ATPase contacts, including the trans-acting Arginine finger that is important for hydrolysis, are lost (fig. S6B). A similar loss of intersubunit contacts has been observed in ClpXP and the ClpAP D2 ring, indicative of a conserved ATP hydrolysis mechanism across the HCLR clade (22–24).
Notably, the ATP1, ATP2, and ATP3 subunits are observed interacting with substrate, whereas the ATP′ subunit, similarly to the ADP-bound subunits, is disengaged from substrate (Figs. 1A and 2C). This indicates that our reconstruction represents a conformational intermediate where the ATP′ subunit is likely trapped in a posthydrolysis ADP-Pi state that has released its grip on substrate. It is possible that this intermediate is a result of perturbing the native chemical environment through introduction of a Walker B mutation. However, the Walker B Lon slowly hydrolyzes ATP (fig. S1E), suggesting that the ATP′ subunit is in a physiologically relevant conformation, and the displacement of this subunit from the spiral staircase is required for phosphate release. Regardless, these findings indicate that, like in other AAA+ proteins, coexisting nucleotide states in the substrate-bound structure of Lon correlate with domain positions that produce the observed closed spiral staircase.
In stark contrast to our substrate-bound structure of Lon, the six subunits in the substrate-free conformation of Lon form a continuous left-handed spiral (Fig. 1D). We observe density that unambiguously corresponds to ADP molecules in the binding pockets of the lower five subunits in the staircase and substantially weaker nucleotide density in the uppermost subunit. This weaker density likely corresponds to an averaging of empty and occupied binding pockets (Fig. 1D and fig. S8A). Similar to the ADP-bound subunits in our substrate-bound Lon reconstruction, the pore loops of all subunits in our substrate-free conformation are disordered. The crystal structure of the ADP-bound Bacillus subtilis Lon (42) superimposes onto the ADP-bound subunits of our substrate-free structure with a Cα RMSD of 1.1 Å, further supporting our assignment of nucleotide state (figs. S7E and S8B). The density of the uppermost subunit of the spiral is weaker than the ADP-bound subunits, indicative of conformational flexibility. Notably, nucleotide-free subunits in cryo-EM reconstructions of AAA+ proteins are consistently more flexible than nucleotide-bound subunits, further suggesting that this subunit likely represents a mixture of ADP and apo states (8, 13, 15, 17, 18).
Given that prior studies have shown that ADP inhibits Lon activity, we conclude that the left-handed substrate-free configuration represents the Lon off (hereafter referred to as LonOFF) operational mode. Furthermore, the experimental conditions that produced this conformation are consistent with prior biochemical observations that the absence of substrate promotes Lon to adopt an inactive or off state (38, 51). These two distinct states of the Lon AAA+ protease present a unique opportunity to examine the mechanism of conformational switching between operational modes and to identify structural features dictating Lon activity in both conformations.
Nucleotide-dependent movements of the ATPase domains are transmitted to adjacent domains
The ATPase domain is located at the center of the Lon polypeptide, and it is flanked by an NTD and a C-terminal protease domain. Our structures reveal the spatial relationships between the nucleotide states of ATPases and these neighboring domains. Akin to many other AAA+ proteins, the NTD of Lon has been shown to be involved in substrate recruitment and recognition (52). This ~300-residue domain contains an N-terminal β sheet subdomain connected to a long α helix, followed by a three-helix bundle known as the NTD3H (42, 53). While we do not observe density for the β sheet and long α helix in any of the N termini, the NTD3Hs all appear to be stably ordered in both the open and closed states (Fig. 2, A and B, and fig. S3). The NTD3Hs of Lon were proposed to be flexibly attached to the ATPase domain, given that these domains are connected by a 12-residue linker (42). However, our structures show that the NTD3Hs are rigidly positioned above the ATPase domains and tightly incorporated into the staircase organization (Fig. 2, A and B, and fig. S3). As a result, the NTD3H closely mirrors the behavior of the ATPase domain, following a left- and right-handed spiral in the open and closed states, respectively (Figs. 1 and 2, A and B). Interactions between the ATPase domain and the NTD3H suggest that the NTD3H functions as a rigid extension of each ATPase domain. Accordingly, in the LonOFF state, the NTD3Hs do not appear to directly contact each other, forming a ~30-Å-wide pore with an open seam, but upon transitioning to the substrate-engaged and proteolytically active form of Lon (hereafter referred to as LonENZ), these domains form a pore approximately 15 Å in diameter immediately above the substrate-bound central channel of the ATPases (Fig. 2B).
While we do not observe substrate density extending into the NTD3H staircase (Fig. 2, A and B), residue M284 is positioned at the interdomain interface within a helix turn at the entrance to the NTD3H pore of the LonENZ conformer (Fig. 2A, shown in dark blue). This uniquely positions this residue such that it could simultaneously be involved in substrate interactions and stabilizing the pore-like organization of the NTD3Hs of the closed state (Fig. 2B). We tested the relevance of this methionine for Lon activity by introducing an M284A mutation. We confirmed that the mutant is competent in forming a substrate-bound hexamer by size-exclusion chromatography (fig. S1B). However, this mutation significantly reduced substrate-stimulated ATPase and protease activity, implicating M284 in potentially stabilizing the closed, active conformation (fig. S1, G and H). The functional relevance of this pore formation is underscored by previous biochemical studies, indicating that substrate degrons simultaneously contact both the central pore and allosteric sites at positions flanking the pore (38). These allosteric contacts were proposed to stabilize the LonENZ state (38), and the ring formed by the NTD3H is ideally positioned to fulfill this role, potentially through combined substrate interactions and stabilization of the closed hexamer ring.
The C-terminal protease domains in the LonOFF state adopt a helical organization that follows the left-handed helical organization of the ATPases (Fig. 1, C and D). However, unlike the NTDs, the protease domains depart from the spiraling organization of the ATPase domains in the LonENZ state and, instead, assemble as a planar sixfold symmetric proteolytic ring. The ability for the ATPase and protease domains within a single protomer to adopt these varied positions in regard to one another appears to be accommodated by a flexible ~13-residue interdomain linker containing a strictly conserved glycine residue located at the base of the small ATPase subdomain (G580; fig. S9A). This residue is analogous to the glycine linker between the ATPase and protease domains of classical AAA+ proteases, shown to be critical for substrate translocation (13, 54). Consistent with an important role for this glycine residue in the mechanism of substrate translocation, incorporating a G580L mutation in Lon diminishes HspQ- and Y2853-stimulated ATPase rates by 52 and 64%, respectively, and degradation of these substrates by 26 and 33%, respectively (fig. S1, G and H).
The protease domains undergo a notable pivoting motion as they transition from the spiral organization of the LonOFF state to the planar LonENZ state (fig. S9B). This pivoting motion and adoption of sixfold symmetry is associated with, and likely a result of, an interprotease charged network that is established in the LonENZ state (fig. S9C). These observations, combined with the fact that isolated protease domains crystallize as sixfold symmetric rings (37, 55), suggest that the organization of the protease domains in the LonENZ state is more energetically favorable than in the spiraling LonOFF configuration. Thus, the open organization of the protease domains in LonOFF is likely enforced by the ATPase domains, and the transition from a spiraling to planar organization has important functional implications for proteolytic activity. The ADP-bound state of Lon was shown to be incapable of cleaving small peptides and that protease activity required binding, but not hydrolysis, of ATP (56). This and other studies collectively indicate that the protease activity of Lon requires both ATP and substrate binding in the ATPase domains (38, 46), suggesting that these combined interactions trigger an allosteric activation of the protease domain. The two conformational states we present here define the nucleotide- and substrate-dependent rearrangements that allosterically shift the Lon protease from an open, inactive state to the closed, active state.
The proteolytic active site is autoinhibited in the LonOFF conformation and activated during the substrate-induced rearrangement
In the LonENZ conformation, the protease active sites are sequestered in a protected, sixfold symmetric protease chamber. Folded substrates can only be degraded after ATPase-driven unfolding and shuttling into the protease chamber, where they can bind in the proteolytic active sites. Lon uses a serine-lysine catalytic dyad formed by S679 and K722 to perform proteolysis of unfolded substrate peptides. Numerous models for substrate binding and hydrolysis have been proposed to explain the structural basis of this proteolytic activity (37, 41, 55, 57). In our active, substrate-bound LonENZ structure, these catalytic residues are positioned orthogonal to each other within a cavity formed at the interprotomer interface of the hexamer (Fig. 3, A and B, and fig. S10A). K722 is positioned on an α helix that is proximal to the ATPase-protease interface, while S679 is located on a loop including residues 673 to 677 that extends toward the central channel of the hexamer (Fig. 3B and fig. S10A). Within the context of the oligomerized protease, the six S679-containing loops within the hexamer organize into a ring-like assembly, forming a series of grooves (Fig. 3, A and B) that have been implicated in substrate binding (57). A crystal structure of E. coli Lon bound to bortezomib, a known Lon inhibitor, showed how this ligand engages in β sheet–like interactions within this binding groove (57). It is likely that an unfolded substrate would become positioned in proximity of the catalytic dyad through similar interactions (Fig. 3E and movie S2).
Fig. 3. Lon proteolytic active site forms cleft for substrates in LonENZ and is autoinhibited in LonOFF.
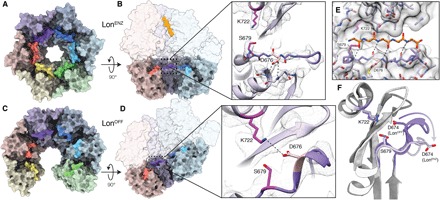
(A) An axial view of the chamber of the sixfold symmetric protease ring from the substrate-bound LonENZ structure is shown as a molecular surface with subunits are colored as in Fig. 1, with the six catalytic serine-containing loops (673 to 677) emphasized using a more saturated color. These loops organize into a ring-like assembly, generating a series of substrate-binding grooves. (B) A cutaway side view of the substrate-bound structure shown as a molecular surface. A close-up view of the cryo-EM density of the proteolytic active site is shown to the right as a gray mesh, with the atomic model showing the catalytic dyad (magenta) and serine-containing loop (purple stick representation). The serine-containing loop is likely stabilized in this extended conformation, in part, by hydrogen bonding between D676 (pink) and the peptide backbone of a nearby loop (light purple). (C) An axial view of the chamber of the protease ring from the LonOFF structure is shown and colored as in (A). The serine-containing loops are no longer interacting because of the separation of the protease domains in this conformation. (D) A cutaway side view of the substrate-free structure showing the location of the protease active sites in the open, exposed proteolytic chamber. A close-up view of the cryo-EM density of the proteolytic active site is shown as in (B), showing that in LonOFF, the catalytic serine-containing loop adopts a 310 helix that sterically occludes the proteolytic active site. D676 and K722 are within hydrogen-bonding distance, further limiting cleavage by the catalytic dyad. (E) A five–amino acid polyalanine peptide (orange) was modeled into the substrate-binding groove of LonENZ (represented using a transparent space-filling representation of the atomic model), based on the position of bortezomib covalently bound to S679 (PDB: 4YPM) (57). This demonstrates how an unfolded peptide substrate putatively docks into the active site for proteolytic cleavage by Lon protease. (F) The rearrangement of the serine-containing loop during the transition from LonOFF (dark gray/purple) to LonENZ (light gray/purple). This rearrangement is also shown in movie S2.
In contrast to the privileged sequestered protease chamber observed in our LonENZ structure (Fig. 3, A and B), the inactive LonOFF conformational state contains a large opening at the interface between its highest and lowest subunits, exposing its proteolytic active sites to the external environment (Figs. 1C and 3C). The accessibility of the protease active site afforded by this opening could allow for promiscuous degradation of folded or unfolded protein substrates. However, Lon protease activity is known to be inhibited in the conditions used to produce our LonOFF cryo-EM structure (38). Thus, a key question is how is Lon proteolytic activity suppressed in the LonOFF conformational state?
Comparing the protease domains of our LonENZ and LonOFF structures revealed the mechanism by which Lon regulates proteolytic activity. In the LonOFF conformer, the S679-containing loop folds into a 310 helix that is positioned in close proximity to the active site, sterically blocking access of substrates to the catalytic dyad (Fig. 3, D and F, and fig. S10B). Furthermore, an aspartic acid residue within this helix (D676), which is conserved from bacteria to humans, appears to hydrogen bond with K722, inhibiting the catalytic dyad by suppressing abstraction of a proton from the S679 hydroxyl (Fig. 3, D and F, and fig. S10B). Upon substrate binding and transition to the proteolytically active state, symmetrization of the protease domains causes the 310 helix within the S679-containing loop to unfold and extend toward the neighboring protease domain (Fig. 3, B and F, and movie S2). The extended conformation of this loop may be stabilized through van der Waals contacts and hydrogen bonds with conserved residues V633, P678, and E706 in the neighboring subunit (movie S2) (57). As the S679-containing loop switches to this extended conformation, the autoinhibiting aspartic acid residue is repositioned to stabilize an extended form of this loop by hydrogen bonding with the backbone atoms of residues E632 and V633 (movie S2). This extended conformation forms a binding cleft into which unfolded substrates could be positioned in proximity of the active site residues for cleavage (37).
Collectively, these results indicate that Lon proteolytic activity is autoinhibited in LonOFF in the absence of substrate and ATP to prevent nonspecific proteolysis, as predicted by previous biochemical studies (38, 46). However, this autoinhibition is relieved upon substrate binding in the ATPase domains while simultaneously generating a substrate-binding cleft in the protease domains that positions substrates for cleavage. The substrate-modulated access to proteolytic active sites we observe here in Lon, a covalently linked AAA+ protease, offers an alternative regulatory mechanism to the gating mechanisms used by noncovalently associated AAA+ proteases such as ClpX, ClpA, and the 26S proteasome (8, 22–24, 58). These gating mechanisms likely evolved to prevent nonspecific degradation of substrates by making the proteolytic active sites accessible exclusively in the context of substrate engagement and translocation.
Substrate translocation occurs through an HCLR-specific allosteric mechanism
The organization of our LonENZ is generally consistent with other cryo-EM structures of substrate-bound AAA+ protein translocases (7–24, 58). This supports a conserved substrate translocation mechanism among AAA+ protein translocases across diverse evolutionary clades. ATP-dependent quality control proteases in the classical clade display conservation of an intersubunit signaling (ISS) motif that senses the nucleotide state of the neighboring subunit and transmits ATP-dependent conformational changes to substrate-interacting pore loop residues within the central channel to drive substrate translocation process (13). However, this ISS motif is not present in Lon or any other HCLR clade AAA+ protein (3, 59). Independently, superimposing either the large or the small ATPase subdomains of the six Lon subunits in our LonENZ structure showed an average Cα RMSD of 0.81 and 0.44 Å, respectively (Fig. 4A). Thus, nucleotide-induced rigid-body motions of the subdomains must be the main drivers of translocation in Lon, and these motions must occur in the absence of rearrangements in secondary structure. Although cryo-EM structures of other ATPases in the HCLR clade have been determined (7, 9, 15, 16, 18, 22–24, 58, 60), it remains unclear how a sequential ATPase mechanism might be coordinated in these AAA+ rings.
Fig. 4. The PS1βH motif of Lon connects pore loop 1 to adjacent nucleotide-binding pockets.
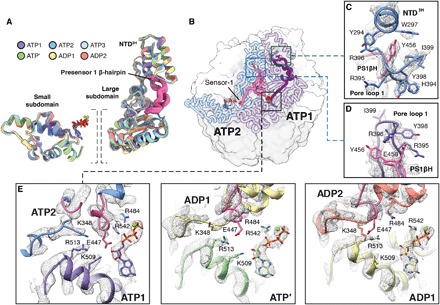
(A) Individual units of the AAA+ domains from the LonENZ structure are aligned and shown overlaid using a ribbon representation. The two units are (i) the small ATPase subdomains (left) and (ii) the NTD3H and large subdomain. Each subunit is colored according to the same color scheme assigned in Fig. 1. Superimposing these subdomains shows an average Cα RMSD of 0.81, indicating that these regions individually move as rigid units throughout the hydrolysis cycle. (B) ATP1 and ATP2 subunits are highlighted using a coil representation in the context of Lon structures (smoothed surface representation, ATPase domains are white, and protease domains are gray). The PS1βHs are colored purple and pink in ATP1 and ATP2, respectively. Sensor-1 at the C-terminal base of the PS1βH is denoted in ATP2. Close-ups of the regions enclosed by the boxes are shown in (C) and (D). (C) Y456, at turn of the PS1βH, stabilizes the NTD3H through proximal interactions with conserved aromatic residues Y294 and W297. (D) Pore loop 1 interactions with substrate are stabilized through interactions between E458 in the PS1βH and two tandem arginine residues in pore loop 1. (E) Remodeling cis and trans subunit interactions in the nucleotide-binding pocket drives sequential ATP hydrolysis cycle and stepwise substrate translocation. In the nucleotide-binding pocket of two adjacent ATP-bound subunits (e.g., ATP1 and ATP2; left), nucleotide is stabilized by interactions with the arginine finger (R484) in trans and sensor-2 (R542) in cis. Furthermore, a bridging glutamate residue (E447) at the N-terminal base of the PS1βH motif engages a cluster of basic residues within the nucleotide-binding pocket, stabilizing the intersubunit interface. In the ATP′ nucleotide-binding pocket (middle), this organization is disrupted upon E447 being retracted from the nucleotide-binding pocket upon nucleotide hydrolysis, causing the subunit to compress and bringing sensor-2 and other motifs involved in ATP hydrolysis (i.e., Walker A and Walker B motifs) in the proximity of the bound nucleotide. The ADP1 (right) and ADP2 (not shown) nucleotide-binding pockets reveal a similar organization to that observed in ATP′, indicating that nucleotide exchange is necessary to “reset” the hydrolysis cycle.
Intriguingly, HCLR AAA+ proteins contain a presensor-1 β hairpin (PS1βH) insertion that is the defining element of the HCLR clade and has been shown to be critically important for substrate translocation (61). Our cryo-EM map was of sufficient resolution to reveal how the PS1βH runs laterally along the intersubunit interface of Lon, with the turn of the hairpin positioned in close proximity to pore loop 1 (Fig. 4, B to D, and fig. S3). A conserved tyrosine residue (Y456) within the PS1βH turn inserts into a pocket at the interface of the NTD3H and pore loop 1 and is stabilized by aromatic interactions with conserved tryptophan (W297) and tyrosine (Y294) residues (Fig. 4C). In addition, a conserved glutamate residue (E458) from PS1βH interacts with two tandem arginine residues (R395 and R396) that are immediately adjacent to the conserved pore loop 1 aromatic residue (Y398) (Fig. 4D). This indicates that the PS1βH likely serves a role in stabilizing the substrate-engaged organization of the pore loops as Lon subdomains progress through the hydrolysis cycle. Consistent with this, disruptions in this stabilization with an E458A mutation impaired degradation of the Lon substrates Y2853 and HspQ but did not substantially affect ATPase activity (fig. S1, G to H).
In addition to stabilizing pore loop 1 within the protomer, the PS1βH is structured to sense or influence the nucleotide state both within the subunit and in the preceding neighboring protomer. Previous studies suggest that the PS1βH allosterically communicates nucleotide state to neighboring protomers (61), although the detailed mechanism for this allostery was unclear. We show that the C-terminal end of the PS1βH continues into the β4 strand of the canonical AAA+ fold that contains the sensor-1 residue (N473) involved in ATP hydrolysis within the protomer (Fig. 4B). At the other end of the PS1βH, a highly conserved glutamate residue (E447) extends toward the nucleotide-binding pocket of the counterclockwise neighboring subunit, serving as an “acidic bridge” between subunits (Fig. 4E and fig. S11). In ATP1 to ATP3, this glutamate interacts with a patch of basic residues within the adjacent small subdomain that includes the sensor-2 residue R542 (Fig. 4E). However, within the ATP′, ADP1, and ADP2 nucleotide-binding pockets, this trans-acting glutamate bridge is retracted (Fig. 4E and fig. S11). This observation suggests that the PS1βH responds to nucleotide hydrolysis and is involved in displacing ADP-bound protomers from the hexamer. Accordingly, these three subunits are not engaged with the polypeptide substrate. We confirmed the functional relevance of this acidic bridge in the Lon mechanochemical cycle by introducing an E447A mutation, which caused defects in both substrate-induced ATPase (57 and 72% reductions for HspQ- and Y2853-stimulated ATPase rates, respectively) and proteolytic activity (45 and 48% reductions for HspQ and Y2853 degradation rates, respectively; fig. S1, G to H).
Our results indicate that the conserved PS1βH insertion likely functions as a structural entity linking cis- and trans-acting nucleotide state sensors to the substrate translocation elements in the absence of an ISS motif for the HCLR clade. Sequence and structural alignments of the PS1βH in Lon homologs and other HCLR clade AAA+ domains, including HslUV, ClpX, RuvB, and the D2 domains of HSP104 and ClpB, reveal strict conservation of this bridging element and its organization (figs. S12 to S13). We thus posit that the allosteric mechanism that drives around-the-ring hydrolysis in Lon is conserved throughout the HCLR clade.
Rearrangement of LonOFF to LonENZ engages substrate
Having established the structural basis for nucleotide-dependent rearrangements and substrate translocation in LonENZ, we next examined how substrate and ATP binding might induce LonOFF to transition to the active conformation. All the subunits of the left-handed helix in LonOFF are trapped in an ADP-bound configuration with the exception of the topmost subunit, which has an exposed nucleotide-binding pocket (fig. S8A). ATP likely binds at this topmost site, but the trans-acting motifs required to stabilize nucleotides within the binding pocket are more than 40 Å away, at the opposite end of the spiral. Thus, ATP binding is unlikely to result in hydrolysis, as hydrolysis in the absence of a trans-acting arginine finger is inefficient (50). Given that Lon activation depends on both ATP and a protein substrate (38, 46), pore loop interactions with the substrate likely triggers the rearrangement from LonOFF to LonENZ.
Our LonENZ structure shows how the PS1βH establishes coordination between nucleotide state and pore loop intercalation into substrate. On the basis of the nucleotide-associated conformations observed in the LonENZ structure, ATP and substrate binding by the topmost subunit of LonOFF would lead to a rigid-body domain rotation that exposes the nucleotide-binding site of its counterclockwise neighbor for nucleotide exchange. The motions that reorganize the intersubunit nucleotide-binding pocket for nucleotide exchange in LonOFF during this transition are likely to be identical to the subunit motions involved in the processive translocation mechanism observed for LonENZ, where simultaneous ATP and substrate binding introduces a rearrangement that positions a subunit at the uppermost position of the spiral staircase, thereby enabling subsequent nucleotide exchange in the neighboring ADP-bound subunit (Fig. 5, A and B). ATP binding in this second nucleotide pocket within LonOFF would establish intersubunit interactions mediated by the PS1βH and the trans-acting arginine finger of the first subunit. The domain rotation associated with ATP binding in the second topmost subunit would enable nucleotide exchange in its counterclockwise neighbor, initiating a domino effect in which a total of four subunits would exchange nucleotide sequentially (Fig. 5A and movie S3). As each subunit binds ATP, the associated domain rotations would progressively reconstitute the ATP-bound staircase until the LonENZ configuration (with two remaining ADP-bound subunits) is attained. It would not be possible for all six subunits to successively exchange ADP for ATP, as this would require continued progression toward a right-handed open spiral, which is sterically prevented by the position of the ADP-bound subunits (movie S3). Simultaneous with the reorganization of the ATPase domains, the neighboring protease domains assemble into a C6-symmetric conformation, activating the proteolytic active sites for substrate cleavage. In agreement with this model for substrate engagement, biochemical assays indicate that addition of substrate induces ADP release (51).
Fig. 5. Summary of the mechanism of substrate translocation in Lon protease.
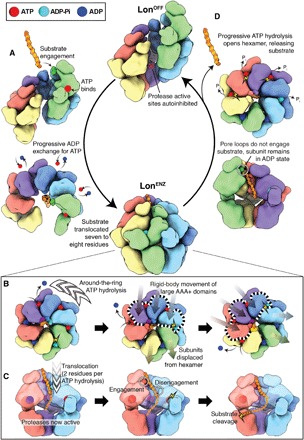
(A) In the absence of substrate, Lon that is fully ADP-bound adopts a left-handed, open lock washer configuration with autoinhibited protease active sites. Simultaneous ATP and substrate binding to the uppermost protomer initiates the reorganization toward the proteolytically active LonENZ conformer. Subunits then progressively exchange ADP for ATP in a counterclockwise manner. The LonENZ conformer is achieved upon binding a total of four ATP molecules, finalized with one hydrolysis event to close the ring. Hydrolysis occurs in the subunit that had previously been at the top of the spiral (green) when it reaches the lowest position of the ATPase spiral within the closed hexamer. The rearrangement results in a seven to eight-residue translocation of the substrate peptide, which is now threaded through the center of the central ATPase channel. (B) The LonENZ conformation is competent for hand-over-hand translocation, and the protease domains form an enclosed sixfold symmetric ring and with active protease sites. Around-the-ring ATP hydrolysis drives substrate translocation, with the pore loops of the ATP-bound subunits engaging with substrate (substrate-interacting pore loops shown using space-filling representation). ATP binding and hydrolysis at opposite sides of the ring result in rigid-body movements of the three upper-most ATP-bound subunits (dotted outline), leading to two-residue translocation steps toward the protease. (C) Cutaway view showing progression of substrate along the central axis, being translocated two amino acids per hydrolysis event, and substrate binding within the active site for cleavage. As in (B), substrate-interacting pore loops shown using space-filling representation. Substrate is directed to the protease, where it can be positioned for cleavage. A summary of the LonENZ mechanochemical cycle is shown in movie S1. (D) Inability of the pore loop 1 of an ADP-bound subunit to bind substrate either due to reaching the terminus of a substrate or encountering a tightly folded domain will trigger a return to the LonOFF conformation. The remaining ATP- and substrate-bound subunits will continue around-the-ring hydrolysis and translocation, but each will remain in the ADP-bound conformation after nucleotide hydrolysis. Remaining nonproteolyzed substrate peptides are released. A summary of the entire conformational cycle is shown in movie S3.
Lon activation was previously shown to be a two-step process (62), the first involving the formation of a Lon:ATP:substrate complex and the second corresponding to an ATP hydrolysis–dependent conformational change in the complex (Fig. 5, A and B) (38, 46). This indicates that the LonOFF to LonENZ transition not only depends on ATP and substrate binding but also on an ATP hydrolysis event. We speculate that this first hydrolysis event occurs after four nucleotide exchanges have occurred during the transition from LonOFF to LonENZ. The motions associated with these four nucleotide exchange events would position the first ATP-bound subunit (which was previously positioned at the top of the LonOFF helix) at the lowest position within the reconfigured hexamer. This lowermost subunit would be positioned in close proximity of the trans-acting finger of the clockwise neighboring subunit, establishing the appropriate chemical environment for ATP hydrolysis. This ATP hydrolysis event would complete the transition to the hexameric LonENZ conformation with substrate fully threaded through the central pore (Fig. 5, A to C, and movie S3).
Notably, this mechanism enables the substantial conformational rearrangement required for Lon activation to occur through a single ATP hydrolysis event. This progressive closure model also maximizes substrate translocation, as the substrate–pore loop interactions initiated at the topmost subunit of the LonOFF spiral become positioned at the bottom of the staircase in LonENZ (Fig. 5A). Thus, in the absence of any other forces, this single ATP hydrolysis event would result in an effective translocation of seven to eight residues and robust capture of substrate within the central channel of Lon (Fig. 5, B and C, and movie S3). This is consistent with the requirement of an unfolded tail (or degron) of approximately 10 residues for processing of Lon substrates (38). Furthermore, formation of the closed-ring LonENZ conformer around the engaged substrate promotes organization of the protease domains into an activated sixfold-symmetric ring and the assembly of the NTD3H pore above the central ATPase channel. This extensive network of substrate-induced reorganizations would trap the enzyme in the substrate-bound LonENZ conformation for as long as it remains bound to substrate. As in the LonOFF conformer, ADP release is dictated by the conformational rearrangements associated with substrate and ATP binding of the clockwise neighboring subunit, providing a molecular explanation for the previous observation that ADP release is the rate-limiting step of substrate processing in Lon (63). This enforces a sequential, one-at-a-time ATP hydrolysis cycle where hand-over-hand translocation directs the unfolded substrate, two amino acids for every ATP hydrolysis event, into the enzymatically activate degradation chamber, consistent with other ATPases (Fig. 5, B and C, and movie S1) (7–24, 44, 45, 47, 48).
Model for substrate disengagement from LonENZ
In the LonENZ conformation, the enzyme will continue to translocate peptide using a sequential mechanism as long as unfolded substrate can be threaded through the central pore (Fig. 5, B and C). Lon will likely return to the LonOFF conformation when the substrate has been fully processed or when the enzyme encounters a tightly folded protein domain. In either scenario, the pore loops of the ADP2 subunit will be unable to engage a region of unfolded substrate (Fig. 5D). Having released ADP but unable to assume a position at the top of the right-handed staircase, this subunit becomes trapped in a nucleotide-free state. The counterclockwise adjacent subunit (ADP1) would likely then progress to the ADP2 state, allowing phosphate release from its neighboring ATP′ subunit, which would then assume an ADP-bound state (Fig. 5D). This would, in turn, cause its counterclockwise neighbor to hydrolyze ATP, leading to a domino effect where the subunits would sequentially fire without releasing nucleotide (Fig. 5D and movie S3). Given the correlation between ATP hydrolysis and substrate disengagement by the pore loops, adoption of the ADP state in each subunit would result in release of substrate. Simultaneously, the domain rotations associated with the ADP-bound state of the ATPases would cause each subunit to shift from its position in the right-handed ATP spiral (observed for LonENZ) to a register in the left-handed ADP staircase (observed for LonOFF; Fig. 5D). Sequential hydrolysis in all four subunits would render Lon in an ADP-bound left-handed staircase that is disengaged from substrate, reproducing the organization of our LonOFF state (Fig. 5D and movie S3). This suggests that Lon will remain in the fully ADP-bound LonOFF state until the next substrate-binding event, given the requirement for simultaneous substrate and ATP binding by the uppermost subunit of this conformer to initiate progressive nucleotide exchange and a return to the LonENZ conformer.
Notably, this transition from the closed LonENZ state to inactive LonOFF state involves successive ATP hydrolysis events that are not limited by ADP release, which was shown to be rate-limiting for ATPase activity (29). Thus, this transition may be associated with fast, sequential ATP hydrolysis. Since each hydrolysis event at the lowermost ATP-bound subunit would result in a downward rigid-body motion of the remaining ATP-bound subunits, each event would impose a pull on the substrate before disengagement. Thus, the transition from LonENZ to LonOFF would lead to successive, near-synchronous, mechanical-pulling events on a substrate.
DISCUSSION
The two conformational states, LonOFF and LonENZ, observed in this study provide a structural framework to understand decades of biochemical and biophysical studies on Lon, informing on the mechanisms of substrate engagement, translocation, and release for this central AAA+ protease and likely other AAA+ protein translocases.
The enzymatically active LonENZ conformer demonstrates a conservation of a sequential hydrolysis and substrate translocation mechanism now proposed for numerous other AAA+ proteins, underscoring how AAA+ proteases from distantly related clades use distinct structural elements to power a broadly conserved mode of substrate conveyance. Meanwhile, the open-form complex, which we ascribe to the proteolytically inactive LonOFF state, sheds light on a largely unanswered question in the field: How are diverse protein substrates engaged and threaded into the central channel of a closed AAA+ hexamer? Our structures outline the molecular infrastructure and mechanisms involved in grasping a degron and subsequently encircling it for efficient translocation.
On the basis of interpolations between our LonENZ and LonOFF structures, a substrate-processing model emerges for Lon that appears to be consistent with biochemical studies of the related HCLR AAA+ ATPase ClpXP. We propose that the transition between the LonOFF and LonENZ supplies the force needed to drive the initial pull or denaturation event, after which a slower, one-at-a-time ATP hydrolysis cycle commences, resulting in a constant stepwise translocation of the unfolded polypeptide powered by sequential ATP hydrolysis. Local increases in substrate resistance to translocation might initiate a return to the LonOFF conformer, which is associated with a successive firing of subunits that is not limited by ADP release. This near-synchronous firing of up to four ATPase subunits would provide numerous opportunities for local unwinding of the substrate such that two residues could become accessible to the pore loops of the uppermost nucleotide-free subunit. Binding by these uppermost pore loops could result in a reengagement of substrate, triggering a return to the processive LonENZ mode. However, if these events fail to make substrate available for the pore loops of the topmost subunit to rebind substrate, then all four subunits would hydrolyze ATP, resulting in a transition to the substrate-disengaged, ADP-bound LonOFF spiral.
The transitions between the two observed conformational configurations of Lon would give rise to ATP hydrolysis bursts of different sizes, providing numerous opportunities for local unwinding events to encourage substrate reengagement and continued translocation. Hard-to-unfold substrates would resist these energetic pulls and are subsequently released. Consistent with this model, both Lon and ClpXP were shown to undergo multiple cycles of substrate binding and release before unfolding stable domains (26, 30, 38, 49, 64). In line with this multimodal ATP hydrolysis and translocation model, previous observations of ClpX showed that small translocation steps resulting from a single ATP hydrolysis event are primarily followed by small steps, whereas bigger translocation steps corresponding to near-synchronous firing in several subunits were typically followed by bigger steps (26). We propose that the speed at which substrate can be reengaged at the top of the staircase enables these enzymes to adjust their operational mode to the irregularity of the thermodynamic stability, topology, and sequence characteristics of the different protein substrates processed by Lon and other AAA+ protein translocases. This model could thus explain the partially probabilistic behaviors observed for AAA+ protein translocases, whereas DNA AAA+ translocases, which move along a much more rigid and periodic track of double-stranded DNA, operate through a strictly sequential mode (65).
The core mechanistic features defined here appear to be conserved in distantly related AAA+ protein unfoldases. For instance, HSP104, an AAA+ disaggregase that contains two tandem AAA+ domains (one classical clade and one HCLR clade), presents an almost identical open lock washer organization with a left-handed helical pitch when disengaged from substrate (fig. S14) (60). Meanwhile, substrate-bound structures of HSP104 present a right-handed helix, supporting a model whereby a sequential ATP hydrolysis cycle powers stepwise substrate translocation (9). Substrate engagement introduces an analogous conformational change within the ATPase hexamer of the 26S proteasome, whereupon the AAA+ subunits transition from a translocation-incompetent conformation to one that is capable of progressive substrate translocation (8, 66, 67). Notably, this rearrangement involves the movement of the uppermost subunit within the staircase to the lowest position, in a motion previously implicated as a potential mechanism by which substrates are captured and positioned within the central ATPase channel (67). It thus appears that a conformational switch involving a substrate-induced reorganization of the staircase upon substrate engagement is a common theme among these enzymes. While future studies will be required to determine the degree of conservation of the mechanisms underlying substrate engagement and release in different AAA+ proteins, the core molecular principles presented here are likely conserved across AAA+ protein unfoldases.
MATERIALS AND METHODS
Protein expression and purification
E. coli strain BL21 star (DE3) was used to express recombinant proteins. Cells were grown in Lysogeny Broth (5 g of yeast extract, 10 g of tryptone, and 5 g of NaCl per liter) supplemented with kanamycin (50 μg/ml). Cells were cultured at 37°C with shaking at 250 rpm. We have previously described the cloning and expression of Y. pestis lon, hspQ, and y2853 genes (46). The pET28b-lon plasmid was used to overexpress Yersinia Lon protein in E. coli strain BL21 star (DE3). Cells were cultured in LB containing kanamycin (50 μg/ml) at 37°C to optical density at 600 nm (OD600) of 0.5. Protein expression was induced by 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Protein overexpression was carried out for 16 hours at 16°C. Cells were harvested by centrifugation at 3700g and resuspended in buffer A [50 mM KHPO4 (pH 7), 1 mM EDTA, 1 mM dithiothreitol (DTT), and 10% glycerol]. After sonication, cleared cell lysate was prepared by centrifugation at 30,000g. Activated and buffer A–equilibrated P11-cellulose resin was added to the cleared cell lysate to allow Lon binding. The column was washed with buffer A to remove unbound proteins, and bound Lon protein was eluted in 10 ml of elution buffer B [400 mM KHPO4 (pH 7), 1 mM EDTA, 1 mM DTT, and 10% glycerol]. Lon protein was further purified on a SOURCE 15Q ion-exchange column using buffer C [50 mM tris (pH 7.5), 50 mM KCl, and 1 mM DTT]. Bound Lon was eluted using a 20 column-volume linear gradient (0 to 100%) of buffer D [50 mM tris (pH 7.5), 1 M KCl, and 1 mM DTT]. Fractions containing Lon protease were pooled, concentrated, and loaded on S300 gel filtration column in buffer E [50 mM tris (pH 7.5), 100 mM KCl, 10 mM MgCl2, 1 mM DTT, and 20% glycerol]. Aliquots of purified Lon were flash-frozen and stored at −80°C. The pET28b-lon plasmid served as a template for generating lon mutants using the quick-change site-directed mutagenesis approach. Each Lon mutant protein was expressed and purified as described above.
HspQ and Y2853 were purified using a combination of Ni-NTA affinity, ion exchange, and size-exclusion chromatography steps. BL21 star (DE3) harboring pET28b-hspQ plasmid was grown in LB containing kanamycin (50 μg/ml), and protein expression was induced with 1 mM IPTG at OD600 of 0.5. Cultures were allowed to grow for 3 hours while shaking. Harvested cells were resuspended in lysis buffer [50 mM tris (pH 8), 1 M NH4Cl, 2 mM β-mercaptoethanol (β-ME), and 10 mM imidazole]. After sonication, buffer-equilibrated Ni-NTA beads were added to cleared cell lysates. After 1-hour end-to-end rocking at 4°C, unbound proteins were removed, the beads were extensively washed, and the bound proteins were eluted using a step elution with lysis buffer containing 250 mM imidazole. HspQ protein containing fractions were combined, buffer exchanged to buffer F [50 mM tris (pH 8), 50 mM KCl, and 2 mM β-ME], loaded on a SOURCE 15Q column, and eluted using a 20 column-volume linear gradient (0 to 100%) of buffer G [50 mM tris (pH 8), 1 M KCl, and 2 mM β-ME]. Fractions containing HspQ were pooled, concentrated, and loaded on a Superdex 75 column equilibrated in buffer H [50 mM tris (pH 8), 50 mM KCl, and 2 mM β-ME]. Protein aliquots containing 10% glycerol were flash-frozen and stored at −80°C.
In vitro proteolysis assay
Each in vitro proteolysis assay reaction was carried out in Lon activity buffer [50 mM tris-HCl (pH 8), 100 mM KCl, 10 mM MgCl2, 1 mM DTT, and 10% glycerol] and ATP regeneration system [16 mM creatine phosphate, creatine kinase (0.32 mg/ml), and 4 mM ATP]. Reactions contained 100 nM Lon hexamer (Lon6) and 10 μM each. All reaction components except ATP regeneration system were mixed and incubated at 37°C. ATP regeneration system, prewarmed at 37°C, was added to initiate the reaction. Aliquots at specific time were mixed with 2X SDS sample buffer to terminate the reaction. Reaction products were resolved by electrophoresis on 15% tris-Tricine gels, scanned using a LI-COR Odyssey scanner, and quantified using the Image Studio software. Fraction of the substrate remaining was estimated, and the data were normalized to creatine kinase as a loading control. Three biological repeats were performed for each Lon mutant, the data were fit to a straight line, and the slope was extracted to calculate the rate of substrate degradation. GraphPad Prism software was used for data analysis. Mean and SEM were calculated by performing column statistics.
In vitro ATP hydrolysis assay
Coupled ATP hydrolysis assay was carried out in Lon activity buffer [50 mM tris-HCl (pH 8), 100 mM KCl, 10 mM MgCl2, 1 mM DTT, and 10% glycerol]. Reactions contained 100 nM Lon6, 1 mM reduced form of nicotinamide adenine dinucleotide (NADH), lactate dehydrogenase (10 U/ml), 20 mM phosphoenol pyruvate, pyruvate kinase (10 U/ml), and 2 mM ATP (Key Resources Table). Lon and other reaction components were warmed separately at 30°C. Reactions were initiated by adding Lon, and NADH disappearance was monitored at 340 nm. Three biological repeats were performed for each Lon mutant and the indicated substrate protein, the data were fit to a straight line, and the slope were extracted to calculate ATPase activation rates. GraphPad Prism software was used for data analysis. Mean and SEM were calculated by performing column statistics.
Sample preparation for electron microscopy
LonE424Q was diluted to a concentration of 0.95 mg/ml in 50 mM tris (pH 8), 75 mM KCl, 10 mM MgCl2, 1 mM tris(2-carboxyethyl)phosphine (TCEP), and 1 mM ATP. Four microliter of the sample was applied onto 300-mesh R1.2/1.3 UltrAuFoil Holey Gold Films (Quantifoil) that had been previously plasma-treated for 30 s using a 15-mA current operating under atmospheric gases using a glow discharger (Electron Microscopy Sciences). Excess sample was blotted away for 4 s using Whatman no. 1 filter paper and vitrified by plunge-freezing into a liquid ethane slurry cooled by liquid nitrogen using a manual plunger in a 4°C cold room whose humidity was raised to 95% using a humidifier. LonWT was diluted to a concentration of 17 mg/ml in 50 mM tris (pH 8), 75 mM KCl, 10 mM MgCl2, 1 mM TCEP, and 1 mM ATP, and samples were prepared for cryo-EM analyses using the same procedures used for LonE424Q.
EM data acquisition
For LonE424Q, cryo-EM data were collected on a Thermo Fisher Scientific Talos Arctica transmission electron microscope operating at 200 kV using parallel illumination conditions (68). Micrographs were acquired using a Gatan K2 Summit direct electron detector, operated in electron-counting mode applying a total electron exposure of 51 e−/Å2 as a 44-frame dose-fractionated movie during a 11-s exposure. The Leginon data collection software (69) was used to collect 4071 micrographs at ×36,000 nominal magnification (1.15 Å/pixel at the specimen level) with a nominal defocus range of −0.8 to −1.3 μm. Stage movement was used to target the center of four 1.2-μm holes for focusing, and an image shift was used to acquire high-magnification images in the center of each of the four targeted holes.
Likewise, for LonWT, Cryo-EM data were collected on a Thermo Fisher Scientific Talos Arctica transmission electron microscope operating at 200 keV using parallel illumination conditions (68). Micrographs were acquired using a Gatan K2 Summit direct electron detector, operated in electron counting mode applying a total electron exposure of 50 e−/Å2 as a 58-frame dose-fractionated movie during an 11.6-s exposure. The Leginon data collection software (69) was used to collect 1864 micrographs at ×36,000 nominal magnification (1.15 Å/pixel at the specimen level) with a nominal defocus range of −0.8 to −1.5 μm. Images for LonWT were collected using a similar strategy used for LonWB.
Image processing
For LonE424Q, real-time preprocessing was performed during cryo-EM data collection using the Appion processing environment (70). Micrograph frames were aligned using MotionCor2 (71), and contrast transfer function (CTF) parameters were estimated with CTFFind4 (72). Approximately 100,000 particles were selected from a subset of micrographs using a difference of Gaussian–based automated particle picker (73). Particles were extracted using a box size of 128 pixels, and the stack was binned by a factor of 2 for reference-free two-dimensional (2D) alignment using an iterative multivariate statistical analysis with multireference alignment in Appion. 2D classes representing orthogonal views of the Lon complex were used for template-based particle selection with FindEM (74). A stack of 1,176,205 particles was created using a 256 pixel box size, which was scaled down by a factor of 4 using RELION 1.4 (75) for initial processing. An ab initio model was created in cryoSPARC (76), low pass filtered to 60 Å, and used as an initial model for 3D refinement of particles in RELION 2.0 (77). These particles refined to a reported resolution of 9.5 Å as estimated by Fourier shell correlation (FSC) using a cutoff of 0.143, but the resolution was anisotropic, and the reconstruction exhibited artifacts from preferred orientation. The particles from this reconstruction were then sorted by classification without alignment into four classes, two of which, accounting for 65.9% of particles, displayed high-resolution features that did not contain anisotropic resolution artifacts. The 788,445 particles from these classes were merged and refined. The x and y shifts from this refinement were used to reextract centered, unbinned particles using a box size of 256 pixels. The reextracted particles were then refined and postprocessed to produce a reconstruction with an estimated resolution of 3.7 Å at an FSC cutoff of 0.143. 2D classification was used at this point to filter out false-particle picks and noise, resulting in 749,413 particles that were further refined to an estimated overall resolution of 3.6 Å. The cryo-EM density of the two seam subunits was poorly resolved in this reconstruction, so a soft-edged 3D mask was generated to encompass the step subunit for focused classification with three classes, resulting one class containing ~10% of the particles that contained higher resolution step subunits and an overall reported resolution of 3.4 Å by FSC at 0.143. CTF refinement was performed to estimate per-particle defocus values using RELION 3.0 (78). The x and y image shifts applied during data acquisition were used to group images for beam tilt estimation. Refining with local defocus and beam-tilt estimation improved the overall reported resolution of our reconstruction to 3.0 Å by FSC at 0.143. Focused refinement of the masked step subunits and NTDs improved the quality of the map in these regions to a reported resolution of 3.4 Å by FSC at 0.143. A final composite map of the focused and nonfocused refinements was generated for atomic model building and refinement using the “vop max” operation in University of California, San Francisco (UCSF) Chimera (79).
For LonWT, on-the-fly preprocessing—including motion correction, CTF estimation, particle picking, and sorting—was performed during cryo-EM data collection using the Warp-processing environment (80). Approximately 412,719 particles were selected from a subset of micrographs using BoxNet automated particle picker. Particles were extracted using a box size of 256 pixels, and the stack was exported to cryoSPARC v2.11.0 (76) for reference-free 2D classification. The 287,859 particles were selected from 2D classes and used to create an ab initio reconstruction. The resulting 3D reconstruction was used as an initial model for 3D heterogenous refinement with five classes. Two of the best classes representing 140,506 of the particles were used for a final 3D homogeneous refinement. The particles refined to a reported resolution of 3.8 Å as estimated by FSC using a cutoff of 0.143.
Atomic model building and refinement
A homology model was generated using the crystal structure of Lon as a starting model using SWISS-MODEL (81). This initial model was split into ATPase and protease domains and rigid body docked into the density of each of the subunits using UCSF Chimera (79). Real-space refinement of the docked structures and ab initio model building were performed in Coot (82). The seam ATPase subunits and flexible linker regions were modeled ab initio, and a seven amino acid polyalanine peptide, as well as ATP, ADP, and magnesium cofactor molecules, were built into the density corresponding to substrate and nucleotide. Further refinement of the full hexameric atomic model was performed using real-space refinement in PHENIX (83). This refined model served as a starting point to generate 100 models in Rosetta, and the top five scoring models were selected for further refinement in Phenix and Coot using the multimodel pipeline developed by Herzik et al. (84). UCSF Chimera and ChimeraX (85) were used to visualize the structure and to generate the figures. Only the top-scoring model is included in the figures and deposited to the PDB.
Individual subunits from the LonE424Q atomic model were docked into the density of each of the subunits of the LonWT map using UCSF Chimera. Real-space refinement of the docked structures and ab initio model building were performed in Coot (82). ADP molecules were built into the density corresponding to nucleotide in five of the subunits. Further refinement of the full hexameric atomic model was performed using real-space refinement in PHENIX (83). Similar to LonE424Q, this refined model of LonWT served as a starting point to generate 100 models in Rosetta, and the top five scoring models were selected for further refinement in Phenix and Coot using the multi-model pipeline developed by Herzik et al. (84). UCSF Chimera and ChimeraX (79, 85) were used to visualize the structure and to generate the figures. Only the top-scoring model is included in the figures and deposited to the PDB.
Supplementary Material
Acknowledgments
We thank J.C. Ducom at Scripps Research High Performance Computing and C. Bowman at Scripps Research for computational support, and B. Anderson at the Scripps Research Electron Microscopy Facility for microscopy support. We thank C. Sandate, A. Hernandez, M. Herzik, and M. Hirschi for assistance in atomic model building and refinement and members of the Lander and Wiseman laboratories at Scripps Research for helpful discussions. Funding: M.S. is supported by the NSF Graduate Research Fellowship Program. G.C.L. is supported by the Pew Charitable Trusts as a Pew Scholar, an Amgen Young Investigator award, and by the NIH DP2EB020402. A.W.K. is supported by NIH AI-127533. R.L.W. is supported by the NIH NS095892. G.C.L. and R.L.W. are supported by NIH AG061697. Computational analyses of EM data were performed using shared instrumentation funded by NIH S10OD021634 to G.C.L. Author Contributions: M.S. performed all cryo-EM analyses, structure determination, and model building and refinement. M.S. and C.P. performed mechanistic interpretation and wrote the manuscript. A.A., N.P., and E.A. constructed, expressed, and purified proteins, and performed biochemical experiments. All authors contributed to the experimental design and editing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The EM density maps and top atomic models obtained using a multimodel pipeline by Herzik et al. (53) for the LonENZ and LonOFF conformations have been deposited at the Electron Microscopy Data Bank and PDB under accession numbers EMDB: 20133 and 21009 and PDB: 6ON2 and 6 V11, respectively. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaba8404/DC1
REFERENCES AND NOTES
- 1.Erzberger J. P., Berger J. M., Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Hanson P. I., Whiteheart S. W., AAA+ proteins: Have engine, will work. Nat. Rev. Mol. Cell Biol. 6, 519–529 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Iyer L. M., Leipe D. D., Koonin E. V., Aravind L., Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 146, 11–31 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Sauer R. T., Baker T. A., AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 80, 587–612 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Gates S. N., Martin A., Stairway to translocation: AAA+ motor structures reveal the mechanisms of ATP-dependent substrate translocation. Protein Sci. 29, 407–419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puchades C., Sandate C. R., Lander G. C., The molecular principles governing the activity and functional diversity of AAA+ proteins. Nat. Rev. Mol. Cell Biol. 21, 43–58 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cooney I., Han H., Stewart M. G., Carson R. H., Hansen D. T., Iwasa J. H., Price J. C., Hill C. P., Shen P. S., Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science 365, 502–505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de la Peña A. H., Goodall E. A., Gates S. N., Lander G. C., Martin A., Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis–driven translocation. Science 362, eaav0725 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gates S. N., Yokom A. L., Lin J., Jackrel M. E., Rizo A. N., Kendsersky N. M., Buell C. E., Sweeny E. A., Mack K. L., Chuang E., Torrente M. P., Su M., Shorter J., Southworth D. R., Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han H., Monroe N., Sundquist W. I., Shen P. S., Hill C. P., The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. eLife 6, e31324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Monroe N., Han H., Shen P. S., Sundquist W. I., Hill C. P., Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. eLife 6, e24487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puchades C., Ding B., Song A., Wiseman R. L., Lander G. C., Glynn S. E., Unique structural features of the mitochondrial AAA+ protease AFG3L2 reveal the molecular basis for activity in health and disease. Mol. Cell 75, 1073–1085.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Puchades C., Rampello A. J., Shin M., Giuliano C. J., Wiseman R. L., Glynn S. E., Lander G. C., Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358, eaao0464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ripstein Z. A., Huang R., Augustyniak R., Kay L. E., Rubinstein J. L., Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 6, e25754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizo A. N., Lin J., Gates S. N., Tse E., Bart S. M., Castellano L. M., DiMaio F., Shorter J., Southworth D. R., Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase. Nat. Commun. 10, 2393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Twomey E. C., Ji Z., Wales T. E., Bodnar N. O., Ficarro S. B., Marto J. A., Engen J. R., Rapoport T. A., Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 365, eaax1033 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White K. I., Zhao M., Choi U. B., Pfuetzner R. A., Brunger A. T., Structural principles of SNARE complex recognition by the AAA+ protein NSF. eLife 7, e38888 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu H., Lupoli T. J., Kovach A., Meng X., Zhao G., Nathan C. F., Li H., ATP hydrolysis-coupled peptide translocation mechanism of Mycobacterium tuberculosis ClpB. Proc. Natl. Acad. Sci. U.S.A. 115, E9560–E9569 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beck F., Unverdorben P., Bohn S., Schweitzer A., Pfeifer G., Sakata E., Nickell S., Plitzko J. M., Villa E., Baumeister W., Förster F., Near-atomic resolution structural model of the yeast 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 109, 14870–14875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S., Wu J., Lu Y., Ma Y.-B., Lee B.-H., Yu Z., Ouyang Q., Finley D. J., Kirschner M. W., Mao Y., Structural basis for dynamic regulation of the human 26S proteasome. Proc. Natl. Acad. Sci. U.S.A. 113, 12991–12996 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Śledź P., Unverdorben P., Beck F., Pfeifer G., Schweitzer A., Förster F., Baumeister W., Structure of the 26S proteasome with ATP-γS bound provides insights into the mechanism of nucleotide-dependent substrate translocation. Proc. Natl. Acad. Sci. U.S.A. 110, 7264–7269 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fei X., Bell T. A., Jenni S., Stinson B. M., Baker T. A., Harrison S. C., Sauer R. T., Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. eLife 9, e52774 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez K. E., Rizo A. N., Tse E., Lin J., Scull N. W., Thwin A. C., Lucius A. L., Shorter J., Southworth D. R., Conformational plasticity of the ClpAP AAA+ protease couples protein unfolding and proteolysis. bioRxiv, 820209 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ripstein Z. A., Vahidi S., Houry W. A., Rubinstein J. L., Kay L. E., A processive rotary mechanism couples substrate unfolding and proteolysis in the ClpXP degradation machinery. eLife 9, e52158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aubin-Tam M.-E., Olivares A. O., Sauer R. T., Baker T. A., Lang M. J., Single-molecule protein unfolding and translocation by an ATP-fueled proteolytic machine. Cell 145, 257–267 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cordova J. C., Olivares A. O., Shin Y., Stinson B. M., Calmat S., Schmitz K. R., Aubin-Tam M.-E., Baker T. A., Lang M. J., Sauer R. T., Stochastic but highly coordinated protein unfolding and translocation by the ClpXP proteolytic machine. Cell 158, 647–658 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maillard R. A., Chistol G., Sen M., Righini M., Tan J., Kaiser C. M., Hodges C., Martin A., Bustamante C., ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145, 459–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin A., Baker T. A., Sauer R. T., Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Aliaga P., Ramirez L., Kim F., Bustamante C., Martin A., Substrate-translocating loops regulate mechanochemical coupling and power production in AAA+ protease ClpXP. Nat. Struct. Mol. Biol. 23, 974–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen M., Maillard R. A., Nyquist K., Rodriguez-Aliaga P., Pressé S., Martin A., Bustamante C., The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell 155, 636–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker M. J., Tatsuta T., Langer T., Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 3, a007559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee I., Suzuki C. K., Functional mechanics of the ATP-dependent Lon protease- lessons from endogenous protein and synthetic peptide substrates. Biochim. Biophys. Acta 1784, 727–735 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinti M., Gibellini L., Nasi M., De Biasi S., Bortolotti C. A., Iannone A., Cossarizza A., Emerging role of Lon protease as a master regulator of mitochondrial functions. Biochim. Biophys. Acta 1857, 1300–1306 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Quiros P. M., Langer T., Lopez-Otin C., New roles for mitochondrial proteases in health, ageing and disease. Nat. Rev. Mol. Cell Biol. 16, 345–359 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Lee I., Berdis A. J., Suzuki C. K., Recent developments in the mechanistic enzymology of the ATP-dependent Lon protease from Escherichia coli: Highlights from kinetic studies. Mol. Biosyst. 2, 477–483 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Culp E., Wright G. D., Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 70, 366–377 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Botos I., Melnikov E. E., Cherry S., Tropea J. E., Khalatova A. G., Rasulova F., Dauter Z., Maurizi M. R., Rotanova T. V., Wlodawer A., Gustchina A., The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J. Biol. Chem. 279, 8140–8148 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Gur E., Sauer R. T., Degrons in protein substrates program the speed and operating efficiency of the AAA+ Lon proteolytic machine. Proc. Natl. Acad. Sci. U.S.A. 106, 18503–18508 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park S. C., Jia B., Yang J. K., Van D. L., Shao Y. G., Han S. W., Jeon Y. J., Chung C. H., Cheong G. W., Oligomeric structure of the ATP-dependent protease La (Lon) of Escherichia coli. Mol. Cells 21, 129–134 (2006). [PubMed] [Google Scholar]
- 40.Vieux E. F., Wohlever M. L., Chen J. Z., Sauer R. T., Baker T. A., Distinct quaternary structures of the AAA+ Lon protease control substrate degradation. Proc. Natl. Acad. Sci. U.S.A. 110, E2002–E2008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin C. C., Su S. C., Su M. Y., Liang P. H., Feng C. C., Wu S. H., Chang C. I., Structural insights into the allosteric operation of the Lon AAA+ protease. Structure 24, 667–675 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Duman R. E., Löwe J., Crystal structures of Bacillus subtilis Lon protease. J. Mol. Biol. 401, 653–670 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Botos I., Loutos G. T., Wu W., Cherry S., Ghirlando R., Kudhaev A. M., Tropea J. E., Gustchina A., Wlodawer A., Cryo-EM structure of substrate-free E. coli Lon protease provides insights into the dynamics of Lon machinery. Curr. Res. Struct. Biol. 1, 13–20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ho C. M., Beck J. R., Lai M., Cui Y., Goldberg D. E., Egea P. F., Zhou Z. H., Malaria parasite translocon structure and mechanism of effector export. Nature 561, 70–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sandate C. R., Szyk A., Zehr E. A., Lander G. C., Roll-Mecak A., An allosteric network in spastin couples multiple activities required for microtubule severing. Nat. Struct. Mol. Biol. 26, 671–678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puri N., Karzai A. W., HspQ functions as a unique specificity-enhancing factor for the AAA+ Lon protease. Mol. Cell 66, 672–683.e74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim Y. C., Snoberger A., Schupp J., Smith D. M., ATP binding to neighbouring subunits and intersubunit allosteric coupling underlie proteasomal ATPase function. Nat. Commun. 6, 8520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith D. M., Fraga H., Reis C., Kafri G., Goldberg A. L., ATP binds to proteasomal ATPases in pairs with distinct functional effects, implying an ordered reaction cycle. Cell 144, 526–538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gur E., Sauer R. T., Recognition of misfolded proteins by Lon, a AAA(+) protease. Genes Dev. 22, 2267–2277 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Besche H., Tamura N., Tamura T., Zwickl P., Mutational analysis of conserved AAA+ residues in the archaeal Lon protease from Thermoplasma acidophilum. FEBS Lett. 574, 161–166 (2004). [DOI] [PubMed] [Google Scholar]
- 51.Menon A. S., Goldberg A. L., Binding of nucleotides to the ATP-dependent protease La from Escherichia coli. J. Biol. Chem. 262, 14921–14928 (1987). [PubMed] [Google Scholar]
- 52.Wohlever M. L., Baker T. A., Sauer R. T., Roles of the N domain of the AAA+ Lon protease in substrate recognition, allosteric regulation and chaperone activity. Mol. Microbiol. 91, 66–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li M., Gustchina A., Rasulova F. S., Melnikov E. E., Maurizi M. R., Rotanova T. V., Dauter Z., Wlodawer A., Structure of the N-terminal fragment of Escherichia coli Lon protease. Acta Crystallogr. D Biol. Crystallogr. 66, 865–873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bieniossek C., Niederhauser B., Baumann U. M., The crystal structure of apo-FtsH reveals domain movements necessary for substrate unfolding and translocation. Proc. Natl. Acad. Sci. U.S.A. 106, 21579–21584 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rotanova T. V., Botos I., Melnikov E. E., Rasulova F., Gustchina A., Maurizi M. R., Wlodawer A., Slicing a protease: Structural features of the ATP-dependent Lon proteases gleaned from investigations of isolated domains. Protein Sci. 15, 1815–1828 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patterson-Ward J., Tedesco J., Hudak J., Fishovitz J., Becker J., Frase H., McNamara K., Lee I., Utilization of synthetic peptides to evaluate the importance of substrate interaction at the proteolytic site of Escherichia coli Lon protease. Biochim. Biophys. Acta 1794, 1355–1363 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Su S. C., Lin C. C., Tai H. C., Chang M. Y., Ho M. R., Babu C. S., Liao J. H., Wu S. H., Chang Y. C., Lim C., Chang C. I., Structural basis for the magnesium-dependent activation and hexamerization of the Lon AAA+ protease. Structure 24, 676–686 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Gatsogiannis C., Balogh D., Merino F., Sieber S. A., Raunser S., Cryo-EM structure of the ClpXP protein degradation machinery. Nat. Struct. Mol. Biol. 26, 946–954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang C. W., Lee S., Tsai F. T. F., Structural elements regulating AAA+ protein quality control machines. Front. Mol. Biosci. 4, 27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yokom A. L., Gates S. N., Jackrel M. E., Mack K. L., Su M., Shorter J., Southworth D. R., Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat. Struct. Mol. Biol. 23, 830–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Heuck A., Schitter-Sollner S., Suskiewicz M. J., Kurzbauer R., Kley J., Schleiffer A., Rombaut P., Herzog F., Clausen T., Structural basis for the disaggregase activity and regulation of Hsp104. eLife 5, e21516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patterson-Ward J., Huang J., Lee I., Detection and characterization of two ATP-dependent conformational changes in proteolytically inactive Escherichia coli Lon mutants by stopped flow kinetic techniques. Biochemistry 46, 13593–13605 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vineyard D., Patterson-Ward J., Berdis A. J., Lee I., Monitoring the timing of ATP hydrolysis with activation of peptide cleavage in Escherichia coli Lon by transient kinetics. Biochemistry 44, 1671–1682 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Kenniston J. A., Baker T. A., Sauer R. T., Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc. Natl. Acad. Sci. U.S.A. 102, 1390–1395 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Enemark E. J., Joshua-Tor L., Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 (2006). [DOI] [PubMed] [Google Scholar]
- 66.Dong Y., Zhang S., Wu Z., Li X., Wang W. L., Zhu Y., Stoilova-McPhie S., Lu Y., Finley D., Mao Y., Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matyskiela M. E., Lander G. C., Martin A., Conformational switching of the 26S proteasome enables substrate degradation. Nat. Struct. Mol. Biol. 20, 781–788 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Herzik M. A. Jr., Wu M., Lander G. C., Achieving better-than-3-Å resolution by single-particle cryo-EM at 200 keV. Nat. Methods 14, 1075–1078 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suloway C., Pulokas J., Fellmann D., Cheng A., Guerra F., Quispe J., Stagg S., Potter C. S., Carragher B., Automated molecular microscopy: The new Leginon system. J. Struct. Biol. 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 70.Lander G. C., Stagg S. M., Voss N. R., Cheng A., Fellmann D., Pulokas J., Yoshioka C., Irving C., Mulder A., Lau P. W., Lyumkis D., Potter C. S., Carragher B., Appion: An integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Voss N. R., Yoshioka C. K., Radermacher M., Potter C. S., Carragher B., DoG Picker and TiltPicker: Software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Roseman A. M., FindEM—A fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 145, 91–99 (2004). [DOI] [PubMed] [Google Scholar]
- 75.Scheres S. H., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Kimanius D., Forsberg B. O., Scheres S. H., Lindahl E., Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. eLife 5, e18722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J., Lindahl E., Scheres S. H., New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 80.Tegunov D., Cramer P., Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 16, 1146–1152 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., Schwede T., SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herzik M. A. Jr., Fraser J. S., Lander G. C., A multi-model approach to assessing local and global cryo-EM map quality. Structure 27, 344–358.e43 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cardone G., Heymann J. B., Steven A. C., One number does not fit all: Mapping local variations in resolution in cryo-EM reconstructions. J. Struct. Biol. 184, 226–236 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prisant M. G., Williams C. J., Chen V. B., Richardson J. S., Richardson D. C., New tools in MolProbity validation: CaBLAM for CryoEM backbone, UnDowser to rethink “Waters”, and NGL viewer to recapture online 3D graphics. Protein Sci. 29, 315–329 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barad B. A., Echols N., Wang R. Y., Cheng Y., DiMaio F., Adams P. D., Fraser J. S., EMRinger: Side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaba8404/DC1