

Single-cell dual RNA sequencing reveals giant virus infection dynamics in a marine bloom-forming alga.
Abstract
The discovery of giant viruses infecting eukaryotes from diverse ecosystems has revolutionized our understanding of the evolution of viruses and their impact on protist biology, yet knowledge on their replication strategies and transcriptome regulation remains limited. Here, we profile single-cell transcriptomes of the globally distributed microalga Emiliania huxleyi and its specific giant virus during infection. We detected profound heterogeneity in viral transcript levels among individual cells. Clustering single cells based on viral expression profiles enabled reconstruction of the viral transcriptional trajectory. Reordering cells along this path unfolded highly resolved viral genetic programs composed of genes with distinct promoter elements that orchestrate sequential expression. Exploring host transcriptome dynamics across the viral infection states revealed rapid and selective shutdown of protein-encoding nuclear transcripts, while the plastid and mitochondrial transcriptomes persisted into later stages. Single-cell RNA-seq opens a new avenue to unravel the life cycle of giant viruses and their unique hijacking strategies.
INTRODUCTION
Nucleocytoplasmic large DNA viruses (NCLDVs) are the largest viruses known today in both genome and virion size. They infect most major lineages of eukaryotes across diverse habitats (1–4), especially in the marine environment (5, 6). The discovery of these giant viruses revolutionized our understanding of the evolution of viruses and their arms race with protist hosts, yet our knowledge of their infection strategies and transcriptome regulation remains limited. Among the NCLDVs of special ecological importance are members of the family Phycodnaviridae, which infect a wide range of key algal hosts (1). The calcifying eukaryotic alga Emiliania huxleyi forms massive annual blooms in the oceans that have a profound impact on the carbon and sulfur biogeochemical cycles (7). E. huxleyi blooms are frequently terminated by a large double-stranded DNA (dsDNA) virus—coccolithovirus (E. huxleyi virus, EhV) (8), which regulates nutrient cycling and carbon export to the deep ocean (9, 10). This host-virus model provides a trackable system for understanding viral life cycle strategies and host responses.
High-throughput bulk RNA sequencing (RNA-seq) has been used for whole-genome expression profiling of NCLDVs during infection, shedding light on gene prediction, transcript structure, and changes in metabolic pathways (11–13). However, bulk RNA-seq profiles average gene expression levels across many cells, whereas infection states can be variable among single cells. To overcome this limitation, single-cell RNA-seq (scRNA-seq) approaches have been developed to probe the transcriptomes of individual cells in a highly parallel manner. These methods have substantially advanced our understanding of various developmental and immunological processes (14, 15), including host-virus interactions in mammalian systems (16, 17). Here, we use scRNA-seq to investigate the transcriptomic profiles of viral and host genomes in EhV infection at the single-cell level. Specifically, we aim to have a comprehensive understanding of the temporal dynamics and regulation of viral genome expression and the associated changes in the entire cellular transcriptome.
RESULTS AND DISCUSSION
Cell-to-cell variability in overall viral expression
To characterize the temporal dynamics and regulation of viral and host transcriptomes, we infected E. huxleyi CCMP2090 cultures with the lytic virus EhV201 (Fig. 1A, table S1, and fig. S1) (13, 18). Individual cells were isolated into 384-well plates by fluorescence-activated cell sorting (FACS) and processed using the MARS-seq protocol (19), which uses cellular barcodes to tag RNA molecules from individual cells and enables transcript quantification by using unique molecular identifiers (UMIs). By mapping reads to the reference E. huxleyi transcriptome and to the EhV201 genome, we were able to profile both viral and host transcripts in each individual cell. The mean of total UMI counts of host or viral transcripts in a single cell is 187.7, while the median is 148.5. These numbers reflect the relatively low RNA content of E. huxleyi cells, which are only 3 to 5 μm in diameter for naked cells (20).
Fig. 1. Transcriptome profiling of EhV infection in E. huxleyi at single-cell resolution.
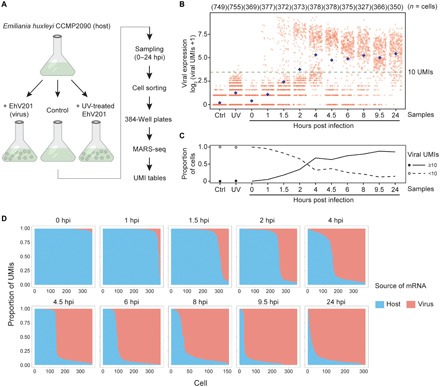
(A) The experimental setup for using scRNA-seq to study EhV infection of E. huxleyi. Individual cells were isolated using FACS during a time course of 0 to 24 hours post infection (hpi). The single-cell transcriptomes were sequenced using the MARS-seq protocol, which allows absolute quantification of viral and host transcripts by tagging each individual cell and each transcript with cellular barcodes and UMIs, respectively. (B) Highly heterogeneous total viral UMI counts among single cells across samples. Each red dot represents an individual cell, and each blue diamond represents the mean of all cells in each column. A threshold of 10 UMIs (fig. S3) highlights the bimodality of the overall viral expression and excludes cells with low- or noise-level viral UMIs, as in mock infection by UV-deactivated EhV (UV; 4 and 6 hpi) and control (Ctrl) samples. (C) The proportion of cells with at least 10 viral UMIs (active viral expression) increased rapidly during early hours of infection and approached 90% at 9.5 hpi. (D) Cliff diagrams represent the relative mRNA levels of host and virus across the infection time course. The cells (columns) are sorted by the relative proportions of host and viral mRNA, showing the sharp decrease in host mRNA relative to viral mRNA (“cliff”) and the within-population heterogeneity through time.
We compared the total expression levels of all viral genes in 5179 cells from infected cultures, control cultures, and cultures mock infected by ultraviolet (UV)–inactivated viruses (Fig. 1B). While the average viral gene expression increased over time of infection, high variability in viral expression levels between single cells was observed despite a high virion-to-cell ratio (multiplicity of infection 5:1) that was used to reduce the possible variation due to encounter rates. Throughout the first 10 hours of infection, two coexisting groups of cells were observed—one with clear detection of active viral expression (≥10 viral UMIs) and the other with viral transcript abundance at the level of noise (cf. control or mock infection). The observed bimodal distribution of viral transcripts implies the existence of an all-or-none switch that rapidly turns on viral expression (Fig. 1B). The proportion of cells with at least 10 viral UMIs increases from 0% to nearly 90% within 10 hours (Fig. 1C). The mRNA pool in each individual cell was mostly dominated by either host or viral transcriptome, with very few cells in intermediate states, forming a sharp decline in host-virus transcript ratio (Fig. 1D and fig. S2), indicating that EhV massively transforms the cellular transcriptome by taking over almost the entire mRNA pool (fig. S3).
Mapping the continuum of viral infection states
To further explore the variation among infected single cells, we applied the MetaCell method using the k-nearest neighbor graph partitions (21) for organizing single cells into cohesive groups (metacells) of cells with similar viral gene expression profiles. Rather than grouping cells into discrete clusters, the two-dimensional (2D) projection of the k-nearest neighbor graphs revealed a continuum of infected cells, where the seven metacells defined by the algorithm correspond to different phases of infection (Fig. 2A). The infected cells were spread along an infection continuum, reaching maximum heterogeneity at 6 hours post infection (hpi) (Fig. 2B). In contrast, cells pretreated with cycloheximide (1 μg/ml), an inhibitor of eukaryotic translation (22), were mainly localized in the initial phase (metacell 1) of infection at 6 hpi (Fig. 2B). This suggests that newly synthesized proteins are required for later viral transcription programs, but not for the initial phase. The continuum of cells allows us to map infection progression on a pseudotemporal scale of 0 to 1 that was calculated as the relative distance to the origin (earliest state) of the continuum (Fig. 2A), providing a new dimension to determine infection states of individual cells within heterogeneous populations.
Fig. 2. The continuum of infection states across individual cells.
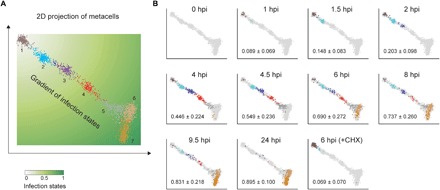
(A) A 2D projection of 2072 single infected cells (dots) with active viral expression (≥10 viral UMIs) that constitute seven metacells (in different colors). The cells form a continuum of infection states that can be quantified by the relative distance to the initial infection state. (B) Distribution of cells from each sampling time point along the infection continuum, with light gray dots representing cells from the other time points. Numbers are means ± SD of the infection states as defined in (A). CHX, cycloheximide treatment before infection. The infection index, a proxy for infection states, (A) of an individual cell was calculated as the ratio of the distance of this cell from the upper-left origin to the distance between the origin and the lower-right end. A color gradient was painted on the basis of the scale of the infection indices between 0 and 1.
Highly resolved viral gene expression programs
By reordering cells on the basis of the newly defined infection states, we found that viral genes were largely organized in five discrete kinetic classes of genes herein termed as immediate-early (73 genes), early (136), early-late (49), late 1 (22), and late 2 (40), which formed partially overlapping sequential waves of expression (Fig. 3 and table S2). Among the 447 predicted coding sequences (CDSs) in EhV201, 320 can be unambiguously assigned to one of the five distinct kinetic classes. Of the other 127 genes, 36 were barely detectable (<20 UMIs in total across all cells in Fig. 3; table S2) and are not shown in Fig. 3, while the others generally have low (<100 UMIs in total) or sparse expression levels with class assignment undetermined (16), early-undetermined (17; clustered with immediate-early, early, and early-late), or late-undetermined (58; clustered with late 1 and late 2) (Fig. 3 and table S2). Most genes within each of the five discrete classes show highly similar expression profiles, but outliers do exist. For example, three immediate-early genes (AET98025.1, AET98171.1, and AET98238.1) were semiconstitutively expressed even at later infection states.
Fig. 3. Resolving viral genes into kinetic classes across infection states of single cells.
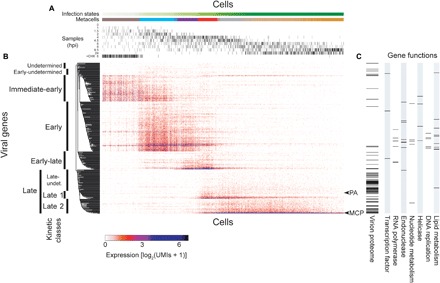
(A) Cells (columns) are ordered by the pseudotemporal scale (infection indices) as defined in Fig. 2, with hpi of each culture sample marked for each cell. (B) Viral genes (rows) are divided into kinetic classes based on the hierarchical clustering of their expression profiles across the individual cells, which are ordered by their infection indices the same way as in (A). (C) Presence in the EhV201 virion proteome, as well as categories of predicted functions, is indicated for each gene ordered the same way as in (B). See table S2 for protein identifiers (IDs) and detailed annotations of the viral genes. PA, packing ATPase; MCP, major capsid protein; undet., undetermined.
We also observe the correlation between the expression dynamics and known functions of genes. The early kinetic class encompasses most of the genes involved in information processing and metabolism, including DNA replication (DNA polymerase delta subunit, proliferating cell nuclear antigen, and DNA topoisomerase II), nucleotide metabolism [ribonucleoside-diphosphate reductase, dUTP (deoxyudirine triphosphate) pyrophosphatase, and thymidylate synthase], and lipid metabolism (Fig. 3C). In addition to the unique virus-encoded sphingolipid biosynthesis enzymes dihydroceramide synthase, dihydroceramide desaturase, sphingosine 1-phosphate phosphatase, and serine palmitoyltransferase (13, 23–25), fatty acid elongase and sterol desaturase are also in the early class (Fig. 3C). The early genes further encode four RNA polymerase II subunits (RPB1, RPB2, RPB3, and RPB6), which probably enable transcription independent of the host machinery. Two small RNA polymerase II subunits (RPB5 and RPB10) are encoded in the early-late class. Together with three transcription factor genes in the early-undetermined, early, and early-late, the RNA polymerase genes may be key regulators of the kinetic classes. Most of the genes assigned to the late 1 and late 2 kinetic classes have unknown functions, but two genes are noteworthy: the packaging adenosine triphosphatase (ATPase) for packing viral DNA into virions (late 1 class) and the major capsid protein, which is the main component of the virion capsid (late 2 class).
To further characterize the viral protein-encoding genes, we purified virions from lysed cultures and analyzed their protein composition using liquid chromatography–mass spectrometry (LC-MS). In total, 76 virus-encoded genes had at least 10 peptide-spectrum matches (table S3) and were shown as present in virion proteome in Fig. 3C. Comparisons with the kinetic class assignments suggest that most components of EhV virions are encoded by the late classes (late 1, late 2, and late-undetermined). Although many early genes have high mRNA levels, only 3 (with unknown functions) out of 136 early genes were detected in the virion proteome. The packaging ATPase encoded by a late 1 gene was also absent in the proteomic data (table S3). These observations clearly suggest that EhV uses transcriptional kinetic classes to temporally regulate production of virion components in a highly selective manner.
To elucidate the regulatory mechanisms underlying the viral gene kinetic classes, we looked for enriched sequence motifs de novo in the promoter regions [±100 base pair (bp) from the first base of the start codon] of genes belonging to different kinetic classes (Fig. 4A and table S4). The most enriched motif in the promoter region of immediate-early genes ends mostly at the ATG start codon (positions 12 to 14) and was previously identified as a putative promoter element associated with initial expression in another EhV strain (26). We also revealed short, highly enriched elements, including a 6-bp motif around the ATG of early genes and a 7-bp motif mostly upstream of the start codon in the early-late genes. An AT-rich 20-bp motif was detected, with a degenerated center, usually immediately upstream of the start codon of late 1, late 2, and other late genes. We further found that these putative promoter elements are found to be conserved across EhV genomes (fig. S4), which share most of their gene contents (fig. S5 and table S5). The late motif bears notable similarity to the late promoter element of Mimivirus, a distantly related NCLDV that infects amoebae, which is composed of two AT-rich decamers separated by a degenerated tetramer and is also present in the genome of the Mimivirus virophage (11). Although genes in most kinetic classes are scattered throughout the EhV201 genome, the immediate-early genes are mainly concentrated in two genomic regions (Fig. 4B), as also seen in the strain EhV86 (26). Such organization could facilitate the rapid activation of the immediate-early class once the host cell is infected, allowing transcription initiation of the entire cascade of the various kinetic classes. Together, our findings of newly defined viral kinetic classes are strongly supported by the discovery of potential regulatory mechanisms that are conserved across EhV genomes.
Fig. 4. Predicted promoter elements and genomic organizations of viral kinetic classes.
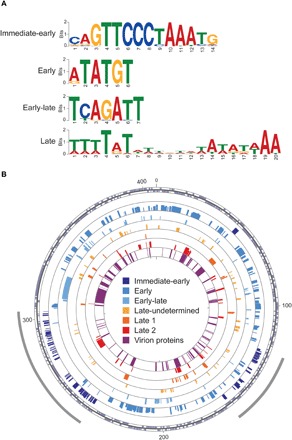
(A) Enriched motifs in the promoter regions of viral genes in the newly defined kinetic classes. (B) Distribution of genes in each kinetic class (circle) on the assembled genome of EhV201. The outermost circle indicates the direction of transcription. The heights of genes in each kinetic class are proportional to their log2-transformed expression levels. The innermost circle marks the presence in the proteome of EhV201 virions. Immediate-early genes are mostly clustered in two genomic regions (gray arcs). Numbers indicate sequence lengths in kilobase.
Host transcriptional profiles along infection progression
Last, our single-cell dual RNA-seq approach allows quantification of not only transcripts encoded by the viral genome but also respective host transcriptomes including those of the mitochondria and chloroplasts (Fig. 5). As depicted in Fig. 1D, there was a sharp shift of the cellular transcriptome from host-encoded to virus-encoded transcripts. Reduction in host transcripts has also been observed at the single-cell level for the non-NCLDV dsDNA Herpesvirus (27), but EhV causes more extensive host shutdown at a faster pace. By mapping the expression levels across infection states, we found differential shutdown of nucleus-encoded and organelle-encoded genes (Fig. 5A). Compared with the control (0.12 UMIs per transcript per cell), mock-infected (0.12), and infected culture with fewer than 10 viral UMIs (0.10), most nuclear transcripts were rapidly shut down at the onset of active viral expression, leaving only 0.02 UMI per transcript per cell in the early phase of infection. On the contrary, levels of the mitochondrial and chloroplast transcripts at the same stage of infection were at similar or slightly higher levels compared with the other cultures. Moreover, the two types of organelles show distinct expression patterns, with mitochondrial transcripts persisting at lower yet relatively stable levels throughout the infection progression and chloroplast transcript levels that were high in the beginning but then mostly soon declined (Fig. 5, B and C).
Fig. 5. Virus-induced host mRNA shutdown as a function of infection states.
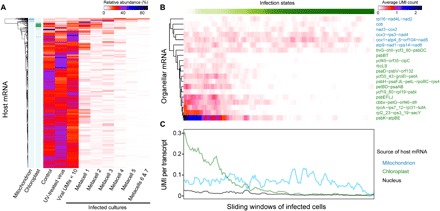
(A) Levels of host transcripts across treatments and infection states. Single E. huxleyi cells from cultures infected with EhV are divided on the basis of overall viral UMI counts and metacell grouping (Fig. 2A). Protein-encoding genes with at least 100 UMIs were included. For each gene, the average UMI count was calculated for each category, and the sum of the average values of all categories was normalized to 100. Transcripts were then hierarchically clustered on the basis of the normalized values. Organelle-encoded transcripts are indicated on the left. (B and C) Abundance of organellar transcripts along the infection progression. Cells with at least 10 viral UMIs are ordered according to the infection states as determined by the MetaCell analysis (Figs. 2 and 3). Sliding window averages were calculated for each transcript (B) or mean of all transcripts (C) with a window size of 50 cells and steps of 10. Polycistronic transcripts in mitochondria (blue) and chloroplasts (green) (B) are named with their first and last genes, as well as some genes in between, according to the organellar genome annotations (40, 41).
Despite the rapid shutdown of most nuclear transcripts, 106 transcripts still had a relative abundance of more than 10% for the bulk average of cells assigned to metacell 1, the early phase of infection progression (Fig. 5A). Analyses of the Gene Ontology (GO) (28) terms assigned to these transcripts (table S6) suggest that they are relatively enriched (P < 0.05) in GO terms that include mitochondrial protein complex, mitochondrial membrane part, monosaccharide metabolic process, and nucleotide metabolic process (table S7). Notable annotations from the Kyoto Encyclopedia of Genes and Genomes (KEGG) (29) include NADH (reduced form of nicotinamide adenine dinucleotide) dehydrogenase subunits NDUFV2 (KEGG number K03943) and NDUFA13 (K11353), as well as succinate dehydrogenase iron-sulfur subunit SDHB (K00240), which are nucleus-encoded components of mitochondrial protein complexes involved in the tricarboxylic acid (TCA) cycle and oxidative phosphorylation. This suggests that the expression of both the nuclear (Fig. 5A) and mitochondrial genes [e.g., nad1-6 encoding NADH dehydrogenase subunits (Fig. 5B)] was maintained at a high level to sustain these key mitochondrial metabolic processes. Other putative gene products are TOM40 (K11518), part of the mitochondrial outer membrane complex that imports proteins, phosphoglycerate kinase (PGK; K00927), a major glycolytic enzyme, and phosphoglucomutase (PGM; K01835), which channels glucose to glycolysis or the pentose phosphate pathway. While most nuclear genes are shut down, the high expression of these genes likely prepares the cell to produce enough energy for viral protein synthesis, DNA replication, lipid biosynthesis, virion assembly, and other processes. The high metabolic demand of EhV and other giant viruses with large burst size points to high dependence of viruses on their host metabolic state for optimal replication (13, 30). Previous single-cell analyses revealed down-regulation of most of the examined E. huxleyi metabolic genes in the stationary phase, concomitant with low viral gene expression, as compared with infected exponential phase cells (31).
The delayed shutdown of chloroplast-encoded genes suggests the need for active chloroplasts in early stages of infection, which is in agreement with the recent finding that viral infection depends on light in both the coccolithophore E. huxleyi (32) and the pelagophyte alga Aureococcus anophagefferens (33). Consistent with protein immunoblot analysis showing significant decrease in the level of RbcL [RuBisCO (ribulose-1,5-bisphosphate carboxylase-oxygenase) large subunit] compared with PsbD (photosystem II protein D2) at 24 hpi (32), the transcript encoding PsbD persisted until the late phase of infection while most transcripts, including the one for RbcL, were shut down earlier (Fig. 5B). This transcriptional difference among chloroplast-encoded genes could be a possible mechanism to divert photosynthetic energy to nucleotide synthesis via the pentose phosphate pathway (32), which coincides with the high level of phosphoglucomutase despite the systemic shutdown of nuclear transcripts. Although it is unclear how genes are regulated differently between the chloroplast and mitochondrial genome, the organellar membranes might play a role in preventing transcript shutdown at least early during infection. Unlike mRNA transcripts that were either quickly shut down or maintained, ribosomal RNA (rRNA) transcripts, especially that comprising cytosolic ribosomes, increased in quantities until the middle stage of infection (fig. S6), which could suggest a corresponding boost in translation.
scRNA-seq has proven to be a very powerful tool for comprehensive understanding of complex host-virus dynamics, disentangling cells from different sampling times and infection states. Using this approach, we dissected the transcriptomic dynamics and regulation of viral infection in a globally important microalga and showed rapid selective shutdown of host genes induced by an NCLDV. The comprehensive high-resolution mapping of infection states and kinetic classes allowed identification of conserved regulatory sequences, which likely orchestrate the sequential expression programs during the viral life cycle. Functional analyses of nuclear, chloroplast, and mitochondrial transcripts along the infection progression further shed light on the potential strategies used to optimize viral replication. The applicability of scRNA-seq provides new resolution to track host-virus dynamics and holds great potential for quantification of active viral infection in natural populations and identification of additional ecologically important host-virus systems. This, in turn, will facilitate the assessment of the impact of viruses on the function and composition of microbial food webs in the marine environment.
MATERIALS AND METHODS
Algal culture and viral stock
The coccolithophore E. huxleyi strain CCMP2090 was grown in K/2 (34) filtered seawater medium at 18°C with an irradiance of 100 μmol photons m−2 s−1 and a 16:8–hour light-dark cycle. The viral strain EhV201 (35) was propagated on CCMP2090 culture until the culture lysed to clearance, and viral lysate was filtered using a 0.45-μm PVDF (polyvinylidene difluoride) Stericup-HV vacuum filtration system (Merck). We used the plaque assay as previously described (36) to quantify infectious virions, and filtered viral lysate generally contained ~108 infectious virions/ml. Cultures at the exponential phase (~106 cells/ml−1) were used for viral infection at a high multiplicity of infection (five infectious virions to one cell) at 2 hours after the light cycle began. Algal cells and virion particles were enumerated using an Eclipse flow cytometer analyzer (iCyt) during an infection time course (fig. S1). UV-inactivated virus was obtained by exposing viral lysate in a plastic petri dish to UV in a transilluminator system for 15 min. For inhibition of protein synthesis, cycloheximide was added to culture for a final concentration of 1 μg/ml immediately before viral infection.
Single-cell isolation by FACS
Single cells were isolated into individual wells in 384-well plates on a FACSAria II or III sorter (BD Biosciences) using a 488-nm laser for excitation and a nozzle size of 100 μm. Cells were identified on the basis of chlorophyll red autofluorescence (663 to 737 nm) and separated under the “Single Cell” mode for maximal purity.
Massively parallel scRNA-seq
The scRNA-seq method is based on the MARS-seq2.0 protocol (19). Before sorting, each 384-well plate was prepared using a Bravo liquid handling platform (Agilent) to transfer into each well a 2-μl lysis buffer containing 2 or 4 nM reverse transcription (RT) primers with cellular and molecular (UMI) barcodes. Plates with sorted cells were immediately frozen on dry ice and later stored at −80°C. After thawing, the plates were heated to 95°C for 3 min to evaporate the lysis buffer, cooled, added RT reagent mix, and placed in a Labcycler (SensoQuest) for RT reaction. The unused primers were then degraded with Exonuclease I (NEB). cDNA from each set of wells with 384 (one pool per plate) or 192 (two pools per plate) unique cellular barcodes were then combined into pools, which underwent second-strand synthesis and in vitro transcription to amplify the sequences linearly. The RNA products were fragmented and ligated with single-stranded DNA adapters containing pool-specific barcodes. A second round of RT was carried out, followed by polymerase chain reaction with primers containing Illumina adapters to construct DNA libraries for paired-end sequencing on a NextSeq or MiniSeq machine (Illumina).
Read processing and mapping to reference sequences
The FASTQ files were processed using the analytical pipeline of MARS-seq2.0 (19), which mapped the reads to viral and host reference sequences and demultiplexed them based on the pool, cellular, and molecular barcodes. At least four reads per UMI were obtained per cell. For the virus, the predicted CDSs in the EhV201 genome sequence (JF974311) (37) were used as reference. For the host, an integrated transcriptome reference of E. huxleyi (38) was used, where both forward and reverse (labeled with “_rev”) strands of each sequence were included for mapping because the CDS directionality was often unknown. In addition to the nuclear sequences, the transcriptome reference contains chloroplast and mitochondrial transcripts, which were identified by the basic local alignment search tool (BLAST) (39) searches against the respective organellar genome sequences (40, 41).
Viral and host transcript abundance
To avoid empty wells or those with low transcript capture during library preparation, wells with fewer than 20 total UMIs (including all viral and host transcripts) were removed for most downstream analyses. Wells with more than 1000 transcripts were also removed to prevent wells with doublet or multiplet cells from confounding single-cell infection state analyses. The vast majority (>99%) of control or infected E. huxleyi cells had fewer than 1000 total UMIs (mean and median UMI counts, 187.7 and 148.5), which reflects their low RNA content due to the small cell size [3 to 5 μm in diameter for naked cells (20)].
Cliff diagrams of viral and host mRNA
A cliff diagram was plotted for each time point after infection (Fig. 1D). The total viral mRNA transcript counts were calculated by summing up all UMIs assigned to virus-encoded CDSs. The total host mRNA counts were all host UMIs except for those assigned to the three transcript sequences corresponding to nuclear, mitochondrial, and chloroplast rRNA genes. For each sampling time point, the cells were ordered by the ratio of host to all (host + virus) mRNA from high to low. Cells with fewer than 20 total UMIs were excluded from the plots to avoid sampling biases.
Doublet assay and in silico simulations
The EhV201 viral genome has a much lower GC content (40.46%) (37) than the E. huxleyi host genome (65%) (42). This difference in GC content and other factors might bias the capture and amplification of viral and host transcripts during MARS-seq and might lead to the either-or dominance of host and viral mRNA (Fig. 1D). To test if the presence of viral mRNA has effects on the quantification of host mRNA transcripts and vice versa, we conducted a doublet assay. Half of the doublet assay plate consisted of single cells sorted from infected culture at 8 hpi, and the other half plate consisted of two cells per well of which one cell was sorted from infected culture at 8 hpi and the other cell from control culture (table S1). Cliff diagrams were then plotted for each half of the plate (fig. S2, A and B). If the viral mRNA does not interfere with host mRNA quantification, wells with cells isolated from both the infected and the control cultures should show host and viral mRNA UMI counts that are additive, as if the two cellular transcriptomes were sequenced separately and merged together. To simulate this in silico, we randomly sampled sequenced single-cell transcriptomes from the control culture plate with the closest time point to 8 hpi (table S1) using the sample function in R, and they were combined one-by-one with those from the 8-hpi half plate with single cells. Cliff diagrams were plotted for each of the eight simulations (fig. S2, C to J).
MetaCell analysis of infected cells
The MetaCell package is an unbiased approach for cell grouping based on sparse single-cell expression profiles without enforcing any global structure (21). To account for the small number of UMIs per cell and to use more viral genes for grouping infected cells, we used more inclusive criteria for gene marker selection (T_tot = 20, T_top3 = 1, T_szcor = −0.01, T_vm = 0.2, and T_niche = 0.05). A total of 178 variable genes were selected as marker genes for constructing k-nearest neighbor graphs with K = 150, followed by coclustering with bootstrapping based on 1000 iterations of resampling 75% of the cells and an approximated target minimum metacell size of 80. Unbalanced edges were filtered with K = 40 and α = 3.
After plotting single cells in a 2D projection and identifying the directionality of infection progression (Fig. 2), we quantified the infection states of individual cells based on the relative distance to the beginning of the progression (i.e., upper-left). The ad hoc origin was defined as having x and y coordinates of the leftmost and the highest cells, respectively, and the ad hoc end as having those of the rightmost and the lowest cells, respectively. The infection index of an individual cell was calculated as the ratio of its distance from the origin to the distance between the origin and the end. A color gradient was painted on the basis of the scale of the infection indices between 0 and 1.
Hierarchical clustering of genes
We used hierarchical clustering to group viral genes with similar expression patterns across infected cells. Viral genes with fewer than 20 UMIs in total across all the cells with active viral expression were excluded to avoid spurious clusters or groupings that are not well supported. The pheatmap package in R was used for hierarchical clustering of UMI counts in log scale, using Pearson correlation as distance measure and the “average” agglomeration method. The cluster tree of viral genes was manually curated by branch swapping using Archaeopteryx (43) to order the genes by kinetic classes and by expression levels.
Annotations of EhV201 genome
A comprehensive annotation table was prepared for EhV201 genes by ordering them according to the manually curated cluster tree (Fig. 3B). The sequence descriptions were based on the GenBank record (JF974311), with modifications based on relevant publications (44, 45), BLAST searches against the nr database (46), and the nucleocytoplasmic virus orthologous groups (2).
Virion proteome composition
Purified EhV201 virion samples were lysed using an 8 M urea buffer and subjected to tryptic digestion followed by LC-MS analysis on a Q Exactive HF instrument (Thermo Fisher Scientific). We used Byonic version 2.10.5 (Protein Metrics) as the software to process the data and to search against the viral CDS database (table S3). In total, 76 viral CDSs had at least 10 peptide-spectrum matches and were shown as present in virion proteome in Fig. 3.
Sequence motif analyses of viral genomes
A 200-bp region surrounding the predicted start codon ATG (A of ATG is base 101) was extracted for each CDS in the EhV201 genome using the Extract Genomic DNA tool of Galaxy (47). Promoter motif discovery was performed on the positive strand of these regions for genes in different kinetic classes using MEME Suite version 5.0.5 (48), Genomatix Genome Analyzer CoreSearch (Intrexon Bioinformatics), and Improbizer (https://users.soe.ucsc.edu/~kent/improbizer/improbizer.html), with the number of motifs to detect set to four. While all three programs agreed on the motifs detected, the results of the MEME analyses are used in this study, where the most common motifs of lengths 6 to 20 are shown (Fig. 4A, fig. S4, and table S4). To test if the short 6-bp sequence motif in early genes is significantly enriched, a differential analysis was performed for early genes against all late (late 1, late 2, and late-undetermined combined) using DREME (49) (table S4).
Circular visualization of viral genes
The UMI counts of individual viral genes were summed over all cells showing active viral expression (≥10 viral UMIs). The log2 values of these total UMI counts were visualized on the 407-kb genome sequence of EhV201 (JF974311) using CCT (CGView Comparison Tool) (50), with a ring for each kinetic class and the height proportional to the log-transformed expression value. Another ring shows the presence/absence of each gene product in the virion proteome (Fig. 4B).
Comparative analyses of other EhV genomes
The best BLAST hits in the CCT analyses were used to find the homologous proteins of the various kinetic classes in other EhV genomes. DNA surrounding the ATG of those genes was extracted for each strain with the GetFastaBed tool in Galaxy (47). Promoter analyses were run on the sequences of the promoter regions of three additional strains (EhV207, EhV86, and EhV202) individually as described above and with all four together (fig. S4 and table S4). These three strains were chosen as representatives of three major clades of EhV strains (51). A CCT plot was drawn to show the sequence similarity between EhV201 and all 12 other EhV strains with whole genome sequences (fig. S5).
Nuclear and organellar transcripts
Host gene expression patterns were analyzed for different categories of single cells, including control culture, mock infected culture, infected culture with fewer than 10 viral UMIs, and different metacells of infected cells with at least 10 viral UMIs. Transcript sequences with fewer than 100 total UMIs across all the cells were excluded to avoid lowly expressed ones, leaving 720 unique transcripts in total (Fig. 5A). The average UMI count was calculated for each transcript in each category, and the sum of the average values of all categories was normalized to 100. Hierarchical clustering of host transcripts was done using pheatmap as described above on the basis of the normalized values. The sliding windows of infected cells (window size = 50 cells, step = 10 cells) were generated for mRNA transcripts from the nuclear, mitochondrial, and plastid genomes (Fig. 5, B and C) and the major transcript from each genome that encodes rRNA (fig. S6) using the rollapply function of the zoo package in R.
Functional analyses of nuclear transcripts
Nucleus-encoded transcripts in Fig. 5A were annotated with GO identifiers (28), as previously described (38). GO term enrichment analyses of transcripts with a relative abundance of more than 10% (Fig. 5A) were conducted using the classicFisher method with a nodeSize of 10 as implemented in topGO version 2.34.0 (52), with P values listed in table S7. Identifiers of KEGG (29) were assigned to the transcripts with KEGG Automatic Annotation Server (53) based on the BBH (bi-directional best hit) method (nucleotide) with the default list of reference organisms.
Supplementary Material
Acknowledgments
We thank N. Stern-Ginossar, A. Nachshon, M. Shnayder, and E. Koh for help with MARS-seq and cycloheximide experiments; R. Avraham, D. Hoffman, S. H. Avivi, G. Rosenberg, A. Solomon, and the Weizmann Institute Advanced Sequencing Technologies Unit of the Life Sciences Core Facilities for assistance with Illumina sequencing; Y. Levin and M. Kupervaser from the de Botton Institute for Protein Profiling of the Nancy and Stephen Grand Israel National Center for Personalized Medicine for assistance with proteomic analyses; and all Vardi laboratory members for fruitful discussions. Funding: This work was supported by the European Research Council Consolidator Grant (CoG) (VIROCELLSPHERE consolidator grant no. 681715; A.V.) and the European Molecular Biology Organization Long-Term Fellowship (ALTF 1172-2016; C.K.). Author contributions: C.K., U.S., S.R., and A.V. designed the study, with input from A.S.-P. and A.T.; U.S. and C.K. prepared the cell cultures; C.K. and U.S. performed MARS-seq with assistance of A.S.-P.; C.K. analyzed the MARS-seq data with assistance of A.S.-P. and A.T.; D.S. performed the virion proteome experiment; S.B.-D. analyzed the motifs and compared the EhV genomes, with input from C.K.; C.K. prepared the figures, with input from U.S., S.B.-D., S.R., and A.V.; C.K., S.R., and A.V. wrote the manuscript, with input from all authors; and A.V. supervised the study. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. scRNA-seq data were deposited in the Gene Expression Omnibus with the accession number GSE135429. The R code for processing data and generating figures is available at https://github.com/chuanku-lab/EhVEmiliania_scRNAseq.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaba4137/DC1
REFERENCES AND NOTES
- 1.Van Etten J. L., Meints R. H., Giant viruses infecting algae. Annu. Rev. Microbiol. 53, 447–494 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Yutin N., Wolf Y. I., Raoult D., Koonin E. V., Eukaryotic large nucleo-cytoplasmic DNA viruses: Clusters of orthologous genes and reconstruction of viral genome evolution. Virol. J. 6, 223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilhelm S. W., Coy S. R., Gann E. R., Moniruzzaman M., Stough J. M. A., Standing on the shoulders of giant viruses: Five lessons learned about large viruses infecting small eukaryotes and the opportunities they create. PLOS Pathog. 12, e1005752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulz F., Yutin N., Ivanova N. N., Ortega D. R., Lee T. K., Vierheilig J., Daims H., Horn M., Wagner M., Jensen G. J., Kyrpides N. C., Koonin E. V., Woyke T., Giant viruses with an expanded complement of translation system components. Science 356, 82–85 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Carradec Q., Pelletier E., Silva C. D., Alberti A., Seeleuthner Y., Blanc-Mathieu R., Lima-Mendez G., Rocha F., Tirichine L., Labadie K., Kirilovsky A., Bertrand A., Engelen S., Madoui M.-A., Méheust R., Poulain J., Romac S., Richter D. J., Yoshikawa G., Dimier C., Kandels-Lewis S., Picheral M., Searson S., Coordinators T. O., Jaillon O., Aury J.-M., Karsenti E., Sullivan M. B., Sunagawa S., Bork P., Not F., Hingamp P., Raes J., Guidi L., Ogata H., de Vargas C., Iudicone D., Bowler C., Wincker P., A global ocean atlas of eukaryotic genes. Nat. Commun. 9, 373 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gregory A. C., Zayed A. A., Conceição-Neto N., Temperton B., Bolduc B., Alberti A., Ardyna M., Arkhipova K., Carmichael M., Cruaud C., Dimier C., Domínguez-Huerta G., Ferland J., Kandels S., Liu Y., Marec C., Pesant S., Picheral M., Pisarev S., Poulain J., Tremblay J.-É., Vik D., Coordinators T. O., Babin M., Bowler C., Culley A. I., de Vargas C., Dutilh B. E., Iudicone D., Karp-Boss L., Roux S., Sunagawa S., Wincker P., Sullivan M. B., Marine DNA viral macro- and microdiversity from pole to pole. Cell 177, 1109–1123.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown C. W., Yoder J. A., Coccolithophorid blooms in the global ocean. J. Geophys. Res. 99, 7467–7482 (1994). [Google Scholar]
- 8.Wilson W. H., Tarran G. A., Schroeder D., Cox M., Oke J., Malin G., Isolation of viruses responsible for the demise of Emiliania huxleyi bloom in the English Channel. J. Mar. Biol. Assoc. United Kingdom 82, 369–377 (2002). [Google Scholar]
- 9.Laber C. P., Hunter J. E., Carvalho F., Collins J. R., Hunter E. J., Schieler B. M., Boss E., More K., Frada M., Thamatrakoln K., Brown C. M., Haramaty L., Ossolinski J., Fredricks H., Nissimov J. I., Vandzura R., Sheyn U., Lehahn Y., Chant R. J., Martins A. M., Coolen M. J. L., Vardi A., DiTullio G. R., Van Mooy B. A. S., Bidle K. D., Coccolithovirus facilitation of carbon export in the North Atlantic. Nat. Microbiol. 3, 537–547 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Sheyn U., Rosenwasser S., Lehahn Y., Barak-Gavish N., Rotkopf R., Bidle K. D., Koren I., Schatz D., Vardi A., Expression profiling of host and virus during a coccolithophore bloom provides insights into the role of viral infection in promoting carbon export. ISME J. 12, 704–713 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Legendre M., Audic S., Poirot O., Hingamp P., Seltzer V., Byrne D., Lartigue A., Lescot M., Bernadac A., Poulain J., Abergel C., Claverie J.-M., mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Res. 20, 664–674 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blanc G., Mozar M., Agarkova I. V., Gurnon J. R., Yanai-Balser G., Rowe J. M., Xia Y., Riethoven J.-J., Dunigan D. D., Van Etten J. L., Deep RNA sequencing reveals hidden features and dynamics of early gene transcription in Paramecium bursaria chlorella virus 1. PLOS ONE. 9, e90989 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenwasser S., Mausz M. A., Schatz D., Sheyn U., Malitsky S., Aharoni A., Weinstock E., Tzfadia O., Ben-Dor S., Feldmesser E., Pohnert G., Vardi A., Rewiring host lipid metabolism by large viruses determines the fate of Emiliania huxleyi, a bloom-forming alga in the ocean. Plant Cell 26, 2689–2707 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolodziejczyk A. A., Kim J. K., Svensson V., Marioni J. C., Teichmann S. A., The technology and biology of single-cell RNA sequencing. Mol. Cell 58, 610–620 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Tanay A., Regev A., Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zanini F., Robinson M. L., Croote D., Sahoo M. K., Sanz A. M., Ortiz-Lasso E., Albornoz L. L., Rosso F., Montoya J. G., Goo L., Pinsky B. A., Quake S. R., Einav S., Virus-inclusive single-cell RNA sequencing reveals the molecular signature of progression to severe dengue. Proc. Natl. Acad. Sci. U.S.A. 115, E12363–E12369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russell A. B., Trapnell C., Bloom J. D., Extreme heterogeneity of influenza virus infection in single cells. eLife 7, e32303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheyn U., Rosenwasser S., Ben-Dor S., Porat Z., Vardi A., Modulation of host ROS metabolism is essential for viral infection of a bloom-forming coccolithophore in the ocean. ISME J. 10, 1742–1754 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keren-Shaul H., Kenigsberg E., Jaitin D. A., David E., Paul F., Tanay A., Amit I., MARS-seq2.0: An experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat. Protoc. 14, 1841–1862 (2019). [DOI] [PubMed] [Google Scholar]
- 20.Barak-Gavish N., Frada M. J., Ku C., Lee P. A., DiTullio G. R., Malitsky S., Aharoni A., Green S. J., Rotkopf R., Kartvelishvily E., Sheyn U., Schatz D., Vardi A., Bacterial virulence against an oceanic bloom-forming phytoplankter is mediated by algal DMSP. Sci. Adv. 4, eaau5716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baran Y., Bercovich A., Sebe-Pedros A., Lubling Y., Giladi A., Chomsky E., Meir Z., Hoichman M., Lifshitz A., Tanay A., MetaCell: Analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 20, 206 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider-Poetsch T., Ju J., Eyler D. E., Dang Y., Bhat S., Merrick W. C., Green R., Shen B., Liu J. O., Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 6, 209–217 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson W. H., Schroeder D. C., Allen M. J., Holden M. T. G., Parkhill J., Barrell B. G., Churcher C., Hamlin N., Mungall K., Norbertczak H., Quail M. A., Price C., Rabbinowitsch E., Walker D., Craigon M., Roy D., Ghazal P., Complete genome sequence and lytic phase transcription profile of a Coccolithovirus. Science 309, 1090–1092 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Vardi A., Van Mooy B. A. S., Fredricks H. F., Popendorf K. J., Ossolinski J. E., Haramaty L., Bidle K. D., Viral glycosphingolipids induce lytic infection and cell death in marine phytoplankton. Science 326, 861–865 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Ziv C., Malitsky S., Othman A., Ben-Dor S., Wei Y., Zheng S., Aharoni A., Hornemann T., Vardi A., Viral serine palmitoyltransferase induces metabolic switch in sphingolipid biosynthesis and is required for infection of a marine alga. Proc. Natl. Acad. Sci. U.S.A. 113, E1907–E1916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen M. J., Forster T., Schroeder D. C., Hall M., Roy D., Ghazal P., Wilson W. H., Locus-specific gene expression pattern suggests a unique propagation strategy for a giant algal virus. J. Virol. 80, 7699–7705 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wyler E., Franke V., Menegatti J., Kocks C., Boltengagen A., Praktiknjo S., Walch-Rückheim B., Bosse J., Rajewsky N., Grässer F., Akalin A., Landthaler M., Single-cell RNA-sequencing of herpes simplex virus 1-infected cells connects NRF2 activation to an antiviral program. Nat. Commun. 10, 4878 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The Gene Ontology Consortium , The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanehisa M., Goto S., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenwasser S., Ziv C., van Creveld S. G., Vardi A., Virocell metabolism: Metabolic innovations during host–virus interactions in the ocean. Trends Microbiol. 24, 821–832 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Rosenwasser S., Sheyn U., Frada M. J., Pilzer D., Rotkopf R., Vardi A., Unmasking cellular response of a bloom-forming alga to viral infection by resolving expression profiles at a single-cell level. PLOS Pathog. 15, e1007708 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thamatrakoln K., Talmy D., Haramaty L., Maniscalco C., Latham J. R., Knowles B., Natale F., Coolen M. J. L., Follows M. J., Bidle K. D., Light regulation of coccolithophore host-virus interactions. New Phytol. 221, 1289–1302 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Gann E. R., Gainer P. J., Reynolds T. B., Wilhelm S. W., Influence of light on the infection of Aureococcus anophagefferens CCMP 1984 by “a giant virus”. PLOS ONE. 15, e0226758 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keller M. D., Seluin R. C., Claus W., Guillard R. R. L., Media for the culture of oceanic ultraphytoplankton. J. Phycol. 23, 633–638 (1987). [Google Scholar]
- 35.Allen M. J., Martinez-Martinez J., Schroeder D. C., Somerfield P. J., Wilson W. H., Use of microarrays to assess viral diversity: From genotype to phenotype. Environ. Microbiol. 9, 971–982 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Schleyer G., Shahaf N., Ziv C., Dong Y., Meoded R. A., Helfrich E. J. N., Schatz D., Rosenwasser S., Rogachev I., Aharoni A., Piel J., Vardi A., In plaque-mass spectrometry imaging of a bloom-forming alga during viral infection reveals a metabolic shift towards odd-chain fatty acid lipids. Nat. Microbiol. 4, 527–538 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nissimov J. I., Worthy C. A., Rooks P., Napier J. A., Kimmance S. A., Henn M. R., Ogata H., Allen M. J., Draft genome sequence of the Coccolithovirus Emiliania huxleyi virus 202. J. Virol. 86, 2380–2381 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schatz D., Rosenwasser S., Malitsky S., Wolf S. G., Feldmesser E., Vardi A., Communication via extracellular vesicles enhances viral infection of a cosmopolitan alga. Nat. Microbiol. 2, 1485–1492 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T. L., BLAST+: Architecture and applications. BMC Bioinformatics. 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sánchez-Puerta M. V., Bachvaroff T. R., Delwiche C. F., The complete plastid genome sequence of the haptophyte Emiliania huxleyi : A comparison to other plastid genomes. DNA Res. 156, 151–156 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Sánchez-Puerta M. V., Bachvaroff T. R., Delwiche C. F., The complete mitochondrial genome sequence of the haptophyte Emiliania huxleyi and its relation to heterokonts. DNA Res. 11, 1–10 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Read B. A., Kegel J., Klute M. J., Kuo A., Lefebvre S. C., Maumus F., Mayer C., Miller J., Monier A., Salamov A., Young J., Aguilar M., Claverie J.-M., Frickenhaus S., Gonzalez K., Herman E. K., Lin Y.-C., Napier J., Ogata H., Sarno A. F., Shmutz J., Schroeder D., de Vargas C., Verret F., von Dassow P., Valentin K., Van de Peer Y., Wheeler G.; Emiliania huxleyi Annotation Consortium, Dacks J. B., Delwiche C. F., Dyhrman S. T., Glöckner G., John U., Richards T., Worden A. Z., Zhang X., Grigoriev I. V., Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 499, 209–213 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Han M. V., Zmasek C. M., phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics. 10, 356 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mirzakhanyan Y., Gershon P. D., Multisubunit DNA-dependent RNA polymerases from vaccinia virus and other nucleocytoplasmic large-DNA viruses: Impressions from the age of structure. Microbiol. Mol. Biol. Rev. 81, e00010–e00017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Assarsson E., Greenbaum J. A., Sundström M., Schaffer L., Hammond J. A., Pasquetto V., Oseroff C., Hendrickson R. C., Lefkowitz E. J., Tscharke D. C., Sidney J., Grey H. M., Head S. R., Peters B., Sette A., Kinetic analysis of a complete poxvirus transcriptome reveals an immediate-early class of genes. Proc. Natl. Acad. Sci. U.S.A. 105, 2140–2145 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.NCBI Resource Coordinators , Database resources of the national center for biotechnology information. Nucleic Acids Res. 44, D7–D19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Grüning B. A., Guerler A., Hillman-Jackson J., Hiltemann S., Jalili V., Rasche H., Soranzo N., Goecks J., Taylor J., Nekrutenko A., Blankenberg D., The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey T. L., Williams N., Misleh C., Li W. W., MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bailey T. L., DREME: Motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grant J. R., Arantes A. S., Stothard P., Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics 13, 202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nissimov J. I., Pagarete A., Ma F., Cody S., Dunigan D. D., Kimmance S. A., Allen M. J., Coccolithoviruses: A review of cross-kingdom genomic thievery and metabolic thuggery. Viruses 9, 52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.A. A. Rahnenfuhrer, topGO: Enrichment analysis for gene ontology. R package version 2.38.1. (2019).
- 53.Moriya Y., Itoh M., Okuda S., Yoshizawa A. C., Kanehisa M., KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaba4137/DC1