

A prodrug hydrogelator serves as an effective immune booster for checkpoint blocker–based cancer immunotherapy.
Abstract
Immune checkpoint blockers (ICBs) have shown great promise at harnessing immune system to combat cancer. However, only a fraction of patients can directly benefit from the anti–programmed cell death protein 1 (aPD1) therapy, and the treatment often leads to immune-related adverse effects. In this context, we developed a prodrug hydrogelator for local delivery of ICBs to boost the host’s immune system against tumor. We found that this carrier-free therapeutic system can serve as a reservoir for extended tumoral release of camptothecin and aPD1 antibody, resulting in an immune-stimulating tumor microenvironment for boosted PD-1 blockade immune response. Our in vivo results revealed that this combination chemoimmunotherapy elicits robust and durable systemic anticancer immunity, inducing tumor regression and inhibiting tumor recurrence and metastasis. This work sheds important light into the use of small-molecule prodrugs as both chemotherapeutic and carrier to awaken and enhance antitumor immune system for improved ICBs therapy.
INTRODUCTION
Cancer immunotherapy, especially immune checkpoint blockers (ICBs) targeting the programmed cell death protein 1 (PD-1) pathway, has gained prominence because of its high clinical efficacy (1, 2). PD-1 ligands (PD-L1 and PD-L2) expressed on the surface of tumor cells and antigen-presenting cells engage PD-1+ T cells, resulting in T cell apoptosis, anergy, and exhaustion (3, 4). Blocking these interactions between PD-1 and its ligands with anti–PD-1 antibody (aPD1) leads to restoration of T cell function and long-term antitumor immune response (5, 6). Despite its promising potential, current aPD1 therapy benefits a relatively small fraction of patients, exhibiting only a 10 to 30% treatment response rate (7, 8). This is partially because patients characterized by insufficient infiltration of tumor antigen–specific T cells (primarily CD8+ T cells) and low tumoral expression of PD-L1 respond poorly to ICBs (9, 10). Furthermore, systemic administration as a conventional means results in off-target binding of the antibodies to normal tissues, compromising the efficacy of aPD1 therapy and inducing severe immune-related side effects (11, 12). Consequently, to fully realize the potential of ICBs, approaches are needed to elicit an immune-stimulating tumor microenvironment and to sensitize tumors to aPD1 with minimal off-target adverse events.
Recent studies have suggested that chemotherapy can initiate an antitumor immune response owing to immunogenic cell death (ICD) (13, 14). Tumors undergoing ICD trigger infiltration of tumor-associated T cells and promote an immunogenic tumor phenotype, revealing a promising strategy for combination immunotherapy (7, 15, 16). Systemic chemotherapy may impair bone marrow cell production and subsequently affect the number and activation state of resident immune cells (17, 18); in contrast, local chemotherapy, which can promote activation of tumor-associated dendritic cells and expansion of effector T cells (Teffs), potentiates the antitumor immune response (18, 19). Therefore, we hypothesized that local chemotherapy has the potential to activate the immune system in a manner synergistic with a targeted aPD1 therapy.
Hydrogels are highly appealing “smart” drug delivery systems that allow site-specific delivery of various bioactive agents, with a potential for controlled and sustainable release (20–23). For local delivery, off-target–associated adverse events are minimized, while drug bioavailability is increased (24). More specifically, supramolecular hydrogels are formed by the physical cross-linking of filamentous assemblies, derived of peptide amphiphile–building units (25, 26). These supramolecular hydrogels use responsive solution-to-hydrogel (sol-gel) phase transitioning, permitting their direct injection into the target sites with minimal nonsurgical invasiveness (27, 28). Recently, direct conjugation of therapeutic agents onto the peptides has been shown to convert the drug into an effective hydrogelator, capable of creating a “self-delivery” hydrogel (29, 30). In addition, the ability of hydrogels to respond to the tumor microenvironment allows for better controlled release kinetics and improved therapeutic efficacy (19, 31).
Here, we report on the use of a drug-based supramolecular hydrogelator for local delivery of ICBs and demonstrate the high potency of this two-component system for enhanced immune stimulation and tumor regression (Fig. 1A). Our findings reveal that this prodrug hydrogelator can serve as a reservoir for long-term, responsive release of both camptothecin (CPT) and aPD1 within the tumor microenvironment, so as to induce an immune-stimulating phenotype and prompt robust PD-1 blockade immune response, leading to 100% tumor regression for all treated mice in both GL-261 brain cancer and CT 26 colon cancer models.
Fig. 1. Schematic and characterization of in situ formed P-NT–aPD1 hydrogel.

(A) Schematic illustration of localized CPT and aPD1 delivery using an in situ formed supramolecular hydrogel to attain bioresponsive drug release and tumor microenvironment regulation. (B) Representative transmission electron microscopy (TEM) images of diCPT-PLGLAG-iRGD nanotubes (P-NT). Scale bar, 100 nm. (C) The circular dichroism (CD) spectrum of the diCPT-PLGLAG-iRGD nanotubes solution. (D) Photographs of the sol-gel transition of P-NT upon the addition of phosphate-buffered saline (PBS). (E) Degradation profiles of diCPT-PLGLAG-iRGD in the presence or absence of glutathione (GSH) (10 mM). Data are given as means ± SD (n = 3). (F) In vitro cytotoxicity studies of free CPT and diCPT-PLGLAG-iRGD toward GL-261 brain cancer cells. IC50, median inhibitory concentration. (G) Inhibition of tumor spheroid growth was evaluated following treatment with free CPT or P-NT. Spheroids treated with drug-free Dulbecco’s modified Eagle’s medium were used as the blank control. Scale bar, 500 μm. (H) The degradation profiles of 200 μM diCPT-PLGLAG-iRGD solutions incubated in the presence or absence of matrix metalloproteinase 2 (MMP-2; 2 μg/ml). Data are given as means ± SD (n = 3). (I) Cumulative release profiles of CPT prodrugs (including diCPT-PLGLAG-iRGD and diCPT-PLG) and (J) aPD1 from P-NT–aPD1 hydrogels incubated in PBS with or without MMP-2. Data are given as means ± SD (n = 3). Photo credit: Feihu Wang, Johns Hopkins University.
RESULTS
Characterization of the bioresponsive hydrogel
We first synthesized the amphiphilic prodrug, diCPT-PLGLAG-iRGD, by conjugating a hydrophilic iRGD [a peptide known to facilitate tumor tissue penetration of anticancer agents (32)] to two hydrophobic CPT molecules through a matrix metalloproteinase 2 (MMP-2) responsive linker (PLGLAG peptide) (fig. S1, A to C) (33, 34). The two CPT moieties were attached to the PLGLAG peptide through a reducible disulfanyl-ethyl carbonate (etcSS) linker that forms disulfide bonds with the cysteine residues of the peptide sequence. This CPT prodrug spontaneously assembles into supramolecular nanotubes (P-NTs) with tens of micrometers in length in aqueous environments (Fig. 1B). Corresponding circular dichroism (CD) data showed a negative peak around 222 nm, indicative of forming highly ordered intermolecular hydrogen bonding, and a series of absorptions between 260 and 400 nm that are attributed to CPT packing in a highly ordered fashion (Fig. 1C). We found that increasing the concentration of the P-NT solution or adding either phosphate-buffered saline (PBS) or cell medium could induce immediate formation of a self-supporting hydrogel (Fig. 1D and movie S1). This is due to the addition of couterions and/or biomolecules from PBS or cell medium that promote nanotube-nanotube interactions through charge screening, enabling a greater degree of entanglement and subsequent hydrogel formation. Moreover, the P-NT gelation behavior and mechanical properties were assessed using a rheometer (fig. S2A). Addition of 10× PBS at 180 s resulted in a rapid increase in the storage modulus (G′), with the crossover point (G′ > G″) occurring at ~183 s (3 s after PBS injection), indicative of forming a supramolecular hydrogel. This quick sol-gel transition suggests that this CPT prodrug gelator can be used to deliver aPD1 by simply mixing the antibodies with the P-NTs before gelation (Fig. 1A). To prepare aPD1-encapsulated P-NT hydrogel, a therapeutic dose of aPD1 (50 μg) was mixed with a P-NT solution (150 μg of CPT in 30 μl of H2O). A hydrogel was formed immediately after the addition of PBS to the mixture (fig. S2B). Confocal imaging with Cy3-labeled aPD1 suggests spatially uniform distribution of aPD1 across the hydrogel (fig. S2, C and D).
To determine the release of free CPT from the diCPT-PLGLAG-iRGD prodrug, 10 mM glutathione (GSH) was used to mimic intracellular reductive conditions (35, 36). GSH is a cancer-relevant reducing agent that breaks the etcSS disulfide linker to release the parent CPT (fig. S3). We found that GSH-induced cleavage of diCPT-PLGLAG-iRGD occurred rapidly, with 95% of CPT prodrug degraded within 1 hour (Fig. 1E), suggesting that the diCPT-PLGLAG-iRGD prodrug can quickly supply bioactive CPT in the intracellular GSH reductive environment. In the absence of GSH, the drug conjugate concentration decreased only 4% over 24 hours as a result of hydrolysis. In addition, the CPT prodrug and P-NT hydrogel exhibited superior cytotoxicity against GL-261 brain tumor cells and three-dimensional tumor spheroids, respectively (Fig. 1, F and G, and fig. S4).
We used a PLGLAG peptide as a bioresponsive linker in our CPT prodrug design because it is cleavable by MMP-2, an enzyme known to be overexpressed in the tumor extracellular matrix (33, 34). MMP-2 was selected as the cleavage enzyme due to its association with the progression of malignant tumors and high expression in various tumor types including GL-261 brain tumors (fig. S5). To test the cleavability of the PLGLAG spacer, we incubated diCPT-PLGLAG-iRGD with the MMP-2 enzyme and analyzed the resultant products using high-performance liquid chromatography (HPLC) and mass spectrometry (MS). It is apparent that a new peak, identified as the cleaved product LAG-iRGD (fig. S6), was eluted at a retention time of 2.9 min. Monitoring the changes of this peak over time shows that diCPT-PLGLAG-iRGD is quickly degraded by MMP-2, with 54% cleaved after 8 hours (Fig. 1H). Transmission electron microscopy (TEM) and CD (fig. S7) studies confirmed further that MMP-2 accelerates the degradation of the P-NT hydrogel by cutting and shortening these supramolecular nanotubes. The release of CPT and aPD1 from the P-NT–aPD1 hydrogel was subsequently investigated in the presence of MMP-2. As expected, an accelerated release was observed in MMP-2–treated hydrogels. CPT prodrugs displayed a sustained, linear release from the P-NT hydrogel (Fig. 1I), whereas aPD1 showed a much faster release rate, with 53% released within 10 days in the presence of MMP-2 (Fig. 1J). The nonlinear release profile displayed by aPD1 is characteristic of physically encapsulated proteins within a supramolecular hydrogel. The release rate is concentration dependent and largely determined by the protein diffusion parameters and hydrogel breakdown, often showing an initial burse release that quickly drops over time.
Supramolecular hydrogel extends local retention and release of aPD1 antibody
The in vivo formation of gels after injection and their subsequent degradation were then evaluated in C57BL/6 mice. After subcutaneous injection into the backs of mice, a solution containing 7.2 mM diCPT-PLGLAG-iRGD was observed to undergo a sol-gel transition within 5 min (Fig. 2A). This in situ formed P-NT hydrogel displayed a nearly linear degradation profile, as determined by the mass loss method, with ~78% degraded within 45 days (Fig. 2B). To assess the potential of this prodrug hydrogel to act as a drug delivery scaffold, we compared tumoral injections of aPD1 solutions and aPD1-encapsulating P-NT hydrogels. Three days following injection of free (CPT + aPD1), we found that Cy5.5-labeled aPD1 fluorescence appeared throughout the whole body and in major organs (Fig. 2C and fig. S8, A and B). After 7 days, the fluorescence signal was not observed in tumors, indicating that aPD1 rapidly leaked out of the tumor tissue (Fig. 2D). In stark contrast, for P-NT–aPD1–treated mice, almost all aPD1 was contained within the tumor tissue, and no fluorescence was detected in other parts of the bodies (Fig. 2, C and D, and fig. S8, A and B). These results indicate that the P-NT hydrogel increases local retention of the therapeutic agents within the tumor tissue, reducing drug leakage and off-site accumulation. Furthermore, in vivo imaging system (IVIS) imaging revealed no fluorescence at the periphery of the tumors treated with free (CPT + aPD1) (Fig. 2D), whereas P-NT–aPD1–treated tumors displayed strong fluorescence throughout the whole tumor. These results were also confirmed by confocal imaging of tumor tissue sections (Fig. 2E and fig. S8C). The release profiles of aPD1 and CPT were quantitatively assessed using IVIS and HPLC, respectively. For free drug–treated mice, more than 90% of fluorescence was lost within 7 days after injection (Fig. 2F), while for P-NT–aPD1–treated mice, a strong fluorescence signal could still be detected 15 days after injection, with approximately 29.2% remaining in the tumor tissue. Consistently, CPT exhibited a steady release profile in the P-NT–aPD1–treated group, with 53.6% liberated within 15 days, in contrast to a rapid release in the free drug–treated group, where no drug was detectable in the tumor site after 15 days (Fig. 2G). To confirm whether iRGD can induce tumor tissue penetration of anticancer drugs, we designed and synthesized a control prodrug, diCPT-PLGLAG-iRGRD with a scrambled peptide sequence (fig. S9). This prodrug also formed a hydrogel (defined as P-NT–Sham) under physiological condition. Both P-NT–Sham–treated and P-NT–treated tumors displayed very strong CPT fluorescence in the tumor center (fig. S10). However, no blue fluorescence was detected at the periphery of tumors treated with P-NT–Sham hydrogel. This is in sharp contrast with the P-NT–treated mice, where distinct CPT fluorescence was observed even at the edges of the tumor.
Fig. 2. P-NT–aPD1 hydrogel enhances local retention and prolongs in vivo release of aPD1.
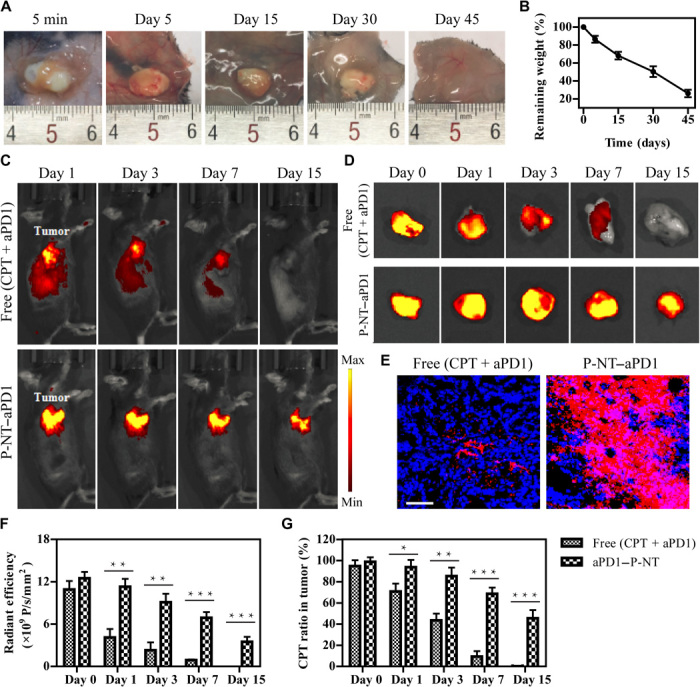
(A) In vivo gel formation and retention after subcutaneous injection of P-NT solution in the back of C57BL/6 mice. (B) In vivo degradation profile of the P-NT hydrogel over time, as determined by the mass loss method. (C) Fluorescence IVIS imaging of the local retention and distribution of aPD1-Cy5.5 in mice, administered in solution form and with the P-NT hydrogel. Experiments were repeated three times. (D) Fluorescence imaging of tumor tissues and (E) tumor sections of GL-261 brain tumor–bearing mice after tumoral injections of free (CPT + aPD1) or P-NT–aPD1. Red, Cy5.5-labeled aPD1; blue, 4′,6-diamidino-2-phenylindole–stained nuclei. Scale bar, 200 μm. (F) Quantification of the in vivo retention profile of Cy5.5-aPD1 and (G) CPT. Statistical significance was calculated using a two-sided unpaired t test. Data are given as means ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. Photo credit: Feihu Wang, Johns Hopkins University.
P-NT–aPD1 hydrogel elicits a robust antitumor immunity
To assess the immune response of each treatment, we euthanized all mice at day 25 after tumor implantation and then analyzed tumor-infiltrating lymphocytes (TILs) and tumor cells using flow cytometry (Fig. 3A). We designed and used diC12-PLGLAG-iRGD as a therapeutic-free hydrogel (fig. S11). Our results suggest that this “empty” hydrogel (E-Gel) had no important effects on TILs and tumor cells (Fig. 3). P-NT treatment stimulated the production of type I interferons (IFNs) and chemokine CXCL10 (fig. S12, A to C). Type I IFNs are known to propagate dendritic cells activation and lead to antitumor T cell response, whereas CXCL10 is believed to facilitate the recruitment of Teffs to the tumor site (22, 37). Consistent with these findings, we also observed that the proportion of CD103+ dendritic cells and CD3+, CD4+, and CD8+ T cells in the P-NT–treated tumor tissue was substantially augmented (Fig. 3, B to F, and fig. S12D). This observation shows that the P-NT hydrogel alone (no aPD1) could induce infiltration of T cells. When aPD1 was loaded into the E-Gel [aPD1(L) (no CPT)], an increased percentage of T cells were also observed within the tumor. This suggests that, as expected, the aPD1 alone can block the interactions between PD-1 and its ligands, resulting in increased T cell survival. The addition of aPD1 to P-NT (P-NT–aPD1) increased the percentage of CD8+ Teffs in tumors by 4.1- and 1.5-fold, relative to that of untreated and aPD1(L)-treated mice, respectively. Furthermore, P-NT treatment moderately reduced the percentage of FoxP3+CD4+ regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs; Gr-1+CD11b+CD45+ cells) compared to the E-Gel (Fig. 3, G and H, and fig. S13), whereas the combined P-NT–aPD1 treatment significantly decreased the ratio of Tregs and MDSCs. We also found that both P-NT and aPD1(L) treatment increased the frequency of PD-1+CD8+ T cells in the tumor microenvironment compared to those untreated mice (Fig. 3I and fig. S14). The PD-1+CD8+ T cell response was most evident in tumors treated with dual P-NT–aPD1 therapy. Similar effects were also observed in CD4+ T cells (Fig. 3J). Thus, the combined P-NT–aPD1 treatment provoked a potent antitumor immune response, as evidenced by the significantly decreased percentage of PD-L1–expressing CD45− cells in P-NT–aPD1–treated tumors (Fig. 3K). These results led us to conclude that P-NT–aPD1 treatment elicits an immune-stimulating tumor microenvironment in mice.
Fig. 3. Local delivery aPD1 by CPT prodrug hydrogel elicits a robust antitumor immunity.
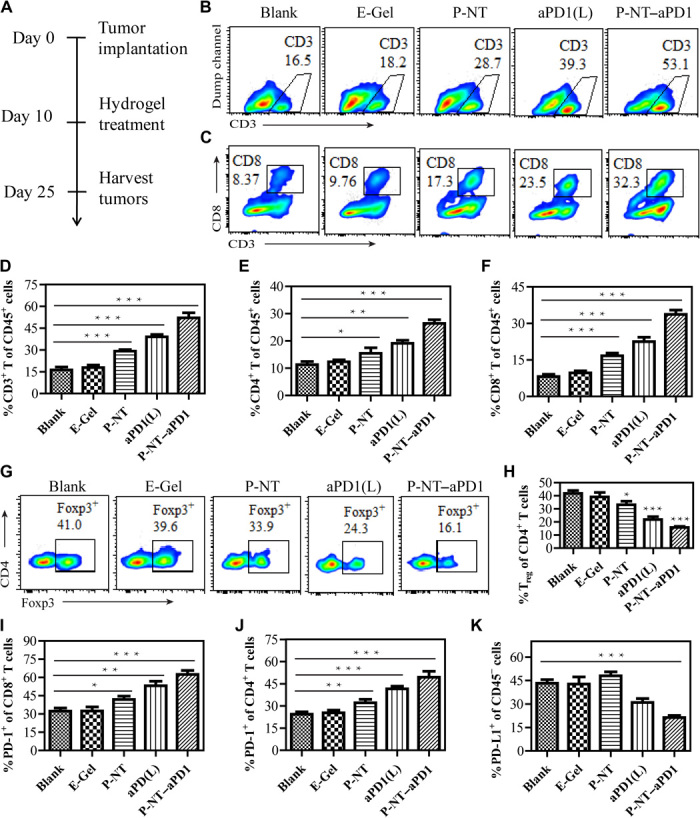
(A) Experimental schedule: GL-261 brain cancer cells were implanted into the right flank of mice on day 0. Mice were intratumorally injected on day 10 with P-NT, aPD1-loaded diC12-PLGLAG-iRGD (aPD1-L), or P-NT–aPD1 hydrogels, with diC12-PLGLAG-iRGD hydrogel (E-Gel) used as a drug-free control. Flow cytometric analysis was performed on lymphocytes extracted from the tumor on day 25. (B) Representative flow cytometric analysis images and (D) relative quantification of CD3+ T cell within the tumor by different treatment groups. (C) Representative flow cytometric images and (F) relative quantification of CD8+ T cell infiltration within the tumor by different treatment groups. (E) Quantification of CD4+ T cell that infiltration within the tumor in different treatment groups. (G) Representative flow cytometric analysis images and (H) relative quantification of Foxp3+CD4+ T cells (Tregs). (I) The percentages of PD-1–expressing CD8+ T cells and (J) PD-1–expressing CD4+ T cells after different treatments. (K) The percentage of PD-L1–expressing CD45− cells after different treatments. Statistical significance was calculated using a two-sided unpaired t test. Data are given as means ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
P-NT–aPD1 elicits complete regression of established GL-261 brain tumors
To evaluate the synergistic antitumor effects of the P-NT–aPD1 hydrogel, we used a subcutaneous GL-261 brain tumor model (Fig. 4A). Tumor burden was monitored and quantified using bioluminescence signals and caliper measurements. The in situ formed gels were injected into the tumor when its volume reached ~100 to 150 mm3 at day 10. The E-Gel had no tumor inhibition effect (fig. S15, A and B). P-NT–treated mice showed a delay in tumor growth (Fig. 4, B and C). Although aPD1(L) monotherapy was not sufficient to control tumor burden, tumor suppression was more pronounced in aPD1(L)-treated mice than those treated with P-NT. P-NT–aPD1 combination therapy resulted in the most effective tumor recession (Figs. 4C and 5D), with all P-NT–aPD1–treated tumors fully regressed and 100% mouse survival at 100 days (Fig. 4E). In contrast, free (CPT + aPD1) treatment exhibited worse tumor growth inhibition than P-NT–aPD1 treatment, even when a much higher dose of aPD1 (150 μg versus 50 μg in P-NT–aPD1) was given over three administrations. This study confirms that the P-NT–aPD1 hydrogel has an enhanced synergistic antitumor effect. In addition, mouse body weight, serum biochemistry, and blood cell count indicated no significant difference following P-NT–aPD1 treatment, indicating that localized P-NT–aPD1 did not induce obvious side effects (fig. S15C and table S1). Residual tumors were collected on day 25 after tumor implantation, and cells were subsequently analyzed using flow cytometry to investigate immune cell subset changes in response to different treatments. P-NT–aPD1–treated mice exhibited the highest frequencies of CD3+, CD4+, and CD8+ T cells among all the treatment groups (Fig. 4, F and G, and fig. S15D). The percentage of CD8+ Teffs in the P-NT–aPD1–treated mice was 1.6-fold higher than the aPD1(L)-treated group and 1.9-fold higher than that of the free (CPT + aPD1)–treated mice. Moreover, the ratio of tumor-infiltrating CD8+ Teffs to Tregs was substantially increased after P-NT–aPD1 treatment (Fig. 4H). Collectively, these results suggest that P-NT–aPD1 can effectively suppress tumor growth in mice by eliciting a robust, T cell–mediated, antitumor immune response.
Fig. 4. Intratumoral injection of P-NT–aPD1 elicits complete regression of established GL-261 brain tumors.

(A) Experimental schedule: GL-261 brain cancer cells were implanted into the right flanks of mice on day 0. Mice were intratumorally (it.) injected on day 10 with free (CPT + aPD1), P-NT, aPD1-loaded diC12-PLGLAG-iRGD [aPD1(L)], or P-NT–aPD1 solutions. In the free (CPT + aPD1) group, treatment was administered three times (on days 10, 17, and 24). (B) The in vivo bioluminescence images of the GL-261 tumors on day 35 and (C) day 60. (D) Average tumor growth kinetics of different treatment groups; growth curves were plotted until the first mouse death. Data are given as means ± SD (n = 10 for P-NT–aPD1–treated group and n = 5 for other groups). (E) Survival curves corresponding to different treatment groups. (F) Quantification of CD3+ T cells and (G) CD8+ T cells infiltrating within the tumor between different treatment groups. (H) Ratios of the tumor-infiltrating CD8+ Teffs to Tregs in the tumors of different treatment groups. Statistical significance was calculated using a two-sided unpaired t test. Data are given as means ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Fig. 5. Intratumoral delivery of P-NT–aPD1 induces T cell memory against tumor.
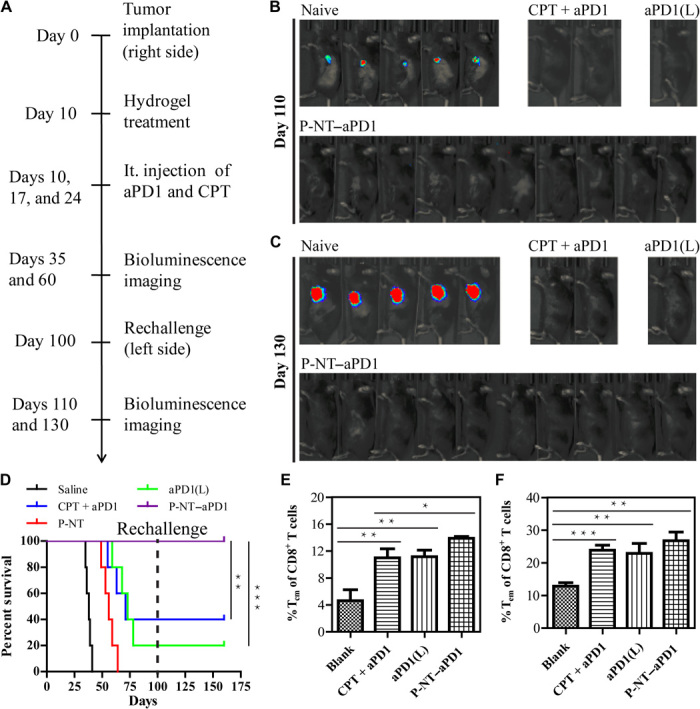
(A) Mice that considered long-term survival from all treatment groups were rechallenged on the opposite flank in an attempt to develop new tumors. (B) The in vivo bioluminescence imaging of the GL-261 tumors was observed on day 110 and (C) on day 130. (D) Survival curves for naive and rechallenged mice from different treatment groups. Statistical significance was calculated via the log-rank (Mantel-Cox) test. (E) The percentage of CD8+ Tcm cells and (F) CD8+ Tem cells in splenocytes of the naive and rechallenged mice. Statistical significance was calculated using a two-sided unpaired t test. Data are given as means ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
P-NT–aPD1 induces durable immune response
To assess whether P-NT–aPD1 therapy could induce a memory response, we rechallenged mice that displayed long-term survival from all previous treatment groups with GL-261 cells on the opposite flank on day 100, with naive mice used as controls (Fig. 5A). No tumor growth was observed in the free (CPT + aPD1), aPD1(L), and P-NT–aPD1 groups, as evidenced by the absence of bioluminescence signal of GL-261 cells (Fig. 5, B and C). In contrast, all naive mice developed large tumors and died within 40 days of cancer cell implantation (Fig. 5C and fig. S15E). These results reveal that a memory response upon tumor recognition occurred in all surviving mice treated with aPD1. It is noteworthy that the 100% overall survival of the P-NT–aPD1–treated mice was significantly higher than that of any other treatment group (Fig. 5D). Sixty days after tumor rechallenge, spleens were collected to analyze memory immune cells. The percentages of CD8+CD44highCD62Lhigh central memory T (Tcm) cells and CD8+CD44highCD62Llow effector memory T (Tem) cells were both elevated in the long-term survival mice (Fig. 5, E and F). It is notable that P-NT–aPD1–treated mice showed higher percentages of CD8+ Tcm (14.3%) and Tem (27.1%) cells than the saline (4.6 and 12.8%, respectively), CPT + aPD1 (10.9 and 23.7%, respectively), and aPD1(L) (11.2 and 22.8%, respectively) groups. These results suggest that a durable and robust T cell memory response was generated by local delivery of P-NT–aPD1 hydrogel.
Systemic antitumor immunity induced by localized P-NT–aPD1 therapy
Next, we tested whether localized P-NT–aPD1 therapy induces systemic antitumor immunity against GL-261 tumors. An orthotopic glioblastoma tumor model was established in the left cortical surface to mimic tumor metastasis. On day 6, the primary tumors were locally treated with P-NT–aPD1 hydrogels (Fig. 6, A and B). We observed that not only was the primary tumor growth substantially inhibited by the P-NT–aPD1 therapy but also the distant intracranial glioma development was suppressed (Fig. 6C). In contrast, all untreated mice died within 25 days as a result of orthotopic glioma (fig. S15F). The P-NT–aPD1 therapy significantly prolonged survival time and completely eliminated tumors in 40% of mice (Fig. 6D), indicating that localized P-NT–aPD1 therapy effectively suppresses the progression of both primary and “distant tumors.” Consistent with these results, the frequency of CD8+ T cells and the ratio of tumor-infiltrating Teff to Treg increased in tumors treated with P-NT–aPD1 (Fig. 6, E and F). These treatment outcomes suggest that localized P-NT–aPD1 therapy induced a protective systemic immune response against GL-261 tumors.
Fig. 6. P-NT–aPD1 treatment induces systemic antitumor immune response.
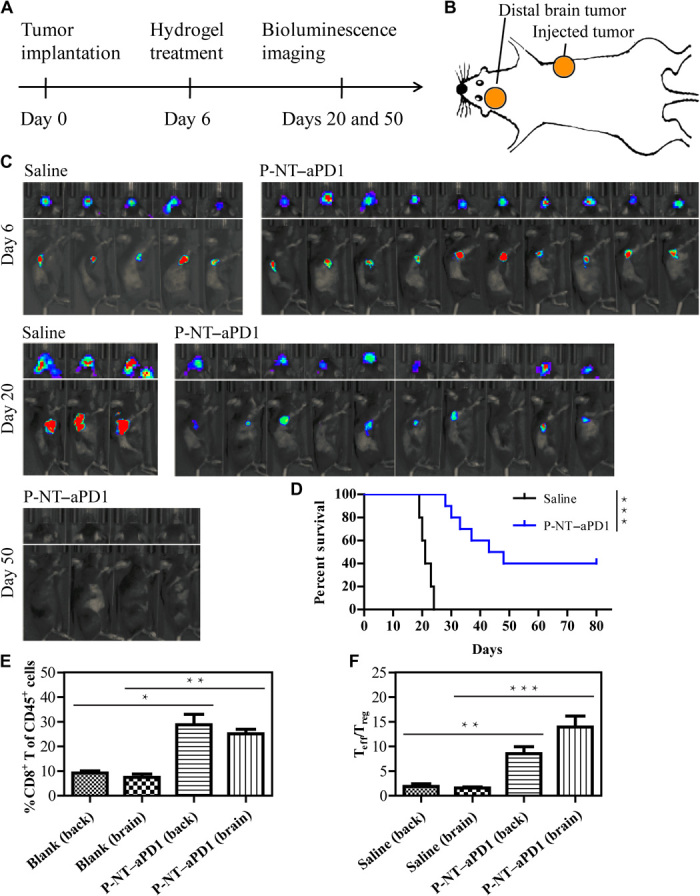
(A) Experimental scheme: Mice were implanted with GL-261 cells in the right back and left cortical surface, and then primary tumors were locally treated with P-NT–aPD1 on day 6. In vivo bioluminescence imaging of the tumors was observed at scheduled time points. (B) Tumors on the right flank were locally treated with P-NT–aPD1 hydrogel, while intracranial gliomas were designated as distant tumors and were left untreated. (n = 10 for P-NT–aPD1–treated group and n = 5 for saline group). (C) In vivo bioluminescence imaging of the GL-261 tumors in response to local P-NT–aPD1 hydrogel treatment. (D) Survival curves corresponding to saline and P-NT–aPD1–treated mice. Statistical significance was calculated via the log-rank (Mantel-Cox) test. (E) Quantification of CD8+ T cells infiltrating within the tumors of the two treatment groups. (F) Ratios of the tumor-infiltrating Teff to Treg in the tumors of the treatment groups. Statistical significance was calculated using a two-sided unpaired t test. Data are given as means ± SD (n = 3). *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Efficacy in a colon cancer model
To ascertain the broad application potential of the P-NT–aPD1 chemoimmunotherapeutic hydrogel, we evaluated CT 26 colon cancer–bearing BALB/c mice. When tumor volume reached 100 to 150 mm3 at day 8, a solution containing P-NT and aPD1 was injected into the mice to from P-NT–aPD1 hydrogels in situ (fig. S16A). Consistent with our findings in the brain tumor model, mice treated with P-NT–aPD1 showed noticeable tumor growth inhibition (fig. S16B). One hundred percent of the mice treated with P-NT–aPD1 survived for at least 130 days and developed antitumor immune memory (fig. S16C). These results corroborate the high antitumor efficacy of P-NT–aPD1 hydrogel in cancer immunotherapy.
DISCUSSION
Previous reports have shown that tumors undergoing ICD can elicit infiltration of tumor associated T effector cells and promote an immune-stimulating tumor phenotype (7, 15, 16). Our results suggest that the CPT prodrug hydrogel offers an effective and universal strategy to kill cancer cells while simultaneously eliciting antitumor immunity. We found an increased population of CD3+, CD4+, and CD8+ T cells (including PD-1+CD8+ T cells) in the tumor tissue upon P-NT hydrogel treatment. Moreover, P-NT substantially reduced the percentage of immune suppressor cells such as Tregs and MDSCs. These findings reveal that localized P-NT treatment has the potential to sensitize tumors to ICBs therapy.
The local delivery of aPD1 by a P-NT solution resulted in the formation of P-NT–aPD1 hydrogel within the injection site. Our results confirmed that this in situ formed P-NT–aPD1 hydrogel can notably enhance tumoral drug retention, serving as a reservoir for sustained release of both CPT and aPD1. We found that P-NT–aPD1 treatment can lead to an increase in the percentage of Teffs and in the ratio of Teff to Treg and induced a robust antitumor immune response. In both GL-261 brain cancer and CT 26 colon cancer models, 100% tumor regression was observed, revealing the most robust survival benefit among all the treatment groups. This is in stark contrast with the 40% survival of mice treated with free (CPT + aPD) at a much higher aPD1 dose. In this sense, P-NT–aPD1 essentially induces an immune-stimulating tumor microenvironment in mice. Our results also revealed that local administration of P-NT–aPD1 could induce durable T cell memory and robust systemic antitumor immunity, which should be useful for fighting tumor recurrence and potential metastasis. Collectively, these results led us to draw a conclusion that the combination of local chemotherapy with ICBs enhances antitumor immune response and substantially improves the overall response rate.
In addition to the observed therapeutic efficacy, the reported system also lowers the aPD1 dosage and minimizes systemic exposure. In previous studies, aPD1 has shown immune-related side effects with multiple systemic administrations (12, 38, 39). To overcome this challenge, Hubbell and coworkers (40) have designed a matrix-binding checkpoint inhibitor conjugate to enhance intratumoral retention of anti-PD–L1 and to reduce systemic drug exposure. The Gu Lab attempted to use microneedle patches and injectable hydrogels to locally deliver aPD1 antibody to tumor sites (19, 41). These strategies can improve therapeutic outcome and, to some extent, avoid the side effects associated with systemic administration. In the case here, a single dose (50 μg versus 200 μg for three intraperitoneal injections) of aPD1-hydrogel could generate the desired therapeutic effect. Our results revealed that aPD1 can be hardly detected in major organs on day 7 after treatment (fig. S8, A and B), thereby reducing systemic toxicity and possible off-target inflammation. Moreover, the use of a prodrug hydrogelator to deliver aPD1 eases concerns about any possible short- or long-term toxicities of synthetic or natural drug carriers (42), representing a simple yet effective means to achieve combination chemo- and immunotherapy with translation potential. The incorporation of an MMP-2 responsive substrate and the iRGD segment in the peptide design may have contributed to the improved treatment efficacy and reduced side effects.
In summary, we have developed a combined chemoimmunotherapy strategy based on a prodrug hydrogelator to boost immunity against cancer. This in situ formed P-NT–aPD1 hydrogel can serve as a therapeutic reservoir for intratumoral release of both CPT and aPD1 over a long period of time. It increases the frequency of Teffs and reduces the population of immune suppressor cells, so as to provoke a robust antitumor immune response. The long-term memory T cell and systemic immune response, as a result of the P-NT–aPD1 treatment, suggests that this platform could potentially be used to treat tumor recurrence and metastasis.
MATERIALS AND METHODS
Experimental design
The objective of this study was to develop an in situ formed, bioresponsive hydrogel for local delivery of aPD1 and CPT for enhanced cancer chemoimmunotherapy. The antitumor efficacy was evaluated in GL-261 and CT 26 tumor models. Mice were randomized to different treatment groups based on tumor size and body weight. Animals from varying groups were imaged to assess tumor progression, tracked to create survival curves, and rechallenged with tumors to assess immune memory. To assess the phenotype of the TME, mice were euthanized at various time points. Sample sizes were selected on the basis of our previous experimental experience. Body weight and tumor size were measured every 2 days, and mice were euthanized when tumor volume exceeded 1 cm3 or when body weight loss exceeded 20%. Blinding was not performed. All experiments were repeated at least three times.
Animals
Female BALB/c and C57BL/6 mice (8 to 10 weeks) were purchased from Charles River Laboratories. Animal experiments were performed in accordance with the animal protocol approved by the Animal Care and Welfare Committee at the Johns Hopkins University.
Synthesis of diCPT-PLGLAG-iRGD
The peptide C2K-GPLGLAG-cyl[CRGDRGPDC] was synthesized using an AAPPTEC Focus XC synthesizer via the standard Fmoc solid-phase technique. The purity and identity of the peptide were confirmed by HPLC and matrix-assisted laser desorption/ionization–time-of-flight MS, respectively. CPT-etcSS-Pyr was synthesized using the same method as previously reported (35). Then, C2K-GPLGLAG-cyl[CRGDRGPDC] (100 mg, 51 μmol) was dissolved in 5 ml of N2-purged dimethyl sulfoxide solution of CPT-etcSS-Pyr (115 mg, 204 μmol) and allowed to react overnight. The solution was purified by reversed-phase HPLC, and the product was lyophilized to obtain diCPT-PLGLAG-iRGD. The identity of diCPT-PLGLAG-iRGD was confirmed by electrospray ionization MS.
Self-assemble of diCPT-PLGLAG-iRGD
For preparation of self-assembled diCPT-PLGLAG-iRGD nanotubes (P-NT), diCPT-PLGLAG-iRGD conjugate was directly dissolved in deionized water at 500 μM and aged for 24 hours at room temperature. The structure was then characterized by TEM (Technai 12 TWIN).
Hydrogel formation assay
Sol-gel transition experiments were performed by adding 20 μl of 10× PBS to 180 μl of 7.2 mM P-NT solution. For preparation of aPD1-loaded hydrogel (P-NT–aPD1), 50 μg of aPD1 was added to the P-NT solution (150 μg of CPT). After vortexing, the mixture was incubated at 37°C for 30 min. PBS (10×) was then added to the P-NT–aPD1 solution to induce the formation of a hydrogel. Distribution of aPD1 in the hydrogel was further characterized using confocal microscopy (Zeiss LSM 510). For in vitro release studies and confocal imaging, aPD1 was labeled with Cy3. For in vivo imaging, Cy5.5-labeled aPD1 was used.
GSH responsive drug release
Briefly, 20 mM GSH and 400 μM diCPT-PLGLAG-iRGD stock solutions were prepared in water. Equal volumes of GSH and diCPT-PLGLAG-iRGD solutions were then mixed to reach a final GSH concentration of 10 mM and the mixtures were incubated at 37°C. Samples were then taken at predetermined time points (0, 0.5, 1, 2, 4, 8, 12, and 24 hours) and quantified using HPLC.
MMP-2 responsive drug release
MMP-2 recombinant protein (4 μg/ml) and 400 μM diCPT-PLGLAG-iRGD stock solutions were prepared in water. Equal volumes of MMP-2 and diCPT-PLGLAG-iRGD solutions were then mixed and incubated at 37°C for the desired period of time (0, 0.5, 1, 2, 4, 8, 12, and 24 hours). Samples were then quantified using HPLC.
aPD1 and CPT release from P-NT–aPD1 hydrogel
The in vitro hydrogel (P-NT–aPD1) release study was performed at 37°C in the presence or absence of MMP-2 (2 μg/ml). The quantity of aPD1 (labeled with Cy3) released was determined using a fluorescence spectrophotometer, whereas CPT release was quantified using HPLC.
In vivo hydrogel degradation
One hundred microliters of 7.2 mM P-NT solution was injected into the back of C57BL/6 mice. The mice were then euthanized at predetermined time points (1 hour and 5, 15, 30, and 45 days). The remaining hydrogel in each mouse was photographed, and the amount of drug that persisted within the hydrogel was determined using HPLC.
In vivo drug release from P-NT–aPD1 hydrogel
To evaluate the in vivo release of aPD1 and CPT, free (CPT + aPD1) or P-NT–aPD1 solution was injected into the tumors. Fluorescence imaging of Cy5.5-aPD1 was monitored by an IVIS spectrum imaging system (PerkinElmer). At predetermined time points, mice were euthanized, and major organs and tumors were collected and imaged. Bioluminescence images were analyzed using Living Image software (PerkinElmer). The amount of CPT in the tumors was measured by HPLC. Harvested tumors were also snap-frozen, cut into micrometer sections, and stained with 4′,6-diamidino-2-phenylindole. The slides were then imaged using a confocal microscope (Zeiss LSM 510).
In vivo tumor models and treatment
For the subcutaneous GL-261–luc tumor model, 2.5 × 106 cells were inoculated on the right flanks of female C57BL/6 mice. After 10 days (tumor volume had reached 100 to 150 mm3), mice were randomly assigned to five groups (n = 5 to 10). Then, the mice were intratumorally injected with saline (30 μl on days 10, 17, and 24), free (CPT + aPD1) [(50 μg of CPT + 50 μg of aPD1)/30 μl on days 10, 17, and 24], diCPT-PLGLAG-iRGD NT solution (P-NT; 150 μg of CPT/30 μl single injection on day 10), aPD1-loaded diC12-PLGLAG-iRGD NF [aPD1(L); 50 μg of aPD1/30 μl single injection on day 10], or P-NT–aPD1 [(150 μg of CPT + 50 μg of aPD1)/30 μl single injection on day 10]. For rechallenge studies, mice with long-term survival from all treatment groups were inoculated with 2.5 × 106 GL-261–luc cells on their opposite flank to develop new tumors.
To assess the systemic antitumor effects of P-NT–aPD1, mice were implanted with GL-261–luc cells in the both right back and left brain. Briefly, 2.5 × 106 cells were inoculated on the right flanks of female C57BL/6 mice. The orthotopic glioblastoma tumors were established by implanting 5 × 104 GL-261–luc cells into the left frontal lobe. The injection site was 2.5 mm lateral and 0.5 mm anterior from the bregma and 2.5 mm deep from the outer border of the cranium. Mice were imaged using an IVIS spectrum imaging system (PerkinElmer) to evaluate tumor growth. The tumor take rate was 100%. After 6 days, tumors on the right flank were treated with P-NT–aPD1 hydrogel (n = 10), while orthotopic gliomas were designated as distant tumors and did not receive any treatment. Control mice (n = 5) were treated with saline.
The tumor size and body weight were measured every 2 days. The tumor volume was calculated by the formula: V = (major axis) × (minor axis)2/2. The mice were also monitored using an IVIS spectrum imaging system (PerkinElmer). Mice were euthanized when tumor volume exceeded 1000 mm3 or when body weight loss surpassed 20%.
Flow cytometry
Flow cytometry was performed using a FACSCanto II instrument (BD Biosciences). Data were analyzed with FlowJo software (Tree Star). Antibodies against CD45 (30-F11), CD4 (RM4-5), CD8 (53-6.7), CD3 (17A2), CD44 (IM7), CD62L (MEL-14), Foxp3 (FJK-16 s), CD11b (ICRF44), Gr-1 (RB6-8C5), PD-1 (29F.1A12), and PD-L1 (10F.9G2) were purchased from BioLegend or eBioscience.
In vivo bioluminescence and imaging
Tumor growth in mice was observed using bioluminescence imaging. Ten minutes after intraperitoneal injection of d-luciferin (150 μg/g), mice were anesthetized and imaged using an IVIS spectrum imaging system (PerkinElmer). Bioluminescence images were analyzed using Living Image software.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software 5. Data are presented as means ± SD. The two-tailed unpaired t test was used to determine statistical significance between two treatment groups. Survival was plotted using a Kaplan-Meier curve and assessed by a log-rank (Mantel-Cox) test.
Supplementary Material
Acknowledgments
We thank M. Lim and J. Fu from the Johns Hopkins University School of Medicine for sharing the GL-261–luc and CT 26 cells, respectively. We also thank Q. Huang and Y. Guan for support relating to cryosection. Funding: The work was supported by the Johns Hopkins Discovery Award. Author contributions: F.H.W. and H.C. participated in the conception and overall design of the study. F.H.W., D.X., H.S., W.J.Z., X.R.S., R.W.C., Z.Y.W., W.B.D., and H.W. performed the experiments and collected the data. F.H.W., H.S., and H.W. synthesized and characterized the diCPT-PLGLAG-iRGD. F.H.W., D.X., and F.W. performed the flow cytometry study. F.H.W., D.X., F.W., and H.C. analyzed and interpreted the data. F.H.W., D.X., M.K.M., R.C., R.O., F.W., and H.C. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz8985/DC1
REFERENCES AND NOTES
- 1.Ferris R. L., Blumenschein G. Jr., Fayette J., Guigay J., Colevas A. D., Licitra L., Harrington K., Kasper S., Vokes E. E., Even C., Worden F., Saba N. F., Iglesias Docampo L. C., Haddad R., Rordorf T., Kiyota N., Tahara M., Monga M., Lynch M., Geese W. J., Kopit J., Shaw J. W., Gillison M. L., Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 375, 1856–1867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaufman H. L., Russell J., Hamid O., Bhatia S., Terheyden P., D'Angelo S. P., Shih K. C., Lebbe C., Linette G. P., Milella M., Brownell I., Lewis K. D., Lorch J. H., Chin K., Mahnke L., von Heydebreck A., Cuillerot J.-M., Nghiem P., Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 17, 1374–1385 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boussiotis V. A., Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 375, 1767–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou W., Wolchok J. D., Chen L., PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 8, 328rv4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buchbinder E. I., Hodi F. S., Melanoma in 2015: Immune-checkpoint blockade—Durable cancer control. Nat. Rev. Clin. Oncol. 13, 77–78 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Killock D., Lung cancer: Anti-PD-1 therapy in the frontline. Nat. Rev. Clin. Oncol. 13, 715 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Kuai R., Yuan W., Son S., Nam J., Xu Y., Fan Y., Schwendeman A., Moon J. J., Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv. 4, eaao1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robert C., Schachter J., Long G. V., Arance A., Grob J. J., Mortier L., Daud A., Carlino M. S., McNeil C., Lotem M., Larkin J., Lorigan P., Neyns B., Blank C. U., Hamid O., Mateus C., Shapira-Frommer R., Kosh M., Zhou H., Ibrahim N., Ebbinghaus S., Ribas A.; KEYNOTE-006 investigators , Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Gajewski T. F., Schreiber H., Fu Y.-X., Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hegde P. S., Karanikas V., Evers S., The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 22, 1865–1874 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Ishihara J., Ishihara A., Sasaki K., Lee S. S., Williford J. M., Yasui M., Abe H., Potin L., Hosseinchi P., Fukunaga K., Raczy M. M., Gray L. T., Mansurov A., Katsumata K., Fukayama M., Kron S. J., Swartz M. A., Hubbell J. A., Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl. Med. 11, eaau3259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topalian S. L., Hodi F. S., Brahmer J. R., Gettinger S. N., Smith D. C., McDermott D. F., Powderly J. D., Carvajal R. D., Sosman J. A., Atkins M. B., Leming P. D., Spigel D. R., Antonia S. J., Horn L., Drake C. G., Pardoll D. M., Chen L., Sharfman W. H., Anders R. A., Taube J. M., McMiller T. L., Xu H., Korman A. J., Jure-Kunkel M., Agrawal S., McDonald D., Kollia G. D., Gupta A., Wigginton J. M., Sznol M., Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu J. Q., Liu X., Liao Y.-P., Wang X., Ahmed A., Jiang W., Ji Y., Meng H., Nel A. E., Breast cancer chemo-immunotherapy through liposomal delivery of an immunogenic cell death stimulus plus interference in the IDO-1 pathway. ACS Nano 12, 11041–11061 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Pfirschke C., Engblom C., Rickelt S., Cortez-Retamozo V., Garris C., Pucci F., Yamazaki T., Poirier-Colame V., Newton A., Redouane Y., Lin Y. J., Wojtkiewicz G., Iwamoto Y., Mino-Kenudson M., Huynh T. G., Hynes R. O., Freeman G. J., Kroemer G., Zitvogel L., Weissleder R., Pittet M. J., Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity 44, 343–354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang T. H., Mao C. P., Lee S. Y., Chen A., Lee J. H., Kim T. W., Alvarez R. D., Roden R. B., Pardoll D., Hung C. F., Wu T. C., Chemotherapy acts as an adjuvant to convert the tumor microenvironment into a highly permissive state for vaccination-induced antitumor immunity. Cancer Res. 73, 2493–2504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nolan E., Savas P., Policheni A. N., Darcy P. K., Vaillant F., Mintoff C. P., Dushyanthen S., Mansour M., Pang J. B., Fox S. B.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer (kConFab), Perou C. M., Visvader J. E., Gray D. H. D., Loi S., Lindeman G. J., Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 9, eaal4922 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lombardi G., Rumiato E., Bertorelle R., Saggioro D., Farina P., Della Puppa A., Zustovich F., Berti F., Sacchetto V., Marcato R., Amadori A., Zagonel V., Clinical and genetic factors associated with severe hematological toxicity in glioblastoma patients during radiation plus temozolomide treatment: A prospective study. Am. J. Clin. Oncol. 38, 514–519 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Mathios D., Kim J. E., Mangraviti A., Phallen J., Park C. K., Jackson C. M., Garzon-Muvdi T., Kim E., Theodros D., Polanczyk M., Martin A. M., Suk I., Ye X., Tyler B., Bettegowda C., Brem H., Pardoll D. M., Lim M., Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 8, 370ra180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C., Wang J., Zhang X., Yu S., Wen D., Hu Q., Ye Y., Bomba H., Hu X., Liu Z., Dotti G., Gu Z., In situ formed reactive oxygen species-responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med. 10, eaan3682 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Chen Q., Wang C., Zhang X., Chen G., Hu Q., Li H., Wang J., Wen D., Zhang Y., Lu Y., Yang G., Jiang C., Wang J., Dotti G., Gu Z., In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat. Nanotechnol. 14, 89–97 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Li J., Mooney D. J., Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 1, 16071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park C. G., Hartl C. A., Schmid D., Carmona E. M., Kim H. J., Goldberg M. S., Extended release of perioperative immunotherapy prevents tumor recurrence and eliminates metastases. Sci. Transl. Med. 10, eaar1916 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Zhao F., Ma M. L., Xu B., Molecular hydrogels of therapeutic agents. Chem. Soc. Rev. 38, 883–891 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Singh A., Peppas N. A., Hydrogels and scaffolds for immunomodulation. Adv. Mater. 26, 6530–6541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haines-Butterick L., Rajagopal K., Branco M., Salick D., Rughani R., Pilarz M., Lamm M. S., Pochan D. J., Schneider J. P., Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. U.S.A. 104, 7791–7796 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar V. A., Shi S., Wang B. K., Li I. C., Jalan A. A., Sarkar B., Wickremasinghe N. C., Hartgerink J. D., Drug-triggered and cross-linked self-assembling nanofibrous hydrogels. J. Am. Chem. Soc. 137, 4823–4830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sleep E., Cosgrove B. D., McClendon M. T., Preslar A. T., Chen C. H., Sangji M. H., Perez C. M. R., Haynes R. D., Meade T. J., Blau H. M., Stupp S. I., Injectable biomimetic liquid crystalline scaffolds enhance muscle stem cell transplantation. Proc. Natl. Acad. Sci. U.S.A. 114, E7919–E7928 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan C., Altunbas A., Yucel T., Nagarkar R. P., Schneider J. P., Pochan D. J., Injectable solid hydrogel: Mechanism of shear-thinning and immediate recovery of injectable β-hairpin peptide hydrogels. Soft Matter 6, 5143–5156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chakroun R. W., Wang F., Lin R., Wang Y., Su H., Pompa D., Cui H., Fine-tuning the linear release rate of paclitaxel-bearing supramolecular filament hydrogels through molecular engineering. ACS Nano 13, 7780–7790 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Lock L. L., Li Y., Mao X., Chen H., Staedtke V., Bai R., Ma W., Lin R., Li Y., Liu G., Cui H., One-component supramolecular filament hydrogels as theranostic label-free magnetic resonance imaging agents. ACS Nano 11, 797–805 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalafatovic D., Nobis M., Son J., Anderson K. I., Ulijn R. V., MMP-9 triggered self-assembly of doxorubicin nanofiber depots halts tumor growth. Biomaterials 98, 192–202 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Sugahara K. N., Teesalu T., Karmali P. P., Kotamraju V. R., Agemy L., Greenwald D. R., Ruoslahti E., Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 328, 1031–1035 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang T., Olson E. S., Nguyen Q. T., Roy M., Jennings P. A., Tsien R. Y., Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. U.S.A. 101, 17867–17872 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng Z.-H., Kopecek J., Enhancing accumulation and penetration of HPMA copolymer-doxorubicin conjugates in 2D and 3D prostate cancer cells via iRGD conjugation with an MMP-2 cleavable spacer. J. Am. Chem. Soc. 137, 6726–6729 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheetham A. G., Ou Y. C., Zhang P., Cui H., Linker-determined drug release mechanism of free camptothecin from self-assembling drug amphiphiles. Chem. Commun. 50, 6039–6042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang F., Huang Q., Wang Y., Shi L., Shen Y., Guo S., NIR-light and GSH activated cytosolic p65-shRNA delivery for precise treatment of metastatic cancer. J. Control. Release 288, 126–135 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Koshy S. T., Cheung A. S., Gu L., Graveline A. R., Mooney D. J., Liposomal delivery enhances immune activation by STING agonists for cancer immunotherapy. Adv. Biosyst. 1, 1600013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boutros C., Tarhini A., Routier E., Lambotte O., Ladurie F. L., Carbonnel F., Izzeddine H., Marabelle A., Champiat S., Berdelou A., Lanoy E., Texier M., Libenciuc C., Eggermont A. M., Soria J. C., Mateus C., Robert C., Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 13, 473–486 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Johnson D. B., Balko J. M., Compton M. L., Chalkias S., Gorham J., Xu Y., Hicks M., Puzanov I., Alexander M. R., Bloomer T. L., Becker J. R., Slosky D. A., Phillips E. J., Pilkinton M. A., Craig-Owens L., Kola N., Plautz G., Reshef D. S., Deutsch J. S., Deering R. P., Olenchock B. A., Lichtman A. H., Roden D. M., Seidman C. E., Koralnik I. J., Seidman J. G., Hoffman R. D., Taube J. M., Diaz L. A. Jr., Anders R. A., Sosman J. A., Moslehi J. J., Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med. 375, 1749–1755 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishihara J., Fukunaga K., Ishihara A., Larsson H. M., Potin L., Hosseinchi P., Galliverti G., Swartz M. A., Hubbell J. A., Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci. Transl. Med. 9, eaan0401 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Wang C., Ye Y., Hochu G. M., Sadeghifar H., Gu Z., Enhanced cancer immunotherapy by microneedle patch-assisted delivery of anti-PD1 antibody. Nano Lett. 16, 2334–2340 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Peppas N. A., Khademhosseini A., Make better, safer biomaterials. Nature 540, 335–337 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/18/eaaz8985/DC1